
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reshapable magnetic particles for morphology-controlled soft systems†
Sarah
Schyck‡
 *,
Nitin
Rajendra Madam
and
Laura
Rossi
*
*,
Nitin
Rajendra Madam
and
Laura
Rossi
*
Department of Chemical Engineering, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: l.rossi@tudelft.nl
First published on 18th March 2025
Abstract
Spherical polymeric particles are essential in a wide range of applications, from fundamental self-assembly to bioseparation technologies, with well-established synthetic methodologies. However, incorporating functional components into polymeric matrices to enhance their properties remains a significant challenge, especially when uniformity is required. In this study, we introduce a simple and versatile emulsion evaporation method to fabricate magnetic-loaded polymeric microparticles with exceptional malleability. These composite particles maintain their magnetic functionality while being reshaped into ellipsoids through mechanical stretching. This scalable and straightforward approach offers precise control over particle morphology, offering broad potential for applications in soft robotics, drug delivery, and other magnetically responsive systems.
Introduction
Functional microparticles embedded with inorganic materials find broad applications, from cosmetics1 to optics2 and biotechnology.3–5 Of particular interest are magnetic particles, due to their ability to be manipulated by external magnetic fields.5–8 Several approaches have been developed for the preparation of magnetic polymeric particles. A common method involves polymerizing a monomer in the presence of magnetic nanoparticles,5 such as via dispersion or (mini-)emulsion-based polymerization. These processes typically produce two-phase systems, where polystyrene and magnetic nanoparticles form distinct phases, resulting in Janus-like or core–shell structures depending on the polymerization method.6,9–13 Additional challenges such as phase separation, nanoparticle aggregation, and low nanoparticle loading significantly limit the effectiveness of these techniques.Physical-based methods provide viable alternatives to polymerization. For instance, Ugelstad et al.14,15 developed a technique that involves swelling polymeric microparticles with an organic solvent to allow diffusion of reagents and direct nucleation of inorganic nanoparticles throughout the polymer matrix. In another approach, magnetic micron-size particles are embedded in a polymer matrix by exploiting opposite surface charges and using a plasticizer to physically trap the magnetic particles.16 Similarly, magnetic nanoparticles can be embedded onto the surface of a polymeric particle with opposite surface charge.17,18
Emulsion droplet evaporation has emerged as a promising technique to prepare polymeric microparticles. This method involves evaporation of solvent from emulsion droplets containing dissolved polymers, producing robust polymeric microparticles.19 When paired with block copolymers, microparticles with different morphologies can be formed under controlled evaporation conditions and surfactant selection.20 Adjustment of the emulsification method and evaporation time can also produce dimpled and crumpled microparticles, systems with higher surface areas compared to their spherical counterpart.21 This is advantageous for specific applications such as drug and cargo delivery.22 The most promising synthetic methods primarily yield spherical polymeric particles, while anisotropic shapes, such as ellipsoids, donuts, discs, and rods are generally limited to techniques that lack scalability.23
An alternative to produce anisotropic shapes is the thermal stretching method, developed by Ho et al.,24,25 where polymeric films containing polystyrene microspheres are heated above the glass transition temperature of the film and stretched, resulting in ellipsoidal particles with controlled axial ratios. The stretching of the particle is in part due to the fact that the polymer is uncrosslinked allowing for easier deformation. Champion et al.26 refined this technique to achieve a wide variety of shapes, including rods, disks, and even UFO-like structures. More recently, Lo et al.27,28 further simplified this technique, generating ellipsoids with sharp or blunt ends by simply stretching the particle embedded films with a weight linked to a binder clip. Despite these advances in particle synthesis, achieving precise control over both nanoparticle loading percentage and overall particle shape remains challenging.
In this work, we introduce a straightforward two-step synthesis technique to expand the design space for colloidal particles. Using uncrosslinked polystyrene, and cobalt ferrite nanoparticles, we employ an emulsion evaporation method to produce a variety of magnetic-loaded composite microparticles. These particles are embedded into PVA films and stretched to achieve controlled anisotropic shapes, demonstrating the versatility of this approach in the preparation of anisotropic colloid with controlled magnetic properties.
Methods
Materials
Styrene (with 4-tert-butylcatechol as a stabilizer, ≥99%), dibenzoyl peroxide (BPO, >75%), cobalt(II) chloride hexahydrate (CoCl2·6H2O, ≥95%), iron(III) chloride hexahydrate (FeCl3·6H2O, ≥98%), iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O, ≥98%), sodium hydroxide (NaOH, ≥98%), hydrochloric acid (HCl, 37%), nitric acid (HNO3, 60%), oleic acid (90%), sodium lauryl sulfate solution (SDS, 10%), poly(vinyl alcohol) (PVA, 145![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mW), and tetrahydrofuran (THF, 99.5%) were used as received and purchased from Sigma Aldrich or TCI Europe.
000 mW), and tetrahydrofuran (THF, 99.5%) were used as received and purchased from Sigma Aldrich or TCI Europe.
Chemical synthesis
Polystyrene was synthesized by free radical polymerization of styrene monomers.29 Styrene (27 g), toluene (45 mL), and BPO (0.28 g) were combined in a 100 mL round bottom flask, deoxygenated with argon for 30 minutes, and heated to 80 °C for 37 hours. Polystyrene was obtained at 84% conversion (determined by 1H NMR). The product was diluted with 30 mL chloroform and precipitated in 500 mL ice-cold methanol, then dried under vacuum to achieve 99% purity (Fig. S1, ESI†).Magnetic cobalt ferrite nanoparticles were prepared via a co-precipitation method.30,31 CoCl2·6H2O (2.40 g) was dissolved into a 5 mL aqueous solution of 7.5% HCl, and 5.50 g of FeCl3·6H2O was dissolved in 40 mL of water. The two solutions were briefly stirred together and then rapidly added to 200 mL of boiling NaOH (1 M) solution where a black precipitate was quickly formed. The mixture was vigorously stirred with a magnetic stirrer at 100 °C for 1 hour under reflux. The dispersion was cooled to room temperature and washed via magnetic sedimentation once. The particles were redispersed in 50 mL of water, and 30 mL of 2 M HNO3 was added. Under stirring, 30 mL of a 0.35 M Fe(NO3)3·9H2O solution was added to the particle suspension and heated at 100 °C for 1 hour. The dispersion was cooled again to room temperature, washed thrice via magnetic separation, and redispersed into 50 mL of water (Fig. S1, ESI†). Surface modification was achieved by adding 15 mL oleic acid dropwise to the dispersion, stirring for 18 hours. Surface modification was confirmed upon the successful phase transfer of the cobalt ferrite particles to the oil phase. The aqueous phase was removed using a separatory funnel, and the particles were washed three times with ethanol through magnetic separation. Excess ethanol was then removed, and the particles were dried overnight at 80 °C to yield a powder.
Evaporation-based particle formation
Loaded magnetic polystyrene microparticles were prepared by emulsion evaporation.32 In a typical synthesis, 1 mL of 0.1% SDS solution was mixed with 50 μL DCM solution containing 1.5–10 wt% dissolved polystyrene and 0–1 wt% dispersed cobalt ferrite. The mixture was vortexed for 30 seconds, sonicated up to 5 minutes until it appeared milky white, and diluted with 3 mL water. The dispersion was placed under a vacuum on a rotary evaporator at 40 °C for 25 minutes to remove DCM. Particles were washed thrice via gravitational sedimentation overnight and redispersed in 1 mL of water. Plasticization of the particles was employed to eliminate voids within their structure. To 1 mL of the prepared particles, 3 mL of a 40% THF solution was added. The mixture was gently agitated by hand for approximately 20 seconds, diluted with 20 mL of water, and subsequently washed using magnetic separation or centrifugation.Stretching
Films with embedded polymeric particles were prepared following a modified method from ref. 27. A 20 mL aqueous dispersion containing 0.01 wt% (loaded) polystyrene particles was heated to 80 °C. At temperature, 1 g of PVA was added and magnetically stirred until the PVA dissolved completely, typically 1 hour. The solution sat without stirring for at least 3 hours to remove bubbles. Inside a round 8.5 cm plastic Petri dish, 10 mL of the solution was poured and placed on a level surface overnight for the water to evaporate. The films prepared as described resulted in films with thicknesses around 50 μm devoid of bubbles and lumps. After drying, PVA films were cut into strips (3.5 × 1 cm2) and stretched to specific draw ratios using a Universial Testing Machine (Instron 3365) at 140 °C (Fig. S2, ESI†). Uniform stretching was confirmed by marking films every 0.25 cm before stretching. Films stretched under a magnetic field were first inserted into the machine and clamped with two neodymium magnets placed to apply a magnetic field perpendicular to the stretching direction. Stretched films were cut, rinsed with IPA, and submerged in a 20% v/v IPA–water mixture at 60 °C to dissolve the PVA. The now dispersed stretched particles were washed via centrifugation at 13000 rpm for 1 hour and redispersed in distilled water.
Material characterization
Transmission electron microscopy (TEM) measurements were performed on a JEOL JEM-1400 plus TEM at 120 kV. Samples were prepared by placing 5 μL of a diluted particle dispersion onto carbon-coated copper grids. Scanning electron microscopy (SEM) measurements were performed on a JEOL JSM-6010LA with secondary electron, back-scattered electron detectors, and energy dispersive X-ray spectroscopy (EDX) capabilities. Samples were prepared by drying 5 μL of a diluted particle dispersion onto a cut silica wafer. Electron images and elemental distribution maps were collected. Particle (and droplet) size distributions were calculated from the microscopy images using a custom python script. Particle dynamics were followed using an inverted optical microscope (Nikon Eclipse E600pol) equipped with a 50× objective and a CCD camera. Optical microscopy samples were prepared inside flat rectangular capillaries (Vitrocom 0.05 × 1.00 mm) and sealed with wax at each end to a microscope slide. A uniform magnetic field was applied to the sample with a custom magnetic set-up (Fig. S2, ESI†). Image analysis was preformed with ImageJ Fiji.33 We fit images of the stretched particles to an ellipse with a major axis, a, and minor axis, b, and determine the axial ratio by . The reported angle is defined as the angle between the major axis of the ellipses and a line parallel to the x-axis of the image.
. The reported angle is defined as the angle between the major axis of the ellipses and a line parallel to the x-axis of the image.
Results and discussion
Magnetic polystyrene microparticles were synthesized using an emulsion evaporation method. In this process, polystyrene (PS) and cobalt ferrite nanoparticles were emulsified and subsequently dried to form solid particles. As illustrated in Fig. 1A, the process involves three main stages: (i) mixing of the dichloromethane (DCM) solvent phase containing PS and cobalt ferrite nanoparticles with the aqueous surfactant solution, (ii) emulsification to encapsulate nanoparticles and PS within DCM droplets, and (iii) evaporation of the solvent to solidify the droplets into particles. Two mechanisms are proposed for particle formation during solvent evaporation within an emulsion.34,35 In both, the polymer migrates to the droplet interface, forming a region of high polymer concentration. As the solvent evaporates, the polymer at the interface begins to aggregate and solidify, creating a shell. This shell formation occurs either through a receding mechanism, where the polymer layer contracts inward with the evaporating solvent, or through a stationary mechanism, where the shell remains at the original interface while the solvent diffuses outward through the polymer layer.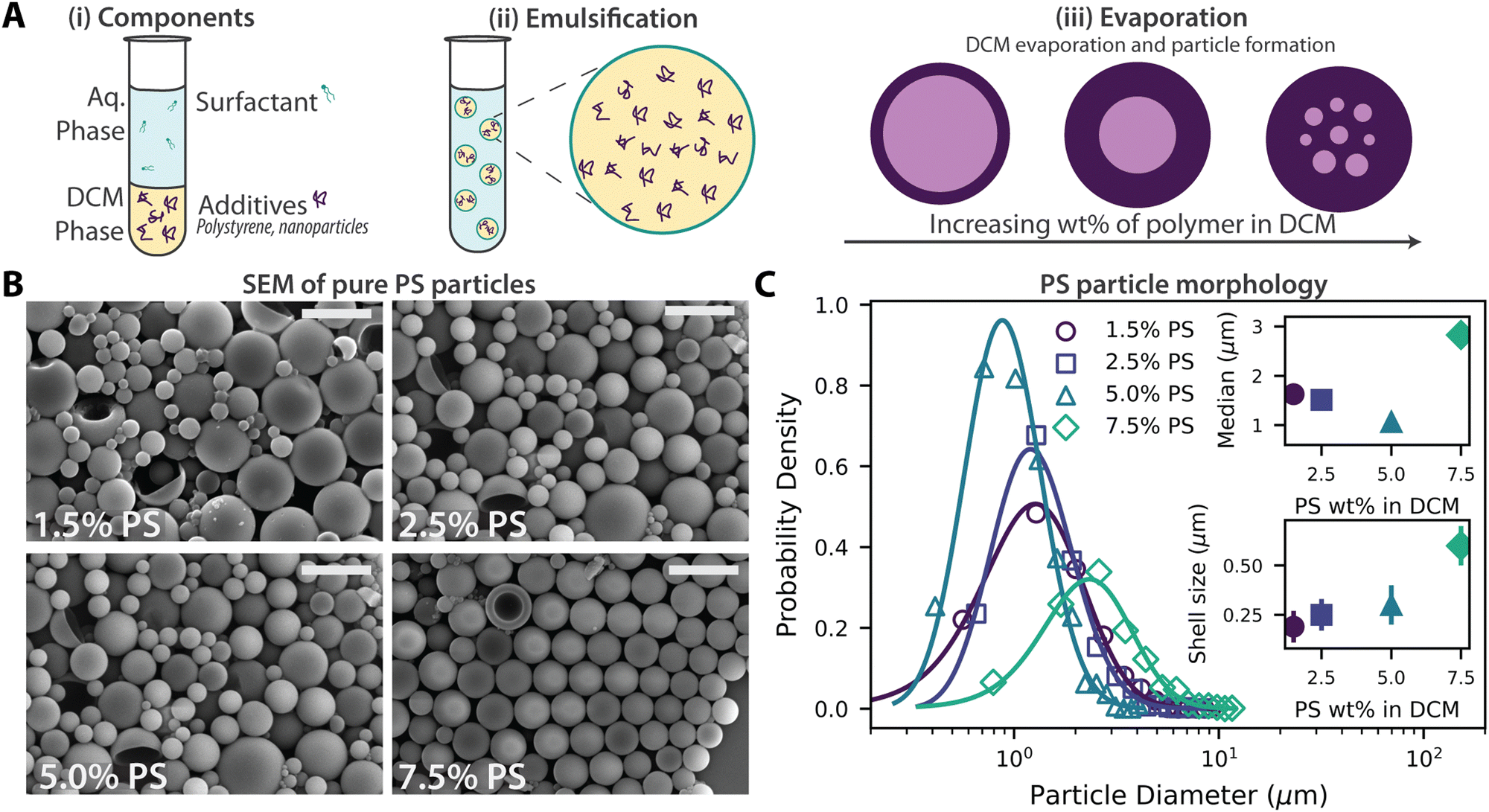 | ||
| Fig. 1 Preparation and morphology of magnetic polystyrene microparticles via the emulsion evaporation method. (A) Schematic of the emulsion evaporation process: (i) initial components include polystyrene (PS), cobalt ferrite nanoparticles, and a surfactant solution. (ii) Emulsification results in PS droplets with encapsulated nanoparticles. (iii) Evaporation produces particles with thinner shells at low PS% and thicker shells at higher PS wt%. (B) SEM images of particles formed from varying PS concentrations: 1.5 wt%, 2.5 wt%, 5.0 wt%, and 7.5 wt%. (C) Particle size distributions for each PS concentration, with insets showing the median particle size and corresponding shell thickness. | ||
To assess the baseline effects of the emulsion evaporation method without the influence of magnetic nanoparticles, we prepared PS microparticles at various initial PS concentrations (1.5, 2.5, 5.0, and 7.5 wt%) in DCM. Higher concentrations (≥10 wt%) produced viscous polymer solutions that failed to fully emulsify, resulting in large, phase-separated polystyrene regions. Conversely, lower concentrations (<1.5 wt%) did not yield sufficient amount of particles. Within the specified range, spherical particles were easily prepared. SEM images in Fig. 1B show representative samples. Generally, most particles exhibited spherical shapes, with occasional bucket-like and dimpled or donut-like morphologies, with sizes ranging from 0.3 to 15 μm. The proportion of non-spherical particles decreased as the initial PS concentration increased. For example, at 1.5 wt% PS, 21% of particles had non-spherical shapes, whereas this number decreases to 2% at 7.5 wt% PS, based on a sample size of over 500 particles. The buckling observed at lower PS concentrations is likely caused by significant pressure differences between the inside and the outside of the particles during drying under vacuum.36,37 Similar shell formation phenomena have been observed in two-polymer systems of PS and PMMA, where dented and dimpled microparticles were observed.38,39 It should be noted that the surfactant type and concentration also strongly influence particle morphology. Previous work by Wang et al.36 demonstrated that altering the surfactant type and concentration could produce unique PMMA particle shapes, such as acorns and ellipsoids. In particular, using SDS consistently produced particles with spherical or dimpled morphologies. Fig. 1C shows the size distributions of microparticles synthesized at different initial PS concentrations: 1.5, 2.5, 5.0, and 7.5 wt%. The distributions, analyzed using log-normal fits,40 revealed median diameters between 1 and 3 μm, with dispersities ranging from 45% to 57%. The wide range of particle sizes reflects the inherent variability of the emulsion-based process37 and, in specific, the broad distribution of droplet sizes generated during sonication-based emulsification, as shown in Fig. S3–S6 (ESI†). The formation of shells (microcapsules) during preparation, is evident in the SEM images. By examining partially encapsulated or broken particles, we estimate the shell thickness to increase with initial PS concentrations, ranging from 0.19 ± 0.08 μm to 0.6 ± 0.1 μm.
To investigate the effect of nanoparticle encapsulation on the composite particles, we prepared samples in the presence of cobalt ferrite (CoFe) nanoparticles. Fig. 2A–E shows SEM images of particles prepared with 2.5 wt% PS in DCM and PS/CoFe nanoparticle weight ratios of 50:1, 25:1, 15:1, 10:1, and 5:1. These ratios correspond to theoretical CoFe loading values of 2–20%. Across all samples, most particles appear spherical, although a small number of bucket-like and dimpled morphologies were also observed. Fig. 2F shows the probability density plots of particle diameters for different loading ratios. As the PS/CoFe ratio decreases (i.e., increasing CoFe content), we observe a decrease in particle diameters. For instance, the 5:1 sample has a median diameter of 1.06 μm with 56% dispersity, suggesting lower size uniformity. In contrast, the 50:1 sample shows a median diameter of 3.22 μm, and a significantly higher dispersity of 84%. As we increase the CoFe content in the particles, as shown in Fig. 3A, more surface features appear. At a 10:1 PS/CoFe ratio, CoFe nanoparticles aggregated into chains and clusters, visible as bright white spots and spiderweb-like structures on the particle in SEM images. This suggests that as the CoFe loading increases, also interactions between nanoparticles are more favorable, leading to clusters on the microparticle surface. At 5:1 ratios and up, we notice the presence of a small number of particles with crumpled textures (<2% of particles), likely due to nanoparticle clusters disrupting the shell uniformity during evaporation. At a 2.5:1 ratio, these aggregates are particularly pronounced, leading to stripe-like features spanning the surface of the particle. Similarly, previous studies on alginate microparticles loaded with drugs such as antioxidants and anti-metabolites observed the formation of dimples, wrinkles, cracks, and overall particle shrinkage at higher loading levels.41–44 These effects were attributed to the presence of amphiphilic surfactants in their systems, which is similar to the one used here. Thus, the observed changes in microparticle morphology may be attributed to two contributing factors: (1) enhanced aggregation of CoFe nanoparticles at higher loadings and (2) interfacial interactions with CoFe and the surfactants present during the synthesis.
 | ||
| Fig. 2 Influence of magnetic cobalt ferrite nanoparticles on composite particle morphology. (A)–(E) Representative SEM images of polystyrene particles with an initial concentration of 2.5 wt% polystyrene in dichloromethane (DCM) at polystyrene-to-cobalt ferrite (PS/CoFe) nanoparticle ratios of 50:1, 25:1, 15:1, 10:1, and 5:1, respectively. (F) Probability density plot of particle diameters corresponding to the samples shown in panels (A)–(E). | ||
 | ||
| Fig. 3 Morphology of polystyrene microparticles loaded with cobalt ferrite. (A) Scanning electron microscopy (SEM) images of particle surfaces at polystyrene-to-cobalt ferrite ratios of 15:1, 10:1, 5:1, and 2.5:1. Scale bars represent 1 μm. (B) Energy-dispersive X-ray spectroscopy (EDS) analysis of particles prepared at a 2.5:1 polystyrene-to-cobalt ferrite ratio, with corresponding back-scattered electron composition (BEC) images and elemental maps for carbon, iron, and cobalt. Scale bars represent 20 μm. | ||
To investigate the uniformity of the cobalt ferrite distribution within the microparticles, we utilize an SEM that detects back-scattered electrons (BSE) and is equipped with energy-dispersive X-ray spectroscopy (EDS). In back-scattered electron composition (BEC) images, changes in apparent intensity correspond to variations in material density, with darker regions indicating lower density and brighter regions indicating higher density. This, combined with EDS, allows us to determine the elemental composition of the samples, as shown in Fig. 3B for a 2.5:1 PS/CoFe sample. In the elemental maps, the PS matrix is identified in the carbon signal, while the cobalt ferrite nanoparticles appear in the iron and cobalt maps. Since the sample is deposited on a silica wafer, particle edges are clearly visible across all maps. The elemental maps confirm that cobalt ferrite nanoparticles are evenly distributed throughout the PS matrix rather than aggregating into distinct, phase-separated domains. However, we observe significant density variations in the BEC-SEM image that are not reflected in the elemental maps, indicating that these density differences arise from voids within the composite particles rather than variations in cobalt ferrite concentration. Additional EDS maps for 100:1 and 10:1 PS/CoFe samples are provided in Fig. S7 (ESI†), further confirming the cobalt ferrite distribution across samples even at low concentrations.
To remove internal voids and ensure the particles are solid, we added tetrahydrofuran (THF) as a plasticizer to the dispersion. The addition of THF causes the particles to swell, allowing the polymer strands to minimize the interfacial energy with the surrounding medium,27 thereby forming uniform spherical particles. After diluting the dispersion and removing the THF, the particles deswell into uniform spheres. Non-destructive BEC-SEM imaging was used to explore the internal morphology of the particles. Fig. 4A shows PS/CoFe particles before and after THF treatment. The bright areas in the SEM image likely correspond to CoFe nanoparticle aggregates, as iron and cobalt are heavier elements compared to the carbon and oxygen atoms in the polymer. Before THF treatment, 94% of particles with diameters >3 μm displayed hollow cores, visible as darker centers compared to the edges in the SEM images. Some examples are highlighted with white arrows in the image. Post-THF treatment, the overall particle size decreases and darker cores are observed in less than 5% of the particles. Darker patches seen post-treatment are mostly gaps between particles formed during SEM sample preparation. Fig. 4B shows size distributions for a series of samples with varying initial PS concentrations before and after THF treatment. The data reveals a dramatic narrowing of the size distribution after plasticization, with no particles exceeding 2.4 μm in diameter. The mean particle diameter of plasticized samples ranges from 1.16 μm to 1.35 μm, representing a reduction of 40–50% compared to pre-plasticized samples. Size dispersity also decreases significantly, with a reduction of up to 45% in some cases. For instance, the sample with an initial 7.5 wt% PS concentration shows the most pronounced change, with a mean diameter reduction from 2.42 μm to 1.21 μm (50.2% reduction) and a dispersity decrease from 56.9% to 36.7%. Similarly, the 1.5 wt% sample exhibits a diameter reduction of 43.8%, with the dispersity dropping from 49.2% to 40.9%. These results confirm that THF processing effectively reduces particles size and homogenizes the size distribution. Statistical analysis using Kolmogorov–Smirnov (K–S) tests yielded p-values <0.0001, supporting the hypothesis that plasticization results in both smaller and more uniform particles.
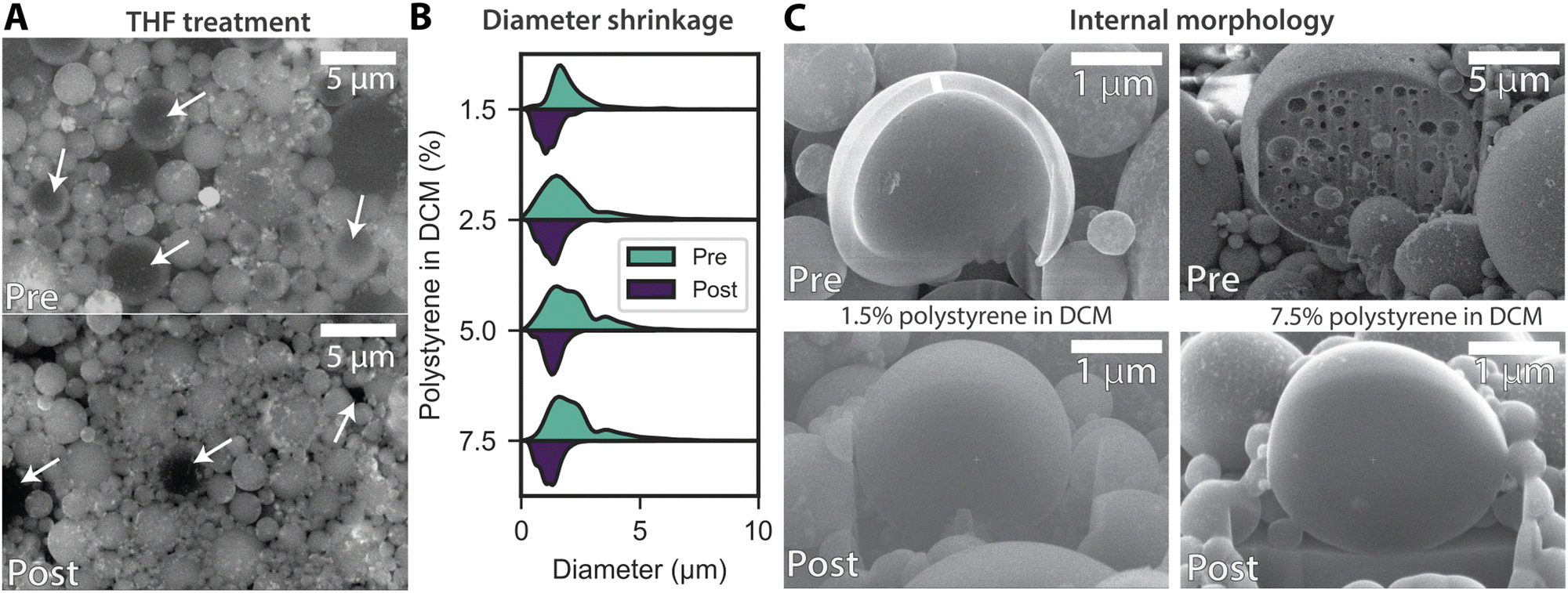 | ||
| Fig. 4 Densification of particles through void removal using a plasticizer. (A) Back-scattered electron composition (BEC) and scanning electron microscopy (SEM) images of polystyrene (PS) and cobalt ferrite particles in a 10:1 ratio, starting with an initial PS concentration of 2.5% in dichloromethane (DCM), shown before and after plasticization with tetrahydrofuran (THF). (B) Violin plot illustrating the particle size distribution changes before and after THF treatment at a 10:1 PS-to-cobalt ferrite ratio. (C) Focused ion beam (FIB)-cut SEM images revealing internal particle structures at low (1.5%) and high (7.5%) initial PS concentrations in DCM, both before and after plasticization. | ||
The presence and subsequent removal of hollow structures in the PS/CoFe particles were confirmed by slicing and imaging selected particles using a focused ion beam (FIB)-equipped SEM. Fig. 4C shows cross-sections of particles with a 10:1 polymer-to-nanoparticle ratio before and after THF treatment at low (1.5%) and high (7.5%) initial PS concentrations in DCM. Prior to the THF treatment, the particles exhibited hollow cores, with a shell of about 0.21 μm for 1.5% initial PS concentration, and a multi-void structure, with an average void diameter of 0.51 μm, for 7.5% initial PS content. After THF treatment, the voids and shell morphologies were no longer present in all samples, indicating that the observed decrease in particle diameter corresponds to the elimination of internal voids.
To further modify the particle morphology, PS spheres treated with THF were embedded in polyvinyl alcohol (PVA) films, which were stretched under controlled conditions. To perform this procedure, PVA films loaded with 10:1 PS/CoFe particles were dried, clamped, and subjected to stretching 140 °C for 25 minutes. During stretching, the PS particles deformed along with the PVA film, owing to their glass transition temperature of approximately 100 °C. After the stretching process, the particles were recovered by selectively dissolving the PVA film in an isopropanol–water solution. The stretching process was controlled to achieve specific draw ratios (DRs), defined as the final film length divided by the initial film length. Fig. 5B shows SEM of particles stretched to draw ratios of 1.5, 2, and 3. We clearly observe the presence of ellipsoidal particles with sharp tips throughout the samples. This is in contrast with previous reports by Lo et al.,27 in which sharp-tipped ellipsoids were only observed when using a pre-annealing step. Without pre-annealing, only blunt-tipped particles formed. Since the driving effect of the thermal stretching process stems from the differences in glass transition temperature of PS and PVA,24,27 we speculate that the molecular weight of our PS (40000) and PVA (145000) polymers is the main cause of the formation of sharp-tipped particles without an annealing step due to the slower deformation of the PVA film under stretching. We characterized the particle dimensions by fitting the particles to 2D ellipses with a major and minor axis length defined by  where 2a and 2b is the length of the major and minor axis, respectively. Measuring the axial size of the particles and their distribution, we obtain similar dispersity (<40%) to the post-THF treated particles.
where 2a and 2b is the length of the major and minor axis, respectively. Measuring the axial size of the particles and their distribution, we obtain similar dispersity (<40%) to the post-THF treated particles.
 | ||
| Fig. 5 Stretching of composite magnetic polystyrene particles. (A) Schematic of the stretching process: magnetic polystyrene particles embedded in a polyvinyl alcohol (PVA) film are stretched vertically at 140 °C to specific draw ratios. This deformation results in ellipsoidal particles as the polystyrene stretches along with the PVA film. (B) SEM images showing the morphology of composite particles at draw ratios (DR) of 1.5, 2, and 3. (C) Plot of draw ratio versus the calculated and measured axial ratios of the stretched particles. (D) Schematic of aligning the magnetic nanoparticles when the microparticles are embedded within the PVA film. (E) Optical micrographs of magnetically treated microparticles in the absence and presence of an applied magnetic field. (F) Alignment of the particle's minor axis with the applied magnetic field direction (90°), derived from the analysis of the optical microscope movie. The particle angles relative to the field direction were used to calculate the cosine similarity, which was normalized to the range [0, 1], where 1 indicates perfect alignment with the magnetic field and 0 indicates complete misalignment. | ||
For PS particles, Lo et al.27,28 showed that the draw ratio, n can be closely correlated to the resultant axial ratio by  . To test this prediction, we calculated the axial ratio
. To test this prediction, we calculated the axial ratio  for draw ratios of 1.5, 2, and 3, yielding measured values of 1.53 ± 0.36, 3.14 ± 0.85, and 5.48 ± 2.12, respectively. These results align reasonably well with the theoretically predicted axial ratios of 1.84, 2.83, and 5.20, as shown in Fig. 5C.
for draw ratios of 1.5, 2, and 3, yielding measured values of 1.53 ± 0.36, 3.14 ± 0.85, and 5.48 ± 2.12, respectively. These results align reasonably well with the theoretically predicted axial ratios of 1.84, 2.83, and 5.20, as shown in Fig. 5C.
We also observe an increase in dispersity with increasing draw ratio, with DR = 3 exhibiting a dispersity of 38%. At higher draw ratios, larger deviations arise due to the deformation of large PS particles, which, because of their size, can fracture or break during the washing phase. Similar work on stretched polystyrene spheres45 reported dispersities of up to ∼24% for the aspect ratio of ellipsoids prepared from monodisperse spherical polystyrene particles (<5% dispersity). Given the already high dispersity (>50%) of our starting spherical particles, the dispersities observed in the stretched particles remain within an acceptable range (Fig. S8, ESI†).
For PS particles loaded with magnetic nanoparticles, such as CoFe, the application of a magnetic field during stretching should result in an overall magnetization of the particles.46 To confirm this hypothesis, we stretched the films under a static magnetic field, as depicted in Fig. 5D. By attaching two neodymium magnets to the film, we applied a magnetic field perpendicular to the stretching direction. Stretching under a magnetic field produced sharp-tipped ellipsoids, confirming that the presence of the magnets and the nanoparticles does not hinder the stretching process.
To assess the magnetization of the PS/CoFe microparticles, we first prepared stretched films with a 10:1 PS/CoFe ratio and a draw ratio DR = 2 under a magnetic field applied perpendicular to the stretching direction. We then re-dispersed the particles in water to create an aqueous sample for optical microscopy. A low magnetic field (3.5 mT) was applied for 20 seconds along the positive y-direction. Fig. 5E shows optical microscope images of the magnetic ellipsoids in the absence and presence of this applied field. Without the field, the particles are randomly oriented. However, when the field is applied, all particles align their minor axes parallel to the field direction.
We tracked the effect of the magnetic field by measuring the angle, θ, of the particles' minor axes with respect to a line parallel to the x-axis, indicated by the white arrows in the images. In Fig. 5F, we plot the angular alignment of the particles with respect to the direction of the magnetic field, where a value of 1 corresponds to perfect alignment. When the magnetic field is applied at 5 seconds, all particles rotate such that their minor axes align with the field. This demonstrates a preferential orientation of magnetic moments parallel to the minor axis of the particle, confirming that the stretching process induces a permanent dipole moment aligned with the short axis.
To further examine alignment dynamics, we analyzed time-resolved optical microscopy images of particles under repeated field application. Fig. S9 and S10 (ESI†) show frame-by-frame tracking of particle rotation upon field activation and deactivation, respectively. The ellipsoids align within 0.5 ± 0.2 s, limited by the frame rate. When the field is turned off after 17 s, relaxation occurs over 0.8 ± 0.2 s, a process most clearly observed in larger particles. These timescales are comparable to those reported for magnetically actuated ellipsoids in low Reynolds number environments, where alignment and relaxation depend on a combination of Brownian motion, viscous damping, and residual magnetic torques.47 Additionally, as shown in Fig. S11 (ESI†), we performed cyclic field application experiments on the two largest particles that remained in-plane throughout the observation period. These particles showed consistent realignment with the applied field when activated and returned to a misaligned state when the field was removed. Interestingly, most particles appeared to reorient to a preferred direction (10°) upon field removal, possibly due to residual stray magnetic fields. These could originate from nearby laboratory equipment or the Earth's magnetic field, as this experiment was not designed to explore zero-field conditions.
For PS/CoFe particles stretched in the absence of a magnetic field, no preferential alignment of microparticles is observed when exposed to low magnetic fields later (Fig. S12, ESI†). These results demonstrate that stretching in a magnetic field successfully imparts a permanent dipole moment despite some dispersity in particle shape and size. Prior studies have shown that polydispersity influences magnetic relaxation behavior, with larger aspect ratio particles exhibiting slower alignment dynamics.48 Alternatively, incorporating external field-driven actuation, as demonstrated in controlled navigation studies,47 may allow for precise reorientation of polydisperse microparticles, mitigating alignment issues in self-assembly applications.
While our study is a proof of concept, future work could explore how variations in aspect ratio dispersity affect the long-term magnetic response of these microparticles. Additionally, tuning the magnetic directionality during the stretching phase could enable greater control over the final alignment of the dipole moments. By modifying the orientation of the applied magnetic field, either through custom set-ups to hold stationary magnets or a rotatable solenoid, it would be possible to direct the magnetic axes of the particles along desired orientations rather than being constrained by a perpendicular field configuration. Furthermore, the resulting particle shape could be tailored by stretching in multiple planes, as demonstrated in prior work on anisotropic particle fabrication.26 Together, these approaches would enable the fabrication of customizable microparticles with tunable shape and magnetic directionality.
These results highlight the versatility of the magnetic alignment process during particle elongation and complement previous methods for anisotropic particle fabrication. For example, Güell et al.49 produced Janus-like ellipsoids using a non-uniform magnetic field applied during stretching in a solvent bath. Their solvent-based approach was particularly effective for commercial magnetic polystyrene spheres, which are coated with polymer layers that hinder adhesion during thermal stretching.50,51 In contrast, our method leverages the thermal plasticization of PS/CoFe particles in a PVA film, enabling the formation of sharp-tipped ellipsoids with high magnetic loadings. This approach provides a scalable and efficient route for fabricating magnetically anisotropic particles with tunable shapes and magnetic properties, opening avenues for applications in advanced colloidal assemblies and responsive materials.
Conclusion
In this study, we developed a general and versatile method to produce magnetic polystyrene microparticles embedded with varying weight percentages of cobalt ferrite nanoparticles. This technique involves creating a solvent-in-water emulsion where the solvent phase (DCM) contains various concentrations of polystyrene and cobalt ferrite nanoparticles, while the continuous phase is an aqueous SDS solution. To eliminate porosity and improve size distributions, we employed a plasticizer to condense the structure while maintaining the desired magnetic loading. Additionally, embedding the particles in a polyvinyl alcohol (PVA) film and thermally stretching them produced anisotropic shapes.Future applications could leverage the inherent magnetic properties of these composite particles for remote manipulation. We showed that by aligning magnetic nanoparticles within a polymer matrix during stretching induces a net magnetization, with the magnetic response depending on factors like magnetocrystalline anisotropy,46,49 which governs magnetic moments reorientations and relaxation behaviors. Cobalt ferrite nanoparticles, with their long Néel relaxation time,52 are particularly promising in this regard, offering stable magnetic retention that could be advantageous in creating permanently magnetized active materials.
Currently, controlling the orientation of magnetic particles is achieved on the millimeter scale through 3D printing, where magnetic fields applied at the extruder nozzle align particles in specific configurations.53–56 Such approaches have been used to create millimeter-sized robots capable of controlled locomotion.57–59 However, scaling these capabilities down to the micrometer level remains challenging due to fabrication limitations. Alternative lithographic techniques have been explored to induce magnetic alignment on a smaller scale, with methods such as confining a photopolymer in a mold under a magnetic field to create micro-actuators.60 In contrast, we show that our method offers a new avenue for producing uncrosslinked, anisotropic magnetic particles embedded in a solid matrix. This opens possibilities for microscale systems, such as soft robotics, where anisotropically aligned magnetic particles could enable precise and responsive movement. Similarly, these particles hold potential in targeted drug delivery, where magnetic guidance could direct them to specific sites in the body. As research progresses, embedding techniques like ours may help overcome microfabrication constraints, paving the way for the development of remotely controlled, magnetically responsive materials at the microscale.
Author contributions
S. S.: conceptualization, methodology, investigation, and writing – original draft. N. R. M.: validation. L. R.: resources, writing – review & editing, and supervision.Data availability
The data that support the findings of this study are openly available in 4TU.ResearchData at https://doi.org/10.4121/f0c6a28c-9f22-430c-8c43-f2199150192b, ref. 61.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Duco Bosma for help characterizing loaded polystyrene with various SEM techniques as well as Hozanna Miro for assistance with FIB measurements and Reece Lewis for assistance with PS synthesis and characterization.Notes and references
- M. J. Murray and M. J. Snowden, Adv. Colloid Interface Sci., 1995, 54, 73–91 CrossRef CAS.
- T. Nisisako, T. Torii, T. Takahashi and Y. Takizawa, Adv. Mater., 2006, 18, 1152–1156 CrossRef CAS.
- O. Yassine, C. P. Gooneratne, D. Abu Smara, F. Li, H. Mohammed, J. Merzaban and J. Kosel, Biomicrofluidics, 2014, 8, 034114 CrossRef CAS.
- H. Zhang and M. E. Meyerhoff, Anal. Chem., 2006, 78, 609–616 CrossRef CAS PubMed.
- B. Zhang, J. Xing and H. Liu, Front. Chem. Eng. China, 2007, 1, 96–101 CrossRef CAS.
- P. Tempesti, M. Bonini, F. Ridi and P. Baglioni, J. Mater. Chem. A, 2014, 2, 1980–1984 RSC.
- C. Baresel, V. Schaller, C. Jonasson, C. Johansson, R. Bordes, V. Chauhan, A. Sugunan, J. Sommertune and S. Welling, Heliyon, 2019, 5, e02325 Search PubMed.
- Q. Liu, L. Wang, A. Xiao, J. Gao, W. Ding, H. Yu, J. Huo and M. Ericson, J. Hazard. Mater., 2010, 181, 586–592 CAS.
- H. Baharvand, Colloid Polym. Sci., 2014, 292, 3311–3318 CAS.
- W. Wichaita, D. Polpanich, C. Kaewsaneha, K. Jangpatarapongsa and P. Tangboriboonrat, Colloids Surf., B, 2019, 184, 110557 CrossRef CAS PubMed.
- M. M. Rahman, F. Montagne, H. Fessi and A. Elaissari, Soft Matter, 2011, 7, 1483–1490 CAS.
- S. Braconnot, M. M. Eissa and A. Elaissari, Colloid Polym. Sci., 2013, 291, 193–203 CrossRef CAS.
- H. Zhang, Q. Zhang, B. Zhang and F. Guo, J. Magn. Magn. Mater., 2009, 321, 3921–3925 CrossRef CAS.
- J. Ugelstad, L. Söderberg, A. Berge and J. Bergström, Nature, 1983, 303, 95–96 CrossRef.
- J. Ugelstad, P. Stenstad, L. Kilaas, W. S. Prestvik, R. Herje, A. Berge and E. Hornes, Blood Purif., 1993, 11, 349–369 CAS.
- T. Hueckel, G. M. Hocky and S. Sacanna, Nat. Rev. Mater., 2021, 6, 1053–1069 CrossRef CAS.
- Y.-H. Li, T. Song, J.-Q. Liu, S.-J. Zhu and J. Chang, J. Mater. Chem., 2011, 21, 12520–12528 CAS.
- T.-H. Chung and W.-C. Lee, React. Funct. Polym., 2008, 68, 1441–1447 CAS.
- K. H. Ku, J. M. Shin, H. Yun, G.-R. Yi, S. G. Jang and B. J. Kim, Adv. Funct. Mater., 2018, 28, 1802961 Search PubMed.
- S.-J. Jeon, G.-R. Yi and S.-M. Yang, Adv. Mater., 2008, 20, 4103–4108 CAS.
- S. Liu, R. Deng, W. Li and J. Zhu, Adv. Funct. Mater., 2012, 22, 1692–1697 CAS.
- M. Iqbal, N. Zafar, H. Fessi and A. Elaissari, Int. J. Pharm., 2015, 496, 173–190 CAS.
- J. H. Kim, T. Y. Jeon, T. M. Choi, T. S. Shim, S.-H. Kim and S.-M. Yang, Langmuir, 2014, 30, 1473–1488 CAS.
- C. C. Ho, R. H. Ottewill, A. Keller and J. A. Odell, Polym. Int., 1993, 30, 207–211 CAS.
- C. C. Ho, A. Keller, J. A. Odell and R. H. Ottewill, Colloid Polym. Sci., 1993, 271, 469–479 CAS.
- J. A. Champion, Y. K. Katare and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11901–11904 CrossRef CAS.
- Y.-C. Lo, H.-F. Tseng, Y.-J. Chiu, B.-H. Wu, J.-W. Li and J.-T. Chen, Langmuir, 2018, 34, 8326–8332 CrossRef CAS PubMed.
- Y.-C. Lo, Y.-J. Chiu, H.-F. Tseng and J.-T. Chen, Langmuir, 2017, 33, 12300–12305 CrossRef CAS PubMed.
- G. M. Burnett, G. G. Cameron and S. N. Joiner, J. Chem. Soc., Faraday Trans. 1, 1973, 69, 322–327 RSC.
- F. A. Tourinho, R. Franck and R. Massart, J. Mater. Sci., 1990, 25, 3249–3254 CrossRef CAS.
- J. Wagner, T. Autenrieth and R. Hempelmann, J. Magn. Magn. Mater., 2002, 252, 4–6 CrossRef CAS.
- L. Rossi, S. N. Schyck and N. Madam, Nanoparticle-Loaded Polystyrene Colloids, IPC No. A61K, A61L, C08L, A61P, C09J, C08J, H01F, Priority date 6 Apr 2023, Priority No. WO2024/210746, 2024.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- W.-I. Li, K. W. Anderson and P. P. Deluca, J. Controlled Release, 1995, 37, 187–198 CrossRef CAS.
- M. Li, O. Rouaud and D. Poncelet, Int. J. Pharm., 2008, 363, 26–39 CAS.
- Y. Wang, B.-H. Guo, X. Wan, J. Xu, X. Wang and Y.-P. Zhang, Polymer, 2009, 50, 3361–3369 CAS.
- A. Loxley and B. Vincent, J. Colloid Interface Sci., 1998, 208, 49–62 CAS.
- N. Saito, Y. Kagari and M. Okubo, Langmuir, 2006, 22, 9397–9402 Search PubMed.
- M. Okubo, N. Saito and T. Fujibayashi, Colloid Polym. Sci., 2005, 283, 691–698 Search PubMed.
- J. C. Thomas, J. Colloid Interface Sci., 1987, 117, 187–192 Search PubMed.
- A. R. Ahmady, K. Razmjooee, V. Nazar and S. Saber-Samandari, Mater. Chem. Phys., 2023, 294, 126982 CAS.
- M. Dhanka, C. Shetty and R. Srivastava, Int. J. Biol. Macromol., 2018, 110, 294–307 Search PubMed.
- Z. G. Cadena-Velandia, J. C. Montenegro-Alarcón, X. Marquínez-Casas and C. E. Mora-Huertas, J. Drug Delivery Sci. Technol., 2020, 56, 101558 Search PubMed.
- P. B. O'Donnell and J. W. McGinity, Adv. Drug Delivery Rev., 1997, 28, 25–42 Search PubMed.
- S. Trevenen and P. J. Beltramo, J. Colloid Interface Sci., 2021, 583, 385–393 CrossRef CAS.
- S. van Berkum, P. D. Biewenga, S. P. Verkleij, J. H. B. A. van Zon, K. W. M. Boere, A. Pal, A. P. Philipse and B. H. Erné, Langmuir, 2015, 31, 442–450 CrossRef CAS PubMed.
- H. M. Gauri, R. Patel, N. S. Lombardo, M. A. Bevan and B. Bharti, Small, 2024, 20, 2403007 CrossRef CAS PubMed.
- A. O. Ivanov and P. J. Camp, Phys. Rev. E, 2023, 107, 034604 CrossRef CAS.
- O. Güell, F. Sagués and P. Tierno, Adv. Mater., 2011, 23, 3674–3679 CrossRef.
- L. Gabrielson and M. J. Folkes, J. Mater. Sci., 2001, 36, 1–6 CrossRef CAS.
- H. Kim, J. L. Bauer, P. A. Vasquez and E. M. Furst, J. Phys. D: Appl. Phys., 2019, 52, 184002 CrossRef CAS.
- C. R. Stein, M. T. S. Bezerra, G. H. A. Holanda, J. André-Filho and P. C. Morais, AIP Adv., 2018, 8, 056303 CrossRef.
- Y. Wang, R. Deng, L. Yang and C. D. Bain, Lab Chip, 2019, 19, 3077–3085 CAS.
- J. Zhang, Z. Ren, W. Hu, R. H. Soon, I. C. Yasa, Z. Liu and M. Sitti, Sci. Rob., 2021, 6, eabf0112 Search PubMed.
- H. Song, J. Spencer, A. Jander, J. Nielsen, J. Stasiak, V. Kasperchik and P. Dhagat, J. Appl. Phys., 2014, 115, 17E308 Search PubMed.
- M. Gao, M. Kuang, L. Li, M. Liu, L. Wang and Y. Song, Small, 2018, 14, 1800117 Search PubMed.
- M. H. D. Ansari, V. Iacovacci, S. Pane, M. Ourak, G. Borghesan, I. Tamadon, E. Vander Poorten and A. Menciassi, Adv. Funct. Mater., 2023, 33, 2211918 CAS.
- T. Xu, J. Zhang, M. Salehizadeh, O. Onaizah and E. Diller, Sci. Rob., 2019, 4, eaav4494 Search PubMed.
- W. Hu, G. Z. Lum, M. Mastrangeli and M. Sitti, Nature, 2018, 554, 81–85 CAS.
- J. Kim, S. E. Chung, S.-E. Choi, H. Lee, J. Kim and S. Kwon, Nat. Mater., 2011, 10, 747–752 CAS.
- S. Schyck, N. R. Madam and L. Rossi, Data underlying the article: Reshapable Magnetic Particles for Morphology-Controlled Soft Systems, 2025, Version 1. 4TU.ResearchData. dataset, DOI:10.4121/f0c6a28c-9f22-430c-8c43-f2199150192b.v1.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sm00061k |
| ‡ Present address: Faculty of Aerospace Engineering, Delft University of Technology, Kluyverweg 1, Delft, 2629 HS, The Netherlands. E-mail: s.n.schyck@tudelft.nl |
| This journal is © The Royal Society of Chemistry 2025 |