
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advances in the catalysis of reduction of nitroaromatics and its mechanism: a tutorial review
Wenjie Guo,
Yisha Zheng,
Wenlong Xiang and
Yanhui Zhang *
*
College of Chemistry, Chemical Engineering and Environment, Fujian Province Key Laboratory of Morden Analytical Science and Separation Technology, Minnan Normal University, Zhangzhou, 363000, P.R. China. E-mail: zhangyh@mnnu.edu.cn; Tel: +86 596 2591445
First published on 16th December 2024
Abstract
This paper reviews the research on the conversion of nitroaromatics to aminoaromatics through catalysis. The traditional catalytic technology employs iron powder as a catalyst, which tends to generate a large amount of solid waste and water pollutants, making it the focus of research for improvement. The current research status of the catalytic reduction of nitroaromatic reactions is introduced, highlighting the main catalytic mechanism currently employed and the work conducted by our group in furthering this research. The current status and limitations of the catalytic reduction of nitroaromatic reactions are discussed. Additionally, the possibility of modulating the reaction pathway and the future development of the reaction are explored.
Sustainability spotlightNitroaromatic compounds, widely used as essential chemical intermediates in numerous industrial applications, pose significant environmental and health risks due to their carcinogenic, mutagenic, and teratogenic properties. These compounds are among the most persistent pollutants, presenting major challenges for environmental remediation. Traditional methods for their removal, such as adsorption, ozonation, bioremediation, and electrochemical processes, are often inefficient, particularly for large-scale applications. Recent advancements in room-temperature catalytic reduction offer a promising alternative, primarily due to its effectiveness and the relatively low toxicity of the resulting product, aminophenol (AP), which is a valuable fine chemical. The comprehensive utilization of industrial waste water has attracted tremendous interest in recent times. Therefore, exploring related reduction techniques including recycling of wastes containing harmful substances within aqueous ecosystems is not only the most elemental environmental problem but also crucial for economic performance. Traditional methods for nitroaromatic reduction often involve the use of toxic reagents and high-energy-consuming processes, which pose significant environmental hazards. The review identifies important gaps in the current understanding, such as the precise role of hydrogen sources in the reduction process, and emphasizes the need for further exploration in this area. The development of highly efficient, sustainable catalysts is crucial for optimizing room-temperature catalytic reduction technologies, which not only address environmental concerns related to hazardous nitroaromatic compounds but also contribute to the broader challenge of industrial wastewater management. These advancements have the potential to improve both the economic viability and environmental sustainability of industrial processes, particularly in the context of wastewater recycling and pollution reduction. |
Introduction
Among organic pollutants, nitro-phenolic compounds are considered to be some of the main pollutants causing water pollution due to their high solubility and stability in water.1,2 However, they occupy an important place in chemical production and are of great use in the fields of medicine, dyes, and rubber.3,4 Due to their toxicity and carcinogenicity, it is necessary to degrade them into less toxic compounds.5,6 The amine compounds obtained from the reduction of aromatic nitro compounds are relatively less toxic and represent important fine chemicals.7–11 With the development of arylamine derivatives for medical anticancer applications, the quest for environmentally friendly and efficient methods to reduce nitroaromatic hydrocarbons to arylamines has also gained considerable attention.12–14 Scientists have proposed many methods for reducing nitrophenols, including catalytic hydrogenation,15,16 metal reduction,17–19 hydrazine hydrate reduction,19–21 alkali sulfide reduction,22 electrochemical reduction,23–25 catalytic hydrogen transfer,26–28 photocatalytic reduction,29–33 glucose reduction,34 and enzyme-catalyzed reduction.35Generally, the traditional industrial methods of preparing aromatic amines from nitroaromatics include (1) reduction of nitroaromatics using sulfide bases, with commonly used sulfide bases being Na2S, NaHS, Na2S2, etc.;36,37 (2) using nitroaromatics as the raw material, with iron powder employed for reduction in an acidic medium. The reaction solution is neutralized, washed, and distilled to obtain the finished product;38,39 (3) nitroaromatics or their derivatives are reduced in a fluidized bed vessel by gas phase hydrogenation in the presence of RANEY® Ni or an active Cu catalyst. The reaction product is then condensed and distilled under reduced pressure to obtain the desired product.40,41 There are various methods for treating nitroaromatic pollutants in water, and catalytic technology meets the dual requirements of achieving both economic and environmental benefits through wastewater treatment, owing to its simplicity, high efficiency, and low cost.42–47 However, the traditional catalyst, iron powder, tends to produce a large amount of iron sludge during the reaction process, which is not easy to dispose of. Therefore, catalyst preparation becomes crucial for improving efficiency.48
The main catalytic techniques for the green reduction of nitroaromatics are room temperature catalysis and photocatalysis. Catalyst activation or electron transfer becomes challenging in room temperature reactions alone. Therefore, NaBH4 is often used as the hydrogen source and electron donor, which greatly restricts its popularity in catalytic reactions due to the difficulties in long-term preservation at room temperature and the limited supply of NaBH4.49 In contrast, photocatalytic technology does not require the participation of a reducing agent and uses the proton solvent as the hydrogen source, thereby eliminating the need for an additional hydrogen source and overcoming the drawbacks associated with NaBH4. The catalytic reduction reaction involves the transfer of nitroaromatics and hydrogen reduction, highlighting the importance of the hydrogen source and electron transfer in exploring the mechanism. Additionally, further research focuses on the reuse potential of the products and by-products.
In this paper, we review the research progress on the reduction of nitroaromatics within the framework of catalytic nitroaromatic reduction–amination reactions, focusing on the catalytic mechanisms.
Main catalytic mechanism
Precious metal catalysts
The currently widely accepted mechanism, proposed by Haber50 in 1898 based on experiments with the electrochemical reduction of nitrobenzene, is divided into two pathways, the “one-step hydrogenation” and the “condensation reaction”, as depicted in Fig. 1.51 The direct route involves N-phenylhydroxylamine as an intermediate, while the indirect route involves the oxidation of azobenzene as an intermediate. In this process, the reductive dehydration of aniline first produces nitrobenzene. Nitrosobenzene can react with hydrogen to first produce the phenylhydroxylamine intermediate, which is then reduced to aniline. Alternatively, it can undergo a condensation reaction with the resulting phenylhydroxylamine to produce azobenzene oxide. In this case, the oxidized azobenzene can be further reduced to aniline through the intermediate process of azobenzene. | ||
| Fig. 1 Two reaction routes of the reduction of nitrobenzene. Reprinted with permission from ref. 51. Copyright 2018, American Chemical Society. | ||
Gelder and co-workers52 improved the hydrogenation reaction mechanism of nitroaromatics and their derivatives, building on the mechanism proposed by Haber, as depicted in Fig. 2. Based on the low rate of aniline production from the hydrogenation of nitrosobenzene and the observation of different kinetic isotope effects, they concluded that nitrosobenzene is not an intermediate of the nitrobenzene hydrogenation reaction. Furthermore, in the reduction reaction of nitrobenzene with hydrogen, the initial product was found to be oxidized azobenzene, resulting from the condensation reaction, rather than phenylhydroxylamine. Notably, no aniline production could be detected when nitrobenzene was not completely converted to oxidized azobenzene. Through further studies, Gelder and colleagues proposed the presence of another surface intermediate, Ph–N(OH) (a). During the hydrogenation of nitrobenzene, Ph–N(OH) (a) reacts with adsorbed hydrogen; while during the reaction of nitrobenzene, Ph–N(OH) (a) reacts with itself to undergo dehydration and produce azobenzene oxide.
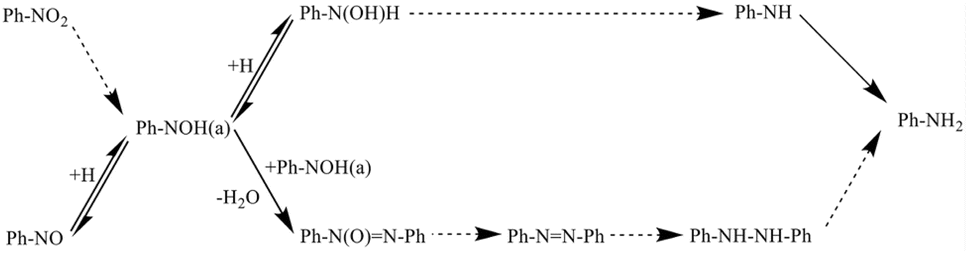 | ||
| Fig. 2 Mechanism of nitrobenzene hydrogenation proposed by Gelder. Reprinted with permission from ref. 52. Copyright 2005, Royal Society of Chemistry. | ||
Makaryan and Savchenko,53 by loading Ir, Pt, and Pd on carbon as catalysts, proposed a new argument for the mechanism of catalytic hydrogenation of nitroaromatics. They argued that the conversion of aromatic nitro compounds to aniline occurs through the disproportionation of the corresponding hydroxylamine rather than via hydrogenation. Studer and colleagues54 addressed this disproportionation mechanism creatively by using catalysts such as iron salts and vanadium salts in solution to promote the disproportionate reaction of hydroxylamine and nitroso intermediates, thereby preventing the accumulation of hydroxylamine. On the other hand, Visentin and co-workers,55 utilizing calorimetry, FTIR-ATR (attenuated total reflection), and gas absorption signals with Pd/C as the catalyst, concluded that the findings of Makaryan and Savchenko were consistent with a direct hydrogenation route, without significant hydroxylamine accumulation.
Corma and his team56 proposed the reaction pathway for the hydrogenation reaction of aromatic nitro compounds on Au/TiO2, which is shown in Fig. 3. The concentration of nitrosobenzene decreased during the reaction, while the concentrations of phenylhydroxylamine and aniline gradually increased. This observation suggests that under the influence of hydrogen, nitroaromatics undergo dehydration and reduction to form nitrosobenzene compounds. Subsequently, these compounds are further reduced to phenylhydroxylamine through reaction with hydrogen. Finally, phenylhydroxylamine undergoes hydrogenation to yield aniline. The reaction pathway was inferred from in situ FTIR experiments involving nitrobenzene and nitrosubstituted benzene. Corma and his colleagues proposed that the reaction may not proceed through nitrosubstituted benzene as an intermediate, but rather directly reduce nitrobenzene to phenylhydroxylamine. This direct reduction pathway contributes to aniline production, aligning with the findings of Gelder and his team. These observations suggest that nitrobenzene may not serve as an intermediate product in the hydrogenation of nitrobenzene.
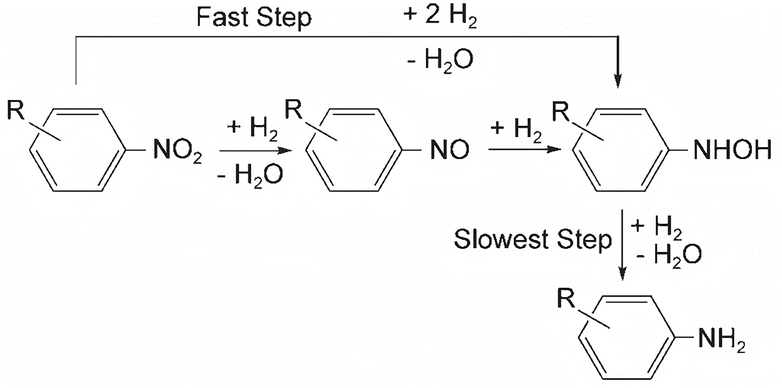 | ||
| Fig. 3 Hydrogenation pathways of aromatic nitro compounds on Au/TiO2. Reprinted with permission from ref. 56. Copyright 2007, Wiley. | ||
Electronic transmission pathways
In the reduction of nitroaromatics, the choice of catalyst directly influences the reaction efficiency, selectivity, and the formation of by-products. Sodium borohydride is a commonly used strong reducing agent that effectively provides hydrogen atoms.57 It is stable under mild conditions and has a wide range of applications. Its low reactivity and easily controllable hydrogen by-products make it safer for practical use. Aminoborane, on the other hand, is a mild reducing agent with good selectivity and operates under mild conditions.58 However, it is prone to hydrolysis, so moisture should be avoided during its use. Alcohol-based reductants are easy to handle and have low toxicity, but they exhibit weaker reducing power and are less efficient than sodium borohydride.59,60 Formic acid is used in acidic environments with mild reaction conditions, but its reducing power is limited, often requiring high temperatures or catalysts to enhance its effectiveness.61 Hydrazine hydrate is an efficient reducing agent that provides hydrogen atoms.62 The hydrogen bonds in the hydrazine hydrate molecule contribute to its stability. However, during the reaction, hydrazine hydrate preferentially dissociates the N–H bond to form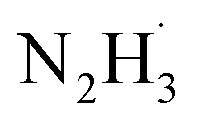 and H˙. After several N–H bond cleavages, hydrazine decomposes into nitrogen and hydrogen, making it relatively environmentally friendly.63 Although hydrazine hydrate is highly efficient, it should be used with caution, as excessive temperatures and acidic conditions can trigger its decomposition. Additionally, it is somewhat toxic, necessitating careful handling.
and H˙. After several N–H bond cleavages, hydrazine decomposes into nitrogen and hydrogen, making it relatively environmentally friendly.63 Although hydrazine hydrate is highly efficient, it should be used with caution, as excessive temperatures and acidic conditions can trigger its decomposition. Additionally, it is somewhat toxic, necessitating careful handling.
During the catalyzed reaction, both nitroaromatic compounds and reducing agents are adsorbed onto the surface of the catalyst. Specifically, the electrons from NaBH4 are transferred to the nitroaromatic molecules, facilitating the reaction.64 The rate of this electron transfer process is strongly influenced by the surface potential of the catalyst, particularly that of the metal nanoparticles.65,66 Metal nanoparticles with a higher surface potential can support a stronger electron flow, thereby accelerating the transfer of electrons from NaBH4 to the nitroaromatic compounds.
The ability of metal nanoparticles to accelerate electron transfer primarily stems from their unique electronic structure, particularly the properties of their atomic valence and Fermi energy levels. First, the atomic valence of a metal determines the number and reactivity of electrons in the outermost electron shell. Metals with higher atomic valence generally have more electrons, making it easier for them to interact with external substances. In catalytic reactions, these additional electrons are more likely to participate in the electron transfer process. For example, on the surface of metal nanoparticles, metals with higher atomic valence can provide more electrons, enabling stronger interactions with adsorbed reactants (e.g., NaBH4 and nitroaromatics), thus facilitating electron transfer. Additionally, the Fermi energy level of a metal represents the highest energy state of its electrons, reflecting the electron distribution within the material. In redox reactions, the electron transfer process is often governed by the Fermi energy level. Metals with higher Fermi energy levels can more readily donate or accept electrons, accelerating the reaction. When the Fermi energy level is high, electrons on the metal surface are more easily detached and transferred to neighboring reactants, thereby enhancing the overall reaction rate.67 Therefore, when selecting a catalyst, both the Fermi energy level and surface potential of the metal are critical factors for accelerating the reaction.
Overall, the role of metal nanoparticles in catalytic reactions extends beyond providing catalytic sites; they also enhance electron mobility and accelerate the transfer of electrons from NaBH4 to nitroaromatics through their surface potential, atomic valence, and Fermi energy levels. These properties determine the catalysts' activity and efficiency and are key factors to consider when designing effective catalysts.
In bimetallic or polymetallic catalysts, the synergistic effects between metals—encompassing both electronic and geometric effects—play a key role in the catalytic process.68 The electronic effect refers to the transfer of electrons between metal atoms. In bimetallic catalysts, there are typically metal atoms with high activity, where the outer electrons are more readily involved in the reaction. These highly active metal atoms can transfer electrons to neighboring, less active metal atoms through additional electrons in their outer orbitals. In this process, the higher-activity metal atoms donate electrons to the lower-activity metal atoms, creating an electron-rich region.69 This electron redistribution enhances the activity of the catalytic reaction, particularly by facilitating the transfer of electrons from the adsorbed reductant to the reactants, which is crucial for the reaction's success. On the other hand, geometric effects refer to how the morphology, size, and arrangement of metals influence catalytic performance. The catalyst's geometry determines how reactants interact with the metal surface, which in turn impacts the reaction efficiency. For example, metal nanoparticles, with their large specific surface area, provide more active sites compared to bulk metals, thereby enhancing reactant adsorption and increasing the reaction rate.
In bimetallic catalytic systems, a typical example is that nitroaromatic molecules are more readily adsorbed on Au atoms, while NaBH4 tends to adsorb on Pt.70 In this system, Au atoms transfer electrons to neighboring Pt atoms due to their higher electron density, resulting in electron enrichment on the Pt surface. As a result, the electron-enriched Pt surface is more likely to accept electrons from NaBH4, thereby facilitating the electron transfer process. This electron transfer between metals not only accelerates the reaction rate but also enhances the overall activity of the catalyst.
Liu et al.71 introduced a novel approach to investigate the catalytic reduction of nitroaromatics by Ag nanoparticles (NPs) in the presence of NaBH4. In their study, they proposed that silver nanoparticles play a more complex role in the reaction, not only acting as conventional catalysts but also enhancing reaction efficiency through a unique catalytic mechanism. First, when silver nanoparticles are immersed in a solution containing NaBH4, NaBH4 undergoes a decomposition reaction that produces a large amount of H2. They suggested that this process accelerates hydrogen generation by adsorbing BH4− onto the surface of the silver nanoparticles. The adsorption of BH4− lowers the energetic barrier for the reaction, facilitating hydrogen release. The generated H2 molecules then interact with the surface of the silver nanoparticles, specifically exchanging electrons with the d-orbitals of the silver atoms. Silver atoms, rich in d-orbital electrons, donate electrons to the H2 molecule via antibonding molecular orbitals. This interaction reduces the bonding level of the H2 molecule, activating it. Specifically, the molecular orbitals of the H2 molecule change, and the antibonding molecular orbitals receive electrons from the silver d-orbitals, making the H2 molecule more susceptible to cleavage, thereby generating highly reactive hydrogen atoms. Additionally, the nitro (–NO2) group coordinates with the silver nanoparticles. The oxygen atom in the nitro group interacts with the unoccupied orbitals of silver through its electron-rich region, transferring electrons from the nitro group's oxygen to the d-orbitals of the silver atoms. The silver nanoparticles provide a channel for electron transfer through their surface-rich electrons, facilitating the reduction of nitroaromatics. Electrons are transferred from the nitro group to the hydrogen molecule, further promoting its activation. In this way, silver nanoparticles act as mediators of electron transfer during the catalytic process, accelerating the transfer of electrons from the nitro group to hydrogen. Ultimately, the highly activated hydrogen atoms react to form metal hydrides.
Our group's main work and its extensions
Building upon the aforementioned mechanism, our research group conducted a series of explorations to enhance the catalytic efficiency of the reduction of nitroaromatics. Initially, we employed room temperature catalysis to investigate the loading of Au–Pd bimetallic catalysts. We synthesized Au–Pd/RGO, taking into account the unique two-dimensional electronic structure and properties of reduced graphene oxide (RGO). As the field progressed towards metal–organic framework (MOFs) catalysts with high crystallinity, we shifted our focus to these materials. Subsequently, we synthesized a range of iron-based MOFs, specifically MIL materials, and assessed their catalytic performance in the reduction of nitroaromatics. Furthermore, we explored a photocatalytic approach using CdS-ATP photocatalysts. This approach harnesses the energy of visible light to facilitate the reduction of nitroaromatics under milder conditions. Our research not only deepens the understanding of the reduction reaction of nitroaromatics but also provides valuable insights for investigating other catalytic reactions.Metal-supported catalysts
Carriers typically possess a large specific surface area, providing more catalytic sites and helping to disperse nanoscale catalyst particles. This prevents aggregation during the reaction process, thereby maintaining the catalyst's high efficiency.72 Additionally, the interfacial effect of the carrier strengthens the interaction between the catalyst and the reactants, attracting more reactant molecules to adsorb onto the catalyst surface. This enhanced adsorption not only improves catalyst activity but also facilitates the effective transfer of photogenerated carriers, further boosting catalytic efficiency. Graphene, as a carrier, possesses excellent electron transport properties, which help increase the electron transport rate of the bimetallic catalyst.73–79 Additionally, the synergistic effect of the bimetallic catalyst enhances its catalytic activity. Moreover, graphene exhibits high mechanical strength and chemical stability, effectively preventing the exfoliation or deactivation of the bimetallic catalyst and prolonging its service life.Zhang's group80 developed a simple and environmentally friendly one-step method to prepare palladium (Pd) nanoparticles loaded on graphene oxide (GO). In this method, Pd nanoparticles were uniformly anchored on the surface of graphene oxide, preventing aggregation and ensuring high exposure of active sites. The uniformly distributed palladium nanoparticles provided sufficient channels for electron transfer, further facilitating the efficient progress of the reaction. More importantly, the small size of the palladium nanoparticles effectively lowered the reaction's potential barrier, accelerating the electron transfer rate and, consequently, the reduction reaction. In the reaction, NaBH4, acting as a reducing agent in an aqueous, basic environment, rapidly captured hydrogen ions (H+) from the nitrophenol molecule, leading to the formation of nitrophenol salts. This process promotes smooth hydrogenation.
After investigating the selective catalytic reduction of p-nitroaniline to p-phenylenediamine (PPD) using Au–Pd/RGO, our group81 found that Au–Pd/RGO exhibited the highest catalytic activity compared to Pd/RGO and Au/RGO, primarily due to its smaller particle size. Our investigation revealed that BH4− and p-nitroaniline diffused from the aqueous solution onto the surface of the noble metal, as displayed in Fig. 4. The 2D-RGO effectively dispersed the noble metal, providing more active sites and enhancing electron transfer efficiency. Additionally, the p–p stacking interaction exhibited high adsorption capacity, facilitating the adsorption of BH4− and p-nitroaniline onto the noble metal surface. Consequently, BH4− transfers electrons to the noble metal, which then catalyzes the reduction of p-nitroaniline to PPD by transferring electrons to it.
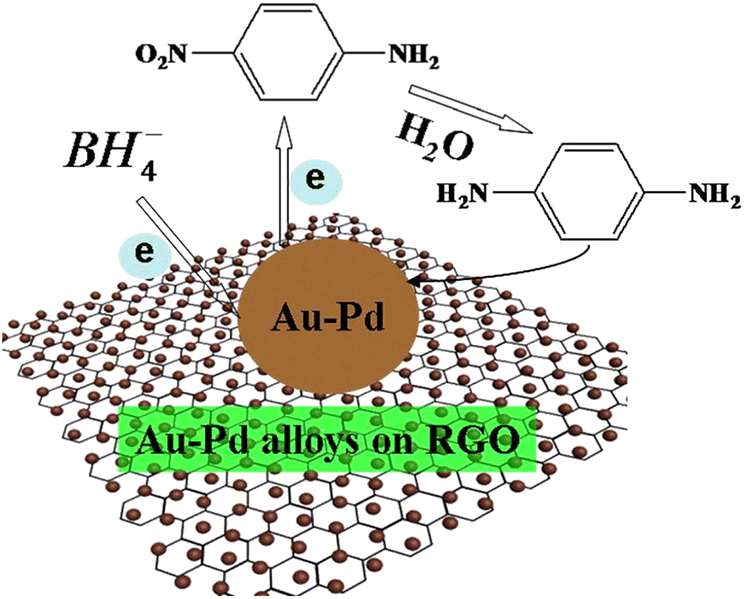 | ||
| Fig. 4 Proposed schematic mechanism for the catalytic selective reduction of 4-nitroaniline to p-phenylenediamine over Au–Pd/RGO under ambient conditions. Reprinted with permission from ref. 79. Copyright 2018, Elsevier. | ||
The importance of an actual hydrogen source for understanding the interfacial reaction mechanism from –NO2 to –NH2 is often overlooked in research, where the reductant NaBH4 is generally considered as the hydrogen source. First, BH4− is activated on the catalytic metal surface, producing adsorbed H species. This mechanism has been applied to most of the p-NP catalytic reduction reactions on metal catalysts, such as Ag NPs, Ag–Pd bimetallic alloys, and CuNPs. However, as a reaction medium for catalytic reduction, H2O can also provide hydrogen atoms or protons for the reaction. Wang and co-workers82 observed the important role of H2O in the reduction of hydrides. Although both the reducing agent BH4− and the solvent H2O can contribute hydrogen to form the same reduction products, the hydrogen transfer pathways of BH4− and H2O are different: one originates from metal–H adsorbed at the interface, and the other arises from protons in the solvent. Zhao and co-workers83 conducted deuterium isotope experiments using heavy water (D2O) and sodium borohydride deuteride (NaBD4). Based on the results of Raman, Fourier transform infrared (FTIR), and 1H nuclear magnetic resonance (NMR) spectroscopy, it was concluded that the hydrogen atoms used for the conversion of –NO2 to –NH2 originate from the solvent protons.
Metal–organic framework materials (MOFs)
MOFs have a very high specific surface area and can provide a large number of active sites, improving the efficiency of the catalyst. The pore size and pore structure of MOFs can be precisely controlled by synthetic methods to achieve selective adsorption and conversion of reactant molecules. These numerous advantages make their application prospects very promising.84–87Furthermore, after investigating the catalytic reduction of p-nitrophenol with simultaneous hydrogen production using MIL-101(Fe)/SiO2, our group88 found that the number of active sites on the surface decreased after SiO2 loading on MIL-101(Fe). Unexpectedly, MIL-101(Fe)/SiO2 exhibited better catalytic performance for aromatic reactions. Nevertheless, the enhanced catalytic activity is not primarily attributed to the increase in surface area. Simultaneously, the excellent alkali resistance of MIL-101(Fe)/SiO2 opens up new avenues for the application of such materials. The mechanism of the catalytic reaction was then proposed: the adsorption of NaBH4 on the unsaturated metal coordination site surface facilitates the dissociation of NaBH4 to release H-donating electrons. An oxidation reaction between NaBH4 and MIL-101(Fe) resulted in the transfer of electrons to the surface of the coordinated Fe. Subsequently, these electrons interacted with the p-NP compounds adsorbed on the catalyst's surface, leading to the transfer of electrons to p-NP and its reduction to p-aminophenol, as displayed in Fig. 5.
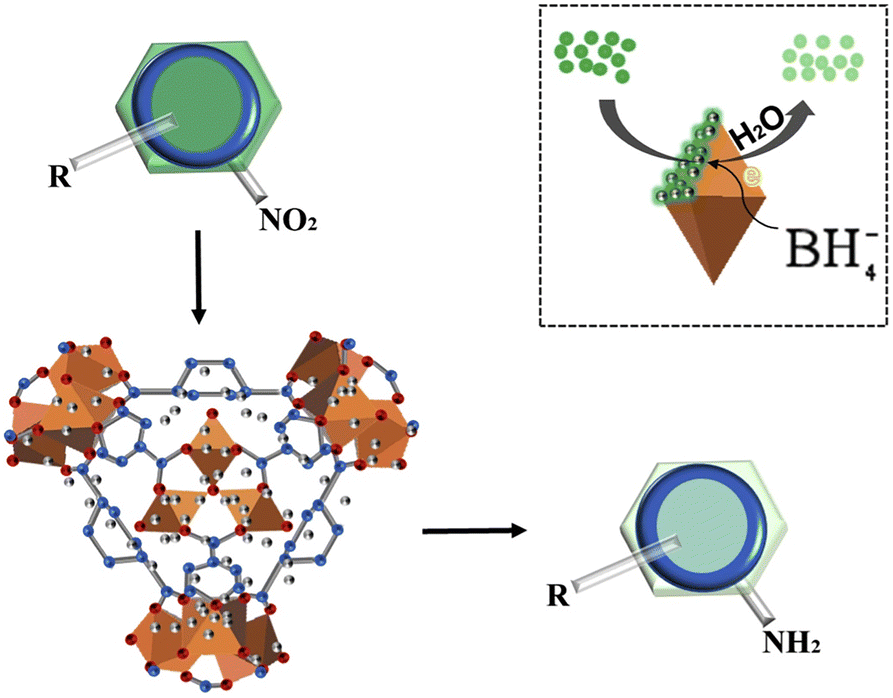 | ||
| Fig. 5 Schematic illustration of the proposed mechanisms of the conversion of NP to AP by MIL-101(Fe)/SiO2. Reprinted with permission from ref. 88. Copyright 2020, Elsevier. | ||
Building upon the aforementioned research, our group89 conducted a study on hydrogen production through the catalytic reduction of nitro-organics using MIL-101(Fe)-based composites. We further optimized the catalytic reaction mechanism as follows: upon the addition of MIL-101(Fe) catalysts, the adsorption of NaBH4 on the unsaturated metal coordination site surface facilitates the dissociation of NaBH4 to release H-donating electrons. The hydrogen atoms adsorbed from BH4− either combine with OH− to form H2O or with neighboring borohydride ions to generate H2. Subsequently, an oxidation reaction takes place between NaBH4 and MIL-101(Fe), resulting in the transfer of electrons from the Fe surface to the reaction center of the nitroxide. A source of hydrogen in water is then utilized to reduce p-NP hydride to p-AP, followed by the acquisition of protons from water by –NO2, leading to its conversion into p-aminophenol ions through multiple intermediates, as shown in Fig. 6.
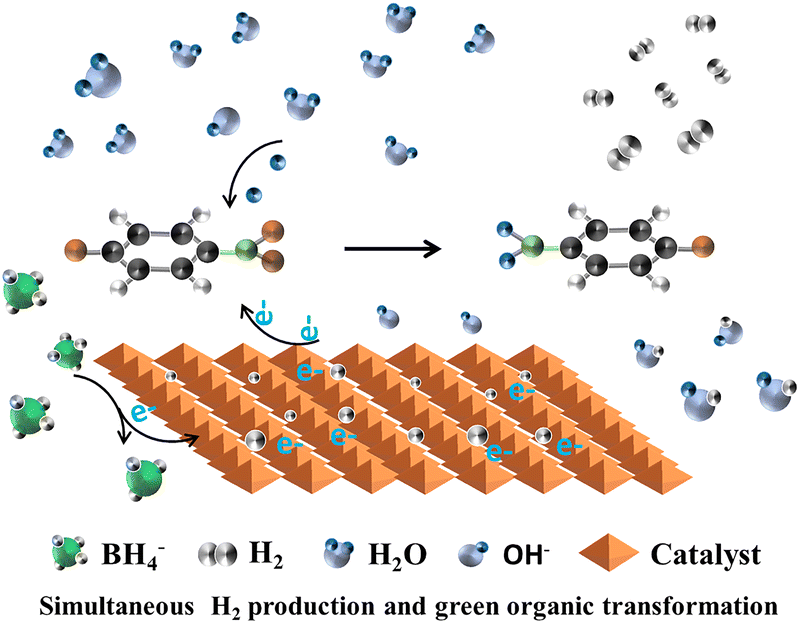 | ||
| Fig. 6 Schematic illustration of the proposed mechanism of simultaneous H2 production and green organic transformation. Reprinted with permission from ref. 89. Copyright 2021, Royal Society of Chemistry. | ||
Transition metal oxides
Transition metal oxides are widely used in chemical research due to their unique electronic structure and tunable redox properties, particularly in catalytic reactions where they demonstrate excellent performance. Taking Mn2O3 as an example, this transition metal oxide not only exhibits good catalytic activity but also offers strong stability and environmental friendliness. The advantage of Mn2O3 lies in its multiple oxidation states (Mn2+ and Mn3+),90 which allow it to deliver excellent catalytic performance in a variety of reactions. In particular, Mn2O3 plays a key role in the oxygen reduction reaction, where it effectively regulates electron transfer and enhances catalytic efficiency.Han's group91 synthesized Mn2O3 nanorods using a calcination method. It was demonstrated that the higher proportion of lattice oxygen in Mn2O3 nanorods significantly promotes surface electron transfer, leading to excellent catalytic performance, particularly in the reduction of nitroaromatics. As shown in Fig. 7, during the reaction, NaBH4 molecules first adsorb onto the catalyst surface and decompose to form NaB(OH)4 and active hydrogen (Mn–H−). These active hydrogens then attack the positively charged nitrogen in the nitro group of the nitro compound, forming intermediate (1). At this stage, the negatively charged oxygen in the nitro compound absorbs protons (H+) from the solvent, producing intermediate (2). As the reaction progresses, active hydrogen further attacks these intermediates, undergoing rearrangement and dehydration, ultimately leading to the rapid generation of aromatic amines. During the catalytic process, the adsorbed oxygen in the Mn2O3 material may have some adverse effects on the reduction reaction, as its oxidizing nature depletes active hydrogen, inhibiting the reaction's efficiency. However, the oxygen adsorbed in the Mn2O3 nanorods has a high lattice energy, which effectively prevents excessive oxidation and reduces the consumption of active hydrogen. Moreover, the lattice oxygens enhance surface electron transfer, further accelerating the reduction reaction.
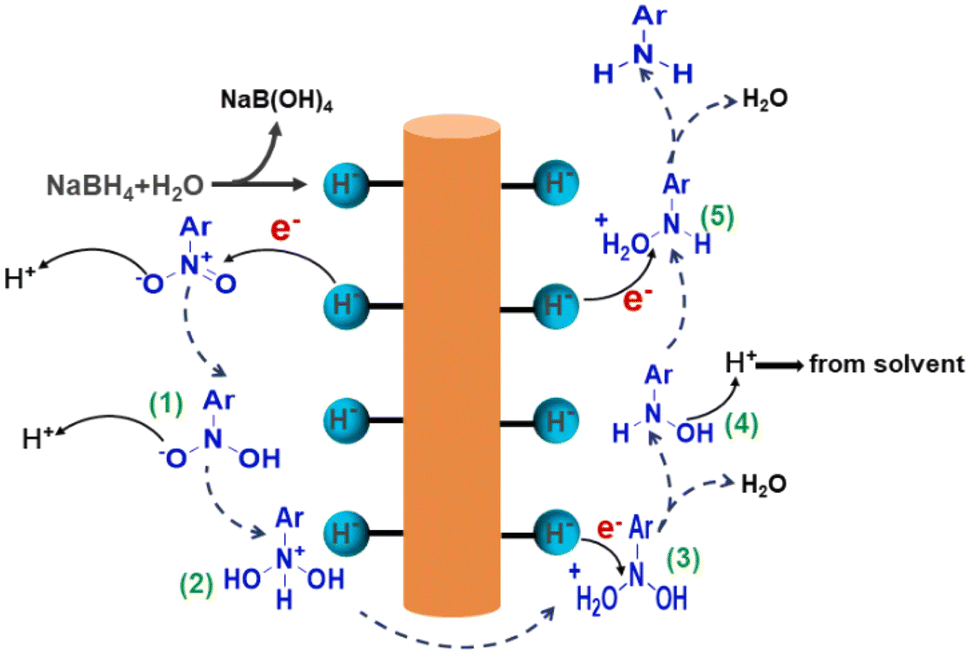 | ||
| Fig. 7 Plausible mechanism of reduction of nitroarenes catalyzed by Mn2O3 nanorods. Reprinted with permission from ref. 91. Copyright 2024, Elsevier. | ||
Non-metal catalysts
The structure of non-metallic catalysts is typically based on elements such as carbon, nitrogen, and sulfur, which interact with reactants through coordination or chemical bonding. Unlike metallic catalysts, non-metallic catalysts do not rely on electron transfer from metal sites; instead they promote reactions through intermolecular interactions (e.g., hydrogen bonding, electrostatic interactions, etc.).92 Specific active centers on the catalyst (e.g., acid–base groups, lone electron pairs, π-electrons, etc.) enhance their interaction with reactants. Non-metallic catalysts are abundant, inexpensive, and environmentally friendly, as they do not introduce metal contamination. Moreover, they offer better selectivity in certain reactions, effectively control reaction pathways, and reduce by-product formation.Chen's group93 successfully demonstrated the reduction of nitroaromatics using phosphorus-doped carbon nanotubes (P-CNTs) as metal-free catalysts. The P-CNT catalysts play a key role in the adsorption and activation of hydrogen molecules due to their unique structure and properties. Specifically, the phosphorus atoms on the surface of P-CNTs influence the electron distribution of the carbon nanotubes, leading to the accumulation of localized charges and the formation of Lewis-like acid–base pairs (FLP mechanism). This acid–base pair facilitates the hydrogenation of nitroaromatics by polarizing hydrogen molecules and dissociating them into hydrogen ions (H+) and hydride ions (H−), generating a highly active hydrogen source. The acidic and basic sites of the P-CNT catalysts work together to enhance the activation efficiency of H2, enabling smooth nitroaromatic reduction reactions under mild conditions. Additionally, this synergistic effect effectively inhibits the formation of by-products and ensures reaction selectivity. The P-CNT catalysts exhibit good adaptability to nitroaromatics with different functional groups, selectively reducing the nitro group to an amino group without the need for additional steps. For example, P-CNTs can selectively hydrogenate nitro groups on nitroaromatics containing functional groups such as cyano and acyl, with minimal by-product generation. Notably, the P-CNT catalyst maintains efficient catalytic performance even for difficult-to-reduce nitro compounds, such as dinitrobenzene and iodonitrobenzene. It selectively converts these compounds to the corresponding amino aromatics without dehalogenation or other undesired reactions. This makes P-CNTs an ideal catalyst for the efficient and highly selective hydrogenation of nitroaromatics.
Semiconductor composite photocatalysts
Compared with precious metals, metals, and bimetals, photocatalysis offers mild reaction conditions with few by-products and high energy utilization, aligning well with the principles of green chemistry. Additionally, catalysts predominantly consist of semiconducting materials or composites, known for their stability and renewability. This technology has wide-ranging application prospects and potential, emerging as a hot topic of research in recent years.94After investigating the catalytic reduction of p-NP by CdS-ATP, our group95 found that ATP (natural attapulgite, with the ideal molecular formula Al2Mg2Si8O20(OH)2(OH2)4·4H2O) possesses a porous structure that provides numerous micro-reactive sites, effectively accelerating the adsorption of pollutants. The dispersed deposition of CdS particles on the surface of ATP carriers enhances resistance to photocorrosion. Additionally, the CdS–20%ATP composite material, with its lower impedance, promotes interfacial electron transfer, prolongs the lifetime of photo-generated charge carriers, and thereby increases the photocatalytic reaction rate.
A possible photocatalytic reduction mechanism is also proposed: upon exposure to simulated solar light, electrons in the valence band of CdS undergo excitation and transition to the conduction band, transforming into photogenerated electrons. The low charge transfer resistance of the composite material effectively transfers electrons produced by CdS to ATP quickly, prolonging the lifetime of photogenerated carriers. During the reduction process, photogenerated holes are captured by the hole sacrificial agent ammonium formate, effectively preventing the recombination of electron–hole pairs, while photogenerated electrons reduce p-NP to generate p-AP, as shown in Fig. 8.
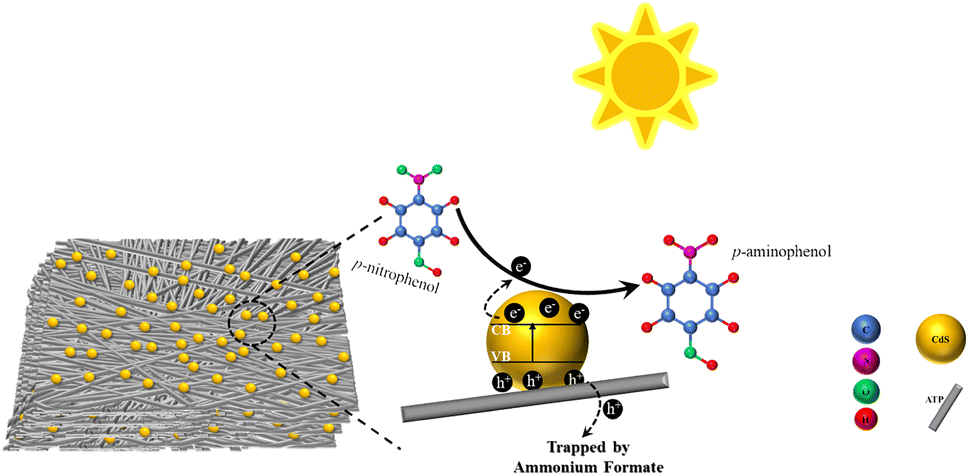 | ||
| Fig. 8 Possible mechanism of photocatalytic reduction of p-NP under simulated solar light. Reprinted with permission from ref. 95. Copyright 2021, Royal Society of Chemistry. | ||
Quantum dots
Quantum dots are among the common nanomaterials known to possess a band gap that varies with size. Scientists have discovered that modifying wide-bandgap semiconductors with narrow-bandgap semiconductor quantum dots can lead to rapid charge transfer through the swift diffusion of charge carriers at the interface, thereby significantly enhancing photocatalytic activity. To the best of our knowledge, CdS is a crucial narrow-bandgap semiconductor with a notable quantum size effect, featuring excellent absorption properties for visible light and demonstrating superior photocatalytic activity.96,97By coupling InOOH with CdS, Wang's group98 synthesized CdS–In heterojunction semiconductors, which enhance the separation efficiency of photo-generated charge carriers, leading to an increase in available electrons. It was also observed that optimizing the photodeposition time could result in the effective dispersion of CdS QDs on InOOH and an excellent response to visible light, thus enhancing activity. Based on this, the CdS–In-120 catalysts were prepared (where 120 refers to 120 minutes of irradiation with a 254 nm UV lamp prior to centrifugation), and their performance was investigated. The decreased PL intensity observed in CdS–In-120 suggests that the recombination of photogenerated charge carriers is impeded by the heterojunction formed between CdS QDs and InOOH. Explicitly, a possible mechanism for the photocatalytic reduction of nitroaromatics by CdS–In-120 was proposed, as shown in Fig. 9. Upon exposure to visible light, CdS QDs are excited to create electron–hole pairs. By utilizing the appropriate band alignment between CdS QDs and InOOH, as well as a well-defined interface structure, electron transfer from the conduction band (CB) of CdS QDs to that of InOOH is achieved, significantly enhancing the efficiency of photo-generated charge carrier separation. Meanwhile, the addition of HCOONH4 as a hole scavenger under a N2 atmosphere allows the photogenerated holes in CdS–In-120 to be captured, thereby lowering the likelihood of electron–hole complex formation and providing sufficient photogenerated electrons for the reaction. This effectively prevents the oxidation reaction of nitroaromatics.
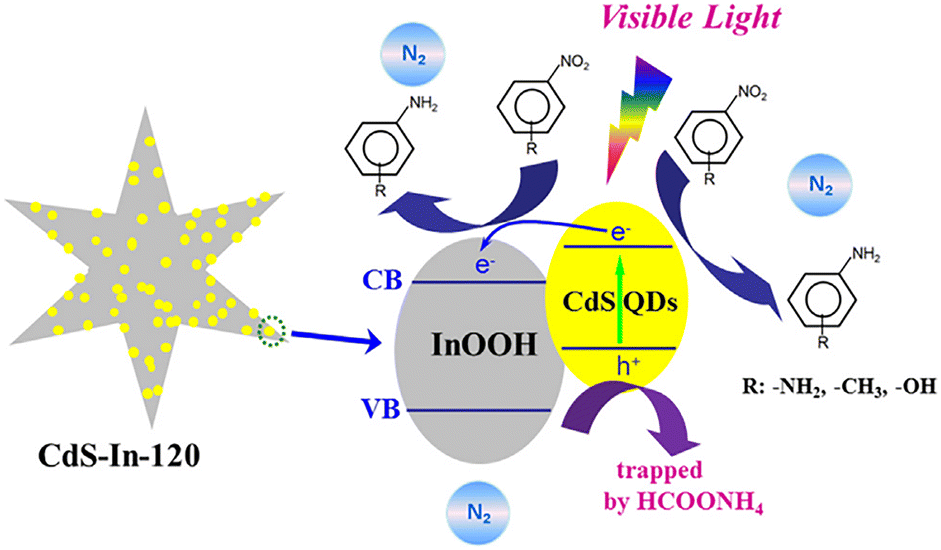 | ||
| Fig. 9 Mechanism proposed for the visible light-induced photocatalytic reduction of nitro aromatics using CdS–In-120. Reprinted with permission from ref. 98. Copyright 2021, Elsevier. | ||
Under room temperature catalytic conditions, different catalysts using NaBH4 as a reducing agent exhibit significant differences in their performance in reduction reactions, as shown in Table 1. Metal–carrier type catalysts exhibited the highest reduction efficiency within a short time, owing to the small size of the metal nanoparticles and the use of appropriate carrier materials. This suggests that both the size of the nanoparticles and the design of the carrier play a crucial role in the catalytic reaction. Fine metal nanoparticles enhance the exposure of active sites, while the carrier material ensures the uniform dispersion of nanoparticles, which in turn improves substrate adsorption and reactivity. Other types of materials also offer unique advantages. MOF materials promote electron transfer through the effective dispersion of unsaturated metal sites. Although their reaction efficiency is relatively low, their structural design holds significant potential. On the other hand, transition metal oxide catalysts promote surface electron transfer through the involvement of lattice oxygen. Despite requiring longer reaction times, they maintain a reduction efficiency of up to 99%, and their inexpensive raw materials make them promising for practical applications.
| Catalytic types | Catalyst types | Catalyst | Active site | Structure–activity relationship | Reduction efficiency | Reaction settings | Ref. |
|---|---|---|---|---|---|---|---|
| Room temperature catalysis (reductant: NaBH4) | Metal–support catalysts | Pd/GO | Pd NPs | Small-sized palladium nanoparticles can effectively reduce the reaction barrier, while the support GO provides an environment for the uniform anchoring of the Pd NPs | 99% (p-NP, 10 min) | p-NP: 1 mmol, catalyst: 20 μL, NaBH4: 1.2 mmol | 80 |
| Au–Pd/RGO | Au–Pd NPs | The small size of NPs helps expose more active sites and p–p stacking interactions contribute to enhancing substrate adsorption | 99% (4-NA, 4 min) | 4-NA: 10 ppm, catalyst: 5 mg, NaBH4: 8 mg (a total of 160 mL of solution) | 81 | ||
| MOFs | MIL-101(Fe)/SiO2 | Unsaturated metal sites on the Fe surface | Dispersion of unsaturated metal sites promotes effective adsorption and facilitates electron transfer | 94% (p-NP, 4 min) | p-NP: 10 ppm, catalyst: 10 mg, NaBH4: 60 mg (a total of 60 mL of solution) | 88 | |
| Transition metal oxides | Mn2O3 | Mn2O3 | A high proportion of lattice oxygen can facilitate the transfer of surface electrons | 99% (HNBA, 20 min) | HNBA: 10 μL (0.1 M), catalyst: 1 mg, NaBH4: 2 mL (0.1 M) | 91 | |
| Visible light catalysis (hole scavenger: ammonium formate) | Semiconductor composites | CdS–20%ATP | CdS | The porous structure of ATP provides abundant micro-reaction sites for the reaction, and the uniform dispersion of CdS particles enhances the material's resistance to photo-corrosion | 93% (p-NP, 10 min) | p-NP: 10 ppm, catalyst: 0.1 g, AF: 0.1 g (a total of 120 mL of solution) | 95 |
| QDs | CdS–In-120 | CdS QDs | The heterojunction structure significantly reduces the recombination of photogenerated charge carriers | 99% (p-NP, 40 min) | p-NP: 20 ppm, catalyst: 30 mg, AF: 30 mg (a total of 80 mL of solution) | 98 |
Semiconductor composites and quantum dot catalysts exhibit unique structural properties. The uniform dispersion of CdS particles enhances photocorrosion resistance, while quantum dot catalysts optimize the dispersion of photogenerated carriers through photodeposition techniques. Although the reaction time is relatively long (40 minutes), the advantage of photocatalytic reactions lies in their ability to utilize renewable light sources to drive the reaction, thereby avoiding the need for large quantities of chemical reductants such as NaBH4, which are commonly used in traditional chemical reduction reactions. This characteristic aligns with the sustainable development principles of green chemistry. A key future direction for green chemistry may involve the optimization of catalyst nanostructures and the design of carrier materials. Specifically, the synthesis of catalysts with efficient electron transfer properties could further enhance reaction rates and selectivity. Therefore, when designing catalysts, it is crucial to consider not only the selection of suitable active sites but also the electronic properties, carrier design, and stability of the catalysts in practical applications.
Summary and outlook
In conclusion, although the reduction of nitroaromatics can be achieved under mild conditions using strong reducing agents and a large number of catalysts, the high cost, operational complexity, and extensive catalyst use limit the scalability of this method for industrial production. Future research should focus on optimizing catalysts and exploring alternatives, particularly the development of greener, low-cost, and efficient catalytic systems. For example, metal-free catalysts such as phosphorus-doped carbon nanotubes (P-CNTs) can effectively promote the activation and dissociation of hydrogen under mild reaction conditions, enabling efficient reduction of nitroaromatics with good potential for industrial application. These catalysts not only eliminate the need for strong reducing agents and excess catalysts, but also demonstrate excellent selectivity and high efficiency. Additionally, catalytic systems based on Lewis acid–base pairs show great promise. These novel catalysts can efficiently activate hydrogen and achieve selective reduction of nitroaromatics through precise modulation of surface electrons. Although these approaches are still in the research and development stage, they represent more environmentally friendly and sustainable reduction strategies, with the potential to overcome the limitations of existing technologies. Overall, while the nitroaromatic reduction reaction has made significant progress as a probe reaction for MOF materials and quantum dot materials in the past two decades, further optimization of catalyst design, cost reduction, and improvements in the stability and reproducibility of the catalytic process are still necessary to address challenges in practical applications. These directions undoubtedly offer a broad prospect for future research.In summary, the catalytic efficiency and stability of catalysts can be effectively improved by selecting suitable carriers for loading the catalysts and doping the carriers with heteroatoms. Although significant research has been conducted in recent years to develop greener, environmentally friendly, stable, and cost-effective efficient catalysts, the understanding of the reduction process of nitroaromatics remains incomplete. Additionally, the presence of by-products during the reaction and the effects of related additives are unclear. For instance, does the use of different hole-trapping agents in the photocatalytic reaction have varying effects on different types of catalysts, directly impacting the rate of recombination of electron–hole pairs, reaction kinetics, and product selectivity? Will further treatment be required to separate the catalysts from the water? If different products are formed, could different reaction pathways be regulated by adjusting the surface properties of the catalysts, optimizing reaction conditions, and controlling the generation of reaction intermediates and the conversion process? Therefore, future research should focus on the following aspects: (1) Further strengthening the study of reaction by-products and their reaction pathways. Regulation of product selectivity can be achieved for different reaction paths and products by adjusting the surface properties of the catalyst, optimizing reaction conditions, controlling the generation of reaction intermediates, and the conversion process. (2) Modulating reaction pathways to increase the generation of target products. (3) Optimization of the reaction process for green, recyclable, easily recoverable, and large-scale preparation. (4) Investigating the relationship between different additive auxiliaries, nitroaromatics, and catalysts. Further exploration of the mechanism of different types of hole-trapping agents on various catalysts, including their impact on photogenerated carrier utilization efficiency, electron–hole pair recombination rate, reaction kinetics, and product selectivity. (5) Exploring the application of electrocatalytic technology in the reduction of nitroaromatics to achieve an efficient reaction using the electron transfer process. This will help expand future studies from exploring the fundamental aspects of the nitroaromatic reduction process to its potential impact in different areas, providing new ideas and methods for studying the reaction mechanism and industrial production of aromatic nitro compounds.
Data availability
Data availability is not applicable to this article as no new data were created or analyzed in this study.Author contributions
Wenjie Guo: investigation, writing – original draft. Yisha Zheng: validation, project administration. Wenlong Xiang: conceptualization. Yanhui Zhang: resources, writing – review & editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (Grant No. 21703094), the Natural Science Foundation of Fujian Province (Grant No. 2019J01743), and the Natural Science Foundation of Zhangzhou Municipality (Grant No. ZZ2023J11).Notes and references
- J. Tiwari, P. Tarale, S. Sivanesan and A. Bafana, Environ. Sci. Pollut. Res., 2019, 26, 28650–28667 CrossRef CAS PubMed.
- S. Payamifar and A. P. Marjani, Appl. Organomet. Chem., 2023, 37, e7287 CrossRef CAS.
- S. U. Rehman, M. Siddiq, H. Al-Lohedan and N. Sahiner, Chem. Eng. J., 2015, 265, 201–209 CrossRef.
- Y. S. Keum and Q. X. Li, Chemosphere, 2004, 54, 255–263 CrossRef CAS.
- M. Stiborova, Chem. Listy, 2002, 96, 784–791 CAS.
- P. Kovacic and R. Somanathan, J. Appl. Toxicol., 2014, 34, 810–824 CrossRef CAS PubMed.
- C. V. Rode, M. J. Vaidya and R. V. Chaudhari, Org. Process Res. Dev., 1999, 3, 465–470 CrossRef CAS.
- D. Hu, Y. Zhou and X. Jiang, Natl. Sci. Rev., 2022, 9, nwab156 CrossRef CAS.
- X. Liu, W. Chen, J. Zou, L. Ye and Y. Yuan, ACS Sustainable Chem. Eng., 2022, 10, 6988–6998 CrossRef CAS.
- F. Lu, K. Lu, G. Zhao, S. Zhou, B. He, Y. Zhang, J. Xu, Y. Li, X. Liu and L. Chen, RSC Adv., 2022, 12, 19869–19874 RSC.
- B. Wang, Z. Deng and Z. Li, J. Catal., 2020, 389, 241–246 CrossRef CAS.
- H. Nawaz, J. Zhang, W. Tian, K. Jin, R. Jia, T. Yang and J. Zhang, J. Hazard. Mater., 2020, 387, 121719 CrossRef CAS PubMed.
- M. Kang, H. Zhou, D. Tang, X. Chen, Y. Guo and N. Zhao, RSC Adv., 2019, 9, 42474–42480 RSC.
- R. J. West, L. Euskirchen, A. Neuhahn, S. Seneker and M. W. Spence, Toxicol. Ind. Health, 2022, 38, 544–555 CrossRef CAS.
- S. H. M. Abe, V. C. P. Silva, A. F. Bakuzis, G. B. Stelzer and M. J. Jacinto, Environ. Nanotechnol. Monit. Manag., 2024, 21, 100921 CAS.
- T. Huang, Y. Fu, Q. Peng, C. Yu, J. Zhu, A. Yu and X. Wang, Appl. Surf. Sci., 2019, 480, 888–895 CrossRef CAS.
- L. R. Shultz, L. Hu, K. Preradovic, M. J. Beazley, X. Feng and T. Jurca, ChemCatChem, 2019, 11, 2590–2595 CrossRef CAS.
- Y. R. Mejía and N. K. R. Bogireddy, RSC Adv., 2022, 12, 18661–18675 RSC.
- J. Zhang, G. Lu and C. Cai, Catal. Commun., 2016, 84, 25–29 CrossRef CAS.
- P. L. Gkizis, M. Stratakis and I. N. Lykakis, Catal. Commun., 2013, 36, 48–51 CrossRef CAS.
- D. Nandi, S. Siwal, M. Choudhary and K. Mallick, Appl. Catal., A, 2016, 523, 31–38 CrossRef CAS.
- J. Liu, Q. Gui, Z. Yang, Z. Tan, R. Guo and J. C. Shi, Synthesis, 2013, 45, 943–951 CrossRef CAS.
- X. Wang, M. Li, S. Yang, X. Bai and J. Shan, Microchem. J., 2022, 179, 107473 CrossRef CAS.
- S. Wang, T. Zhang, L. Jia, P. Yang, P. He, F. Xiao, P. Zhou, Y. Wang and X. Wang, Microchem. J., 2023, 186, 108252 CrossRef CAS.
- A. F. Baye, H. A. Bandal and H. Kim, Environ. Res., 2024, 246, 118071 CrossRef CAS.
- D. Yin, J. Zhang, W. Li and Y. Fu, Catal. Lett., 2021, 151, 1902–1910 CrossRef CAS.
- M. Ding, B. Q. Shan, B. Peng, J. F. Zhou and K. Zhang, Phys. Chem. Chem. Phys., 2022, 24, 7923–7936 RSC.
- N. Mei and B. Liu, Int. J. Hydrogen Energy, 2016, 41, 17960–17966 CrossRef CAS.
- Y. Liu, Y. Liu, Y. Xu, Q. He, R. Yin, P. Sun and X. Dong, J. Colloid Interface Sci., 2022, 608, 2088–2099 CrossRef CAS PubMed.
- J. W. Gregory, Y. Gong, Y. Han, S. Huband, R. I. Walton, V. Hessel and E. V. Rebrov, Catal. Today, 2023, 418, 114145 CrossRef CAS.
- A. P. Devi, D. K. Padhi, P. M. Mishra and A. K. Behera, J. Ind. Eng. Chem., 2022, 108, 118–129 CrossRef CAS.
- J. C. Bernardes, D. Müller, E. Latocheski, J. B. Domingos, T. Fey and C. R. Rambo, Ceram. Int., 2022, 48, 15946–15950 CrossRef CAS.
- S. Patar, S. Konwer, T. Chetia, B. K. Bhuyan and L. J. Borthakur, ACS Appl. Nano Mater., 2023, 6, 6567–6580 CrossRef CAS.
- M. Kumar, U. Sharma, S. Sharma, V. Kumar, B. Singh and N. Kumar, RSC Adv., 2013, 3, 4894–4898 RSC.
- P. R. Race, A. L. Lovering, R. M. Green, A. Ossor, S. A. White, P. F. Searle, C. J. Wrighton and E. I. Hyde, J. Biol. Chem., 2005, 280, 13256–13264 CrossRef CAS PubMed.
- S. Abramavicius, A. G. Petersen, N. S. Renaltan, J. Prat-Duran, R. Torregrossa, E. Stankevicius, M. Whiteman and U. Simonsen, Front. Pharmacol, 2021, 12, 613989 CrossRef CAS.
- J. Wang, Y. Zhang, J. Diao, J. Zhang, H. Liu and D. Su, Chin. J. Catal., 2018, 39, 79–87 CrossRef CAS.
- W. J. Liu, T. T. Qian and H. Jiang, Chem. Eng. J., 2014, 236, 448–463 CrossRef CAS.
- Y. Tang, H. Huang, Y. Peng, Q. Ruan, K. Wang, P. Yi, D. Liu and C. Zhong, Chin. J. Chem., 2017, 35, 1091–1097 CrossRef CAS.
- P. Zhou, L. Jiang, F. Wang, K. Deng, K. Lv and Z. Zhang, Sci. Adv., 2017, 3, e1601945 CrossRef.
- X. Li, Y. Tan, Z. Liu, J. Su, Y. Xiao, B. Qiao and Y. Ding, J. Catal., 2022, 416, 332–343 CrossRef CAS.
- A. Corma and P. Serna, Science, 2006, 313, 332–334 CrossRef CAS PubMed.
- A. Corma and P. Serna, Nat. Protoc., 2006, 1, 2590–2595 CrossRef CAS PubMed.
- G. Yue, Y. Yu, S. Li, H. Li, S. Gao, Y. Wang, W. Guo, N. Wang, X. Li and Z. Cui, Small, 2023, 19, 2207918 CrossRef CAS.
- J. Zhao, L. Fang, J. Fu, J. Wang, Q. Jiang, T. Li and J. Huang, Mol. Catal., 2022, 531, 112705 CrossRef CAS.
- Y. Peng, Z. Geng, S. Zhao, L. Wang, H. Li, X. Wang, X. Zheng, J. Zhu, Z. Li and R. Si, Nano Lett., 2018, 18, 3785–3791 CrossRef CAS PubMed.
- M. Zhang, Q. Zhang, Y. Lu, Y. Zhao, D. Zhang and T. Huang, ACS Appl. Nano Mater., 2021, 4, 13995–14003 CrossRef CAS.
- Y. Wang, Q. Li, P. Zhang, D. O'Connor, R. S. Varma, M. Yu and D. Hou, J. Colloid Interface Sci., 2019, 539, 161–167 CrossRef CAS PubMed.
- T. Q. Yang, T. Y. Ning, B. Peng, B. Q. Shan, Y. X. Zong, P. Hao, E. Yuan, Q. Chen and K. Zhang, Catal. Sci. Technol., 2019, 9, 5786–5792 RSC.
- F. Haber, Z. Elektrochem., 1898, 4, 506–514 CrossRef CAS.
- C. Liao, B. Liu, Q. Chi and Z. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 44421–44429 CrossRef CAS.
- E. A. Gelder, S. D. Jackson and C. M. Lok, Chem. Commun., 2005, 4, 522–524 RSC.
- J. A. Makaryan and V. I. Savchenko, Stud. Surf. Sci. Catal., 1993, 75, 2439–2442 CrossRef.
- M. Studer, S. Neto and H. U. Blaser, Top. Catal., 2000, 13, 205–212 CrossRef CAS.
- F. Visentin, G. Puxty, O. M. Kut and K. Hungerbühler, Ind. Eng. Chem. Res., 2006, 45, 4544–4553 CrossRef CAS.
- A. Corma, P. Concepción and P. Serna, Angew. Chem., 2007, 119, 7404–7407 CrossRef.
- C. Wang, Q. Wang, F. Fu and D. Astruc, Acc. Chem. Res., 2020, 53, 2483–2493 CrossRef CAS PubMed.
- S. Lau, D. Gasperini and R. L. Webster, Angew. Chem., Int. Ed., 2021, 60, 14272–14294 CrossRef CAS.
- Z. H. Farooqi, M. W. Akram, R. Begum, W. Wu and A. Irfan, J. Hazard. Mater., 2021, 402, 123535 CrossRef CAS.
- J. Wu and C. Darcel, J. Org. Chem., 2020, 86, 1023–1036 CrossRef PubMed.
- C. Wang, J. Li, F. Zhang, Y. Zhao and T. Xiao, Int. J. Hydrogen Energy, 2024, 51, 1286–1305 CrossRef CAS.
- M. Lauwiner, P. Rys and J. Wissmann, Appl. Catal., A, 1998, 172, 141–148 CrossRef CAS.
- F. Zhao, S. Nie, L. Wu, Q. Yuan and X. Wang, Adv. Mater., 2023, 35, 2303672 CrossRef CAS PubMed.
- K. Zhang, J. M. Suh, J. W. Choi, H. W. Jang, M. Shokouhimehr and R. S. Varma, ACS Omega, 2019, 4, 483–495 CrossRef CAS PubMed.
- S. Dutta, S. Sarkar, C. Ray, A. Roy, R. Sahoo and T. Pal, ACS Appl. Mater. Interfaces, 2014, 6, 9134–9143 CrossRef CAS.
- W. Zhang, F. Tan, W. Wang, X. Qiu, X. Qiao and J. Chen, J. Hazard. Mater., 2012, 217, 36–42 Search PubMed.
- K. Poonia, T. T. H. Nguyen, P. Singh, T. Ahamad, S. Thakur, V. H. Nguyen and P. Raizada, Mol. Catal., 2024, 561, 114166 CrossRef CAS.
- D. R. Petkar, B. S. Kadu and R. C. Chikate, RSC Adv., 2014, 4, 8004 RSC.
- J. Li, T. Cai, Y. Feng, X. Liu, N. Wang and Q. Sun, Sci. China Chem., 2024, 67, 2911–2917 CrossRef CAS.
- G. Fu, L. Ding, Y. Chen, J. Lin, Y. Tang and T. Lu, CrystEngComm, 2014, 16, 1606 RSC.
- X. Liu, H. Cheng and P. Cui, Appl. Surf. Sci., 2014, 292, 695–701 CrossRef CAS.
- M. I. Din, R. Khalid, Z. Hussain, J. Najeeb, A. Sahrif, A. Intisar and E. Ahmed, RSC Adv., 2020, 10, 19041–19058 RSC.
- D. Deng, K. S. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218–230 CrossRef CAS PubMed.
- X. Fan, G. Zhang and F. Zhang, Chem. Soc. Rev., 2015, 44, 3023–3035 RSC.
- V. Georgakilas, J. N. Tiwari, K. C. Kemp, J. A. Perman, A. B. Bourlinos, K. S. Kim and R. Zboril, Chem. Rev., 2016, 116, 5464–5519 CrossRef CAS PubMed.
- G. Zhao, X. Li, M. Huang, Z. Zhen, Y. Zhong, Q. Chen, X. Zhao, Y. He, R. Hu and T. Yang, Chem. Soc. Rev., 2017, 46, 4417–4449 RSC.
- H. Cheng, Y. Huang, G. Shi, L. Jiang and L. Qu, Acc. Chem. Res., 2017, 50, 1663–1671 CrossRef CAS PubMed.
- Y. Zhang, Z. Chen, S. Liu and Y. J. Xu, Appl. Catal., B, 2013, 140, 598–607 CrossRef.
- Y. Zhang, N. Zhang, Z. R. Tang and Y. J. Xu, Phys. Chem. Chem. Phys., 2012, 14, 9167–9175 RSC.
- K. Zhang, K. Hong, J. M. Suh, T. H. Lee, O. Kwon, M. Shokouhimehr and H. W. Jang, Res. Chem. Intermed., 2019, 45, 599–611 CrossRef CAS.
- Y. Zhang, F. Gao and M. L. Fu, Chem. Phys. Lett., 2018, 691, 61–67 CrossRef CAS.
- C. Wang, W. Zou, J. Wang, Y. Ge, R. Lu and S. Zhang, New J. Chem., 2017, 41, 3865–3871 RSC.
- Y. Zhao, R. Li, P. Jiang, K. Zhang, Y. Dong and W. Xie, J. Phys. Chem. C, 2019, 123, 15582–15588 CrossRef CAS.
- B. Tan, Y. Luo, X. Liang, S. Wang, X. Gao, Z. Zhang and Y. Fang, Ind. Eng. Chem. Res., 2019, 58, 2983–2990 CrossRef CAS.
- D. Wang, R. Huang, W. Liu, D. Sun and Z. Li, ACS Catal., 2014, 4, 4254–4260 CrossRef CAS.
- Q. Wang and D. Astruc, Chem. Rev., 2019, 120, 1438–1511 CrossRef.
- L. Yuan, C. Zhang, Y. Zou, T. Bao, J. Wang, C. Tang, A. Du, C. Yu and C. Liu, Adv. Funct. Mater., 2023, 33, 2214627 CrossRef CAS.
- Y. Huang, H. Lin and Y. Zhang, J. Solid State Chem., 2020, 283, 121150 CrossRef CAS.
- Y. Huang, L. Xie, K. Zhuo, H. Zhou and Y. Zhang, New J. Chem., 2021, 45, 3120–3127 RSC.
- L. G. Ghanem, D. M. Sayed, N. Ahmed, M. Ramadan and N. K. Allam, Langmuir, 2021, 37, 5161–5171 CrossRef CAS.
- Y. Wang, X. Y. Ye and G. Z. Han, Colloids Surf., A, 2024, 682, 132869 CrossRef CAS.
- D. Wu, D. Han, W. Zhou, S. Streiff, A. Y. Khodakov and V. V. Ordomsky, Catal. Rev., 2024, 66, 640–686 CrossRef CAS.
- X. Chen, Q. Shen, Z. Li, W. Wan, J. Chen and J. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 654–666 CrossRef CAS PubMed.
- Y. Zhang, N. Zhang, Z. Tang and Y. Xu, ACS Sustainable Chem. Eng., 2013, 1, 1258–1266 CrossRef CAS.
- R. Ma, L. Xie, Y. Huang, K. Zhuo, J. Xu and Y. Zhang, RSC Adv., 2021, 11, 27003–27010 RSC.
- X. Song, A. Li, Z. Cai, X. Chen, W. Dai and X. Fu, Appl. Surf. Sci., 2023, 623, 157004 CrossRef CAS.
- Y. Dong, P. Ji, X. Xu, R. Li, Y. Wang, K. P. Homewood, X. Xia, Y. Gao and X. Chen, Energy Environ. Mater., 2023, 3, e12643 Search PubMed.
- P. Wang, X. Zhou, Y. Shao, D. Li, Z. Zuo and X. Liu, J. Colloid Interface Sci., 2021, 601, 186–195 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |