
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel model complexes to mimic carbon monoxide dehydrogenase reactions
Changho
Yoo
 *ad,
Jonghoon
Choi
*b and
Yunho
Lee
*c
*ad,
Jonghoon
Choi
*b and
Yunho
Lee
*c
aDepartment of Chemistry, Ulsan National Institute of Science and Technology, Ulsan 44919, Republic of Korea. E-mail: cyoo@unist.ac.kr; Tel: +82 52 217 2694
bDepartment of Chemistry Education, Chonnam National University, Gwangju 61186, Republic of Korea. E-mail: jonghoon92@jnu.ac.kr; Tel: +82 62 530 2492
cDepartment of Chemistry, Seoul National University, Seoul 08826, Republic of Korea. E-mail: yunhochem@snu.ac.kr; Tel: +82 2 880 6653
dGraduate School of Carbon Neutrality, Ulsan National Institute of Science and Technology, Ulsan 44919, Republic of Korea
First published on 13th December 2024
Abstract
Biological CO2/CO interconversion catalyzed at the Ni/Fe heterobimetallic active site of anaerobic carbon monoxide dehydrogenases (CODHs) offers important insights for the design of efficient and selective synthetic catalysts for CO2 capture and utilization (CCU). Notably, this organometallic C1 interconversion process is mediated at a three-coordinate nickel site. Extensive research has been conducted to elucidate the redox and structural changes involved in substrate binding and conversion. The CO2-bound structure of CODH, in particular, has inspired many synthetic studies aimed at exploring key questions, concerning the choice of metal, the role of the unique iron (Feu), and the geometry and oxidation states of both Ni and Feu, as well as CO2/CO exchange mechanism. A better understanding of CODH chemistry promises to reveal and uncover fundamental principles for small molecule activation of first-row transition metal complexes. This mini-review focuses on three key aspects: (1) the coordination environment of the Ni centre in CODH, (2) bioinorganic Ni model systems that provide insight into the biological CO2/CO interconversion at the CODH active site, and (3) recent advances in CODH-inspired catalysis for selective CO2-to-CO conversion.
 Changho Yoo | Changho Yoo is an Assistant Professor in the Department of Chemistry at the Ulsan National Institute of Science and Technology (UNIST). He earned his BS and PhD degrees from the Korea Advanced Institute of Science and Technology (KAIST) in 2011 and 2016 under Prof. Yunho Lee. He conducted postdoctoral research with Prof. Alexander J. M. Miller at the University of North Carolina at Chapel Hill (2017–2020). He worked as a senior researcher at the Korea Research Institute of Chemical Technology (2020–2023) before joining UNIST in August 2023. His research focuses on bioinorganic and organometallic chemistry for sustainable catalysis. |
 Jonghoon Choi | Jonghoon Choi received his PhD degree in the department of chemistry at Korea Advanced Institute of Science and Technology (KAIST) under the supervision of Prof. Yunho Lee in 2020. Then, he was a postdoctoral research associate in Seoul National University (2020–2021) in the same group, followed by a second postdoctoral research position at Humboldt Universität zu Berlin (2021–2022) under the supervision of Prof. Christian Limberg. In 2022, Jonghoon Choi began his independent career as an Assistant Professor at Chonnam National University. |
 Yunho Lee | Yunho Lee obtained M.S. and PhD degrees from the Johns Hopkins University at Baltimore under the guidance of professor Kenneth D. Karlin. In 2007, he started postdoctoral work with professor Jonas C. Peters at the Massachusetts Institute of Technology and the California Institute of Technology. In 2010, Yunho returned to Korea and started his independent career at KAIST. In spring 2020, he moved his research group to Seoul and currently he is a professor in the Department of Chemistry, SNU. His primary research interest focuses on the small molecule activation inspired by the bioorganometallic chemistry of various metalloenzymes. |
1 Introduction
Carbon dioxide conversion is a highly active and multidisciplinary research field, essential for establishing a sustainable chemical industry due to CO2 being a major byproduct of energy production and petrochemical processes.1 Moreover, CO2 plays a critical role in the global carbon cycle (GCC), which is significantly impacted by anthropogenic activities. To restore balance to the GCC, it is crucial to control atmospheric CO2 concentration and develop various methods to convert CO2 into useful chemicals without further CO2 emissions.2–5 Catalytic CO2 conversion to value-added chemicals, such as formic acid, cyclic carbonates and polycarbonates, has gained considerable attention in recent years. This approach not only helps to reduce the atmospheric greenhouse gas as part of the carbon capture and utilization (CCU) strategies but also supports the development of a sustainable, carbon-neutral chemical industry.Among the value-added products, carbon monoxide (CO) is one of the most important industrial feedstocks. To efficiently generate CO from CO2, numerous transition metal catalysts have been developed. However, many systems face challenges with selectivity and high operating potentials for electrocatalysis.6–9 In this context, the selective and efficient conversion of CO2 to CO, catalyzed by carbon monoxide dehydrogenase (CODH) enzymes, is particularly appealing, because it offers a blueprint for developing transition metal-based CO2 conversion catalysts.10–13 The enzyme employs nickel in the active site, which is incorporated within an iron–sulfur cluster, and it catalyzes reversible interconversion between CO2 and CO under ambient conditions. According to the recent X-ray crystallographic data, the Ni ion is coordinated by three sulfur donors as a part of the Fe/S cluster.14–19 Considering nickel coordination, two distinct geometries are proposed to form during the CO2 conversion, as illustrated in Fig. 1.
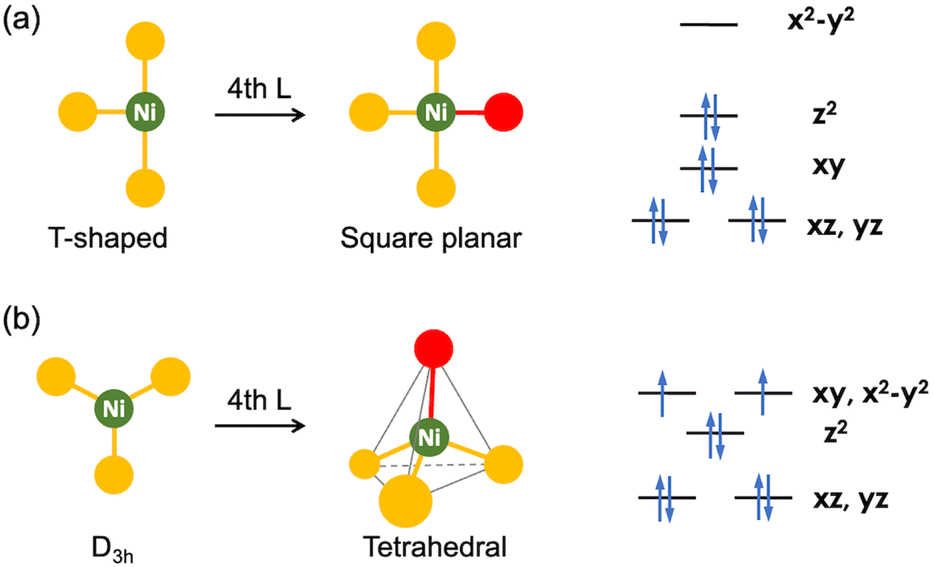 | ||
| Fig. 1 Two distinct Ni structures: (a) T-shaped and (b) D3h, each supported by three donors. Their reaction with a fourth ligand yields a 4-coordinate species (left) and their Ni(II) electronic structures (right). | ||
Ligand binding at a three-coordinate nickel centre results in the formation of either a square planar or tetrahedral nickel species, with each geometry associated with a specific spin state (Fig. 1). The square planar geometry generally results in a ligand field that favours a low-spin (S = 0) ground state of Ni(II), while the tetrahedral geometry favours a high-spin (S = 1) ground state.20,21 While distortions away from these two ideal geometries influence the energy of the frontier orbitals, a more significant distortion is required to observe spin state change.21 In CODH, structural and spectroscopic studies suggest that the nickel centre predominantly adopts a low-spin state throughout the entire catalytic cycle, consistent with the structural data revealing a nearly ideal square planar geometry around Ni.14,22–24 Initial studies, however, proposed the possibility of a tetrahedral, high-spin Ni(II) states.15,24–26 Thus, it is crucial to examine the structures of CO2- and CO-bound Ni species in the different states of CODH to understand how nature facilitates this process. Key questions that arise in understanding CO2 conversion are: (a) how does CODH enable the catalytic reaction to proceed along a low-energy pathway and (b) why are nickel and iron specifically employed for COx conversion (x = 1 or 2). By designing synthetic Ni complexes that mimic the coordination environment of the Ni site and certain COx-bound intermediate species, one can develop organonickel catalysis for selective CO2 conversion.
In this review, we will discuss the geometry of the nickel ion as revealed by CODH X-ray structural data and relate these findings to synthetic model complexes. Nickel catalysis in CODH operates as a part of an Fe/S cluster, a component that plays an essential role in both nickel's geometry and electron transfer (ET) processes and cannot be overlooked. Recent studies have explored the structural modelling of the NiFe4S4 cluster summarized in other review articles.13,27–29 Here, we focus on both structural and functional model compounds, particularly those containing Ni, which bind and mediate the interconversion between CO2 and CO.
2 Nickel coordination in CODH
Nickel is employed by a bifunctional metalloenzyme, known as carbon monoxide dehydrogenase (CODH)/acetyl CoA synthase (ACS), to facilitate biological organometallic reactions.10 These reactions include the interconversion of CO2 to CO at the Ni/Fe site of CODH and C–C and C–S coupling reactions at the single nickel centre in ACS. The X-ray structural data of CODH reported by the Dobbek group reveal that its active site consists of an open cubane Fe–S cluster equipped with a Ni ion, forming a [NiFe4S4] core.16 Two bridging sulfide donors and one cysteine thiolate ligand accommodate a Ni centre, which can form a 4-coordinate nickel species by accepting a fourth ligand, such as CO2 or CO, as illustrated in Fig. 1. The XRD structural data of the intermediate state Cred2-CO2 exhibits a CO2 molecule is coordinated to the Ni ion via a Ni–C bond. One of the O atoms coordinates to the unique Fe center, to give a Ni-μ-CO2-κC:κO-Fe binding mode, as depicted in Fig. 2.14,16 In 2015, an atomic-resolution structure of the CO2-bound state was reported, showing a Ni–C distance of 1.805 Å, the Fe–O distance of 2.030 Å, and the O–C–O angle of 117.2°, indicating its sp2 hybridisation.14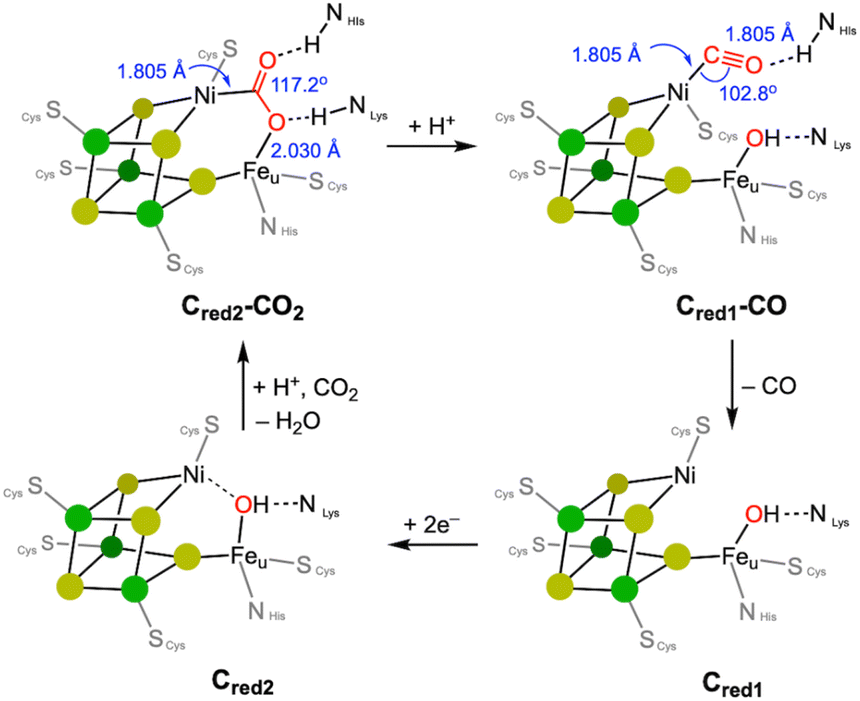 | ||
| Fig. 2 Proposed catalytic cycle of the CODH. Structural parameters are based on the X-ray crystallographic data. | ||
A notable change in the nickel's geometry occurs in the Cred2-CO2 state, which clearly shows a more flattened structure, approaching square planar geometry. Both ∠S–Ni–S of ∼166° and ∠S–Ni–C of 175° are significantly larger than those in the Cred1-CO state; ∠S–Ni–S of 128–144° and ∠S–Ni–C of 121–120°, as shown in Table 1. The corresponding angles for both Cred1 and Cred2 states fall between these states. Enzymes often utilise steric factors and/or secondary coordination sphere, such as H-bonding, to control the metal's geometry, vide infra. And CODH also exhibits H-bonding with bound CO2, CO and OH ligands.10,30 It is difficult, however, to determine whether these geometrical changes are due to the oxidation state changes accordingly affecting its electronic structure, as depicted in Fig. 1 or are influenced by other factors such as H-bonding, as shown in Fig. 2. Nevertheless, this observation suggests that the NiS3 core is fairly flexible and that a planar geometry may play a key role in the CO2 conversion.
| Cred1 | Cred2 | Cred2-CO2 | Cred2-CO2 | Cred1-CO | Cred1-COa | |
|---|---|---|---|---|---|---|
| a Two C-clusters were found over two distinct positions. | ||||||
| d Ni–S [Å] | 2.07 | 2.03 | 2.23 | 2.198 | 2.2 | 2.3, 2.3 |
| 2.14 | 2.16 | 2.09 | 2.329 | 2.3 | 2.3, 2.4 | |
| d Ni–SCys [Å] | 2.17 | 2.17 | 2.10 | 2.111 | 2.3 | 2.3, 2.3 |
| d Ni–C/O [Å] | 2.72 | 2.70 | 1.96 | 1.805 | 2.0 | 1.7 |
| d Ni–Fe [Å] | 2.85 | 2.87 | 2.76 | 3.2 | 3.5/3.6 | |
| d CO [Å] | — | — | 1.25 | 1.298 | 1.2 | 1.2 |
| 1.26 | 1.316 | |||||
| ∠S–Ni–S [deg] | 108.6 | 109.6 | 98.4 | 103.4 | 110.2 | 103.1 |
| ∠S–Ni–SCys [deg] | 93.9 | 93.6 | 93.2 | 90.6 | 90.8 | 91.2 |
| 157.4 | 156.8 | 168.3 | 166.0 | 143.5 | 128.5 | |
| ∠S–Ni–C/O [deg] | 78.8 | 78.7 | 82.3 | 100.8 | 98.6 | 94.3 |
| 151.8 | 148.2 | 171.7 | 174.9 | 125.8 | 120.6 | |
| PDB | 3B53 | 3B51 | 3B52 | 4UDX | 3CF4 | 1OAO |
| Ref. | 16 | 16 | 16 | 14 | 15 | 17 |
The binding mode of CO2 is particularly interesting, as it may be related to the selective CO generation. Generally, a metal hydride species reacts with CO2 to form a metal formate species by generating a strong C–H bond, a well-established mechanism with various transition metal catalysts.31–33 However, an abnormal insertion mechanism, where CO2 inserts into the Ni–H bond to form a nickel hydroxycarbonyl species, has been proposed for the CODH reaction, vide infra.26 In the case of low-valent metal species that do not involve a metal–hydride bond, direct CO2 coordination can occur leading to the formation of a metal–COO species, as observed at the Ni site in CODH. Both nickel(I) and nickel(0) states are considered responsible for CO2 binding, although the reduction potential for the nickel(I/0) couple may be too negative for the biological systems.13,16,34–36 The Cred2 state generated below −500 mV is responsible for reacting with CO2.37 Considering the Ni(II)–Fe(II) ground state, the two-electron reduction of CO2 to form a Ni(II)–COO–Fe(II) species may involve: (a) fast electron transfer from the Fe/S cluster, or (b) activation of a chemical bond such as Ni–H or Ni–Fe.
After CO2 is introduced to the active site, protonation occurs, breaking the C–O bond and forming a nickel(II)–CO species. According to biological studies, the CO ligand should be dissociated from the Ni site at this stage, as the π-back-bonding from a Ni(II) ion is weaker compared to lower oxidation states such as Ni(I) and Ni(0).38–40 Typically, low-coordinate metal–CO species are fairly stable, which can lead to the deactivation or poisoning of metal catalysts.41,42 A notable example is the stability of nickel(0) tetracarbonyl, which plays a key role in the Mond process for producing pure Ni.43 Strong coordination of CO makes it difficult to dissociate from Ni, but it can be weaker when nickel is in its higher oxidation states due to the poor back-bonding. This suggests that a redox process should be coupled with the CO dissociation from Ni, vide infra. The Cred1-CO species generated by addition of CO(g) to the Cred1 state at −100 mV, exhibits five IR vibrational peaks ranging from 1900 to 2074 cm−1, corresponding to Ni–CO stretching vibrations.44 The X-ray crystal structure of CODH shows CO binding to the nickel ion in a bent fashion with the Ni–C–O angle of 103°,15 as shown in Fig. 2. Although it is uncertain if this structure is directly involved in the catalytic cycle, it suggests that hydrogen bonding may promote this unusual bent Ni–CO coordination, which could be crucial for CO release. The multiple CO vibrations and unusual CO binding make it challenging to fully understand the CO and CO2 exchange mechanism at the CODH active site.
3 Ni–CO2 structural models
3.1 Ni–CO2 adducts
The Ni–CO2 model compounds present how CO2 binds at the nickel center. These studies provided valuable structural and spectroscopic information about CO2-bound nickel complexes, as shown in Scheme 1. The first nickel–CO2 complex Ni(PCy3)2(CO2) (1a) was reported by Aresta and coworkers in 1975.45 Analogous Ni(PR3)2(CO2) compounds (1b and 1c) were subsequently reported.46,47 In these complexes, the low-coordinate nickel ion is supported by two P donors, allowing both C and O atoms of CO2 to coordinate to the metal center via an η2 binding mode. Following these initial reports, many other transition metal–CO2 compounds were synthesized and examined.48,49 The elucidation of the CODH active site structures reported in the 2000s further heightened interest in CO2-bound nickel compounds. In the 2010s, nickel–CO2 compounds supported by monodentate (1d) and bidentate (2) alkyl phosphine ligands were reported by the Johnson and the Hillhouse groups, respectively.50,51 More recently, a similar L2Ni(CO2) complex supported by an N-heterocyclic carbene ligand (3) was also reported by the Roesler group in 2021.52 These η2 complexes, in which CO2 binds to the Ni(0) centre, clearly demonstrate activation of the coordinated CO2 moiety (dC–O = 1.22–1.28 Å, νCO2 = 1695–1740 cm−1) compared to the free CO2 molecule (dC–O = 1.16 Å, νCO2 = 2344 cm−1).53,54 Among the L2Ni-η2-CO2 compounds, the degree of CO2 activation varies slightly depending on the electron-donating ability of the supporting ligands, see Table 2. The NHC compound 3 exhibits a higher degree of CO2 activation (dC–O = 1.283(4) Å, νCO2 1695 cm−1) compared to the bisphosphine compounds (dC–O = 1.22–1.27 Å, νCO2 = 1721–1740 cm−1), due to the stronger electron donation of the NHC donor. In 2024, the Limberg group reported a Ni-η2-CO2 complex (4) supported by an anionic β-diketiminate ligand.55 The use of this anionic ligand renders the nickel centre more electron-rich, resulting in the highest level of CO2 activation observed (dC–O = 1.333(3) Å, νCO2 1627 cm−1).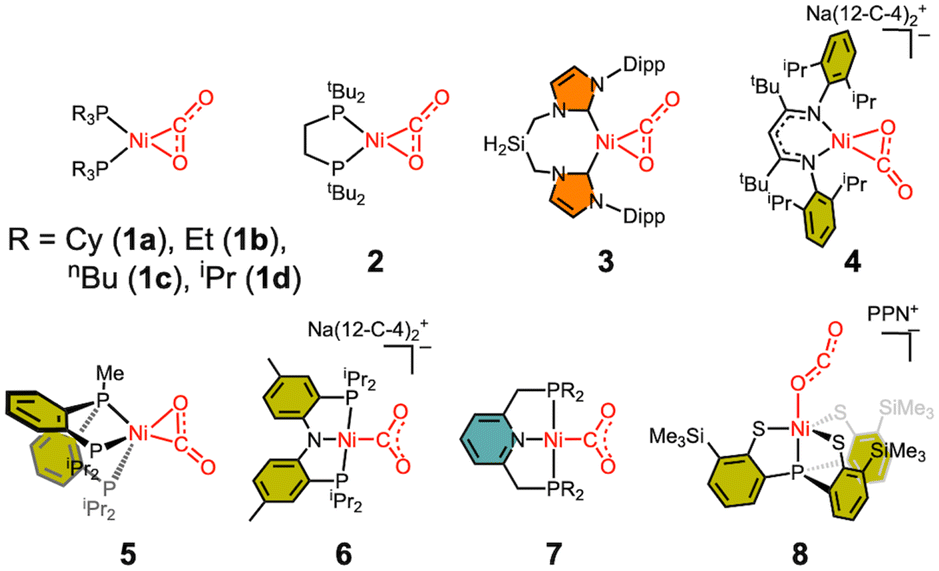 | ||
| Scheme 1 Structurally characterized Ni–CO2 adducts. | ||
| Binding mode | d (Ni–C) | d (Ni–O) | d (C–O) | d (C–O′) | ∠O–C–O′ | ν CO2asym | |
|---|---|---|---|---|---|---|---|
| 1a | η2-κC,O | 1.84 | 1.99 | 1.22 | 1.17 | 133 | 1740 |
| 1d | η2-κC,O | 1.842(3) | 1.932(2) | 1.265(4) | 1.211(4) | 136.7(4) | 1721 |
| 2 | η2-κC,O | 1.868(2) | 1.904(2) | 1.266(3) | 1.200(3) | 138.0(2) | 1724 |
| 3 | η2-κC,O | 1.828(3) | 1.949(2) | 1.283(4) | 1.218(4) | 134.6(3) | 1695 |
| 4 | η2-κC,O | 1.834(2) | 1.924(2) | 1.333(3) | 1.172(3) | 132.1(2) | 1627 |
| 5 | η2-κC,O | 1.904(1) | 2.191(1) | 1.252(2) | 1.218(2) | 135.1(1) | 1682 |
| 6 | η1-κC | 1.911(2) | 2.614(1) | 1.248(2) | 1.247(2) | 128.4(2) | 1620 |
| 7 | η1-κC | 1.950(3) | 2.721(1) | 1.254(3) | 1.244(3) | 129.3(3) | — |
| 8 | η1-κO | — | 2.028(3) | 1.132(6) | 1.240(7) | 171.7(7) | 2177 |
Considering the coordination geometry of the CODH nickel site, the nickel complexes supported by tridentate ligands are particularly intriguing and have been reported relatively recently. In 2014, the Lee group described a Ni–CO2 compound (5) supported by a neutral pincer-type PPP ligand.56 Adding one more donor to the L2Ni core significantly influences CO2 binding and activation. Although 5 adopts an η2 binding mode, the Ni–O bond (2.191(1) Å) is more elongated than in other analogous complexes (1.90–1.99 Å), due to the presence of an additional donor trans to the CO2 ligand. Compound 5 exhibits a higher degree of CO2 activation compared to those of 1–3 as evidenced by a lower νCO2 of 1682 cm−1 and a longer C–O distance of 1.252(2) Å with an O–C–O angle of 135.1°. Computational analysis reveals that its HOMO displays an antibonding character between Ni and O atoms of CO2, supporting its weaker interaction. Interestingly, while all 4-coordinate Ni(CO2) adducts (1–4) show planar structures with two donors and both C and O atoms of CO2 coordinated to Ni, compound 5 exhibits CO2 binding orthogonally to Ni. Because of this orthogonal binding mode, the interaction between Ni and O weakens. This observation suggests that having three sulfur donors as seen in CODH may be critical for optimal CO2 activation. Upon the initial coordination of CO2 to a nickel site, the oxygen atom should be removed from the nickel ion to form a Fe–O bond, as seen in the structure of Cred2-CO2. The addition of a Lewis acid such as borane induces an immediate reaction of 5 forming a Ni(II)-μ-COO-BAr3 adduct, as discussed later.
Further exploration of planar and anionic pincer ligands led to the elongation of a Ni–O bond, resulting in a novel Ni-η1-CO2 binding mode. Our group also reported the first example of a Ni-η1-CO2-κC binding mode by employing a diphosphinoamido (PNP) ligand (6).57 X-ray structural data revealed that 6 does not possess any Ni–OCO2 bond. Supported by the planar PNP ligand, the nickel center adopts a square planar geometry with two anionic N and C donors aligned trans to each other in the xy plane. This configuration leads to repulsive interaction between the filled dπ orbitals of Ni and two O atoms of the bound CO2 ligand. The structural parameters of 6 suggest that the CO2 moiety can be assigned as carbonite (CO22−), a 2e− reduced form of CO2.58 A similar Ni-η1-CO2-κC compound (7) was prepared from the CO2 reaction of a nickel hydride species reported by the Milstein group.59 The metal–ligand cooperation of the lutidine-based PNP ligand is essential for facilitating Ni–C bond formation. The structural parameters of 7 are similar to those in 6, reinforcing the idea that the use of a planar ligand is crucial for enhancing CO2 activation.
As recognized from earlier research involving uranium,60 a rare example of an oxygen-bound Ni-η1-CO2-κO species (8) was reported by the Liaw group in 2016.61 Compound 8, which exhibits a Ni-η1-CO2-κO binding mode, stands out from other examples due to its unique binding mode and relatively low degree of CO2 activation. The O–C–O angle of 171.7(7)° indicates minimal activation of CO2, Table 2. The bond angles and distances of the CO2 moiety are closely aligned with those observed in the free CO2 molecule. Although the XAS study suggests 8 possesses a Ni(III)–(CO2˙−) character, the bond lengths, angle and stretching frequency clearly indicate that CO2 activation is relatively low compared to other Ni–CO2 compounds, likely due to the limited reducing power of Ni(II) in this example.
3.2 Ni–CO2–LA (Lewis acid) adducts
The CO2-bound structure of CODH reveals that the CO2 moiety interacts with both Feu and Lys563 through one O atom, while another interaction occurs with His93, as shown in Fig. 2.14,16 These interactions with Fe and protein residues are believed to contribute significantly to CO2 activation.30,62 The influence of such interactions can be inferred from model studies. To replicate similar interactions with a bound CO2 moiety, nickel complexes with Lewis acids such as proton, borane, alkali cation, and transition metal have been synthesized using various approaches. In 2013, our group firstly reported a nickel hydroxycarbonyl (9a) species, a proton adduct of a nickel CO2 compound, Scheme 2.63 With a PNP ligand, 9a was prepared through CO insertion into the corresponding nickel hydroxyl compound. A similar nickel hydroxycarbonyl species (9b) was also prepared using an acridane-based PNP ligand, showing comparable physical parameters, see Table 3.39 Another nickel hydroxylcarbonyl compound 10 was also prepared by carbonylation reported by the Wendt group in 2018.64 The Schneider group reported the synthesis of 11 from the reaction of a nickel hydride species with CO2 under photochemical conditions, vide infra.65 All hydroxycarbonyl species (9–11) exhibit a Ni(II)–CO22− character.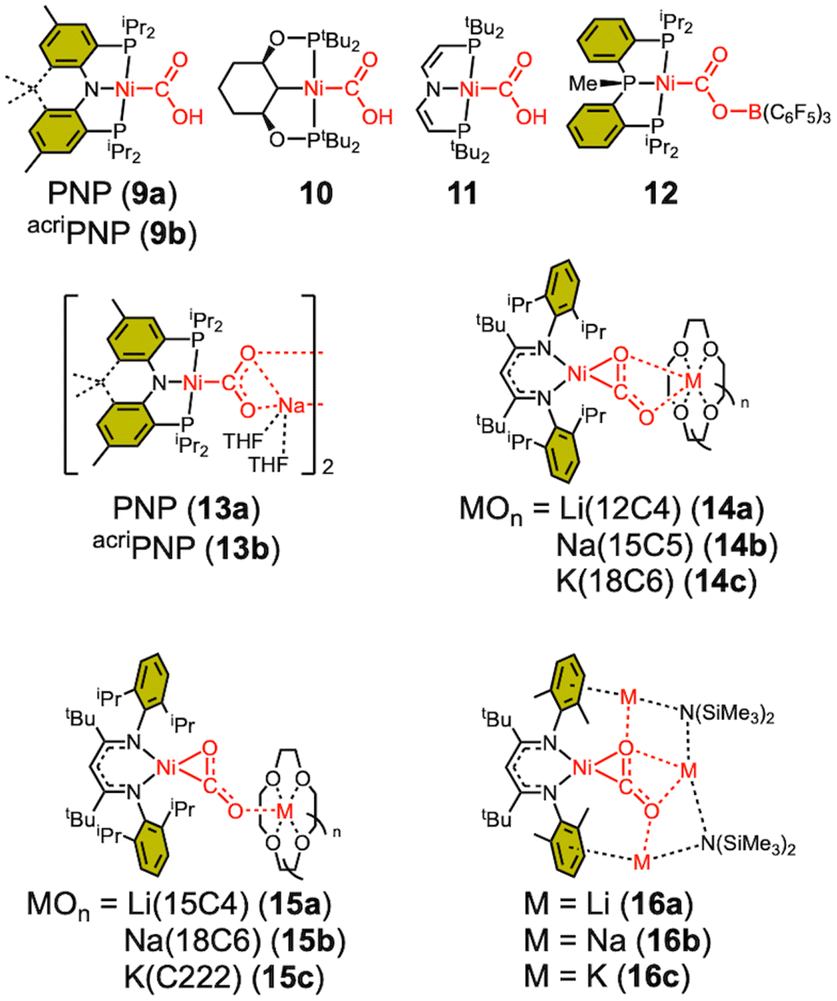 | ||
| Scheme 2 Selected Ni–CO2–LA adducts. | ||
| LA | Binding mode | d (Ni–C) | d (C–O) | d (C–O′) | ∠O–C–O′ | ν CO2asym | |
|---|---|---|---|---|---|---|---|
| 9a | H | μ2-κC:κO | 1.866(2) | 1.313(3) | 1.269(3) | 119.6(2) | 1582 |
| 9b | H | μ2-κC:κO | 1.859(1) | 1.338(2) | 1.244(2) | 119.3(1) | 1579 |
| 10 | H | μ2-κC:κO | 1.899(4) | 1.299(5) | 1.297(5) | 117.5(4) | — |
| 11 | H | μ2-κC:κO | 1.854(2) | 1.299(3) | 1.274(3) | 119.5(2) | 1584 |
| 12 | B | μ2-κC:κO | 1.923(3) | 1.340(4) | 1.223(4) | 122.9(3) | 1639 |
| 13a | Na | μ3-κC:κO,O′:κO | 1.882(1) | 1.271(1) | 1.260(1) | 124.0(1) | ∼1602 |
| 13b | Na | μ3-κC:κO,O′:κO | 1.889(2) | 1.262(2) | 1.261(2) | 123.5(2) | 1533 |
| 14a | Li | μ2-κC,O:κO′:κO′′ | 1.828(4) | 1.291(6) | 1.192(6) | 127.8(5) | 1630 |
| 14b | Na | μ2-κC,O:κO′:κO′ | 1.855(2) | 1.281(3) | 1.209(3) | 133.2(2) | 1600 |
| 14c | K | μ2-κC,O:κO′:κO′ | 1.890(6) | 1.231(9) | 1.22(1) | 144.0(8) | 1621 |
| 15a | Li | μ2-κC,O:κO′:κO′ | 1.799(3) | 1.263(4) | 1.216(4) | 131.6(3) | 1621 |
| 16a | Li | μ4-κC,O:κO,O′:κO:κO | 1.786(4) | 1.275(5) | 1.235(5) | 128.0(4) | 1616 |
As a borane adduct, a Ni–COO–B(C6F5)3 species (12) was prepared from addition of B(C6F5)3 to a Ni-η2-CO2 compound (5), reported in 2015.56 Due to the support from a neutral PPP ligand, the level of CO2 activation is relatively weak compared to other examples, see Table 3. This can be attributed not only to the charge effect (anionic vs. neutral donor as the central moiety) but also to the planarity of the pincer ligand. The PPP ligand is not ideally suited to accommodate a square planar geometry, as evidenced by ∠P–Ni–P of 154.43(4)° in 12. Compound 5 and 12 offer a direct comparison between Ni–CO2 and Ni–CO2–LA adducts. The incorporation of Lewis acidic borane significantly alters the degree of CO2 activation. Specifically, the C–O distance is elongated from 1.252(2) Å to 1.340(4) Å and νCO2 values shift from 1682 cm−1 to 1639 cm−1. These changes indicate that upon addition of borane, the nickel-bound CO2 is clearly converted to carbonite (CO22−), as shown in Fig. 3. The negative charge on the oxygen atom is stabilized by the Lewis acidic borane, resulting in a shift in the binding mode from η2 to μ2-κC:κO. This transformation highlights how both structural changes and the Lewis acid effect can induce a two-electron transfer from Ni(0) to the bound CO2. As anticipated, structural change to a square planar geometry increases the energy of the dx2−y2 orbital. If its energy surpasses that of π* orbital of CO2, carbonite formation occurs, as depicted in Fig. 3. Similar nickel carbonite adducts with alkali metal cations are also known. Our group reported sodium nickel-carbonite adducts (13a and 13b) through the deprotonation of corresponding hydroxycarbonyl species.39,57,63 Interestingly, the Limberg group recently reported a series of Ni–CO2–LA adducts with Li, Na and K ions in the presence of polyether co-ligands (14 and 15), synthesized via deprotonation of corresponding formate species.55,66,67Table 3 summarises the physical parameters for selected Ni–CO2–LA adducts.
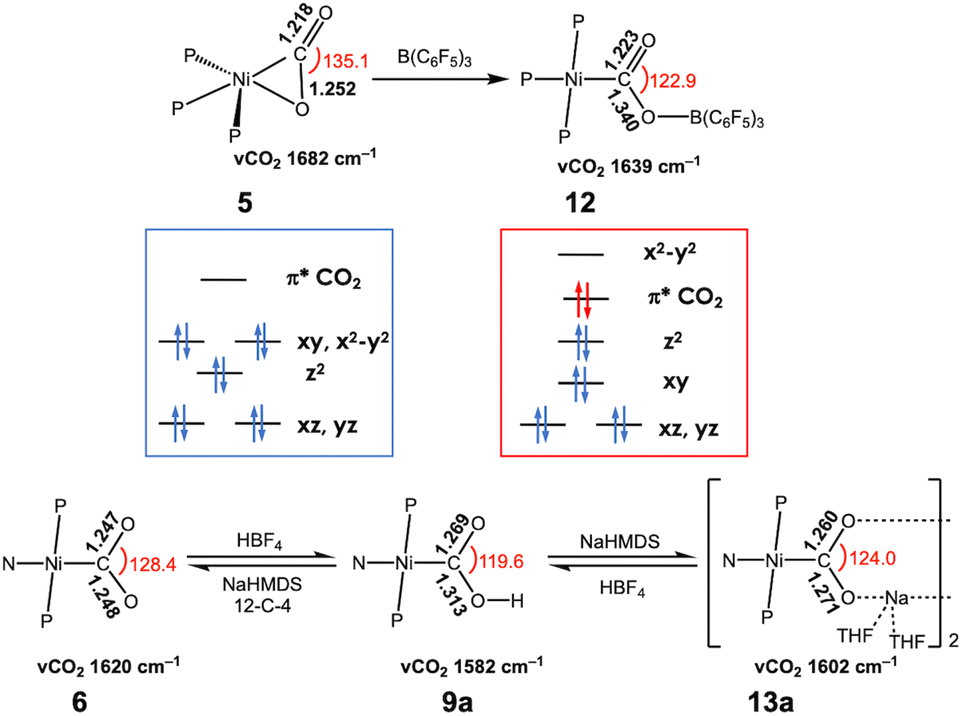 | ||
| Fig. 3 Comparison of Ni–CO2 and Ni–CO2–LA adducts. | ||
A comparison between 6, 9a, and 13a further highlights the influence of proton and alkali metal cation on CO2 activation, as shown in Fig. 3. The addition of a proton notably increases the C–O distance of the protonated oxygen atom from 1.247(2) to 1.313(3) Å. The other C–O bond also elongates slightly from 1.248(2) to 1.269(3) Å, likely due to the H-bonding interactions within a dimeric solid-state structure. Similarly, the sodium cation in 13a promotes CO2 activation, with C–O bonds elongated to 1.260(1) and 1.271(1) Å. Both C–O bonds exhibit comparable lengths as the sodium cation interacts with both O atoms in a dimeric form. The C–O bond elongation and IR shift (∼1602 cm−1) in 13a are less significant than in 9a, this is presumably due to the lower Lewis acidity of sodium cations. These examples underscore how Lewis acid, such as proton, borane, and alkali metals, can stabilize carbonite (CO22−), thereby enhancing CO2 activation.
The Limberg group systematically investigated the impact of the distance, number and nature of cations by preparing a series of Ni–CO2–LA adducts with different cations and polyether co-ligands (14–16).55,66,67 A comparison of cations (14a–c) with the same μ2-κC,O:κO′:κO′′ binding mode reveals that the degree of CO2 activation follows the trend Li > Na > K based on stretching frequencies and 13C NMR chemical shifts of CO2 of 170.34 (14a), 168.58 (14b) and 167.59 (14c) ppm. This trend can be attributed to the higher Lewis acidity of lighter ions. Furthermore, the spatial separation, which varies with a different co-ligands, was also evaluated. Increasing the size of the macrocycle results in a detachment of the cation from the Ni–CO2 in 15a–c, leading to weaker CO2 activation. Conversely, increasing the number of cation interactions enhances CO2 activation observed in 16a–c.
3.3 Bimetallic Ni–CO2–TM (transition metal) adducts
Only a limited number of bimetallic Ni–CO2–TM species have been reported in the literature (Scheme 3). In 2007, the Sadighi group reported a bimetallic Ni–CO2 compound (17) supported by an NHC ligand, where CO2 is bound as a bridging ligand between two nickel ions in a μ-η2,η2 fashion.68 Although the binding mode of CO2 in 17 differs significantly from that observed in the CODH chemistry, 17 can still be considered as an early example of a dinuclear nickel CO2 adduct. In 2013, our group reported a dinickel Ni–CO2–Ni compound (18a), which adopts a μ-κC:κO binding mode, reminiscent of that of CO2 in the CODH active site.63 This species was synthesized by reacting 9a with (PNP)NiOH. A similar compound (18b) with the acriPNP ligand was synthesized by reacting a (acriPNP)Ni(I) species with CO2.69 The degree of CO2 activation in 18a is comparable to that in 9a, as indicated by the C–O distances (1.296(3) and 1.240(3) Å), the O–C–O angle of 123.7(2)° and the CO2 asymmetric stretching frequency of 1518 cm−1. A similar dinickel compound 19 was prepared by the Schneider group through a photo-induced reaction of a nickel hydride species with CO2.65 Mechanistic studies revealed that the Ni(I) species is formed during the photochemical reaction. The bridging CO2 moiety in 19, where both O atoms are coordinated to a single nickel ion, differs from that in 18, likely due to the flexibility of the PNP ligand in 19, which permits the de-coordination of one of the P donors.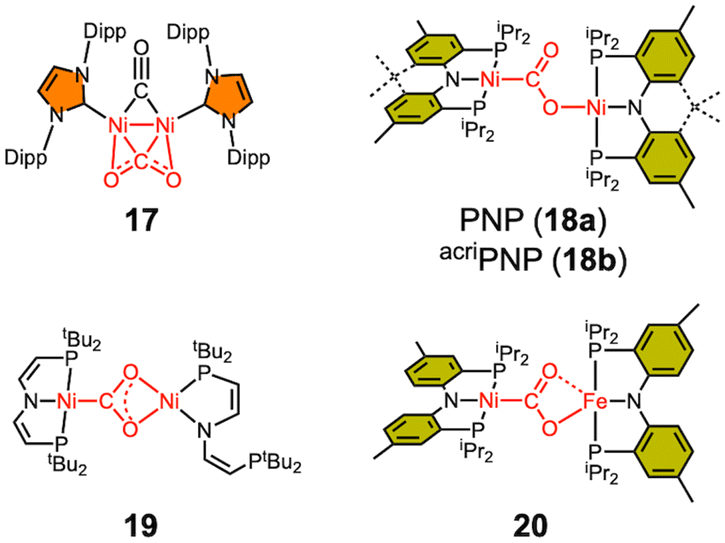 | ||
| Scheme 3 Reported Ni–CO2–TM adducts. | ||
Considering the CODH active site, only one example of a Ni–CO2–Fe adduct has been reported. Our group reported a Ni–CO2–Fe compound (20), which was prepared from condensation between 9a and (PNP)FeOH.57 Its C–O distances are 1.269(2) and 1.289(2) Å, which are consistent with those in other bimetallic CO2 adducts, see Tables 3 and 4. This indicates that the iron ion acts as a typical Lewis acid. However, this does not necessarily imply that the CO2 binding and activation involving structural and redox changes are solely related to Ni. To address such a question, Ni–Fe bimetallic complexes have previously been synthesized and studied,70 but none exhibited CODH-like activity. A redox-active Fe ion likely plays a crucial role in CO2 activation, a hypothesis that remains to be evaluated.
| TM | CO2 binding mode | d (Ni–C) | d (C–O) | d (C–O′) | ∠O–C–O′ | ν CO2asym | |
|---|---|---|---|---|---|---|---|
| a The CO2 moiety was disordered over two distinct positions. b Asymmetric unit cell contains two molecules of 18. | |||||||
| 17 | Ni | μ2-κC:κO:κC:κO′ | 1.952(2) | 1.255(2) | 1.257(2) | 133.4(1) | 1630 |
| 18a | Ni | μ2-κC:κO | 1.888(2) | 1.296(3) | 1.240(3) | 123.7(2) | 1518 |
| 18b | Ni | μ2-κC:κO | 1.94(2)a | 1.34(2)a | 1.22(1)a | 122(1)a | 1511 |
| 1.94(1)a | 1.24(2)a | 1.24(1)a | 121(1)a | ||||
| 19 | Ni | μ2-κC:κO,O′ | 1.875(3)b | 1.292(3)b | 1.280(3)b | 114.0(2)b | 1584 |
| 1.869(3)b | 1.291(3)b | 1.285(3)b | 114.1(2)b | ||||
| 20 | Fe | μ2-κC:κO,O′ | 1.858(1) | 1.289(2) | 1.269(2) | 116.5(1) | 1510 |
3.4 Lessons from model compounds
The structural and spectroscopic data offer valuable insights into CO2 binding and its level of activation. While η2-CO2 compounds (1–5) exhibit a lower degree of CO2 activation assigned as Ni(0)–(CO2), other bridging CO2 compounds (9–20) possessing a single Ni–C bond present a higher degree of CO2 activation, which can be assigned as Ni(II)–CO22−. This suggests that the CO2-bound active site of CODH can be conceptualized as a Ni(II)–(CO22−)–Fe species based on its binding mode and structural parameters. A series of nickel model systems disclose the design principles underlying the CODH active site. Comparisons between monodentate (1a–d), bidentate (2–4), and tridentate ligands (5–7) indicate that the tridentate ligands are more effective than two-donor systems in CO2 activation. The presence of an additional donor increases electron density at the nickel center, thereby facilitating further reduction of CO2. Moreover, the geometry imposed by the ligand plays a crucial role. As illustrated in Fig. 1 and 3, three donors create a situation where a Ni dx2−y2 orbital increases in energy relative to other d orbitals. This allows for electron transfer to the CO2 π* orbital. When the ligand supports a square planar environment (6 and 7), the donors can direct more electron density toward the Ni dx2−y2 orbital, enabling bond formation with CO2 π* orbital (Fig. 4). Through this bonding interaction, two electrons are transferred from Ni to CO2, reducing it to a carbonite (CO22−) form. Furthermore, as previously discussed, three donors can create an electronic structure that weakens the bond between Ni and O of bound CO2. The Lewis acid interaction (12, 13 and 14) not only promotes inner-sphere electron transfer from the Ni ion during the initial stage of CO2 binding but also stabilizes the negative charge on the carbonite oxygen by accepting electron density through its empty orbital. A comprehensive evaluation of the model Ni–CO2–LA compounds listed in Tables 2 and 3 reveals that the ancillary ligand plays a more significant role in CO2 activation than the Lewis acid. Thus, the degree of CO2 activation is primarily governed by the choice of the ancillary ligand, while the Lewis acid serves a supplementary role.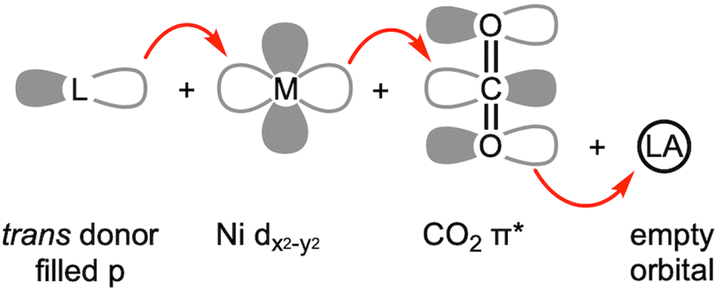 | ||
| Fig. 4 Frontier orbitals involved in CO2 activation. | ||
4 Reactivity of model Ni–CO2 compounds
4.1 Formation of Ni–CO2 adducts
In CODH, the Cred2 state is responsible for CO2 binding, leading to the formation of the Ni–COO–Fe species. The Cred2 state is generally thought to involve a low-valent nickel ion, such as Ni(0) or Ni(I), but there remains ongoing debate regarding its oxidation state. Alternative possibilities, such as Ni(I) or Ni(II)–H species, have also been proposed.26,34–36 A critical question remains: which nickel oxidation state is capable of reacting with CO2 under biological conditions.13,16,34–36 To address this, various synthetic efforts have led to the preparation of several Ni–CO2 compounds, offering chemical insights into the proposed CO2 activation mechanisms.Aresta's first Ni–CO2 compound (1) was synthesized by CO2 addition to (PCy3)Ni(N2). Other Ni(0)-η2-CO2 compounds have also been prepared from Ni(0) precursors possessing a coordinating N2 or a solvent molecule. The five-coordinate Ni-η2-CO2 compound 5 was prepared from {(PPMeP)Ni0}2(μ-N2). Although the degree of CO2 activation in 5 is low, the addition of a Lewis acid leads to the formation of 12 possessing a Ni(II)–CO22− moiety. This may mimic the initial CO2 binding at the CODH nickel center and subsequent stabilization with Feu. As a notable example, our group reported the first instance of CO2 binding at a Ni(0)–CO species (A) with the expulsion of CO, as illustrated in Fig. 5a.39 By employing a rigid acriPNP ligand, Ni(0)–CO rapidly reacts with CO2 at room temperature, leading to the selective generation of a Ni(II)–carbonite species 13b. In contrast, a (PPMeP)Ni(0)–CO species supported by a neutral PPMeP ligand does not react with CO2 due to thermodynamic reasons (ΔG = +20 kcal mol−1). However, the corresponding (PPMeP)Ni(CO2) species (5) can be formed from the corresponding Ni(0)–N2 species.40 This demonstrates that the structural preference of the tridentate ligand supporting either square planar or tetrahedral geometry influences the thermodynamics of the reaction.
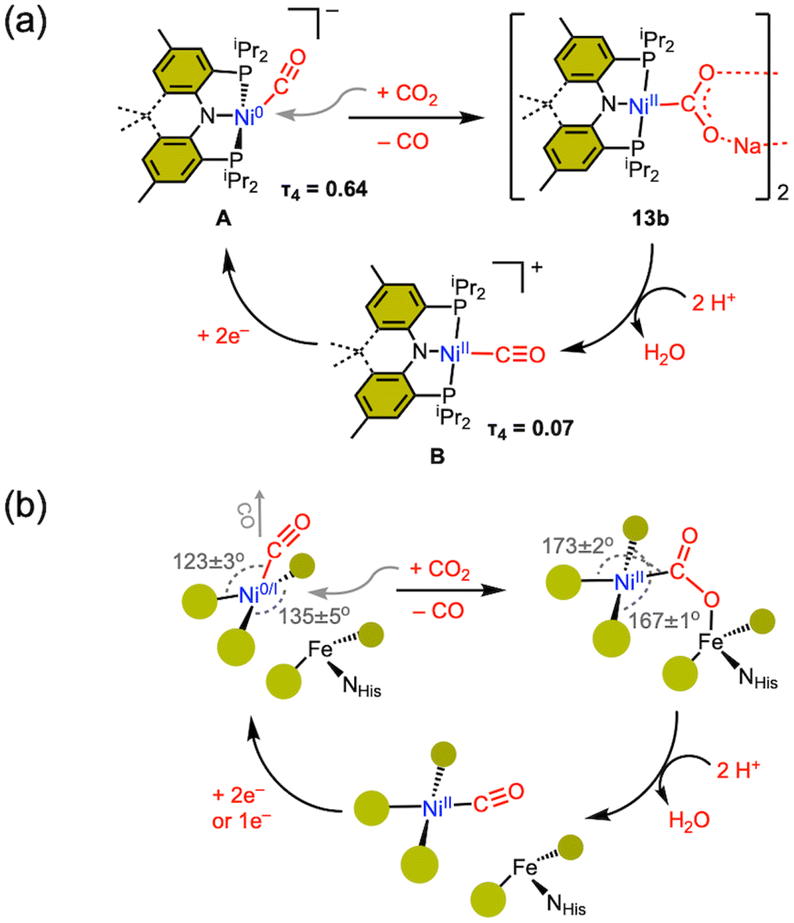 | ||
| Fig. 5 (a) Synthetic cycle for CO2 to CO in the (acriPNP)Ni system. (b) Proposed CODH mechanism based on the model study; bond angles are based on Table 1. | ||
The reaction of Ni(I) with CO2 has been recently investigated. In 2017, our group reported a metalloradical species (acriPNP)Ni(I), that reacts with CO2 to produce the dinickel carbonite compound {(acriPNP)Ni}2(μ-CO2) (18b) via cooperative binuclear reduction.69 A similar reaction involving Ni(I) leading to the formation of dinickel carbonite 19 was also reported by the Schneider group.65 Both reactions of Ni(I) with CO2 occur almost instantaneously at room temperature, indicating that two Ni(I) species cooperatively undergo a single-electron transfer pathway with a low activation barrier for both binding and reduction of CO2. In CODH, a comparable reaction route may be facilitated by electron transfer from the Fe/S cluster or Feu to a Ni(I) ion during the CO2 binding.
Several recent studies have highlighted these reactions involving a nickel-hydride species and CO2 can lead to unusual transformation to give a nickel-carbonite species, referred to as “abnormal” insertion (Scheme 4a). These reactions, however, typically require photolysis, strong base, or metal–ligand cooperative transformation.59,65–67 In 2013, our group reported that the thermolysis of (PNP)Ni–COOH (9a) resulted in producing a nickel hydride species with ∼35% yield.63 Although the reverse reaction was not observed with (PNP)NiH, this suggests the potential for abnormal insertion based on the microscopic reversibility. In 2018, the Schneider group provided the first instance of abnormal insertion forming a hydroxycarbonyl species 11 under photochemical conditions.65 This study demonstrates that to access a nickel carbonite species, the transformation of a nickel hydride species requires high energy light (λ > 305 nm) to overcome a 35 kcal mol−1 energy barrier. In contrast, the formation of formate proceeds with a relatively lower barrier of 25 kcal mol−1. Mechanistic studies revealed that the irradiation promotes N–H reductive elimination to form a Ni(0) species, which subsequently reacts with CO2 to produce 11. The Limberg group reported deprotonation of a nickel-formate species to generate a corresponding nickel carbonite (14–16).66,67 Since formate can be readily generated by normal CO2 insertion into Ni–H, this result implies that CO2 may react with a Ni–H bond at the active site. Another approach was presented by the Milstein group, reporting the formation of a Ni(II)-η1-CO2-κC compound (7) from the reaction of nickel(II) hydride with CO2via metal–ligand cooperation.59 Hydride transfer involving aromatization and dearomatization of the lutidine-based PNP ligand enables the generation of the Ni–CO2 compound from nickel hydride.
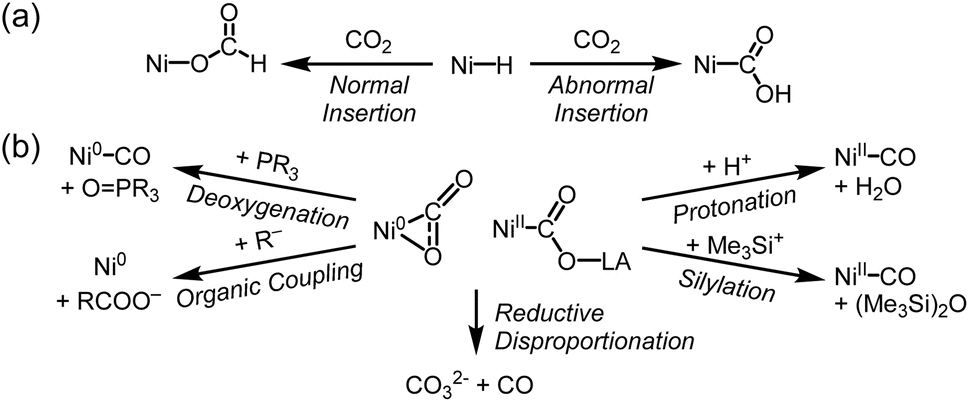 | ||
| Scheme 4 (a) Normal and abnormal insertion of CO2 into nickel hydride species. (b) Common reactions found in Ni–CO2 adducts. | ||
4.2 Conversion of Ni–CO2 to Ni–CO
The reactivity of Ni–CO2 adducts is summarized in Scheme 4b. Early studies on Ni(0)-η2-CO2 compounds reported the deoxygenation of CO2 using internal or external phosphine, resulting in the formation of Ni(0)–CO and phosphine oxide.50,51,59 Aresta's complex 1a was employed as a Ni(0) precursor and catalyst for organic coupling reactions.71,72 The Dong group also reported a CO2 coupling reaction with an organozinc reagent to produce carboxylic acid.73 Reductive disproportionation has been observed, where two CO2 molecules are converted to CO32− and CO via electron transfer.45,65,66,68,74To mimic the CODH reaction, protonation of Ni–CO2 compounds was investigated. Protonation of 1a was attempted with PhSH. Although (PCy3)2Ni(CO)2 was obtained, a nickel hydride species was formed accompanied by the elimination of CO2 at room temperature.75–77 In contrast, protonation of Ni(II)–CO2 compounds yields Ni(II)–CO in high yield without any evidence of hydride formation. Our group reported that the protonation of a hydroxycarbonyl species (9a and 9b) produced Ni(II)–CO and H2O in high yield of >90%.63 This result is somewhat distinctive compared to the result of (PPMeP)Ni(CO2) (5). Due to the weak activation of CO2, 5 is converted back to {(PPMe)Ni0}2(μ-N2) under a N2 atmosphere. Protonation of another Ni(II) carbonite compound (acriPNP)NiCOO–Na (13b) also produces Ni(II)–CO in high yield of 93% via a hydroxycarbonyl intermediate. As a related example, silylation of a Ni(II)–CO2–Li compound (16a) also gave Ni–CO.67 These studies demonstrate that the oxidation state and the degree of CO2 activation significantly influence reactivity.
4.3 CO elimination and completion of the cycle
As the final step of CO2 conversion, the product CO should be eliminated from the nickel center. According to the current proposed mechanism, CO should be removed from a Ni(II) state, due to the poor back donation to the CO ligand.38–40 At lower oxidation states, such as Ni(I) or Ni(0), in general, a nickel carbonyl species do not react with CO2, as back-donation stabilizes the Ni–CO species. As expected, (PPMeP)Ni–CO does not react with CO2.40 However, the CO2 reaction of {(PNP)Ni0(CO)}− revealed immediate transformation to various products, including carbamate, carbonate, and carbonyl species.78 After this undesired result, our group successfully increased the selectivity of the CO2 reaction with a nickel(0)–CO species by employing a structurally rigidified ligand.39 The newly designed acriPNP ligand (acriPNP− = 4,5-bis(diisopropylphosphino)-2,7,9,9-tetramethyl-9H-acridin-10-ide) effectively accommodates a square planar nickel(II) center, revealing unusual structural features due to its rigid backbone.69 Interestingly, two-electron reduction effectively alters the geometry of [(acriPNP)Ni0–CO]− (A), causing the CO ligand to shift towards an axial position. This change opens the binding site for CO2, as depicted in Fig. 5a. This structural change facilitates the selective generation of a Ni(II)–carbonite species (13b) from the CO2 reaction of A with the expulsion of CO. This process could be closely related to the CODH mechanism and the positioning of both CO and CO2 channels, as illustrated in Fig. 5b.17,79–81 After the CO2 binding to the nickel center, subsequent protonation can cleave the C–O bond to give a Ni(II)–CO species (B). A two-electron reduction of Ni(II)–CO completes a closed synthetic cycle for CO2 reduction to CO at a single nickel center, as shown in Fig. 5a.Based on the (acriPNP)Ni scaffold, the synthetic COCH2 reduction cycle encompasses the following features, as illustrated in Fig. 5a. (1) Reduction of the nickel center systematically alters the position of the CO ligand (from equatorial to axial), thereby opening a CO2 binding site. (2) CO liberation occurs upon CO2 coordination to a nickel(0) center, which oxidises Ni and reduces the back-bonding to CO. (3) Structural change of Ni(II) to a square planar geometry, along with the cationic Lewis acid interaction facilitate the formation of a Ni(II)–carbonite species. (4) Protonation of the carbonite moiety cleaves the C–O bond to produce a Ni–CO species. Finally, (5) reduction of nickel–CO may occur at a more positive reduction potential, approximately −1.2 V vs. Fc/Fc+. These findings offer chemical insights that may help elucidate the organonickel chemistry of the CODH active site. As depicted in Fig. 5b, we may propose an alternative CODH mechanism. The coordination geometry about the nickel center in CODH aligns fairly well with the nickel species generated with an (acriPNP)Ni scaffold, as illustrated in Fig. 5. In particular, the S–Ni–S angles differ significantly between Ni–CO and Ni–CO2, as listed in Table 1. The reduction potential of the nickel site, which is connected to a Fe–S cluster, is particularly considered. Although a bound CO ligand generally reduces the reactivity of a Ni–CO species, it can significantly and positively shift the reduction potential, aiding electron transfer to the nickel center. In the case of an (acriPNP)Ni scaffold, the reduction potential of a Ni(II)–CO species is ∼1 V more positive than that of (acriPNP)NiII–Cl. This demonstrates the influence of a π-acidic CO ligand compared to a π-basic X-type ligand in the reduction of a nickel(II) complex. Thus, we propose one or two electron reduction of the Ni ion should occur when CO is bound, as illustrated in Fig. 5b. Given the complex nature of the CODH active site, applying this model directly to its mechanism may be challenging. However, both reduction potential and the positioning of the CO/CO2 channels should be carefully considered. Interestingly, the mechanism suggested by our model study is in line with recent proposals based on calculations and kinetic studies with CODH.34,82,83
5 CODH functional models
There are numerous examples of electrocatalytic CO2 conversion based on transition metals.6–9 Some systems have adopted strategies inspired from CODH, such as hydrogen bonding in the secondary coordination sphere and bimetallic approaches.6,84,85 These include not only molecular catalysts but also heterogeneous catalytic systems and photocatalysis, which are beyond the scope of this review. This section focuses on molecular nickel catalysts for the electrocatalytic CO2 reduction. Seminal works by the DuBois,86 Lehn87 and Kubiak groups88,89 have demonstrated electrocatalytic CO2 conversion to CO, involving the formation of a M-COO− intermediate, reminiscent of CODH chemistry. Only the DuBois group's work exhibits a bimetallic pathway. Most of the nickel electrocatalytic systems discussed below and highlighted in Scheme 5 proceed through the reductive transformation of CO2 to CO via the formation of a nickel metallacarbonite species, as a proposed intermediate.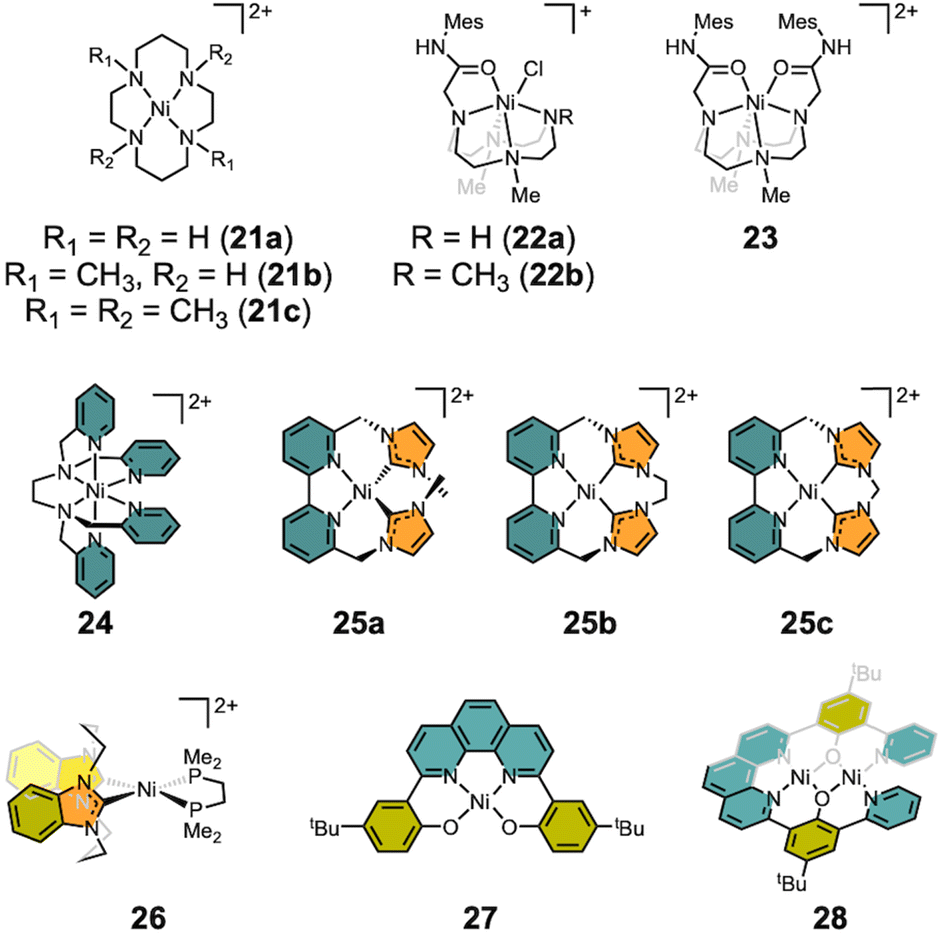 | ||
| Scheme 5 Functional models of CODH. | ||
Nickel catalysts supported by macrocyclic ligands were explored early on. In 1984, the Sauvage group investigated the electrocatalytic CO2 reduction using Ni(cyclam)Cl2 (21a) (cyclam: 1,4,8,11-tetra-azacyclotetradecane), as shown in Scheme 5.90 The catalysis was carried out at −1.0 V (vs. NHE) in water yielding selective CO production with a current efficiency of 99% and TOF = 18 h−1. A unique feature of this system is that the active nickel catalyst adsorbs onto the Hg working electrode, whereas 21a tends to promote hydrogen evolution in the aqueous solution. When the same reaction was conducted using a glassy carbon electrode, lower faradaic efficiencies (FE) were observed due to hydrogen evolution.91 In 2023, the Cowan group incorporated the pulsed electrolysis, with asymmetric anodic pulse ranging from 40 ms to 1 s, throughout electrolysis on 21a. This approach resulted in a 4-fold increase in selectivity for CO production (CO/H2 = 2.42 ± 0.10). XPS analysis on the glassy carbon electrode surface suggested that standard electrolysis causes catalyst degradation through cyclam loss and the deposition of Ni(0) carbonyl species on the electrode surface enhances hydrogen evolution.92 Similarly, the Rosenthal group reported electrocatalytic CO2 reduction using a nickel(II) cyclen complex (22, cyclen: 1,4,7,10-tetraazacyclododecane).93 The catalytic reaction was conducted in MeCN achieving a FE of up to 78 ± 5% with [Ni(TrMCyMes)(Cl)]Cl (22b) (TrMCyMes: 1,4,7-trimethyl-10-acetamideMes-1,4,7,10-tetraazacyclododecane) at −1.95 V vs. SCE (−2.33 V vs. Fc/Fc+).94 The electrocatalytic process undergoes two-electron reduction coupled with the chloride extrusion, which is presumably a prerequisite for CO2 coordination. The importance of an unmasked coordination site was highlighted by modifying TrMCyMes to 1,7-dimethyl-4,10-bis(acetamideMes)-1,4,7-10-tetraazacyclododecane (DMCy2Mes).94 Under the same conditions at −1.95 V (vs. SCE, −2.33 V vs. Fc/Fc+), [Ni(DMCy2Mes)](PF6)2 (23) exhibited a low catalytic faradaic efficiency of 24 ± 4%, as the two amide arms interfere with CO2 coordination.
In 2018, the Machan group reported the electrocatalytic CO2 reduction mediated by [Ni(TPEN)](PF6)2 (24) (TPEN = N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine).95 Using foot-of-the-wave (FOWA) analysis, the divalent nickel catalyst demonstrated a TOFmax of 7.72 × 108 s−1 in the presence of 2.50 M phenol as a proton source, a remarkable result compared to other systems exhibiting TOFs ranging from 4 to 47.5 s−1. Compared to 22 and 23, the weaker pyridine donors in 24 enable a milder operation potential of −2.05 V, compared to the stronger aliphatic amine donor (−2.33 V vs. Fc/Fc+). Unfortunately, 24 undergoes significant degradation to Ni(CO)4, likely due to the weak field donors within the TPEN ligand, leading to ligand loss during the catalysis. Consequently, CO scavenger 21c is necessary to prolong the catalyst lifetime.
In 2018, Panetier and Jurss reported electrocatalytic CO2 reduction using a series of nickel complexes supported by bipyridyl-N-heterocyclic carbene donors (25), as shown in Scheme 5.96 These nickel complexes were prepared with the ligands generated from the reaction of 6,6′-bis(bromomethyl)-2,2′-bipyridine alkylated with 1-methylimidazole (25a) or diimidazoles (25b and 25c). Structural analysis of 25a–c reveals that both 25b and 25c adopt a square planar geometry (τ4 = 0.11 and 0.08, respectively), while 25a exhibits noticeable distortion at the Ni center (τ4 = 0.26) due to the two methyl groups. All nickel complexes display high FE of 98–99% at approximately −2.4 V vs. Fc/Fc+. However, the selectivity and TOF for CO generation increase from 5% (25a) to 87% (25c) and from <1 s−1 (25a) to 47.5 s−1 (25c), respectively. According to DFT analysis, the two-electron reduced species of 25 can exist as an open-shell singlet species as Ni(I)-L˙− for 25a or Ni(II)-L2˙− for 25c. Further reduction of a Ni(I) center may lead to the formation of Ni(II)–H in the presence of a proton.
In 2019, the Kubiak group reported the square planar nickel(II) complex [(bis-NHC)Ni(dmpe)](PF6)2 (26) by employing both bis-NHC ligand (bis-NHC = 3,3′-bis(1,3-propanediyl)dibenzimidazolin-2,2′-diylidene) and dmpe (1,2-bis(dimethylphosphino)ethane).97 Prior to this work, nickel complexes supported by two diphosphine ligands were explored revealing the formation of nickel hydrides. The hydricity of these complexes was insufficient to effectively reduce CO2.98,99 By incorporating carbene donors, however, a concerted two-electron reduction of the nickel center was achieved at a relatively positive reduction potential of −1.87 V vs. Fc/Fc+. Consequently, 26 demonstrated electrocatalytic CO2 reduction at an operating potential of −1.75 V, which is more positive than that of 25a–c, but it suffered from a relatively low CO production with a FE of 25%, due to substantial hydrogen evolution (FEH2 = 55%). Interestingly, the combination of phosphine and carbene donors creates a unique electronic environment around the nickel, enhancing its reducing power for CO2 reduction. After recognizing that nickel complexes with both aliphatic and aromatic nitrogen donors (21–24) could catalyze the conversion of CO2 to CO, the nickel catalyst was further optimized by introducing carbene donors, as seen in 25a–c, which showed improved performance. Due to the strong σ-donation, however, the operating potentials for CO2 reduction in these complexes were too negative, ranging from −2.2 V to −2.4 V vs. Fc/Fc+. Incorporation of a bis-phosphine ligand in 26 finally shifted the operating potential positively by over 400 mV, making it more favorable for CO2 reduction.
More recently, the Zhang group employed both mono- and dinuclear nickel complexes (27 and 28) supported by a redox-active 1,10-phenanthroline backbone (H2bphpp: 2,9-bis(5-tert-2-hydroxy-3-pyridylphenyl)-1,10-phenanthroline and H2hbpp: 2,9-bis(5-tert-butyl-2-hydroxyphenyl)-1,10-phenanthroline).100 Both species exhibited ∼90% of FE to give CO production when operating at −2.20 V vs. Fc/Fc+ in DMF. Notably, the electrocatalytic current and TOF of the dinuclear Ni2(bphpp) species (28) are approximately 5 times greater than those for mononuclear Ni(hbpp) (27) (∼2.8 mA and 20.5 s−1 for 28vs. ∼0.5 mA and 4.4 s−1 for 27). These results suggest that the additional nickel ion within 28 plays a crucial role in the catalytic CO2 reduction process, facilitating disproportionation to yield both CO and CO32−.
6 Conclusions
This review examines the current status and recent advancements in the field of CODH model chemistry to enhance our understanding of the fundamental chemical principles underlying enzymatic organometallic transformations. Notably, the CO2-bound form of Ni,Fe-CODH features a Ni–COO–Fe metallocarbonite moiety, which involves a two-electron reduction process. The active site of CODH features three sulfur donors that support a Ni center, enabling it to shuttle between T-shaped and tetrahedral geometries upon CO and CO2 binding. As a part of a Fe–S cluster, the unique iron (Feu) site facilitates electron transfer and stabilizes the reduced CO2 substrate through Lewis acid–base interaction. This bimetallic Ni, Fe-mediated transformation presents an intriguing approach for the selective conversion of CO2 to CO under ambient conditions, thus inspiring further synthetic model studies. In particular, Ni–CO2–TM complexes and related nickel electrocatalysts have been highlighted. It is noteworthy that Ni plays a crucial role in the biological CO2/CO conversion, potentially due to the stability of a Ni–C bond, which likely drives selectivity towards CO generation over the formate generation, as a competing two-proton, two-electron reduction process. To achieve CO2 reduction at low overpotential, the incorporation of secondary coordination spheres, Lewis acid interactions, and changes in the nickel's geometry may be essential. One of the most enigmatic aspects of CODH chemistry is the storage of two reducing equivalents within the Ni–Fe/S cluster before CO2 binding. This may arise from Ni–H or Ni–Fe bonds, warranting further research on heterometallic Ni–CO2–TM model compounds to uncover the role of heterometals such as Feu. Future model studies varying metal geometry and electronic structure will not only provide critical chemical insights into the understanding of CODH active site chemistry but also contribute to the development of advanced (electro)catalysts for selective CO generation.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Y. C., J. C. and Y. L. are prepared the manuscript. All authors contributed to the discussion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Research Founda-tion of Korea (2018R1A5A1025208, 2020R1A2C3007364, 2022M3C1A3092056 to Y. L., 2024M3J5A1023902 to C. Y., RS-2023-00240996 to J. C.) and by Chonnam National University (2024-0405-01 to J. C.).Notes and references
- CO2 Emissions in 2022, IEA, IEA, Paris, 2023 Search PubMed.
- International Energy Agency, Putting CO2 to Use: Creating Value From Emissions, OECD, 2019 Search PubMed.
- Y. Yang and J.-W. Lee, Chem. Sci., 2019, 10, 3905–3926 RSC.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS.
- Carbon Dioxide as Chemical Feedstock, ed. M. Aresta, Wiley, 1st edn, 2010 Search PubMed.
- E. Boutin, L. Merakeb, B. Ma, B. Boudy, M. Wang, J. Bonin, E. Anxolabéhère-Mallart and M. Robert, Chem. Soc. Rev., 2020, 49, 5772–5809 RSC.
- F.-Y. Gao, R.-C. Bao, M.-R. Gao and S.-H. Yu, J. Mater. Chem. A, 2020, 8, 15458–15478 RSC.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef PubMed.
- C. Costentin, M. Robert and J.-M. Savéant, Acc. Chem. Res., 2015, 48, 2996–3006 Search PubMed.
- M. Can, F. A. Armstrong and S. W. Ragsdale, Chem. Rev., 2014, 114, 4149–4174 CrossRef.
- M. Inoue, I. Nakamoto, K. Omae, T. Oguro, H. Ogata, T. Yoshida and Y. Sako, Front. Microbiol., 2019, 9, 3353 CrossRef PubMed.
- H. S. Shafaat and J. Y. Yang, Nat. Catal., 2021, 4, 928–933 CrossRef.
- S. H. Newman-Stonebraker, T. J. Gerard and P. L. Holland, Chem, 2024, 10, 1655–1667 Search PubMed.
- J. Fesseler, J.-H. Jeoung and H. Dobbek, Angew. Chem., Int. Ed., 2015, 54, 8560–8564 CrossRef PubMed.
- W. Gong, B. Hao, Z. Wei, D. J. Ferguson, T. Tallant, J. A. Krzycki and M. K. Chan, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9558–9563 CrossRef PubMed.
- J.-H. Jeoung and H. Dobbek, Science, 2007, 318, 1461–1464 CrossRef PubMed.
- C. Darnault, A. Volbeda, E. J. Kim, P. Legrand, X. Vernède, P. A. Lindahl and J. C. Fontecilla-Camps, Nat. Struct. Mol. Biol., 2003, 10, 271–279 CrossRef.
- C. L. Drennan, J. Heo, M. D. Sintchak, E. Schreiter and P. W. Ludden, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11973–11978 CrossRef PubMed.
- H. Dobbek, V. Svetlitchnyi, L. Gremer, R. Huber and O. Meyer, Science, 2001, 293, 1281–1285 CrossRef.
- J. Cirera, P. Alemany and S. Alvarez, Chem.–Eur. J., 2004, 10, 190–207 CrossRef PubMed.
- J. Ghannam, T. Al Assil, T. C. Pankratz, R. L. Lord, M. Zeller and W.-T. Lee, Inorg. Chem., 2018, 57, 8307–8316 CrossRef PubMed.
- L. C. Lewis, J. A. Sanabria-Gracia, Y. Lee, A. J. Jenkins and H. S. Shafaat, Chem. Sci., 2024, 15, 5916–5928 RSC.
- V. J. DeRose, J. Telser, M. E. Anderson, P. A. Lindahl and B. M. Hoffman, J. Am. Chem. Soc., 1998, 120, 8767–8776 CrossRef.
- C. Y. Ralston, H. Wang, S. W. Ragsdale, M. Kumar, N. J. Spangler, P. W. Ludden, W. Gu, R. M. Jones, D. S. Patil and S. P. Cramer, J. Am. Chem. Soc., 2000, 122, 10553–10560 CrossRef.
- Z. Hu, N. J. Spangler, M. E. Anderson, J. Xia, P. W. Ludden, P. A. Lindahl and E. Münck, J. Am. Chem. Soc., 1996, 118, 830–845 CrossRef.
- P. Amara, J.-M. Mouesca, A. Volbeda and J. C. Fontecilla-Camps, Inorg. Chem., 2011, 50, 1868–1878 CrossRef PubMed.
- A. Majumdar, Dalton Trans., 2014, 43, 12135–12145 RSC.
- D. Evans, Coord. Chem. Rev., 2005, 249, 1582–1595 CrossRef.
- S. Groysman and R. H. Holm, Biochemistry, 2009, 48, 2310–2320 CrossRef PubMed.
- S. T. Stripp, B. R. Duffus, V. Fourmond, C. Léger, S. Leimkühler, S. Hirota, Y. Hu, A. Jasniewski, H. Ogata and M. W. Ribbe, Chem. Rev., 2022, 122, 11900–11973 CrossRef PubMed.
- K. M. Waldie, A. L. Ostericher, M. H. Reineke, A. F. Sasayama and C. P. Kubiak, ACS Catal., 2018, 8, 1313–1324 CrossRef.
- K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2018, 118, 372–433 CrossRef.
- W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef PubMed.
- R.-Z. Liao and P. E. M. Siegbahn, Inorg. Chem., 2019, 58, 7931–7938 CrossRef.
- R. Breglia, F. Arrigoni, M. Sensi, C. Greco, P. Fantucci, L. De Gioia and M. Bruschi, Inorg. Chem., 2021, 60, 387–402 CrossRef PubMed.
- D. W. N. Wilson, M. S. Fataftah, Z. Mathe, B. Q. Mercado, S. DeBeer and P. L. Holland, J. Am. Chem. Soc., 2024, 146, 4013–4025 CrossRef PubMed.
- V. C.-C. Wang, M. Can, E. Pierce, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2013, 135, 2198–2206 CrossRef PubMed.
- C. Yoo, S. Oh, J. Kim and Y. Lee, Chem. Sci., 2014, 5, 3853–3858 Search PubMed.
- D. Sahoo, C. Yoo and Y. Lee, J. Am. Chem. Soc., 2018, 140, 2179–2185 CrossRef.
- K. Lee, J. Choi, P. M. Graham and Y. Lee, Bull. Korean Chem. Soc., 2022, 43, 222–226 CrossRef CAS.
- S. A. Akhade, W. Luo, X. Nie, N. J. Bernstein, A. Asthagiri and M. J. Janik, Phys. Chem. Chem. Phys., 2014, 16, 20429–20435 RSC.
- J. D. Froehlich and C. P. Kubiak, J. Am. Chem. Soc., 2015, 137, 3565–3573 CrossRef PubMed.
- P. W. Jolly and G. Wilke, in The Organic Chemistry of Nickel, Elsevier, 1974, pp. 1–32 Search PubMed.
- J. Chen, S. Huang, J. Seravalli, H. Gutzman, D. J. Swartz, S. W. Ragsdale and K. A. Bagley, Biochemistry, 2003, 42, 14822–14830 CrossRef PubMed.
- M. Aresta, C. F. Nobile, V. G. Albano, E. Forni and M. Manassero, J. Chem. Soc., Chem. Commun., 1975, 636–637 RSC.
- M. Aresta and C. F. Nobile, J. Chem. Soc., Dalton Trans., 1977, 708–711 RSC.
- A. Dohring, P. W. Jolly, C. Kruger and M. J. Romão, Z. Naturforsch., B, 1985, 40, 484–488 CrossRef.
- A. Paparo and J. Okuda, Coord. Chem. Rev., 2017, 334, 136–149 CrossRef.
- D. H. Gibson, Chem. Rev., 1996, 96, 2063–2096 CrossRef PubMed.
- J. S. Anderson, V. M. Iluc and G. L. Hillhouse, Inorg. Chem., 2010, 49, 10203–10207 CrossRef PubMed.
- R. Beck, M. Shoshani, J. Krasinkiewicz, J. A. Hatnean and S. A. Johnson, Dalton Trans., 2013, 42, 1461–1475 RSC.
- B. M. Puerta Lombardi, C. Gendy, B. S. Gelfand, G. M. Bernard, R. E. Wasylishen, H. M. Tuononen and R. Roesler, Angew. Chem., Int. Ed., 2021, 60, 7077–7081 CrossRef PubMed.
- CRC Handbook of Chemistry and Physics, ed. W. M. Haynes, CRC Press, 95th edn, 2014 Search PubMed.
- F. A. Van Broekhuizen, I. M. N. Groot, H. J. Fraser, E. F. Van Dishoeck and S. Schlemmer, Astron. Astrophys., 2006, 451, 723–731 CrossRef CAS.
- S. Wolff, V. Pelmenschikov, R. Müller, M. Ertegi, B. Cula, M. Kaupp and C. Limberg, Chem.–Eur. J., 2024, 30, e202303112 CrossRef CAS PubMed.
- Y.-E. Kim, J. Kim and Y. Lee, Chem. Commun., 2014, 50, 11458–11461 RSC.
- C. Yoo and Y. Lee, Chem. Sci., 2017, 8, 600–605 RSC.
- A. Paparo and J. Okuda, J. Organomet. Chem., 2018, 869, 270–274 CrossRef.
- D. Oren, Y. Diskin-Posner, L. Avram, M. Feller and D. Milstein, Organometallics, 2018, 37, 2217–2221 CrossRef.
- I. Castro-Rodriguez, H. Nakai, L. N. Zakharov, A. L. Rheingold and K. Meyer, Science, 2004, 305, 1757–1759 CrossRef.
- T.-W. Chiou, Y.-M. Tseng, T.-T. Lu, T.-C. Weng, D. Sokaras, W.-C. Ho, T.-S. Kuo, L.-Y. Jang, J.-F. Lee and W.-F. Liaw, Chem. Sci., 2016, 7, 3640–3644 RSC.
- U. Terranova, JBIC, J. Biol. Inorg. Chem., 2021, 26, 617–624 CrossRef PubMed.
- C. Yoo, J. Kim and Y. Lee, Organometallics, 2013, 32, 7195–7203 CrossRef.
- K. J. Jonasson, A. H. Mousa and O. F. Wendt, Polyhedron, 2018, 143, 132–137 CrossRef.
- F. Schneck, J. Ahrens, M. Finger, A. C. Stückl, C. Würtele, D. Schwarzer and S. Schneider, Nat. Commun., 2018, 9, 1161 CrossRef PubMed.
- P. Zimmermann, S. Hoof, B. Braun-Cula, C. Herwig and C. Limberg, Angew. Chem., Int. Ed., 2018, 57, 7230–7233 CrossRef PubMed.
- P. Zimmermann, D. Ar, M. Rößler, P. Holze, B. Cula, C. Herwig and C. Limberg, Angew. Chem., Int. Ed., 2021, 60, 2312–2321 CrossRef PubMed.
- C. H. Lee, D. S. Laitar, P. Mueller and J. P. Sadighi, J. Am. Chem. Soc., 2007, 129, 13802–13803 CrossRef.
- C. Yoo and Y. Lee, Angew. Chem., Int. Ed., 2017, 56, 9502–9506 CrossRef CAS PubMed.
- D. Huang and R. H. Holm, J. Am. Chem. Soc., 2010, 132, 4693–4701 CrossRef CAS.
- M. Aresta, A. Dibenedetto, E. Quaranta, M. Lanfranchi and A. Tiripicchio, Organometallics, 2000, 19, 4199–4207 CrossRef CAS.
- F. D'Accriscio, A. Ohleier, E. Nicolas, M. Demange, O. Thillaye Du Boullay, N. Saffon-Merceron, M. Fustier-Boutignon, E. Rezabal, G. Frison, N. Nebra and N. Mézailles, Organometallics, 2020, 39, 1688–1699 CrossRef.
- C. S. Yeung and V. M. Dong, J. Am. Chem. Soc., 2008, 130, 7826–7827 CrossRef CAS PubMed.
- P. Mastrorilli, G. Moro, C. F. Nobile and M. Latronico, Inorg. Chim. Acta, 1992, 192, 189–193 CrossRef CAS.
- M. Aresta, E. Quaranta and I. Tommasi, J. Chem. Soc., Chem. Commun., 1988, 450–452 RSC.
- M. Aresta, R. Gobetto, E. Quaranta and I. Tommasi, Inorg. Chem., 1992, 31, 4286–4290 CrossRef CAS.
- I. Tommasi, M. Aresta, P. Giannoccaro, E. Quaranta and C. Fragale, Inorg. Chim. Acta, 1998, 272, 38–42 CrossRef CAS.
- C. Yoo and Y. Lee, Inorg. Chem. Front., 2016, 3, 849–855 RSC.
- T. I. Doukov, T. M. Iverson, J. Seravalli, S. W. Ragsdale and C. L. Drennan, Science, 2002, 298, 567–572 CrossRef CAS PubMed.
- T. I. Doukov, L. C. Blasiak, J. Seravalli, S. W. Ragsdale and C. L. Drennan, Biochemistry, 2008, 47, 3474–3483 CrossRef CAS PubMed.
- P. Wang, M. Bruschi, L. De Gioia and J. Blumberger, J. Am. Chem. Soc., 2013, 135, 9493–9502 CrossRef CAS PubMed.
- J. Ruickoldt, Y. Basak, L. Domnik, J.-H. Jeoung and H. Dobbek, ACS Catal., 2022, 12, 13131–13142 CrossRef CAS.
- J. Heo, C. R. Staples, C. M. Halbleib and P. W. Ludden, Biochemistry, 2000, 39, 7956–7963 CrossRef CAS PubMed.
- P. Gotico, Z. Halime, W. Leibl and A. Aukauloo, ChemPlusChem, 2023, 88, e202300222 CrossRef CAS PubMed.
- A. W. Nichols and C. W. Machan, Front. Chem., 2019, 7, 397 CrossRef CAS PubMed.
- B. D. Steffey, C. J. Curtis and D. L. DuBois, Organometallics, 1995, 14, 4937–4943 CrossRef.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1984, 328–330 RSC.
- K. S. Ratliff, R. E. Lentz and C. P. Kubiak, Organometallics, 1992, 11, 1986–1988 CrossRef CAS.
- M. D. Sampson, A. D. Nguyen, K. A. Grice, C. E. Moore, A. L. Rheingold and C. P. Kubiak, J. Am. Chem. Soc., 2014, 136, 5460–5471 CrossRef CAS PubMed.
- M. Beley, J.-P. Collin, R. Ruppert and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1984, 1315–1316 RSC.
- J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2012, 51, 3932–3934 CrossRef.
- F. Greenwell, B. Siritanaratkul, P. K. Sharma, E. H. Yu and A. J. Cowan, J. Am. Chem. Soc., 2023, 145, 15078–15083 CrossRef PubMed.
- T. Qiu, G. P. A. Yap and J. Rosenthal, ACS Appl. Energy Mater., 2019, 2, 8560–8569 CrossRef.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef.
- L. E. Lieske, A. L. Rheingold and C. W. Machan, Sustain. Energy Fuels, 2018, 2, 1269–1277 RSC.
- X. Su, K. M. McCardle, J. A. Panetier and J. W. Jurss, Chem. Commun., 2018, 54, 3351–3354 RSC.
- A. L. Ostericher, T. M. Porter, M. H. Reineke and C. P. Kubiak, Dalton Trans., 2019, 48, 15841–15848 RSC.
- D. E. Berning, B. C. Noll and D. L. DuBois, J. Am. Chem. Soc., 1999, 121, 11432–11447 CrossRef.
- C. J. Curtis, A. Miedaner, W. W. Ellis and D. L. DuBois, J. Am. Chem. Soc., 2002, 124, 1918–1925 CrossRef PubMed.
- Y. Xiao, F. Xie, H.-T. Zhang and M.-T. Zhang, JACS Au, 2024, 4, 1207–1218 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |