
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of a cross-chain bridging cryptand†‡
Takafumi Yashima,
Ryoga Hori ,
Taisei Maruyama,
Kohta Nakashima,
Hiroki Fujihara,
Masaya Naito,
Shinobu Miyagawa and
Yuji Tokunaga*
,
Taisei Maruyama,
Kohta Nakashima,
Hiroki Fujihara,
Masaya Naito,
Shinobu Miyagawa and
Yuji Tokunaga*
Department of Materials Science and Engineering, Faculty of Engineering, University of Fukui, Bunkyo, Fukui 910-8507, Japan. E-mail: tokunaga@u-fukui.ac.jp
First published on 10th January 2025
Abstract
We present two methodologies for synthesizing cross-chain bridging cryptand 1 that incorporates tri- and tetra(ethylene glycol) linkers (methods B and C). Method B involves the synthesis through the intramolecular cross-linking of C2-symmetric crown ether 3. While this method does not substantially reduce the lengthy reaction steps compared to the previous approach, it improves the overall yield of cryptand 1a containing three tri(ethylene glycol) linkers and allows for the creation of a new form of a cross-chain bridging cryptand 1c with one distinct and two identical linkers. However, method C entails a synthesis accomplished through a triple-linking reaction in a single step. This method offered a streamlined synthesis of cryptand 1. The crucial triple linking reaction produced cross-chain bridging cryptand 1 as the major isomer and the corresponding linear regioisomer 6 as the minor isomer. Moreover, we observed the interconversion of enantiomers of cryptand 1c, which contains a 28-membered macrocycle, under chiral high-performance liquid chromatographic (HPLC) analytical conditions (with a half-life of 20.7 min at room temperature). Finally, X-ray crystallography confirmed the cross-chain bridging structure in the two chemically equivalent chains in the solid state of cryptand 1a.
Introduction
Many molecularly chiral architectures have been developed from unique structural systems that lack classical chiral elements.1 These include increasingly complex molecular arrangements, such as calixarene host families,2 their analogs,3–7 helicenes,8 other distorted aromatics,8c,d,9–12 Möbius strips,13 and others.8d,14,15 These inherently chiral architectures are structurally intriguing, and their chiroptical properties have been extensively studied.8b,16–20The development of a cross-chain bridging structure represents an intriguing approach for inducing molecular chirality in the absence of asymmetric centers. Figure-of-eights are representative examples of creating such chiral systems.8d,21,22 For a chiral figure-of-eight structure to be effective, it must incorporate rigid functional groups to inhibit the interconversion of its enantiomers. Walba and colleagues have successfully synthesized a chiral Möbius strip featuring a cross-chain bridging arrangement, demonstrating the induction of chirality without requiring rigid linkers.23 Currently, molecular knots and links with multiple and intricately entangled structures have been synthesized through transition metal templating and/or thermodynamically controlled methodologies,24 with one such system being used as an asymmetric catalyst in organic reactions.25
In a previous work, we synthesized a cross-chain bridging cryptand 1 bearing three oligo(ethylene glycol) linkers (Fig. 1).26 The cryptand exhibits C2 symmetry, and its cross-chain bridging structure is formed through triple linking at different positions of two benzene moieties, with two of the three cross-chain linkers being chemically equivalent. Cryptand 1a, which contains three tri(ethylene glycol) linkers and a 25-membered macrocycle, demonstrates molecular chirality. However, the conversion of the enantiomers of the larger cryptand 1b, which features long linkers, occurred at room temperature (Fig. S1‡) owing to the large 31-membered macrocycle in 1b failing to sufficiently inhibit the flipping of the benzoate moiety. Consequently, this type of cross-chain bridging cryptand can act as both chiral and dynamic chiral hosts, allowing for variation in the size of the macrocycles.
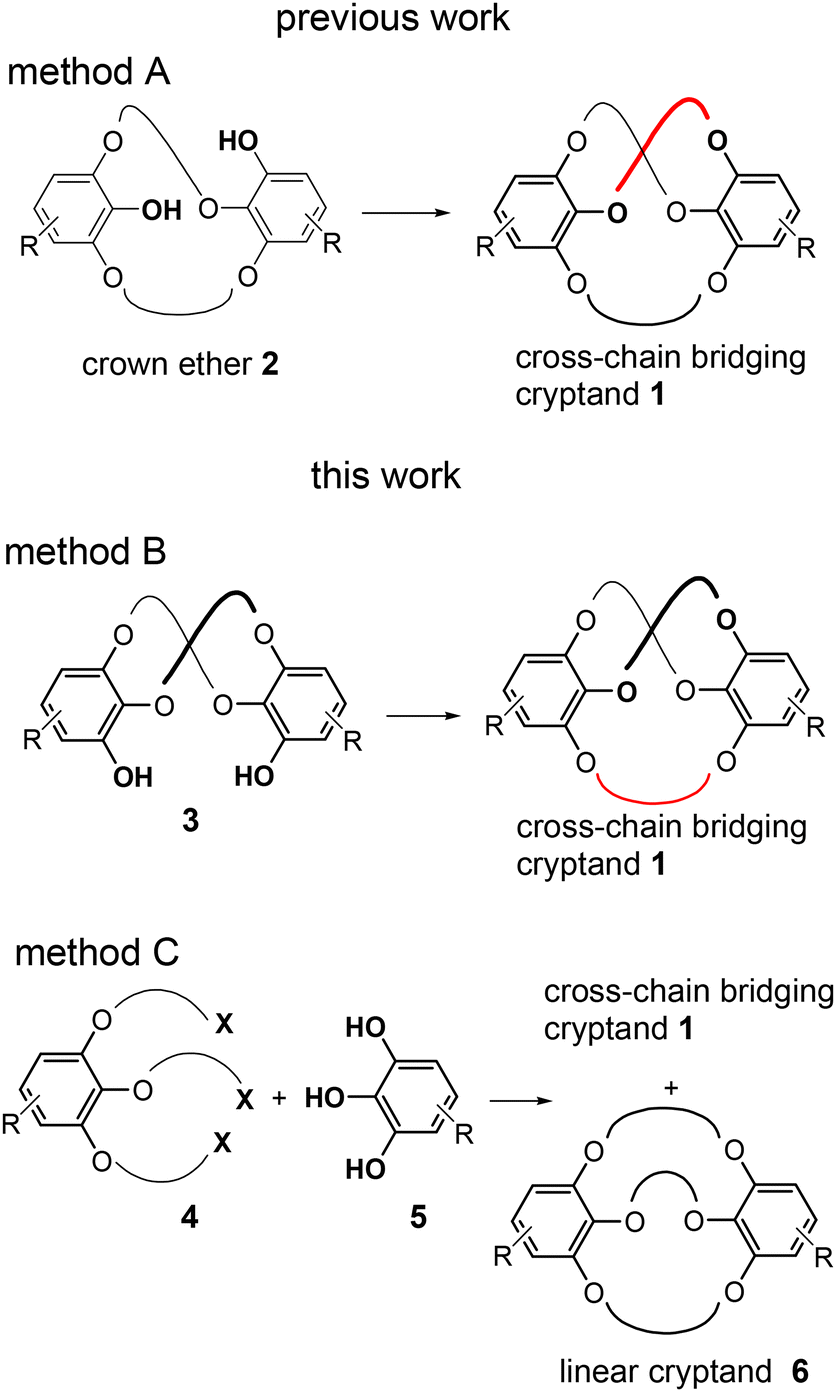 | ||
| Fig. 1 Synthetic strategies for cross-chain bridging cryptand 1. | ||
In a previous study, the cross-chain bridging cryptand was synthesized through the intramolecular cross-linking of the unsymmetric crown ether 2 (Fig. 1, method A). The primary advantage of method A is that it yields cryptand 1 as a single product when the intramolecular cross-linking reaction is performed. However, this method requires extensive synthetic steps, resulting in relatively low total yields of the cryptand, particularly owing to the low yield associated with the synthesis of crown ether 2. Additionally, this approach is limited to the production of cross-chain bridging cryptands featuring three identical linkers for creating C2-symmetric cryptands.
Here, we present two new methodologies for the synthesis of cross-chain bridging cryptand 1 (Fig. 1, methods B and C).27 In method B, the intramolecular cross-linking of C2-symmetric crown ethers 3 is a key step, while method C involves performing the triple linking reaction in a single step. Although method B does not considerably reduce the number of lengthy reaction steps, it increases the synthetic yield of the small cryptand. Furthermore, this method allows for the incorporation of a distinct linker different from the other two, enabling the synthesis of a new type of cross-chain bridging cryptand featuring two types of linkers. However, method C offers a short-step synthesis of cryptand 1, with the final triple linking step producing cross-chain bridging cryptand 1 as the major isomer alongside the corresponding linear regioisomer 6. Additionally, we confirmed through X-ray crystallography that cryptand 1a, bearing three tri(ethylene glycol) units as linkers, exhibits a cross-chain bridging structure formed by two chemically equivalent linkers.
Results and discussion
Synthesis of cryptand 1 by linking the C2-symmetric crown ethers 3 (method B)
We used 3-O-benzyl ethyl gallate (7)26 as the starting material for synthesizing the C2-symmetric crown ethers 3 (Scheme 1). Alkylation of compound 7 and the corresponding tosylates 8, which contain tri- and tetra(ethylene glycol) units, produced the oligoethers 9. To synthesize larger crown ethers (10b and 11b), diol 9b was converted into ditosylate 9c. Macrocyclization of either dichloride 9a or ditosylate 9c with ethyl gallate 7 resulted in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of C2-symmetric crown ethers 10 and σ-symmetric crown ethers 11, obtained in moderate yields. These mixtures were subsequently converted into the corresponding deprotected crown ethers 3 and 12 as additional mixtures. C2-symmetric and σ-symmetric small crown ethers 3a and 12a were successfully isolated through chromatographic separation. The 1H NMR spectra of crown ethers 3a and 12a indicated their symmetric structures. Specifically, the spectrum of 12a, which features two sets of chemically equivalent protons in the ethylene glycol units (Fig. 2), showed two singlet signals for the ethylene glycol protons at 3.77 and 3.69 ppm. In contrast, the 1H NMR spectrum of 3a did not exhibit any singlet signals from the ethylene glycol units. The separation of crown ethers 3b and 12b, which contain long linkers, was achieved through crystallization. After the deprotection of the benzyl groups in the mixture of 10b and 11b, the crude product was treated with MeOH, affording a solid that consisted of a 93:7 mixture of 3b and 12b in 44% yield. Further washing of this mixture with EtOH resulted in the isolation of C2-symmetric crown ether 3b as a single product with a yield of 25%. σ-Symmetric crown ether 12b was obtained as a solid from the first filtrate after treatment with iPrOH. At this stage, the structures of the isomers were not elucidated through 1H NMR spectroscopy. Finally, we established their structures following the conversion of 3b and 12b into the cross-chain bridging and linear cryptands 1b and 6b, respectively (as described below).
:1 mixture of C2-symmetric crown ethers 10 and σ-symmetric crown ethers 11, obtained in moderate yields. These mixtures were subsequently converted into the corresponding deprotected crown ethers 3 and 12 as additional mixtures. C2-symmetric and σ-symmetric small crown ethers 3a and 12a were successfully isolated through chromatographic separation. The 1H NMR spectra of crown ethers 3a and 12a indicated their symmetric structures. Specifically, the spectrum of 12a, which features two sets of chemically equivalent protons in the ethylene glycol units (Fig. 2), showed two singlet signals for the ethylene glycol protons at 3.77 and 3.69 ppm. In contrast, the 1H NMR spectrum of 3a did not exhibit any singlet signals from the ethylene glycol units. The separation of crown ethers 3b and 12b, which contain long linkers, was achieved through crystallization. After the deprotection of the benzyl groups in the mixture of 10b and 11b, the crude product was treated with MeOH, affording a solid that consisted of a 93:7 mixture of 3b and 12b in 44% yield. Further washing of this mixture with EtOH resulted in the isolation of C2-symmetric crown ether 3b as a single product with a yield of 25%. σ-Symmetric crown ether 12b was obtained as a solid from the first filtrate after treatment with iPrOH. At this stage, the structures of the isomers were not elucidated through 1H NMR spectroscopy. Finally, we established their structures following the conversion of 3b and 12b into the cross-chain bridging and linear cryptands 1b and 6b, respectively (as described below).
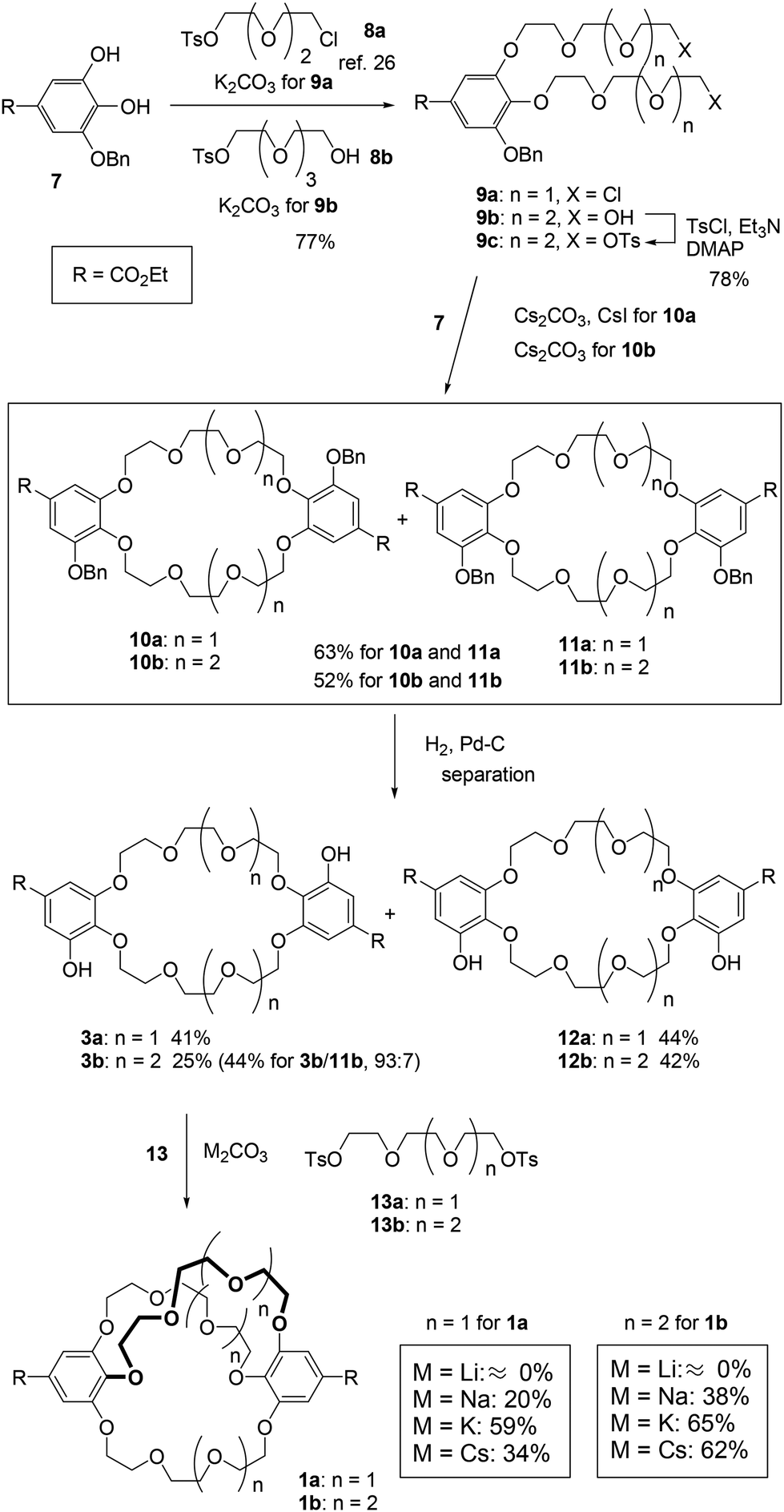 | ||
| Scheme 1 Method B: synthesis of cross-chain bridging cryptand 1. | ||
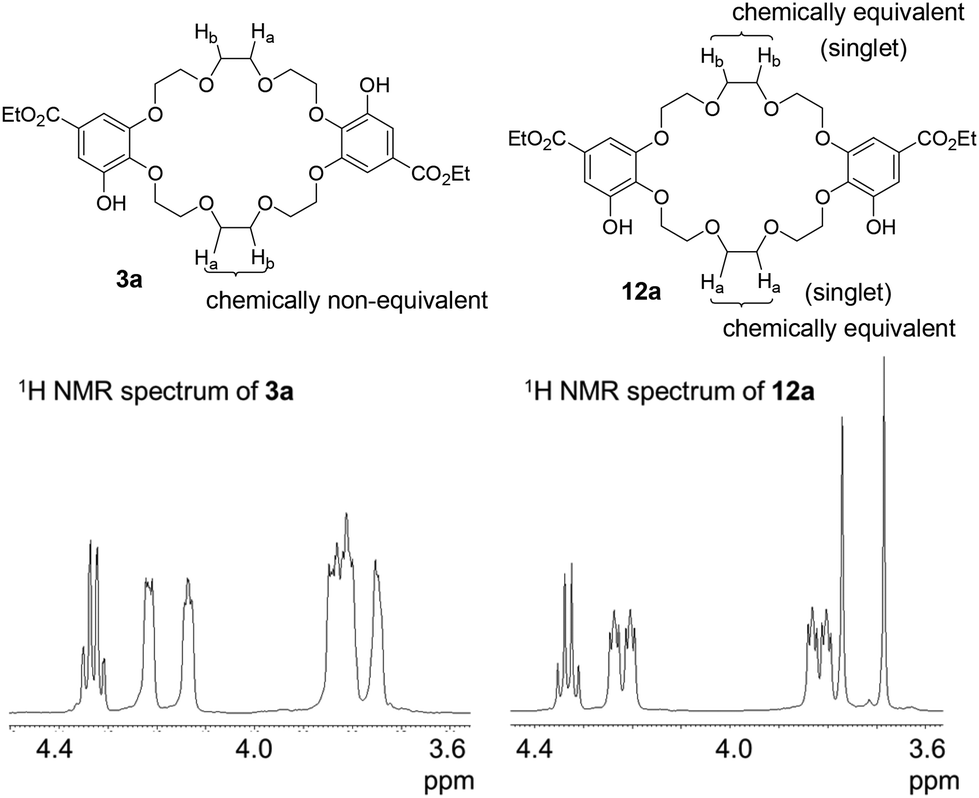 | ||
| Fig. 2 Structures and 1H NMR spectra (600 MHz, CDCl3) of crown ethers 3a and 12a. | ||
The synthesis of the cross-chain bridging cryptand 1 from 3 and the corresponding ditosylate 13 was investigated using four carbonates as bases and templates (Scheme 1). K2CO3 proved to be the most effective base for producing the small cross-chain bridging [4.4.4]cryptand 1a with a yield of 59%.28 The cross-linking of crown ether 3b led to the formation of the large [5.5.5]cryptand 1b, achieved using K2CO3 and Cs2CO3 with a good yield. To evaluate the products of the cross-linking reactions, the crude products were analyzed by gel permeation chromatography (GPC) (Fig. S2 and S3‡).
When Li2CO3 was used as a base for the synthesis of 1, only crown ethers 3 were recovered because the combination of the crown ethers and the carbonate salt resulted in the formation of an insoluble complex(es). After the workup of the reaction between 3b and 13b, only the peak corresponding to 3b was present in the chromatogram (Fig. S3a for 3b‡). In contrast, when using Na2CO3, the peaks of cryptand 1 appeared prominently alongside other distinct peaks in the chromatograms. For the synthesis of the small [4.4.4]cryptand 1a, a non-negligible peak was observed before the peak for 1a with both K2CO3 and Cs2CO3. The cyclization of two molecules of crown ether 3a and ditosylate 13a likely yielded the larger macrocycle.29 Conversely, peaks for the large [5.5.5]cryptand 1b were detected in the chromatograms when K2CO3 and Cs2CO3 were used. The template effect of the potassium ion is more effective than that of the cesium ion for synthesizing the small cryptand 1a, while both potassium and cesium ions may serve as effective templates for the synthesis of the large cryptand 1b. Upon purification of the cryptand using GPC, we confirmed that the 1H NMR spectra matched those previously reported.26
Synthesis of the cross-chain bridging cryptand 1c possessing two different linkers
Next, we synthesized [5.5.4]cryptand 1c, which contains two tetra(ethylene glycol) linkers and one tri(ethylene glycol) linker, to demonstrate that method B allows the construction of a cryptand with two different types of linkers (Scheme 2). The macrocyclization reaction involving the large C2-symmetric crown ether 3b and tri(ethylene glycol) ditosylate 13a, using K2CO3 and Cs2CO3, yielded [5.5.4]cryptand 1c in 52% yield under both conditions.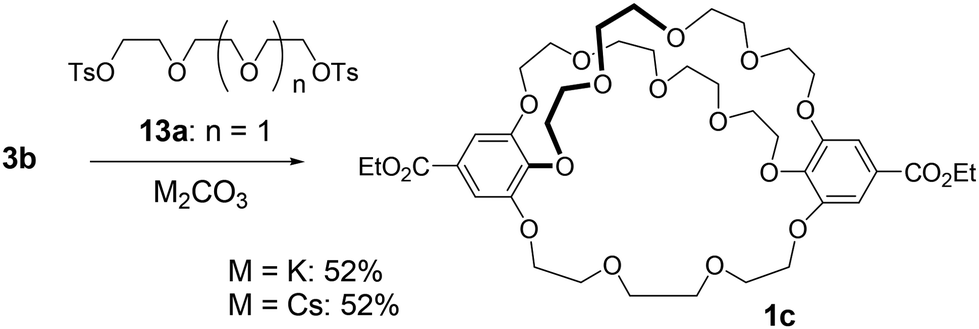 | ||
| Scheme 2 Method B: synthesis of cross-chain bridging cryptand 1c. | ||
The mass spectrum (MALDI) of cryptand 1c showed a peak at m/z 849.3499 (calcd 849.3515), which corresponds to a complex with the sodium cation ([M + Na]+) (Fig. S10‡). The NMR spectra of 1c in C6D6 indicate that the cryptand possesses a C2 symmetric structure. In the 13C NMR spectrum of 1c, ten carbon signals from the ethylene glycol units were observed at 73.1, 71.4, 71.3, 71.1, 71.0, 70.9, 69.8, 69.7, 69.6, and 69.1 ppm; one signal appeared to overlap, although eleven signals would be expected theoretically (Fig. S5‡). In the 1H NMR spectrum, several signals overlapped, yet many proton signals from the ethylene glycol units were split into two (Fig. 3 and S4‡). This splitting is attributed to the desymmetrization of the crown ether moiety in the cryptand, caused by cross-chain bridging. These observations suggest that the interconversion of the enantiomers of the 28-membered 1c does not proceed or occurs slowly on the NMR spectroscopic time scale, as indicated by the similar 1H NMR spectral features of the previously reported 25-membered [4.4.4]cryptand 1a.26 The interconversion likely involves the rotation of the benzoate moiety (Fig. S1‡), but the 28-membered crown macrocycle lacks sufficient space for the rotation of ethyl benzoate in the NMR spectroscopic time scale at room temperature. The proton and carbon signals of 1c were assigned using 2D NMR spectroscopy (Fig. S6–S9‡).
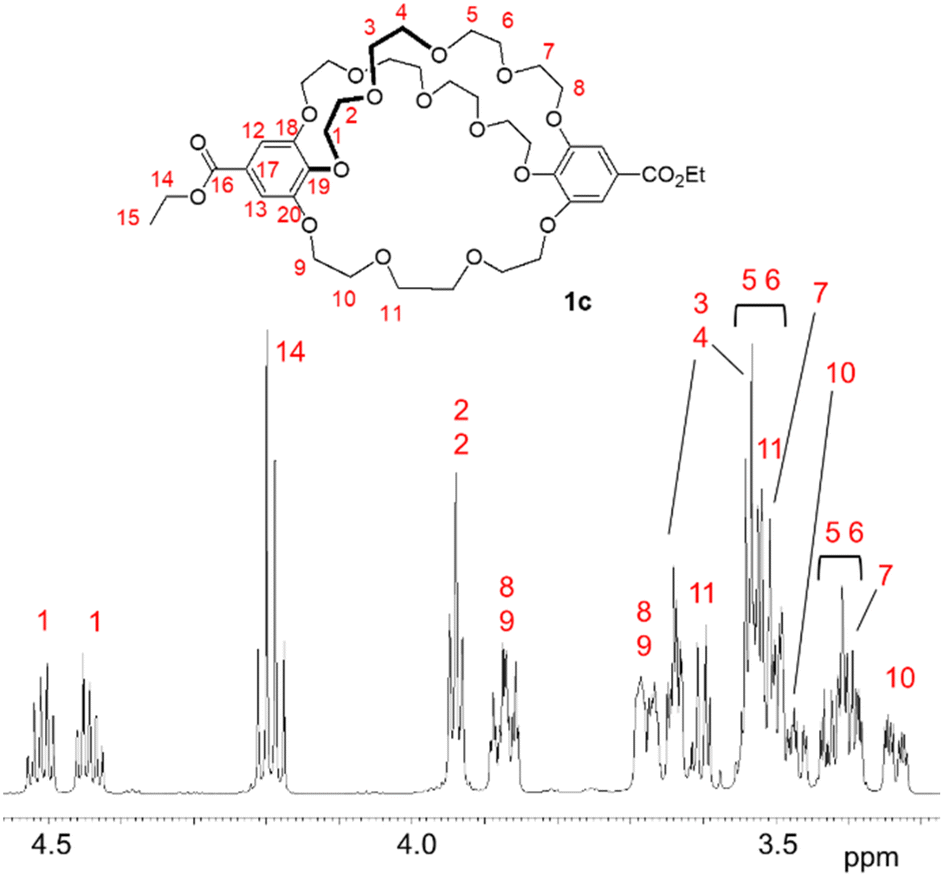 | ||
| Fig. 3 Partial 1H NMR spectrum (600 MHz, C6D6) of cryptand 1c. | ||
Synthesis of linear cryptand 6 by linking σ-symmetric crown ethers 12
To synthesize linear cryptand 6 before method C and to confirm the structures of the crown ethers (3b and 12b), which could not be determined by NMR spectroscopy (as described above), we linked the σ-symmetric crown ethers 12 with the ditosylates 13 using Cs2CO3 as a base (Scheme 3).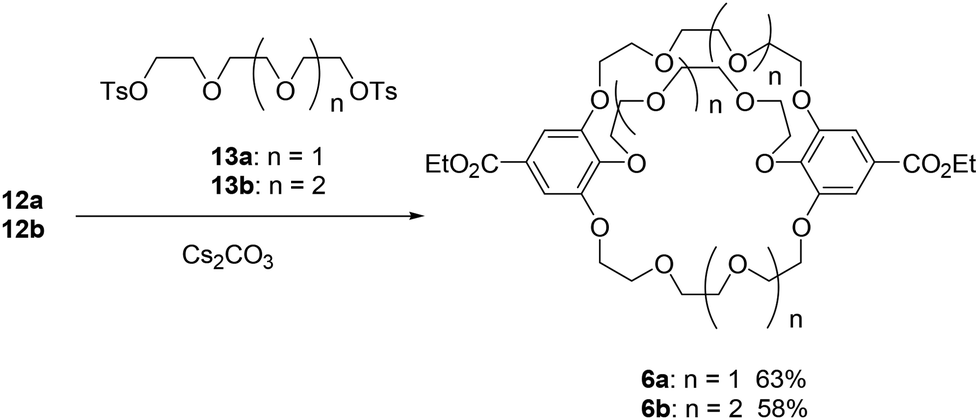 | ||
| Scheme 3 Synthesis of linear cryptand 6. | ||
The mass spectra (MALDI) of the small [4.4.4]cryptand 6a (derived from 12a and 13a) and the large [5.5.5]cryptand 6b (derived from 12b and 13b) exhibit peaks at m/z 761.2994 (calcd 761.2991) and 893.3775 (calcd 893.3778), respectively, corresponding to the sodium ion complexes ([M + Na]+) (Fig. S14 for 6a and Fig. S18 for 6b‡). The 1H NMR spectra of both cryptands were in line with the highly symmetric structures of the linear cryptand. For instance, six sets of signals for ethylene glycol protons were identified at 4.17 (8H), 4.14 (4H), 3.96 (4H), 3.89 (8H), 3.80 (8H), and 3.79 ppm (4H). Additionally, two singlet signals (3.80 and 3.79 ppm) were observed in the 1H NMR spectrum of 6a (Fig. S11‡). The signal pattern and the integration confirmed the structure of linear cryptand 6a. Likewise, the 1H NMR spectrum of 6b also confirmed the structure of the linear cryptand (Fig. S15‡). All proton signals were assigned through 2D NMR experiments performed on linear cryptand 6 (Fig. S12 and S13 for 6a and Fig. S16 and S17 for 6b‡).
The isomeric structures of the crown ethers (3 and 12) and the cryptands (1 and 6) were confirmed through chemical conversion and NMR analysis of the products.
Short-step synthesis of cryptand 1 by triple linking in one step (method C)
First, we synthesized trichloride 14 through the alkylation of ethyl gallate (5) and chlorotosylate, which was essential for the key triple linking reaction (Scheme 4). However, the triple linking of 14 with ethyl gallate (5) was inconsistent, preventing the successful production of cryptands 1a and 6a owing to the generation of insoluble complexes during the process. To enhance the solubility of the substrates and intermediates, we then produced tritosylates 4 for the triple linking reaction by alkylating ethyl gallate (5) with the monotosylate of tri- and tetra(ethylene glycol) and subsequently tosylating the triols 15. As mentioned earlier, potassium and cesium ions act as efficient templates during the final linking steps (the conversion of crown ethers 3 to cryptands 1) in method B. The triple linkage of tritosylates 4 with ethyl gallate (5) was performed using K2CO3 and Cs2CO3 as bases and templates, respectively. Following GPC and high-performance liquid chromatographic (HPLC) separation, we successfully isolated cross-chain and linear cryptands 1 and 6. The 1H NMR spectra of all isolated cryptands (1 and 6) matched those of the products obtained through the abovementioned methods. The isolated yields for both the cross-chain bridging and linear cryptands from the triple-linking reactions ranged from 24% to 32% across all experiments (Table 1). Notably, the yields of cross-chain bridging cryptand 1 consistently surpassed those of linear cryptand 6, and K2CO3 provided slightly higher yields for both cryptands compared to Cs2CO3.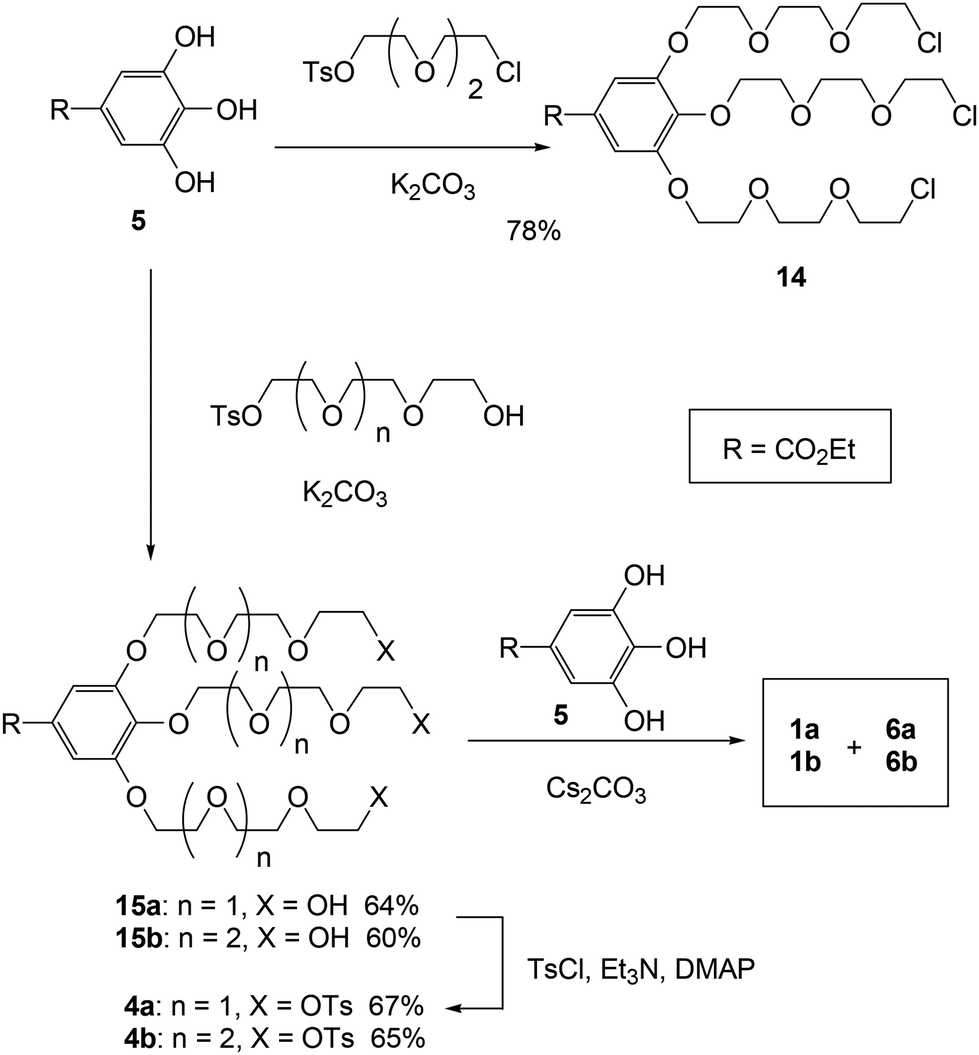 | ||
| Scheme 4 Method C: synthesis of cross-chain bridging cryptand 1 and linear cryptand 6. | ||
| K2CO3 isolated yield | Cs2CO3 isolated yield | |
|---|---|---|
| [4.4.4]cryptand | 1a: 17% | 1a: 15% |
| 6a: 15% | 6a: 10% | |
| [5.5.5]cryptand | 1b: 17% | 1b: 15% |
| 6b: 13% | 6b: 9% |
The detailed processes of the triple linking reaction between tosylate 4 and ethyl gallate 5 are shown in Fig. 4. In the initial linking step, the hydroxy group at the 4-position of ethyl gallate primarily undergoes deprotonation owing to the electron-withdrawing effect of the ethoxy carbonyl group. Consequently, the phenoxide anion is likely to statistically attack the tosyloxy alkyl groups located at the 3- (5-) and 4-positions in tosylate 4, resulting in a 2:1 mixture of intermediates A and B. The subsequent intramolecular linking of intermediate A is expected to yield the cross-chain bridging cryptand 1 through intermediates C and D, which correspond to the key precursors from methods B and A, respectively. In contrast, only linear cryptand 6 is expected to be formed from the second and third intramolecular linking of intermediate B via intermediate E. Consequently, the triple linking reaction may statistically yield a 2:1 mixture of the cross-chain bridging cryptand 1 and linear cryptand 6, assuming that only the 4-hydroxy group of ethyl gallate is deprotonated and that the second and third linking reactions occur equally. However, the observed experimental selectivity of the triple linking reactions was lower than this statistical ratio (2:1). Previous studies on each linking reaction involving the analogs of the intermediates (methods A and B) indicated that the macrocyclization of the A-analog produced the D-analog in a poor yield (method A). As a result, the proportion of linear cryptand 6 may increase relative to the cross-chain product. Additionally, a minor first alkylation at the 3-hydroxy group of ethyl gallate could further enhance the ratio of linear cryptand 6. The detailed minor processes of the triple linking reaction are provided in the ESI (Fig. S19‡).
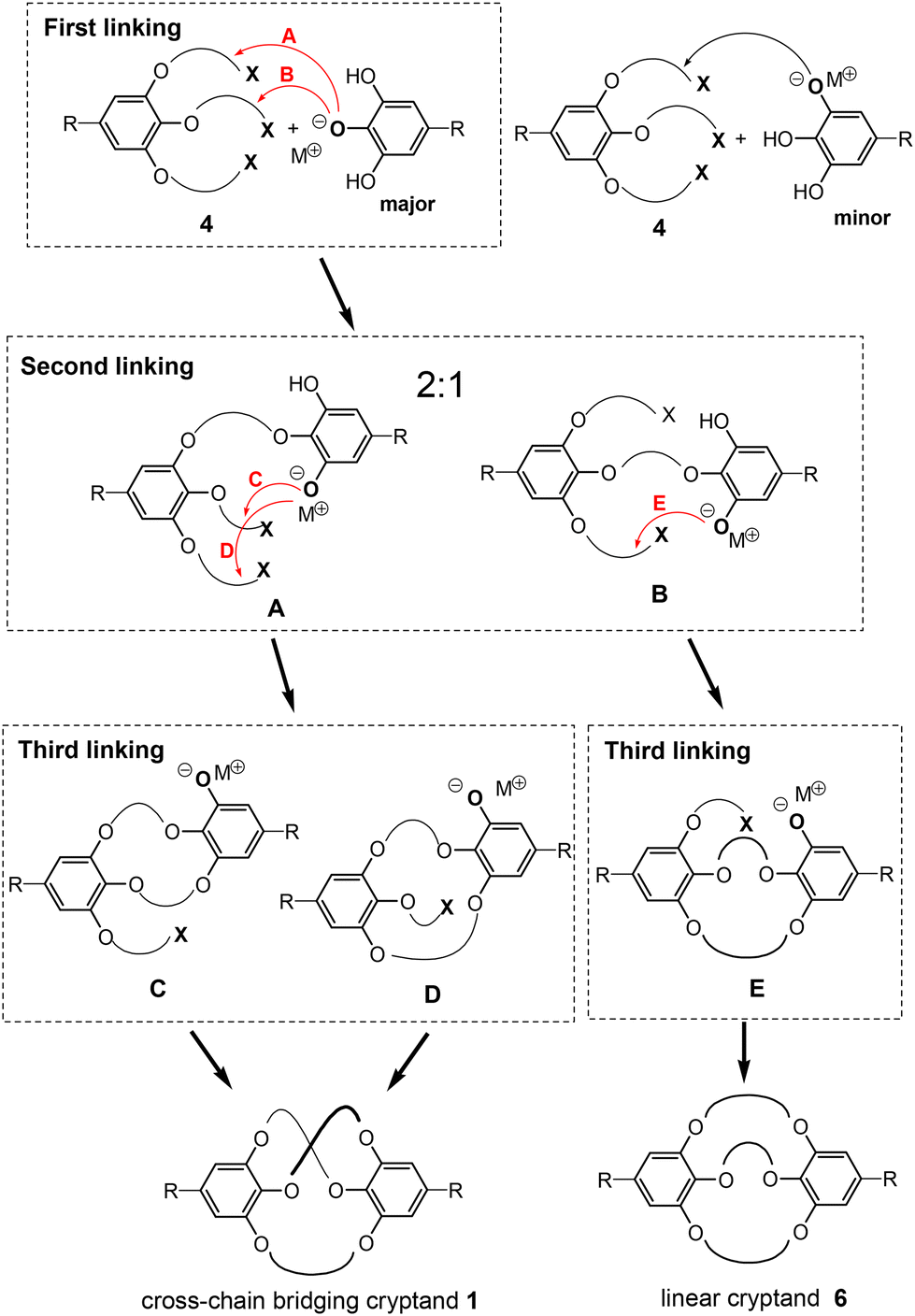 | ||
| Fig. 4 Key steps in the triple-linking reaction between tosylate 15 and ethyl gallate. | ||
The isolated yields of both cryptands may align with the yields observed from the two macrocyclization reactions of method B (first macrocyclization: 63% and 52% yields for the synthesis of 1:1 mixtures of crown ethers 10 and 11a in the presence of Cs2CO3; second macrocyclization: 34–65% yields for the synthesis of cryptands 1 and 6 in the presence of K2CO3 or Cs2CO3). The overall isolated yields (24–32%) are a result of the moderate yields achieved in the two macrocyclization reactions.
Total yields and steps for the synthesis of the cross-chain bridging cryptand 1 using three methods
Table 2 shows the total yields and number of steps for the three methods used in the synthesis of cross-chain bridging cryptand 1. The new methods (B and C) considerably enhanced the yields for synthesizing the small cross-chain bridging cryptand 1a, with method B showing the most improvement. Conversely, method A yields the best results for preparing the larger cryptand 1b. Though method C allows for a shorter step-synthesis of cryptand 1, it requires HPLC and GPC for the isolation of each cryptand.Chiral HPLC analysis of cryptand 1c: observation of racemization
The 25-membered cryptand 1a is chiral, whereas the racemization of the 31-membered cryptand 1b occurred at room temperature under NMR spectroscopic conditions. As mentioned above, the interconversion of the 28-membered cryptand 1c was not detected by NMR spectroscopy. Subsequently, we investigated the chiral HPLC separation of the enantiomers of 1c using CHIRALPAK IG-3 as the stationary phase (Fig. S20‡). The chromatogram displayed the typical sharp peaks for both enantiomers of 1c, along with a plateau peak located between these peaks, indicating that the racemization of 1c was occurring slowly under the HPLC conditions. Additionally, changes in flow rate affected the ratio of the peak areas, which was dependent on the retention time. The two usual peaks and the plateau peak were attributed to the nonracemized and racemized forms of 1c, respectively. A plot of ln[(peak area of enantiomers)/(total area)] versus time (t) produced a straight line, allowing us to determine a racemization rate constant k of 5.57 × 10−4 (s−1) and a half-life (τ) of 20.7 min at room temperature from the first-order plot (Fig. 5).30,31 While the racemization reaction is too rapid to isolate both enantiomers of 1c, increasing the steric bulk of the benzoate groups in 1c is likely to inhibit racemization because this reaction may occur through the rotation of the benzoate moiety.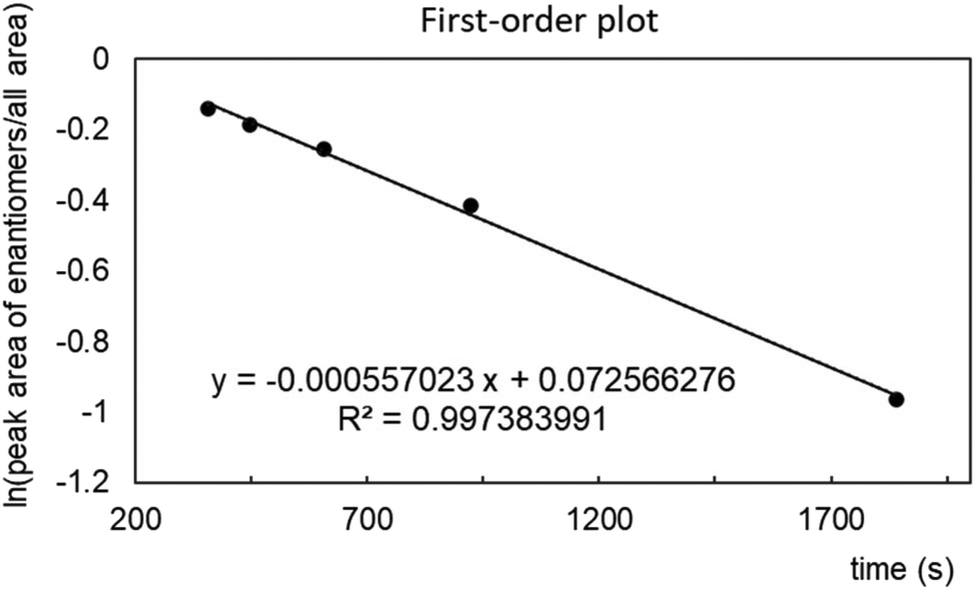 | ||
| Fig. 5 First-order plot depicting the racemization of cryptand 1c under HPLC conditions. | ||
In this study, considering the racemization rate of the cryptand, we only show the ring member of macrocycles. However, the length of the crossing-chain linkage should affect the rate because the length influences the flexibility and precise size of the macrocycle even though the ring member is identical. Further experimental studies should be needed to elucidate the effects of the cross-chain linkage on the racemization rate.
Crystal structure of the cross-chain bridging cryptand 1a
In our previous work, we successfully crystallized a 1:1 complex of the (−)-[4.4.4]cryptand 1a with NH4PF6.26 X-ray crystallography analysis indicated that two chemically equivalent tri(ethylene glycol) linkers were entangled in the solid, and the absolute configuration of the complex was established. In this study, we obtained a single crystal for analysis by recrystallizing (±)-[4.4.4]cryptand 1a in the absence of any additional species, using slow evaporation of a 1:1 acetone/hexane mixture at room temperature. The crystal structure revealed that 1a adopted an approximately C2-symmetric conformation, with two benzene rings arranged in parallel (Fig. 6 and S21, Table S1‡). As expected, the two chemically equivalent tri(ethylene glycol) chains became cross-linked with each other, a structure resembling that of the (−)-1a and NH4PF6 complex in the solid state. A pair of enantiomers assembled to form a crystal lattice featuring six CH–O intermolecular interactions between the CH groups and the oxygen in the ethylene glycol chains, alongside four CH–π intermolecular interactions between the ethylene glycol CH groups and the phenyl groups (Fig. S21a‡). One enantiomer aligned along the a axis, resulting in a hetero-columnar crystal, with two crown cavities of 1a oriented perpendicularly along the a axis (Fig. S21b‡).
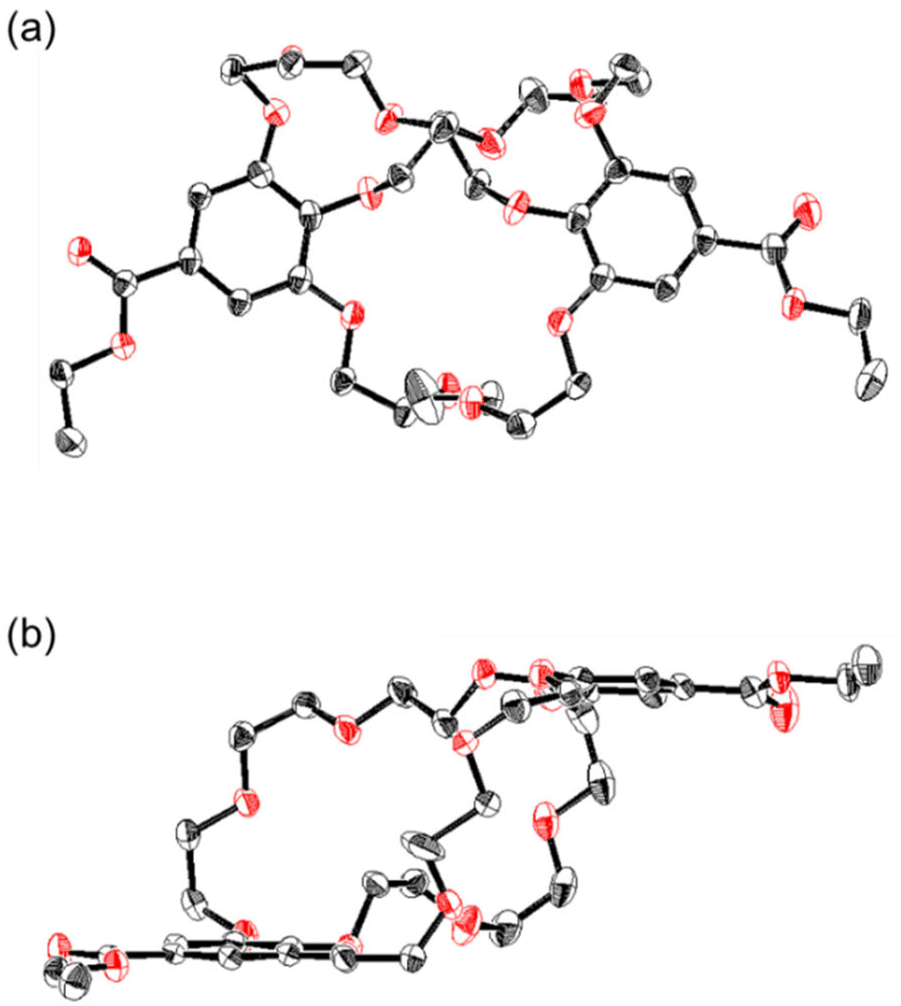 | ||
| Fig. 6 ORTEP representation of the solid-state structure of cryptand 1a. (a) Overhead view and (b) side view. Thermal ellipsoids are shown at the 50% probability level. For clarity, hydrogen atoms and disordered oxyethylene chains are omitted. Black: carbon; red: oxygen. | ||
Conclusions
We explored two new methodologies (methods B and C) for synthesizing cryptand 1. In method B, we produced the cross-chain bridging cryptand 1 through the linkage of C2-symmetric crown ethers 3. This approach increased the overall yield and reduced the number of steps for synthesizing [4.4.4]cryptand 1a, using K2CO3 as a base and template for the final cross-linking step. However, the total yield of [5.5.5]cryptand 1b did not surpass that of the earlier methodology (method A). Notably, method B allowed the synthesis of a new type of cross-chain bridging cryptand 1c, which incorporates two tetra(ethylene glycol) linkers and one tri(ethylene glycol) linker. This is a new type of cross-chain bridging cryptand featuring various linkers and a C2-symmetric structure. Furthermore, the isomeric σ-symmetric crown ethers generated during the initial macrocyclization were transformed into linear cryptand 6 to elucidate the structures of the crown ethers. The triple linking method was investigated for synthesizing cross-chain bridging cryptand 1 from tritosylate 4 and ethyl gallate (method C). The final triple linking step predominantly yielded cross-chain bridging cryptand 1 as the major isomer, while the corresponding linear regioisomer 6 appeared as the minor isomer. Both cross-chain bridging cryptand 1 and linear cryptand 6 could be isolated using GPC and HPLC separation techniques. This methodology could considerably reduce the total number of synthetic steps required for preparing cryptand 1. Chiral HPLC analysis of cryptand 1c, which features a 28-membered macrocycle, demonstrated the interconversion of enantiomers, with a half-life of 20.7 min at room temperature. Finally, X-ray crystal analysis confirmed the cross-chain bridging structure of the chemically equivalent chains in cryptand 1a.Author contributions
T. Y., R. H., T. M., K. N., and H. F.: investigation, data curation, and formal analysis, M. N.: investigation, data curation, and analysis of X-ray crystallography, S. M.: investigation, data curation, and analysis of mass spectrometry, and Y. T.: conceptualization, supervision, and writing – original draft.Data availability
The data that support the findings of this study are available in the ESI‡ of this article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by JSPS KAKENHI [JP20K05461 (YT) and JP24K08392 (YT)] and Izumi Science and Technology Foundation.References
- A. Szumna, Inherently Chiral Concave Molecules—from Synthesis to Applications, Chem. Soc. Rev., 2010, 39, 4274–4285 RSC.
- (a) V. Böhmer, D. Kraft and M. Tabatabai, Inherently Chiral Calixarenes, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 19, 17–39 CrossRef; (b) Y.-S. Zheng and J. Luo, Inherently Chiral Calixarenes: A Decade's Review, J. Inclusion Phenom. Mol. Recognit. Chem., 2011, 71, 35–56 CrossRef CAS; (c) G. E. Arnott, Inherently Chiral Calixarenes: Synthesis and Applications, Chem. – Eur. J., 2018, 24, 1744–1754 CrossRef CAS PubMed.
- T. Ogoshi, T. Yamagishi and Y. Nakamoto, Pillar-Shaped Macrocyclic Hosts Pillar[n]arenes: New Key Players for Supramolecular Chemistry, Chem. Rev., 2016, 116, 7937–8002 CrossRef CAS PubMed.
- X.-N. Han, Y. Han and C.-F. Chen, Pagoda[4]arene and i-Pagoda[4]arene, J. Am. Chem. Soc., 2020, 142, 8262–8269 CrossRef CAS PubMed.
- G.-W. Zhang, P.-F. Li, Z. Meng, H.-X. Wang, Y. Han and C.-F. Chen, Triptycene-Based Chiral Macrocyclic Hosts for Highly Enantioselective Recognition of Chiral Guests Containing a Trimethylamino Group, Angew. Chem., Int. Ed., 2016, 55, 5304–5308 CrossRef CAS PubMed.
- A. Collet, Cyclotriveratrylenes and Cryptophanes, Tetrahedron, 1987, 43, 5725–5759 CrossRef CAS.
- T. Brotin and J.-P. Dutasta, Cryptophanes and Their Complexes–Present and Future, Chem. Rev., 2009, 109, 88–130 CrossRef CAS PubMed.
- Selected reviews of helicenes: (a) Y. Shen and C. F. Chen, Helicenes: Synthesis and Applications, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS PubMed; (b) K. Dhbaibi, L. Favereau and J. Crassous, Enantioenriched Helicenes and Helicenoids Containing Main-Group Elements (B, Si, N, P), Chem. Rev., 2019, 119, 8846–8953 CrossRef CAS PubMed; (c) H. V. Anderson, N. D. Gois and W. A. Chalifoux, New Advances in Chiral Nanographene Chemistry, Org. Chem. Front., 2023, 10, 4167–4197 RSC; (d) M. Rickhaus, M. Mayor and M. Juricek, Strain-Induced Helical Chirality in Polyaromatic Systems, Chem. Soc. Rev., 2016, 45, 1542–1556 RSC.
- Y. Segawa, H. Ito and K. Itami, Structurally Uniform and Atomically Precise Carbon Nanostructures, Nat. Rev. Mater., 2016, 1, 15002 CrossRef CAS.
- M. Saito, H. Shinokubo and H. Sakurai, Figuration of Bowl-Shaped π-Conjugated Molecules: Properties and Functions, Mater. Chem. Front., 2018, 2, 635–661 RSC.
- I. R. Marquez, S. Castro-Fernandez, A. Millan and A. G. Campana, Synthesis of Distorted Nanographenes Containing Seven- and Eight-Membered Carbocycles, Chem. Commun., 2018, 54, 6705–6718 RSC.
- Y. Zhang, S. H. Pun and Q. Miao, The Scholl Reaction as a Powerful Tool for Synthesis of Curved Polycyclic Aromatics, Chem. Rev., 2022, 122, 14554–14593 CrossRef CAS PubMed.
- Selected reviews and examples of Möbius strips: (a) R. Herges, Topology in Chemistry: Designing Möbius Molecules, Chem. Rev., 2006, 106, 4820–4842 CrossRef CAS PubMed; (b) S. Nishigaki, Y. Shibata, A. Nakajima, H. Okajima, Y. Masumoto, T. Osawa, A. Muranaka, H. Sugiyama, A. Horikawa, H. Uekusa, H. Koshino, M. Uchiyama, A. Sakamoto and K. Tanaka, Synthesis of Belt- and Möbius-Shaped Cycloparaphenylenes by Rhodium-Catalyzed Alkyne Cyclotrimerization, J. Am. Chem. Soc., 2019, 141, 14955–14960 CrossRef CAS PubMed; (c) J. Yuan, Y. Song, X. Li, J. Xie, S. Dong and K. Zhu, A Tubular Belt and a Möbius Strip with Dynamic Joints: Synthesis, Structure, and Host−Guest Chemistry, Org. Lett., 2021, 23, 9554–9558 CrossRef CAS PubMed; (d) S. Wang, J. Yuan, J. Xie, Z. Lu, L. Jiang, Y. Mu, Y. Huo, Y. Tsuchido and K. Zhu, Sulphur-Embedded Hydrocarbon Belts: Synthesis, Structure and Redox Chemistry of Cyclothianthrenes, Angew. Chem., Int. Ed., 2021, 60, 18443–18447 CrossRef CAS PubMed; (e) Y. Segawa, T. Watanabe, K. Yamanoue, M. Kuwayama, K. Watanabe, J. Pirillo, Y. Hijikata and K. Itami, Synthesis of a Möbius Carbon Nanobelt, Nat. Synth., 2022, 1, 535–541 CrossRef CAS; (f) B. Yao, X. Liu, T. Guo, H. Sun and W. Wang, Molecular Möbius Strips: Twist for a Bright Future, Org. Chem. Front., 2022, 9, 4171–4177 RSC.
- Y. Segawa, A. Yagi, K. Matsui and K. Itami, Design and Synthesis of Carbon Nanotube Segments, Angew. Chem., Int. Ed., 2016, 55, 5136–5158 CrossRef CAS PubMed.
- S. Sato, A. Yoshii, S. Takahashi, S. Furumi, M. Takeuchi and H. Isobe, Chiral Intertwined Spirals and Magnetic Transition Dipole Moments Dictated by Cylinder Helicity, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13097–13101 CrossRef CAS PubMed.
- H. Lu and N. Kobayashi, Optically Active Porphyrin and Phthalocyanine Systems, Chem. Rev., 2016, 116, 6184–6261 CrossRef CAS PubMed.
- H. Tanaka, Y. Inoue and T. Mori, Circularly Polarized Luminescence and Circular Dichroisms in Small Organic Molecules: Correlation between Excitation and Emission Dissymmetry Factors, ChemPhotoChem, 2018, 2, 386–402 CrossRef CAS.
- T. Mori, Chiroptical Properties of Symmetric Double, Triple, and Multiple Helicenes, Chem. Rev., 2021, 121, 2373–2412 CrossRef CAS PubMed.
- M. Hasegawa, Y. Nojima and Y. Mazaki, Circularly Polarized Luminescence in Chiral π-Conjugated Macrocycles, ChemPhotoChem, 2021, 5, 1042–1058 CrossRef CAS.
- Y. Zhang, J. Guan, L. Luo, X. Han, J. Wang, Y. Zheng and J. Xu, Chiral Twisted Molecular Carbons: Synthesis, Properties, and Applications, Interdiscip. Mater., 2024, 3, 453–479 CAS.
- Selected reviews and examples of figure-of-eight: (a) M. Stępień, N. Sprutta and L. Latos-Grażyński, Figure Eights, Möbius Bands, and More: Conformation and Aromaticity of Porphyrinoids, Angew. Chem., Int. Ed., 2011, 50, 4288–4340 CrossRef PubMed; (b) S. Saito and A. Osuka, Expanded Porphyrins: Intriguing Structures, Electronic Properties, and Reactivities, Angew. Chem., Int. Ed., 2011, 50, 4342–4373 CrossRef CAS PubMed; (c) K. Mitsuno, T. Yoshino, I. Gupta, S. Mori, S. Karasawa, M. Ishida and H. Furuta, Doubly N-Confused [36]Octaphyrin(1.1.1.1.1.1.1.1): Isomerization, Bis-Metal Coordination, and Topological Chirality, Angew. Chem., Int. Ed., 2017, 56, 14252–14256 CrossRef CAS PubMed; (d) K. Senthilkumar, M. Kondratowicz, T. Lis, P. J. Chmielewski, J. Cybińska, J. L. Zafra, J. Casado, T. Vives, J. Crassous, L. Favereau and M. Stępień, Lemniscular [16]Cycloparaphenylene: A Radially Conjugated Figure-Eight Aromatic Molecule, J. Am. Chem. Soc., 2019, 141, 7421–7427 CrossRef CAS PubMed; (e) J. Oniki, T. Moriuchi, K. Kamochi, M. Tobisu and T. Amaya, Linear [3]Spirobifluorenylene: An S-Shaped Molecular Geometry of p-Oligophenyls, J. Am. Chem. Soc., 2019, 141, 18238–18245 CrossRef CAS PubMed; (f) M. Krzeszewski, H. Ito and K. Itami, Infinitene: A Helically Twisted Figure-Eight [12]Circulene Topoisomer, J. Am. Chem. Soc., 2022, 144, 862–871 CrossRef CAS PubMed; (g) J. Malinčík, S. Gaikwad, J. P. Mora-Fuentes, M.-A. Boillat, A. Prescimone, D. Häussinger, A. G. Campaña and T. Šolomek, Circularly Polarized Luminescence in a Möbius Helicene Carbon Nanohoop, Angew. Chem., Int. Ed., 2022, 61, e202208591 CrossRef PubMed; (h) R. Yoshina, J. Hirano, E. Nishimoto, Y. Sakamoto, K. Tajima, S. Minabe, M. Uyanik, K. Ishihara, T. Ikai, E. Yashima, T. Omine, F. Ishiwari, A. Saeki, J. Kim, J. Oh, D. Kim, G. Liu, T. Yasuda, H. Shinokubo and N. Fukui, Inner-Bond-Cleavage Approach to Figure-Eight Macrocycles from Planar Aromatic Hydrocarbons, J. Am. Chem. Soc., 2024, 146, 29383–29390 CrossRef CAS PubMed.
- Double helicate has been developed as a chiral host, see: N. Ousaka, S. Yamamoto, H. Iida, T. Iwata, S. Ito, Y. Hijikata, S. Irle and E. Yashima, Water-Mediated Deracemization of a Bisporphyrin Helicate Assisted by Diastereoselective Encapsulation of Chiral Guests, Nat. Commun., 2019, 10, 1457 CrossRef PubMed.
- (a) D. M. Walba, R. M. Richards and R. C. Haltiwange, Total Synthesis of the First Molecular Möbius Strip, J. Am. Chem. Soc., 1982, 104, 3219–3221 CrossRef CAS; (b) D. M. Walba, J. Simon and F. Harary, Topicity of Vertices and Edges in the Möbius Ladders: A Topological Result with Chemical Implications, Tetrahedron Lett., 1988, 29, 731–734 CrossRef CAS.
- Selected reviews of molecular knots, links, and the related compounds: (a) O. Lukin and F. Vögtle, Knotting and Threading of Molecules: Chemistry and Chirality of Molecular Knots and Their Assemblies, Angew. Chem., Int. Ed., 2005, 44, 1456–1477 CrossRef CAS PubMed; (b) R. S. Forgan, J.-P. Sauvage and J. F. Stoddart, Chemical Topology: Complex Molecular Knots, Links, and Entanglements, Chem. Rev., 2011, 111, 5434–5464 CrossRef CAS PubMed; (c) J.-P. Sauvage and D. B. Amabilino, The Beauty of Knots at the Molecular Level, Top. Curr. Chem., 2012, 323, 107–126 CrossRef CAS PubMed; (d) S. D. P. Fielden, D. A. Leigh and S. L. Woltering, Molecular Knots, Angew. Chem., Int. Ed., 2017, 56, 11166–11194 CrossRef CAS PubMed; (e) W.-X. Gao, H.-J. Feng, B.-B. Guo, Y. Lu and G.-X. Jin, Coordination-Directed Construction of Molecular Links, Chem. Rev., 2020, 120, 6288–6325 CrossRef CAS PubMed; (f) T. Sawada and M. Fujita, Folding and Assembly of Metal-Linked Peptidic Nanostructures, Chem, 2020, 6, 1861–1876 CrossRef CAS; (g) Z. Ashbridge, S. D. P. Fielden, D. A. Leigh, L. Pirvu, F. Schaufelberger and L. Zhang, Knotting Matters: Orderly Molecular Entanglements, Chem. Soc. Rev., 2022, 51, 7779–7809 RSC.
- G. Gil-Ramírez, S. Hoekman, M. O. Kitching, D. A. Leigh, I. J. Vitorica-Yrezabal and G. Zhang, Tying a Molecular Overhand Knot of Single Handedness and Asymmetric Catalysis with the Corresponding Pseudo-D3-Symmetric Trefoil Knot, J. Am. Chem. Soc., 2016, 138, 13159–13162 CrossRef.
- H. Fujihara, M. Naito, T. Yashima, Y. Okada, N. Kobayashi, S. Miyagawa, H. Takaya and Y. Tokunaga, Synthesis of Cross-Chain Bridging Cryptands and Induction of Molecular Chirality, Org. Lett., 2023, 25, 8959–8964 CrossRef CAS PubMed.
- We reported a part of this study as a patent: Y. Tokunaga and M. Naito, Optically Active compound and Method for Producing the Same, Coordination Compound Containing Optically Active Compound, Cyclic Compound, as Well as Intermediate Compounds, JP2021130627A, 2021.
- The number of oxygen atoms in each bridge is used for the abbreviated nomenclature of this type of cryptand in this paper.
- The products will be reported elsewhere.
- O. Trapp, G. Schoetz and V. Schurig, Determination of Enantiomerization Barriers by Dynamic and Stopped-Flow Chromatographic Methods, Chirality, 2001, 13, 403–414 CrossRef CAS PubMed.
- T. Kimura, S. Miyagawa, H. Takaya, M. Naito and Y. Tokunaga, Locking the Dynamic Axial Chirality of Biphenyl Crown Ethers through Threading, Chem. – Asian J., 2020, 15, 3897–3903 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Dr Frank Würthner on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Synthetic procedures, analytical data, and a cif file for the single-crystal X-ray analysis. CCDC 2392777. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo02330g |
| This journal is © the Partner Organisations 2025 |