
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOvercoming bottlenecks towards complete biocatalytic conversions and complete product recovery
Roland Wohlgemuth *abc
*abc
aFaculty of Chemistry, Lodz University of Technology, 90-537 Lodz, Poland. E-mail: roland.wohlgemuth.1@p.lodz.pl
bSKB, Swiss Coordination Committee Biotechnology, 8021 Zurich, Switzerland
cESAB, European Society of Applied Biocatalysis, 1000-Brussels, Belgium
First published on 14th November 2024
Abstract
Biocatalysis has become an attractive and powerful technology for resource-efficient conversions of starting materials to products because of selectivity, safety, health, environment and sustainability benefits. One of the key success factors for any synthetic method has traditionally been the yield of the product which has been isolated from the reaction mixture after the conversion and purified to the required purity. The conversion economy and the final product recovery, which determine the isolated yield of a product, are therefore also of key importance for biocatalytic processes, from biocatalytic single-step to multi-step reactions and total synthesis. In order to progress towards complete biocatalytic conversions and to aim at completely recovering and isolating the pure product, relevant thermodynamic, kinetic and other constraints leading to incomplete biocatalytic conversions and incomplete product recovery need to be identified and overcome. The methods and tools for overcoming various types of bottlenecks are growing and can provide valuable guidance for selecting the most suitable approaches towards the goal of achieving 100% yield of the isolated pure product for a specific biocatalytic conversion.
Introduction
The concept of ideal synthesis, which has evolved over the past decades, is of much interest for synthetic innovation and improvements towards complete and selective conversion in a single operation.1–4 As practical synthesis in the real world is also affected by many other relevant considerations, such as safety, health and environmental aspects, the early focus on the economy of materials, processes and reaction yields1 has been widened towards sustainable chemistry. The molecular economy aspects of synthesis, such as the atom, step, redox and conversion economy of reactions,5–9 the ratio of waste per product, as measured by the E-factor,10 provide important criteria for guiding the search for the efficiency and sustainability of synthesis. Driving highly selective reactions to full conversions eliminates the need for separating products from structurally similar non-converted substrates, thus contributing to economic and sustainability benefits. Reactions with incomplete conversions not only directly decrease synthetic efficiency and sustainability, but may present challenges in case of precious starting materials, costly and difficult product recovery and purification. Therefore, it is of major interest to identify and overcome important limiting factors which hinder a given reaction system going to completion, such as kinetic, thermodynamic and other constraints. In order to take full advantage of complete conversions, it is also essential to identify and overcome the factors which prevent to completely recover and isolate the product in high quality.Biocatalytic reactions in living systems and in synthesis
Biocatalytic reactions are attractive for ideal synthesis by meeting many criteria of synthetic efficiency and sustainability due to their high selectivity, which enables them to efficiently work together in multistep enzyme-catalyzed processes within biological cells of living systems without the use of protecting groups.11–13 Healthy biological cells and their metabolism require however that biocatalytic reactions are constrained and regulated at several levels in order to keep the complex material and energy flow in balance. Ten constraints for metabolic networks in living systems, such as buffer capacity, solubility and thermodynamic and kinetic constraints, have been identified and quantified for E. coli.14 When biocatalytic processes are used for the preparation of single products, it is of key importance not only to identify but also to overcome constraints in order to progress towards complete conversions and to aim at completely recovering and isolating the pure product.Data on the thermodynamics of reactions
While the thermodynamic aspects of chemical processes with respect to physical process parameters such as heat and mass transfer have been addressed by highly efficient engineering solutions, the involved chemical reactions have received much less attention. New approaches for overcoming the limitations of thermodynamic equilibria of reversible reactions beyond the coupling of reactions with separations, have only recently been described.15 Extensive experimental data on enthalpy of formation and entropy are available for organic compounds and reaction enthalpies and Gibbs free energies of reaction for more than 8000 chemical reactions have been measured and stored in the NIST chemistry webbook.16 Experimental data on Gibbs free energies for biochemical reactions and their dependence on reaction conditions are however more limited.17 Based on the combined molecular knowledge on the thermodynamics of chemical and biochemical reactions, the development of the eQuilibrator database has been very valuable for estimating Gibbs free energies and equilibrium constants of biocatalytic reactions.18Data on the kinetics of biocatalytic reactions
The kinetic aspects of biocatalytic conversions play another key role in reaching the goal of a full conversion of substrates to products. While reactions taking place within a homogeneous single phase are ultimately limited by diffusion, biocatalytic conversions are further constrained by the catalytic efficiency of the utilized biocatalysts. The tremendous and continuously growing amount of experimental data on enzyme properties, which either exist already in the literature or are newly being measured and on its way to be published, may provide a good starting point for bringing the attention to potential kinetic limitations in specific biocatalytic conversions. Experimental enzyme kinetic properties which exist already in the literature have been retrospectively extracted into the BRENDA database,19 while the STRENDA database has been started as the functional equivalent to the structure database PDB in a prospective way, by entering experimental enzyme kinetic datasets as part of the workflow from experiment to publication.20 In addition to the catalytic efficiency of enzymes the diffusion of substrates to the active site of the enzyme and subsequently products out of the enzyme active site influence the overall kinetics. Mass-transfer limitations need to be considered when immobilized enzymes are used or when biocatalytic conversions are performed in more than one phase. In systems consisting of two phases the rates at which substrates and products can transfer from one phase to the other, for example at gas–liquid or aqueous–organic interfaces, are important parameters which also need to be taken into account. In whole cell systems the rates at which substrates and products are transported into and out of the cells can further limit kinetic constraints of biocatalytic conversions.Properties and stabilities of the reaction components
The full conversion of substrates to products can also be influenced by constraints in the properties and stabilities of the reaction components, such as substrates, intermediates or products. Chemically labile compounds may be undergoing non-enzymatic degradation over the time course of biocatalytic conversions under certain process conditions, thus reducing in any case the degree of the intended biocatalytic conversion accordingly. Whether substrates, intermediates or products are undergoing non-enzymatic degradation or are subjected to undesired side-reactions catalyzed by the utilized enzymes, this will reduce in any case the degree of the intended biocatalytic conversion accordingly and prevent completion of the desired reaction. Adequate approaches for overcoming the various bottlenecks are therefore needed.Other important factors for directing the course of the desired biocatalytic substrate-to-product conversions towards completion are the stability and properties of the utilized biocatalysts. A variety of biocatalyst properties, such as purity, main activity, side activities which are relevant for the desired reaction, as well as the biocatalyst stability, which is needed to maintain the catalytic action over the course of the biocatalytic conversion under process conditions, play a decisive role.21
Product recovery and purification
Driving biocatalytic conversions towards completion is however not yet the end in preparative applications, but the starting point for developing efficient downstream operations towards completely recovering and purifying the product obtained in the biocatalytic conversion. A successful final product recovery and purification is not only key for a resource-efficient and sustainable bioprocess, but also of interest for reducing the production costs, because downstream operations are often responsible for a major part of the production costs.Thermodynamic constraints of biocatalytic conversions
The Gibbs free energy of reaction, determined by its reactants, products and reaction conditions such as temperature, pH and ionic strength, represents an important thermodynamic constraint for reversible biocatalytic conversions. It determines the directionality and feasibility of a biocatalytic conversion, whereby the feasibility of the forward conversion of reactants to products is indicated by a negative Gibbs free energy of reaction.22 The thermodynamic equilibrium constant of the biocatalytic conversion cannot be changed by biocatalysts, although biocatalysts can catalyze forward and backward reactions towards reaching equilibrium. The thermodynamic equilibrium constant is however not only affecting the directionality and feasibility of a biocatalytic conversion but represents another constraint regarding the kinetics by limiting the net flux because of the dependence of kinetic parameters on the thermodynamics of the reaction, as described by the Haldane relationship.23–25 The effects of the thermodynamic driving force and the enzyme saturation level by substrate and/or product on the enzyme kinetics are not only of much interest for converting one substrate to one product, but also for conversions of multiple substrates to multiple products, as catalyzed by most enzymes.26Identification of thermodynamic constraints of biocatalytic conversions
Experimental methods for measuring thermodynamic equilibria of biocatalytic reactions are of much interest to obtain precise equilibrium constants under defined reaction conditions.27 Determining Gibbs free energies of reaction as a function of reaction conditions is valuable for identifying thermodynamic constraints and guiding the improvement of biocatalytic conversions, such as the transaminase-catalyzed synthesis of L-valine, L-leucine and L-tert-leucine (see Fig. 1) from the corresponding 2-keto acids.28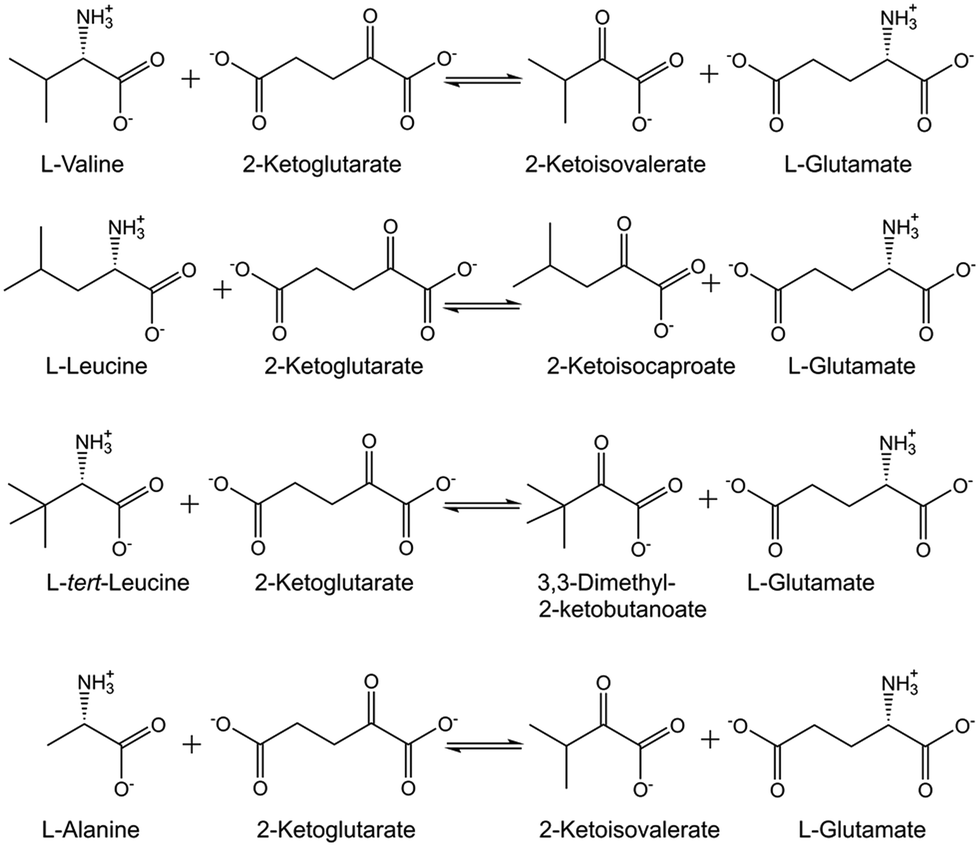 | ||
| Fig. 1 Thermodynamic equilibria for transamination reactions between L-aminoacid/2-ketoglutarate and the 2-ketocarboxylic acid/L-glutamate. | ||
Simple experimental methods for determining the thermodynamic transamination equilibrium between 4-phenyl-2-butylamine/cyclohexanone and 4-phenyl-2-butanone/cyclohexylamine under defined reaction conditions have shown large differences between experimental and predicted values and potential benefits of building a thermodynamic knowledge base for improving computational approaches.29 The experimentally determined molality-based apparent thermodynamic equilibrium constants for the transamination between L-alanine/2-ketoglutarate and pyruvate/L-glutamate (see Fig. 1), catalyzed by alanine aminotransferase, have been demonstrated to depend on reaction conditions.30 The electrolyte perturbed-chain statistical associating fluid theory (ePC-SAFT) has been shown to successfully predict the equilibrium concentrations and how temperature, pH, absolute starting molalities of the L-alanine and 2-ketoglutarate reactants, and the initial reactant molalities ratio impact the thermodynamic equilibrium.30
Overcoming thermodynamic constraints of biocatalytic conversions
Thermodynamic equilibria of some biocatalytic conversions can be highly valuable, such as the triosephosphate isomerase-catalyzed interconversion of dihydroxyacetone phosphate and D-glyceraldehyde-3-phosphate or the isomerization of isopentenyl pyrophosphate to dimethylallylpyrophosphate catalyzed by isopentenylpyrophosphate isomerase. For preparative applications in general however thermodynamic equilibria of biocatalytic conversions are regarded as limitations, although it may turn out that from the experimental investigation of a reaction no thermodynamic constraints exist for a particular biocatalytic conversion under the selected process conditions.31 In case of thermodynamic constraints however, methods to overcome these are highly desirable for achieving high product yields. These methods range from optimized reactants or reaction conditions for a favorable equilibrium constant to increased concentrations of starting materials, and removal of products or byproducts, which are favorable for shifting the thermodynamic equilibria towards the product side. Increasing the starting material concentrations alone can however still lead to incomplete conversions and may make product recovery and purification more challenging.Shifting reversible biocatalytic acyltransfer reactions towards complete acylation has become well established in preparative applications by using an enolester or an acid anhydride as acyldonor, and a non-aqueous medium instead of water.32
Achieving complete conversions despite thermodynamic constraints is essential for the resource efficiency and sustainability of biocatalytic reactions in living organisms and preparative applications. The approaches identified in nature for overcoming thermodynamic limitations inherent to certain substrate to product transformations provide also inspirations for developing biocatalytic conversions which fully utilize the starting materials for a sustainable synthesis of the desired products, even under thermodynamically unfavorable conditions.
A general principle for moving an equilibrium reaction towards complete conversion is the coupling of the reaction with the separation of a product or by-product, and a number of such reactive separations have been developed.33 These separation methods (see Fig. 2) include adsorption, crystallization, distillation, extraction, phase separation, or chromatography. Coupling a biocatalytic reaction with subsequent product removal has also been described as extractive bioconversion or extractive biocatalysis. If the product is not the final product, it is attractive to couplie the equilibrium reaction with other reactions, where an irreversible reaction at the end removes a product in situ and pulls the flowing equilibrium to completion.
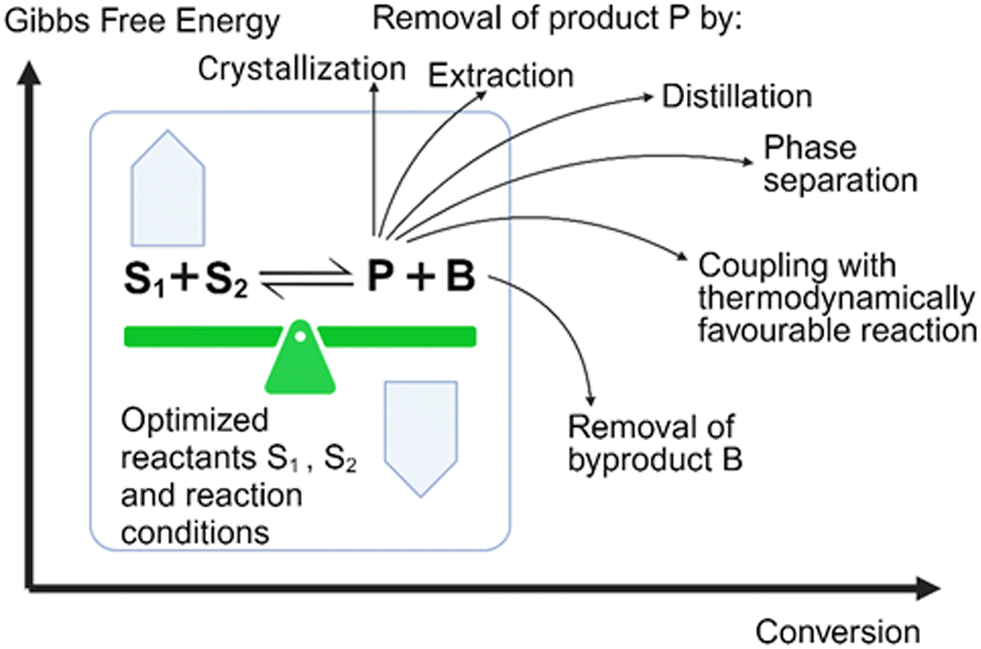 | ||
| Fig. 2 Approaches for overcoming thermodynamic constraints of a biocatalytic conversion in the forward direction by shifting the thermodynamic equilibrium towards the product side, optimizing reactants S1, S2 and reaction conditions, and coupling the biocatalytic conversion with removal from the equilibrium of the product P or the byproduct B. | ||
A simple way of in situ product removal (ISPR) can be realized if the product crystallizes or precipitates selectively under the process conditions, a well established method in the chemical industry and also described as reactive crystallization.34 In the production of nicotinamide from 3-cyanopyridine by nitrile hydratase-catalyzed water addition (see Fig. 3A) the crystallization of nicotinamide enables to overcome the unfavorable Gibbs free energy of reaction and to reach complete conversion at 9 M and 12 M substrate concentration.35 Spontaneous crystallization or precipitation of a product under reaction conditions is an ideal case, which simplifies also product recovery and purification. A shift of the thermodynamic equilibrium reaction of cytidine, in the presence of hydroxylamine, via the uridine intermediate to the product N-hydroxycytidine, has been achieved (see Fig. 3B) by coupling the reaction, for which an engineered cytidine deaminase was developed, with in situ crystallization of N-hydroxycytidine.36 In situ product crystallization was observed due to the substantially lower solubility of N-hydroxycytidine than cytidine and uridine in aqueous solution, by increasing the concentration of the substrate to >100 g L−1 and by lowering the reaction temperature to 4 °C.36 This enabled a scalable biocatalytic conversion of cytidine at high substrate concentration with 90% conversion to highly pure N-hydroxycytidine in 24 h, thus also minimizing the uridine byproduct by shifting its conversion to the product.36
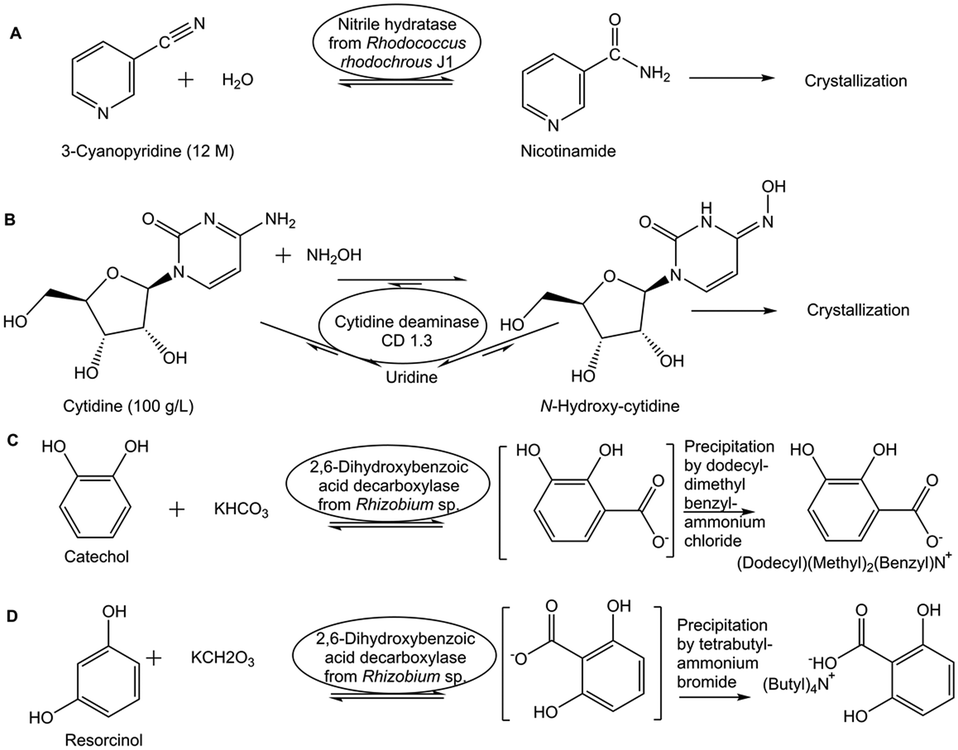 | ||
| Fig. 3 Overcoming thermodynamic constraints by coupling the reaction with in situ product removal using selective crystallization or precipitation. This is illustrated by the nitrile hydratase-catalyzed water addition to 3-cyanopyridine for nicotinamide production (Fig. 3A), the cytidine deaminase-catalyzed conversion of cytidine to N-hydroxycytidine (Fig. 3B), the 2,6-dihydroxybenzoic acid decarboxylase-catalyzed carboxylation of catechol to 2,3-dihydroxybenzoic acid (Fig. 3C), and the 2,6-dihydroxybenzoic acid decarboxylase-catalyzed carboxylation of resorcinol to 2,6-dihydroxybenzoic acid (Fig. 3D). | ||
Shifting thermodynamic equilibrium reactions towards the product side can also be achieved by discovering optimum conditions which decrease product solubility and can induce in situ product crystallization or precipitation.37 This has been demonstrated by the use of carboxylic acids in chiral amine synthesis catalyzed by transaminases38 and by using quaternary ammonium salts in the biocatalytic Kolbe–Schmitt reactions catalyzed by 2,6-dihydroxybenzoic acid decarboxylase. The constraints of the thermodynamic equilibrium in the carboxylation direction39 can be overcome by the addition of quaternary ammonium salts, which by selective product precipitation enabled to shift conversion of the carboxylation reactions of catechol to 2,3-dihydroxybenzoic acid (see Fig. 3C) and of resorcinol to 2,6-dihydroxybenzoic acid (see Fig. 3D), from less than 40% up to 97%.40 The use of a strong anion exchanger for the in situ removal of 2,6-dihydroxybenzoic acid in the biocatalytic carboxylation reaction of resorcinol enabled to increase yields to more than 80% and simplified downstream processing.41
Various in situ removal methods of volatile byproducts have been utilized for moving biocatalytic equilibrium reactions coupled with excess sacrificial substrates towards completion. Biocatalytic ketone reductions involving highly selective and tolerant ketoreductases are well-established and have become methods of first choice. Thereby cofactor regeneration can be achieved by formate/formatedehydrogenase or an isopropanol-based system, with the reductant in large excess and/or carbon dioxide (see Fig. 4A) or acetone (see Fig. 4B) byproduct removal, for pushing the equilibrium towards the chiral alcohol product.42,43 Analogously, the use of isopropylamine as amine donor and/or the acetone byproduct removal have enabled to push transaminase-catalyzed equilibrium reactions towards the chiral amine product (see Fig. 4C and D) using evolved transaminases.44,45 Whether in situ removal of byproducts is really required for a particular biocatalytic reaction depends on its actual Gibbs free energy of reaction. Alkanone reductions with concomitant oxidation of isopropanol to acetone have been characterized as equilibrium reactions,46 but favourable thermodynamics of a ketone reduction with excess isopropanol as reductant may not need acetone removal for completion.47 Other options of driving equilibria towards completion are the use of suitable cosubstrates, or the use of suitable enzymes to convert byproducts. In the first case using the two-carbon donor hydroxypyruvate has been preparatively useful for driving transketolase-catalyzed carbon–carbon bond formation reactions to completion by releasing carbon dioxide as byproduct.48 Experimental investigations of the thermodynamics and kinetics demonstrated that transketolase-catalyzed reactions are initially under kinetic control and over longer time approach equilibrium,49 thus making it interesting to evaluate novel approaches for reaching complete conversions with carbon dioxide release. Examples for the second case include pyruvate decarboxylase catalyzing the release of carbon dioxide from the byproduct pyruvate when L-alanine is used as amino donor for moving transaminase-catalyzed equilibrium reactions to the product side.50 The thermodynamically favourable sucrose phosphorylase-catalyzed reaction for removing the phosphate byproduct has been successfully applied for pulling forward the three subsequent equilibrium reactions from 2-ethynylglyceraldehyde 3-phosphate to islatravir catalyzed by evolved deoxyribose 5-phosphate aldolase, phosphopentomutase and purine nucleoside phosphorylase.51
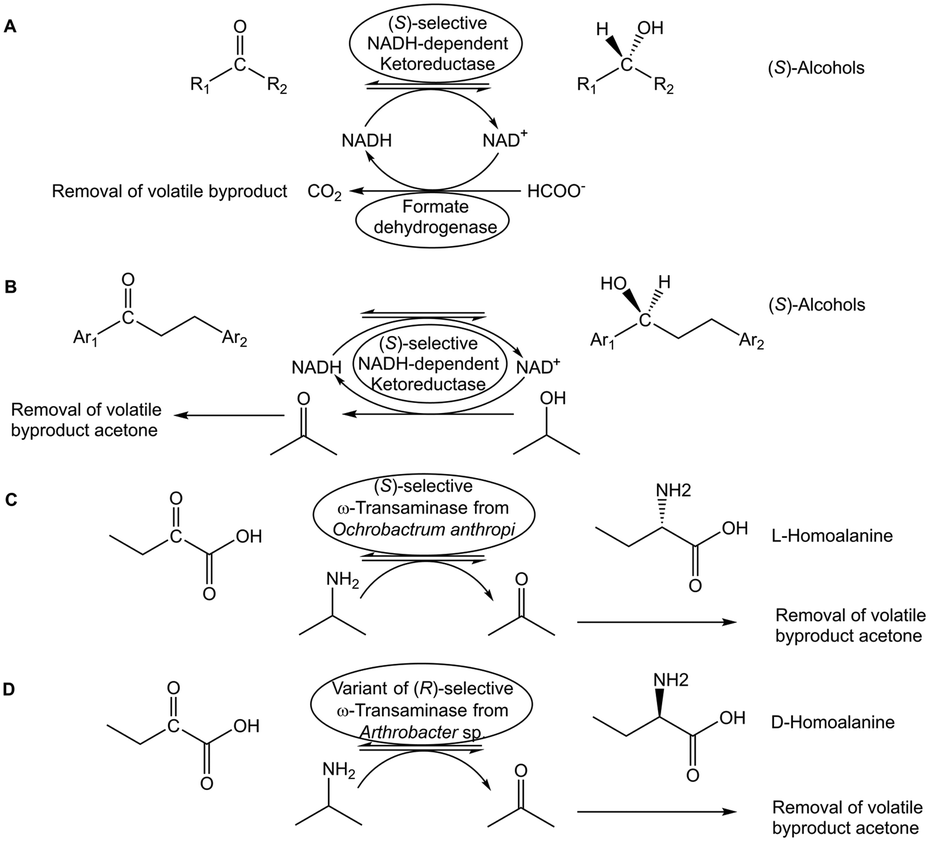 | ||
| Fig. 4 Overcoming thermodynamic constraints by coupling the reaction with in situ removal of volatile byproducts. The carbon dioxide formed from formate in the formatedehydrogenase-catalyzed regeneration of NADH in the reduction of a non-symmetric ketone to the corresponding (S)-alcohol catalyzed by a (S)-selective NADH-dependent ketoreductase enables shifting the equilbrium towards the (S)-alcohol (Fig. 4A). The removal of acetone formed from isopropanol, when the cofactor NADH is regenerated by the same (S)-selective NADH-dependent ketoreductase utilized for the ketone reduction, also allows pushing the equilibrium towards the (S)-alcohol (Fig. 4B). The removal of the volatile byproduct acetone formed from the amine donor isopropylamine can also be used to shift the transamination reaction catalyzed by a (S)-selective transaminase towards L-homo-alanine (Fig. 4C), and to shift the transamination reaction catalyzed by a (R)-selective transaminase towards D-homoalanine (Fig. 4D). | ||
The excellent selectivity and orthogonality of biocatalytic conversions is especially useful for overcoming thermodynamic constraints by coupling within the same reaction space the thermodynamically unfavourable equilibrium reaction with subsequent thermodynamically favourable reactions.52 Various biocatalytic approaches overcome isomerization and epimerization equilibria, for example by coupling in a one-pot procedure the unfavourable aldose–ketose equilibrium reactions with subsequent biocatalytic phosphorylations, as shown by the examples of the biocatalytic conversion of L-arabinose to L-ribulose 1-phosphate in Fig. 5A, and the biocatalytic conversion of D-xylose to D-ribulose 1-phosphate in Fig. 5B.53 The same pot has been used in a subsequent step in the removal of ATP and ADP, followed by dephosphorylations, for the efficient synthesis of the corresponding rare (3S)- and (3R)-ketoses in high yield and purity from easily available monosaccharides.53
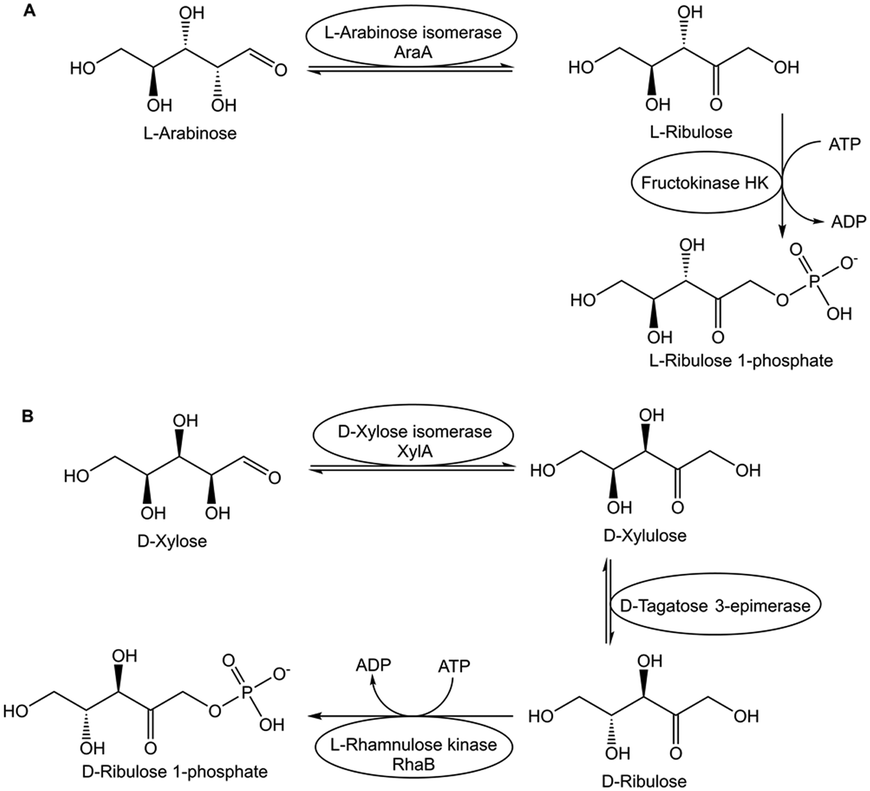 | ||
| Fig. 5 Overcoming thermodynamic constraints by coupling the equilibrium reaction(s) with a subsequent thermodynamically favourable reaction in one pot. The coupling of the isomerization of L-arabinose to L-ribulose catalyzed by L-arabinose isomerase AraA with the fructokinase HK-catalyzed phosphorylation to L-ribulose-1-phosphate overcomes the isomerization equilibrium (Fig. 5A). The two equilibrium reactions of the isomerization of D-xylose to D-xylulose catalyzed by D-xylose isomerase XylA and the epimerization of D-xylulose to D-ribulose catalyzed by D-tagatose 3-epimerase can be overcome by coupling with the L-rhamnulose kinase RhaB-catalyzed phosphorylation of D-ribulose to the final product D-ribulose-1-phosphate (Fig. 5B). | ||
The need for a thermodynamically favorable reaction at the end of a reaction system can be overcome by its coupling with continuous product separation into a high-yield integrated bioprocess, as demonstrated by the D-psicose synthesis from sucrose.54
Kinetic constraints of biocatalytic conversions
The overall biocatalytic reaction rates are not only determined by functional properties and kinetic constants of a given biocatalyst for a specific substrate to product conversion, but also by additional factors. These include the influence of the reaction conditions on biocatalyst properties and stability, concentration, activation or inhibition of the biocatalyst (substrate and/or product inhibition). For certain biocatalysts, possible mass transfer limitations may have to be taken into account, such as substrate transfer to the active site of the biocatalyst and product transfer from the biocatalyst active site. The ultimate constraints for the rates of biocatalytic conversions are given by the rates of diffusion in the reaction medium for the substrates and mass transfer of substrates across phase boundaries. The maximum rate obtainable for a biocatalytic conversion in the forward direction55 depends on the kinetic parameters, which can be described by kcat and KM for the biocatalysts following Michaelis–Menten kinetics, and the Haldane relationship connecting the ratio of the reactions rates of the forward to the backward reaction with thermodynamics.24Identification of kinetic constraints of biocatalytic conversions
The experimental determination of the enzyme functional properties and kinetic constants for a specific enzyme-catalyzed substrate to product conversion has been performed for more than a century.56 A key requirement is the development of suitable enzyme assays measuring the rate at which a product is formed and the utilized enzyme concentration.57 The preparation of enzymes and enzyme substrates, progress in the development of high-information content analytical methods, standardization and miniaturization have facilitated the kinetic characterization of numerous enzymatic reactions in vitro.19 Microfluidic enzyme kinetics has accelerated the throughput by miniaturizing fast and sensitive enzyme assays using fluorogenic substrates, and the development of droplet microfluidics, for example to measuring the Michaelis–Menten constants and inhibition constants of >1500 alkaline phosphatase variants within days.58 As enzyme kinetic parameters such as kcat are however still scarce and require significant efforts for their experimental determination in vitro, both computational and experimental approaches are of much interest for accelerating their acquisition. New approaches for calculating the in vivo maximal catalytic rates of enzymes in a biological cell from omics data are thereby promising, as demonstrated by the comparison of the in vivo catalytic rates of 132 enzymes in E. coli with their corresponding in vitro catalytic rates.59 This is of much interest for increasing the small fraction of kinetically characterized enzymes in biological cells, for example covering less than 10% of the enzymatic reactions in E. coli, S. cerevisiae, A. thaliana or H. sapiens.60 Therefore the systematic measurement of the kinetics of all enzymes in a biological cell is of broad interest.61 With the rapid growth of experimentally determined values of enzyme kinetic parameters, standardizing their reporting and assuring the completeness of the experimental conditions have become important tasks in the workflow from discovery to publication.62 These tasks are supported by the STRENDA guidelines, which provide a highly valuable and widely recommended tool for standardizing the reporting and checking the completeness of enzyme function data across various disciplines and research areas. The retrospective extraction from literature into the BRENDA database19 and the deposition and storage of a growing number of experimental enzyme kinetics datasets in the STRENDA database20 serve to facilitate the early identification of kinetic constraints of biocatalytic conversions. Significant advances have been achieved in the prediction of kcat values for yeast and E. coli enzymes.63,64 Starting from the protein sequence and the structure of the substrate, the prediction accuracy for kcat, KM and kcat/KM has been improved with the unified framework UniKP.65 A major limitation for the prediction of enzyme kinetic parameters by machine learning and computational approaches is however still the small size of experimental datasets. Measuring kinetic parameters under defined reaction conditions remains crucial and analytical methods facilitating high-throughput measurements of enzyme kinetics parameters are of much interest, such as fast microfluidic enzyme analysis using detection by sensitive mass spectrometry66 or by absorbance.67 In a desired sequence of reactions catalyzed by a limited number of isolated enzymes the kinetic parameters and constraints can easily be determined for each enzyme, which enables to overcome specific kinetic bottlenecks.68Overcoming kinetic constraints of biocatalytic conversions
General approaches for overcoming kinetic constraints (see Fig. 6) which hinder biocatalytic conversions to go to completion include enzyme engineering for optimizing enzyme activity and stability for a desired conversion, reaction engineering for optimizing the concentrations, conditions and stabilities of substrates, intermediates and products, and approaches for overcoming enzyme inhibition.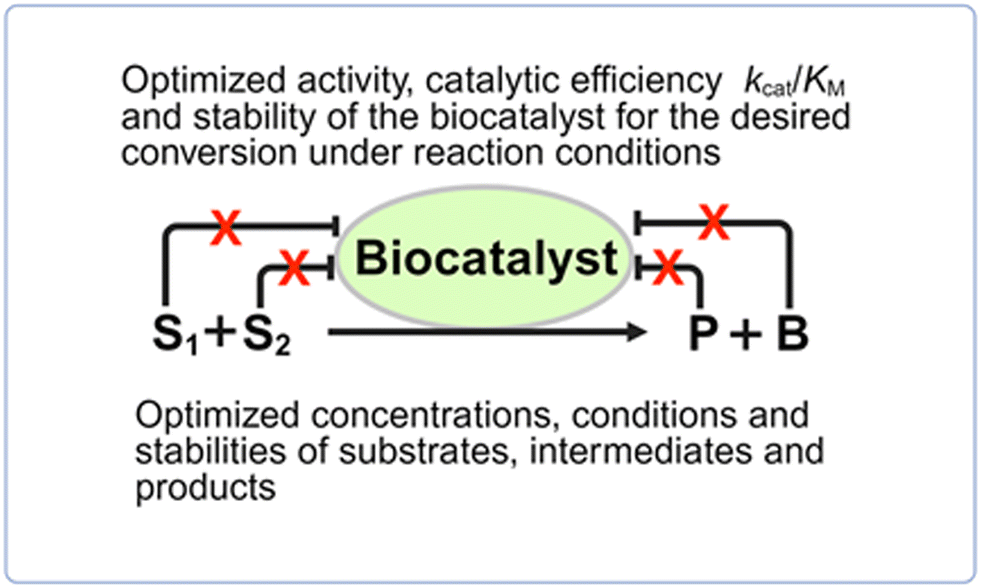 | ||
| Fig. 6 Approaches for overcoming kinetic constraints of biocatalytic conversions, such as inhibition by substrates S1 and S2, product P or byproduct B. The removal of the inhibition is marked by X. | ||
A high ratio of substrate concentration to the Michaelis constant KM is favorable for rapidly reaching complete conversion, if there is no enzyme inhibition by the substrate. As substrate inhibition is however widespread and plays important regulatory roles in biological cells,69 it is not surprising that many enzymes have already been demonstrated to be inhibited by their own substrates. This substrate inhibition is undesirable for synthetic applications of biocatalytic reactions, where enzymes may cease working at high substrate concentrations and thus prevent full conversions.
The methods which have been applied for overcoming the substrate inhibition constraint fall into the categories of enzyme engineering or reaction engineering. As the inhibition constant for a substrate is a property of the enzyme, finding a better enzyme with a higher inhibition constant or ideally no substrate inhibition at all while maintaining or even improving its catalytic efficiency is an attractive option. This can be achieved by searching more suitable enzymes from nature,70,71 or by protein engineering methods.72,73
The inhibition constant Ki of haloketone reductase SsCR for its substrate 2,2′,4′-trichloro-acetophenone has been increased up to 16-fold (see Fig. 7A) by structure-guided engineering of residues on crucial loops, thus enabling complete conversion at 0.1 M substrate concentration due to attenuated substrate inhibition.72 Substrate inhibition has been completely removed in lycopene cyclase (see Fig. 7B) without reducing its enzyme activity by isolating a mutant from screening only 50 variants selected by a combination of structure and phylogenetic information, because no crystal structure was available.73
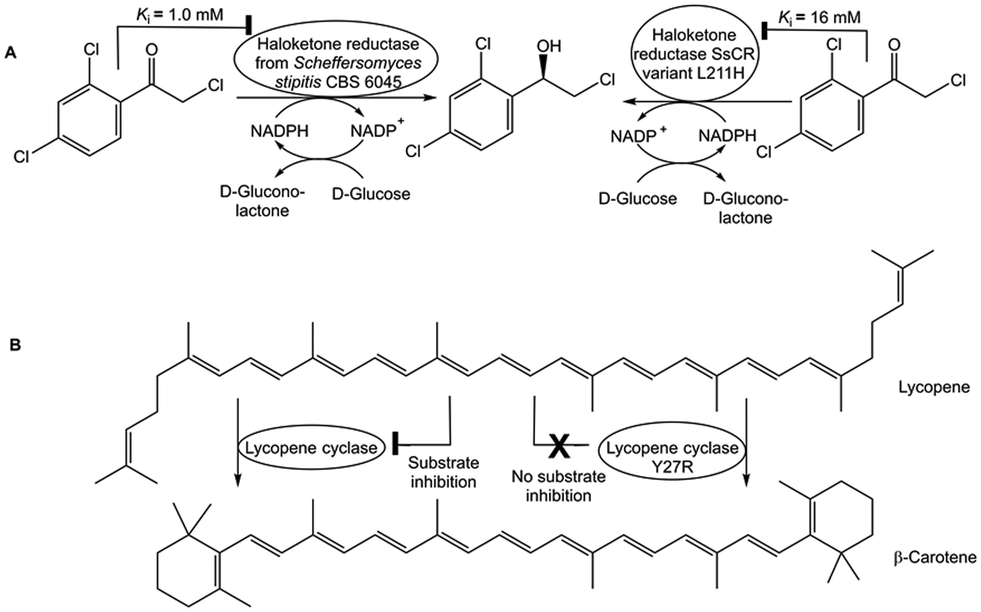 | ||
| Fig. 7 Approaches for overcoming kinetic constraints of substrate inhibition. The inhibition of haloketone reductase SsCR by its substrate 2,2′,4′-trichloro-acetophenone has been attenuated by enzyme engineering and the SsCR variant L211H, which had a 16-fold incerased inhibition constant Ki, enabled complete conversion (Fig. 7A). The lycopene cyclase variant Y27R has been found to catalyze the conversion of lycopene to β-carotene without substrate inhibition and without reduced enzyme activity (Fig. 7B). | ||
Reaction engineering methods are another attractive approach for overcoming a substrate inhibition constraint by keeping the substrate concentration below the level of inhibitory action during the entire biocatalytic conversion, by controlling its concentration to stay always sufficiently below the Ki of the substrate for the enzyme. This controlled substrate feed can be achieved in different ways, such as directly feeding the substrate continuously to the reaction medium at a rate which keeps an optimal substrate concentration in a fed-batch reactor,74 synthesizing the substrate by in situ capping the carboxylic acid group, which inhibits the subsequent biocatalytic oxidation, at an adequately balanced rate,75 or feeding it from an auxiliary second phase.76
Overcoming the product inhibition constraint has already been described long ago for the oxidoreductase-catalyzed oxidations of alcohols by removal, either by physical or chemical methods, of the product as it is formed, or by high substrate concentrations, when the product inhibits competitively and the substrate does not inhibit the enzyme.77
Overcoming the double kinetic constraints of simultaneous substrate and product inhibition requires its reduction or elimination, either by the development of a novel biocatalyst with higher enzyme inhibition constants Ki for substrate and product through screening natural or engineered variants, or by reaction engineering and simultaneously limiting the effective concentration of the substrate and the product to a sublethal or sub-inhibitory level.
The approach of engineering enzymes with reduced enzyme inhibition by substrate and product has been successfully demonstrated by the development of an (S)-selective trans-aminase catalyzing the production of the agrochemical inter-mediate (S)-methoxy-isopropylamine (see Fig. 8A), with 97% conversion and >99% ee at 1.94 M final product concentration, from initial substrate concentrations of 2.08 M methoxyacetone and 2.50 M isopropylamine.78 Directed evolution of a NADP+-dependent 7α-hydroxysteroid dehydrogenase for improved activity and higher tolerance for higher substrate and product concentration (see Fig. 8B) enabled the efficient biocatalytic synthesis of 7-ketolithocholic acid in 99% yield at an increased substrate concentration of 0.1 M chenodeoxycholic acid.79
 | ||
| Fig. 8 Approaches for overcoming double kinetic constraints of simultaneous substrate and product inhibition. Engineering a (S)-selective transaminase with reduced substrate and product inhibition enabled a highly efficient biocatalytic production of (S)-methoxy-isopropylamine with >99% ee and 97% conversion (Fig. 8A). Evolving a 7α-hydroxysteroid dehydrogenase towards tolerating higher substrate and product concentration as well as towards improved activity (Fig. 8B) enabled the efficient biocatalytic oxidation of chenodeoxycholic acid to 7-ketolithocholic acid in 99% yield, whereby the NADPH-cofactor was reoxidized using D-amino acid dehydrogenase and 2-oxobutanoic acid. The use of a solid phase to adsorb substrate and product for overcoming enzyme inhibition is illustrated by Amberlite XAD-7 in the (S)-selective biocatalytic reduction of 3,4-methylenedioxyphenyl acetone (Fig. 8C), and by Optipore L-493 in the biocatalytic asymmetric Baeyer–Villiger oxidation of bicyclo[3.2.0]hept-2-en-6-one (Fig. 8D). | ||
Practical applications of the latter approach for hydrophobic substrates and products have made use of a liquid–solid system where the substrate is adsorbed onto a suitable solid resin, from which it diffuses rapidly into free solution, where after the biocatalytic conversion the product is rapidly bound by the same adsorber, such as Amberlite XAD-7 in the (S)-selective biocatalytic reduction of 3,4-methylenedioxyphenyl acetone (see Fig. 8C),80 and Optipore L-493 in the biocatalytic asymmetric Baeyer–Villiger oxidation of bicyclo[3.2.0]hept-2-en-6-one (see Fig. 8D).81 By the selection of the most suitable adsorber, this resin-based in situ substrate feeding and product removal (SFPR) technology enables to overcome very well substrate and product inhibition constraints.82
Identifying inhibition of an (R)-selective biocatalytic hydroxylation of an indanone, which was caused neither by the substrate nor the product but by a diketone impurity formed by overoxidation (see Fig. 9), was essential for overcoming this constraint by minimizing the formation of this impurity through the addition of 1-octanol in a manufacturing process for belzutifan.83
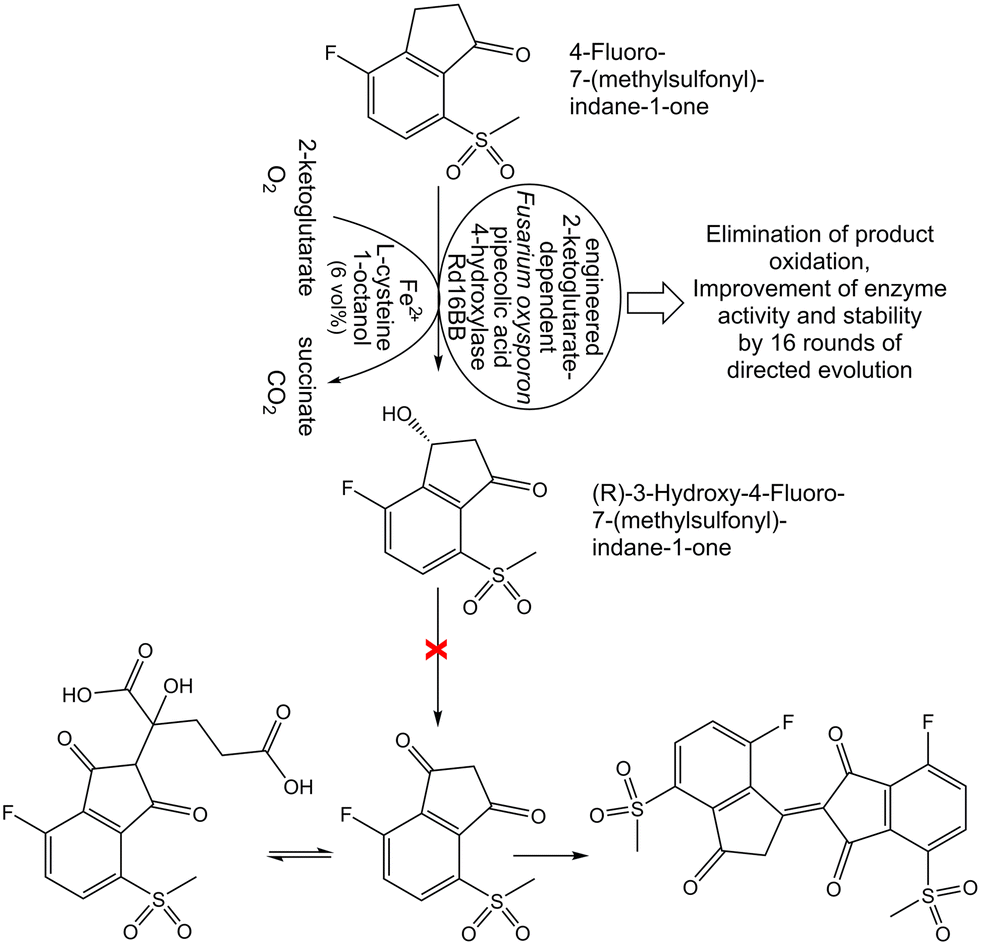 | ||
| Fig. 9 Overcoming constraints of rates of oxygen mass transfer and substrate dissolution, activity, selectivity, solvent tolerance thermal and oxidative stability of the enzyme, product over-oxidation in the (R)-selective biocatalytic 3-hydroxylation of 4-fluoro-7-(methylsulfonyl)-indan-1-one.83–85 | ||
Other constraints of biocatalytic conversions
In addition to identifying and overcoming thermodynamic and kinetic constraints to biocatalytic conversions several other constraint categories can also be of relevance.The identification of possible other constraints and the development of approaches for overcoming these (see Fig. 10) are also relevant for achieving complete biocatalytic conversions and are therefore discussed in the following subsections.
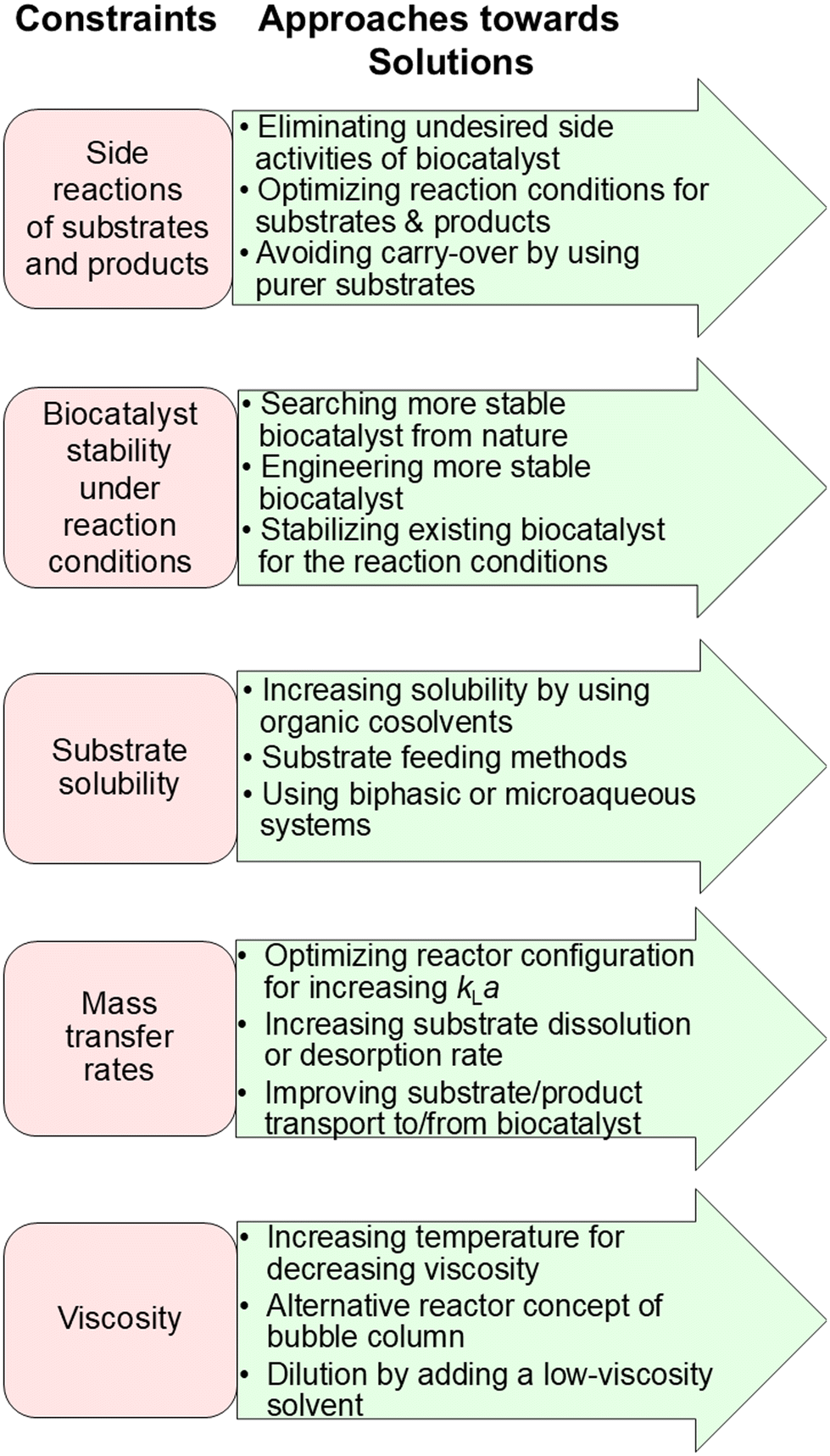 | ||
| Fig. 10 Additional constraints of biocatalytic conversions and approaches towards finding solution for achieving complete conversions. | ||
Identifying and overcoming constraints of side reactions of substrates and products
Any side reactions acting on substrates and products are not only preventing complete conversions to the desired target product but may also complicate product recovery and purification. Analyzing the exact nature and cause of such side reactions is therefore essential for overcoming this constraint. One obvious cause of side reactions can originate from side activities in the preparation of the utilized biocatalyst, which counteract product formation. The removal of relevant side activities by using purified recombinant biocatalysts instead of crude extracts enables complete conversions, such as in the elimination of water from D-gluconate in its biocatalytic conversion to 2-keto-3-deoxy-D-gluconate.86 Biocatalyst preparations obviously need to be analyzed and freed from side activities cleaving the bond whose formation is catalyzed, such as analyzing and removing glycosidase side activities counteracting complete biocatalytic glycosylations catalyzed by glycosyltransferases.87 Not only biocatalytic but also chemical side reactions need to be diminished to reach complete biocatalytic conversions, as demonstrated by the efficient biocatalytic three-step synthesis of cis-(+)-12-oxophytodienoic acid (see Fig. 11), where an intermediate is instable and the product is stereochemically labile.88 Complete conversion at 10 g L−1 substrate concentration has been achieved by optimizing a whole-cell biocatalyst as well as reaction parameters in order to avoid ketol formation of the intermediate and cis–trans isomerization of the product.88 Some side products can be challenging to analyze and identify, for example in the hydroxylation of an indanone catalyzed by a 2-ketoglutarate dependent pipecolic acid 4-hydroxylase the analysis of a diketone overoxidation side product (see Fig. 9), which undergoes further reactions and could be detected by its transient adduct with 2-ketoglutarate.84 This product overoxidation limits not only the degree of conversion to the product but the diketone affects enzyme activity and stability by acting as irreversible enzyme inhibitor and cross-linker. Therefore multiple rounds of directed evolution were required to reduce overoxidation, but also to increase activity and stability of the pipecolic acid 4-hydroxylase.84 Other limitations affecting reaction kinetics, such as the rates of oxygen mass transfer and 4-fluoro-7-(methylsulfonyl)-indan-1-one dissolution, have been investigated by various process analytical technologies.85 Complete conversion has been achieved by overcoming the limitations in a combination of reaction and enzyme engineering, such as eliminating product overoxidation, improving pipecolic acid-4-hydroxylase activity, selectivity and stability (see Fig. 9). This direct hydroxylation process for the belzutifan intermediate (R)-3-hydroxy-4-fluoro-7-(methylsulfonyl)-indan-1-one replaces a sequence of five reaction steps using protecting groups, a rare transition metal and oxidation state manipulations.83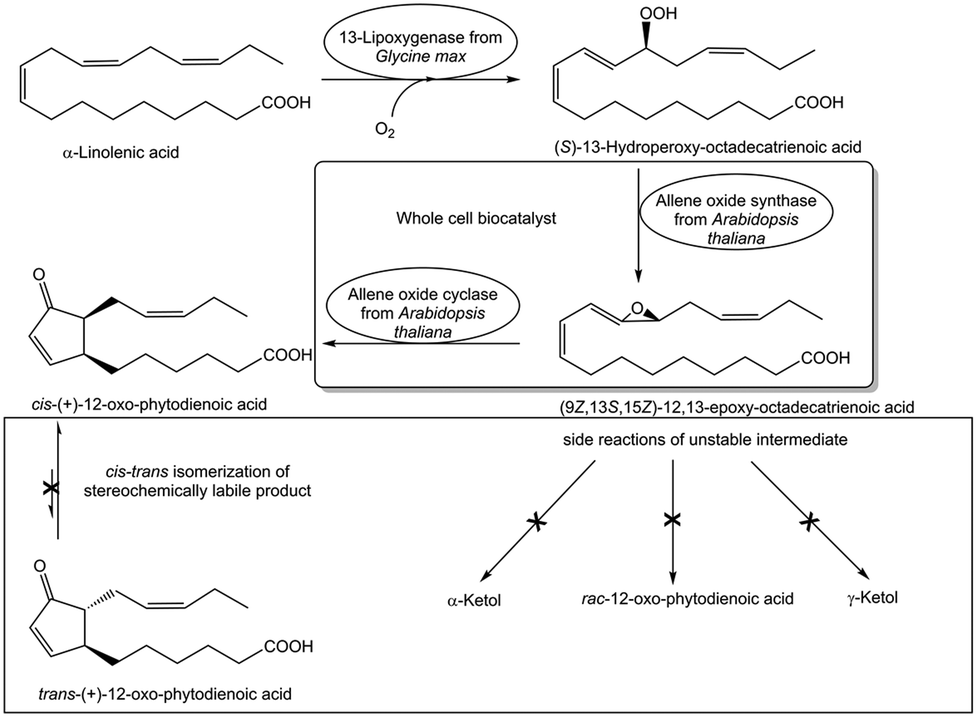 | ||
| Fig. 11 Overcoming constraints of side reactions of an instable intermediate and a stereochemically labile product in the biocatalytic synthesis of cis-(+)-12-oxophytodienoic acid.88 | ||
An obvious additional constraint to complete biocatalytic conversions, which can be easily overcome, is the carry-over to the product of impurities in the utilized substrates. This can become important when aiming at >99.5% overall product yield in multi-step conversions, as for example in template-free biocatalytic stepwise single nucleotide addition to a growing oligonucleotide chain (see Fig. 12) catalyzed by an engineered terminal deoxynucleotidyl transferase.89
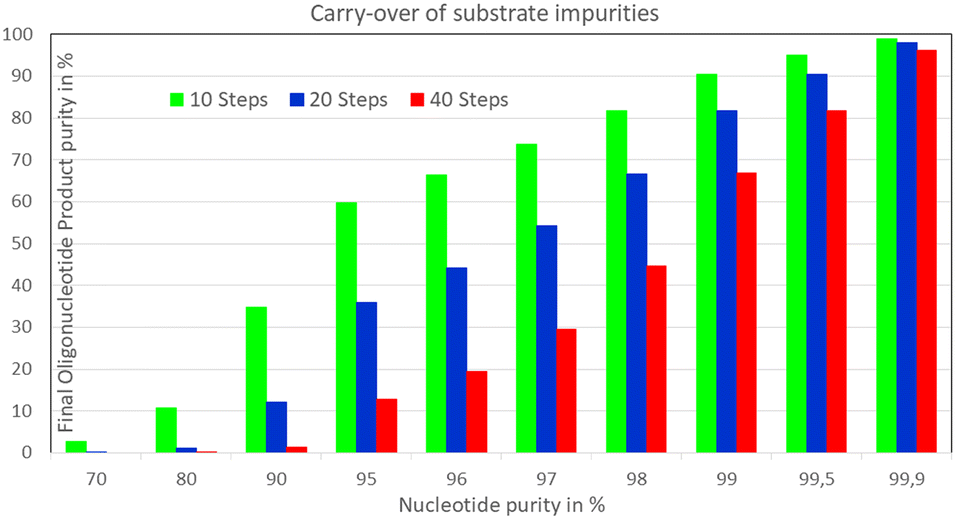 | ||
| Fig. 12 Importance of the purity of the nucleotide substrate on the final oligonucleotide purity in the stepwise single nucleotide addition to a growing oligonucleotide chain and avoiding the carry-over of impurities of the utilized substrates. | ||
Identifying and overcoming stability constraints of biocatalysts, substrates and products under reaction conditions
Another limitation to full conversion can arise when biocatalysts and reaction components are not sufficiently stable under the reaction conditions of the biocatalytic conversion. While it is desirable to identify the exact nature of decomposition reactions of substrates and products, it is a necessity to know their stability under reaction conditions over the course of the reaction. Limited stability of substrates, biocatalysts and products can cause major constraints to complete biocatalytic conversions. Optimal conditions, molecular and engineering approaches need therefore to be identified for overcoming the major stability constraints and for developing the best way forward to complete conversions. The stability analysis of the substrate 2-oxo-4-[(hydroxy)(methyl)phosphinoyl]butyric acid as a function of pH, temperature and initial substrate concentration showed the substrate to be stable at a pH in the range of 7.0 to 7.5, at a temperature of 35 °C and at 300 mM initial concentration.90 This has been key for developing a transamination process catalyzed by γ-aminobutyrate aminotransferase, whereby in situ recycling of the amino donor L-glutamate and continuous substrate feeding overcame the limitations and full conversion and >99.9% ee was achieved (Fig. 13).90 When the required enzymes are not sufficiently stable, active or selective under the conditions which are optimal for the stability of the reaction components, the improvement of enzyme properties through directed evolution has been a key methodology to successfully overcome this constraint by the development of better enzymes which are highly active, selective and stable under optimal reaction conditions, such as temperature, pH and organic solvent.91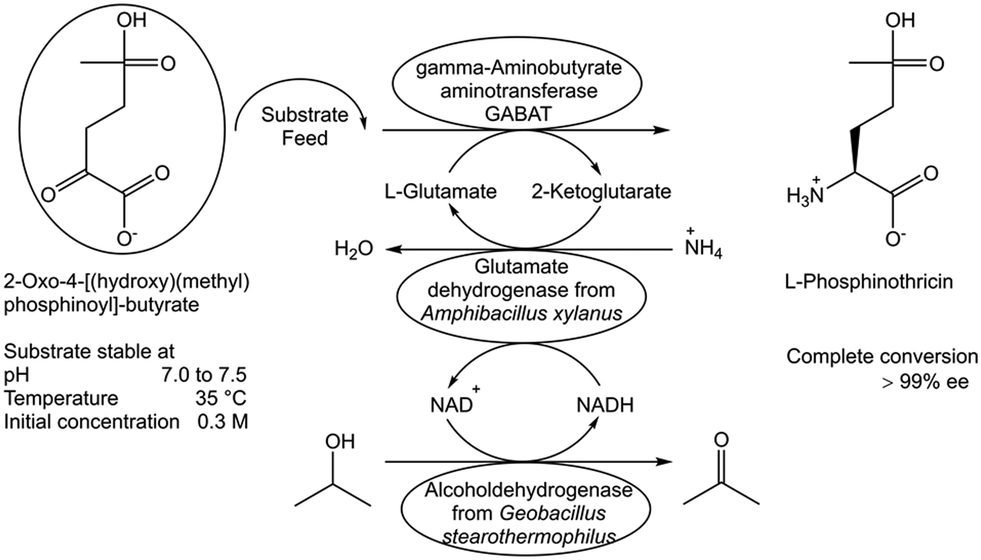 | ||
| Fig. 13 Identifying and overcoming substrate stability constraints in the biocatalytic synthesis of L-phosphinothricin.90 | ||
This is exemplified by the manufacturing of isopropyl (1S,3S)-3-(methylamino)cyclobutane-1-carboxylate, a key chiral intermediate of abrocitinib, an inhibitor of Janus kinase 1.92 The discovery of the highly selective reductive aminase SpRedAm from Streptomyces purpureus catalyzing the biocatalytic reductive amination from the corresponding ketone and methylamine showed this reductive aminase to be highly selective but with a narrow pH and temperature range and a low tolerance for the ketone substrate.92 The initial performance of giving only 0.75% conversion in 24 hours at 100 g L−1 ketone substrate concentration has been improved by enzyme engineering to nearly complete conversion, with greater than 95% conversion in 60 hours to the product with excellent purity.92 An impressive example of optimizing enzyme performance under manufacturing conditions, such as higher substrate concentration, reduced cosubstrate excess, elevated temperature and DMSO tolerance, by eight rounds of directed evolution, has been demonstrated in the biocatalytic reductive amination of a non-native α-keto acid with ethyl (S)-alaninate to the highly pure (S,S)-stereoisomer of a key intermediate in manufacturing a neprilysin inhibitor.93 A trace imine reductase activity of an engineered glucose dehydrogenase Rd1bb was converted into a highly active and selective imine reductase variant, whereby performance has been improved tremendously and the degree of conversion has been increased from 0.4% to 99%, which has been achieved in 3 hours at 50 g L−1 α-keto acid concentration using the evolved variant Rd6bb.93
Identifying and overcoming substrate solubility constraints
A limited substrate solubility in the reaction medium represents another constraint of biocatalytic conversions, as lower substrate concentrations in the reaction phase limit process efficiency and increase thermodynamic and kinetic limitations. The simple substrate feed method for achieving complete conversions at high substrate loads has been successfully demonstrated in biocatalytic dehydrations of aldoximes.94 By adding the substrate n-octanaloxime in seven portions to the potassium phosphate buffer pH 7.0 which contains as cosolvent 10% (v/v) ethanol, the record load of 1 kg substrate per liter has been completely converted (see Fig. 14A) in the aldoxime dehydratase OxdB-catalyzed dehydration to n-octanenitrile.94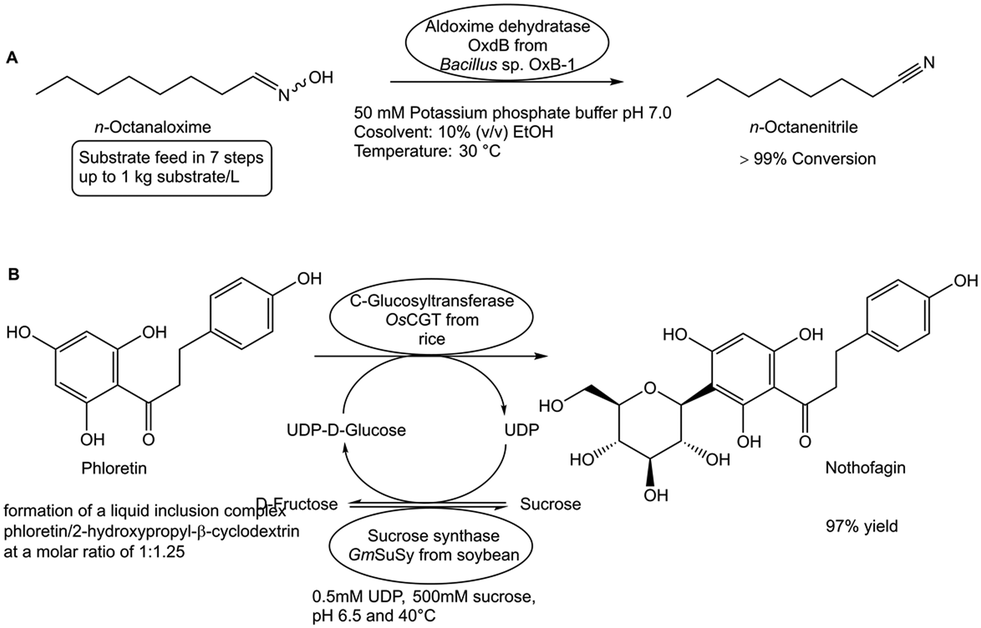 | ||
| Fig. 14 Overcoming substrate solubility constraints is illustrated by two approaches. The substrate feed method has been used in the aldoxime dehydratase OxdB-catalyzed dehydration of n-octanaloxime to n-octanenitrile (Fig. 14A), where the n-octanaloxime is added in seven portions to the aqueous buffer medium containing 10 % (v/v) ethanol, resulting in complete conversion of up to 1 kg L−1 substrate load. The use of suitable β-cyclodextrin solubilizers has been demonstrated to be superior to the cosolvent DMSO in the C-glucosyltransferase-catalyzed complete conversion of phloretin, which is β-D-glucosylated at its 3′-carbon; to nothofagin (Fig. 14B). | ||
The choice of the most suitable reaction medium is of central importance, from aqueous buffer systems for highly water-soluble substrates to the use of compatible organic cosolvents, neat, biphasic and microaqueous systems for substrates with low solubility.95 As the use of organic cosolvents may however negatively affect the degree of a biocatalytic conversion when the substrate concentration is increased, searching for other solubilizing methods which maintain the activity and stability of enzymes has attracted much interest.96 In the biocatalytic one-pot nothofagin synthesis, using a β-D-glucosylation at the 3′-carbon position of phloretin catalyzed by a C-glucosyltransferase and sucrose synthase-catalyzed UDP-glucose formation, 20 mM β-cyclodextrin has been demonstrated to achieve complete conversion at higher substrate concentrations than by using 20% DMSO as cosolvent.97 Moving from β-cyclodextrin to 2-hydroxypropyl-β-cyclodextrin enabled an efficient biocatalytic conversion of phloretin to nothofagin (see Fig. 14B) at 100 g scale by further increasing the phloretin solubility limits for complete conversion to about 120 mM, above which proper mixing of the increasingly viscous fluid becomes a limiting factor.98 A 4-fold higher space–time yield for complete conversion was found when C-glucosyltransferase and sucrose synthase were coimmobilized than when the two enzymes were immobilized individually.99
Identifying and overcoming viscosity constraints
Overcoming limitations in viscosity, whether staying constant, decreasing or increasing during the biocatalytic conversion, can be challenging when increasing the stirrer speed required for efficient mixing or lowering the viscosity by increasing the temperature of the biocatalytic conversion affects system stability. The development of a reactor concept comprising a bubble column is therefore of much interest for overcoming the viscosity limitations in the solvent-free lipase-catalyzed esterifications of reactants such as polyglycerol-3 with lauric acid to polyglycerol-3 laurate in a space–time 3042 g L−1 d−1 and myristylalcohol with myristic acid to myristylmyristate in a space time yield of 6731 g L−1 d−1.100Identifying and overcoming application-specific constraints
The breakthrough of the biocatalytic breakdown and recycling of the most abundant synthetic polyester poly(ethylene terephthalate) catalyzed by an engineered poly(ethylene terephthalate) depolymerase, a quadruple variant of leaf-branch compost cutinase named LCCICCG which gave a conversion of 90%,101 illustrates the importance of identifying and overcoming the constraints to complete biocatalytic depolymerization.102 Polymer reaction surface, polymer crystallinity and recrystallization kinetics near the glass transition temperature, composition of the depolymerized monomers, and high polymer concentration are critical constraints which need to be considered towards complete biocatalytic conversion at industrial large scale.103 Lowering the temperature for the LCCICCG-catalyzed reaction by 4 °C to 68 °C lead to an increased conversion of 98%, as the biocatalytic depolymerization overcomes the reduced poly(ethylene terephthalate) recrystallization kinetics and the enzyme amount was reduced 3-fold.103Product recovery constraints
Reaching full biocatalytic conversion eliminates the need of purifying the product from unreacted substrate and is a necessary but not sufficient condition for achieving a nearly theoretical yield of isolated product in high purity. Product recovery constraints, which can be as diverse as the downstream and purification methods104 used after the completed biocatalytic conversion and are as important as the biocatalytic conversion constraints, should be identified as early as possible and need to be overcome for avoiding valuable product losses. An overview of product recovery constraints and approaches towards overcoming them is shown in Fig. 15. | ||
| Fig. 15 Approaches for overcoming product recovery constraints after biocatalytic conversions. | ||
Product recovery using a simple separation of two different phases, such as liquid–liquid or liquid–solid phases, has been demonstrated to give excellent product recovery yields, when phase separation can be achieved either spontaneously with high product concentrations after full conversion, or by simple standard operations. An ideal product recovery from a biocatalytic reaction by exclusively crystallizing only the product puts up several constraints.
Full conversion is needed at higher substrate concentration for which the corresponding product concentration is above its solubility limit. All other reaction components, individually as well as possible complexes, are required to be kept below their solubility limits. Reaction optimization for overcoming these constraints can lead to highly resource-efficient processes. It may be possible that at a high concentration after complete conversion, the product crystallizes or precipitates spontaneously from the reaction medium.93 In other cases crystallization is only achieved after product extraction, after which filtration, washing and drying of the solid can be performed.92 The biocatalytic conversion of uridine to pseudouridine, catalyzed by four enzymes in one pot, yields a supersaturated solution of the product at 250 g L−1, from which pseudouridine crystallizes (see Fig. 16A).105 A clear separation of phases, with crude n-octanenitrile exclusively located in one phase, has been achieved after completion of the biocatalytic dehydration of n-octanaloxime, when the homogeneous emulsion during the intensely stirred reaction was left without mixing for 5 minutes.94 Product recovery has therefore been simplified tremendously (see Fig. 16B) and a high product recovery yield of 97.5% has been achieved.94
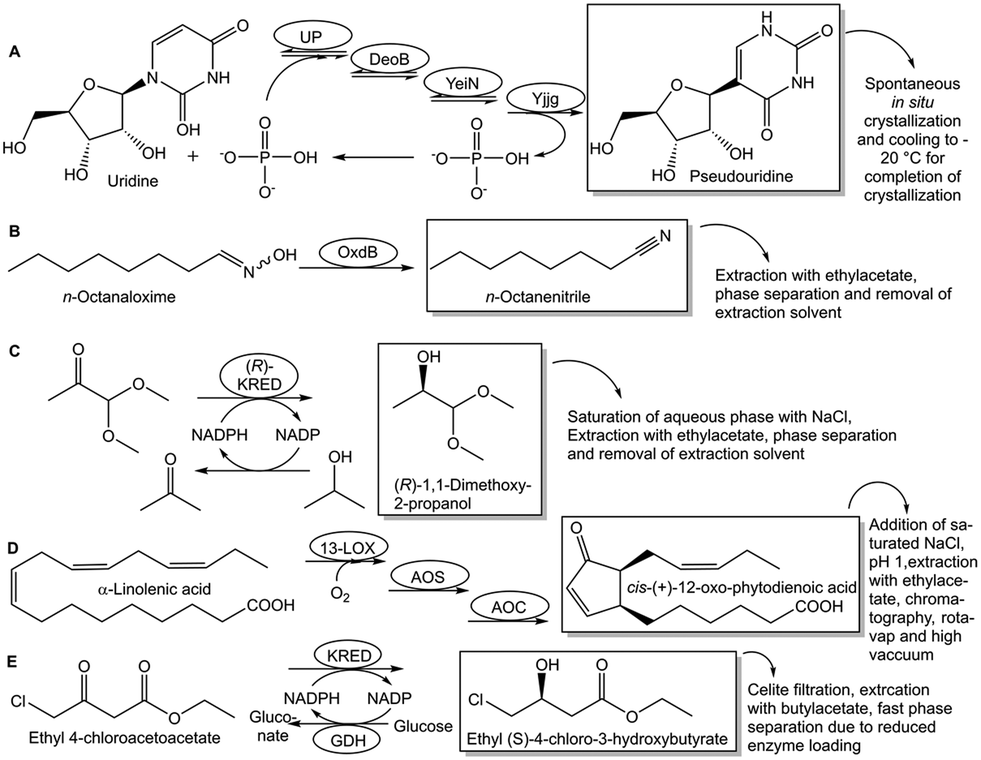 | ||
| Fig. 16 Approaches for overcoming product recovery constraints by simple phase separation. Crystallization from a supersaturated solution of the product is illustrtated by the spontaneous in situ crystallization of pseudouridine, which has been synthesized by biocatayltic conversion of uridine using 4 enzymes in one pot (Fig. 16A). The crystallization has been completed by cooling down to −20 °C. A simple product recovery by a clear phase separation has been achieved for n-octanenitrile after completion of its synthesis by biocatalytic dehydration of n-octanaloxime (Fig. 16B). Ineffective product extraction from aqueous reaction solutions can be overcome by saturating the aqueous phase with sodium chloride, as shown by the complete extraction of (R)-1,1-dimethoxy-2-propanol after completion of the biocatalytic reduction of 1,1-dimethoxy-2-propanone catalyzed by a (R)-selective ketoreductase (Fig. 16C). Adding saturated sodium chloride solution and adapting downstream processing to minimize product degradation has improved product recovery in the whole-cell biocatalytic reaction of α-linoleic acid to the cis-(+)-12-oxo-phytodienoic acid product (Fig. 16D). Product recovery problems and emulsion formation have been overcome by enzyme engineering for higher activity and stability at higher substrate concentrations in the case of the ketoreductase-catalyzed reduction of 4-chloroacetoacetate ethylester to 4-chloro-(3S)-butyrate ethylester (Fig. 16E). | ||
If the bottleneck is the product extraction from an aqueous reaction medium, distribution and partitioning aspects between the phases, emulsion formation and phase separation time need to be considered. Incomplete extraction can be overcome by optimizing the extraction process and repeating the extraction process until all of the product obtained in the biocatalytic conversion is extracted.
An illustration of this type of constraint is the ineffective extraction of the products (R)-1,1-dimethoxy-2-propanol and (S)-1,1-dimethoxy-2-propanol, which have been obtained in two different biocatalytic asymmetric reductions going to full conversions.47 This bottleneck has been overcome by adding sodium chloride for saturating the aqueous reaction solution and performing several extractions so that the pure 1,1-dimethoxy-2-propanol enantiomers could be almost quantitatively recovered (see Fig. 16C) after three extractions.47 An improved work-up process using also saturated sodium chloride solution has been developed for the whole-cell biocatalytic reaction to cis-(+)-12-oxo-phytodienoic acid, which is stereochemically labile, accumulates in cell membranes and has emulsifying properties.88 The improved product recovery procedure (see Fig. 16D) starts with the addition of saturated sodium chloride solution instead of cell separation, and involves then cell disruption by acidification to pH 1, extraction with ethylacetate and rapid drying using a rotary evaporator and high vacuum, in order to avoid undesired product isomerization during long freeze drying.88
Another type of bottleneck can be the formation of emulsions in aqueous–organic systems, due to the high biocatalyst concentrations, which may be overcome by a polishing operation for removing the biocatalysts through filtration, or by decreasing the enzyme concentration using a highly active engineered enzyme. The emulsion formation can significantly hinder and decrease product recovery, as for example in the biocatalytic asymmetric reduction of 4-chloroacetoacetate ethylester to the atorvastatin intermediate 4-chloro-(3S)-butyrate ethylester. Although complete conversion has been achieved in this reduction, both with whole cells or isolated enzyme systems combining a ketoreductase with the glucose/glucosedehydrogenase-system for NADP cofactor regeneration, the product recovery was only 82% for whole cell systems106 and 85% in case of isolated enzymes.107 This was due to problematic product recovery and some emulsion separation after one hour despite complete conversion of the ketoreductase-catalyzed enantioselective reduction of ethyl 4-chloroacetoacetate to ethyl (S)-4-chloro-3-hydroxybutyrate in the initial process.107 This bottleneck was caused by the large loadings of biocatalysts, which were required due to their low activities and which led to emulsion formation and product recovery problems. Enzyme engineering for improving the ketoreductase and glucosedehydrogenase activity and stability while keeping the excellent ketoreductase enantioselectivity has been key for overcoming this bottleneck (see Fig. 16E) by greatly reducing the enzyme concentrations and phase separation time, increasing substrate concentrations, space–time yields and isolated yields.107 The emulsion problems were overcome with the reduced loading of an engineered ketoreductase in the final process leading to phase separation in less than one minute and >95% product recovery yield.107
Outlook
The increasing awareness about the importance of a continuous growth of experimental data on thermodynamic, kinetic and other constraints of biocatalytic conversions, and their standardized description and storage, is very beneficial for a number of advances in different directions of biocatalytic one-step conversions. These range from computational approaches to predict thermodynamic and kinetic parameters, identification of potential bottlenecks to complete biocatalytic conversions at an early stage to the design phase of a biocatalytic conversion. The design principles of nature for achieving highly efficient biocatalytic multi-step conversions in the same reaction space can provide great inspiration for optimized synthetic approaches to overcome bottlenecks along a whole reaction sequence towards complete conversion. The spatial and temporal coupling of multiple biocatalytic reactions in biological cells is also of much interest for developing novel reaction architecture for complete substrate to product conversion. Inventing a novel two-step cascade using engineered ribosyl-1-kinase, uridine phosphorylase, and phosphate recycling mediated by pyruvate oxidase, acetate kinase and catalase, has been key for achieving the quantitative conversion of 5-isobutyryl-D-ribose to ((2R,3S,4R,5R)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)-methylisobutyrate, an intermediate to molnupiravir.108 The design principle that multiple steps in a reaction cascade are catalyzed by a multifunctional biocatalyst, confining the reaction space to the active sites of the enzyme, is also widely distributed in nature.109 The trifunctional propionyl-CoA synthase, which has been shown to isolate the highly reactive, unstable and toxic intermediate acrylyl-CoA from the external environment during the complex and synchronized catalytic reaction mechanism, can be an inspiration for the spatial and temporal control of novel reaction sequences catalyzed by a multifunctional enzyme.110 The multi-functional biocatalyst EneIRED, which catalyzes asymmetric conjugate reduction, imine reduction and reductive amination, has been demonstrated to catalyze the coupling of various α,β-unsaturated carbonyl compounds with amines to chiral amine diastereomers with complete conversion.111 The further exploration of multifunctional biocatalysts and their reaction mechanisms is of much interest towards discovering novel reaction sequences with complete conversion.112Equally important is the attention to limitations in product recovery and a standardized description and storage of experimental data on product properties, such as solubilities, portioning between phases, adsorption.
Guidance and idea generation for designing complete biocatalytic conversions and product recovery is not only important in case of very precious and limited starting materials but also from the perspective of cost- and resource-efficient processes. The development of a similarity-based algorithm for a single-step enzymatic retrosynthesis search offers great opportunities for computer assisted synthesis planning of biocatalytic conversions.113 The extension of experimental data on enzyme functions and kinetics and advances in the predictive power of computational approaches for enzyme functions,114 kinetic parameters115 and machine learning for enzyme engineering116 and their interactions are mutually beneficial for further growth. For the design of complete biocatalytic conversions knowledge of the thermodynamics is key, and the extension of experimental characterization of thermo-dynamic parameters of enzyme-catalyzed reactions will also be beneficial for the predictive power of computational approaches.117 As these thermodynamic data as well as molecular properties of products and substrates are independent of biocatalysis, the expansion of experimental data can benefit from interactions with the chemical sciences. From the proof of feasibility to reaction optimization, reliable quantification of the reaction yield118 is important when keeping in mind from the beginning and throughout development the final goal of complete conversion.
Identifying and overcoming bottlenecks to complete conversions of individual single-step biocatalytic reactions becomes even more important when an increasing number of biocatalytic reactions are performed in a sequence. Bottlenecks in individual reactions or limitations in average yields per reaction step have a decisive influence on final product yields, such as in the D-xylose conversion to 2-ketoglutarate in 5 reaction steps,119 the automated synthesis of complex oligosaccharides using glycosyltransferases,120 or the cell-free synthesis of adenosylcobalamine from 5-aminolevulinic acid in 32 reaction steps.121 Therefore overcoming bottlenecks at each of the biocatalytic reaction steps involved will add up to the overall process improvement, which will be key to the development of economically viable, resource-efficient and sustainable bioprocesses.122
Conclusions
The conversion economy of biocatalytic processes, from biocatalytic single-step to multi-step reactions and total synthesis, and the final product recovery, which determine the isolated yield of a product, are key success factors deserving sufficient attention. The identification and analysis of thermodynamic, kinetic and other constraints of biocatalytic conversions is thereby a starting point for improvements. The growing toolbox of methods how to overcome bottlenecks to complete biocatalytic conversions and complete product recovery can provide useful guidance for selecting the most suitable approaches towards the goal of achieving 100% yield of the isolated pure product for a specific biocatalytic conversion.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Roland Wohlgemuth: conceptualization; formal analysis; methodology; writing – original draft; writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
I would like to thank all my coworkers and cooperation partners.References
- J. B. Hendrickson, J. Am. Chem. Soc., 1975, 97, 5784 CrossRef CAS.
- P. A. Wender and B. L. Miller, Nature, 2009, 460, 197 CrossRef CAS PubMed.
- T. Gaich and P. S. Baran, J. Org. Chem., 2010, 75(14), 4657 CrossRef CAS PubMed.
- P. A. Wender, Nat. Prod. Rep., 2014, 31(4), 433 RSC.
- B. M. Trost, Science, 1991, 254(5037), 1471 CrossRef CAS PubMed.
- P. A. Wender, Tetrahedron, 2013, 69(36), 7529 CrossRef CAS PubMed.
- N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48(16), 2854 CrossRef CAS PubMed.
- T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38(11), 3010 RSC.
- R. Wohlgemuth, Chem. Biochem. Eng. Q., 2017, 31(2), 131–138 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2023, 25(5), 1704 RSC.
- R. Wohlgemuth, New Biotechnol., 2021, 60, 113 CrossRef CAS PubMed.
- A. R. Alcántara, P. Dominguez de Maria, J. A. Littlechild, M. Schürmann, R. A. Sheldon and R. Wohlgemuth, ChemSusChem, 2022, 15(9), e202102709 CrossRef PubMed.
- E. L. Bell, W. Finnigan, S. P. France, A. P. Green, M. A. Hayes, L. J. Hepworth, S. L. Lovelock, H. Niikura, S. Osuna, E. Romero, K. S. Ryan, N. J. Turner and S. L. Flitsch, Nat. Rev. Methods Primers, 2021, 1, 46 CrossRef CAS.
- A. Akbari, J. T. Yurkovich, D. C. Zielinski and B. O. Palsson, Nat. Commun., 2021, 12, 3178 CrossRef CAS PubMed.
- I. S. Metcalfe, B. Ray, C. Dejoie, W. Hu, C. de Leeuwe, C. Dueso, F. R. García-García, C. M. Mak, E. I. Papaioannou, C. R. Thompson and J. S. O. Evans, Nat. Chem., 2019, 11(7), 638–643 CrossRef CAS PubMed.
- National Institute of Standards and Technology (NIST), U.S. Department of Commerce, NIST Chemistry WebBook, NIST Standard Reference Database Number 69, Last update to data: 2023, website: https://webbook.nist.gov/chemistry/.
- R. N. Goldberg, Y. B. Tewari and T. N. Bhat, J. Phys. Chem. Ref. Data, 2007, 36(4), 1347–1397 CrossRef CAS.
- M. E. Beber, M. G. Gollub, D. Mozaffari, K. M. Shebek, A. I. Flamholz, R. Milo and E. Noor, Nucleic Acids Res., 2022, 50, D603–D609 CrossRef CAS PubMed.
- A. Chang, L. Jeske, S. Ulbrich, J. Hofmann, J. Koblitz, I. Schomburg, M. Neumann-Schaal, D. Jahn and D. Schomburg, Nucleic Acids Res., 2021, 49(D1), D498–D508 CrossRef CAS PubMed.
- N. Swainston, A. Baici, B. M. Bakker, A. Cornish-Bowden, P. F. Fitzpatrick, P. Halling, T. S. Leyh, C. O'Donovan, F. M. Raushel, U. Reschel, J. M. Rohwer, S. Schnell, D. Schomburg, K. F. Tipton, M. D. Tsai, H. V. Westerhoff, U. Wittig, R. Wohlgemuth and C. Kettner, FEBS J., 2018, 285(12), 2193 CrossRef CAS PubMed.
- A. S. Bommarius, Annu. Rev. Chem. Biomol. Eng., 2015, 6, 319–345 CrossRef CAS PubMed.
- C. Held and G. Sadowski, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 395–414 CrossRef CAS PubMed.
- G. E. Briggs and J. B. S. Haldane, Biochem. J., 1925, 19(2), 338–339 CrossRef CAS PubMed.
- J. B. S. Haldane, Enzymes, Longmans & Green, London, 1930 Search PubMed.
- E. Noor and W. Liebermeister, Interface Focus, 2024, 14, 20230029 CrossRef PubMed.
- E. Noor, A. Flamholz, W. Liebermeister, A. Bar-Even and R. Milo, FEBS Lett., 2013, 587(17), 2772–2777 CrossRef CAS PubMed.
- R. N. Goldberg, R. T. Giessmann, P. J. Halling, C. Kettner and H. V. Westerhoff, Beilstein J. Org. Chem., 2023, 19, 303–316 CrossRef CAS PubMed.
- Y. B. Tewari, R. N. Goldbergand and J. D. Rozzell, J. Chem. Thermodyn., 2000, 32, 1381–1398 CrossRef CAS.
- P. Tufvesson, J. S. Jensen, W. Kroutil and J. M. Woodley, Biotechnol. Bioeng., 2012, 109(8), 2159–2162 CrossRef CAS PubMed.
- M. Voges, F. Schmidt, D. Wolff, G. Sadowski and C. Held, Fluid Phase Equilib., 2016, 422, 87e98 CrossRef.
- S. J. Novick, N. Dellas, R. Garcia, C. Ching, A. Bautista, D. Homan, O. Alvizo, D. Entwistle, F. Kleinbeck, T. Schlama and T. Ruch, ACS Catal., 2021, 11(6), 3762–3770 CrossRef CAS.
- K. Faber and S. Riva, Synthesis, 1992, 10, 895–910 CrossRef.
- O. Fellechner, M. Blatkiewicz and I. Smirnova, Chem. Ing. Tech., 2019, 91(11), 1522–1543 CrossRef CAS.
- M. A. McDonald, H. Salami, P. R. Harris, C. E. Lagerman, X. Yang, A. S. Bommarius, M. A. Grover and R. W. Rousseau, React. Chem. Eng., 2021, 6, 364–400 RSC.
- T. Nagasawa, C. D. Mathew, J. Mauger and H. Yamada, Appl. Environ. Microbiol., 1988, 54(7), 1766–1769 CrossRef CAS PubMed.
- A. J. Burke, W. R. Birmingham, Y. Zhuo, T. W. Thorpe, B. Zucoloto da Costa, R. Crawshaw, I. Rowles, J. D. Finnigan, C. Young, G. M. Holgate, M. P. Muldowney, S. J. Charnock, S. L. Lovelock, N. J. Turner and A. P. Green, J. Am. Chem. Soc., 2022, 144(9), 3761–3765 CrossRef CAS PubMed.
- D. Hülsewede, L. E. Meyer and J. von Langermann, Chem. – Eur. J., 2019, 25(19), 4871–4884 CrossRef PubMed.
- S. Tiedemann, J. E. Neuburger, A. Gazizova and J. von Langermann, Eur. J. Org. Chem., 2024, 27, e202400068 CrossRef CAS.
- L. Pesci, S. M. Glueck, P. Gurikov, I. Smirnova, K. Faber and A. Liese, FEBS J., 2015, 282(7), 1334–1345 CrossRef CAS PubMed.
- J. Ren, P. Yao, S. Yu, W. Dong, Q. Chen, J. Feng, Q. Wu and D. Zhu, ACS Catal., 2016, 6, 564–567 CrossRef CAS.
- D. Ohde, B. Thomas, P. Bubenheim and A. Liese, Processes, 2023, 12(1), 10 CrossRef.
- T. Stillger, M. Bönitz, M. V. Filho and A. Liese, Chem. Ing. Tech., 2002, 74, 1035–1039 CrossRef CAS.
- G. W. Huisman, J. Liang and A. Krebber, Curr. Opin. Chem. Biol., 2010, 14, 122–129 CrossRef CAS PubMed.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed.
- E. S. Park, J. Y. Dong and J. S. Shin, Org. Biomol. Chem., 2013, 11, 6929–6933 RSC.
- Y. B. Tewari, M. M. Schantz, K. W. Phinney and J. D. Rozzell, J. Chem. Thermodyn., 2005, 37(1), 89–96 CrossRef CAS.
- M. A. K. Vogel, H. Burger, N. Schläger, R. Meier, B. Schönenberger, T. Bisschops and R. Wohlgemuth, React. Chem. Eng., 2016, 1(2), 156–160 RSC.
- R. Wohlgemuth, J. Mol. Catal. B: Enzym., 2009, 61(1–2), 23–29 CrossRef CAS.
- S. R. Marsden, L. Gjonaj, S. J. Eustace and U. Hanefeld, ChemCatChem, 2017, 9(10), 1808–1814 CrossRef CAS PubMed.
- M. Höhne, S. Kühl, K. Robins and U. T. Bornscheuer, ChemBioChem, 2008, 9, 363–365 CrossRef PubMed.
- M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater, S. T. Grosser, H. M. Halsey, G. J. Hughes, J. Jo, L. A. Joyce, J. N. Kolev, J. Liang, K. M. Maloney, B. F. Mann, N. M. Marshall, M. McLaughlin, J. C. Moore, G. S. Murphy, C. C. Nawrat, J. Nazor, S. Novick, N. R. Patel, A. Rodriguez-Granillo, S. A. Robaire, E. C. Sherer, M. D. Truppo, A. M. Whittaker, D. Verma, L. Xiao, Y. Xu and H. Yang, Science, 2019, 366, 1255–1259 CrossRef CAS PubMed.
- R. Abu and J. M. Woodley, ChemCatChem, 2015, 7, 3094–3105 CrossRef CAS.
- L. Wen, K. Huang, M. Wei, J. Meisner, Y. Liu, K. Garner, L. Zang, X. Wang, X. Li, J. Fang, H. Zhang and P. G. Wang, Angew. Chem., Int. Ed., 2015, 54(43), 12654–12658 CrossRef CAS PubMed.
- N. Wagner, A. Bosshart, J. Failmezger, M. Bechtold and S. Panke, Angew. Chem., Int. Ed., 2015, 54(14), 4182–4186 CrossRef CAS PubMed.
- W. W. Cleland, Acc. Chem. Res., 1975, 8(5), 145–151 CrossRef CAS.
- A. Cornish-Bowden, Perspect. Sci., 2015, 4, 3–9 CrossRef.
- J. L. Reymond, Enzyme Assays: High-throughput Screening, Genetic Selection and Fingerprinting, Wiley-VCH Verlag, 2006 Search PubMed.
- C. J. Markin, D. A. Mokhtari, F. Sunden, M. J. Appel, E. Akiva, S. A. Longwell, C. Sabatti, D. Herschlag and P. M. Fordyce, Science, 2021, 373, eabf8761 CrossRef CAS PubMed.
- D. Davidi, E. Noor, W. Liebermeister, A. Bar-Even, A. Flamholz, K. Tummler, U. Barenholz, M. Goldenfeld, T. Shlomi and R. Milo, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(12), 3401–3406 CrossRef CAS PubMed.
- D. Davidi and R. Milo, Curr. Opin. Biotechnol., 2017, 46, 81–89 CrossRef CAS PubMed.
- A. Nilsson, J. Nielsen and B. O. Palsson, Cell Syst., 2017, 5, 538–541 CrossRef CAS PubMed.
- P. Halling, P. F. Fitzpatrick, F. M. Raushel, J. Rohwer, S. Schnell, U. Wittig, R. Wohlgemuth and C. Kettner, Biophys. Chem., 2018, 242, 22–27 CrossRef CAS PubMed.
- D. Heckmann, C. J. Lloyd, N. Mih, Y. Ha, D. C. Zielinski, Z. B. Haiman, A. A. Desouki, M. J. Lercher and B. O. Palsson, Nat. Commun., 2018, 9, 5252 CrossRef CAS PubMed.
- F. Li, L. Yuan, H. Lu, G. Li, Y. Chen, M. K. M. Engqvist, E. J. Kerkhoven and J. Nielsen, Nat. Catal., 2022, 5, 662–672 CrossRef.
- H. Yu, H. Deng, J. He, J. D. Keasling and X. Luo, Nat. Commun., 2023, 14, 8211 CrossRef CAS PubMed.
- N. M. Morato, D. T. Holden and R. G. Cooks, Angew. Chem., Int. Ed., 2020, 59(46), 20459–20464 CrossRef CAS PubMed.
- S. Neun, L. van Vliet, F. Hollfelder and F. Gielen, Anal. Chem., 2022, 94(48), 16701–16710 CrossRef CAS PubMed.
- L. Shen, M. Kohlhaas, J. Enoki, R. Meier, B. Schönenberger, R. Wohlgemuth, R. Kourist, F. Niemeyer, D. van Niekerk, C. Bräsen, J. Niemeyer, J. Snoep and B. Siebers, Nat. Commun., 2020, 11, 1098 CrossRef CAS PubMed.
- M. C. Reed, A. Lieb and H. F. Nijhout, BioEssays, 2010, 32, 422–429 CrossRef CAS PubMed.
- J. H. Ahn, H. Seo, W. Park, J. Seok, J. A. Lee, W. J. Kim, G. B. Kim, K. J. Kim and S. Y. Lee, Nat. Commun., 2020, 11, 1970 CrossRef CAS PubMed.
- M. Tavanti, J. Hosford, R. C. Lloyd and M. J. B. Brown, Green Chem., 2021, 23, 828–837 RSC.
- Y. P. Shang, Q. Chen, A. T. Li, S. Quan, J. H. Xu and H. L. Yu, J. Biotechnol., 2020, 308, 141–147 CrossRef CAS PubMed.
- Y. Ma, N. Liu, P. Greisen, J. Li, K. Qiao, S. Huang and G. Stephanopoulos, Nat. Commun., 2022, 13, 572 CrossRef CAS PubMed.
- C. V. Baldwin, R. Wohlgemuth and J. M. Woodley, Org. Process Res. Dev., 2008, 12(4), 660–665 CrossRef CAS.
- J. H. Sattler, M. Fuchs, F. G. Mutti, B. Grischek, P. Engel, J. Pfeffer, J. M. Woodley and W. Kroutil, Angew. Chem., Int. Ed., 2014, 53, 14153–14157 CrossRef CAS PubMed.
- I. Hilker, M. C. Gutiérrez, R. Furstoss, J. Ward, R. Wohlgemuth and V. Alphand, Nat. Protoc., 2008, 3(3), 546–554 CrossRef CAS PubMed.
- L. G. Lee and G. M. Whitesides, J. Am. Chem. Soc., 1985, 107, 6999–7008 CrossRef CAS.
- G. Matcham, M. Bhatia, W. Lang, C. Lewis, R. Nelson, A. Wang and W. Wu, Chimia, 1999, 53, 584–589 CrossRef CAS.
- B. Huang, Q. Zhao, J. H. Zhou and G. Xu, Appl. Microbiol. Biotechnol., 2019, 103, 2665–2674 CrossRef CAS PubMed.
- J. T. Vicenzi, M. J. Zmijewski, M. R. Reinhard, B. E. Landen, W. L. Muth and P. G. Marler, Enzyme Microb. Technol., 1997, 20, 494–499 CrossRef CAS.
- I. Hilker, R. Wohlgemuth, V. Alphand and R. Furstoss, Biotechnol. Bioeng., 2005, 92(6), 702–710 CrossRef CAS PubMed.
- I. Hilker, V. Alphand, R. Wohlgemuth and R. Furstoss, Adv. Synth. Catal., 2004, 346, 203–214 CrossRef CAS.
- J. Kim, V. Zhang, K. Abe, Y. Qin, D. A. DiRocco, J. P. McMullen, A. Sun, R. Gangam, M. Chow, A. Pitts-McCoy and A. S. Malkani, Org. Process Res. Dev., 2024, 28, 422–431 CrossRef CAS.
- W. L. Cheung-Lee, J. N. Kolev, J. A. McIntosh, A. A. Gil, W. Pan, L. Xiao, J. E. Velásquez, R. Gangam, M. S. Winston, S. Li, K. Abe, E. Alwedi, Z. E. X. Dance, H. Fan, K. Hiraga, J. Kim, B. Kosjek, D. N. Le, N. S. Marzijarani, K. Mattern, J. P. McMullen, K. Narsimhan, A. Vikram, W. Wang, J.-X. Yan, R.-S. Yang, V. Zhang, W. Zhong, D. A. DiRocco, W. J. Morris, G. S. Murphy and K. M. Maloney, Angew. Chem., Int. Ed., 2024, 63(13), e202316133 CrossRef CAS PubMed.
- Y. Qin, K. A. Mattern, V. Zhang, K. Abe, J. Kim, M. Zheng, R. Gangam, A. Kalinin, J. N. Kolev, S. Axnanda, Z. E. X. Dance, U. Ayesa, Y. Ji, S. T. Grosser, E. Appiah-Amponsah and J. P. McMullen, Org. Process Res. Dev., 2024, 28(2), 432–440 CrossRef CAS.
- K. Matsubara, R. Köhling, B. Schönenberger, T. Kouril, D. Esser, C. Bräsen, B. Siebers and R. Wohlgemuth, J. Biotechnol., 2014, 191, 69–77 CrossRef CAS PubMed.
- R. Wohlgemuth, Biocatal. Biotransform., 2008, 26(1–2), 42–48 CrossRef CAS.
- T. L. Guntelmann, K. J. Dietz and H. Gröger, Org. Biomol. Chem., 2024, 22, 5406 RSC.
- X. Lu, J. Li, C. Li, Q. Lou, K. Peng, B. Cai, Y. Liu, Y. Yao, L. Lu, Z. Tian, H. Ma, W. Wang, J. Cheng, X. Guo, H. Jiang and Y. Ma, ACS Catal., 2022, 12(5), 2988–2997 CrossRef CAS.
- H. Zhou, L. Meng, X. Yin, Y. Liu, J. Wu, G. Xu, M. Wu and L. Yang, Appl. Catal., A, 2020, 589, 117239 CrossRef CAS.
- F. H. Arnold, Angew. Chem., Int. Ed., 2019, 58(41), 14420–14426 CrossRef CAS PubMed.
- R. Kumar, M. J. Karmilowicz, D. Burke, M. P. Burns, L. A. Clark, C. G. Connor, E. Cordi, N. M. Do, K. M. Doyle, S. Hoagland, C. A. Lewis, D. Mangan, C. A. Martinez, E. L. McInturff, K. Meldrum, R. Pearson, J. Steflik, A. Rane and J. Weaver, Nat. Catal., 2021, 4, 775–782 CrossRef CAS.
- X. Yi, F. Kleinbeck, C. Ching, L. Boghospor, S. Gomes, O. Alvizo, T. Allmendinger, J. Fell, N. Subramanian, M. Li, R. Garcia, J. Riggins, D. Entwistle, Y. Richter, D. Gschwend, L. Lauener, T. S. Peat, H. Lebhar, T. Schlama and T. Ruch, ACS Catal., 2024, 14(9), 7087–7096 CrossRef CAS.
- A. Hinzmann, S. Glinski, M. Worm and H. Gröger, J. Org. Chem., 2019, 84, 4867–4872 CrossRef CAS PubMed.
- M. M. C. H. van Schie, J.-D. Spöring, M. Bocola, P. Domínguez de María and D. Rother, Green Chem., 2021, 23, 3191 RSC.
- J. R. A. Kincaid, M. J. Wong, N. Akporji, F. Gallou, D. M. Fialho and B. H. Lipshutz, J. Am. Chem. Soc., 2023, 145, 4266–4278 CrossRef CAS PubMed.
- L. Bungaruang, A. Gutmann and B. Nidetzky, Adv. Synth. Catal., 2016, 358, 486–493 CrossRef CAS.
- K. Schmölzer, M. Lemmerer and B. Nidetzky, Biotechnol. Bioeng., 2018, 115(3), 545–556 CrossRef PubMed.
- H. Liu, G. Tegl and B. Nidetzky, Adv. Synth. Catal., 2021, 363, 2157–2169 CrossRef CAS.
- L. Hilterhaus, O. Thum and A. Liese, Org. Process Res. Dev., 2008, 12, 618–625 CrossRef CAS.
- V. Tournier, C. M. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M. L. Desrousseaux, H. Texier, S. Gavalda, M. Cot, E. Guémard, M. Dalibey, J. Nomme, G. Cioci, S. Barbe, M. Chateau, I. André, S. Duquesne and A. Marty, Nature, 2020, 580(7802), 216–219 CrossRef CAS.
- A. Singh, N. A. Rorrer, S. R. Nicholson, E. Erickson, J. S. DesVeaux, A. F. T. Avelino, P. Lamers, A. Bhatt, Y. Zhang, G. Avery, L. Tao, A. R. Pickford, A. C. Carpenter, J. E. McGeehan and G. T. Beckham, Joule, 2021, 5, 2479–2503 CrossRef CAS.
- G. Arnal, J. Anglade, S. Gavalda, V. Tournier, N. Chabot, U. T. Bornscheuer, G. Weber and A. Marty, ACS Catal., 2023, 13, 13156–13166 CrossRef CAS PubMed.
- R. Wohlgemuth, in Comprehensive biotechnology, ed. M. Moo-Young, M. Butler, C. Webb, A. Moreira, B. Grodzinski and Z. F. Cui, Elsevier, Amsterdam, 2nd edn, 2011, vol. 2, pp. 591–601 Search PubMed.
- M. Pfeiffer, A. Ribar and B. Nidetzky, Nat. Commun., 2023, 14, 2261 CrossRef CAS PubMed.
- Z. Q. Liu, J. J. Ye, Z. Y. Shen, H. B. Hong, J. B. Yan, Y. Lin, Z. X. Chen, Y. G. Zheng and Y. C. Shen, Appl. Microbiol. Biotechnol., 2015, 99, 2119–2129 CrossRef CAS PubMed.
- S. K. Ma, J. Gruber, C. Davis, L. Newman, D. Gray, A. Wang, J. Grate, G. W. Huisman and R. A. Sheldon, Green Chem., 2010, 12, 81–86 RSC.
- J. A. McIntosh, T. Benkovics, S. M. Silverman, M. A. Huffman, J. Kong, P. E. Maligres, T. Itoh, H. Yang, D. Verma, W. Pan, H. I. Ho, J. Vroom, A. M. Knight, J. A. Hurtak, A. Klapars, A. Fryszkowska, W. J. Morris, N. A. Strotman, G. S. Murphy, K. M. Maloney and P. S. Fier, ACS Cent. Sci., 2021, 7(12), 1980–1985 CrossRef CAS PubMed.
- C. T. Walsh and B. S. Moore, Angew. Chem., Int. Ed., 2019, 58(21), 6846–6879 CrossRef CAS.
- I. Bernhardsgrütter, B. Vögeli, T. Wagner, D. M. Peter, N. S. Cortina, J. Kahnt, G. Bange, S. Engilberge, E. Girard, F. Riobé, O. Maury, S. Shima, J. Zarzycki and T. J. Erb, Nat. Chem. Biol., 2018, 14, 1127–1132 CrossRef.
- T. W. Thorpe, J. R. Marshall, V. Harawa, R. E. Ruscoe, A. Cuetos, J. D. Finnigan, A. Angelastro, R. S. Heath, F. Parmeggiani, S. J. Charnock, R. M. Howard, R. Kumar, D. S. B. Daniels, G. Grogan and N. J. Turner, Nature, 2022, 604, 86–91 CrossRef CAS.
- T. W. Thorpe, J. R. Marshall and N. J. Turner, J. Am. Chem. Soc., 2024, 146, 7876–7884 CrossRef CAS.
- K. Sankaranarayanan, E. Heid, C. W. Coley, D. Verma, W. H. Green and K. F. Jensen, Chem. Sci., 2022, 13, 6039 RSC.
- G. B. Kim, J. Y. Kim, J. A. Lee, C. J. Norsigian, B. O. Palsson and S. Y. Lee, Nat. Commun., 2023, 14(1), 7370 CrossRef CAS PubMed.
- F. Li, L. Yuan, H. Lu, G. Li, Y. Chen, M. K. Engqvist, E. J. Kerkhoven and J. Nielsen, Nat. Catal., 2022, 5(8), 662–672 CrossRef.
- J. Yang, F.-Z. Li and F. H. Arnold, ACS Cent. Sci., 2024, 10, 226–241 CrossRef CAS PubMed.
- M. E. Beber, M. G. Gollub, D. Mozaffari, K. M. Shebek, A. I. Flamholz, R. Milo and E. Noor, Nucleic Acids Res., 2022, 50(D1), D603–D609 CrossRef CAS PubMed.
- M. A. McDonald, B. A. Koscher, R. B. Canty and K. F. Jensen, Chem. Sci., 2024, 15, 10092 RSC.
- L. Shen, M. Kohlhaas, J. Enoki, R. Meier, B. Schönenberger, R. Wohlgemuth, R. Kourist, F. Niemeyer, D. van Niekerk, C. Bräsen, J. Niemeyer, J. Snoep and B. Siebers, Nat. Commun., 2020, 11, 1098 CrossRef CAS PubMed.
- T. Li, L. Liu, N. Wei, J.-Y. Yang, D. G. Chapla, K. W. Moremen and G.-J. Boons, Nat. Chem., 2019, 11, 229–236 CrossRef CAS PubMed.
- Q. Kang, H. Fang, M. Xiang, K. Xiao, P. Jiang, C. You, S. Y. Lee and D. Zhang, Nat. Commun., 2023, 14, 5177 CrossRef CAS PubMed.
- R. Wohlgemuth, EFB Bioeconomy J., 2021, 1, 100009 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |