
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdding multiple electrons to helicenes: how they respond?
Zheng
Zhou
 ab and
Marina A.
Petrukhina
*a
ab and
Marina A.
Petrukhina
*a
aDepartment of Chemistry, University at Albany, State University of New York, Albany, NY 12222, USA. E-mail: mpetrukhina@albany.edu
bSchool of Materials Science and Engineering, Tongji University, Shanghai 201804, China
First published on 22nd November 2024
Abstract
Helicenes of increasing dimensions and complexity have recently burst into the scene due to their unique structures coupled with interesting chiral, optical, and conducting properties. The helicene-related research has quickly progressed from fundamental curiosity to a diverse range of applications in organic catalysis, optoelectronic devices, chiroptical switches, sensors, and energy storage. The in-depth understanding of electron accepting properties of helicenes should further advance their materials chemistry applications, however, previous reports only relied on spectrocopic and electrochemical studies, while their structural changes weren't extensively discussed. Therefore, we initiated a broad investigation of chemical reduction behaviour of helicenes ranging in size and properties coupled with X-ray diffraction characterization of the reduced products. The responses of helicenes with different structures to the stepwise electron addition were investigated using a combination of X-ray crystallography, spectroscopic methods, and calculations. This study revealed topology- and charge-dependent consequences of chemical reduction ranging from reversible geometry perturbation to irreversible core transformation and site-specific reactivity of helicenes in addition to original alkali metal coordination patterns. This overview is focused on the crystallographically confirmed examples stemming from chemical reduction reactions of different helicenes with alkali metals. The opened discussion should stimulate further exploration of reactivity and complexation of novel π-expanded and heteroatom-doped helicenes based on the revealed structure–property correlations, thus advancing their applications as intriguing new materials.
Introduction
Helicenes are a class of polyaromatic compounds that are comprised of a certain number of consecutive ortho-fused aromatic rings (Scheme 1a).1–3 The sterically induced curvature along the spiral backbone, as described by M- (minus) or P- (plus) configuration, provides helicenes with nonplanarity, inherent chirality, and structural flexibility, as well as tunable chemical and physical properties. Since the pioneering report of helicene synthesis by Meisenheimer in 1903,4 a wide variety of carbohelicenes with different lengths and substituents has been prepared either as racemic or enantioenriched forms.5,6 In the following decades, doping with heteroatoms or combining two or more helicene units enabled the preparation of heterohelicenes (Scheme 1b),7–12 multiple helicenes (Scheme 1c)13–20 and topological helicenes21–25 with modulated helicity and stability/reactivity. Moreover, fusion of additional rings or planar/nonplanar subunits (i.e. pyrene, hexabenzocoronene, and corannulene) to a helicene periphery (Scheme 1d)8,26–39 or backbone (all called π-expanded helicenes, Scheme 1e)40–44 has been broadly used as a strategy to synthesize “hybrid” helicenes with different topological carbon frameworks. Nowadays, helicenes constitute a very extended family of polyaromatic compounds with a diverse range of three-dimensional structures.43,45–54 The development of helicenes has recently grown from a lab curiosity to a practically important field of research, which was triggered by multiple applications of helicenes in fundamental chemistry and materials science, such as organic catalysis, optoelectronic devices, chiroptical switches, sensing applications, energy storage, and more.55–67 | ||
| Scheme 1 Depictions of (a) classical carbohelicenes and selected examples of (b) heterohelicene,1–3 (c) multiple helicene,15 (d) “hybrid” helicene with periphery27 and (e) backbone π-extension.40 | ||
Importantly, the reduction and metalation of helicenes started to emerge as a route to access novel functional molecules and materials for several reasons. First, large π-conjugated helicenes show good electroconductivity and rich redox properties which allows them to serve as good electron acceptors.30,34,36,38,68,69 Second, the spiral nature and core flexibility offer great structural platform for efficient molecular assembly and metal coordination, thus facilitating accommodation of multiple metal ions and also introducing chirality.67,70,71 Third, metal–ligand interactions can be modified by varying the nature of metal centers or functionalization of helicenes, thus providing effective ways to modulate structures and to tune chiroptical and electronic properties.65,72
The chemical reduction of helicenes initiated by Rabinovitz et al. back in 1980s using NMR spectroscopic techniques for studying the smallest helicenes, phenanthrene and its derivatives, revealed disrupted planarity upon a two-electron reduction process.73,74 The above seminal works were followed by a broad investigation of a series of helicenes, revealing the formation of the doubly-reduced products.75 Notably, pure carbo[5]helicene can be reduced to a dianion and further transformed to a new molecule after cyclization,76 while the Cp-fused [5]helicene undergoes a two-step deprotonation to afford a stable dianion.77 For larger [6]helicene and [7]helicene, the degree of helicity in the doubly-reduced species is changed, accompanied by a decreased paratropic effect along the series.78 Recent studies have shown that helicene-like molecules with redox-active functional groups79 or open-shell ground states80 exhibit variable redox processes and intriguing optical and electrochemical properties. These characteristics enable the modulation of aromaticity or spins, making them promising candidates for helical molecular switches.
Despite these interesting results stemming from nuclear magnetic resonance (NMR) or UV-vis absorption monitoring of the reduction behavior in solution, very limited information regarding the structures of the solid-state products has been available, which impeded a better understanding of structural transformations and electronic effects of the reduced species. Over the last decades, only two negatively charged helicenes have been characterized crystallographically by Youngs et al.77 The unsubstituted helicene dianion was formed through cyclization of tribenzocyclyne in the Li-induced reaction. The resulting product has two Li+ ions bound to the opposite sides of different five-membered rings in an η5-fashion (Scheme 2a). In contrast, the Me3Si-substituted helicene was doubly-deprotonated by n-BuLi to form an interesting dimeric complex having two internal Li+ ions sandwiched by two helicene decks in the η5-mode, with the two external Li+ ions being η5-coordinated to the back of the same five-membered rings (Scheme 2b). In both cases, some structural perturbations were observed in the helicene core, including bond alteration and decrease in the helical character. These sparse experimental data on the alkali metal ion binding could have been the limiting factor for the related theoretical investigations. One recent work by Merino et al. can be specifically mentioned here,81 as it pointed out that the alkali metal interactions and stability of complexes are mainly affected by distortion of the helical carbon frameworks, and the metallocene-like structures are favored. In addition, the mobility of alkali metals in complexes was discussed and shown to be metal ion-size dependent and regioselective in different helicenes.
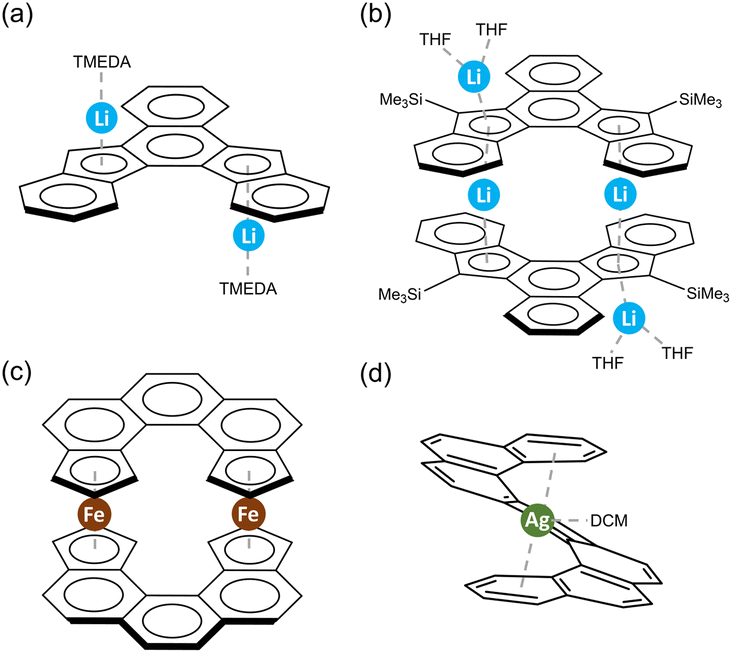 | ||
| Scheme 2 Prior examples of metalation of helicenes with (a and b) alkali metals,77 and (c and d) transition metals.70,82 | ||
In contrast to main group metal complexation, studies of helicenes as π-ligands towards transition metal centers have been dated back to 1970s,82 and the majority of resulting complexes were structurally characterized. Besides the mixed-ligand products, there are mainly two types of helicene-based metal complexes, namely helicenic metallocenes and molecular tweezers.70,83 The η5-helicenic metallocenes are benefited from the embedded cyclopentadienyl (Cp) rings, with a coordination environment similar to that in ferrocene (Scheme 2c), while the η6-helicenic metallocenes are stabilized by the terminal benzene rings. In contrast, the molecular tweezers are only limited to [7]helicene, as two terminal aromatic rings can perfectly sandwich a metal ion (Scheme 2d).70
Although several reviews were dedicated to the metalation of helicenes with main group and transition metals,70,84,85 the structural consequences of electron addition to helicenes and their alkali metal coordination preferences have not been unraveled. Importantly, some recent research shows that helicenes could exhibit advanced performance as an anode material in lithium-ion batteries due to their spiral structures,64,67 thus further emphasizing the importance of understanding their electron accepting and transport behavior. Our recent studies demonstrated remarkable structural deformation and record alkali metal ion intercalation patterns of non-planar molecular nanographenes upon chemical reduction,86,87 both being related to their curved topological carbon frameworks. Therefore, hybrid materials assembled from negatively charged helically twisted nanocarbon π-hosts with intercalated alkali metal ions, which enabled tuning of their electronic, conducting, and other physical properties, should be very attractive targets for their in-depth exploration.
In this review, we summarize the structural and electronic responses of helically twisted polycyclic aromatic hydrocarbons (PAHs) toward multi-electron charging during chemical reduction processes based on the recent X-ray crystallographic and spectroscopic studies. The direct structural comparison of multiple negatively charged states for different helicenes with their original parent frameworks allows us to reveal the outcomes of stepwise electron addition on geometry perturbation or core transformation, as well as on metal binding preferences. This overview of topologically different helicenes should stimulate further discoveries of novel helicene-based metal complexes and the exploration of their performance as advanced conducting and energy-storage materials.
Chemical reduction of mono-helicenes
The successful synthesis of an interesting π-extended PAH consisting of a corannulene bowl fused to a [6]helicene moiety (C76H64, Fig. 1a), has been recently accomplished in Martín group.88 This chiral “hybrid” mono-helicene was selected for the first chemical reduction study with Na metal, which revealed a prompt formation of its doubly-reduced state in solution.89 The resulting product [Na+(18-crown-6)(THF)]2[C76H642−] (Na2-C76H642−) contains an equimolar ratio of the negatively charged P- and M-isomers that are separated from the cationic moiety, as confirmed crystallographically (Fig. 1a). A direct structural comparison with the neutral state allows to evaluate the core response to the two-electron addition. Specifically, the interplanar angle ∠A/F measured at 47.0° in the parent helicene has dramatically enlarged to 80.8° in C76H642−, illustrating a distortion increase of the helicene core upon reduction. The deformation of the C76H64 core is further spread through rings A–F (19.3°–27.7°, torsion angles) and enhanced near rings C and D in the doubly-charged anion (36.6° and 25.6°). Notably, the bowl depth of the corannulene core becomes slightly deeper (0.859(3) Å) in its doubly-reduced form compared to the neutral parent (0.829(4) Å), with a similar trend observed upon reduction of the N-doped bowl.90 The structural deformation upon reduction leads to changes of the electronic structure. Specifically, a broad, red-shifted band and significant downfield shifts of all aromatic signals are observed in the UV-vis absorption and 1H NMR spectra, respectively, indicating charge redistribution over the curved extended π-surface (Fig. 1b). As confirmed by the nucleus-independent chemical shifts (NICS) and anisotropy of induced current density (ACID) calculations,89 an aromaticity reverse was observed on the central five-membered ring (−8.5 ppm to +4.4 ppm) upon two-fold reduction.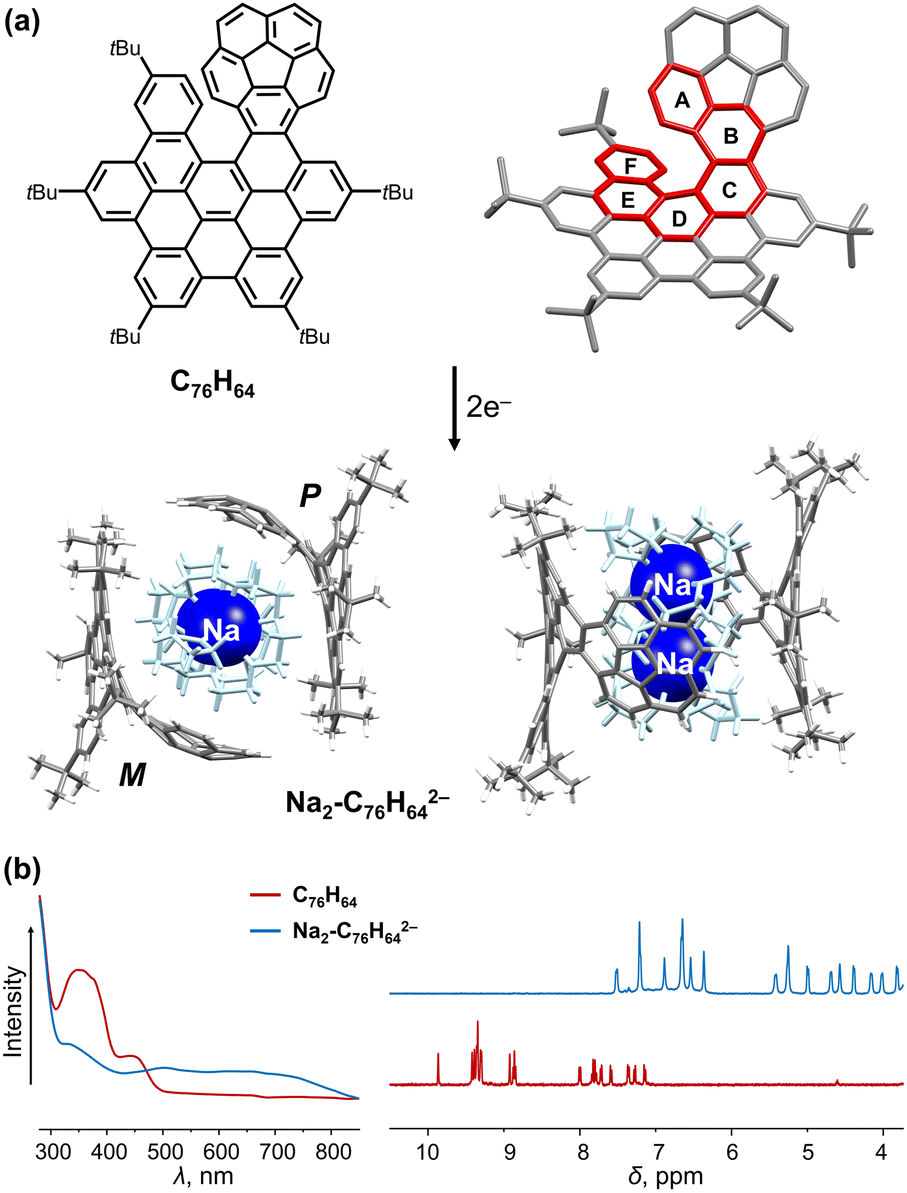 | ||
| Fig. 1 (a) Structure of C76H64 and chemical reduction to afford the dianion with Na+ ions in top and side views. (b) Comparison of UV-vis absorption and 1H NMR spectra of C76H64 and its doubly-reduced product.88,89 | ||
In the solid state, the two isomers with opposite chirality form a cylindrical void – a large internal cavity capable of entrapping a sizable cationic guest, [Na+(18-crown-6)(THF)]2. The resulting host–guest assembly is held by C–H⋯π interactions (2.417(3)–2.743(3) Å) between the complementary cationic and anionic units. Importantly, the C76H642− anion can be reversibly oxidized back to the neutral state, indicating the stability and inherent flexibility of its carbon framework during redox processes.
A great variety of new “hybrid” mono-helicenes featuring π-extended surfaces and intriguing optical properties have been synthesized recently by different research groups,27,30,38 opening this field for further investigations. As helicenes become larger, they tend to exhibit greater electron-accepting abilities, according to electrochemical reduction assessments. Therefore, design and synthesis of novel π-extended helicenes should promote broad exploration of their rich redox properties essential for advancing applications of helicenes as new conducting and energy-storage nanocarbon materials.
Chemical reduction of double-helicenes
Double-helicenes, particularly those with n > 4, can exist in racemic (PP and MM) or meso forms (PM = MP). The twisted double-helicenes are racemic with an intermolecular mirror plane or glide plane found in the crystal lattice, although the chiral forms can be crystallized separately in some rare cases.91 In contrast, the meso double-helicenes (PM = MP) with an intramolecular inversion center are commonly observed. We have investigated both types of double-helicenes to demonstrate their different structural responses to electron addition.We started with investigating the OBO-fused double [5]helicene, C30H16B2O4 (Fig. 2a), a heteroatom-doped double-helicene reported by Müllen group92 and Hatakeyama group93 in the same year. This helicene contains two embedded OBO sites and exhibits excellent stability with a pair of enantiomers (PP and MM). In contrast, the meso isomer was predicted to be thermally unstable.92 The chemical reduction study with Na and K metals revealed that this double [5]helicene readily serves as a two-electron acceptor.94 The resulting doubly-reduced products have been isolated and structurally characterized by single-crystal X-ray diffraction as a solvent-separated ion product [{Na+(18-crown-6)(THF)2}2(C30H16B2O42−)] (Na2-C30H16B2O42−) and a contact-ion complex [{K+(18-crown-6)(THF)2}[{K+(18-crown-6)}(C30H16B2O42−)]] (K2-C30H16B2O42−, Fig. 2a).94 Notably, the use of two different alkali metals allowed to switch the metal binding on and off in the counterion-dependent crystal structures. In the π-complex, one K+ ion is bound to the central six-membered ring of the dianion in a η4-fashion (K⋯C: 3.108(3)–3.360(3) Å), which is in good agreement with the charge distribution observed in the molecular electrostatic potential (MEP) map of the dianion.94 The two-electron acquisition is accompanied by a significant redistribution of the negative charge which is localized in the central six-membered ring of the helicene. This change notably affects the core geometry, as the C–C bond lengths perpendicular to the A–B axis are shortened in both dianions (red highlights), while those along the axis become elongated (green highlights, Fig. 2b). The additional helicenic core twist is illustrated by an increased dihedral angle (planes A/B) from 21.4° in neutral parent to 24.7° in the “naked” dianion, and the core becomes notably more distorted upon direct K+ ion binding (31.8°). Furthermore, the addition of two electrons to C30H16B2O4 is accompanied by distinctive red shift from 300 and 400 nm to 540 and 770 nm in the UV-vis absorption spectra (Fig. 2c). The high-field shifts of the aromatic proton signals are found in the 1H NMR spectra of C30H16B2O42−, particularly in the fjord regions (4.1 ppm). This effect was attributed to the increased electronegativity of the helicene core, where the aromatic protons of the complexed dianion are slightly more deshielded as a result of direct metal coordination in solution. Importantly, this doubly-reduced helicene can be reversibly oxidized back to the neutral state, suggesting potential applications for redox-driven chiroptical switches.
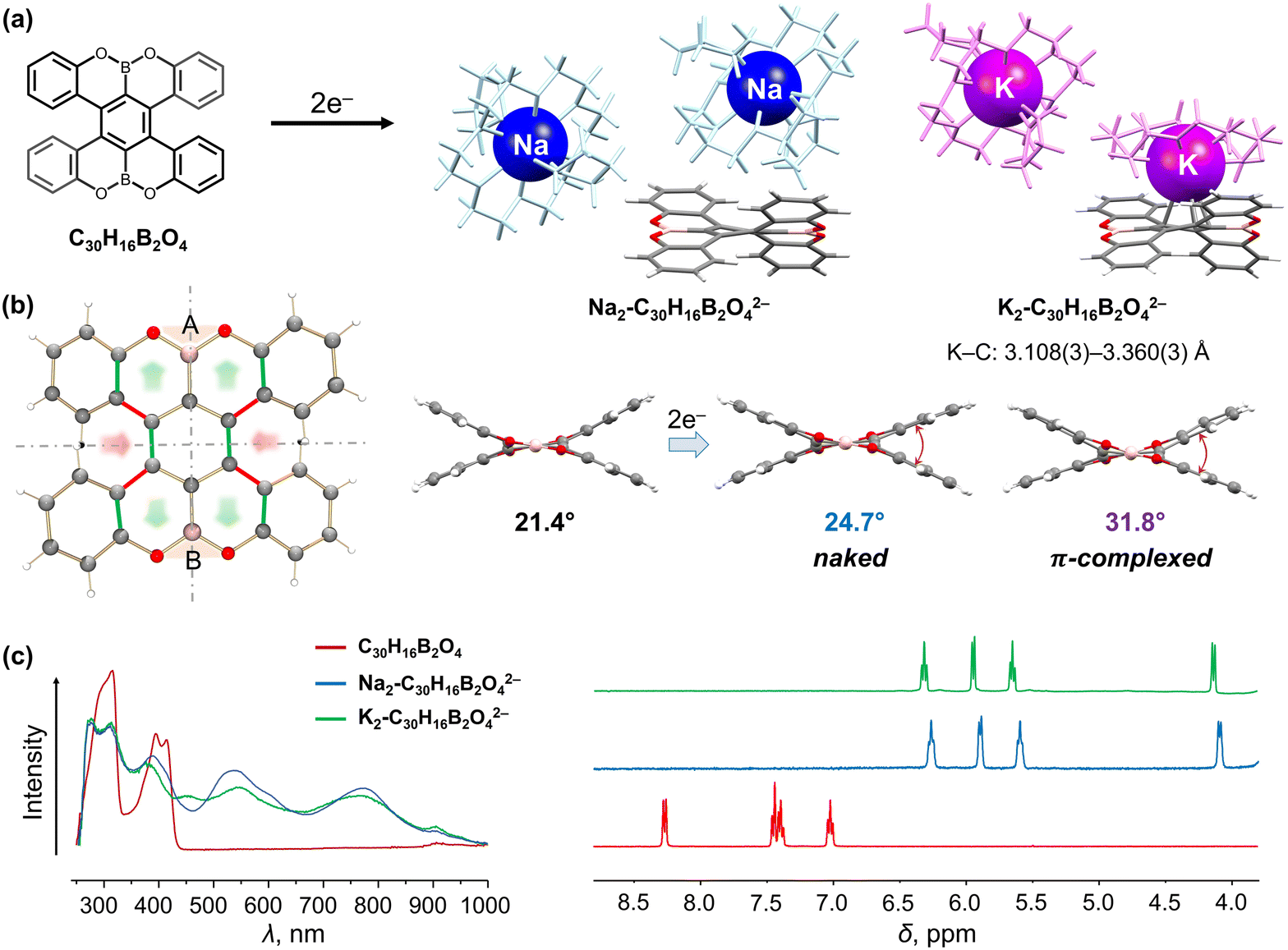 | ||
| Fig. 2 (a) Depiction of C30H16B2O4, chemical reduction to afford its dianions with Na+ and K+ ions. (b) Illustration of structural changes from C30H16B2O4 to its dianions: the bond elongation and shortening is highlighted in green and red, respectively. (c) Comparison of UV-vis absorption and 1H NMR spectra of C30H16B2O4 and its doubly-reduced products.92,94 | ||
In recent years, there has been a substantial increase in both the number and complexity of the heteroatom-doped helicenes.9,10,44,95–97 The inclusion of heteroatoms, such as B, N, S, or O, significantly influences the chemical and electronic structures of the resulting helicenes, leading to interesting fluorescent, light-responsive, electroconductive, or redox properties.10–12,43,44,98 As confirmed through electrochemical studies, complex and doped helicenes, especially those with multiple heteroatoms and π-extended frameworks,44,96 exhibit a pronounced capacity for multi-electron acceptance and storage. Investigation of electron-accepting behavior of various heterohelicenes using chemical reduction methods coupled with crystallographic characterization of the reduced products is a fully open field which should unravel their original responses to multi-electron charging.
As our work94 provided the first structural study of the electron addition effects for double-helicenes, it was then expanded to include a larger double [7]helicene prepared in Müllen group (Fig. 3a).99 Unlike the above double [5]helicene, the selected π-extended double[7]helicene (C66H46, Fig. 3a) exists in the meso form and shows different structural responses to the electron charging. First, double [7]helicene could serve as a multi-electron acceptor undergoing a four-step chemical reduction with alkali metals, accompanied by notable color changes (Fig. 3c).100 For example, the appearance of a green-brown color represents the formation of a transient monoanion (450 and 705 nm), followed by a dark-brown color of the doubly-reduced product (560 nm). The reaction quickly passes through a blueish trianion (469 and 642 nm) to a final purple-brown color (500 nm) corresponding to the tetra-reduced state of C66H46. Second, several products spanning three different reduction states were isolated and characterized by single-crystal X-ray diffraction to reveal the consequences of stepwise electron addition. The doubly-reduced anion was isolated in its “naked” form as a solvent-separated ion product, [{K+(18-crown-6)(THF)}2(C66H462−)] (K2-C66H462−). In addition, the tri- and tetra-reduced states co-crystallized in an equimolar ratio in the mixed-valent product, [{Rb+(18-crown-6)}3(THF)2][{Rb+(18-crown-6)}2(C66H463−)][{Rb+(18-crown-6)}2(C66H464−)] (Fig. 3a).100 In that complex, two Rb+ ions are η4-coordinated to the opposite external benzene rings of the trianion (Rb⋯C, 3.169(5)–3.436(5) Å), while in the tetraanion two Rb+ ions bind to the opposite peripheral benzene rings in an η3-fashion (Rb⋯C, 3.160(5)–3.360(5) Å).
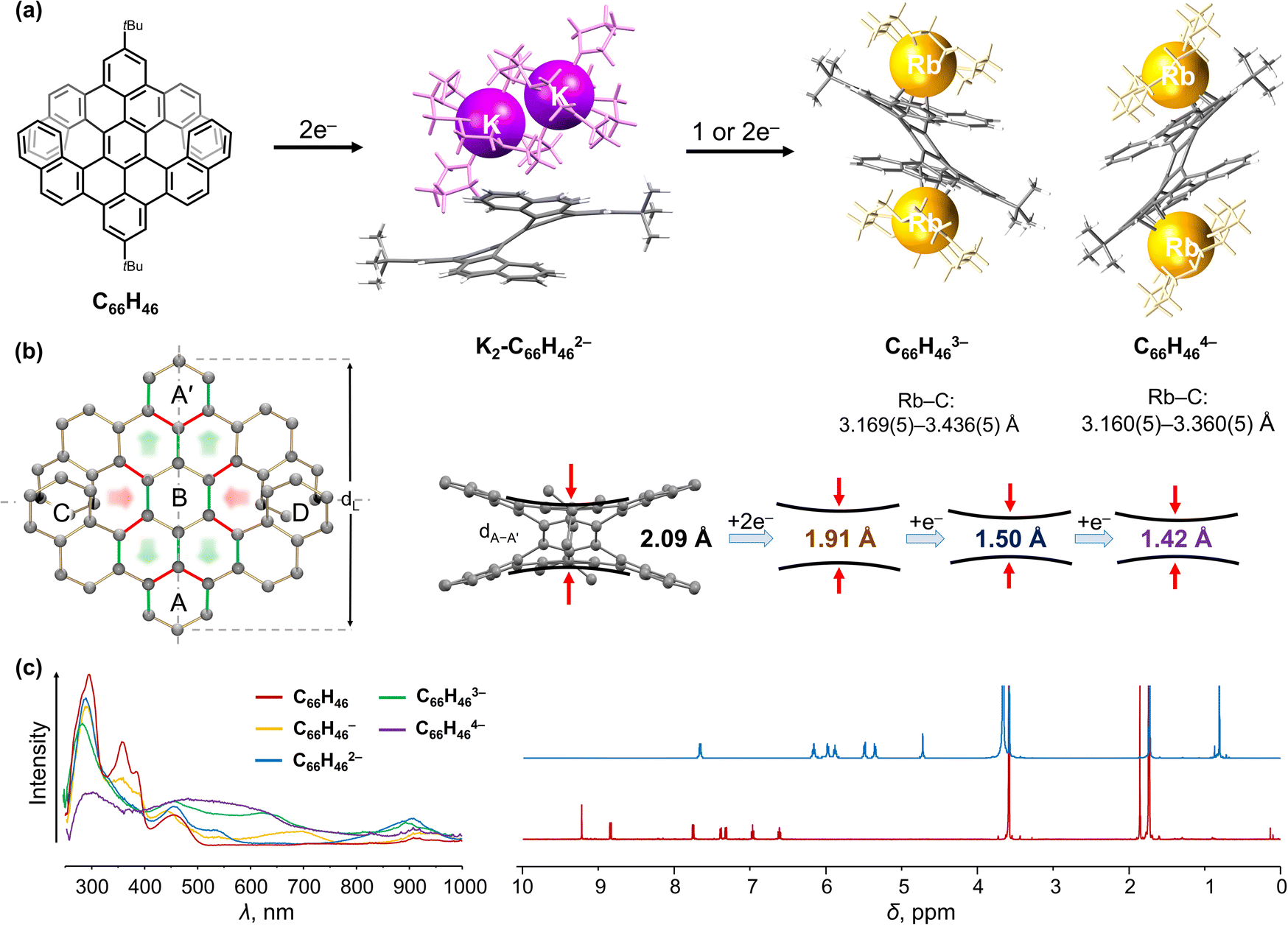 | ||
| Fig. 3 (a) Depiction of C66H46, chemical reduction to afford its dianion with solvent-separated K+ ions and cocrystallized tri-/tetra-anions with coordinated Rb+ ions (additional cationic moieties are omitted). (b) Illustration of structural changes from C66H46 to its anionic forms: the bond elongation and shortening is highlighted in green and red, respectively. (c) Comparison of in situ generated UV-vis absorption spectra of C66H46 and its reduced products, as well as 1H NMR spectra of C66H46 and its doubly-reduced state.99,100 | ||
Upon multi-electron addition, C66H46 exhibits a gradual structural deformation that occurs around the central axis of the helicene core (Fig. 3b). The dihedral angle ∠A/B decreases from 34.3° in the neutral parent to 23.6° in the tetra-reduced state. Notably, the distance between rings A and A′ (dA–A′) (2.090 Å) is shortened in C66H462− (1.913(4) Å) and then further reduced in C66H463− and C66H464− (1.495(13) Å and 1.424(15) Å, respectively). This is accompanied by a slight elongation of the helicene core (dL) from 10.891 Å in C66H46 to 11.222(15) Å in C66H464−. To balance the resulting intramolecular steric hindrance, a helicene core distorts near the peripheral rings C and D with the average of ∠A/C and ∠A/D angles increasing from 16.5° to 20.7°. Notably, the density functional theory (DFT) calculations and 1H NMR spectroscopy supported the deformation observed in the crystal structures.100 In the neutral form, the electrons are mainly localized on rings C and D. In contrast, the negative charge in the reduced products is redistributed to the aromatic core that corresponds to rings A, A′ and B (Fig. 3c), consistent with the largest up-field signal shifts detected in the 1H NMR spectra of C66H462−. Furthermore, the charge distribution in the MEP maps of the triply- and tetra-reduced anions is in agreement with the metal ion binding sites observed in the crystal structure.100 Importantly, the two-electron reduction was proved to be reversible according to the NMR spectroscopic measurements.
The revealed structural flexibility upon reduction and re-oxidation processes points out the potential application of this anti-folded double [7]helicene as an electron-transfer-driven molecular spring. The spring-like behavior of helicenes can potentially furnish functional consequences for their use in electronic, spintronic, piezoelectric and sensing devices. Furthermore, coupling helicity with spin could generate novel responsive and flexible organic materials with interesting magnetic and spintronics applications.
Reactivity of helicenes with ring defects
By doping heteroatoms or fusing π-conjugated subunits, helicenes could gain great stability combined with their spiral geometry. Such helicenes could also exhibit a reversibility of their reduction-induced core changes.94,100 In contrast, introduction of ring defects, mostly five-membered rings, into the helicene framework triggers the reactivity of the carbon core, such as annulation or bond cleavage.76,101Recently, Alabugin group developed a remarkable cascade radical reaction to synthesize π-extended carbon structures.102,103 Starting from alkynes, this approach allowed the preparation of a mono [5]helicene (C31H20, Fig. 4a) and a double [5]helicene (C39H24, Fig. 5a) with an embedded five-membered ring. The presence of the fused five-membered ring introduces a specific deprotonation site in both helicenes, thus enabling the formation of the monoanions prior to chemical reduction. We started the investigation of the racemic mono-helicene which crystallized in a P21/n space group.102 The chemical reaction of the mono-helicene with K and Cs metals in THF proceeds through two distinctive steps (Fig. 4a), and the change of its electronic structures can be monitored by UV-vis absorption spectra (Fig. 4c). As expected, the first step afforded a red solution of the deprotonated product with maximum absorption at 390 and 490 nm, which was isolated and characterized as [{K+(18-crown-6)}(C31H19−)] (K-C31H19−) and [{Cs+(18-crown-6)2}(C31H19−)] (Cs-C31H19−).104 In the crystal structure, the K+ ion binds to a six-membered ring of the C31H19− core in an η2-mode with the K⋯C distances of 3.138(7) and 3.234(7) Å. This coordination site agrees with the MEP map showing the negative charge localization on the five-membered ring and its neighboring six-membered ring. An extra K⋯C contact (3.304(5) Å) found between the K+ ion and the adjacent C31H19− anion results in the propagation of a one-dimensional polymeric structure in the solid state. In contrast, the Cs+ ion in the similar structure remains solvent-separated as {Cs+(18-crown-6)2} to afford a naked C31H19− anion. The loss of proton on the C7 atom (Fig. 4b) leads to a change from sp3- to sp2-hybridization. As a result, the C6–C7 bond becomes significantly shortened to 1.467(7) Å in comparison to the neutral parent (1.527(3) Å). The 1H NMR data reveal a charge redistribution on the mono-helicene core, while it is not significantly affected by metal coordination (Fig. 4c). Upon further reduction, the brownish color of the reaction mixture (λmax = 331 nm) indicates the formation of a new product, which was crystallized as [{K+(18-crown-6)}2(C31H172−)]. As confirmed crystallographically, the dianionic core is transformed via a reductive C–C coupling reaction to form a new planarized radical dianion with g = 2.00127 (Fig. 4c). In the crystal structure, one K+ ion binds to a six-membered ring of C31H172− in an η3-fashion (K⋯C: 3.084(4)–3.383(4) Å), while the second K+ ion is η3-coordinated to the adjacent five-membered ring (K⋯C: 3.057(4)–3.265(5) Å). The geometry of C7 atom remains almost unchanged (169.0° and 1.461(6) Å) compared with the monoanion. The newly formed C22–C27 bond falls in a range of aromatic C–C bonds (1.420(7) Å), resulting in a delocalized π-surface in agreement with the MEP map calculation.104
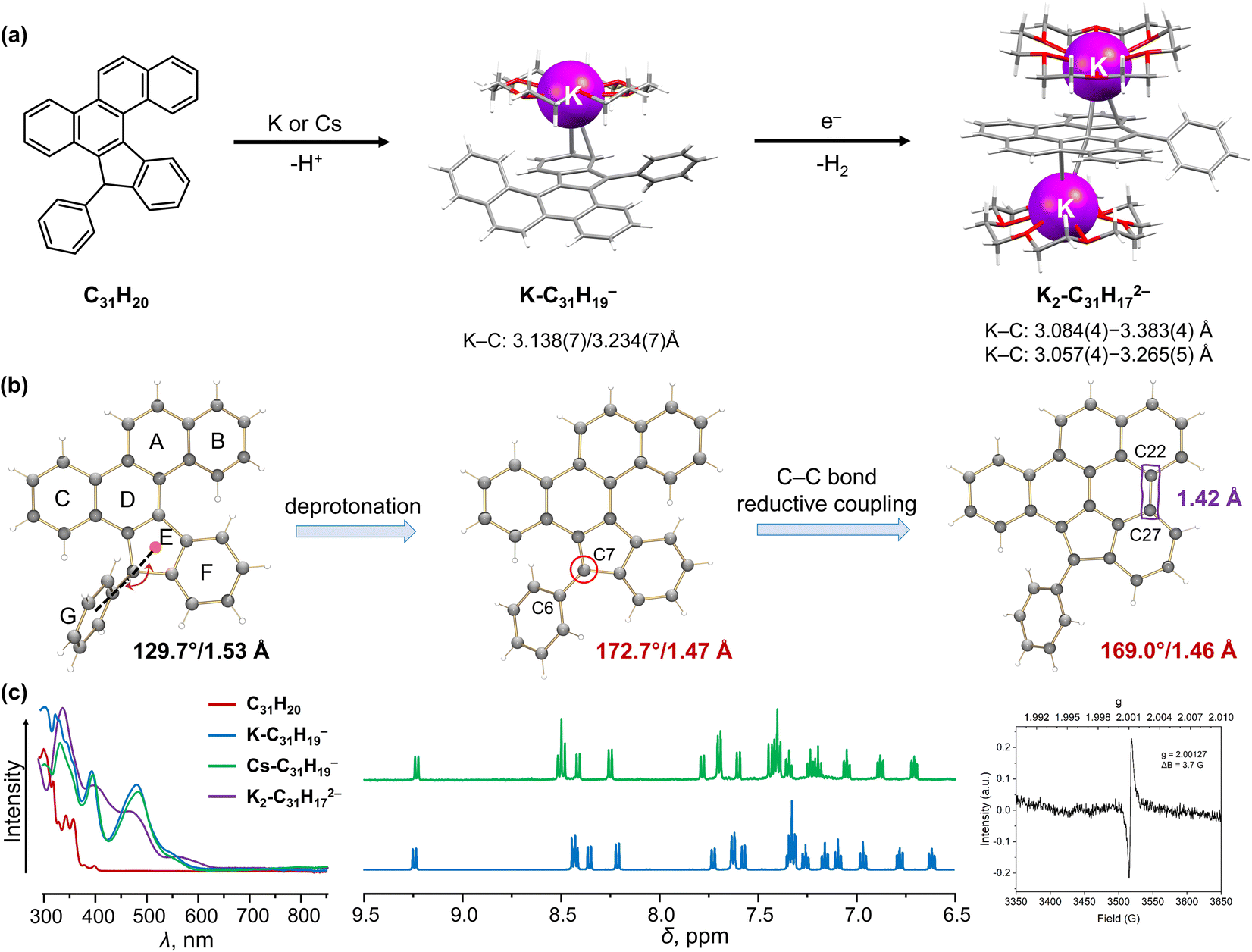 | ||
| Fig. 4 (a) Depiction of C31H20, reaction scheme to afford the deprotonated C31H19− anion and reductively ring-closed C31H172− dianion with coordinated K+ ions. (b) Illustration of structural changes from C31H20 to C31H172−. (c) Comparison of UV-vis absorption and 1H NMR spectra of C39H24 and its anions, along with the EPR spectrum of C31H172−.102,104 | ||
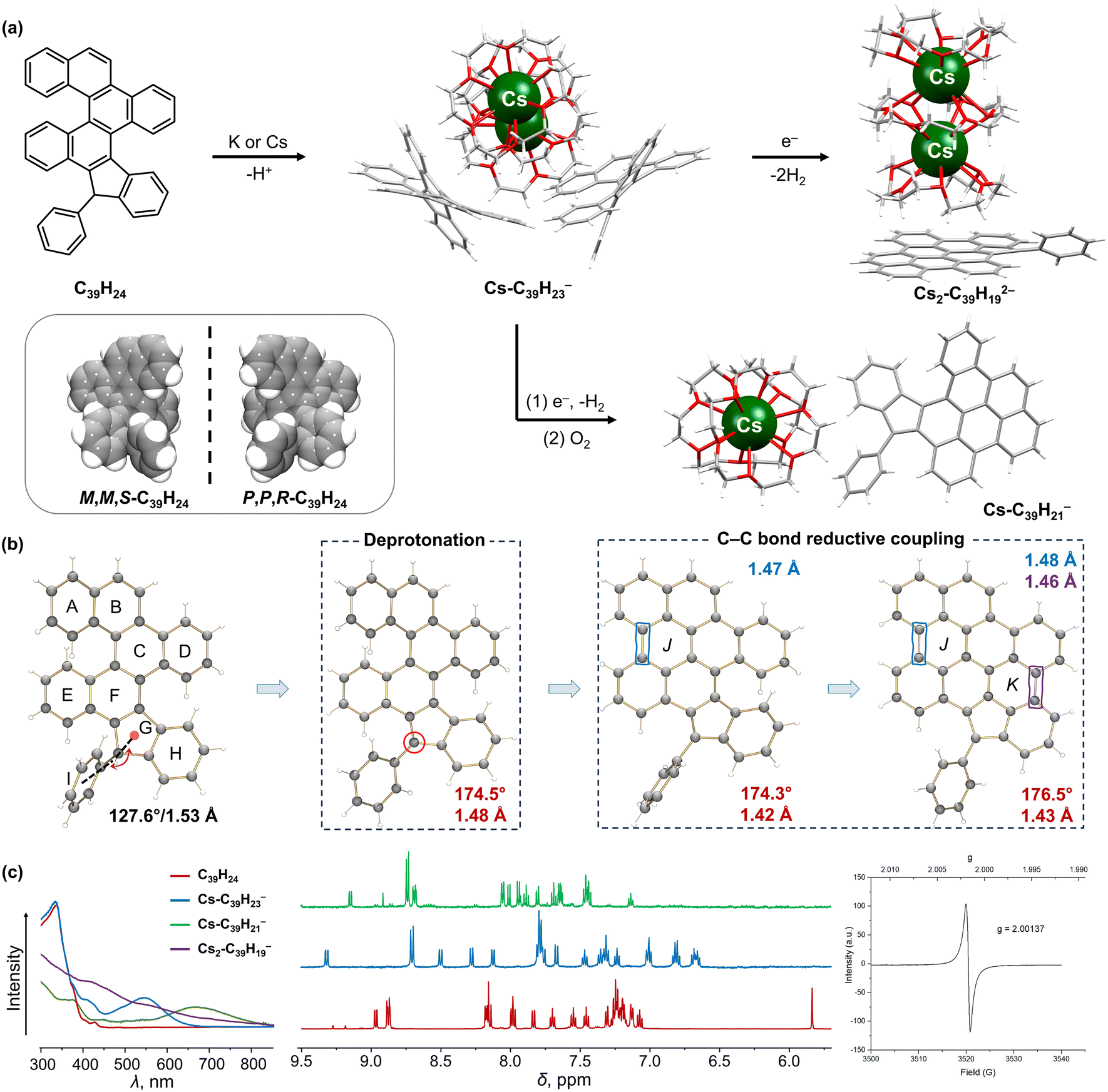 | ||
| Fig. 5 (a) Depiction of C39H24 and its crystallized enantiomers, and reaction scheme to afford the deprotonated C39H23− monoanion, reductively ring-closed C39H192− dianion, and partially ring-closed C39H21− monoanion. (b) Illustration of structural changes from C39H24 to C39H192−. (c) Comparison of UV-vis absorption and 1H NMR spectra of C39H24 and its anions, along with the EPR spectrum of C39H192−.91,102,104 | ||
Next, the reactivity of the double [5]helicene, C39H24, was investigated under similar reaction conditions. Prior to that, crystals of C39H24 of the opposite configurations were isolated and confirmed by X-ray diffraction to belong to the chiral space group P212121, indicating that each crystal contained only one single helicene enantiomer (Fig. 5a).91 Similar to the mono-helicene, the proton of the sp3 C-atom was removed during the first reaction step with Cs metal, affording the red-colored [{Cs2+(18-crown-6)3}(C39H23−)2] product (λmax = 565 nm) lacking direct metal–π interactions. The deprotonation leads to the rearomatization of the π-framework accompanied by deshielding of all aromatic protons, as detected by 1H NMR (Fig. 5c). With an excess of Cs and a prolonged reaction time, the following reduction reaction resulted in the formation of a solvent-separated ion pair [Cs2+(18-crown-6)3][(C39H192−)], with a brown color (431 and 572 nm) and radical behavior (g = 2.00137). Notably, the double helicenic core was further transformed through the double reductive C–C coupling that furnished two additional conjugated rings, leading to the formation of a new C39H192− dianion. As a result, two new C–C bonds were formed C13–C18 and C30–C35 (1.481(13) and 1.456(15) Å, respectively), and the backbone of the new π-extended dianion became nearly planar.
Furthermore, the color change along the reaction pathway indicated the existence of a possible intermediate after the electron injection. The first step of the Cs-induced reaction is deprotonation, accompanied by the appearance of a deep red color, which subsequently turned to red-brown and finally to dark brown. To trap the intermediate, the red-brown reaction solution was briefly exposed to traces of O2 to form a green suspension (λmax = 690 nm), allowing the isolation of a new solvent-separated ion pair, [Cs+(18-crown-6)2][C39H21−] (Fig. 5a).104 Interestingly, the double helicenic core underwent transformation into a mono-helicene through a reductive C–C coupling reaction with the formation of one conjugated six-membered ring based on the newly formed C13–C18 bond (1.47(2) Å). This experimental evidence illustrated that partial oxidation reaction could provide an effective way to transform the relatively unstable transient radical-dianion into a closed-shell monoanion, enabling the entrapment of the reaction intermediate after the first cyclization step. The isolation of this important intermediate allowed to reveal the stepwise reaction mechanism and the first step for π-annulation, and also to experimentally verify the prior mechanistic theoretical predictions.91,104
The X-ray diffraction analysis of the mono- and doubly-annulated products coupled with in-depth DFT calculations91,102,104 demonstrated that reductive cyclization occurs as a consequence of relieving the helicene strain and antiaromaticity, also enabling an increased negative charge delocalization in the final products. In both helicenes, the first step is the deprotonation on the sp3 C-atom to afford the C31H19− or C39H23− anions followed by their reduction. After one electron addition to the mono-helicene, the positive total NICS values of the calculated C31H192− anion and the lower energy of the experimentally observed C31H172− indicate that cyclization occurs after reduction. The creation of a localized antiaromatic hotspot in C31H192− works as a driving force for subsequent relief of antiaromaticity and completion of cyclization process. In contrast, the ring closure in the double-helicene is confirmed as a two-step process, initiated on the remote helicenic site rather than the adjacent one (Fig. 5b). The location of antiaromaticity in the calculated C39H232− anion determines the regioselectivity of the first cyclization step, as confirmed by the crystal structure and 1H NMR of the isolated interrupted C39H21− intermediate.
The ability to confine aromaticity/antiaromaticity to a specific site of the twisted and highly strained π-conjugated framework upon deprotonation/reduction cycles could provide an effective pathway for inducing new core transformations. As a result, this reduction-driven annulation approach could be further explored as a viable alternative to the oxidative Scholl reactions105–110 for the preparation of novel π-extended polyaromatic nanographenes. This approach has been recently expanded to include the stereochemically controlled π-surface annulation of a highly contorted twistacene.111
Reduction-induced hydrogenation of a bilayer helicene
A remarkable example of a giant “hybrid” helicene, with the fusion of two HBC units into a [10]helicene (C138H120), was recently reported by Martín group.27 This rigid and chiral nanographene comprises of two hexa-peri-hexabenzocoronene (HBC) layers, with a nearly aligned AA-stacked conformation and an interlayer distance of 3.6 Å. Unlike the hybrid [6]helicene,89 the two six-membered rings at the helicene backbone exhibit significant deviation from planarity due to the inherent strain, making this highly twisted [10]helicene a very interesting target for reduction studies. The electrochemical study of C138H120 revealed a multi-step oxidation and reduction process, where the oxidized states are relatively stable in line with the DFT calculations,27 but the reduced states tend to be reactive and unstable.112We carried out the first chemical reduction of this large helical bilayer polyarene with K and Rb metals, revealing the formation of a triply-charged species in both cases (Fig. 6). The X-ray diffraction characterization confirmed the isolation of [K+(18-crown-6)(THF)2][{K+(18-crown-6)}2(THF)0.5][C138H1223−] and [Rb+(18-crown-6)2][{Rb+(18-crown-6)}2(C138H1223−)], respectively.112 While the former solvent-separated ion product lacks direct K+ ion binding, in the latter two Rb+ ions wrapped by crown ether directly coordinate to the external surface of the bilayer helicene in the η3-mode (Fig. 6).
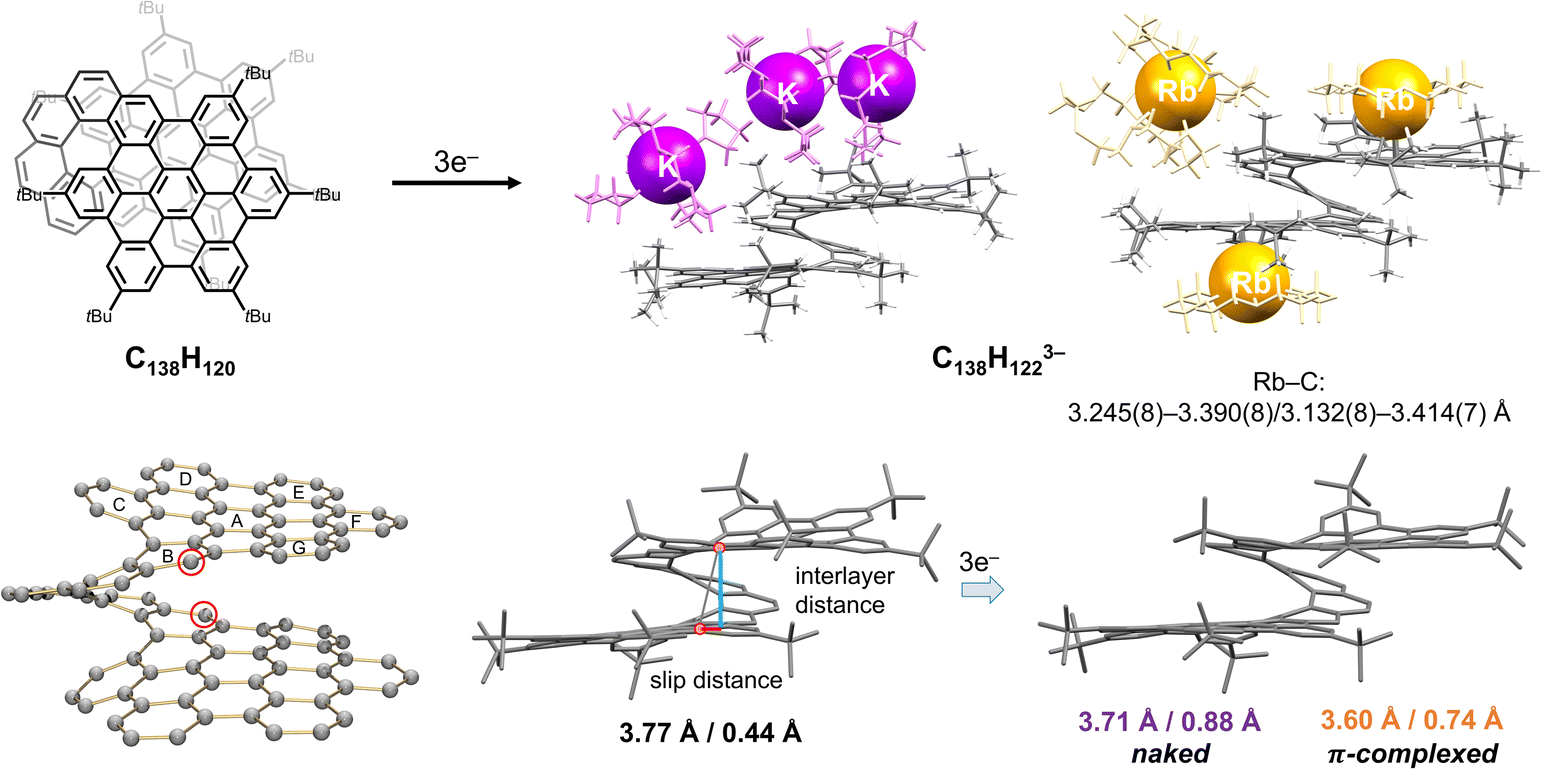 | ||
| Fig. 6 Depiction of C138H120, chemical reduction to afford the C138H1223− anions with K+ and Rb+ ions (naked and complexed, respectively), along with selected geometrical parameters illustrating structural changes.27,105 | ||
A close inspection of carbon backbone revealed the elongation of two C–C bonds with ring distortion on the edge of the [10]helicene core in both trianions coupled with hydrogenation at these C-sites (red circles). The inherent strain of the parent helicene could be the driving force for high propensity of these sites for hydrogenation observed upon reduction. This experimental evidence could now explain the irreversibility of electrochemical reduction.112 Furthermore, the interlayer distance between two HBC units is reduced from 3.774 Å (in neutral parent) to 3.707 Å in the new “naked” trianion isolated with the solvent-separated K+ counterions. The compression is also accompanied by an increase of slippage between two HBC layers from 0.434 Å in C138H120 to 0.884 Å in the doubly-hydrogenated trianion. Moreover, the addition of electrons leads to an increase of nonplanarity in the HBC layer, as illustrated by reduction of the ∠A/B angle from 19.8° in the neutral form to 11.5° in the trianionic species.
In contrast to the K+ ion product with an uncomplexed trianion, the Rb+ ion coordination leads to a less distorted core of the C138H1223− anion. The bilayer structure becomes more compressed (3.603 Å), but the slippage of the HBC blades becomes less evident (0.744 Å). Due to the presence of the two coordinated [Rb+(18-crown-6)] moieties, the ∠A/B angle of 9.2° is reduced compared to that in the K+ product.
The observed site-specificity of hydrogenation was further supported by spin density calculations.112 The first electron added to the LUMO of C138H120 is particularly localized at the carbon atoms on the edge of the helicene backbone of the C138H120− radical-anion. The subsequent hydrogenation/reduction/hydrogenation reactions occur preferentially at these positions leading to the hydrogenated C138H121− and C138H1222− intermediates. After further reduction to the C138H1223− anion, the additional unpaired electron becomes delocalized on the periphery of both HBC blades, thus stabilizing the doubly-hydrogenated triply-reduced species and precluding its further reactivity.
The reduction-induced hydrogenation detected in this system reveals the enhanced chemical reactivity of highly strained π-extended helicenes and opens further studies of their site-specific chemical reactions. The relief of core strain can be used as a tool for directing the functional group placement and for further modulation of properties of helicenic nanocarbon frameworks. As no prior crystallographically confirmed examples of helicene complexes with heavy alkali metals have been reported, the isolated complexes open a new chapter in the organometallic and supramolecular chemistry of strained and helically twisted molecular nanographenes.
Concluding remarks
In summary, we provided the first overview of the structural and electronic responses of various helicenes to multi-electron charging during chemical reduction processes, using the crystallographically characterized products. These examples remain numbered due to difficulties in isolation of the resulting highly reactive and air-sensitive crystalline materials. However, the data reveal a fascinating variation of original core responses of helicenes to gradual electron addition. The direct structural comparison of different reduced states allowed evaluation of the stepwise electron acquisition effect on geometry perturbation or core transformation of helicenes, including their alkali metal coordination preferences. While smaller [5]- and [6]helicenes readily accept up to two electrons, larger [7]- and [10]helicenes can uptake multiple electrons and be isolated in their highly reduced states as alkali-metal complexed products. To accommodate multiple electrons, the helical frameworks built of six-membered rings undergo significant structural deformations and can do that fully reversibly. In contrast, an edge-fused five-membered ring introduces a localized antiaromatic hotspot upon deprotonation/reduction that can facilitate π-annulation reactions, accompanied by strain relief and better negative charge delocalization. Furthermore, imposing a significant in-built strain into a twisted helicene core could facilitate site-specific reactivity which can be utilized for controlled functionalization. The fusion of different π-extended subunits, various heteroatoms, or multiple ring defects into helicenic platforms is rapidly expanding to afford novel helically twisted nanographenes with diverse topologies and intriguing chemical and physical properties. This should provide unique contorted nanocarbons to further explore the role of topology and inherent strain on structural responses and reactivity of helicenes upon electron charging. Furthermore, incorporation of redox-active helicenic building blocks into 2D and 3D covalent- and metal–organic frameworks could provide access to a diverse range of new hybrid and flexible materials with intriguing electronic and structural features. This overview is poised to inspire further discoveries of novel helicene-based π-systems and exploration of their compelling applications as new responsive spin and redox active materials.Data availability
The data that are discussed in this Perspective are available in the ESI of the corresponding articles referenced. All crystallographic data have been deposited at the Cambridge Crystallographic Data Center. These data can be obtained free of charge viahttps://www.ccdc.cam.ac.uk/data_request/cif.Author contributions
Z. Z. and M. A. P. both contributed to the manuscript writing. All illustrations were completed by Z. Z., M. A. P. initiated and supervised this project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of this work from the U. S. National Science Foundation, CHE-2003411 and CHE-2404031, is gratefully acknowledged by M. A. P. We also thank Dr Zheng Wei (UAlbany) for his assistance with X-ray diffraction experiments and ChemMatCARS Sector 15 supported by the National Science Foundation under grant number NSF/CHE-1834750. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. M. A. P. is also very grateful to all collaborators and former group members.Notes and references
- M. Gingras, Chem. Soc. Rev., 2013, 42, 968–1006 RSC.
- M. Gingras, G. Felix and R. Peresutti, Chem. Soc. Rev., 2013, 42, 1007–1050 RSC.
- M. Gingras, Chem. Soc. Rev., 2013, 42, 1051–1095 RSC.
- J. Meisenheimer and K. Witte, Ber. Dtsch. Chem. Ges., 1903, 36, 4153–4164 CrossRef.
- Y. Shen and C.-F. Chen, Chem. Rev., 2012, 112, 1463–1535 CrossRef.
- P. Ravat, Chem.–Eur. J., 2021, 27, 3957–3967 CrossRef PubMed.
- I. Starý and I. G. Stará, Targets Heterocycl. Syst., 2017, 21, 23–53 Search PubMed.
- K. Dhbaibi, L. Favereau and J. Crassous, Chem. Rev., 2019, 119, 8846–8953 CrossRef CAS PubMed.
- P. Liu, X. Bao, J. V. Naubron, S. Chentouf, S. Humbel, N. Vanthuyne, M. Jean, L. Giordano, J. Rodriguez and D. Bonne, J. Am. Chem. Soc., 2020, 142, 16199–16204 CrossRef.
- L. Menduti, C. Baldoli, S. Manetto, M. Bolte, H.-W. Lerner, G. Longhi, C. Villani, E. Licandro and M. Wagner, Angew. Chem., Int. Ed., 2023, 62, e202215468 CrossRef PubMed.
- J.-K. Li, M.-Y. Zhang, L. Zeng, L. Huang and X.-Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202303093 CrossRef.
- W. Zhuang, Y. Liu, Z. Deng, Y. Guo, P. C. Y. Chow, D. L. Phillips, W. Jiang, Z. Wang and J. Liu, Precis. Chem., 2023, 2, 28–39 Search PubMed.
- V. Berezhnaia, M. Roy, N. Vanthuyne, M. Villa, J.-V. Naubron, J. Rodriguez, Y. Coquerel and M. Gingras, J. Am. Chem. Soc., 2017, 139, 18508–18511 CrossRef PubMed.
- Y. Zhu, X. Guo, Y. Li and J. Wang, J. Am. Chem. Soc., 2019, 141, 5511–5517 Search PubMed.
- X. Guo, Z. Yuan, Y. Zhu, Z. Li, R. Huang, Z. Xia, W. Zhang, Y. Li and J. Wang, Angew. Chem., Int. Ed., 2019, 58, 16966–16972 CrossRef PubMed.
- D. Meng, G. Liu, C. Xiao, Y. Shi, L. Zhang, L. Jiang, K. K. Baldridge, Y. Li, J. S. Siegel and Z. Wang, J. Am. Chem. Soc., 2019, 141, 5402–5408 CrossRef PubMed.
- J. Hong, x. Xiao, H. Liu, L. Fu, X.-C. Wang, L. Zhou, X.-Y. Wang, Z. Qiu, X. Cao, A. Narita, K. Muellen and Y. Hu, Chem. Commun., 2021, 57, 5566–5569 RSC.
- X. Xiao, S. K. Pedersen, D. Aranda, J. Yang, R. A. Wiscons, M. Pittelkow, M. L. Steigerwald, F. Santoro, N. J. Schuster and C. Nuckolls, J. Am. Chem. Soc., 2021, 143, 983–991 CrossRef PubMed.
- H. C. Huang, Y. C. Hsieh, P. L. Lee, C. C. Lin, Y. S. Ho, W. K. Shao, C. T. Hsieh, M. J. Cheng and Y. T. Wu, J. Am. Chem. Soc., 2023, 145, 10304–10313 Search PubMed.
- J.-K. Li, X.-Y. Chen, W.-L. Zhao, Y.-L. Guo, Y. Zhang, X.-C. Wang, A. C.-H. Sue, X.-Y. Cao, M. Li, C.-F. Chen and X.-Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202215367 CrossRef PubMed.
- G. Naulet, L. Sturm, A. Robert, P. Dechambenoit, F. Röhricht, R. Herges, H. Bock and F. Durola, Chem. Sci., 2018, 9, 8930–8936 RSC.
- X. Jiang, J. D. Laffoon, D. Chen, S. Pérez-Estrada, A. S. Danis, J. Rodríguez-López, M. A. Garcia-Garibay, J. Zhu and J. S. Moore, J. Am. Chem. Soc., 2020, 142, 6493–6498 CrossRef.
- M. Krzeszewski, H. Ito and K. Itami, J. Am. Chem. Soc., 2021, 144, 862–871 CrossRef.
- Q. Zhou, X. Hou, J. Wang, Y. Ni, W. Fan, Z. Li, X. Wei, K. Li, W. Yuan, Z. Xu, M. Zhu, Y. Zhao, Z. Sun and J. Wu, Angew. Chem., Int. Ed., 2023, 62, e202302266 CrossRef CAS PubMed.
- F. Aribot, A. Merle, P. Dechambenoit, H. Bock, A. Artigas, N. Vanthuyne, Y. Carissan, D. Hagebaum-Reignier, Y. Coquerel and F. Durola, Angew. Chem., Int. Ed., 2023, 62, e202304058 CrossRef CAS.
- C. Li, Y. Yang and Q. Miao, Chem.–Asian J., 2018, 13, 884–894 CrossRef CAS.
- P. J. Evans, J. Ouyang, L. Favereau, J. Crassous, I. Fernandez, J. Perles and N. Martín, Angew. Chem., Int. Ed., 2018, 57, 6774–6779 CrossRef PubMed.
- M. Dračínský, M. Buchta, M. Buděšínský, J. Vacek-Chocholoušová, I. G. Stará, I. Starý and O. L. Malkina, Chem. Sci., 2018, 9, 7437–7446 RSC.
- J. M. Fernández-García, P. J. Evans, S. Filippone, M. Á. Herranz and N. Martín, Acc. Chem. Res., 2019, 52, 1565–1574 CrossRef PubMed.
- M. Milton, N. J. Schuster, D. W. Paley, R. Hernandez Sanchez, F. Ng, M. L. Steigerwald and C. Nuckolls, Chem. Sci., 2019, 10, 1029–1034 RSC.
- J. Urieta-Mora, M. Krug, W. Alex, J. Perles, I. Fernandez, A. Molina-Ontoria, D. M. Guldi and N. Martin, J. Am. Chem. Soc., 2020, 142, 4162–4172 CrossRef PubMed.
- Z. Qiu, C.-W. Ju, L. Frédéric, Y. Hu, D. Schollmeyer, G. Pieters, K. Müllen and A. Narita, J. Am. Chem. Soc., 2021, 143, 4661–4667 CrossRef PubMed.
- M. A. Medel, C. M. Cruz, D. Miguel, V. Blanco, S. P. Morcillo and A. G. Campaña, Angew. Chem., Int. Ed., 2021, 60, 22051–22056 Search PubMed.
- J. Liu, M.-G. Rong and J. Wang, Chem.–Asian J., 2021, 16, 1216–1220 Search PubMed.
- Y. Chen, C. Lin, Z. Luo, Z. Yin, H. Shi, Y. Zhu and J. Wang, Angew. Chem., Int. Ed., 2021, 20, 7796–7801 CrossRef PubMed.
- D. Reger, P. Haines, K. Y. Amsharov, J. A. Schmidt, T. Ullrich, S. Bönisch, F. Hampel, A. Görling, J. Nelson, K. E. Jelfs, D. M. Guldi and N. Jux, Angew. Chem., Int. Ed., 2021, 60, 18073–18081 CrossRef PubMed.
- C. Shen, G. Zhang, Y. Ding, N. Yang, F. Gan, J. Crassous and H. Qiu, Nat. Commun., 2021, 12, 2786 CrossRef PubMed.
- P. Izquierdo-García, J. M. Fernández-García, S. Medina Rivero, M. Šámal, J. Rybáček, L. Bednárová, S. Ramírez-Barroso, F. J. Ramírez, R. Rodríguez, J. Perles, D. García-Fresnadillo, J. Crassous, J. Casado, I. G. Stará and N. Martín, J. Am. Chem. Soc., 2023, 145, 11599–11610 CrossRef.
- J. Tan, X. Xu, J. Liu, S. Vasylevskyi, Z. Lin, R. Kabe, Y. Zou, K. Müllen, A. Narita and Y. Hu, Angew. Chem., Int. Ed., 2023, 62, e202218494 CrossRef PubMed.
- G. R. Kiel, S. C. Patel, P. W. Smith, D. S. Levine and T. D. Tilley, J. Am. Chem. Soc., 2017, 139, 18456–18459 CrossRef PubMed.
- G. R. Kiel, A. E. Samkian, A. Nicolay, R. J. Witzke and T. D. Tilley, J. Am. Chem. Soc., 2018, 140, 2450–2454 CrossRef PubMed.
- W. Fan, T. Matsuno, Y. Han, X. Wang, Q. Zhou, H. Isobe and J. Wu, J. Am. Chem. Soc., 2021, 143, 15924–15929 CrossRef.
- S. Oda, B. Kawakami, Y. Yamasaki, R. Matsumoto, M. Yoshioka, D. Fukushima, S. Nakatsuka and T. Hatakeyama, J. Am. Chem. Soc., 2022, 144, 106–112 CrossRef PubMed.
- P. Karak, S. K. Mandal and J. Choudhury, J. Am. Chem. Soc., 2023, 145, 7230–7241 CrossRef PubMed.
- A. Urbano, Angew. Chem., Int. Ed., 2003, 42, 3986–3989 CrossRef.
- M. Ball, Y. Zhong, Y. Wu, C. Schenck, F. Ng, M. Steigerwald, S. Xiao and C. Nuckolls, Acc. Chem. Res., 2015, 48, 267–276 CrossRef PubMed.
- M. Rickhaus, M. Mayor and M. Juríček, Chem. Soc. Rev., 2016, 45, 1542–1556 RSC.
- J. R. Brandt, F. Salerno and M. J. Fuchter, Nat. Rev. Chem, 2017, 1, 0045 CrossRef.
- I. G. Stará and I. Starý, Acc. Chem. Res., 2020, 53, 144–158 CrossRef PubMed.
- L. Chao, I. A. Stepek, K. E. Yamada, H. Ito and K. Itami, Angew. Chem., Int. Ed., 2021, 60, 23508–23532 CrossRef PubMed.
- X.-Y. Wang, J.-K. Li and X.-Y. Chen, Chin. J. Org. Chem., 2021, 41, 4105–4137 CrossRef.
- C. Duan, J. Zhang, J. Xiang, X. Yang and X. Gao, Angew. Chem., Int. Ed., 2022, 61, e202201494 CrossRef PubMed.
- M.-W. Wang, W. Fan, X. Li, Y. Liu, Z. Li, W. Jiang, J. Wu and Z. Wang, ACS Nano, 2023, 17, 20734–20752 CrossRef PubMed.
- F. Morita, Y. Kishida, Y. Sato, H. Sugiyama, M. Abekura, J. Nogami, N. Toriumi, Y. Nagashima, T. Kinoshita, G. Fukuhara, M. Uchiyama, H. Uekusa and K. Tanaka, Nat. Synth., 2024, 3, 774–786 CrossRef CAS.
- T. W. Bell and N. M. Hext, Chem. Soc. Rev., 2004, 33, 589–598 CAS.
- P. Aillard, A. Voituriez and A. Marinetti, Dalton Trans., 2014, 43, 15263–15278 RSC.
- M. J. Narcis and N. Takenaka, Eur. J. Org Chem., 2014, 21–34 CrossRef.
- J. Bosson, J. Gouin and J. Lacour, Chem. Soc. Rev., 2014, 43, 2824–2840 RSC.
- J. Vacek, J. V. Chocholoušová, I. G. Stará, I. Starý and Y. Dubi, Nanoscale, 2015, 7, 8793–8802 RSC.
- H. Isla and J. Crassous, C. R. Chimie, 2016, 19, 39–49 CrossRef.
- V. Kiran, S. P. Mathew, S. R. Cohen, I. Hernández Delgado, J. Lacour and R. Naaman, Adv. Mater., 2016, 28, 1957–1962 CrossRef PubMed.
- O. Stetsovych, P. Mutombo, M. Švec, M. Šámal, J. Nejedlý, I. Císařová, H. Vázquez, M. Moro-Lagares, J. Berger, J. Vacek, I. G. Stará, I. Starý and P. Jelínek, J. Am. Chem. Soc., 2018, 140, 940–946 CrossRef PubMed.
- F. Lei, L. Weibin, S. Yun and C. Chuanfeng, Chin. J. Org. Chem., 2018, 541–554 Search PubMed.
- S. Oda, T. Shimizu, T. Katayama, H. Yoshikawa and T. Hatakeyama, Org. Lett., 2019, 21, 1770–1773 CrossRef PubMed.
- T. Mori, Chem. Rev., 2021, 121, 2373–2412 CrossRef PubMed.
- Y. Liu, Z. Ma, Z. Wang and W. Jiang, J. Am. Chem. Soc., 2022, 144, 11397–11404 CrossRef PubMed.
- V. Kumar, H. J. Bharathkumar, S. D. Dongre, R. Gonnade, K. Krishnamoorthy and S. S. Babu, Angew. Chem., Int. Ed., 2023, 62, e202311657 CrossRef CAS PubMed.
- M. Mamada, R. Nakamura and C. Adachi, Chem. Sci., 2021, 12, 552–558 RSC.
- J. Bergner, J. Borstelmann, L. M. Cavinato, J. P. Fuenzalida-Werner, C. Walla, H. Hinrichs, P. Schulze, F. Rominger, R. D. Costa, A. Dreuw and M. Kivala, Chem.–Eur. J., 2023, e202303336 Search PubMed.
- N. Saleh, C. Shen and J. Crassous, Chem. Sci., 2014, 5, 3680–3694 RSC.
- P. Jiang, A. S. Mikherdov, H. Ito and M. Jin, J. Am. Chem. Soc., 2024, 146, 12463–12472 CrossRef CAS PubMed.
- W.-L. Zhao, M. Li, H.-Y. Lu and C.-F. Chen, Chem. Commun., 2019, 55, 13793–13803 RSC.
- R. Frim, A. Goldblum and M. Rabinovitz, J. Chem. Soc. Perkin Trans. 2, 1992, 267–274 RSC.
- A. Ioffe, A. Ayalon and M. Rabinovitz, J. Chem. Soc. Perkin Trans. 2, 1994, 1115–1116 RSC.
- A. Weitz and M. Rabinovitz, Synth. Met., 1995, 74, 201–205 CrossRef CAS.
- A. Ayalon and M. Rabinovitz, Tetrahedron Lett., 1992, 33, 2395–2398 CrossRef CAS.
- D. Malaba, A. Djebli, L. Chen, E. A. Zarate, C. A. Tessier and W. J. Youngs, Organometallics, 1993, 12, 1266–1276 CrossRef CAS.
- A. Weitz, PhD Thesis, Hebrew University of Jerusalem, 1997 Search PubMed.
- E. Sidler, R. Hein, D. Doellerer and B. L. Feringa, J. Am. Chem. Soc., 2024, 146, 19168–19176 CrossRef CAS.
- Q. Jiang, H. Tang, Y. Peng, Z. Hu and W. Zeng, Chem. Sci., 2024, 15, 10519–10528 RSC.
- J. Barroso, F. Murillo, G. Martínez-Guajardo, F. Ortíz-Chi, S. Pan, M. A. Fernández-Herrera and G. Merino, Chem.–Eur. J., 2018, 24, 11227–11233 CrossRef CAS PubMed.
- T. J. Katz and W. Slusarek, J. Am. Chem. Soc., 1979, 101, 4259–4267 CrossRef CAS.
- M. Akiyama and K. Nozaki, Angew. Chem., Int. Ed., 2017, 56, 2040–2044 CrossRef CAS.
- R. Benshafrut, E. Shabtai, M. Rabinovitz and L. T. Scott, Eur. J. Org Chem., 2000, 1091–1106 CrossRef CAS.
- I. Aprahamian and M. Rabinovitz, in PATAI'S Chemistry of Functional Groups, 2009 Search PubMed.
- A. V. Zabula, S. N. Spisak, A. S. Filatov, A. Yu. Rogachev and M. A. Petrukhina, Acc. Chem. Res., 2018, 51, 1541–1549 CrossRef CAS PubMed.
- Z. Zhou and M. A. Petrukhina, Coord. Chem. Rev., 2023, 486, 215144 CrossRef CAS.
- J. M. Fernandez-Garcia, P. J. Evans, S. Medina Rivero, I. Fernandez, D. Garcia-Fresnadillo, J. Perles, J. Casado and N. Martín, J. Am. Chem. Soc., 2018, 140, 17188–17196 CrossRef CAS PubMed.
- Z. Zhou, Y. Zhu, J. M. Fernández-García, Z. Wei, I. Fernández, M. A. Petrukhina and N. Martín, Chem. Commun., 2022, 58, 5574–5577 RSC.
- Z. Zhou, Z. Wei, Y. Tokimaru, S. Ito, K. Nozaki and M. A. Petrukhina, Angew. Chem., Int. Ed., 2019, 58, 12107–12111 CrossRef PubMed.
- Z. Zhou, R. K. Kawade, Z. Wei, F. Kuriakose, Ö. Üngör, M. Jo, M. Shatruk, R. Gershoni-Poranne, M. A. Petrukhina and I. V. Alabugin, Angew. Chem., Int. Ed., 2020, 59, 1256–1262 CrossRef.
- X.-Y. Wang, A. Narita, W. Zhang, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 9021–9024 CrossRef.
- T. Katayama, S. Nakatsuka, H. Hirai, N. Yasuda, J. Kumar, T. Kawai and T. Hatakeyama, J. Am. Chem. Soc., 2016, 138, 5210–5213 CrossRef.
- Z. Zhou, X.-Y. Wang, Z. Wei, K. Müllen and M. A. Petrukhina, Angew. Chem., Int. Ed., 2019, 58, 14969–14973 CrossRef.
- X. Zhang, F. Rauch, J. Niedens, R. B. da Silva, A. Friedrich, A. Nowak-Król, S. J. Garden and T. B. Marder, J. Am. Chem. Soc., 2022, 144, 22316–22324 CrossRef PubMed.
- J. Hong, X. Xiao, H. Liu, E. Dmitrieva, A. A. Popov, Z. Yu, M.-D. Li, T. Ohto, J. Liu, A. Narita, P. Liu, H. Tada, X.-Y. Cao, X.-Y. Wang, Y. Zou, K. Müllen and Y. Hu, Chem.–Eur. J., 2022, 28, e202202243 CrossRef.
- J. Full, M. J. Wildervanck, C. Dillmann, S. P. Panchal, D. Volland, F. Full, K. Meerholz and A. Nowak-Król, Chem.–Eur. J., 2023, 29, e202302808 CrossRef PubMed.
- M. Schnitzlein, K. Shoyama and F. Würthner, Chem. Sci., 2024, 15, 2984–2989 RSC.
- Y. Hu, X.-Y. Wang, P.-X. Peng, X.-C. Wang, X.-Y. Cao, X. Feng, K. Müllen and A. Narita, Angew. Chem., Int. Ed., 2017, 56, 3374–3378 CrossRef.
- Z. Zhou, L. Fu, Y. Hu, X. Y. Wang, Z. Wei, A. Narita, K. Müllen and M. A. Petrukhina, Angew. Chem., Int. Ed., 2020, 59, 15923–15927 CrossRef CAS PubMed.
- R. C. Dougherty, J. Am. Chem. Soc., 1968, 90, 5788–5793 CrossRef CAS.
- K. Pati, G. dos Passos Gomes, T. Harris, A. Hughes, H. Phan, T. Banerjee, K. Hanson and I. V. Alabugin, J. Am. Chem. Soc., 2015, 137, 1165–1180 CrossRef CAS.
- I. V. Alabugin and E. Gonzalez-Rodriguez, Acc. Chem. Res., 2018, 51, 1206–1219 CrossRef CAS.
- Z. Zhou, D. T. Egger, C. Hu, M. Pennachio, Z. Wei, R. K. Kawade, Ö. Üngör, R. Gershoni-Poranne, M. A. Petrukhina and I. V. Alabugin, J. Am. Chem. Soc., 2022, 144, 12321–12338 CrossRef CAS PubMed.
- P. Rempala, J. Kroulík and B. T. King, J. Am. Chem. Soc., 2004, 126, 15002–15003 CrossRef CAS.
- P. Rempala, J. Kroulík and B. T. King, J. Org. Chem., 2006, 71, 5067–5081 CrossRef CAS PubMed.
- B. T. King, J. Kroulík, C. R. Robertson, P. Rempala, C. L. Hilton, J. D. Korinek and L. M. Gortari, J. Org. Chem., 2007, 72, 2279–2288 CrossRef CAS PubMed.
- L. Zhai, R. Shukla, S. H. Wadumethrige and R. Rathore, J. Org. Chem., 2010, 75, 4748–4760 CrossRef CAS.
- R. S. Jassas, E. U. Mughal, A. Sadiq, R. I. Alsantali, M. M. Al-Rooqi, N. Naeem, Z. Moussa and S. A. Ahmed, RSC Adv., 2021, 11, 32158–32202 RSC.
- Y. Zhang, S. H. Pun and Q. Miao, Chem. Rev., 2022, 122, 14554–14593 CrossRef CAS PubMed.
- M. Pennachio, Z. Wei, R. G. Clevenger, K. V. Kilway, A. Tsybizova, R. Gershoni-Poranne and M. A. Petrukhina, Org. Chem. Front., 2023, 10, 5823–5833 RSC.
- Z. Zhou, J. M. Fernández-García, Y. Zhu, P. J. Evans, R. Rodríguez, J. Crassous, Z. Wei, I. Fernández, M. A. Petrukhina and N. Martín, Angew. Chem., Int. Ed., 2022, 61, e202115747 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |