
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polyvinyl alcohol-based polarizers for new displays: molecules, processing and properties
Yao Lia,
Jiayu Xieb,
Hong Cheng a,
Xiaoying Weic,
Jie Chena,
Liangpeng Youa and
Wei Chen
*a
a,
Xiaoying Weic,
Jie Chena,
Liangpeng Youa and
Wei Chen
*a
aSchool of Nuclear Science and Technology, National Synchrotron Radiation Laboratory, Anhui Provincial Engineering Laboratory of Advanced Functional Polymer Film, CAS Key Laboratory of Soft Matter Chemistry, University of Science and Technology of China, Hefei 230026, China. E-mail: wc003@ustc.edu.cn
bGuangxi Key Laboratory of Petrochemical Resource Processing and Process Intensification Technology, School of Chemistry and Chemical Engineering, Guangxi University, Nanning 530004, China
cCAS Key Laboratory of Mechanical Behavior and Design of Materials, Department of Modern Mechanics, School of Engineering Science, University of Science and Technology of China, Hefei, 230026, China
First published on 6th March 2025
Abstract
Polarizers are a key component of new display panels (i.e. liquid crystal displays (LCDs) and organic light-emitting diodes (OLEDs)), consisting of a polarizing film, support film, compensation film, and optical clear adhesives between the layers. The key functional layer is the iodine-doped polyvinyl alcohol (PVA) film. The processing of polarizers involves the synthesis of an optical-grade PVA resin, followed by the preparation of highly oriented iodine-doped PVA films, which includes the film casting, iodine doping, boric acid crosslinking, and post-stretching steps. Revealing the multi-scale structure and changes in chain dynamics during processing is crucial for establishing the structure–process–property relationship of PVA-based polarizers. The current work reviews the recent research progress in this direction, primarily including the following: (1) primary chemical structure of PVA, (2) solution casting of PVA films, (3) hierarchical structure and dynamics heterogeneity of plasticized PVA films, (4) formation mechanism of PVA–iodine complexes, and (5) crosslinking mechanism of boric acid in PVA.
 Wei Chen | Wei Chen is a Professor in the School of Nuclear Science and Technology at the University of Science and Technology of China. His primary research interest focuses on developing in situ techniques based on solid-state nuclear magnetic resonance and synchrotron radiation to study polymer processing, with a particular emphasis on polymer optical film systems. |
Introduction
New displays play an important role in our daily life. Driven by diverse application scenarios, convenience in production and daily life, and intelligent engineering technologies, new displays are gradually evolving toward being lighter, thinner, and more flexible while exhibiting higher contrast and higher durability. Currently, the mainstream display panels are primarily thin-film transistor liquid crystal displays (TFT-LCDs) and organic light-emitting diode displays (OLEDs). Fig. 1 illustrates the basic structures of both TFT-LCD and OLED panels, which are composed of multi-layer structures.1–4 Regardless of the type of display, a polarizer is inevitable. | ||
| Fig. 1 Schematic of the panel structure: (a) LCD panel, (b) small- and medium-sized OLED and (c) large-sized OLED. | ||
In LCD panels, two polarizers are required. The bottom polarizer converts the backlight into polarized light, whereas the top polarizer acts as an analyzer to control the brightness and darkness contrasts, thereby achieving an image display. Alternatively, in OLED panels, only a single polarizer is needed to eliminate external environmental reflections and offer high-contrast display. In addition to their impact on display quality, polarizers account for 10% or more of the total cost of a display panel,5 making them a key material in display technology. Therefore, to develop the next generation of large-size, flexible, and thin display panels,6 it is crucial to understand and master the fundamental scientific issues in the processing of polarizers while also analyzing and establishing the relationship between macroscopic properties and microscopic structures.
The most commonly used and best-performing polarizers are H-type polarizers, which use poly(vinyl alcohol) (PVA) as the base material and inorganic iodine as the dichroic substance.7 Polarizers consist of a polarizing film, supporting film, compensation film, and optical clear adhesives (OCA) between the layers. The key material that performs the polarization function is the highly oriented iodine-doped PVA polarizing film, which determines the key optical properties of the polarizer, such as polarization, transmittance, and color tone. Owing to the presence of abundant hydroxyl groups in the side chain of PVA and its crystallinity, PVA films can swell in water for quite a long time without dissolving. Additionally, considering that its melting temperature (∼220 °C) is close to its thermal decomposition temperature (∼230 °C),8 wet processing is preferred, in which water is used as the solvent. As shown in Fig. 2, the main processing steps for polarizers are divided into three stages: pre-processing, middle-processing, and post-processing. Pre-processing involves treating the compensation film (such as tri-acetyl cellulose, TAC film) with an alkaline solution, followed by washing, drying, and winding to reduce its contact angle. Middle-processing involves the stretching and laminating of the PVA film. The PVA film is formed by casting a PVA aqueous solution onto a roll-to-roll line. Subsequently, the resulting PVA film is subjected to a series of steps, including swelling, iodine doping, boric acid (BA) crosslinking, and stretching. During these processes, the originally disordered PVA molecular chains become highly oriented along the machine direction (MD), and the polyiodide ions arrange themselves in an ordered manner. This allows the film to absorb light along MD and transmit light in the tensile direction (TD), perpendicular to MD, resulting in polarized light. After drying, the PVA polarizing film is combined with the pre-treated compensation film (TAC film) through lamination using OCA. Post-processing involves coating OCA and composite release films, among other processes, to produce the final polarizer.
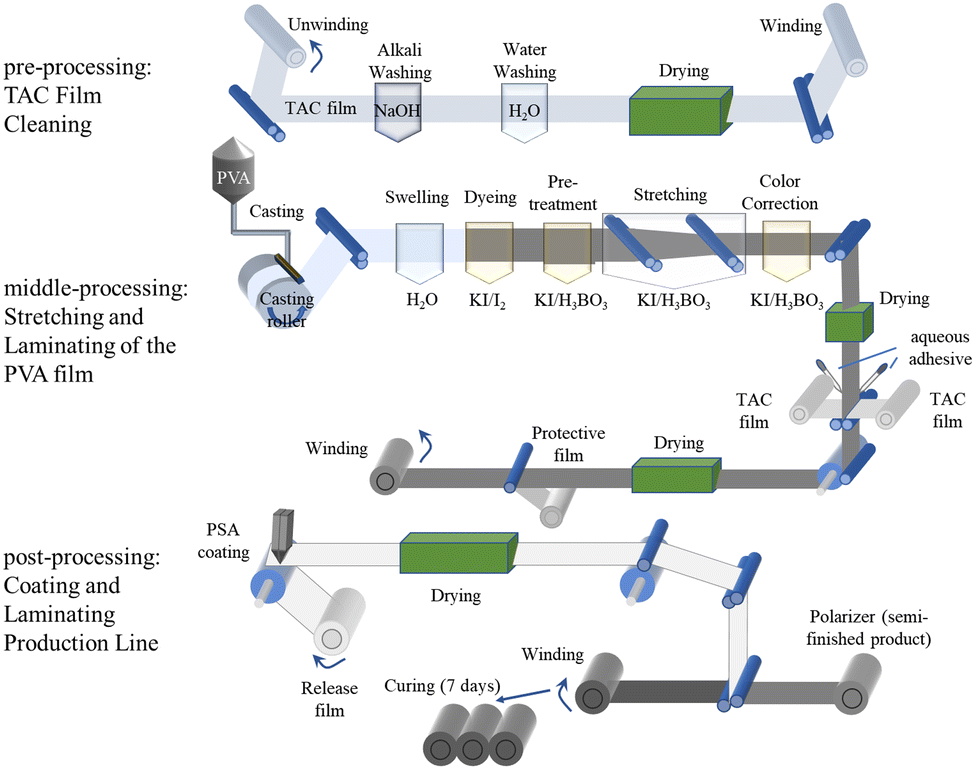 | ||
| Fig. 2 Main steps in the roll-to-roll processing of PVA-based polarizers. | ||
From a scientific viewpoint, the following two aspects are of great interest: (1) PVA film formation mechanism and (2) manipulation of various secondary interactions during processing. As a non-equilibrium process, the competitive and synergistic influence of supercooling and supersaturation on PVA film formation complicates its understanding. Additionally, various phase transitions coexist, including gelation, crystallization and glass transitions. Therefore, advanced analytical techniques with in situ measurement capabilities are required. The precise quantification and control of various secondary interactions during the processing of PVA-based polarizers are prerequisites for producing high-quality polarizers. Secondary interactions, including hydrogen bonding, boronate ester bond, and PVA–iodine complex, are involved during this process. Focusing on the above-mentioned two issues, the current review summarizes the recent developments in understanding the changes in the structure and chain dynamics of PVA-based polarizers during their processing. Firstly, the key chemical structural features of PVA are presented. Later, studies related to the PVA film formation mechanism are summarized, where in situ synchrotron radiation X-ray scattering (SRXS) investigations and skin–core structure studies are summarized. Moreover, various secondary interactions investigated by advanced analytical techniques, such as synchrotron radiation and solid-state nuclear magnetic resonance (SSNMR), are summarized to show the possibility of precisely quantifying the above-mentioned interactions at the molecular level. Finally, our personal perspective on optical polymer processing is presented.
Characteristic chemical structure features of PVA
The characteristic chemical structural features of PVA play a crucial role in the production of polarizing films. At the molecular level, any factors or defect structures that can alter the distribution of hydroxyl groups along the PVA chains, such as the degree of hydrolysis, sequence length, and the content and distribution of 1,2-diol structures, will significantly impact the complexation behavior of PVA and iodine and its orientation during stretching. These factors directly impact the optical performance of PVA-based polarizing films. The molecular weight of PVA as well as its distribution are closely related to the wet processing and the drawability of the as-obtained PVA film.The first synthesis of PVA dates back to 1924, when Haehnel and Herrmann9 obtained PVA solutions by saponifying poly(vinyl ester) using caustic soda solution. In 1937, Haehnel and Berg10 synthesized PVA through transesterification. Currently, the industrial synthesis of PVA involves free-radical polymerization. As illustrated in Fig. 3, this involves initiating the polymerization of vinyl acetate (VAc) to obtain poly(vinyl acetate) (PVAc), which is then hydrolyzed to yield PVA. Radical polymerization reactions typically lack precise control of the chain structure, leading to various structures and defects within the PVA chains (Fig. 4). These structures and their distributions are key determinants of the structural and performance characteristics of polarizing films.
 | ||
| Fig. 3 VAc polymerizes to form PVAc, and the complete hydrolysis of PVAc to PVA. | ||
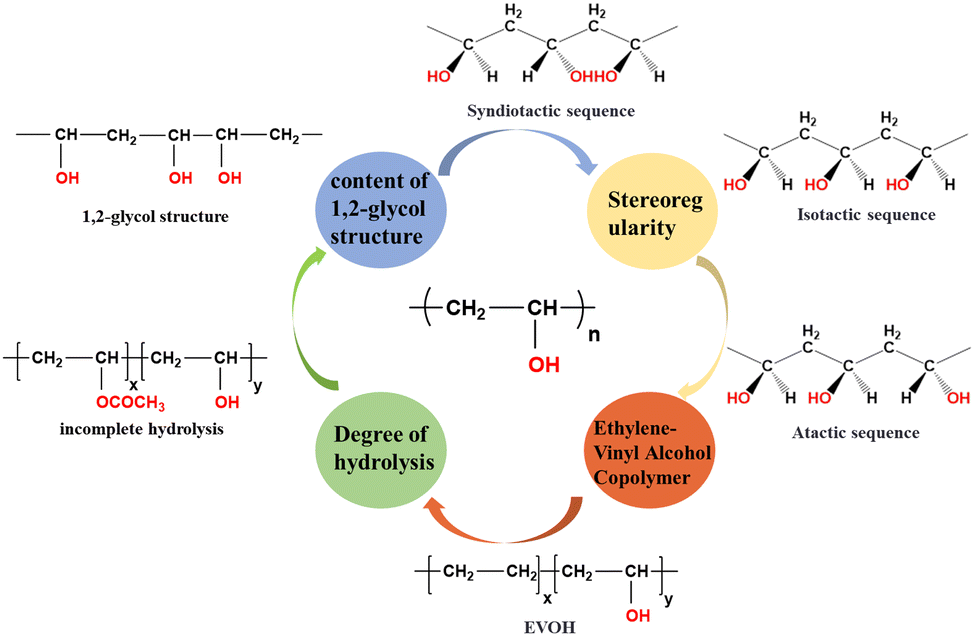 | ||
| Fig. 4 Several key molecular structure features of PVA. | ||
1,2-Diol structures (head-to-head structures)
PVA primarily consists of head-to-tail 1,3-diol structures, where side-chain hydroxyl groups are located on alternating main-chain carbon atoms. However, research has shown that PVA also contains a small amount of 1,2-diol structures (head-to-head connections). Flory et al.11 investigated the relative fraction of 1,2-diol structures in PVA chains by treating PVA with periodic acid (cleaves 1,2-glycol bonds, converting diols to aldehydes or ketones), which selectively attacks 1,2-diol structures. By analyzing the molecular weight of the degradation products, they inferred the relative content of 1,2-diol structures in PVA chains. Their findings indicate that the fraction of 1,2-diol structures is not influenced by the conversion rates, chain initiation, or termination rates, suggesting that these structures arise mainly from the abnormal addition of monomers during chain growth. Additionally, they found that the relative fraction of 1,2-diol structures is primarily dependent on the polymerization temperature. Furthermore, Hirano12 and Mino et al.13 provided evidence for the formation of 1,2-diol structures through selective irradiation-induced backbone cleavage and ceric ion-induced selective oxidation of the PVA backbone, respectively. The presence of 1,2-diol structures can hinder the formation of PVA–I complexes, where when its fraction exceeds a certain threshold, it can prevent the formation of the characteristic blue-violet PVA–I complexes, and the sequences of 1,3-diol structures need to be about 120 units long for effective PVA–I complex formation.14–16Stereoregularity
The stereoregularity of PVA primarily depends on the precursor used. When VAc is used as the precursor, atactic structures are predominantly formed, where the side-chain hydroxyl groups are randomly distributed on either side of the main chain. The use of vinyl ether monomers as precursors facilitates the formation of isotactic structures, whereas vinyl ester monomers tend to produce syndiotactic structures. Syndiotactic-rich PVA can also be prepared through photo-emulsion polymerization and the saponification of vinyl quinine. The stereoregularity of PVA can be estimated using the NMR and infrared spectroscopy techniques. The ratios of isotactic, syndiotactic, and heterotactic triads can be determined by 1H NMR spectroscopy,17,18 while the fraction of pentads can be estimated by 13C NMR spectroscopy. The fraction of dyads can be determined via FTIR, with the absorption peaks at 916 cm−1 and 849 cm−1 corresponding to syndiotactic and isotactic dyads,19 respectively. The syndiotactic structure of PVA more readily forms a complex with iodine ions, and increasing the proportion of syndiotactic structure in PVA facilitates the formation of blue-violet PVA–I complexes.14–16,20 Lower polymerization temperatures yield PVA polymers with higher syndiotacticity.21 Additionally, the stereoregularity of PVA significantly influences its solubility in different solvents, thermal stability, and mechanical properties.17,22–24Degree of hydrolysis
PVA is obtained by the hydrolysis of PVAc, and the degree of hydrolysis (or alcoholysis) significantly impacts the physicochemical properties of PVA. Highly hydrolyzed PVA contains more hydrogen bonds, exhibiting higher crystallinity and better mechanical strength with higher water solubility. In contrast, due to non-crystalline feature of PVAc, low-hydrolyzed PVA shows superior moisture resistance and toughness but reduced thermal stability. Wang et al.25 pointed out that PVA with different degrees of hydrolysis forms distinct complexes with iodine ions. Low-hydrolyzed PVA (88%) forms similar PVA–iodine complexes as fully unhydrolyzed PVAc, suggesting molecular-scale heterogeneity during the hydrolysis of PVAc. After the hydrolysis reaction, some molecules still retain sufficiently long sequences of PVAc.26–28 This implies that not all segments of PVAc chains undergo hydrolysis uniformly, leaving regions that are structurally similar to the original PVAc.Ethylene-vinyl alcohol copolymer (EVOH)
Generally, EVOH consists of hydrophilic vinyl alcohol monomers and hydrophobic ethylene monomers arranged randomly. EVOH is typically prepared by the hydrolysis of ethylene-vinyl acetate copolymers. Ketels et al.29 demonstrated through the 13C NMR spectra of EVOH copolymers that, similar to PVA, EVOH can contain not only 1,3-diol structures but also 1,2-diol structures. Mori et al.30,31 developed methods for preparing sequence-controlled EVOH using group transfer polymerization to synthesize alternating ethylene-vinyl alcohol copolymers. The ethylene content in EVOH can be determined using NMR spectroscopy and dynamic thermogravimetric analysis (TGA). The ethylene content is crucial for determining the structure and properties of EVOH. Pure PVA exhibits excellent gas barrier properties in low-humidity environments due to the crystallinity and dense structure imparted by its side-chain hydroxyl groups. However, the abundance of hydroxyl groups makes pure PVA highly sensitive to water, limiting its application as a gas barrier material. The stereoregularity and sequence structure of EVOH can be studied using NMR techniques. EVOH contains six possible triads including (E, VOH, E), m (E, VOH, VOH), mm (VOH, VOH, VOH), r (E, VOH, VOH), mr, and rr (VOH, VOH, VOH). High-field NMR can be employed to estimate the relative fractions of these six triads.29To achieve the precise design synthesis of PVA, it is essential to focus on the above-mentioned molecular structural parameters. During the PVA film-forming processing, polydispersity can lead to uneven dissolution and even phase separation, resulting in non-uniform micro-nano condensed state structures within the PVA film. Defects such as crystalline spots, dark spots, streaks, and stress concentration points, which negatively impact the high draw ratio and uniform iodine dyeing effects, restrict its further application in downstream polarizing film processing. Therefore, initially, the precise synthesis design and uniformity control of PVA at the molecular scale are critical issues that need to be addressed to advance the manufacturing technology of PVA optical films. Ensuring molecular-level uniformity is the key to overcoming challenges related to dissolution behavior, phase compatibility, and achieving defect-free films with optimal optical properties.
Solution casting of PVA films
In industry, PVA films are typically produced through the casting of PVA aqueous solutions, namely, wet processing. This process begins with dissolving PVA resin, water, glycerol, and surfactants at high temperatures to create a homogeneous, highly concentrated solution. Subsequently, the solution is transported to the casting zone, where it is extruded through a die to form a thin film. Initial shaping takes place on the first casting roll, followed by drying and annealing steps to strengthen the internal structure and enhance the dimensional stability of the film. The casting zone is critical, given that it facilitates the transition from a concentrated solution to a gel film through solvent evaporation and molecular reorganization, involving diffusion, crystallization, and sol–gel transitions.Although extensive studies have been reported on PVA gelation, they primarily focused on low-concentration solutions, where phase transitions are induced by supercooling.32,33 In contrast, the production of PVA films requires high-concentration solutions (∼30 wt%) and elevated temperatures, where phase transitions are driven by supersaturation resulting from solvent evaporation. Alternatively, significant progress has been made in understanding the principles of crystallization and phase separation;32–36 however, systematic studies on the specific underlying mechanisms in PVA film processing remain limited.37–41
The transition from a homogeneous solution to a physical gel film marks the onset of structural heterogeneity. Features such as gel networks, frameworks, and embedded PVA crystals collectively determine the micro- and nano-scale condensed structures.34,42 For the preparation of PVA films in the laboratory, the crystallization-driven gelation of PVA is normally performed under mild conditions, where supersaturation and supercooling promote the formation of smaller, more uniform crystals. However, in industrial casting processes, kinetic factors such as fast solvent evaporation and cooling rates often play a dominant role.34,35,43,44
To capture the PVA film formation process mimicking industry processing conditions, Cheng et al.45 developed an in situ casting device combined with SRXS to investigate the multiscale structural evolution of PVA films under various processing conditions, providing insights into their drying mechanisms (Fig. 5a and b). Using small-angle (SAXS) and wide-angle (WAXS) X-ray scattering, they identified four distinct stages in the drying process of PVA films (Fig. 5c). In the first stage (regime I), water evaporates uniformly, and no crystallization occurs. In the second stage (regime II), the rapid evaporation of water in amorphous regions significantly increases the crystallinity of PVA, leading to a sharp decrease in the long period. During the third stage (regime III), evaporation slows, and the reduction in the long period becomes less pronounced. In the fourth stage (regime IV), macroscopic evaporation ceases, and newly formed lamellae further reduce the long period. In addition to the structural evolution captured through SRXS experiments, the molecular dynamics during the formation of PVA casting films were also examined. Low-field-NMR (LF-NMR) measurements revealed that at higher drying temperatures (e.g., 65 °C), the PVA chains in 10 wt% solutions exhibited greater mobility, predominantly in the amorphous state (Fig. 5d). In contrast, lower temperatures showed distinct multicomponent behavior (Fig. 5e). By fitting the data obtained at various temperatures and concentrations, combined with the SRXS results, a two-dimensional “temperature-concentration” counter map was constructed in this study, identifying the crystallization onset points (Fig. 5f). The results indicated that the chain mobility was the highest at the crystallization onset temperature of 80 °C, followed by 65 °C, and the lowest at 50 °C and 95 °C. These findings elucidate the roles of supercooling and supersaturation in the crystallization and chain dynamics of PVA.
 | ||
| Fig. 5 (a) Schematic of a portable homemade film-casting apparatus. (b) Illustration of the general strategy for the in situ capture of PVA film formation using SRXS. (c) Microstructural evolution of a PVA cast film during drying at 95 °C. (d) MSE-FID curves of 30 wt% PVA at different temperatures of 50 °C, 65 °C, 80 °C, and 95 °C. (e) MSE-FID curves at different PVA weight fractions at 65 °C. (f) Contour plot of the PVA chain mobility in two-dimensional temperature–ωPVA space. Reprinted in part with permission from ref. 45 copyright 2024, the American Chemical Society. | ||
During the casting and drying of PVA films, differences in the solvent evaporation rate and the internal phase transition often result in a distinct skin–core structure. This structure exhibits significant physical and chemical disparities between the outer skin layer and the inner core layer, which critically impact the optical and mechanical properties of the film.46 Rapid solvent evaporation has been shown to drive the separation of crystalline and amorphous phases in PVA, leading to the formation of a skin layer.47,48 This process is closely linked to an increase in solution concentration from water evaporation and a corresponding rise in the glass transition temperature.49,50 When solvent evaporation at the surface outpaces that in the bulk, a glassy layer can develop at the air-solution interface, further impeding solvent removal from the core layer.51 This structural heterogeneity between the skin and core introduces variability in film performance,48,50 as consistently demonstrated in experimental studies.
Fu et al.52 introduced aggregation-induced emission (AIE) fluorescent molecules into PVA solutions to monitor the in situ evaporation process of the solvent. Their study identified two critical fluorescence intensity inflection points at solution concentrations of approximately 25% and 45%, corresponding to gelation and the transition from amorphous to crystalline states, respectively. These findings clarify the phase transition dynamics of PVA solutions during drying and offer crucial experimental evidence for understanding the evolution of the skin–core structure. Li et al.53 investigated the effects of plasticizers on regulating the skin–core structure in PVA films, as illustrated in Fig. 6. They found that plasticizers significantly prolonged the deceleration drying stage and improved the film uniformity by slowing the rapid formation of hydrogen bonding networks. This modulation of the hydrogen bond dynamics effectively suppressed the development of a skin–core structure, thereby enhancing the mechanical properties of the film.
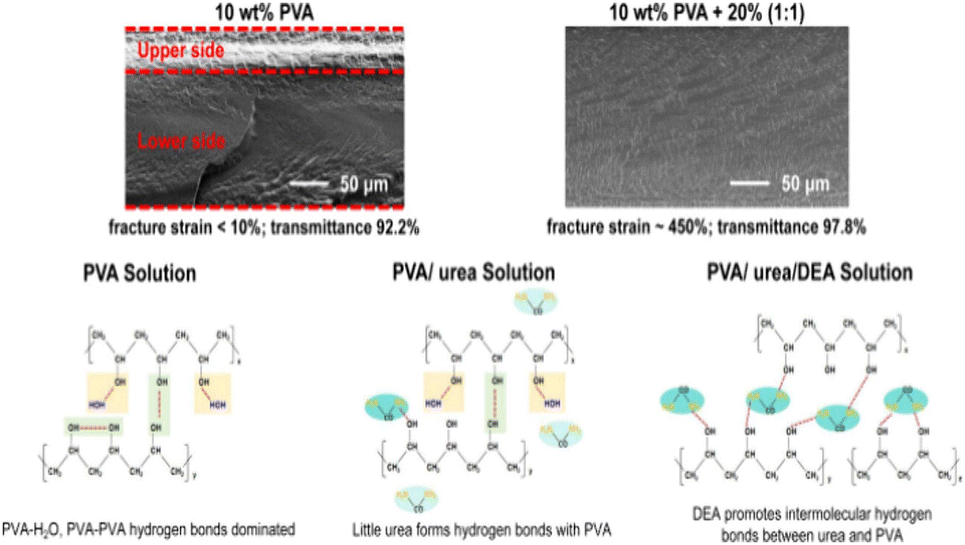 | ||
| Fig. 6 Schematic of the influence of urea and diethanolamine on the microstructure and properties of PVA films. Reprinted in part with permission from ref. 53 copyright 2024, the American Chemical Society. | ||
The impact of initial solution concentration on drying behavior has also been demonstrated experimentally. Higher initial concentrations were found to promote the formation of pronounced skin–core stratification, whereas lower concentrations favored more uniform structures.54 Additionally, researchers observed that under high airflow conditions, films are prone to skin layer formation on the surface, leading to surface defects and compromised mechanical performance by the coating.55
Drying techniques play a crucial role in the development of the skin–core structure. Over the past few decades, numerous experimental, theoretical, and numerical simulation studies have been conducted on the drying processes of polymer films under natural convection, forced convection, and radiation.56–67 Conventional hot air convection drying has been shown to be relatively inefficient, whereas infrared drying, with its higher power density (approximately 6–10 times that of hot air drying), significantly accelerates solvent evaporation, while maintaining the internal structure of the material.68–70 This method not only improves the drying efficiency but also reduces skin layer formation, leading to improved film uniformity.
Building on experimental findings, modeling approaches have been employed to delve deeper into the underlying mechanisms for the formation of the skin–core structure during the drying process of PVA films. Ngui et al.35 developed a mathematical model to predict the isothermal drying kinetics of semicrystalline polymer films, showing that an increase in crystallinity during drying significantly reduces the solvent diffusion rates. This model incorporates the crystallization kinetics, glassy–rubbery transitions, and skin layer formation, enabling the prediction of film thickness, skin layer thickness, solvent removal rate, and crystallinity evolution over time. These predictions showed strong agreement with experimental data.
Wong et al.71 extended this work by developing a mathematical model tailored for multicomponent systems, focusing on the formation of glassy skin layers and their effects on the drying of semicrystalline polymers. They demonstrated that polymers with a high Tg may exhibit rubbery behavior at room temperature in the presence of solvents, transitioning to a glassy state as the solvents are removed. Their model incorporated diffusion-induced crystallization and glassy–rubbery transitions, utilizing the Vrentas-Duda free-volume diffusion theory and crystallization kinetics to predict the drying behaviors in a PVA-water–methanol system. This model accurately reproduced the experimental results.
Given the complexity of skin–core structure formation, different numerical simulation methods offer complementary advantages. Continuum transport models effectively describe the macroscopic mass transfer during solvent evaporation, particularly the dynamic evolution of temperature and concentration fields, but they struggle to capture microscopic structural details. Phase-field models can simulate the interface evolution, making them suitable for studying phase separation and skin–core formation, although they lack detailed molecular descriptions. Molecular simulations provide insights into molecular-level dynamics, such as solvent evaporation, phase separation, and polymer chain motion, but their time and spatial scales limit their applicability to real-world drying processes. Thus, to overcome these limitations, multiscale simulation approaches have emerged as the dominant trend in studying PVA film drying processes. By integrating the strengths of continuum transport models, phase-field models, and molecular simulations, multiscale methods bridge microscopic dynamics and macroscopic drying characteristics. These approaches enable the more accurate prediction of the concentration field, mass transfer parameters, and overall mass transfer efficiency, providing essential theoretical foundations and technical support for optimizing the drying processes and improving the film performance.
Plasticized PVA Films
The molecular chains in PVA are rich in hydroxyl groups, which form numerous inter- and intra-molecular hydrogen bonds, resulting in a significant increase in modulus and a reduction in toughness. Furthermore, the thermal decomposition temperature of PVA (∼230 °C) is close to its melting point (Tm ∼ 220 °C), as mentioned above, and its high viscosity poses challenges for thermoplastic processing, which restricts its applications to some extent. Thus, to improve the flexibility and processability of PVA films, while reducing their hardness and increasing their impact resistance and fracture toughness, plasticizers are often added. Plasticizers alter the material properties of PVA significantly through physical and chemical interactions with the PVA molecular chains. Currently, there are three main theories of plasticization, i.e., the lubrication theory,72–75 gel theory75,76 and free volume theory.75,77 (1) Lubrication theory argues that small-molecule plasticizers fill the space between polymer chains, thereby weakening secondary bonds such as hydrogen bonds and van der Waals forces, which reduces the energy needed for chain movement. In this model, plastics are seen as layered structures, where plasticizers diffuse into and between the polymer chains, decreasing the inter-chain friction. This allows macromolecular chains to glide over each other when the material is bent or stretched. (2) According to the gel theory, plasticized polymers create a three-dimensional network, wherein plasticizer molecules bind to polymer chains through secondary bonding, breaking down the interactions between polymer chains and reducing the number of cross-linking points. (3) Free volume theory indicates that upon cooling from a molten state, the presence of plasticizers increases the free volume within the polymer and enhances the chain mobility. Chandola and Marathe applied this theory to forecast the post-plasticization properties of polyvinyl chloride (PVC) using 25 distinct plasticizers. Their predictions were based on the chemical structures of plasticizers, such as molecular weight, number of end groups, and flexible side chains, and the physical properties of polymers, including viscosity, particularly focusing on changes in Tg.Common plasticizers for PVA include water,78–81 polyols,82–88 amides, alkanolamines, ionic liquids, and urea, with glycerol being the most widely used plasticizer.82,85–88 Xiang et al.82 investigated the effects of stretching temperature and draw ratio on film properties using glycerol as a plasticizer. Their research showed that as the stretching temperature increased, both the Tm and Tc of PVA increased, while the tensile strength and Young's modulus of the PVA films first increased, and then decreased. The tensile strength and Young's modulus reached their maximum values of 197.2% and 470 MPa, respectively, at a temperature of 80 °C. Additionally, as the draw ratio increased, the orientation degree of the PVA molecular chains increased, indicating enhanced chain regularity. Xie et al.89 elucidated the molecular mechanisms by which glycerol exhibits superior plasticizing effects compared to other polyols on PVA using LF-NMR spin diffusion techniques, as follows, (1) different dynamic dimensionality. 1H spin diffusion NMR was used to check the dimensionality from the aspect of chain dynamics. The glycerol-plasticized PVA system has a dynamic dimensionality of 2 (cylindrical structure), whereas others have a dimensionality of 1 (lamellar structure). (2) Chain dynamic heterogeneity in the interphase. With respect to the chain dynamic heterogeneity in the interphase, the glycerol-plasticized PVA shows the most heterogeneous dynamics, with the smallest proportion of the rigid amorphous fraction (RAF) of 0.507 in the interphase (Fig. 7).
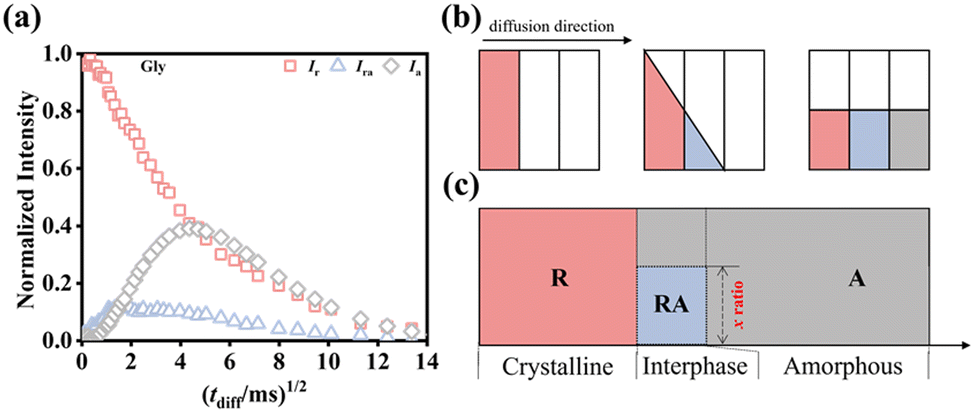 | ||
| Fig. 7 (a) Results of double quantum-spin diffusion (DQ-SD) NMR experiments of glycerol-plasticized PVA film. 1H NMR intensity curves of the three phases at room temperature were established. fx × FID(0, tdiff)/FID(0, tdiff = 0) represents the normalized intensity.90 The normalized (fr, fra, fa) × FID(0, tdiff)/FID(0, tdiff = 0) represents the signal intensity after 1H spin diffusion time tdiff. Note that the FID curve at tdiff = 0 corresponds to the first spin-diffusion data point, fr ∼ 1. The rigid amorphous signal intensity, fra × FID(0, tdiff)/FID(0, tdiff = 0), is usually considered to be part of the rigid. (b) Illustration of the 1H spin diffusion NMR mechanism. (c) Island model (x ratio is the lateral dimension ratio in the interphase). Reprinted with permission from ref. 89 copyright 2024, Elsevier. | ||
In industrial production, to meet the requirements of PVA film service performance under different operating conditions, it is often necessary to adjust the loading amount of plasticizers. Fei et al.91 designed PVA films containing varying amounts of glycerol and investigated their thermal properties, mechanical properties, molecular dynamics and structure. From a thermal perspective (Fig. 8a), as the glycerol content increased, both the crystallization temperature and melting point of the PVA films showed a decreasing trend. In terms of mechanical properties (Fig. 8b), the increase in glycerol content resulted in enhanced toughness of the films, while reducing their modulus. The microstructural changes observed via WAXS and SAXS (Fig. 8c) revealed that the presence of glycerol significantly decreased the crystallinity and increased the size of the amorphous regions, indicating that glycerol effectively influences the amorphous areas of PVA. Furthermore, LF-NMR-based 1H spin diffusion (SD) experiments showed that as the proportion of glycerol increases, the morphology of the PVA films transitions from a lamellar structure (glycerol content below 25 wt%) to a cylindrical structure (glycerol content above 25 wt%). These studies provide detailed insights into the chain dynamics and morphology of PVA-glycerol binary systems.
 | ||
| Fig. 8 (a) Summary of Tm and Tc dependence of the glycerol content and the linear fitting of 1/Tm and 1/Tc. (b) Stress–strain curves of PVA films with different addition ratios of glycerol. (c) Comparison of the crystallinity obtained from WAXS (χc), SAXS (dc/L), and NMR (fc) experiments. Reprinted with permission from ref. 91 copyright 2023, John Wiley and Sons. | ||
Glycerol, as a small-molecule plasticizer, resides in the amorphous regions of PVA. To accurately capture the interphase structure during the sol–gel transition of PVA, Wu et al.92 used plasticizer molecules of different sizes (water and glycerol) as probes to examine the interface structure, employing proton magic-sandwich echo-free induction decay (MSE-FID) NMR and proton double-quantum (DQ) NMR to systematically study the sol–gel transition process of PVA. As shown in Fig. 9, combined with pulsed-field gradient (PFG) NMR, an anomalous phenomenon was observed over time, where instead of decreasing, the mesh size within the system increased somewhat in the later stages of gelation. This phenomenon was interpreted with the gradual formation of a three-phase structure consisting of a “crystalline region/interphase/amorphous region” during the gelation process. It was found that water molecules can penetrate near the crystalline regions, whereas glycerol molecules only reached the vicinity of the interphase. During the evolution of PVA solution crystallization-driven gelation, using water and glycerol, which have two different hydrodynamic radii as probe molecules, the network structure of the interphase was revealed. This directly confirmed the selective plasticization of the two probe molecules in the three-phase structure. Specifically, no plasticizer molecules were present within the crystalline regions; the interphase was selectively plasticized by water molecules; and the flexible amorphous phase was plasticized by both water and glycerol molecules. This directly verified the selective plasticization behavior of the two probe molecules within the three-phase structure and provides a novel method for elucidating the interphase structure of semi-crystalline polymers.
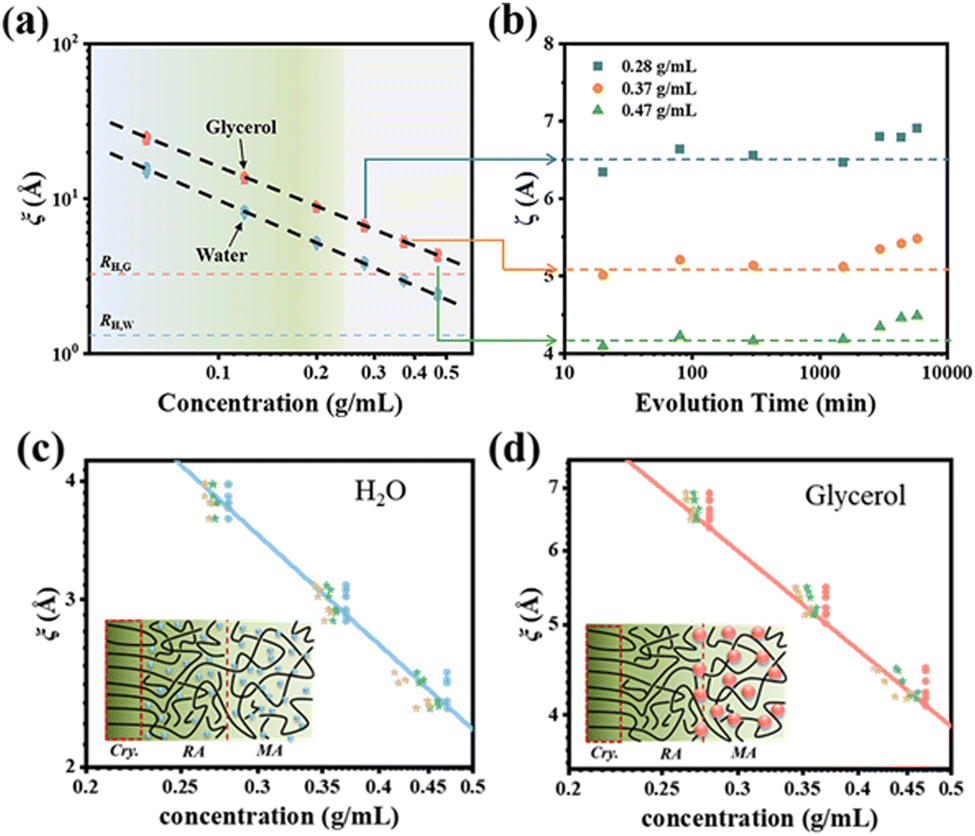 | ||
| Fig. 9 (a) Mesh size ξ as a function of PVA concentration for different evolution times of 20–5780 min obtained from the diffusion data of glycerol (red) and water (blue). (b) Mesh size evolution derived from glycerol diffusion data for the 0.28, 0.37, and 0.47 g mL−1 samples. The mesh size distribution and mechanism diagrams obtained for water (c) and glycerol (d) molecules at different concentrations. Reprinted with permission from ref. 92 copyright 2023, the American Chemical Society. | ||
PVA–iodine complex formation mechanism
The highly oriented polyiodine formed by PVA–iodine complexes is the source of dichroism in polarizing films. Hence, studying the mechanism of PVA–iodine complexes is essential for understanding the optical properties of polarizing films.7,93 The dichroism of polarizing iodine-containing crystals was first observed by Phelps and Herapath94 in 1852, who noticed that these crystals exhibited varying brightness and darkness when overlapped under a microscope. In 1927, Herrmann95 and Staudinger96 independently synthesized PVA and discovered its blue reaction with iodine. In 1928, Land97 dispersed the iodine-containing crystals discovered by Herapath in a viscous nitrocellulose solution and oriented them by extruding them through a narrow slit. The J-type polarizer introduced in 1934 and the H-type polarizer launched by Polaroid in 1938 were both manufactured using this process. The formation of PVA–iodine complexes is influenced by various factors, including the chemical structure of PVA (stereoregularity,98–100 1,2-diol structure,14,98,99 degree of hydrolysis,25,101–104 and chemical modification104,105), and processing parameters (iodine concentration106,107 and swelling conditions108,109). Additionally, the structure of PVA–iodine complexes is intricate, and in-depth studies on their complexation models and mechanisms are crucial for the development of high-performance PVA–iodine-based polarizers.Structure and properties of PVA–iodine complexes at different iodine concentrations
I3− and I5− are the primary forms of iodine ions in iodine complexes. As shown in Fig. 10a, at low iodine concentrations in KI/I2 aqueous solution, I3− ions are preferentially formed due to their relatively stable structure. With an increase in iodine concentration, the number of iodine molecules in the system increases, and I3− further reacts with free iodine molecules to form I5−. However, the situation is entirely different in solid PVA films (Fig. 10b).110 Raman spectra of PVA films dipped in KI/I2 with different iodine concentrations (Fig. 11a) also confirmed that in the iodine-doped PVA film, with an increase in iodine concentration, the predominant form of iodine shifts from I5− to I3−, with an increase in the quantity of I3−,111 as illustrated in Fig. 11b. The iodine concentration is the main factor causing differences in complex formation between different phases of PVA film. At low iodine concentrations, complexation occurs only in the amorphous region, dominated by I5− ions, while the crystalline regions remain PVA crystals;20 at medium and high iodine concentrations, iodine ions invade the PVA crystalline region, forming PVA–I3− complex crystals, and as the iodine concentration further increases, the original PVA crystals gradually disappear.112–114 | ||
| Fig. 10 Schematic of iodine species in (a) iodine solution, (b) solid PVA film, showing its distribution in the amorphous and crystalline regions of a PVA sample dipped in KI/I2 solutions of different concentrations. Reprinted with permission from ref. 110; copyright 2015, the American Chemical Society. | ||
 | ||
| Fig. 11 (a) Raman spectra of PVA films dipped in KI/I2 with different iodine concentrations. (b) Relative fraction of I3− and I5− obtained from peak deconvolution. Reprinted with permission from ref. 115; copyright 2021, the Royal Society of Chemistry. | ||
Polyiodide ions play a key role in polarizing films, making their structural elucidation essential. Many studies aimed to clarify the formation of PVA–iodine complexes in both crystalline and amorphous regions of PVA. In dilute solution, the absorption range of PVA–iodine complexes in the UV absorption spectrum is similar to that of amylose-iodine complexes, both at around 580 nm to 700 nm. Based on this, Zwick et al.116,117 proposed that PVA–iodine complexes possess a helix structure, where the PVA segments form channel-like helices with a period of 12 VAc units, encapsulating linear polyiodide chains. Matsuzawa et al.98 proposed that syndiotactic sequence aggregates in PVA can incorporate iodine ions, where the ions are surrounded by extended PVA chains and stabilized by inter-chain hydrogen bonds. The PVA chains in these aggregates adopt an extended conformation rather than a helical one. These models primarily explain the structure of PVA–iodine complexes in dilute solutions.
In PVA films, at low iodine concentrations (0–0.5 M), when they undergo complexation, I3− and I5− coexist in the amorphous region, with I5− being predominant118–120 (Fig. 14b). I3− typically exists in a linear extended chain state, while I5− can be present in both linear and bent states.121 After the iodine-doped PVA film is stretched and oriented, I3− and I5− do not exist independently within the PVA molecular chains but instead form iodine chains with an average length of about 15 iodine atoms.122 Increasing the orientation degree leads to an increase in the formation of PVA–iodine complexes.123,124 Based on this, Miyasaka et al.15 constructed a structural model of PVA–I complexes in the amorphous region (Fig. 12). Four linear extended PVA chains are stabilized by inter-chain hydrogen bonds spaced about 307 pm apart, forming a planar zigzag conformation that encapsulates the extended polyiodide ions within the PVA–iodine complex. The conformation of PVA chains forming PVA–iodine complexes does not necessarily strictly follow the model; some disordered PVA conformations can also participate in the formation of complexes. This situation is similar to PVA crystallization, where even uncontrolled random PVA from free-radical polymerization can achieve high crystallinity.
 | ||
| Fig. 12 (a) PVA–iodine extended chain model and (b) top view of (a). | ||
Based on the X-ray diffraction patterns of iodine-doped PVA films, Hess et al.125 found that when the iodine concentration exceeds 1.5 × 10−2 mol L−1 or the iodine adsorption content surpasses 12 wt%, new diffraction spots appear along the equatorial direction, indicating that iodine ions begin to enter the crystalline region and form new crystals.126 After stretching, the X-ray diffraction patterns exhibited elongated diffraction signals along the stretching direction (MD), with a repeat spacing of 308 pm,15 which is larger than the repeat spacing of the PVA lattice (250 pm). This suggests that the elongated diffraction signals correspond to PVA–iodine co-crystals, representing a one-dimensional lattice. Herbstein et al.127 calculated that the diffraction peak at 2θ = 9.2° corresponds to the repeat distance of the linear I3− lattice (960 pm), and the arrangement of I3− involves partial distortion. Therefore, the PVA–iodine co-crystal is a PVA–I3− crystal, rather than an I5− form, meaning that as the iodine doping concentration increases, I3− is dominant in the PVA film (Fig. 11b). Tashiro et al.99,128–135 observed the formation of PVA–I3− crystals under different preparation conditions (Fig. 13). Upon entering the PVA crystal lattice, I3− disrupts the hydrogen bonds, causing PVA chain displacement and forming complex I through charge transfer interactions with hydroxyl groups. In complex I, the average occupancy rate of I3− is approximately 0.3, resulting in a superlattice structure with disordered stacking of PVA–iodine pairs and original PVA chains. As the iodine concentration increases further, most of the PVA chains bind with I3−, forming complex II, where the occupancy rate of I3− reaches about 0.7. Through X-ray end diffraction patterns of biaxially oriented samples, it was found that the planar zigzag planes of the original PVA chains are parallel to the rolling planes of the biaxially oriented samples. In complex I, the molecular chains are also parallel to the rolling plane. However, in complex II, PVA chains are not parallel to the rolling plane but are rotated by 38° around the chain axis. It is inferred that iodine ions occupy the original PVA lattice positions, altering the PVA crystal structure through an intercalation mechanism, leading to a transition from a disordered structure to short-range order, and then to long-range order in PVA chains.
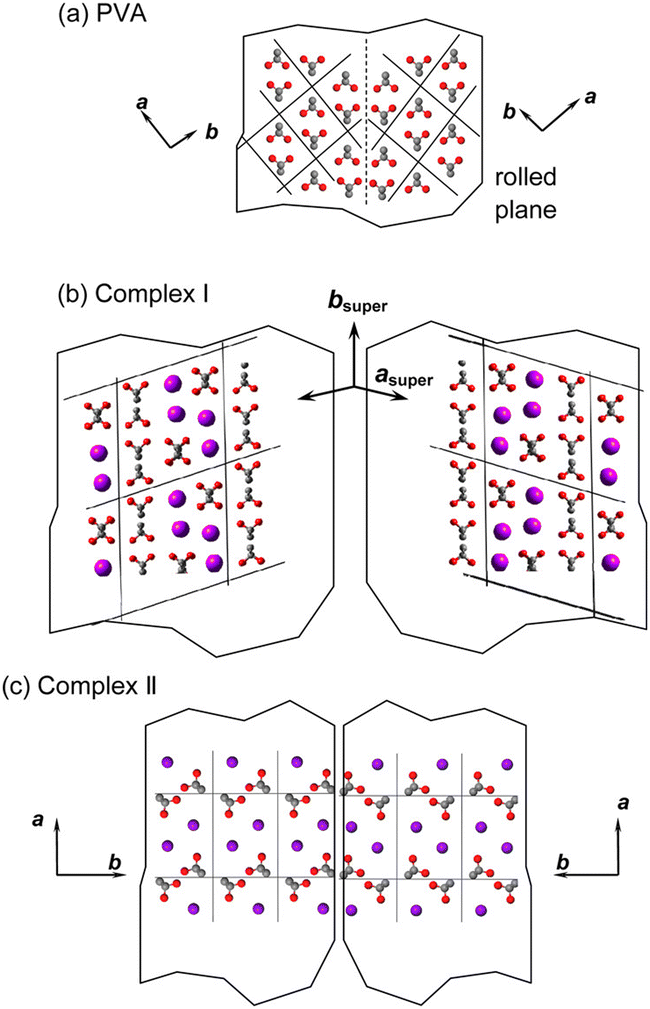 | ||
| Fig. 13 Spatial relation between PVA, complex I and complex II. The twin structures are shown here. (a) PVA: the planar-zigzag planes are parallel to the rolled plane. The intermolecular hydrogen bonds are formed between these chains. (b) Complex I: the PVA chains and PVA–I3 complex form the arrays arranged in a statistically disordered manner. The PVA chains are shifted in the lateral directions from the original positions to create the voids or complex with iodine species. (c) Complex II: the molecular chains rotate about 38° from the original direction when the complex formation is almost completed. Reprinted with permission from ref. 110; copyright 2015, the American Chemical Society. | ||
Based on the influence of iodine concentration on the network structure of the amorphous region in swollen iodine-doped PVA films, Li et al.111 elucidated the formation mechanisms of several types of crystals at different iodine concentrations using the LF-NMR and SRXS techniques. WAXS provided information on the transition of crystal types (Fig. 14a) and divided the structural changes into three regions, as follows: (i) pure PVA crystals at 0 M to 0.1 M; (ii) PVA–I3− complex I at 0.1 M to 1 M; and (iii) PVA–I3− complex II at 1 M to 5 M. The introduction of iodine can reduce the overall chain mobility of the system, but the entanglement density in the amorphous region only significantly decreases when PVA–I3− complex II begins to form at iodine concentrations exceeding 1 M. This is consistent with the inverse trend of the long period L/da/dc with respect to iodine concentration observed in SAXS (Fig. 14b), which reverses after 1 M. Additionally, within the PVA crystalline regions, the molecular chains exhibit no chain motion for ∼100 μs across all three concentration intervals. By combining NMR T2 relaxometry and DQ NMR spectroscopy to quantify the amorphous molecular chain network, the formation mechanism for the PVA–iodine complex is proposed, suggesting that complexation preferentially occurs in the interphase region for the undeformed PVA film (Fig. 14d).
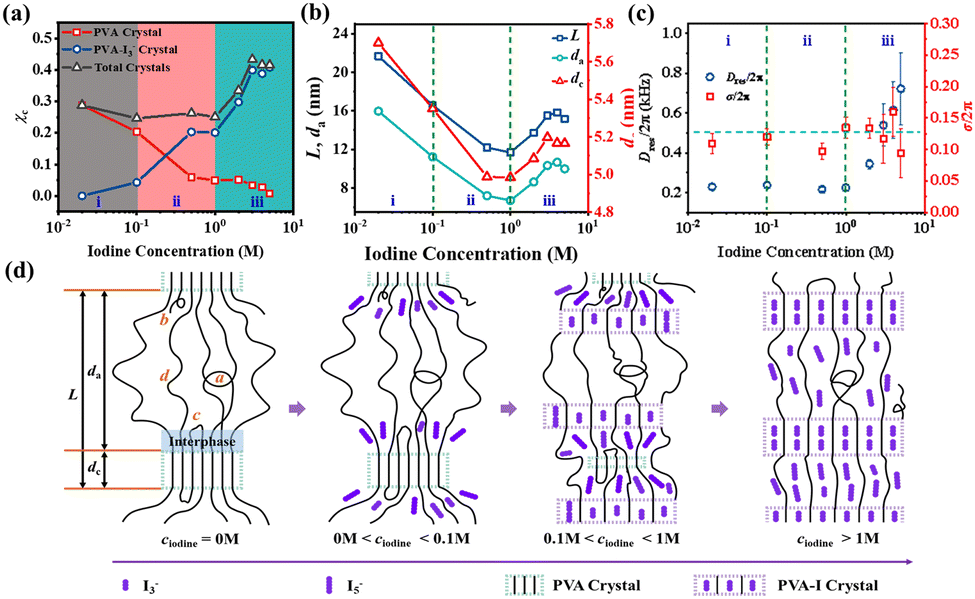 | ||
| Fig. 14 (a) Crystallinity (χc) of the pure PVA crystal, PVA–I3− crystal and total crystals as a function of ciodine. (b) Evolution of L (long period), dc (lamellar thickness) and da (distance between lamellae) as a function of ciodine, (c) change in Dres/2π and σ/2π (distribution of Dres/2π), and (d) formation process of the PVA–iodine complex at different iodine concentrations. Reprinted with permission from ref. 115; copyright 2021, the Royal Society of Chemistry. | ||
Meanwhile, Li et al.132 further analysed the role of iodine doping in the three-phase structural differences of PVA films. The local orientation differences in the molecular chains in the three phases are based on the chemical shift anisotropy (CSA) of nuclear spins. In the crystalline phase, the molecular chains are highly oriented, and a Gaussian distribution curve with a deviation angle β = 1° ± 1° can reconstruct the CH2 peak. In the interphase, approximately 26% of the contribution to the spectrum at 0° comes from chains oriented at an angle of 35° ± 5° relative to the static magnetic field, B0, confirming the disordered all-trans character of the chain conformation in this region (Fig. 15a–d). The amorphous phase exhibits severely broadened spectral lines, indicating an even more random orientation. This also suggests why complexation is more likely to occur in the interphase. By stretching, the conformation entropy differences among the molecular chains in the three phases are reduced, and subsequently the same three-phase selective detection is performed on the oriented samples. It was found that the three-phase spectra of the unoriented iodinated sample (0.5 M) and the oriented sample (0 M–5 M) are essentially consistent. This indicates that the local complexation induced by linear polyiodine ions and the macroscopic orientation induced by stretching have the same effect on the local chemical environment of the PVA chain segments (Fig. 15e).
 | ||
| Fig. 15 (a) Schematic of the orientation of the chain axis and CSA tensors of CH2 (when the stretching direction of the PVA samples was placed at 01 orientation with respect to the B0 field). (b–d) Static three-phase 13C CP spectra measured using the static probe of three phases for the oriented-0 M sample (0 M–5 M) with different orientations relative to the B0 field. (e) Schematic of the effect of macroscopic extension and microscopic complexation on PVA chains. Reprinted with permission from ref. 136; copyright 2022, the Royal Society of Chemistry. | ||
Effect of counter cations on the formation of PVA–iodine complexes
PVA–I complexes exhibit significantly different crystal structures and phase transition behaviors in the presence of various counter cations. Tashiro et al.132 discovered that when PVA films are immersed in iodine solution (KI/I2 aqueous solution) containing different counter cations (such as K+, Na+, Li+, H+, or Cs+), the resulting PVA–iodine complex displays diverse polymorphic forms and phase transition characteristics (Fig. 16). At low iodine concentrations, iodine ions do not affect the PVA crystals. When PVA films are immersed in iodine solutions of (0.5–1 M), iodine ions penetrate and disrupt the original hydrogen bonds between PVA chains, coexisting with PVA crystals to form a superlattice structure known as complex I. As the iodine concentration increases (1–3 M), the planar cooperative rotation of the PVA chains with a zig-zag conformation by 38° results in the formation of complex II. When immersed for sufficient time in a 3 M iodine solution, especially in the presence of H+, the unit cell of complex II undergoes alternating ±a/4 slips along the a-axis, forming the most stable complex III. This slip phenomenon is not observed in the presence of K+, Na+, and Li+. In the case of Na+ and Li+, complex II tends to partially transform into complex III, accompanied by slip phenomena, but this transformation is incomplete, leading to disordered structures or local cell disorder in complex II. In contrast, for K+, no such disorder occurs, where complex II remains the final stable form under K+ conditions. The larger size of K+ likely prevents the slippage of PVA–I structures along the a-axis via I3−, indicating that the cation size is a key factor influencing the transformation from complex II to complex III.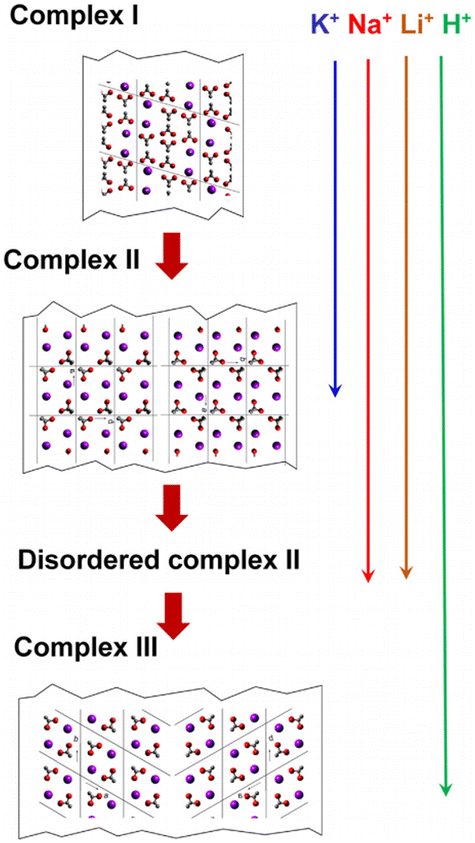 | ||
| Fig. 16 Schematic of the crystal structure changes from complex I to complex II, disordered complex II and complex III. Reprinted with permission from ref. 132 copyright 2016, Elsevier. | ||
Effect of the intrinsic chain structure of PVA on the formation of PVA–iodine complexes
At the molecular level, differences in PVA chain structures significantly impact the formation and properties of PVA–iodine complexes. Studies have shown that variations in the degree of hydrolysis of PVA chains and ethylene content importantly influence the formation mechanism and performance characteristics of PVA–iodine complexes. In PVA chains, two types of monomers exist due to their varying degrees of hydrolysis, VAc and VOH. Within the PVA chains, the VAc units tend to form helical chain structures, whereas the VOH units maintain a planar zigzag conformation. P(VAc-co-VA), with different degrees of hydrolysis, forms a complex with iodine not only through the VOH segments but also the residual VAc segments.Tashiro et al.137 immersed films make from amorphous PVAc into iodine solutions, obtaining two types of crystalline complexes, i.e., α-form and β-form. At low iodine concentrations, the predominantly formed complexes are of the α-form, where the predominant form of iodine ions is I3−. As the iodine concentration increased, the complex gradually transformed into the β-form, where the main form of iodine ions become I5−. However, definitive crystal structures for the α-form and β-form have not been established to date, where two possible structural models have been proposed, i.e., iodine ions sandwiched between helical chains and iodine ions confined within double-helical chains (Fig. 17). Unlike PVA–iodine complexes, which primarily form crystalline complexes via hydrogen bonding interactions between –OH groups and iodine, PVAc-iodine complexes are mainly form through electrostatic interactions. In PVAc, its ester groups (–COO–) carry a negative charge, while the carbon–hydrogen units bear partial positive charges. Therefore, the formation of PVAc–iodine complexes is induced by electrostatic interactions between the positively charged carbon–hydrogen units and negatively charged iodine ions, which are dominant in the process for the generation of PVAc–iodine complexes.
 | ||
| Fig. 17 Models showing the formation of iodine complexes. Reprinted with permission from ref. 137; copyright 2020, the American Chemical Society. | ||
In P(VAc-co-VA) copolymers, inter-chain hydrogen bonds of the OH⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C type exist not only between the VA units but also between VAc and VA units. Tashiro et al.25 found that after immersing the prepared samples in an iodine solution, complexes were not only formed in the crystalline regions but also in the amorphous regions. As shown in Fig. 18, when immersed in a 3 M iodine solution, at a low VA content, the long VAc segments form complexes with the iodine ions. As the VA content increases, PVA–iodine complexes begin to form and the PVA segments gradually lengthen and start forming PVA–iodine complexes, coexisting with PVAc–iodine complexes. This indicates that as the VA content increases, the PVA segments can more effectively interact with iodine ions, leading to the formation of more ordered crystalline structures.
C type exist not only between the VA units but also between VAc and VA units. Tashiro et al.25 found that after immersing the prepared samples in an iodine solution, complexes were not only formed in the crystalline regions but also in the amorphous regions. As shown in Fig. 18, when immersed in a 3 M iodine solution, at a low VA content, the long VAc segments form complexes with the iodine ions. As the VA content increases, PVA–iodine complexes begin to form and the PVA segments gradually lengthen and start forming PVA–iodine complexes, coexisting with PVAc–iodine complexes. This indicates that as the VA content increases, the PVA segments can more effectively interact with iodine ions, leading to the formation of more ordered crystalline structures.
 | ||
| Fig. 18 Formation process of boronate ester bonds between PVA and BA. Reprinted with permission from ref. 25; copyright 2023, Elsevier. | ||
Moreover, the ethylene content in EVOH also influences its complexation structure with iodine ions by affecting the length of its –OH segments. When the ethylene content in EVOH is below 73% (vinyl alcohol content above 27%), its chain structure retains a planar zigzag conformation similar to PVA.138 Under certain conditions, EVOH with a zigzag chain structure can undergo an iodine staining process similar to PVA–iodine complexes, where iodide ions (I3−) form columnar structures and create a crystalline complex via charge transfer interactions with the hydroxyl groups of the VA units in the EVOH chains. The introduction of ethylene alters the original sequence structure and sequence length of the VA units in the PVA chains. Unlike VA units, the ethylene units in EVOH cannot form a complex with iodine ions, where only continuous VA sequences of sufficient length can complex with iodine ions.
Tashiro et al.131 investigated the complexation reactions of films made from four different EVOH samples with varying ethylene contents (0%, 32%, 52%, and 75%) when immersed in iodine solutions. At an iodine concentration of 3 M, EVOH100 (0% ethylene) formed complex II, while EVOH68 (32% ethylene) and EVOH52 (52% ethylene) only formed complex I, and EVOH25 (75% ethylene) did not form any complex. As the ethylene content increased, the corresponding VA content in the chain segments decreased, altering the types of complex formed at the same iodine concentration. When the VA content decreased to a certain level, even immersion in high-concentration iodine solutions failed to form PVA–iodine complexes. Statistical analysis revealed that VA sequences of at least four units are required to form complexes with iodine.
Effect of external processing parameters on the formation of PVA–iodine complexes
In the actual production of polarizing films, the processing steps are extremely complex. Thus, to better understand the impact of various external fields applied during these steps, each external field has been systematically analyzed.106,139,140 Using the WAXS and SAXS techniques, researchers have elucidated the evolution patterns of PVA–iodine complex structures influenced by temperature, stretching, and boric acid concentration.108,109,114,129,141–144 Zhang et al.140 studied the structural evolution of PVA films stretched in water at different temperatures, finding that an increase in temperature led to an expansion of the inter-fibrillar spacing and more perfect stacking of nanofibers. Zhang et al.139 further investigated the fibrillation and complexation reactions of PVA films stretched in KI/I2 solutions of varying concentrations, demonstrating that both iodide ions and the drawing field have synergistic effects on the formation of PVA–iodine complexes, with the drawing field promoting complexation through an entropy reduction effect, while the iodide ion concentration controls the crystallization process of PVA–I3− co-crystals. Ye et al.145 extended this research to study the structural evolution of PVA during stretching in mixed iodine and BA solutions, dividing the deformation process of PVA into elastic, yielding, and hardening stages. At low iodine concentrations, PVA exhibits no obvious yield and behaves like a rubber; as the iodine concentration increases, the crystallinity of PVA–I3− co-crystals decreases; the fractured crystals serve as precursors for PVA-polyiodide complex, which upon further stretching form PVA–I3− co-crystals. An appropriate amount of boric acid promotes the formation of polyiodides, whereas excess inhibits it. Nanofibers lead to strain hardening (Fig. 19).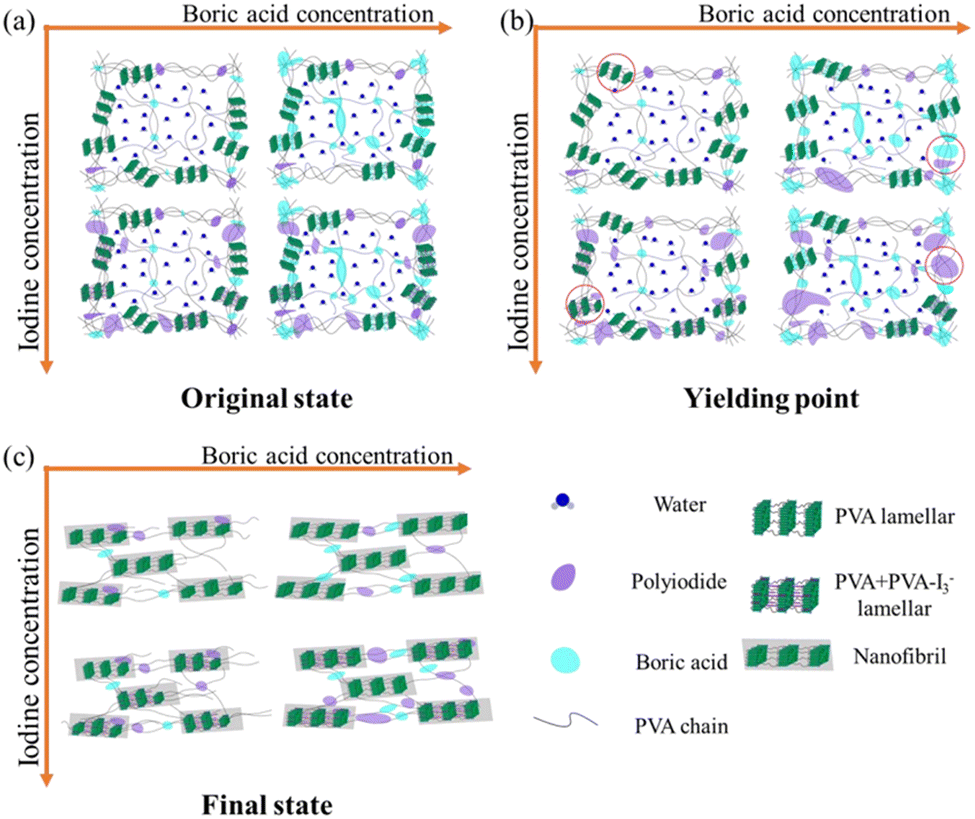 | ||
| Fig. 19 Schematic of the crystal structure changes from complex I to complex II, disordered complex II and complex III. Reprinted with permission from ref. 145; copyright 2021, Elsevier. | ||
Based on previous work, the following findings have been observed: (1) an increase in iodine concentration promotes the formation and periodic arrangement of PVA nanofibers, reducing the onset strain for fibrillation. (2) Elevated temperatures increase the spacing between nanofibers and improve their arrangement, which are beneficial for preparing polarizer materials. (3) An increase in draw ratio leads to PVA crystal melting, nanofiber formation, and periodic arrangement. (4) An appropriate amount of BA aids in polyiodide formation but can inhibit its growth when in excess. These factors collectively influence the evolution of PVA–I3− co-crystal structures, providing a basis for developing high-performance polarizing materials.
In addition to the crystal structure of PVA–iodine complexes, challenges remain related to their formation kinetics pathway. Why does the introduction of iodine species into PVA films result in the formation of an ordered PVA–iodine complex structure? Why does only I3− species exist in the crystalline region rather than I5−? Also, as mentioned above, PVAc can also form crystalline complexes, whereas PVAc is non-crystalline. This suggests that the charged PVA or PVAc chains start to transform from a disordered structure to a locally ordered structure (short-range ordered structure), and finally long-range ordered crystal structure. However, the repulsion force of PVA–iodine complexes due to the Coulomb effect prevents their aggregation, which is opposite in experiment. Thus, efforts to clarify the above-mentioned concerns are required.
Considering the above-mentioned complex crystal structure analysis and crystallization kinetics results,131,135 the formation of PVA–iodine complexes is proposed to be a complexation-driven ordering process, as described below. In the initial stage, in the absence of iodine, the PVA chains adopt a random coil structure, which is characterized by a high conformation entropy-dominated isotropic state, leading to an overall disordered structure.131 Upon introducing KI/I2 solution, iodine ions (such as I3− and I5−) interact with the hydroxyl groups on the PVA chains through charge-transfer interactions,99 resulting in the formation of rigid columnar structures. This complexation significantly reduces the conformation entropy of the PVA chains, inducing preliminary chain alignment in localized regions, corresponding to a transition into a nematic-like state.131 As the iodine concentration further increases, the columnar structures begin to come into contact with each other, leading to closer interactions and a tendency to align orderly.135 The system transitions into a smectic-like phase with long-range orientation and position order. This structural formation is driven by a further reduction in orientation entropy, which is essentially complexation-driven ordering.130 The essence of this process is the increase in rigidity (reduction in conformation entropy, driven by the enthalpy of complexation) and the reduction in translational entropy (due to excluded volume effects), consistent with Maier–Saupe's theory146,147 considering both the anisotropy of the basic unit and inter-unit interaction (Fig. 20). Specifically, the combination of the excluded volume potential induced by the rigid PVA–I complex and inter PVA-l interaction (i.e. hydrogen bonding) drives the phase transition from a disordered phase to ordered phase.148 Additionally, the presence of the ordered phase can enhance the nucleation process.149 Continuous efforts to elucidate this complexation-driven ordering process are still required, especially experimental efforts determine the influence of concentration and temperature as well as flow on the phase transitions among different complexes.
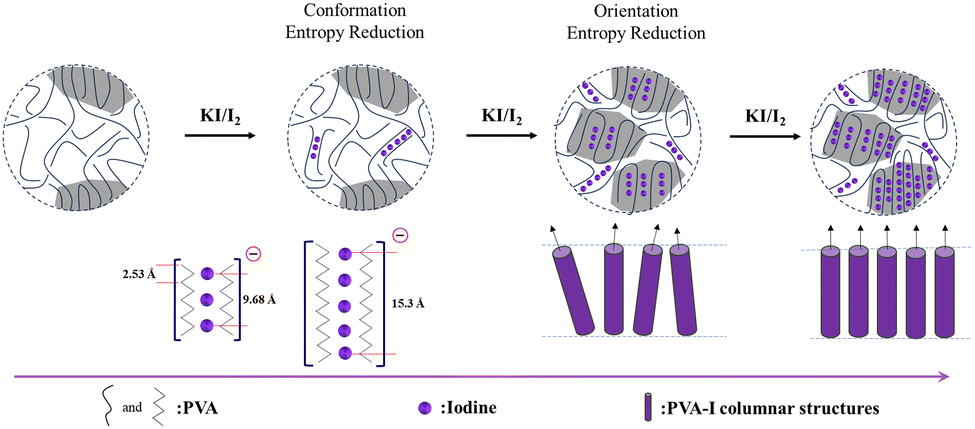 | ||
| Fig. 20 Schematic of complexation-driven ordering. | ||
BA Crosslinked PVA
The use of BA cross-linked PVA is one of the significant methods to enhance the thermal processing properties,142,150 mechanical properties150 and stability105,116,151 of PVA polarizing films. Without BA, the PVA–iodine complexes can remain stable for a certain period, but under practical application conditions and harsh environments such as high temperatures15,152 and high humidity,152 PVA–iodine complexes will gradually degrade, leading to the dissociation of polyiodide ions152 and the failure of polarization effects.153 After adding BA to the PVA–iodine system during the processing of polarizing films, the durability of the polarizer significantly improves.154,155 Structurally, boron atoms adopt sp2 hybridization, and the structural unit of BA is a planar triangular structure. Due to its electron-deficient structure, it often acts as an electron acceptor in aqueous solution, combining with hydroxide ions to form a tetrahedral B(OH)4− structure. This leads to the formation of boronate ester bonds with the hydroxyl groups in PVA (Fig. 21). | ||
| Fig. 21 Formation process of boronate ester bonds between PVA and BA. | ||
Boronate ester bonds are formed through the condensation of BA derivatives (including both organic and inorganic BA) with cis-1,2- or 1,3-diol.156–159 Since 1959, significant progress has been made in the study of boronate ester bonds, when Lorand and Edwards first investigated the complexation equilibrium between BA and polyols in aqueous solutions. They established the association constant, Ka, by tracking changes in pH.160 In solutions with a pH higher than the pKa of BA, the boron atom transitions from an sp2 hybridized planar configuration to an sp3 hybridized tetrahedral configuration, forming hydrophilic five- or six-membered cyclic boronate esters; conversely, under conditions where the solution pH is lower than the pKa of BA, the boronate ester bond dissociates. This reversibility of the boronate ester bond also manifests in its binding affinity, which varies with changes in pH.161,162 For example, molecules with stronger binding affinity (such as monosaccharides) can replace those with weaker binding affinity (such as 1,3-diol), leading to the dissociation and reformation of the boronate ester bond. The boron atom at the center of the boronate ester bond possesses an empty p-orbital, allowing it to combine with Lewis bases that provide lone pairs of electrons, such as nitrogen and oxygen atoms, thereby exhibiting Lewis acidic properties.156,157 For instance, when a nitrogen-containing functional group exists within a molecular chain, charge transfer can occur from the nitrogen atom to the boron atom, forming a directional B–N coordination bond.159
Based on this, there is also a debate regarding the PVA–BA crosslinking structure, including whether it involves 3-coordinated/4-coordinated or mono-cross-linked/di-crosslinked.163–166 The formation of these structures is mainly influenced by the concentration of boric acid solution, with related studies usually proposing structural models within the solution system. In comparison, the situation within the PVA film system is more complex due to its multi-level structures, making it more difficult to elucidate the local cross-linking structure. Tashiro et al.129 reconstructed the crosslinking structure model using model compounds, and through experiments and DFT calculations, they proposed a bridge-like structure model, where two trivalent rings are connected via hydrogen bonds (Fig. 22). This structure effectively restricts the mobility of the molecular chains in the amorphous regions of PVA142 and increases the Tg of the system, thereby enhancing the durability of the PVA film under high temperature and humidity environments, and preventing bending caused by internal stress during processing.129
 | ||
| Fig. 22 Illustration of the PVA–BA cross-linked structure in the PVA amorphous chains. Reprinted with permission from ref. 129; copyright 2014, the American Chemical Society. | ||
Previous work has reported that BA has the ability to inhibit the crystallization of PVA during stretching and promote the formation of PVA–iodine complexes.129,151,167–169 Experiments by Miyazaki et al.143,151 found that BA can promote the orientation of molecular chains in the amorphous region and inhibit stretch-induced crystallization of PVA during stretching. Yang Hun Lee et al.168 discovered that pre-treating PVA films with BA crosslinking can, to some extent, promote the subsequent generation of PVA–iodine complexes. This is due to the restricted effect of cross-linking points allowing slight elongation of the PVA chains, which is favorable for the formation of polyiodide ions. However, some researchers believe that the introduction of BA limits the stretchability of PVA–iodine systems,166 thereby reducing the orientation of the polymer chains and polyiodide ions, and further decreasing the polarization performance. Overall, the role of BA in promoting the generation of dichromatic substances and enhancing force transmission makes the BA cross-linking step indispensable in the processing of polarizing films.129,151,168,169 To achieve the controllable preparation of high-performance polarizing films, understanding how BA affects the mechanical properties of PVA polarizing films is crucial.
Ye et al.106 investigated the structural evolution of PVA during uniaxial stretching at varying concentrations of BA. Using the SAXS and WAXS techniques, they found that an increase in BA concentration can not only promote the earlier emergence of fibrils but also leads to a decrease in fibril content. This study revealed that the stretching process of PVA films at different BA concentrations goes through three stages (Fig. 23). In the first stage, elastic stretching occurs with constant crystallinity, while the interplanar spacing of the (101)/(10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) planes continuously decreases. In the second stage, tensile forces are transmitted to the unit cells, subjecting them to stretching and shearing, which is mainly attributed to the chemical cross-linking points introduced by BA rather than the crystals themselves. In the third stage, when the content of BA is not sufficient enough to maintain the connection of the crystal network, the connection between crystal networks breaks down. Additionally, it was observed that with an increase in BA concentration, the mechanical transition point of PVA films appears at lower strain values, indicating that the addition of BA helps enhance the mechanical properties of PVA.
) planes continuously decreases. In the second stage, tensile forces are transmitted to the unit cells, subjecting them to stretching and shearing, which is mainly attributed to the chemical cross-linking points introduced by BA rather than the crystals themselves. In the third stage, when the content of BA is not sufficient enough to maintain the connection of the crystal network, the connection between crystal networks breaks down. Additionally, it was observed that with an increase in BA concentration, the mechanical transition point of PVA films appears at lower strain values, indicating that the addition of BA helps enhance the mechanical properties of PVA.
 | ||
| Fig. 23 Schematic of the structural evolution of PVA films during stretching in the aqueous solution with different concentrations of BA. Reprinted with permission from ref. 106; copyright 2019, Elsevier. | ||
To further investigate the impact of BA crosslinking on the evolution of both crystalline and amorphous networks in PVA, Li et al.170 utilized SRXS and LF NMR to systematically investigate the effects of BA cross-linking on the physical and chemical cross-linking networks of PVA films, thereby exploring its impact on macroscopic mechanical resilience. As illustrated in Fig. 24b and c, BA influences both the crystalline and amorphous regions of PVA films, leading to reduced crystallinity and structural modifications in the amorphous network. At low and medium BA concentrations, the crystalline network structure of PVA remains largely unchanged; however, at medium concentration, specific crystal planes are selectively disrupted, while the (101)/(10) plane remains stable, contributing to the maintenance of the framework of the crystalline network. At high BA concentrations, the crystallinity and crystal size of PVA significantly decrease, resulting in an increased crosslink density within the amorphous network. Among them, crosslinking at medium BA concentrations (0.1 wt% to 2 wt%) can increase the crosslinking points in the amorphous network, enhancing the strength of the elastic network and further improving the recoverability of the amorphous network in PVA films. Combining the evolution of both the amorphous and crystalline networks (Fig. 24d), based on the Maxwell model, the recovery behavior of crosslinked PVA films at different BA concentrations within the low-strain range was understood.
 | ||
| Fig. 24 (a) Total strain at point A and total strain of the elastic plateau at different cBA, (b) summary of median Dres/2π and σ/2π as a function of cBA, (c) change in L (long period), dc, and da (the distance between two lamellae) as a function of cBA and (d) mathematical model of the PVA film (gel state) in the presence of BA. The destruction of the crystalline lamellae is also illustrated. Reprinted with permission from ref. 170; copyright 2024, the American Chemical Society. | ||
The role of hydrogen bonds in PVA
PVA molecular chains are enriched with hydroxyl groups, which result in the formation of widespread hydrogen bonds.171 The rich hydroxyl groups on the PVA chains enable the formation of a complex hydrogen bond network, comprising intermolecular and intramolecular hydrogen bonds and free hydroxyl groups.172,173 The performance of PVA is intimately associated with these hydrogen bonds. Consequently, researchers have shown great interest in different types of hydrogen bonds, given that they profoundly influence the properties of PVA.174–184 Tubbs et al.185 discovered that hydrogen bonds significantly affect the melting point and crystallinity of copolymers. Partial saponification of PVAc generates ordered, high-melting-point PVA copolymers, while re-acetylated PVA forms disordered, low-melting-point copolymers. Catalytic hydrolysis of PVAc results in a melting point intermediate between the two. Similar melting points indicate similar distributions of crystalline unit sequence lengths. Arai et al.186 suggested that disrupting the continuous intramolecular hydrogen bonds between adjacent hydroxyl groups can improve the solubility of PVA. Li et al.74,181 found that hydrogen bonds play a crucial role in the single-molecule behavior of PVA and its elastic properties, especially in aqueous environments, where hydrogen bonds enable PVA molecules to form complex superstructures, thereby affecting their mechanical properties and dynamic responses. Xie et al.187 used two-dimensional (2D) NMR to study the structural changes in partially hydrolyzed PVA in detail and pointed out that hydrogen bonds between the PVA molecular chains are a key factor determining its mechanical properties. Wang et al.184 proposed that bio-inspired hydrogen bond self-assembly strategies can enhance the strength and toughness of PVA, as well as improve its thermal stability.Additionally, Song et al.183 employed a hydrogen bond cross-linking strategy, and PVA after the hydrogen bond cross-linking treatment exhibited a significantly improved yield strength (approximately 140 MPa), reduced nanoindentation modulus (approximately 22.5 GPa), hardness (approximately 0.5 GPa), and excellent extensibility (about 40%). Compared to uncrosslinked PVA, the material not only possessed superior mechanical properties but also maintained structural stability at high temperatures. Additionally, due to the dynamic reversibility of hydrogen bonds, it exhibited good self-healing capability (Fig. 25). Additionally, Bao et al.188 revealed the impact of intramolecular hydrogen bonds on single-chain elasticity at the single-molecule level using atomic force microscopy (AFM) technology. They discovered that the energy of intramolecular hydrogen bonds in PVA is positively correlated with its degree of alcoholysis (DA), indicating a synergistic enhancement effect under high DA conditions. This synergistic effect may be a key factor in the low water solubility of high DA PVA at room temperature. As the tensile force increases, the PVA supramolecular structure undergoes reversible conformational transitions, shifting from a relaxed state to an over-stretched state (Fig. 26). This suggests that intramolecular hydrogen bonds not only enhance the mechanical properties of PVA but also endow it with unique dynamic response characteristic.
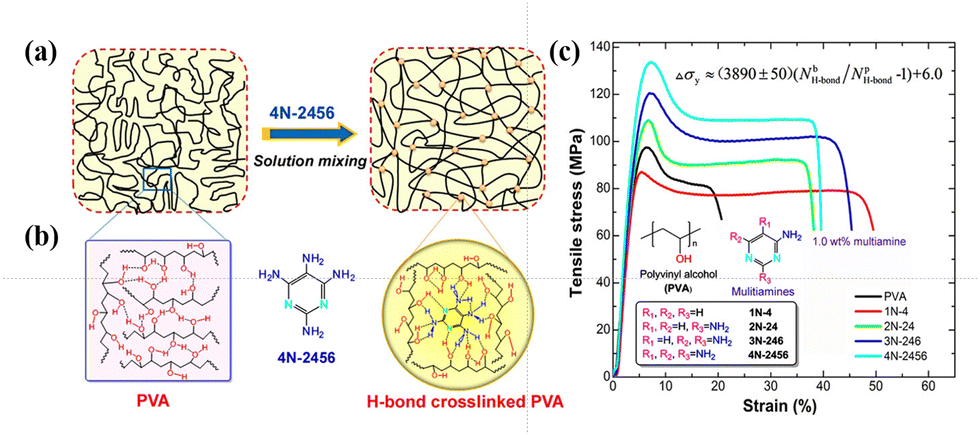 | ||
| Fig. 25 (a) Schematic of interactions for PVA and its cross-linked blends created via a simple solution mixing method. (b) Possible H-bond interactions among PVA chains and PVA with 4N-2456. (c) Typical tensile stress–strain curves (loading level: 1.0 wt%). Reproduced with permission from ref. 183 Copyright 2015, the American Chemical Society. | ||
 | ||
| Fig. 26 (a) Comparison of the F–E curve of PVA-99 in DMSO (blue) with the QM-FRC fitting curve of PE (red dotted line). (b) F–E curves of PVA-80 (orange), PVA-88 (black), and PVA-99 (blue) obtained under vacuum, with the QM-FRC fitting curve of PE shown as a reference (red dotted line). (c) Plots of EDA values versus DA of PVA under vacuum (blue squares) and nonane (red circles). The blue and red curves represent the fitting results according to eqn (1) and (2), respectively. Reprinted in part with permission from ref. 188 copyright 2021, the American Chemical Society. | ||
To comprehensively understand the relationship between hydrogen bonding and the properties of PVA, quantitatively distinguishing different types of hydrogen bonds within PVA remains a challenge. As early as 1997, Horii et al.189 studied the conformation and intramolecular hydrogen bond formation of PVA with varying degrees of regularity using SSNMR. In the 13C-NMR spectra, the –CH carbon resonances of PVA split into three lines due to the presence of two, one, or no intramolecular hydrogen bonds in trimer sequences, indicating that the number of intramolecular hydrogen bonds significantly influences the structure of PVA. Subsequent high-resolution solid-state NMR studies further deepened our understanding of the internal hydrogen-bonding network in PVA.185–188,190 For instance, investigating the probability of intramolecular hydrogen bonding in amorphous, syndiotactic, and highly isotactic PVA films under hydrated conditions through statistical calculations. Specifically, as water content increased, the probability of intramolecular hydrogen bond formation notably increased, especially in crystalline regions, where the probability of intramolecular hydrogen bond formation increased considerably. Moreover, Horii et al.185 also revealed significant changes in the hydrogen-bonding structure of PVA films due to hydration, particularly in mobile regions. Their work showed that the hydration process not only increased the probability of intramolecular hydrogen bond formation but also enhanced the molecular mobility. This enhanced molecular mobility partially disrupted the original hydrogen-bonding network and increased the molar fraction of gauche conformations compared to the dry state. Specifically, the intermolecular hydrogen bonds rapidly broke during the initial stages of hydration, while intramolecular hydrogen bonds gradually decreased over a longer period. This suggests that water molecules preferentially attack the intermolecular hydrogen bonds, leading to faster deuteration exchange. In contrast, even OH protons in crystalline regions that did not participate in hydrogen bonding exhibited extremely slow deuteration exchange rates, indicating that the hydrogen-bonding network in these regions is relatively stable (Fig. 27).
 | ||
| Fig. 27 Plots of the molar fractions of different OH groups against the soaking time for dried A-PVA films soaked in deuterated water for different periods.: total mole fraction of the three OH groups; □: OHinter; ○: OHintra; △: OHf. Reprinted in part with permission from ref. 191; copyright 2001, the American Chemical Society. | ||
During the production process of PVA-based polarizers, hydrogen bonding plays an indispensable role. Miyazaka et al.15 proposed a model for PVA–iodine complexes, where four extended-chain conformations of PVA form hydrogen bonds with each other at a spacing of approximately 307 pm, encapsulating the linear polyiodide ions to create PVA–iodine complexes. Horii et al.151 investigated the role of BA in the formation of unoriented PVA–I composite films and found that BA enhances the mechanical properties of the composite film by forming hydrogen bonds with the hydroxyl groups on the PVA molecular chains. Both BA and iodine interact with PVA through hydrogen bonding, indicating a competitive relationship between them.145,168
Relationship between the performance and structure of polarizers
Further understanding the relationship between the performance of polarizers and their structural and morphological details is particularly important. Firstly, Miyasaka et al.122 confirmed that highly ordered PVA segments can form layered structures with a spacing of 5.2 Å, providing periodic sites for I3− insertion. Miyazaki's research further elucidated how iodine molecules absorbed into PVA films preferentially align along the stretching direction, thereby maximizing light absorption.141 The iodine doping process is crucial for imparting polarization properties to polarizers. When an iodine solution penetrates into PVA films, I3− preferentially diffuse into the amorphous regions.192–195 If there are pores in the adhesive layer, these pores will be enriched with free I3−, potentially leading to bright spots under specific conditions.196 During processing, uniaxial stretching of PVA films results in molecular alignment along the stretching direction, causing the iodine molecules to also preferentially align in this direction, and thus absorbing light from an unpolarized light source.7 Subsequent studies have shown that PVA–iodine systems exhibit a partial crystalline arrangement, with PVA–iodine complexes formed in the amorphous PVA regions.15,122,141 Uniaxial stretching promotes the orderly alignment of the PVA molecular chains along the stretching direction, which not only enhances the polarization effect but also improves the physical strength of the material.7 This ordered alignment is crucial for achieving efficient polarization, given that it ensures maximum light absorption and minimal scattering loss. Finally, BA cross-linking treatment is used to enhance the stability and durability of polarizers. As a cross-linking agent, BA reacts with PVA molecules to form a three-dimensional network structure, effectively inhibiting the iodine desorption rate under humid and hot environmental conditions and improving the weather resistance of polarizers.142,197Summary and perspective
This review focused on addressing the fundamental scientific questions during the processing of PVA-based polarizers, including casting, iodine doping, BA crosslinking, and stretching. By combining techniques such as SXRS and NMR, and in situ devices, the multi-scale condensed micro-nano condensed-state structure and dynamics evolution laws have been elucidated, as follows: (1) primary chemical structure of PVA, (2) solution casting of PVA films, (3) hierarchical structure and dynamic heterogeneity of plasticized PVA films, (4) formation mechanism of PVA–iodine complexes, and (5) crosslinking mechanism of BA in PVA. Accordingly, the structural evolution of PVA films under various processing conditions has been studied, establishing a “structure-processing-performance” relationship. This provides theoretical guidance for designing high-performance polarizing films.Despite the detailed insights into the multi-scale structures and dynamic evolution during the processing of polarizing films, several challenges remain in understanding their behavior under complex operating conditions, such as how does the chain defects, such as VAc and ethylene units, change the PVA crystallization behavior as well as PVA iodine doping mechanism? How does the coordination structure of BA–PVA change under different processing parameters, i.e. boric acid concentration and stretching, and does this change affect the chain dynamics of PVA? Is the PVA–iodine complexation behavior an entropy-driven rather than enthalpy-driven process? During the transformation process from disordered to ordered structures in the PVA–iodine complex model, can the amorphous regions of PVA directly transform into PVA–I3− without going through a crystalline PVA-to-PVA–I3− transition? Answering questions is crucial for optimizing the performance of polarizing films and developing new high-performance materials. Also, answering the above-mentioned questions can help us better understand the ordering process for charged polymer systems, such as polymer electrolytes and bio-macromolecules.
Facing the diversified demands of new display technologies, such as thinness, flexibility, and suitability for multiple applications (e.g., automotive displays, fighter jet displays, and 3D displays) and harsh environments (e.g., high temperature and humidity), polarizing films must evolve towards being lightweight, flexible, offering high contrast, and possessing superior weather resistance. However, challenges remain, including easy damage during their production and difficulty ensuring their uniformity and consistency. Thus, to address these challenges, establishing a “structure–processing–property” relationship is key to developing high-performance polarizing films. Future efforts should focus on the following: (1) development of new OCA: meeting the requirements of high light transmittance and polarization efficiency, and improve weather resistance under high temperature and humidity conditions. (2) Special PVA development: enhancing the draw ratio of PVA films to achieve large-draw-ratio PVA films industrially, meeting the demand for lighter, thinner, and more flexible polarizing films. (3) Azo dye-based polarizing films:198,199 utilizing the abundant hydroxyl groups in PVA to can form strong hydrogen bonds or ionic bonds with azo dyes. Compared to traditional PVA–iodine polarizing films, new dye-based polarizing films exhibit greater stability under high temperatures and UV exposure, offering superior weather resistance. This approach aims to provide a robust foundation for the advancement of polarizing film technology, addressing the current limitations and paving the way for innovative applications in next-generation displays.
Author contributions
Yao Li: conceptualization, investigation, writing – original draft, writing – review and editing, Hong Cheng, Jiayu Xie, Jie Chen and Xiaoying Wei: conceptualization, writing – original draft, Wei Chen: conceptualization, methodology, writing – review & editing, supervision, funding acquisition.Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (52422302 and U20A20256), and Anhui Provincial Natural Science Foundation (2308085UM02 and 2408055UM001).References
- J. Ma, X. Ye and B. Jin, Displays, 2011, 32, 49–57 CrossRef.
- T. Sato, Int. Polym. Sci. Technol., 2012, 39, 15–24 Search PubMed.
- W. Gao, Y. Bi, X. Liu and Z. Xu, Chin. J. Eng. Sci., 2020, 22, 44 Search PubMed.
- H. Zhang, H. Li and H. Zhong, Electron. Technol. Softw. Eng., 2022, 18, 86–89 Search PubMed.
- X. Li, Master's thesis, Southeast University, Nanjing, China, 2020.
- Z. Hong, J. Illumin. Eng., 2021, 32, 4–5 Search PubMed.
- E. H. Land, J. Opt. Soc. Am., 1951, 41, 957 CrossRef.
- R. K. Tubbs, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 4181 CrossRef.
- W. O. Herrmann and W. Haehnel, Chem, Forschungsgemeinschaft, German, Patent DE-OS450286, 1924 Search PubMed.
- W. O. Herrmann, Chem, Forschungsgemeinschaft, German, Patent DE642531, 1937 Search PubMed.
- P. J. Flory and F. S. Leutner, J. Polym. Sci., 1948, 3, 880–890 CrossRef.
- Y. Hirano, Bull. Chem. Soc. Jpn., 1964, 37, 1495–1500 Search PubMed.
- G. Mino, S. Kaizerman and E. Rasmussen, J. Polym. Sci., 1959, 39, 523–529 Search PubMed.
- K. Kikukawa, S. Nozakura and S. Murahashi, Polym. J., 1971, 2, 212–219 Search PubMed.
- K. Miyasaka, in Structure in Polymers with Special Properties, ed. H.-G. Zachmann, Springer, Berlin, Heidelberg, 1993, vol. 108, pp. 91–129 Search PubMed.
- S. Matsuzawa, K. Yamaura and H. Noguchi, Die Makromol. Chem., 1974, 175, 31–41 CrossRef.
- R. Fukae, T. Yamamoto, O. Sangen, T. Saso, T. Kako and M. Kamachi, Polym. J., 1990, 22, 636–637 CrossRef.
- J. R. DeMember, H. C. Haas and R. L. MacDonald, J. Polym. Sci. B Polym. Lett., 1972, 10, 385–389 CrossRef.
- S. Matsuzawa, In Handbook of Thermoplastics, ed. O. Olabisi, Marcel Dekker, New York, 1997, pp. 269–289 Search PubMed.
- Y. Choi and K. Miyasaka, J. Appl. Polym. Sci., 1993, 48, 313–317 Search PubMed.
- K. Imai and M. Matsumoto, J. Polym. Sci., 1961, 55, 335–342 Search PubMed.
- J. H. Choi, W. S. Lyoo, H. D. Ghim and S.-W. Ko, Colloid Polym. Sci., 2000, 278, 1198–1204 CrossRef.
- R. Fukae, T. Yamamoto, Y. Fujita, N. Kawatsuki, O. Sangen and M. Kamachi, Polym. J., 1997, 29, 293–295 Search PubMed.
- I. Sakurada, Polyvinyl Alcohol Fibers, Marcel Dekker, New York, 1985 Search PubMed.
- M. Wang, T. Takahama and K. Tashiro, Polymer, 2023, 285, 126323 Search PubMed.
- S. Hayashi, C. Nakano and T. Motoyama, Kobunshi Kagaku, 1963, 20, 303–311 Search PubMed.
- S. Hayashi, M. Takama and N. Hojo, Makromol. Chem., 1976, 177, 859–870 Search PubMed.
- S. Hayashi, T. Hirai, N. Hojo, H. Sugeta and Y. Kyogoku, J. Polym. Sci. B Polym. Lett. Ed., 1982, 20, 69–73 Search PubMed.
- H. H. T. M. Ketels, J. Polym. Sci. Eng., 1989, 15, 245–260 Search PubMed.
- Y. Mori, H. Sumi, T. Hirabayashi, Y. Inai and K. Yokota, Macromolecules, 1994, 27, 1051–1056 CrossRef.
- T. C. Chung, Polymer, 1991, 32, 1336–1339 CrossRef.
- M. Komatsu, T. Inoue and K. Miyasaka, J. Polym. Sci., Part B:Polym. Phys., 1986, 24, 303–311 Search PubMed.
- K. Kawanishi, M. Komatsu and T. Inoue, Polymer, 1987, 28, 980–984 Search PubMed.
- K. Shibatani, Polym. J., 1970, 1, 348–355 CAS.
- M. O. Ngui and S. K. Mallapragada, Polymer, 1999, 40, 5393–5400 CAS.
- F. Auriemma, C. De Rosa and R. Triolo, Macromolecules, 2006, 39, 9429–9434 CrossRef CAS.
- S. Hayakawa, M. Hanashima and M. Michiwashi, Chin. Pat., CN 109312085 A, 2017 Search PubMed.
- T. Umezaki and K. Takafuji, Chin. Pat., CN 105874364 A, 2014 Search PubMed.
- S. Hayakawa, S. Kitamura, T. Toyota and Y. Teramoto, Chin. Pat., CN 109416424 A, 2017 Search PubMed.
- T. Sanefuji, S. Fujita and T. Kawai, US Pat., US 6833099 B2, 2004 Search PubMed.
- S. Harita and T. Isozaki, US Pat., US 6803411 B2, 2004 Search PubMed.
- T. Kanaya, N. Takahashi, H. Takeshita, M. Ohkura, K. Nishida and K. Kaji, Polym. J., 2012, 44, 83–94 CAS.
- S. Ghoshal, P. Denner, S. Stapf and C. Mattea, Chem. Phys. Lett., 2011, 515, 231–234 CAS.
- M. O. Ngui and S. K. Mallapragada, J. Polym. Sci., Part B: Polym. Phys., 1998, 36, 2771–2780 CAS.
- H. Cheng, Y. Zhao, X. Wei, Q. Zhang, E. Yang, Y. Xiong, J. Zhu, Z. Wu, J. Chen and W. Chen, Macromolecules, 2024, 57, 1569–1580 CAS.
- Y. Li, X. Yu, C. Liu and Y. Han, Macromolecules, 2023, 56, 9175–9188 CAS.
- S. Zeng, L. Li and Q. Wang, Polym. Test., 2023, 126, 108143 CAS.
- L. Keshavarz, M. A. Khansary and S. Shirazian, Polymer, 2015, 73, 1–8 CAS.
- E. Wang, S. Batra and M. Cakmak, Polymer, 2015, 67, 200–207 CrossRef CAS.
- J. Sharma, R. K. Arya and G. D. Verros, Prog. Org. Coat., 2019, 134, 219–225 Search PubMed.
- D. Bouyer, O. M’Barki, C. Pochat-Bohatier, C. Faur, E. Petit and P. Guenoun, AIChE J., 2017, 63, 3035–3047 Search PubMed.
- Z. Fu, W. He, Y. Yao, Z. Qiu, H. Chen, C. Li, K. Wang, Q. Zhang, R. T. K. Kwok, B. Z. Tang and Q. Fu, J. Phys. Chem. Lett., 2022, 13, 9855–9861 CrossRef CAS PubMed.
- Y. Li, X. Liu, D. Lv, S. Hou, X. Yu and Y. Han, Macromolecules, 2024, 57, 6109–6122 CrossRef CAS.
- C. Li, H. Chen, Z. Fu, Q. Zhang, K. Wang and Q. Fu, Polymer, 2021, 224, 123759 CrossRef CAS.
- E. Kumacheva, L. Li, M. A. Winnik, D. M. Shinozaki and P. C. Cheng, Langmuir, 1997, 13, 2483–2489 CrossRef CAS.
- S. Wong, S. A. Altinkaya and S. K. Mallapragada, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 3191–3204 CrossRef.
- A. Tysén, H. Vomhoff and L. Nilsson, Nord. Pulp Pap. Res. J., 2018, 33, 581–591 Search PubMed.
- N. Allanic, P. Salagnac and P. Glouannec, Heat Mass Transfer, 2007, 43, 1087–1095 CrossRef.
- N. Allanic, P. Salagnac, P. Glouannec and B. Guerrier, AIChE J., 2009, 55, 2345–2355 Search PubMed.
- N. Allanic, P. Le Bideau, P. Glouannec and R. Deterre, Heat Mass Transfer, 2017, 53, 223–231 Search PubMed.
- B. S. Dias, J. E. P. Navalho and J. C. F. Pereira, Drying Technol., 2022, 40, 3466–3482 Search PubMed.
- P. Dufour, D. Blanc, Y. Touré and P. Laurent, Drying Technol., 2004, 22, 269–284 Search PubMed.
- P. Dufour, D. Blanc, Y. Touré and P. Laurent, Int. Drying Symp., 2002, 1147–1154 Search PubMed.
- D. Cassidy, A. B. Ebiana, A. Salem and M. Rashidi, Int. J. Adv. Manuf. Technol., 2007, 32, 238–256 Search PubMed.
- R. Dhib, A. D. Broadbent and N. Thérien, Can. J. Chem. Eng., 1994, 72, 894–905 CrossRef.
- R. Dhib, Drying Technol., 2007, 25, 97–105 CrossRef.
- B. Cote, N. Therien and A. D. Broadbent, Text. Res. Inst., 1991, 193–202 Search PubMed.
- P. Shrimal and M. Vinjamur, in Proceedings of the 13th International Society of Coating Science and Technology Symposium, 2006 Search PubMed.
- R. K. Arya and M. Vinjamur, Ind. Eng. Chem. Res., 2009, 48, 10504–10514 Search PubMed.
- R. A. Cairncross, S. Jeyadev, R. F. Dunham, K. Evans, L. F. Francis and L. E. Scriven, J. Appl. Polym. Sci., 1995, 58, 1279–1290 CrossRef CAS.
- S. Wong, S. A. Altinkaya and S. K. Mallapragada, J. Polym. Sci., Part B:Polym. Phys., 2005, 43, 3191–3204 Search PubMed.
- A. J. Kilpatrick, J. Appl. Phys., 1940, 11, 255 Search PubMed.
- F. W. Clark, Chem. Ind., 1941, 60, 225 Search PubMed.
- R. Houwink, Proc. XI Cong. Pure Appl. Chem., London, 1947, 575–583 Search PubMed.
- D. F. Cadogan and C. J. Howick, In Plasticizers. Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, New York, 1996 Search PubMed.
- W. Aiken, T. Alfrey, A. Janssen and H. Mark, J. Polym. Sci., 1947, 2, 178–198 CAS.
- A. Marcilla and M. Beltran, In Handbook of Plasticizers, ed. G. Wypych, Chem Tec Publishing, Toronto, 2004, pp. 107–120 Search PubMed.
- H. Li, W. Zhang, W. Xu and X. Zhang, Macromolecules, 2000, 33, 465–469 CAS.
- R. M. Hodge, G. H. Edward and G. P. Simon, Polymer, 1996, 37, 1371–1376 CAS.
- B. Briscoe, P. Luckham and S. Zhu, Polymer, 2000, 41, 3851–3860 CAS.
- R. M. Hodge, T. J. Bastow, G. H. Edward, G. P. Simon and A. J. Hill, Macromolecules, 1996, 29, 8137–8143 CAS.
- H. Tian, L. Yuan, D. Zhou, J. Niu, H. Cong and A. Xiang, Polym. Adv. Technol., 2018, 29, 2612–2618 CrossRef CAS.
- C. Lv, H. Tian, X. Zhang and A. Xiang, Polym. Test., 2018, 70, 67–72 Search PubMed.
- H. Dai, J. Wang and N. Liu, J. Vinyl Addit. Technol., 2019, 25(S1), E181–E187 Search PubMed.
- T. Liu, X. Peng, Y. Chen, Q. Bai, C. Shang, L. Zhang and H. Wang, Macromol. Rapid Commun., 2018, 39, 1800050 CrossRef PubMed.
- A. Briddick, R. J. Fong, E. F. D. Sabattié, P. Li, M. W. A. Skoda, F. Courchay and R. L. Thompson, Langmuir, 2018, 34, 1410–1418 Search PubMed.
- Z. Fu, S. Guo, C. Li, K. Wang, Q. Zhang and Q. Fu, Phys. Chem. Chem. Phys., 2022, 24, 1885–1895 RSC.
- Z. Fu, Y. Yao, S. Guo, K. Wang, Q. Zhang and Q. Fu, Macromol. Rapid Commun., 2023, 44, 2200296 Search PubMed.
- J. Xie, X. Jin, H. Cheng, W. Chen, W. Yu and L. Wang, Ind. Crops Prod., 2024, 220, 119246 Search PubMed.
- X. Jin, Q. Zhang, Y. Xiong, Z. Wu, H. Cheng, X. Chen and W. Chen, Macromolecules, 2024, 57, 1277–1290 CAS.
- W. Fei, Z. Wu, H. Cheng, Y. Xiong, W. Chen and L. Meng, J. Polym. Sci., 2023, 61, 1959–1970 Search PubMed.
- Z. Wu, Y. Xiong, C. Li, H. Cheng, Y. Li and W. Chen, Macromolecules, 2023, 56, 7113–7124 Search PubMed.
- H. Noguchi, H. Jyodai and S. Matsuzawa, J. Polym. Sci., Part B: Polym. Phys., 1997, 35, 1701–1709 CAS.
- V. K. Walworth, ed. A. J. Woods, N. S. Holliman and G. E. Favalora, History of polarized image stereoscopic display, Burlingame, California, USA, 2013, p. 864804 Search PubMed.
- W. O. Herrmann and W. Haehnel, Ber. Dtsch. Chem. Ges. B, 1927, 60, 1658–1663 Search PubMed.
- H. Staudinger, K. Frey and W. Starck, Ber. Dtsch. Chem. Ges. B, 1927, 60, 1782–1792 Search PubMed.
- E. H. Land and J. S. Friedman, US Pat., US1918848A, 1933 Search PubMed.
- H. Takamiya, Y. Tanahashi, T. Matsuyama, T. Tanigami, K. Yamaura and S. Matsuzawa, J. Appl. Polym. Sci., 1993, 50, 1807–1813 CAS.
- T. Takahama, S. M. Saharin and K. Tashiro, Polymer, 2016, 99, 566–579 CAS.
- K. Imai and M. Matsumoto, J. Polym. Sci., 1961, 55, 335–342 CrossRef CAS.
- S. A. Umoren, I. B. Obot, A. Madhankumar and Z. M. Gasem, J. Adhes. Sci. Technol., 2015, 29, 271–295 Search PubMed.
- S. Hayashi, M. Takama, I. Kaneko and N. Hojo, Makromol. Chem., 1975, 176, 3221–3228 Search PubMed.
- S. Hayashi, Y. Tanabe and N. Hojo, Makromol. Chem., 1977, 178, 1679–1687 CrossRef CAS.
- S. Hayashi, M. Kobayashi, H. Shirai and N. Hojo, Makromol. Chem., 1978, 179, 1397–1401 Search PubMed.
- S. Hayashi, C. Kawamura and M. Takayama, Bull. City Japan, 1970, 43, 537–542 Search PubMed.
- K. Ye, Y. Li, W. Zhang, Q. Zhang, W. Chen, L. Meng, D. Wang and L. Li, Polym. Test., 2019, 77, 105913 Search PubMed.
- Y. Choi and K. Miyasaka, J. Appl. Polym. Sci., 1994, 51, 613–618 Search PubMed.
- T. Miyazaki, A. Hoshiko, M. Akasaka, T. Shintani and S. Sakurai, Macromolecules, 2006, 39, 2921–2929 Search PubMed.
- T. Miyazaki, A. Hoshiko, M. Akasaka, M. Sakai, Y. Takeda and S. Sakurai, Macromolecules, 2007, 40, 8277–8284 Search PubMed.
- K. Tashiro, H. Kitai, S. M. Saharin, A. Shimadzu and T. Itou, Macromolecules, 2015, 48, 2138–2148 Search PubMed.
- Y. Li, J. Yang, H. Cheng, L. Cai, K. Ye, Z. Xia, Q. Zhang, D. Wang and W. Chen, Soft Matter, 2021, 17, 8973–8981 Search PubMed.
- H. Sakuramachi, Y.-S. Choi and K. Miyasaka, Polym. J., 1990, 22, 638–642 Search PubMed.
- Y.-S. Choi, Y. Oishi and K. Miyasaka, Polym. J., 1990, 22, 601–608 Search PubMed.
- Y.-S. Choi and K. Miyasaka, Polym. J., 1991, 23, 977–981 CrossRef.
- Y. Li, J. Yang, H. Cheng, L. Cai, K. Ye, Z. Xia, Q. Zhang, D. Wang and W. Chen, Soft Matter, 2021, 17, 8973–8981 RSC.
- M. M. Zwick, J. Appl. Polym. Sci., 1965, 9, 2393–2424 CrossRef.
- M. M. Zwick, J. Polym. Sci., Part A: Gen. Pap., 1966, 4, 1642–1644 CrossRef.
- F. Inagaki, I. Harada and T. Shimanouchi, Bull. Chem. Soc. Jpn., 1972, 45, 3384–3388 Search PubMed.
- Y. Oishi, H. Yamamoto and K. Miyasaka, Polym. J., 1987, 19, 1261–1268 Search PubMed.
- M. E. Heyde, L. Rimai, R. G. Kilponen and D. Gill, J. Am. Chem. Soc., 1972, 94, 5222–5227 CrossRef.
- S. B. Sharp and G. I. Gellene, J. Phys. Chem. A, 1997, 101, 2192–2197 Search PubMed.
- M. Haisa and H. Itami, J. Phys. Chem., 1957, 61, 817–818 Search PubMed.
- H. Djojosubroto, Y. Tanizaki and T. Hoshi, Bull. Chem. Soc. Jpn., 1970, 43, 3025–3028 CrossRef.
- Y. Oishi and K. Miyasaka, Polym. J., 1987, 19, 331–336 Search PubMed.
- K. Hess, R. Steinmann, H. Kiessig and I. Avisiers, Kolloid-Z., 1957, 153, 128–155 Search PubMed.
- C. D. West, J. Chem. Phys., 1949, 17, 219–220 Search PubMed.
- F. H. Herbstein, M. Kaftory and M. Kapon, Z. Kristallogr. New Cryst. Struct., 1981, 154, 11–30 Search PubMed.
- K. Tashiro, H. Kitai, S. M. Saharin, A. Shimadzu and T. Itou, Macromolecules, 2015, 48, 2138–2148 CrossRef.
- T. Itou, H. Kitai, A. Shimadzu, T. Miyazaki and K. Tashiro, J. Phys. Chem. B, 2014, 118, 6032–6037 CrossRef PubMed.
- K. Tashiro, K. Kusaka, H. Yamamoto and M. Hanesaka, Macromolecules, 2020, 53, 6656–6671 CrossRef.
- S. M. Saharin, T. Takahama, S. Nonogaki, K. Saito and K. Tashiro, Macromolecules, 2015, 48, 8867–8876 CrossRef.
- S. M. Saharin, T. Takahama, S. Nonogaki, K. Saito and K. Tashiro, Polymer, 2016, 89, 81–93 Search PubMed.
- T. Yoshioka, K. Tashiro and N. Ohta, Macromolecules, 2017, 50, 2803–2813 Search PubMed.
- D. Tahara, T. H. Ninh, H. Yamamoto and K. Tashiro, Macromolecules, 2020, 53, 276–287 CAS.
- K. Tashiro and M. Gakhutishvili, Polymer, 2019, 171, 140–148 Search PubMed.
- Y. Li, W. Tao and W. Chen, Soft Matter, 2022, 18, 8974–8982 CAS.
- M. Wang, T. Takahama and K. Tashiro, Macromolecules, 2020, 53, 4395–4406 CAS.
- M. Takahashi, K. Tashiro and S. Amiya, Macromolecules, 1999, 32, 5860–5871 CrossRef CAS.
- R. Zhang, Q. Zhang, Y. Ji, F. Su, L. Meng, Z. Qi, Y. Lin, X. Li, X. Chen, F. Lv and L. Li, Soft. Matter., 2018, 14, 2535–2546 RSC.
- Q. Zhang, R. Zhang, L. Meng, Y. Ji, F. Su, Y. Lin, X. Li, X. Chen, F. Lv and L. Li, Polymer, 2018, 142, 233–243 Search PubMed.
- T. Miyazaki, S. Katayama, E. Funai, Y. Tsuji and S. Sakurai, Polymer, 2005, 46, 7436–7442 CrossRef CAS.
- T. Miyazaki, Y. Takeda, S. Akane, T. Itou, A. Hoshiko and K. En, Polymer, 2010, 51, 5539–5549 Search PubMed.
- T. Miyazaki, Y. Takeda, A. Hoshiko, K. Shimokita and D. Ogomi, Polym. Eng. Sci., 2015, 55, 513–522 CAS.
- T. Miyazaki, N. Miyata, M. Asada, Y. Tsumura, N. Torikai, H. Aoki, K. Yamamoto, T. Kanaya, D. Kawaguchi and K. Tanaka, Langmuir, 2019, 35, 11099–11107 CAS.
- K. Ye, Y. Li, W. Zhang, W. Chen, Q. Zhang, D. Wang and L. Li, Polymer, 2021, 212, 123297 Search PubMed.
- W. Maier and A. Saupe, Z. Naturforsch., A: Phys. Sci., 1958, 13, 564–566 Search PubMed.
- G. R. Luckhurst and C. Zannoni, Nature, 1977, 267, 412–414 CAS.
- K. Tashiro, T. Takahama and M. F. Wang, Polymer, 2021, 233, 124180 CAS.
- R. Cao, F. Peng, C. Nie, Y. Zhang, H. Sun, Z. Liu, T. Xu and L. Li, Macromolecules, 2024, 57, 5979–5990 Search PubMed.
- N. Wang, L. Zhao, C. Zhang and L. Li, J. Appl. Polym. Sci., 2016, 133 CAS , app.43246.
- K. Ohishi, T. Itadani, T. Hayashi, T. Nakai and F. Horii, Polymer, 2010, 51, 687–693 CrossRef CAS.
- R. J. Kostyla and K. J. Abcunas, Proc. SPIE, 1982, 5–12 Search PubMed.
- Y. Ke, PhD dissertation, University of Science and Technology of China, 2021.
- H. Staudinger, K. Frey and W. Starck, Ber. Dtsch. Chem. Ges., 1927, 60, 1782–1792 Search PubMed.
- A. Grasnick, In Basics of Virtual Reality: From the Discovery of Perspective to VR Glasses, Springer Berlin Heidelberg, Berlin, Heidelberg, 2022, pp. 207–288 Search PubMed.
- S. D. Bull, M. G. Davidson, J. M. H. Van Den Elsen, J. S. Fossey, A. T. A. Jenkins, Y.-B. Jiang, Y. Kubo, F. Marken, K. Sakurai, J. Zhao and T. D. James, Acc. Chem. Res., 2013, 46, 312–326 Search PubMed.
- W. L. A. Brooks and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375–1397 CrossRef CAS.
- R. Nishiyabu, Y. Kubo, T. D. James and J. S. Fossey, Chem. Commun., 2011, 47, 1124 RSC.
- L. He, W. Wei, Y. Luo, Y. Xu, B. Zeng, W. Luo, G. Chen, C. Yuan and L. Dai, Chin. Sci. Bull., 2021, 66, 1208–1219 CrossRef.
- J. P. Lorand and J. O. Edwards, J. Org. Chem., 1959, 24, 769–774 CrossRef CAS.
- G. Springsteen and B. Wang, Tetrahedron, 2002, 58, 5291–5300 CrossRef CAS.
- J. Yan, G. Springsteen, S. Deeter and B. Wang, Tetrahedron, 2004, 60, 11205–11209 Search PubMed.
- E. Ruoff, S. Kmiec and A. Manthiram, Adv. Energy Mater., 2024, 2402091 Search PubMed.
- E. J. Shin, W. S. Lyoo and Y. H. Lee, J. Appl. Polym. Sci., 2008, 109, 1143–1149 Search PubMed.
- E. Z. Casassa, A. M. Sarquis and C. H. Van Dyke, J. Chem. Educ., 1986, 63, 57 CrossRef CAS.
- P. M. Williams and P. M. Strack, Limnol. Oceanogr., 1966, 11, 401–404 CrossRef CAS.
- H.-H. Wang, T.-W. Shyr and M.-S. Hu, J. Appl. Polym. Sci., 1999, 74, 3046–3052 CrossRef.
- J. Y. Woo, E. J. Shin and Y. H. Lee, Polym. Bull., 2010, 65, 169–180 Search PubMed.
- B. Bolto, T. Tran, M. Hoang and Z. Xie, Prog. Polym. Sci., 2009, 34, 969–981 Search PubMed.
- Y. Li, Y. Li, R. Cao, J. Xie and W. Chen, Macromolecules, 2024, 57, 6321–6332 Search PubMed.
- G. Tesei, G. Paradossi and E. Chiessi, J. Phys. Chem. B, 2012, 116, 10008–10019 CrossRef PubMed.
- C. Sawatari, Y. Yamamoto, N. Yanagida and M. Matsuo, Polymer, 1993, 34, 956–966 Search PubMed.
- X. Hong, Y. Xu, L. Zou, J. He, J. Zhao and Y. V. Li, J. Appl. Polym. Sci., 2021, 138, 49971 Search PubMed.
- J. M. G. Cowie, I. McEwan, I. J. McEwen and R. A. Pethrick, Macromolecules, 2001, 34, 7071–7075 CrossRef.
- E. Chiellini, A. Corti, G. Del Sarto and S. D’Antone, Polym. Degrad. Stab., 2006, 91, 3397–3406 Search PubMed.
- M. Hua, S. Wu, Y. Ma, Y. Zhao, Z. Chen, I. Frenkel, J. Strzalka, H. Zhou, X. Zhu and X. He, Nature, 2021, 590, 594–599 Search PubMed.
- M. Teodorescu, M. Bercea and S. Morariu, Polym. Rev., 2018, 58, 247–287 Search PubMed.
- S. Moulay, Polym.-Plast. Technol. Eng., 2015, 54, 1289–1319 Search PubMed.
- P. Song, Z. Xu and Q. Guo, ACS Macro Lett., 2013, 2, 1100–1104 Search PubMed.
- W. Xie, Q. Bao, Y. Liu, H. Wen and Q. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 5508–5517 Search PubMed.
- P. Song, J. Dai, G. Chen, Y. Yu, Z. Fang, W. Lei, S. Fu, H. Wang and Z.-G. Chen, ACS Nano, 2018, 12, 9266–9278 Search PubMed.
- P. Song and H. Wang, Adv. Mater., 2020, 32, 1901244 CrossRef PubMed.
- P. Song, Z. Xu, Y. Lu and Q. Guo, Macromolecules, 2015, 48, 3957–3964 Search PubMed.
- S. Wang, L. Lei, Y. Tian, H. Ning, N. Hu, P. Wu, H. Jiang, L. Zhang, X. Luo, F. Liu, R. Zou, J. Wen, X. Wu, C. Xiang and J. Liu, Mater. Horiz., 2024, 11, 2131–2142 RSC.
- R. K. Tubbs, J. Polym. Sci., Part A: Polym. Chem., 1966, 4, 623–629 CrossRef.
- K. Arai, M. Okuzono and T. Shikata, Macromolecules, 2015, 48, 1573–1578 CAS.
- H. Du, T. Zhou, J. Zhang and X. Liu, Anal. Bioanal. Chem., 2010, 397, 3127–3132 CAS.
- Y. Bao, X. Huang, J. Xu and S. Cui, Macromolecules, 2021, 54, 7314–7320 CAS.
- F. Horii, K. Masuda and H. Kaji, Macromolecules, 1997, 30, 2519–2520 CAS.
- H. Li, W. Zhang, X. Zhang, J. Shen, B. Liu, C. Gao and G. Zou, Macromol. Rapid Commun., 1998, 19, 609–612 CAS.
- K. Masuda, H. Kaji and F. Horii, Polym. J., 2001, 33, 356–363 CAS.
- H. Ohgi, H. Yang, T. Sato and F. Horii, Polymer, 2007, 48, 3850–3857 CAS.
- F. Horii, H. Kaji, H. Ishida, K. Kuwabara, K. Masuda and T. Tai, J. Mol. Struct., 1998, 441, 303–311 CAS.
- H. Ohgi, T. Sato, S. Hu and F. Horii, Polymer, 2006, 47, 1324–1332 CAS.
- F. Horii, S. Hu, T. Ito, H. Odani, R. Kitamaru, S. Matsuzawa and K. Yamaura, Polymer, 1992, 33, 2299–2306 CAS.
- Y. Tang, G. Zhu, Y. Li, H. Xu and Z. Liu, J. Fail. Anal. Preven., 2023, 23, 2673–2682 Search PubMed.
- T. Kaiho, Iodine chemistry and applications, John Wiley & Sons, Inc, Hoboken, New Jersey, 2014 Search PubMed.
- W. Zhou and M. Li, China Plast., 2022, 36, 53 Search PubMed.
- W. S. Lyoo, J. H. Yeum, J. M. Park, J. W. Kwak, J. H. Kim, S. S. Kim, B. C. Ji and S. K. Noh, J. Polym. Sci., Part B: Polym. Phys., 2005, 96, 967–974 CAS.
| This journal is © The Royal Society of Chemistry 2025 |