
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorinated catalysts for the oxygen evolution reaction: a comprehensive review of synthesis, structure, and performance
Zhiping
Luo

Department of Chemistry, Physics and Materials Science, Fayetteville State University, Fayetteville, North Carolina 28301, USA. E-mail: zluo@uncfsu.edu
First published on 20th December 2024
Abstract
Electrochemical water splitting is considered one of the most viable, effective, and environmentally friendly approaches for renewable energy conversion and storage. Nevertheless, due to its slow reaction kinetics, the oxygen evolution reaction (OER) at the anode remains a significant challenge. Researchers have discovered that incorporating fluorine into catalysts in the past few years can significantly improve their OER performance. This enhancement is attributed to fluorine's unique characteristic of possessing the highest electronegativity among all the elements. Consequently, fluorine forms highly ionic metal–fluorine bonds, which promote the electrocatalytic reactions necessary for the OER. This approach has led to considerable advancements in catalyst development for OER applications. This review encompasses various types of state-of-the-art fluorinated catalysts, including binary, ternary, and high-entropy transition-metal fluorides, oxyfluorides, fluorinated versions of oxides, (oxy)hydroxides, carbonate hydroxides, carbides, nitrides, phosphides, sulfides, and carbons. Research has shown that fluorine-containing catalysts demonstrate exceptional performance in the OER, with some outperforming industry standards such as IrO2 or RuO2. Incorporating fluorine through doping has emerged as a successful approach to enhance the OER performance of catalysts, significantly decreasing the overpotential and Tafel slope while improving durability. The data indicate that fluorination leads to an average reduction of 21.6% in overpotential and 29.6% in the Tafel slope. When a new OER catalyst is developed, improving its OER performance through fluorination might be worth exploring if this has not been done. The OER performances of these catalysts are closely linked to their synthesis methods and structural characteristics.
1. Introduction
With the rapidly increasing global demand for energy consumption, reliance on traditional fossil fuels has led to an energy crisis due to their gradual depletion and the environmental pollution they cause when burned. Consequently, scientists are actively seeking alternative clean and renewable energy sources. Electrochemical water splitting is considered to be one of the most viable, effective, and eco-friendly approaches for renewable energy conversion and storage.1,2 The hydrogen produced through this process is a clean, renewable, and energy-efficient fuel, offering a way to store renewable energy and overcome its intermittency.3 The water-splitting process involves the oxygen evolution reaction (OER) at the anode and the hydrogen evolution reaction (HER) at the cathode. The OER is particularly critical and kinetically sluggish due to its complex four-electron transfer. Currently, IrO2 and RuO2 are the most effective OER catalysts; however, their high costs limit the scalability of this technology. Thus, finding highly efficient catalysts from earth-abundant materials is of significant scientific and technological importance.Significant progress has been made in identifying efficient OER catalysts, including Group IA compounds (hydrates), Group IVA compounds (carbon-based catalysts and silicates), Group VA pnictogenides (nitrides and phosphides), Group VIA compounds (oxides, sulfides, and selenides), and Group VIIA compounds (fluorides and chlorides).2,4–8 Among them, oxides are the most extensively studied catalysts due to their high performance, wide availability, and lasting durability.
In the early stage of OER catalyst development, metal fluorides did not attract much attention for OER catalysts because of their low conductivity of pure fluorides MxFy.9–11 In recent years, researchers have recognized the merits of fluorine (F) because of its unique features that may have significant potential impacts on the OER process. With the highest electronegativity (3.98) and a similar ionic radius to O ions (F 1.31 Å and O 1.38 Å), fluorine is an ideal n-type dopant,12 which forms weak metal–F bonds compared to metal–O bonds in oxides.12–14 These weak metal–F bonds exhibiting higher ionic character can be readily dissociated in the electrolyte, facilitating surface reconstruction (SR).15,16 For example, a binary cobalt fluoride (CoF2) nanorod catalyst, even without doping, requires only 285 mV overpotential to achieve 10 mA cm−2 current density, outperforming the benchmark IrO2 catalyst, which requires 310 mV in the experiment.9 However, serious corrosion of the fluorides occurred by forming oxidation layers on the surface of the fluorides. The low conductivity and corrosion resistance of fluoride catalysts with low structural stability limit their OER applications.
To improve the OER performance of metal fluorides, several materials design strategies have been proposed, including synthesizing nanostructured morphology,9,15 surface reconstruction,15–17 heteroatomic doping,10,17–19 and hybrid heterocatalysts.20 Nanostructured surfaces expose more active sites to facilitate proton transport and the OER. For instance, a surface-reconstructed NiFe-OH-F-SR catalyst exhibits higher electrical conductivity than the original NiFe-OH-F.15 Following fluoride leaching under OER conditions, the surface undergoes self-reconstruction, resulting in a highly porous and amorphous NiFe oxide structure with a hierarchical arrangement. This transformation leads to a substantial 58-fold enhancement in OER activity at 220 mV. Fe doping in CoF2 nanowires reduces the overpotential at 10 mA cm−2 from 300 mV to 230 mV and Tafel slope from 82 to 59 mV dec−1, with improved stability.18 The doped Fe3+ to this fluoride is thought to create more defects and provide more catalytically active sites, thus improving the OER catalytic activities. The nanosheet structure formed after the stability test favored rapid electron conduction. In KNixCo1−xF3 perovskite fluoride, Ni species enhance electrochemical activity and Co species improve the electronic conductivity and electrochemical stability, leading to superior OER performance compared to RuO2.17 Fe-doped KCo0.8Fe0.2F3 also demonstrates superior OER performance due to the enhanced conductivity of Fe species and the synergistic effect of Fe and Co.19 Additionally, incorporating oxygen into metal fluorides to form oxyfluoride (MxOyFz) can enhance electronic conductivity while preserving key characteristics for the OER.10 As an example, a hybrid heterocatalyst composed of mixed Fe2O3 and FeF2 shows superior OER properties compared to Fe2O3 alone, due to the higher electroconductivity enabled by the coexistence of Fe–O and Fe–F bands.20
Besides using transition-metal (TM) fluorides as effective OER catalysts, another strategy is doping F into existing OER catalysts. For example, Chen et al. introduced F into Co-based catalysts (CoOOH and Co3O4).13 Benefiting from the strong ionicity of weak metal–F bonds, a dynamic migration of F anions from the interior to the surface was observed, significantly enhancing OER activity. The overpotential of F-doped CoOOH was reduced from 370 to 310 mV at 50 mA cm−2, a reduction of 16.2%. For F-doped Co3O4, the overpotential decreased from 460 to 370 mV, a reduction of 19.6%. The Tafel slope was reduced from 83 to 54 mV dec−1 for CoOOH, a reduction of 34.9%, and from 160 to 70 mV dec−1 for Co3O4, a reduction of 56.3%. In another study, the F doping of a NiTiO3/C catalyst reduced the overpotential from 680 mV to 270 mV at 50 mA cm−2, a reduction as high as 60.3%.21 Successful fluorination across various catalysts appears to be a promising strategy for further enhancing the OER activity.
Given the high performance of fluorinated catalysts, this paper comprehensively reviews current fluorinated catalysts for the OER. First, it outlines the benefits of fluorination, such as highly ionic metal–fluorine bonds for the OER, improved electrical conductivity, water wettability, surface reconstruction, reduced energy barriers, optimized band structure, and favorable charge redistribution. The review then details the high performance of specific fluorinated catalysts, including TM fluorides, oxyfluorides, and various fluorinated catalysts. The strategies for fluorinating these catalysts and their structural modulations are discussed, highlighting the significant enhancements in OER properties due to fluorination.
2. Benefits of fluorination
2.1 OER mechanism
To understand the impacts of fluoridation, we first review the OER mechanisms. During the OER process, O2 is formed from H2O on the anode side through four steps involving coupled electron–proton transfer processes:| 2H2O → O2 + 4H+ + 4e− | (1) |
Several OER mechanisms have been proposed in the literature.5,22–24 Here, we present the well-accepted adsorbate evolution mechanism (AEM) and lattice oxygen mechanism or lattice oxygen-participation mechanism (LOM), as illustrated in Fig. 1. In an acidic medium, as shown in Fig. 1a, the AEM involves the following steps:23,24
| Step 1: * + H2O (l) → *OH + H+ + e− | (2) |
| Step 2: *OH → *O + H+ + e− | (3) |
| Step 3: *O + H2O (l) → *OOH + H+ + e− | (4) |
| Step 4: *OOH → * + O2 (g) + H+ + e− | (5) |
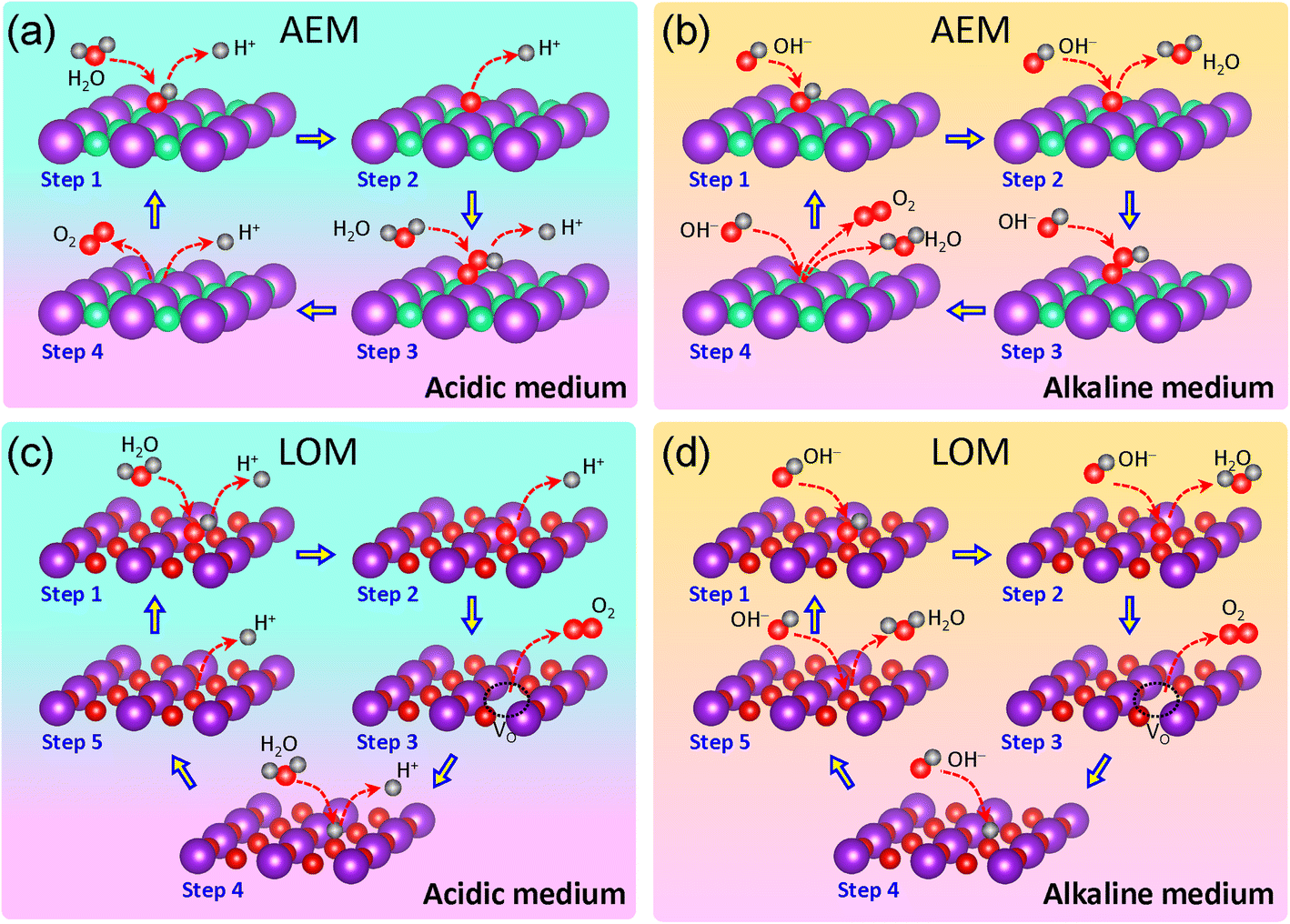 | ||
| Fig. 1 AEM in acidic (a) and alkaline media (b), and LOM in acidic (c) and alkaline media (d). Arrows pointing toward the active catalyst surface indicate reactants, while arrows pointing away from the catalyst surface represent products. Each atomic configuration shown in the figure corresponds to the post-reaction state at each step. Electrons are not depicted in the figure. | ||
In an alkaline medium, as shown in Fig. 1b, the AEM is composed of these reactions:24
| Step 1: * + OH− → *OH + e− | (6) |
| Step 2: *OH + OH− → *O + H2O (l) + e− | (7) |
| Step 3: *O + OH− → *OOH + e− | (8) |
| Step 4: *OOH + OH− → * + O2 (g) + H2O (l) + e− | (9) |
The LOM requires the presence of oxygen in the lattice to form an oxygen vacancy through coupling.25–27 In the acidic medium, as illustrated in Fig. 1c, the LOM can be described by the following reactions:27
| Step 1: M–OL + H2O → M–OLOH + H+ + e− | (10) |
| Step 2: M–OLOH → M–OLO + H+ + e− | (11) |
| Step 3: M–OLO → M–VO + O2 (g) | (12) |
| Step 4: M–VO + H2O → M–OLH + H+ + e− | (13) |
| Step 5: M–OLH → M–OL + H+ + e− | (14) |
In the alkaline medium, as illustrated in Fig. 1d, the LOM reactions are as follow:28,29
| Step 1: M–OL + OH− → M–OLOH + e− | (15) |
| Step 2: M–OLOH + OH− → M–OLO + H2O + e− | (16) |
| Step 3: M–OLO → M–VO + O2 (g) | (17) |
| Step 4: M–VO + OH− → M–OLH + e− | (18) |
| Step 5: M–OLH + OH− → M–OL + H2O + e− | (19) |
Alternative LOM pathways have also been reported in the literature.29,30
It should be noted that the AEM does not require lattice oxygen within the catalyst structure, making it broadly applicable across various catalyst materials, including those without inherent oxygen content, such as pure fluorides, pure or fluorinated carbides, nitrides, phosphides, sulfides, and carbons. In contrast, the LOM specifically involves the active participation of lattice oxygen in the reaction, limiting its applicability to catalysts with accessible lattice oxygen, such as oxyfluorides, fluorinated oxides, (oxy)hydroxides, and carbonate hydroxides. Further discussions of the AEM and LOM are provided in the following sections.
2.2 Impacts of fluorination
Due to fluorine's unique property of having the highest electronegativity, fluorinating catalysts can have several evident impacts on OER performance as follows:(1) Highly ionic metal–fluorine bonds favorable for the OER. Fluorine has the highest electronegativity, which forms high ionic metal–fluorine bonds.13 Tong et al. found that the weak metal–F bond is easy to break, forming active species of oxides or hydroxides and thus improving OER performance.31 Prakash et al. found that the highly polarized M–F (M = Co, Ni) bonds favor the dissociation of the M–F bonds to form a metal hydroxide phase M(OH)2, which can be oxidized into a higher valence oxyhydroxide MOOH, promoting the OER activity.17
(2) Improved electrical conductivity. Fluorination has been found to enhance electrical conductivity in various catalyst materials, which is favorable for OER activities. Wang et al. observed that F-doping in Fe2O3 remarkably increases the charge carrier concentration and conductivity, facilitating rapid charge transfer.32 Xu et al. demonstrated that F-doped F–Ni3S2 exhibits intrinsic conductivity superior to Ni3S2, as shown through the cyclic voltammetry (CV) test.33 Similarly, F-containing PrBaFe2O5+δF0.1 (PBFOF) has higher conductivity than PrBaFe2O5+δ (PBF).34 Density functional theory (DFT) calculations also suggest that F-doping increases the state density of the Ni 3d orbital around the Fermi level, enhancing the conductivity of NiFeOOH.35
(3) Improved water wettability on the catalyst surface. A hydrophilic surface favors contact between reactants and active sites, facilitating the release of O2 gas bubbles and electron transfers during the OER process. Chen et al. measured the water contact angle of nonfluorinated CoOOH at 62.9°, whereas fluorinated CoOOH showed a reduced angle of 44.6°, indicating enhanced hydrophilicity.13 Both samples have a nanosheet morphology, while the fluorinated nanosheets are more hydrophilic for water absorption, boosting O-related intermediate adsorption. Li et al. synthesized surface-smooth Ni3S2 nanoarrays that, after fluorination, became rough due to surface reconstruction.33 The water contact angle changed from 73° for the nonfluorinated sample to 0° for the fluorinated one, significantly improving wettability, as shown in Fig. 2a. The improved wettability contributes to the augmented OER.
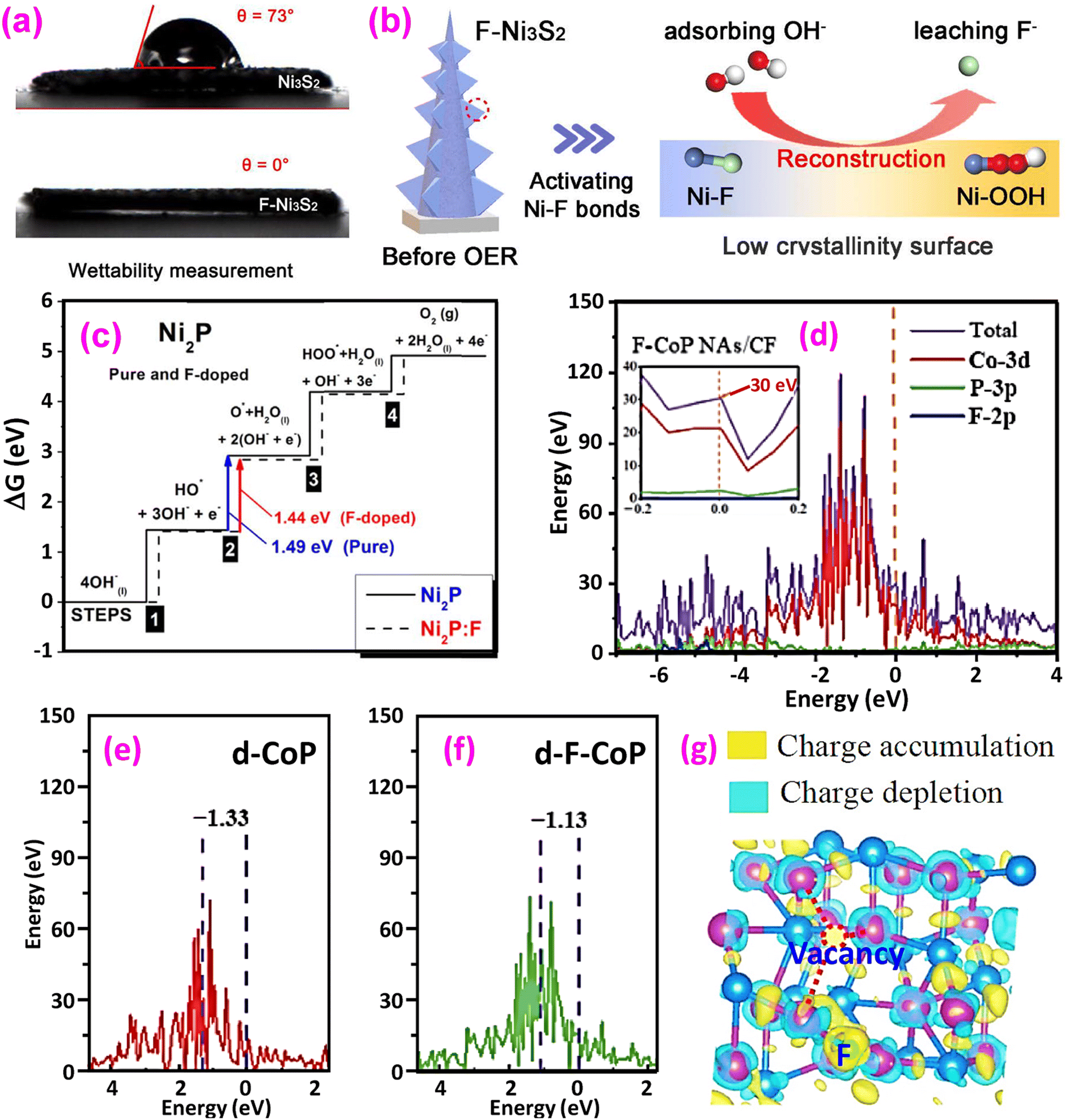 | ||
| Fig. 2 Impacts of fluorination. (a) The fluorinated sample exhibits a hydrophilic nature with good wettability (bottom), while the nonfluorinated sample displays a hydrophobic nature, with a droplet remaining on the surface and (b) F-induced surface reconstruction during the OER. Reproduced from ref. 33 with permission from Elsevier, copyright 2021. (c) Calculated Gibbs free energy of the intermediate reactions for pure and F-doped Ni2P. Reproduced from ref. 36 with permission from IOP Publishing, copyright 2021. (d) DOS of fluorinated CoP; (e and f) projected DOS for the d-bands of pristine and fluorinated CoP, respectively and (g) charge density difference distribution. Reproduced from ref. 37 with permission from Springer Nature, copyright 2022. | ||
(4) Promoted surface reconstruction. Surface reconstruction plays a crucial role in the OER process.38,39 Since the high electronegativity of F causes weakened metal–F bonding in the electrolyte, SR is a notable feature of fluorinated catalysts. Hu et al. incorporated F− into NiFe hydroxide (NiF–OH–F) nanosheet arrays for the OER.15 Following the OER testing, they noted SR triggered by fluorine leaching under OER conditions, and a highly mesoporous and amorphous NiFe oxide structure with a hierarchical arrangement is formed after the surface reconstruction. Notably, the surface reconstructed product NiFe–OH–F-SR showed enhanced OER activity with a reduced overpotential of 181 mV at 10 mA cm−2, compared to the 243 mV of NiFe–OH–F. Li et al. also identified SR in fluorinated Ni3S2 (Fig. 2b).33 It was found that Ni–F bonds are easily activated with F loss under OER conditions, producing highly active Ni–OOH species to accelerate the rate-determining step (RDS, *O → *OOH in Fig. 1). SR has also been reported in other systems.15,37,40–47
(5) Reduced energy barriers to the OER. The reactions described in eqn (2)–(19) require external overpotential to proceed. Fluorination has been shown to reduce these energy barriers, as demonstrated using DFT calculations. For example, Kumta et al. calculated the Gibbs free energies of pure and F-doped Ni2P, as shown in Fig. 2c.36 For both materials, the RDS is Step 2, the largest of the other elementary steps contributing to the OER. However, F-doping reduced this energy from 1.49 eV to 1.44 eV, thus reducing the overpotential and improving the electrocatalytic activity. Other examples are reported in the literature, and some are discussed in the following sections.
(6) Optimized band structure. DFT calculations have revealed optimized electronic band structures of fluorinated materials. The density of states (DOS) of fluorinated CoP is shown in Fig. 2d.37 DOS density at the Fermi level is intimately connected with the material's electrical conductivity. It is found that the DOS of F-CoP (30 eV) is significantly higher than that of CoP (16 eV) at the Fermi level, indicating a rapid charge-transfer process for accelerated electrocatalytic reactions. Furthermore, the d-band center of the 3d Co element is calculated using
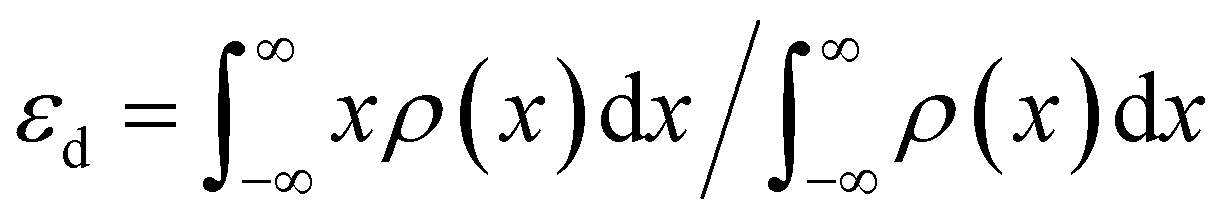 | (20) |
It is found that εd = −1.33 eV for CoP (Fig. 2e), while it is upshifted to εd = −1.13 eV for F-CoP (Fig. 2f). The d-band center is used to predict the adsorption of small molecules, and the upshift induces less electron filling in the antibonding states for strengthened OH− adsorption and improved OER performance. Kumta et al. revealed by calculation that the d-band center of pure Ni2P is −1.84 eV, while for F-doped Ni2P0.67F0.33, its d-band center upshifts to −1.45 eV.36 Zhao et al. calculated the d-band center of NiOOH/NiF2 (εd = −2.341 eV), Ni0.5Co0.5OOH/(NiCo)F2 (εd = −2.177 eV), and CoOOH/CoF2 (εd = −1.717 eV), respectively.46 Although the last material has the highest εd, the optimum OER is found for the second material containing Ni and Co. The hypothesis suggests that a highly elevated d-band center, which results in powerful adsorption, actually hinders the subsequent evolution reaction by increasing the energy barrier of the RDS. The bimetallic fluoride has the balanced d-band center for the OER, consistent with the free energy calculation that the bimetallic fluoride has the lowest energy (1.49 eV) for forming *O, the RDS in the OER. F atoms are observed as electron traders, facilitating electron transfer from CoF2 to Ru sites and inducing charge accumulation at the interface.48
(7) Favorable charge redistribution. DFT calculations have revealed charge redistribution by fluorination, which benefits the OER. The charge density difference of F-doped CoP is shown in Fig. 2g.37 Charge accumulation is found on the F site and near the P vacancy, which favors H capture electrons, improving the HER. Nevertheless, the Co site experiences a reduction in charge, which increases the likelihood of OH losing electrons, thereby promoting the OER. In the NiCo bimetallic fluoride, the Co site exhibits more significant charge deficiency than individual Ni or Co fluorides, suggesting the presence of higher valence metal sites that enhance surface reconstruction and OER kinetics.46
It should be noted that these impacts, as mentioned earlier, of fluorination are indeed frequently interconnected with the electronic properties induced by the fluorination process.
2.3 Enhancement of OER activities by fluorination
F-containing catalysts have exhibited high OER performance; some surpass the benchmark catalysts, IrO2 or RuO2. Composed of earth-abundant elements, these catalysts are promising candidates for future applications. In particular, the F-doping has been demonstrated to be an effective strategy for boosting the OER of catalysts. With F-doping, both the overpotential and Tafel slopes can be significantly reduced, as shown in Fig. 3 for different types of catalysts, according to the literature results summarized in this review. From the available literature data, the overpotential is reduced by an average of 21.6%, and the Tafel slope is reduced by 29.6%.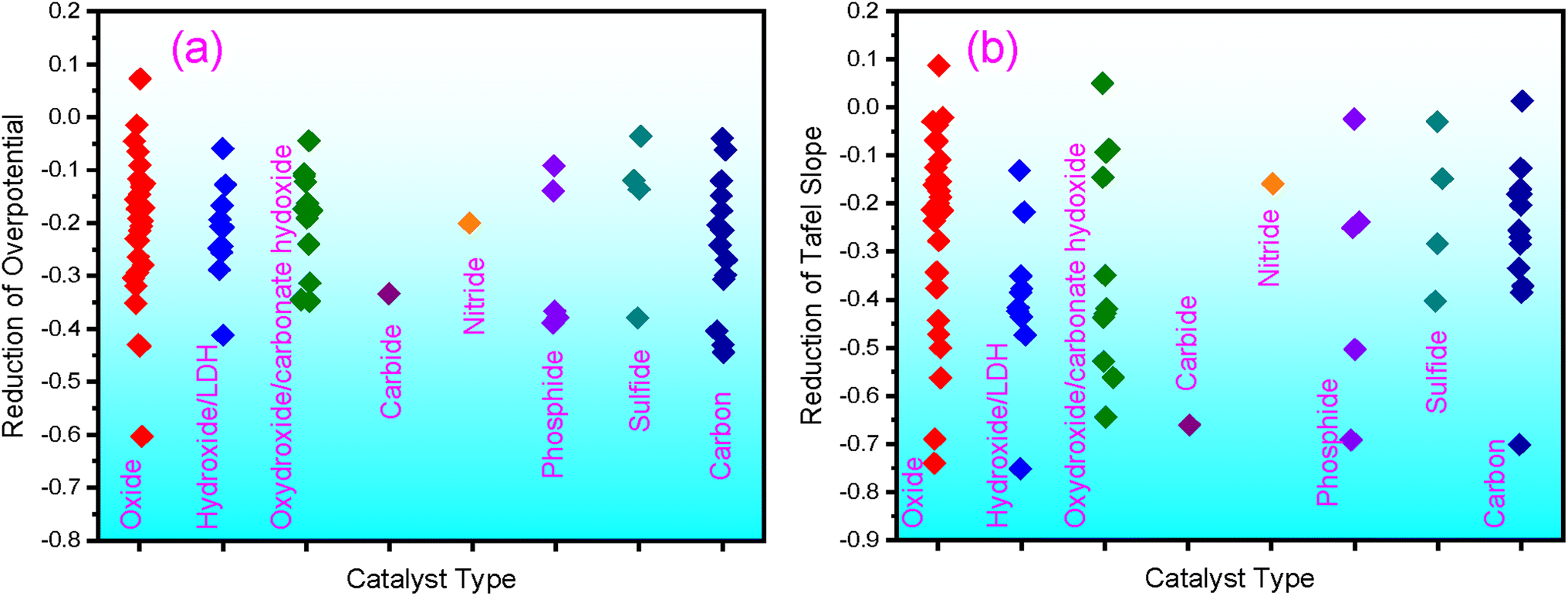 | ||
| Fig. 3 Reduction of overpotential (a) and the Tafel slope (b) by F doping, as summarized from literature results. | ||
In the following sections, we specifically demonstrate the enhancement of the OER by fluorination in various types of catalysts, including transition-metal fluorides (Section 3); fluoride-oxides (Section 4); (oxy)hydroxides, carbonate hydroxides, and their derived fluorides (Section 5); carbides, nitrides, phosphides, and sulfides (Section 6); and carbon-based catalysts (Section 7). The role of F in these catalysts will be highlighted.
3. Transition-metal fluorides
Although noble-metal-based electrocatalysts have been considered the most potent for the OER, their high costs and limited structural stability in both acidic and alkaline electrolytes restrict their practical applications.49 Transition metal-based catalysts, composed of earth-abundant elements, offer lower costs than noble-metal-based catalysts and can achieve higher structural stability. These TM-based catalysts provide diverse structures with various coordination environments and oxidation states, allowing for tunable OER activities through doping.5 Notably, some TM-based catalysts have even outperformed noble-metal-based catalysts.TM fluoride catalysts have also been developed, and their performances are listed in Table 1 (data estimated in this study are indicated with an asterisk). These catalysts include binary, ternary, and high-entropy fluorides (HEFs) containing multiple elements.
| Catalyst | Overpotential at specific current density | Tafel slope (mV dec−1) | Tested durability | Electrolyte | Source |
|---|---|---|---|---|---|
| a (*) Estimated in this study. | |||||
| Binary fluorides | |||||
| Ni-Co-F-1,1 | 300 mV@10 mA cm−2 | 77 | 10 h@15 mA cm−2* | 1 M KOH | 50 |
| CoF2 | 285 mV@10 mA cm−2 | 58 | 12 h@10 mA cm−2 | 1 M KOH | 9 |
| Fe-doped CoF2 | 230 mV@10 mA cm−2 | 59 | 96 h@84 mA cm−2 | 1 M KOH | 18 |
| CoF2 | 300 mV@10 mA cm−2 | 82 | — | 1 M KOH | |
| Ni-doped FeF2 | 275 mV@10 mA cm−2 | 47 | 25 h@10 mA cm−2* | 1 M KOH | 51 |
| Ni-doped Fe MOF | 350 mV@10 mA cm−2* | 81 | — | 1 M KOH | |
| ZIF-FeCo-F-300 | 250 mV@10 mA cm−2 | 51.2 | 20 h@10 mA cm−2* | 1 M KOH | 52 |
| ZIF-FeCo-MOF | 360 mV@10 mA cm−2 | 96.5 | — | 1 M KOH | |
| CoNi ZIF/CoFe-PBA-F-300 | 250 mV@10 mA cm−2 | 47.4 | 14 h@12 mA cm−2* | 1 M KOH | 53 |
| CoNi ZIF/CoFe-PBA | 320 mV@10 mA cm−2 | 65.9 | — | 1 M KOH | |
| CoF2/FeF3 | 252 mV@10 mA cm−2 | 52.1 | 10 h@10 mA cm−2* | 1 M KOH | 54 |
| Fe-CoF2/NF | 210 mV@10 mA cm−2 | 32.2 | 53 h@10 mA cm−2* | 1 M KOH | 55 |
| Fe-Ni-F-350 | 277 mV@10 mA cm−2 | 45 | 24 h@10 mA cm−2 | 1 M KOH | 56 |
| Fe-Co-F-400 | 250 mV@10 mA cm−2 | 38.3 | 10 h@10 mA cm−2* | 1 M KOH | 57 |
| Co0.66Fe0.33F2 | 260 mV@10 mA cm−2 | 54 | 50 h@10 and 50 mA cm−2 | 1 M KOH | 58 |
| Co0.67Fe0.33F2 | 270 mV@10 mA cm−2 | 63.5 | 12 h@270 mV | 1 M KOH | 59 |
| NiF5% (NiF2·4H2O) | 188 mV@10 mA cm−2 | 44 | 100 h@46 mA cm−2 | 1 M KOH | 60 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| Ternary fluorides | |||||
| KNCF82 (KNi0.8Co0.2F3) | 310 mV@10 mA cm−2 | 49 | 20 h@10 mA cm−2 | 1 M KOH | 17 |
| KNFF2@NF (KNi0.8Fe0.2F3) | 258 mV@100 mA cm−2 | 43.7 | 100 h@100 mA cm−2 | 1 M KOH | 16 |
| KCo0.80Fe0.20F3 | 254 mV@10 mA cm−2 | 37.5 | 40 h@10 mA cm−2 | 1 M KOH | 19 |
| ZIF-67@CoFe-PBA-F-250 | 243 mV@10 mA cm−2 | 34 | 12 h@10 mA cm−2 | 1 M KOH | 61 |
| KCo0.5Fe0.5F3@NF | 118 mV@10 mA cm−2 | 19.63 | 130 h@10 mA cm−2 | 1 M KOH | 62 |
| NFCF ((NH4)3FexCo1−xF6) | 243 mV@10 mA cm−2 | 65.4 | 100 h@100 mA cm−2 | 1 M KOH | 63 |
| NaCo0.4Fe0.3Ni0.3F3 | 265 mV@10 mA cm−2 | 49 | 100 h@10 mA cm−2 | 1 M KOH | 64 |
|
|||||
| High-entropy fluorides | |||||
| K(MgMnFeCoNi)F3 | 369 mV@10 mA cm−2 | 61 | 10 h@10 mA cm−2 | 1 M KOH | 65 |
| K0.8Na0.2(MgMnFeCoNi)F3 | 314 mV@10 mA cm−2 | 55 | 10 h@10 mA cm−2 | 1 M KOH | |
| (CuNiFeCoZnMnMg)F2 | 292 mV@10 mA cm−2 | 39 | 6 h@10 and 50 mA cm−2 | 1 M KOH | 66 |
| Al86Ni6Co2Y5Cu0.5Fe0.5 HEF | 261 mV@10 mA cm−2 | 50 | 25 h@33 mA cm−2* | 1 M KOH | 67 |
| Al86Ni6Co2Y5Cu0.5Fe0.5 HEO | 293 mV@10 mA cm−2 | 88 | — | 1 M KOH | |
| K(CoMnFeNiCr)F3 | 242 mV@10 mA cm−2 | 114.57 | 24 h@10 mA cm−2 | 1 M KOH | 68 |
3.1 Binary MF2 and MF3 fluorides
The binary fluorides discussed here are of MF2 and MF3 types, corresponding to divalent M2+ and trivalent M3+, respectively. Here, M may represent one or more transition metals.Binary fluorides such as CoF2, FeF2, MnF2, NiF2, and ZnF2 possess a rutile structure.69 The structure of CoF2, shown in Fig. 4a, is a tetragonal structure with a space group of P42/mnm and lattice constants of a = 0.469 nm and c = 0.318 nm. In the structure, highly distorted CoF6 octahedra share edges along the c-axis. The trifluorides MF3 (M = Fe, Co, Ru, Rh, Pd, and Ir) possess a rhombohedral structure with a space group of R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c.70 The binary fluorides have been reported in the following forms.
c.70 The binary fluorides have been reported in the following forms.
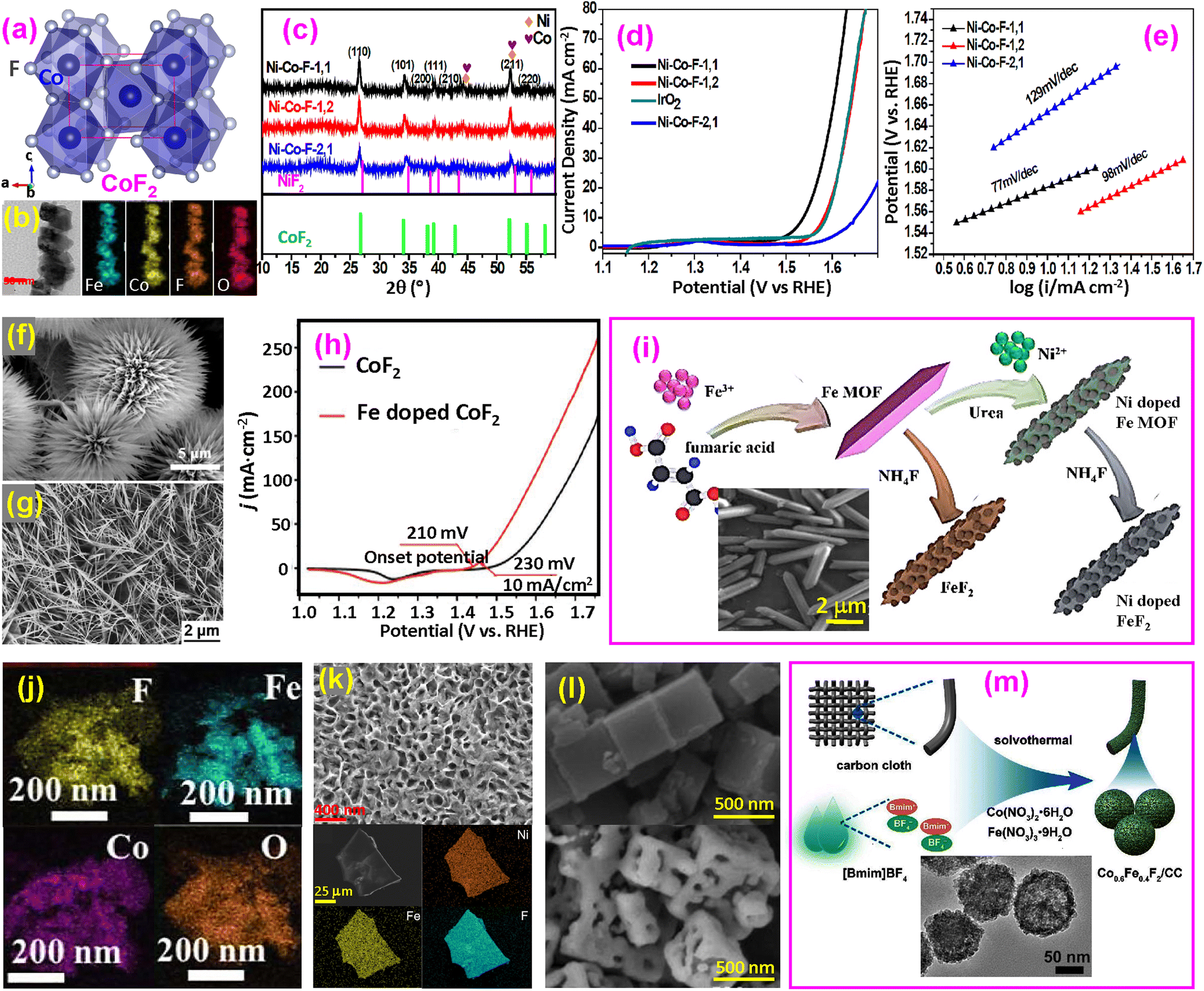 | ||
| Fig. 4 Binary fluoride OER catalysts. (a) Structure of CoF2; (b and c) TEM and XRD of Ni–Co–F; (d and e) LSV and Tafel plots of Ni–Co–F. Reproduced from ref. 50 with permission from the Royal Society of Chemistry, copyright 2018; (f) SEM image of cobalt carbonate hydroxide hydrate for producing CoF2 nanorods. Reproduced from ref. 9 with permission from Elsevier, copyright 2020. (g) SEM image of Fe-doped CoF2 nanowires and (h) LSV curves for CoF2 and Fe-doped CoF2. Reproduced from ref. 18 with open access. (i) Synthesis of Ni-doped FeF2 nanorods, with an inset of the SEM image of Fe MOF. Reproduced from ref. 51 with permission from the Royal Society of Chemistry, copyright 2020. (j) TEM elemental maps of CoF2 and FeF2 irregular nanoparticles. Reproduced from ref. 52 with permission from Elsevier, copyright 2022. (k) SEM (upper) and TEM (bottom) images of Fe–Ni–F nanosheets. Reproduced from ref. 56 with permission from Elsevier, 2024. (l) SEM images of Fe-Co-PBA (upper) and Fe–Co–F (bottom) nanocubes. Reproduced from ref. 57 with permission from Elsevier, copyright 2019. (m) Synthesis of Co–Fe–F nanospheres with an inset of the TEM image. Reproduced from ref. 59 with permission from Elsevier, copyright 2022. | ||
(a) Nanorods and nanowires. Nanorods or nanowires offer direct electron pathways along their lengths, which improves electron conductivity and reduces resistance. Their one-dimensional structure helps create more accessible active sites and enhances interaction with electrolytes. Transition-metal binary fluorides were first used for OER catalysts by the Feng group in 2018.50 A simple one-step microwave method was used to synthesize materials with Ni/Co ratios of 1:1, 1:2, and 2:1, denoted as Ni-Co-F-1,1, Ni-Co-F-1,2, and Ni-Co-F-2,1, respectively. The synthesized one-dimensional (1D) nanorods are shown in the transmission electron microscope (TEM) images in Fig. 4b. The X-ray diffraction (XRD) patterns confirm the rutile-type single phase (Fig. 4c). In performance71–74 it was found that the Ni-Co-F-1,1 catalyst with Co/Fe = 1/1 has the highest current density, even exceeding that of the benchmark catalyst IrO2, as shown in the linear scan voltammetry (LSV) curve in Fig. 4d.50 The overpotential of the Ni-Co-F-1,1 sample is 300 mV at a current density of 10 mA cm−2, which is lower than that of all other samples, including IrO2 (340 mV). The high catalytic activity may be due to the highest Co2+/Co3+ ratio and the lowest Ni2+/Ni3+ ratio. The Tafel plots are shown in Fig. 4e, where the Ni-Co-F-1,1 catalyst exhibits the lowest Tafel slope (77 mV dec−1), consistent with the higher catalytic current density. This Ni-Co-F-1,1 catalyst outperforms the benchmark catalyst IrO2.
Other 1D fluorides were also prepared. Feng and coworkers synthesized nanorod-shaped cobalt carbonate hydroxide hydrate as a precursor (Fig. 4f) on Ti foil through a hydrothermal route.9 This precursor was converted to quasi-single-crystalline CoF2 nanorods through fluorination by placing NH4F in front of the precursor at 350 °C for 2 h under a nitrogen stream. Dong and colleagues synthesized Fe-doped Co(OH)2 nanowires on nickel foam (NF), which were subsequently converted to Fe-doped CoF2 by fluorination (Fig. 4g).18 To reach 10 mA cm−2 current density, the overpotential of Fe-doped CoF2 is only 230 mV, significantly lower than that of pristine CoF2, which requires 300 mV (Fig. 4h). The Fe-doped CoF2 shows a Tafel slope of 59 mV dec−1, compared to 82 mV dec−1 of CoF2. Fe-doping is thought to create defects and provide more active sites, improving the OER activity of CoF2.
Ni-doped FeF2 nanorods were synthesized by forming a Fe metal–organic framework (MOF), followed by fluorination using NH4F (Fig. 4i).51 The Ni doping reduced the overpotential of FeF2 by approximately 63 mV (estimated in this study). It decreased the Tafel slope from 78 to 47 mV dec−1.
(b) Irregular nanoparticles. Nanoparticles have high surface-to-volume ratios, providing abundant active sites for the OER. Fluorides with irregular shapes are also reported in the literature. Starting from preparing an FeCo-based zeolitic imidazolate framework (ZIF) by a simple coprecipitation method, the products were subsequently carbonized and fluorinated to obtain CoF2 and FeF2 nanoparticles (Fig. 4j).52 The hybrid catalyst only needed 250 mV overpotential, compared to 360 mV of pristine ZIF-FeCo MOF, to reach 10 mA cm−2. This clearly shows that fluorination significantly improves the OER.
(c) Nanoflakes and nanosheets. Nanoflakes and nanosheets maximize the exposure of active sites, offering a high density of accessible catalytic sites on broad and flat surfaces, facilitating ion transport along their planes. Two-dimensional (2D) porous Co–Fe nanoflakes were synthesized by a hydrothermal reaction, followed by low-temperature fluorination at 320 °C for 2 h.54 The nanoflakes contain mixed CoF2 and FeF3, requiring only 252 mV overpotential at 10 mA cm−2, with a low Tafel slope of 52.1 mV dec−1. Recently, Fe–Ni–F nanosheets containing mixed NiF2 and FeF2 were synthesized by directly annealing metal acetate precursors with NH4F in a tube furnace at 300–400 °C for 2 h (Fig. 4k).56 The prepared Co–Fe–F nanosheets surpass the single-metal fluorides FeF2 and NiF2 and the benchmark RuO2.
(d) Nanocubes and microcubes. Nanocubes and microcubes can expose specific crystal facets for the OER, with large surface areas and good structural stability. Porous Fe–Co–F nanocubes were prepared from a Fe–Co Prussian blue analogue (PBA).57 First, Fe-Co-PBA nanocubes were formed by coprecipitation at room temperature (Fig. 4l, upper) and subsequently, the PBA nanocubes were fluorinated using NH4F to form porous fluoride nanocubes (Fig. 4l, bottom). The synthesized materials contain a mixture of FeF2, CoF2, and ternary perovskite K(CoFe)F3 (K is from a reactant K3FeC6N6 used for coprecipitation). The overpotential is 250 mV at a current density of 10 mA cm−2, with a low Tafel slope of 38.3 mV dec−1 and excellent catalytic stability for 10 h in long-term water electrolysis. With such a porous nanostructure, the electrochemical surface area is enhanced, resulting in increased exposure of the active sites and the construction of metal oxide layers over the catalyst surface. Another way to synthesize cubes is to use an MOF as a template, resulting in a larger size of about 3 μm.58 Among a series of CoxFe1−xF2, Co0.66Fe0.33F2 shows the best performance (260 mV overpotential at 10 mA cm−2 and a 54 mV dec−1 Tafel slope) due to its large number of reactive sites and low charge transfer resistance.
(e) Nanospheres. Nanospheres maximize surface area relative to their volume, providing uniform active sites for the OER with good structural stability and resistance to aggregation. Using an ionic liquid-assisted solvothermal route, Co–Fe–F nanospheres were synthesized on the surface of carbon cloth (CC) fibers (Fig. 4m).59 Among samples with varied Co/Fe ratios, Co0.67Fe0.33F2 shows the best performance compared to Co0.8Fe0.2F2 or CoF2 due to F− leaching and increased surface areas. This conclusion is consistent with that of the Co–Fe–F microcubes.58
3.2 Ternary and ternary-substituted fluorides
A large number of ternary and quaternary transition-metal fluorides have been reported, including common types such as AxByF3, AxByF4, AxByF5, AxByF6, AxByCzF6, AxByCzF7, and AxByCzF9, where A represents NH4, an alkali or an alkaline earth metal and B and C represent transition metals.75–80 However, only a limited number of ternary fluorides, particularly those in the perovskite category, have been explored for OER applications.Perovskites exhibit diverse structures, such as ABO3-type single perovskite oxides, AA′BB′O6-type double perovskite oxides, and An+1BnO3n+1-type layered perovskite oxides.81–83 Perovskite-related TM-based catalysts have attracted significant interest from the scientific community for the OER due to their low cost, earth abundance, and excellent tunable electrochemical properties.84–88 The following fluoride perovskites have been reported for the OER.
(a) Simple perovskites. The representative ternary perovskite fluoride is ABF3,89,90 as shown in the structural model of KCoF3 in Fig. 5a. Prakash et al. prepared this fluoride in nanoparticle form with varied Ni/Co ratios via a solvothermal method.17 They found that when Ni/Co = 8/2, the KNi0.8Co0.2F3 (KNCF82) catalyst shows an overpotential of 310 mV at 10 mA cm−2, with a low Tafel slope of 49 mV dec−1, outperforming the benchmark precious RuO2 catalyst (Fig. 5b–d). Another study reports K(CoFe)F3 nanoparticles synthesized by a hydrothermal method with varied Co/Fe ratios.19 It was found that when Co/Fe = 0.8/0.2, the KCo0.80Fe0.20F3 catalyst has the lowest overpotential of 254 mV@10 mA cm−2 and a Tafel slope value of 37.5 mV dec−1. It is speculated that Co and the appropriate amount of Fe may produce a synergistic effect to enhance the OER performance.
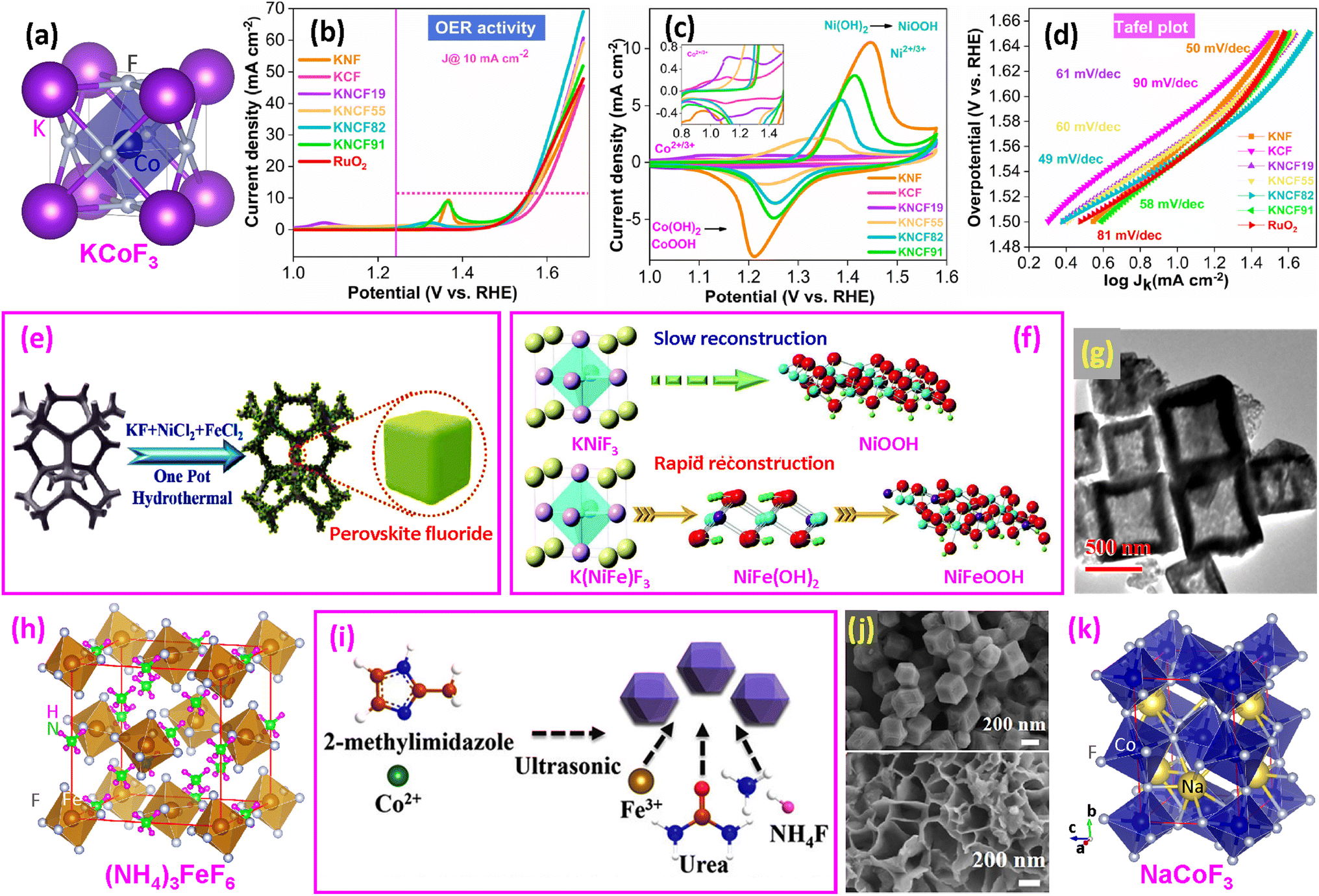 | ||
| Fig. 5 Ternary fluoride OER catalysts. (a) Structure of ternary KCoF3 perovskite; (b) LSV curves; (c) preoxidation peak (Co2+/3+ and Ni2+/3+); and (d) Tafel plots. Reproduced from ref. 17 with permission from American Chemical Society, copyright 2021. (e and f) Synthesis of KNi1−xFexF3 on nickel foam and surface reconstruction during the OER. Reproduced from ref. 16 with open access; (g) TEM image of ZIF-67@CoFe-PBA nanoboxes. Reproduced from ref. 61 with permission from Elsevier, copyright 2021. (h) Structure of (NH4)3FeF6. (i) Synthesis of (NH4)3FexCo1−xF6 and (j) SEM images of ZIF-67 (upper) and NFCF (bottom). Reproduced from ref. 63 with permission from American Chemical Society, copyright 2022. (k) Structure of NaCoF3. | ||
Zhao and coworkers synthesized KNi1−xFexF3 nanocubes with varying Ni/Fe ratios on nickel foam (NF) (Fig. 5e).16 They found that Fe significantly promoted self-reconstruction and efficiently reduced the energy barriers of the OER. The KNi0.8Fe0.2F3@NF electrocatalyst delivered an overpotential of 258 mV to afford 100 mA cm−2, with excellent durability for 100 h. The Tafel slope was as low as 43.7 mV dec−1. In situ Raman spectroscopy identified γ-NiOOH formation from KNiF3@NF when the potential reaches 1.43 V, while for K(NiFe)F3@NF, Ni(OH)2 formed at 1.13 V and then γ-NiOOH at 1.43 V. The reconstruction mechanism is shown in Fig. 5f. The DFT calculations confirm that Fe incorporation alters the OER mechanism and reduces the energy barrier. It is believed that Fe doping below 25% is the optimum level.
Using ZIF-67 as a template, CoFe-PBA nanoboxes were obtained (Fig. 5g).61 Subsequent fluorination etching produced KCoF3 and KFeF3 nanoparticles on the walls of the nanobox. Among the samples fluorinated between 150–350 °C for 2 h, the catalyst processed at 250 °C showed the best OER performance, with an overpotential of 243 mV to reach 10 mA cm−2. Recently, KCo1−xFexF3 nanoparticles on NF were synthesized by a hydrothermal method.62 It was reported that KCo0.5Fe0.5F3@NF requires only an overpotential of 118 mV to reach 10 mA cm−2, with the Tafel slope as low as 19.63 mV dec−1.
(b) Double perovskites and others. A study related to ternary perovskites involved (NH4)3FexCo1−xF6 (NFCF),63 which possesses a double-perovskite cubic structure (Fig. 5h). First, ZIF-67 was synthesized (Fig. 5i), and then (NH4)3FexCo1−xF6 was synthesized by a solvothermal route (Fig. 5j).63 It was found that the NFCF electrocatalyst required an overpotential of only 243 mV@10 mA cm−2, which is approximately 27 mV lower than that of the commercial RuO2. The Tafel slope value is 65.40 mV dec−1, indicating rapid electrocatalytic kinetics with a high transfer coefficient. DFT calculations reveal that the Fe-doped NFCF electrocatalyst has a lower energy barrier from OOH* to O2 than the non-doped fluoride, and the co-existence of Co and Fe in NFCF is beneficial for weakening the OH* adsorption energy during OER catalysis.
A recent study examined the B-site substitution of NaCo1−2xFexNixF3 with three elements.64 They optimized the Co/Fe/Ni ratio to be 4/3/3, which exhibited a low overpotential of 265 mV cm−2 at a current density of 10 mA and outstanding electrochemical stability after 100 h of continuous electrocatalysis. The dual substitution of Fe and Ni atoms was observed to produce higher-valence Co3+ ions and generate more active Fe3+ species. It should be noted that NaCoF3 does not have a simple cubic structure like KCoF3. Its structure is orthorhombic with a Pnma (no. 62) space group and lattice parameters of a = 0.5603 nm, b = 0.7793 nm, and c = 0.5420 nm (Fig. 5k).91
3.3 High-entropy fluorides
In some of the previously mentioned fluoride examples, a metal cation site is occupied by two elements. More elements can be added to form high-entropy (HE) materials, which are synthesized by mixing five or more elements in equal or nearly equal atomic ratios. HE materials are usually characterized by distinguished effects, including sluggish diffusion and cocktail effects to conventional materials.92 HE materials have received significant attention in OER research;92–94 however, studies on HE fluorides for the OER are still limited compared to other HE materials (Table 1).In 2020, the Dai group proposed ABF3-type high-entropy perovskite fluorides (HEPFs) as a new platform for the OER.65 The synthesis process is shown in Fig. 6a. K and Na are selected to replace the A site, while seven metal ions (Mg2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+, and Zn2+) are chosen to replace the B site in ABF3. To achieve thorough mixing, the metal sulfates and acetates underwent ball milling to achieve full mixing of the metal source. Subsequently, the mixture was introduced into a boiling KF solution to produce rapid precipitation of HEPFs. It was found that when both A and B sites were replaced with K0.8Na0.2(MgMnFeCoNi)F3, the catalyst showed the lowest overpotential and Tafel slope, which were lower than those of only the B site replaced by K(MgMnFeCoNi)F3 (Fig. 6b).
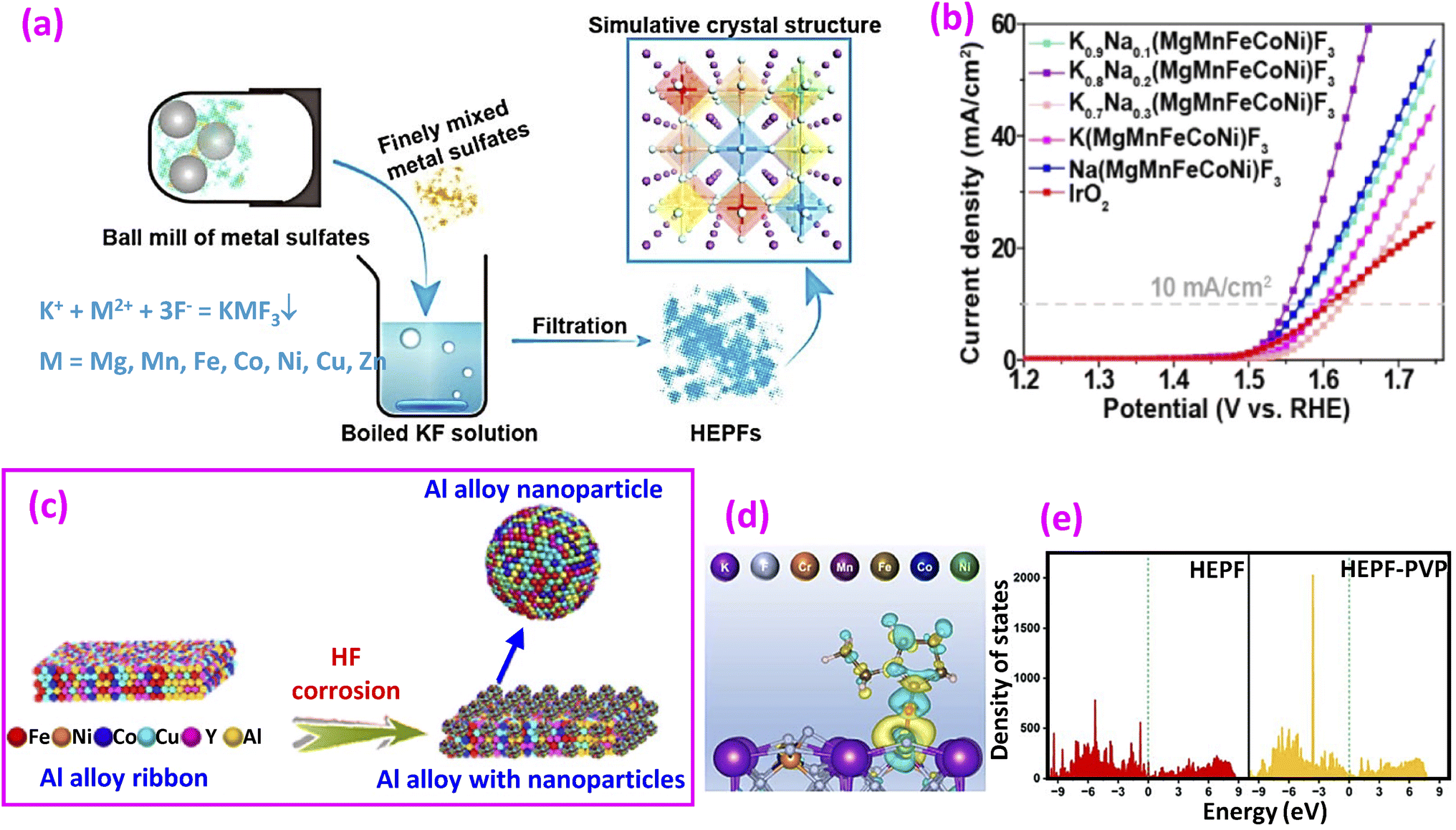 | ||
| Fig. 6 (a) Synthesis of high-entropy ABF3-type perovskite fluorides and (b) LSV curves. Reproduced from ref. 65 with permission from American Chemical Society, copyright 2020. (c) Synthesis of HE alloys from Al alloy ribbons. Reproduced from ref. 67 with permission Elsevier, copyright 2024. (d) Differential charge density of HEPF-PVP, where yellow and blue contours represent electron accumulation and depletion, respectively, and (e) DOS of HEPF and HEPF-PVP. Reproduced from ref. 68 with permission from the Royal Society of Chemistry, copyright 2023. | ||
Breitung and colleagues subsequently synthesized rutile-type HEFs with an AF2 structure by a mechanochemical route.66 Commercially available difluorides MnF2, FeF2, CoF2, NiF2, CuF2, ZnF2, and MgF2 were mixed through a dry, long-term high-energy milling process conducted at 500 rpm for 48 h in a WC jar, and the samples were prepared in an Ar atmosphere. The configurational entropy values are expressed as
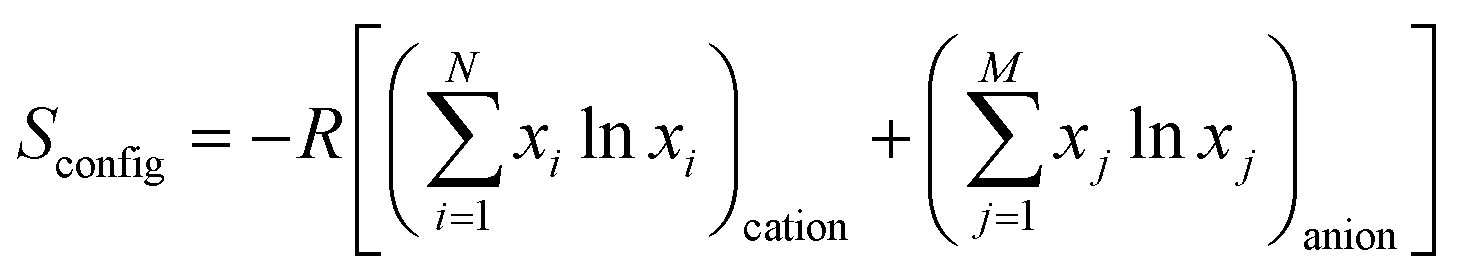 | (21) |
Another approach to preparing HEF involves using HF corrosion of melt-spun HE Al alloy ribbons, as proposed recently (Fig. 6e).67 First, a cast ingot of Al86Ni6Co2Y5Cu0.5Fe0.5 was prepared by arc melting of pure metals, followed by melt spinning to obtain rapidly cooled ribbons. After HF corrosion of the ribbons, surface oxidation yields HE oxide (HEO), while surface fluorination produces HEF. A comparative study showed that HEF has a reduced overpotential compared to HEO by 32 mV and a reduced Tafel slope by 38 mV dec−1.
Although HEPFs possess entropy-dominated structural stability,65 they are considered to have difficulties in restructuring because of their robust electron structure and the high bonding strength of the metal–fluorine bond.68 Hao et al. proposed a strategy to enhance dynamic reconstruction by ligand modification using polyethylene pyrrolidone (PVP).68 Several metal chlorides, nitrate, fluoride and PVP were mixed by a conventional solvothermal treatment at 180 °C for 6 h to produce HEPF. DFT calculations reveal that a PVP molecule enhances the charge concentration at the F site (Fig. 6c), which aids in lowering the bond energy required for reconstruction. The DOS shown in Fig. 6d demonstrates an electron-rich condition that promotes electron transfer. Among similar materials, PVP achieved a record-low overpotential of 242 mV at 10 mA cm−2. However, its Tafel slope of 114.57 mV dec−1 remains higher compared to other catalysts.
4. Fluoride-oxide catalysts
It is known that pure fluorides exhibit poor electroconductibility due to the high ionicity of metal–F bonds, which hinders their catalytic activity. However, the intrinsically low conductivity of metal fluorides can be improved by introducing metal–O bonds.95 Metal fluoride-oxide phases demonstrate higher electroconductivity and electrochemical robustness due to the coexistence of metal–O and metal–F bonds.20,96,97 This fluoride-oxide system has garnered significant attention as a potential class of OER catalysts. It may be categorized as oxyfluoride, various fluorinated oxides, and fluoride/oxide heterocatalysts. The term “oxyfluoride”, also referred to as “fluoride oxide” or “oxide fluoride” in the literature, denotes a compound containing both O and F, with either defined or undefined stoichiometry. “Fluorinated oxide,” on the other hand, refers to an oxide that has undergone fluorination. The term “fluoroxide” is seldom used in the OER research community. Although these terms may overlap in definition, we follow the classification from relevant literature sources.4.1 Oxyfluorides
Oxyfluoride compounds contain both O and F atoms, as shown in Table 2. Yang and colleagues fabricated a NiFe oxyfluoride (NiFeOF) holey film, as depicted in Fig. 7a, by electrochemical deposition of a 2 μm NiFe thin film onto a stainless-steel substrate.98 This was followed by substrate removal and anodization of the free-standing thin film to obtain a porous amorphous NiFeOF structure with an interconnected crystalline NiFe alloy framework. The Ni-15 at% Fe catalyst requires only 295 mV overpotential to reach 10 mA cm−2. DFT analysis verified that the nickel atom situated on the oxide surface functions as the primary site for the OER. Among the materials studied, NiFeOF demonstrated the most favorable energetics for facilitating the OER process. Yamada et al. electrodeposited both Ni and Fe onto a piece of pure Ni plate, followed by anodization to form a nanoporous fluoride-rich Fe-doped Ni oxyfluoride layer, which was then converted into an amorphous or poorly crystalline Fe-doped Ni hydroxide film.100| Catalyst | Overpotential at specific current density | Tafel slope (mV dec−1) | Tested durability | Electrolyte | Source |
|---|---|---|---|---|---|
| a (*) Estimated in this study. | |||||
| Ni0.85Fe0.15OF | 295 mV@10 mA cm−2 | 38 | 2.78 h@10 mA cm−2 | 1 M KOH | 98 |
| TaO2F/gC | 360 mV@10 mA cm−2 | 75 | 120 h@10 mA cm−2 | 1 M KOH | 99 |
| Ta2O5/CC | 530 mV@10 mA cm−2 | 112 | — | 1 M KOH | |
| NiCoFO | 350 mV@10 mA cm−2 | 23 | 10 h@1.58 V | 1 M KOH | 95 |
| Ni-11.8 at% Fe | 260 mV@10 mA cm−2 | 53 | 24 h@50 mA cm−2 | 1 M KOH | 100 |
| NiFe2F4.4O1.8 | 270 mV@10 mA cm−2 | 46 | 270 h@10 mA cm−2 | 1 M KOH | 10 |
| MnFeF5(H2O)2 | 492 mV@10 mA cm−2 | 156 | >5 h@10 mA cm−2 | 0.5 M H2SO4 | 101 |
| MnFe2F8(H2O)2 | 480 mV@10 mA cm−2 | 153 | 13 h@10 mA cm−2 | 0.5 M H2SO4 | |
| MnFeF4.6O0.2 | 500 mV@10 mA cm−2 | 195 | 11 h@10 mA cm−2 | 0.5 M H2SO4 | |
| MnFe2F5.8O1.1 | 515 mV@10 mA cm−2 | 175 | 20 h@10 mA cm−2 | 0.5 M H2SO4 | |
| Co0.5Fe0.5O0.5F1.5 | 220 mV@10 mA cm−2 | 27 | 648 h@10–1000 mA cm−2 | 1 M KOH | 102 |
| Pb3Fe2O5F2 | 640 mV@1 mA cm−2* | — | 3 h@1.7 V | 0.1 M K3PO4 | 103 |
| FeVNbTiZrOF-CC-40 | 348 mV@10 mA cm−2 | 110.3 | 48 h@10 mA cm−2 | 1 M KOH | 104 |
| CoFeOF/NF | 236 mV@10 mA cm−2 | 46.35 | 24 h@10 mA cm−2 | 1 M KOH | 105 |
| CoOF/NF | 320 mV@10 mA cm−2 | 64.21 | 24 h@10 mA cm−2 | 1 M KOH | |
| FeOF/NF | 290 mV@10 mA cm−2 | 58.72 | 24 h@10 mA cm−2 | 1 M KOH | |
| NCoFO/CC-60 | 230 mV@10 mA cm−2 | 62 | 150 h@10 mA cm−2 | 1 M KOH | 106 |
| FeFFIVE-1-Ni | 286 mV@10 mA cm−2 | 86 | 30 h@1.6 V (71% retention) | 1 M KOH | 107 |
| Co0.25Ni0.25Fe0.5O0.5F1.5 | 290 mV@10 mA cm−2 | — | — | 1 M KOH | 108 |
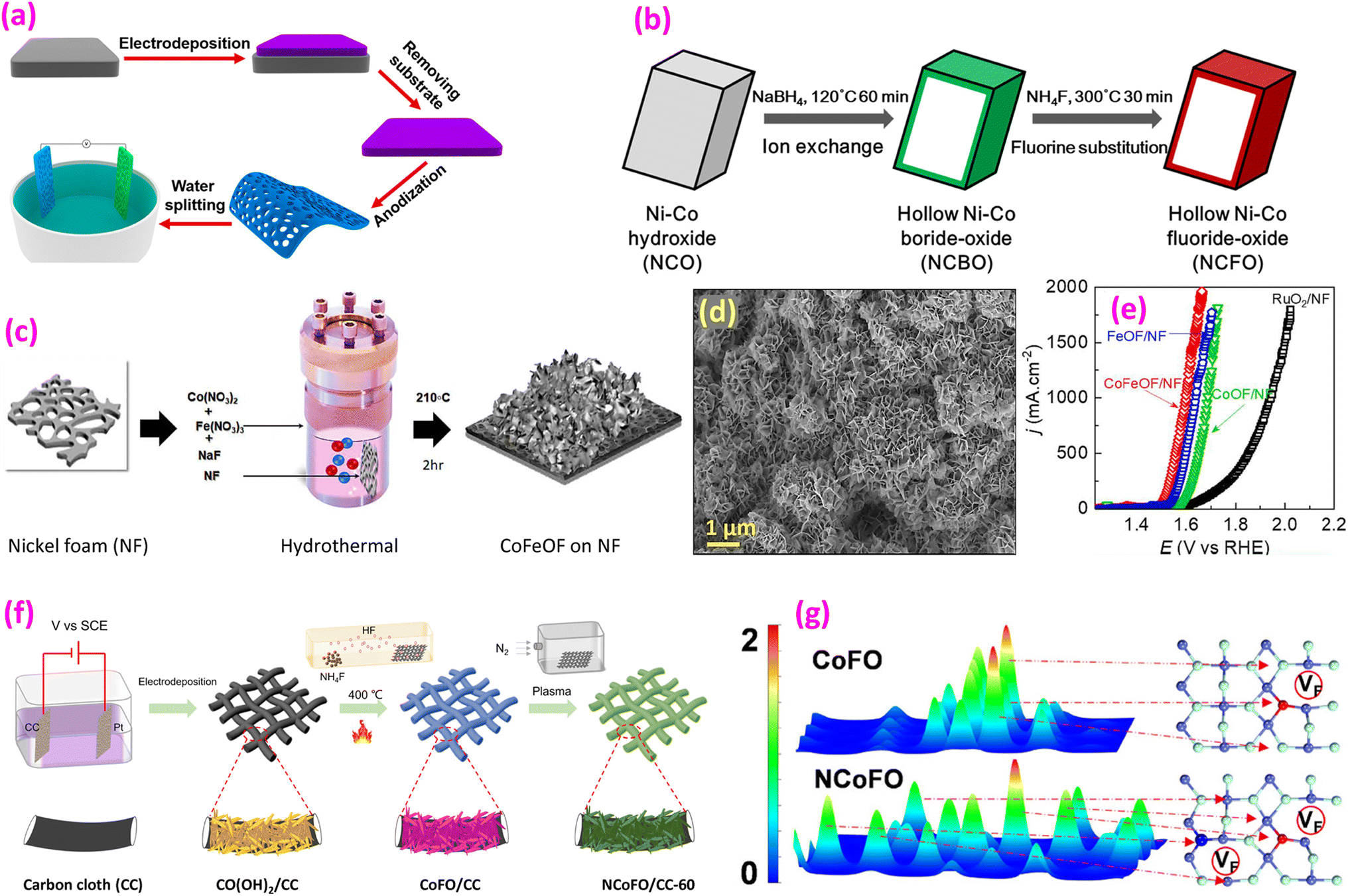 | ||
| Fig. 7 Oxyfluoride catalysts. (a) Fabrication of a NiTiOF holy film. Reproduced from ref. 98 with permission from American Chemical Society, copyright 2017. (b) Synthesis process from NCO through NCBO to NCFO. Reproduced from ref. 95 with permission from American Chemical Society, copyright 2019. (c) Synthesis of a CoFeOF film; (d) SEM image of CoFeOF nanosheets and (e) LSV polarization curves. Reproduced from ref. 105 with permission from Elsevier, copyright 2024. (f) Synthesis of NiCoFO@CC and (g) electron density difference calculated using DFT. Reproduced from ref. 106 with open access. | ||
Kornienko and coworkers synthesized hydrated NiFe fluorides, NiFe2F8(H2O)2 and CoFe2F8(H2O)2, via a simple microwave-assisted solvothermal synthesis.10 Subsequent calcination produced amorphous oxyfluorides NiFe2F4.4O1.8 and CoFe2F6.6O0.7, respectively, with the former catalyst showing superior performance. The same group also prepared Mn–Fe-based fluorinated materials that converted to oxyfluorides after heating in the air.101 The amorphous oxyfluorides were evaluated in H2SO4 acidic media, demonstrating high performance. A stable compound, TaO2F, supported on graphitized carbon (gC), was synthesized using a simple ion-adsorbed method.99 This catalyst exhibited an overpotential of 170 mV lower than that of an unfluorinated Ta2O5/CC catalyst at 10 mA cm−2. A recent study by the same group on the Co0.5Fe0.5O0.5F1.5 oxyfluoride catalyst demonstrated superior performance compared to most known Co-based catalysts.102
Han et al. synthesized a nickel–cobalt fluoride oxide (NCFO) hollow nanoprism through three steps: Ni–Co hydroxide formation, followed by ion exchange, and finally fluorination (Fig. 7b).95 The synthesized catalyst exhibited 350 mV overpotential at 10 mA cm−2 with an exceptionally low Tafel slope of 23 mV dec−1.
Pan et al. dealloyed a melt-spun Zr-based amorphous ribbons in HF and H2O2 corrosive media.104 After in situ chronopotentiometric surface reconstruction, a core–shell structure was obtained. The catalyst showed only 4.6% loss after the 48-h test, compared to RuO2, which had a 43.2% loss. The unique core–shell configuration facilitates improved ion movement and material transport while increasing the number of active sites at the interface between the electrolyte and electrode, thereby enhancing OER performance. DFT calculations suggest that the presence of F contributes to a higher concentration of surface-active sites characterized by an unsaturated electronic configuration. Maeda et al. synthesized 2D perovskite oxyfluoride Pb3Fe2O5F2, which features two layers of perovskite blocks interleaved with a PbF block layer.103 This 2D catalyst exhibits nearly eight times the activity of its three-dimensional (3D) bulk counterpart, PbFeO2F, in the oxygen evolution reaction. A recent study evaluated the ABX2Y type oxynitride, oxyfluoride, and nitrofluoride perovskites for solar water splitting.109 Four catalysts (BaInO2F, InSnO2N, CsPbO2F, and LaNbN2O) were identified as potential photocatalysts, with BaInO2F oxyfluoride showing the lowest calculated overpotential.
Recently, CoFeOF nanosheets on NF were prepared using metal nitrates and NaF (as an F source) through a hydrothermal route, followed by calcination at 210 °C for 2 h (Fig. 7c).105 Nanosheets, 8–10 nm thick, were grown vertically on NF (Fig. 7d). CoFeOF/NF demonstrated superior functionality compared to FeOOF/NF or CoOF/NF (Fig. 7e), performing effectively even in seawater. Another approach involved electrodepositing Co(OH)2 on CC, followed by fluorination at 400 °C using NH4F as the F source and subsequent N2 plasma treatment (Fig. 7f).106 Among the tested materials, N-doped CoFO/CC demonstrated superior performance in both the OER and HER compared to CoFO/CC and Co3O4/CC. Computational studies using DFT revealed that the CoF bond in N-doped CoFO is elongated relative to pure CoF2 and CoFO, attributed to the lower electronegativity of N in comparison to F. In the case of CoFO, electron localization occurs on F atoms adjacent to O, whereas in N-doped CoFO, electrons from F tend to shift towards N and Co atoms (Fig. 7g). N-doping also created more F vacancies, significantly optimizing the electronic structure. FeOF with NiF2 nanosheets was also reported via a solvothermal reaction using pyrazine, followed by calcination in air. This calcination introduces O into the catalyst lattice. DFT simulations revealed that O incorporation led to an increased concentration of electronic states near the Fermi level, suggesting improved electron conductivity. Additionally, the energy barrier for the rate-limiting step in *OOH formation decreased from 2.26 eV to 2.02 eV, resulting in enhanced OER performance.
4.2 Fluorinated oxides
Metal oxides, including both precious metal oxides and non-precious metal oxides (primarily transition-metal oxides), have been widely used as catalysts for the OER.5,49,110–112 Among these, earth-abundant transition-metal oxides have garnered significant attention for the OER due to their high activity, enhanced stability, and low cost.49 The fluorination of these metal oxides has been reported and is summarized in Table 3. The following fluorinated oxides have been studied for the OER.| Catalyst | Overpotential at specific current density | Tafel slope (mV dec−1) | Tested durability | Electrolyte | Source |
|---|---|---|---|---|---|
| a (*)Estimated in this study. | |||||
| Fluorinated MO | |||||
| F–CoO/CC | 237.7 mV@10 mA cm−2 | 68 | 60 h@10 mA cm−2 | 1 M KOH | 113 |
| CoO/CC | 349 mV@10 mA cm−2 | 89 | — | 1 M KOH | |
| Fe, F–NiO | 215 mV@10 mA cm−2 | 67.5 | 100 h@10 mA cm−2 | 1 M KOH | 114 |
| Fe–NiO | 230 mV@10 mA cm−2 | 79.5 | — | 1 M KOH | |
| F–NiO | 266 mV@10 mA cm−2 | 136.3 | — | 1 M KOH | |
| NiO | 270 mV@10 mA cm−2 | 146.4 | — | 1 M KOH | |
|
|||||
| Fluorinated MO 2 | |||||
| (Ir0.30Sn0.35Nb0.35)O2:10F | 290 mV@10 mA cm−2* | 77 | 22.2 h@1.65 V | 1 N H2SO4 | 115 |
| (Ir0.30Sn0.35Nb0.35)O2 | 350 mV@10 mA cm−2* | 98 | 22.2 h@1.65 V | 1 N H2SO4 | |
| (Mn0.7Ir0.3)O2:10F | 210 mV@10 mA cm−2* | 65 | 24 h@1.45 V | 1 N H2SO4 | 116 |
| (Mn0.7Ir0.3)O2 | 220 mV@10 mA cm−2* | 67 | — | ||
| (Mn0.8Ir0.2)O2:10F | 200 mV@10 mA cm−2 | 38 | 24 h@1.45 V | 1 N H2SO4 | 117 |
| (Mn0.8Ir0.2)O2 | 240 mV@10 mA cm−2 | 46 | 24 h@1.45 V | ||
| (Mn0.8Nb0.2)O2:10F | 680 mV@10 mA cm−2 | 371.17 | 24 h@1.9 V | 1 N H2SO4 | 118 |
| (Mn0.8Nb0.2)O2 | 770 mV@10 mA cm−2 | 385.36 | 24 h@1.9 V | ||
| IrOx/F–TiO2 | 272 mV@10 mA cm−2 | 53.1 | 10 h@1.55 V | 0.5 M H2SO4 | 119 |
| IrOx/TiO2 | 322 mV@10 mA cm−2 | 63.3 | 10 h@1.55 V | 0.5 M H2SO4 | |
| RuOx/F–TiO2 | 252 mV@10 mA cm−2 | 50.3 | 10 h@10 mA cm−2 | 0.5 M H2SO4 | 120 |
| RuOx/TiO2 | 295 mV@10 mA cm−2 | 62.8 | 10 h@10 mA cm−2 | ||
| PbO2–F–Fe–Co | 307 mV@10 mA cm−2 | 188 | 156 h@2.5 mA cm−2* | 0.5 M/1 M H2SO4 | 121 |
| F-TMO | 330 mV@50 mA cm−2 | 30 | 336 h@500 mA cm−2 | 1 M KOH | 122 |
| TMO | 380 mV@50 mA cm−2 | 60 | — | ||
|
|||||
| Fluorinated ABO 4 | |||||
| F–NiMoO4 | 188 mV@50 mA cm−2 | 33.8 | 85 h@50 mA cm−2 | 1 M KOH | 41 |
| NiMoO4 | 290 mV@50 mA cm−2 | 130.0 | |||
| F–CoMoO4−x-2@GF | 256 mV@10 mA cm−2 | 64.4 | 20 h@1.54 V | Alkaline solution | 123 |
| CoMoO4−x@GF | 368 mV@10 mA cm−2 | 81.8 | |||
|
|||||
| Fluorinated A 2 B 2 O 7 | |||||
| YRuO-PVDF2 | 235 mV@10 mA cm−2 | 41 | 10 h@10 mA cm−2 | 0.5 M H2SO4 | 124 |
| YRuO | 296 mV@10 mA cm−2 | 46 | 10 h@10 mA cm−2 | ||
|
|||||
| Fluorinated spinel oxides | |||||
| F–Co3O4/NF | 370 mV@50 mA cm−2 | 70 | — | 1 M KOH | 13 |
| Co3O4/NF | 460 mV@50 mA cm−2 | 160 | — | ||
| Co3O3.87F0.13 | 430 mV@10 mA cm−2 | 56 | — | 0.1 M KOH | 125 |
| Co3O3.87□0.13 | 440 mV@10 mA cm−2 | 56 | — | ||
| Co3O4 | 520 mV@2.6 mA cm−2 | 64 | — | ||
| F0.2-V-Co3O4-350 | 323 mV@10 mA cm−2 | 75.9 | — | 1 M KOH | 126 |
| ZnCo2O4−xFx/CNTs | 440 mV@50 mA cm−2 | 59.2 | 13 h@10 mA cm−2 | 1 M KOH | 127 |
| ZnCo2O4−x/CNTs | 560 mV@50 mA cm−2* | 106.3 | 2 h@10 mA cm−2* | ||
| ZnCo2O4-F | 472 mV@10 mA cm−2 | 65.1 | 30 h@1.65 V | 1 M KOH | 128 |
| ZnCo2O4 | 440 mV@10 mA cm−2 | 59.9 | 30 h@1.65 V | ||
| F–Co/CoFe2O4@NC | 280 mV@10 mA cm−2 | 49.7 | 50 h@1.51 V | 1 M KOH | 129 |
| Co/CoFe2O4@NC | 320 mV@10 mA cm−2 | 58.0 | — | ||
| F–MnCo2O4 | 210 mV@10 mA cm−2 | 69.5 | 100 h@10 mA cm−2 | 0.1 M HClO4 | 130 |
| MnCo2O4 | 370 mV@10 mA cm−2 | 131.6 | |||
|
|||||
| Fluorinated perovskite oxides | |||||
| SrCoO2.85−dF0.15 | 380 mV@10 mA cm−2 | 60 | 20 h@1.60 V | 1 M KOH | 14 |
| SrCoO3−d | 434 mV@10 mA cm−2 | 71 | — | ||
| F-Ba0.5Sr0.5Co0.8Fe0.2O3−δ | 280 mV@10 mA cm−2 | 102.65 | 100 h@15 mA cm−2* | 1 M KOH | 131 |
| Ba0.5Sr0.5Co0.8Fe0.2O3−δ | 342 mV@10 mA cm−2 | 119.73 | <20 h@5 mA cm−2* | ||
| (Ba0.5Sr0.5Co0.8Fe0.2O3−d)3/4[KM(II)F3]1/4 | 345 mV@10 mA cm−2 | — | 20 h@10 mA cm−2 | 1 M KOH | 132 |
| Ba0.5Sr0.5Co0.8Fe0.2O3−d | 450 mV@10 mA cm−2 | — | 20 h@10 mA cm−2 | ||
| F0.2–LaCoO3 | 390 mV@10 mA cm−2 | 114.1 | 120 h@2 mA cm−2 (ZAB) | 1 M KOH | 133 |
| LaCoO3 | 530 mV@10 mA cm−2 | 182.8 | 70 h@2 mA cm−2 (ZAB) | ||
| BaFe0.8Co0.2O2F | 400 mV@10 mA cm−2 | 52.8 | — | 1 M KOH | 134 |
| BaFe0.8Co0.2O3 | 440 mV@10 mA cm−2 | 80.3 | — | ||
| F–NiTiO3/C | 270 mV@10 mA cm−2 | 81.2 | 18 h@28 mA cm−2 | 1 M KOH | 21 |
| NiTiO3/C | 680 mV@10 mA cm−2 | 112.4 | 18 h@18 mA cm−2 | ||
| F-La0.6Sr0.4Co0.2Fe0.8O3−d | 350 mV@10 mA cm−2* | 100 | — | 0.1 M KOH | 135 |
| La0.6Sr0.4Co0.2Fe0.8O3−d | 400 mV@10 mA cm−2* | 123* | — | ||
| LSNF-OF | 308.1 mV@10 mA cm−2 | 61.13 | 5 h@10 mA cm−2 | 1 M KOH | 136 |
| LSNF | 381.2 mV@10 mA cm−2 | 62.47 | — | ||
| LSNF-MF | 486.4 mV@10 mA cm−2 | 67.99 | 5 h@10 mA cm−2 | ||
|
|||||
| Fluorinated mixed oxides | |||||
| FeCo-OF | 285 mV@10 mA cm−2 | 95.7 | 12 h@10 mA cm−2 | 1 M KOH | 44 |
| FeCo–O | 370 mV@10 mA cm−2 | 145.9 | 12 h@10 mA cm−2 | 1 M KOH | |
| Fe–OF | 570 mV@10 mA cm−2 | 174.6 | — | 1 M KOH | |
| Fe–O | 1000 mV@10 mA cm−2 | 562.5 | — | 1 M KOH | |
| Co–OF | 330 mV@10 mA cm−2 | 156.8 | — | 1 M KOH | |
| Co–O | 467 mV@10 mA cm−2 | 168.6 | — | 1 M KOH | |
(1) Fluorinated MO. In the MO oxide, the metal M is divalent. Pure cobalt monoxide (CoO) exhibits HER activity due to its low electron conductivity (∼1 × 1012 Ω·m).113 However, after doping CoO nanowires with aliovalent F− anions, superior HER and OER activities are observed. The F ions increase the charge on the Co site, weakening H* adsorption and also narrowing the band gap, improving electron conductivity. As illustrated in Fig. 8a, Fe-doped Fe–Ni(OH)2 hollow flower-spheres are synthesized via a solvothermal method, which is then converted to a Fe-doped NiO cubic structure through calcination and finally to Fe and F-codoped NiO by fluorination.114 Comparison of the OER activities of different catalysts shows that F doping significantly reduces overpotentials (Fig. 8b) and Tafel slopes (Table 3). DFT calculations reveal that the RDS energy barrier is reduced from 2.23 eV (NiO) to 1.92 eV (Fe–NiO) and 1.87 eV (Fe, F–NiO), while the band gap decreases from 2.27 eV (NiO) to 2.09 eV (Fe, F–NiO).
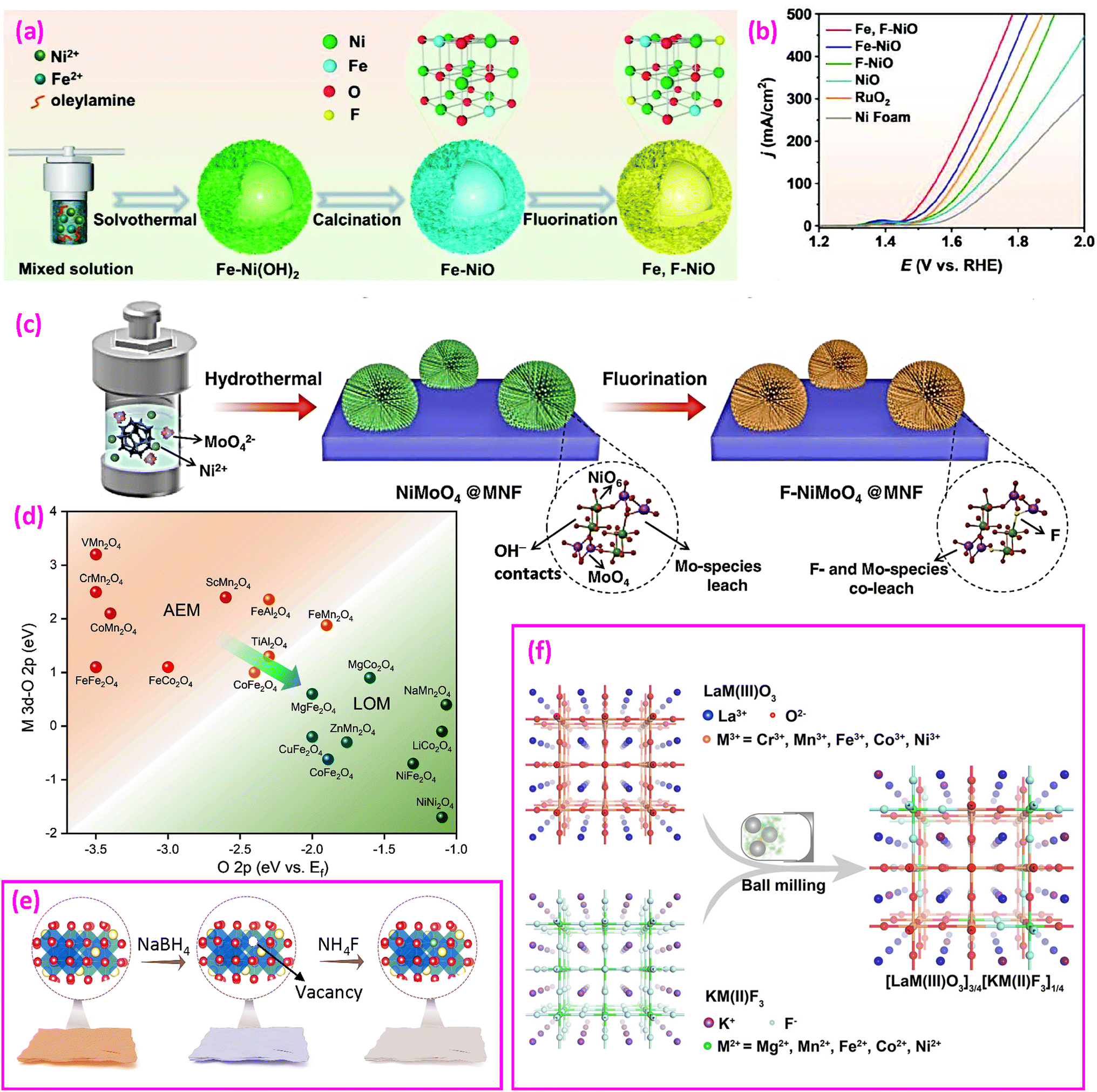 | ||
| Fig. 8 Fluorinated oxides for the OER. (a) Synthesis of Fe and F co-doped NiO hollow flower-spheres and (b) LSV polarization curves. Reproduced from ref. 114 with permission from John Wiley and Sons, copyright 2023. (c) Synthesis of F-doped NiMoO4 on MoNi foam. Reproduced from ref. 41 with permission from American Chemical Society, copyright 2021. (d) Relationship between AEM/LOM and relative band centers and (e) synthesis of ZnCo2O4−x and ZnCo2O4−xFx. Reproduced from ref. 127 with permission from John Wiley and Sons, copyright 2023. (f) Synthesis of a perovskite oxide-fluoride solid solution. Reproduced from ref. 132 with permission from John Wiley and Sons, copyright 2021. | ||
(2) Fluorinated MO2. F has been used to dope MO2 compounds such as (Ir, Sn, Nb)O2,115 TiO2,119,120,137 (Mn, Ir)O2,116,117 (Mn, Nb)O2,118 and PbO2.121 Given the reputation of IrO2 as a highly effective catalyst, researchers have explored alloying Ir with alternative metals to decrease the overall cost of the catalyst.115 With the additions of Sn and Nb, although Ir0.25Sn0.375Nb0.375O2 shows reduced activities, F doping at a level of 10 wt% significantly improves its performance, even surpassing that of IrO2. With the addition of Mn, either Mn0.7Ir0.3O2 or Mn0.8Ir0.2O2 outperforms IrO2, while F doping further enhances their OER performance.116,117 DFT calculations show that the d-band center of the F-doped material is close to that of pure IrO2, suggesting an improvement in overall catalytic activity.116
To stabilize the MnO2 surface for the OER in acidic environments, transition metals are added. The Kumta group demonstrated that adding 20% Nb reduces the overpotential (at 10 mA cm−2) in H2SO4 acid from 900 to 770 mV, and further incorporation of F reduces it to 680 mV.118
Fluorinated TiO2 has been used to enhance the development of OER catalysts. IrOx on F–TiO2 exhibits significantly improved OER activities over IrOx on undoped TiO2 in an acidic solution, likely due to the higher electron conductivity of fluorinated TiO2, improved dispersion, and suppression of Ir dissolution.119 The same group also developed RuOx on a F–TiO2 support, which outperforms RuOx on undoped TiO2.120 DFT calculations suggest that F ions facilitate proton transfer, accelerating the O–O coupling steps.138
(3) Fluorinated ABO4. Chai et al. synthesized NiMoO4 nanorods on nanospheres on MoNi foam (MNF) using a hydrothermal method, followed by fluorination, as shown in Fig. 8c.41 The F-doped materials showed superior OER performance, as detailed in Table 3. F–NiMoO4 is amorphous, and the strong ionicity of the metal–F bonds suggests that F species in the amorphous layer can migrate and leach into the alkaline electrolyte, facilitating the adsorption of reaction intermediates.
F has also been doped into layered oxides. CoMoO4 nanosheets were synthesized on graphite felt (GF) and fluorinated using NH4F at 400 °C.123 F-doped CoMoO4 demonstrated a 112 mV lower overpotential and 17.4 mV dec−1 lower Tafel slope compared to its non-fluorinated counterpart. The F-doped CoMoO4 outperforms RuO2@GF, whose overpotential is 284 mV@10 mA cm−2 and Tafel slope is 76.6 mV dec−1. The F-doped material is enriched in O vacancies and has an optimized electronic configuration of active sites for the OER.
(4) Fluorinated A2B2O7. The pyrochlore oxide A2B2O7 exhibits high chemical and structural stability during the OER.139 A sol–gel method was used to synthesize Y2Ru2O7−d nanoparticles, followed by calcination with polyvinylidene fluoride (PVDF) powders.124 OER testing in 0.5 M H2SO4 showed that both the overpotential and Tafel slope were reduced by fluorination (Table 3). F doping introduces O vacancies, promoting OER kinetics. DFT calculations support the LOM of the OER.
(5) Fluorinated spinel oxides. Liu et al. analyzed the electronic structure of several spinel oxides and proposed their possible AEM or LOM for the OER, as shown in Fig. 8d.127 They synthesized F-doped ZnCo2O4−xFx nanosheets via a two-step route, as illustrated in Fig. 8e. First, oxygen vacancies (VO) in ZnCo2O4 (denoted as ZnCo2O4−x) are created by NaBH4 reduction, followed by fluorination using NH4F to form ZnCo2O4−xFx. The fluorinated catalyst reduces the overpotential at 50 mA cm−2 from 570 mV (estimated in this study) to 440 mV (a 22.8% reduction) and the Tafel slope from 106.3 to 59.2 mV dec−1 (a 44.3% reduction). This approach fills lattice O vacancies on the surface with F ions, strengthening the hybridization of Co 3d and O 2p for the LOM. However, a study showing opposite results was also reported, where F doping had a slight negative impact, raising the overpotential from 440 to 472 mV at 10 mA cm−2 (a 7% increase) and the Tafel slope from 59.9 to 65.1 mV dec−1 (an 8.7% increase).128
The oxide Co3O4 also possesses a spinel structure. DFT calculations indicate that the F-doped Co3O4 (100) surface is active for the OER.140 Mesoporous Co3O3.87□0.13 (□ represents VO) reduces the overpotential of Co3O4 from 520 mV to 440 mV at 10 mA cm−2, while F-doped Co3O3.87F0.13 further reduces it to 430 mV.125
(6) Fluorinated perovskite oxides. Perovskite oxides have been widely studied as catalysts for the OER, and fluorination of these oxides has drawn significant attention.14,21,131–136,141 The perovskite oxide Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF) is considered one of the most effective OER catalysts.142–145 Xiong et al. synthesized F-doped F-BSCF for the OER.131 As O2− is partially replaced by F−, this doping reduces the Co(III) and Fe(III) species to lower oxidation states, activating surface O to highly oxidative O2−/O− for the OER. F-BSCF reduces the overpotential of BSCF from 342 to 280 mV at 10 mA cm−2 and the Tafel slope from 119.73 to 102.65 mV dec−1. The fluorinated oxide demonstrated prolonged stability for over 100 h without significant changes in composition.
In 2021, the Dai group proposed a perovskite oxide-halide solid solution concept, experimentally merging perovskite oxide and perovskite halide into a single lattice phase through high-energy ball milling (Fig. 8f).132 They optimized a combination of ¾ of BSCF with ¼ of KM(II)F3 to achieve an overpotential of 345 mV at 10 mA cm−2, significantly lower than the 450 mV overpotential of BSCF without F.
Besides the fluorination of simple cubic perovskites, it was shown that fluorination of a hexagonal Sr2Co2O5 phase produces a cubic SrCoO2.85−dF0.15 perovskite, with improved OER activities (Table 3).14 According to DFT calculations, F doping and the resulting structural transition improve electronic conductivity and increase the number of reactive O species of O22− and O− for the OER.
Fluorination of Ruddlesden–Popper (R–P) perovskite oxides with their layered structure has also been reported.136,146 La1.2Sr0.8Ni0.6Fe0.4O4+δ (LSNF) was synthesized via a sol–gel method and subsequently fluorinated by calcination at 370 °C with PVDF to obtain microfluorinated LSNF (LSNF-MF).136 Finally, LSNF-MF was calcined at a high temperature of 900 °C, resulting in fully fluorinated perovskite oxyfluoride La1.2Sr0.8Ni0.6Fe0.4O4+δ Fy (LSNF-OF). LSNF-MF reduced the overpotential at 10 mA cm−2 from 484 mV to 377 mV (estimated in this study), while LSNF-OF further reduced it to 308.1 mV. LSNF-OF is considered a triple-conductive oxide (conductive for H+, O2− and electrons).
(7) Fluorinated mixed oxides. The fluorination of mixed oxides has also been reported in the literature. Recently, FeCo–O bimetallic oxide was synthesized by calcinating Fe and Co nitrates at a relatively low temperature of 400 °C, resulting in mixed Co3O4 and CoFe2O4 phases, both of which belong to the spinel oxide type.44 For comparison, Fe–O and Co–O oxides are also prepared. Fluorination significantly reduced the overpotentials and Tafel slopes of all FeCo, Fe–O, and Co–O materials (as shown in Table 3), with a more pronounced effect on the bimetallic oxide. The authors believe that the synergistic effect of Fe and Co bimetals enhances electron transport, and the fluorine dopant reduces charge density in Fe and Co phases, facilitating OH− adsorption.
4.3 Fluoride/oxide heterocatalysts
It is known that metal oxides have demonstrated high performances in the OER,5,49,110,111 and so have metal fluorides, though these fluorides contain multiple metal elements, as mentioned in Section 3. In Sections 4.1 and 4.2, we demonstrated that the addition of oxygen to fluorides, either in the form of oxyfluorides or fluorinated oxides, can further improve OER performance compared to pristine materials. But what about a heterocatalyst combining both fluoride and oxides? Such fluoride/oxide heterocatalysts have been reported, as summarized in Table 4.| Catalyst | Overpotential at specific current density | Tafel slope (mV dec−1) | Tested durability | Electrolyte | Source |
|---|---|---|---|---|---|
| IFONFs-45 | 260 mV@10 mA cm−2 | 45 | 8.33 h@40 mA cm−2 | 1 M KOH | 20 |
| NiCo2O4/NiO/CoF2@mC700 | 240 mV@10 mA cm−2 | 78 | 40 h@10 mA cm−2 | 0.1 M KOH | 147 |
| CFO-RH400 | 230 mV@10 mA cm−2 | 68.7 | 45 h@10 mA cm−2 | 1 M KOH | 148 |
| Co/Co3O4/CoF2@NSC-CC | 310 mV@10 mA cm−2 | 88 | 1000 h@10 mA cm−2 (ZAB) | 0.1 M KOH | 149 |
| Co/Co3O4@NC | 430 mV@10 mA cm−2 | 134 | — | ||
| Co3O4–CoF2 | 169 mV@10 mA cm−2 | 63.5 | 72 h@10–100 mA cm−2 | 1 M KOH | 150 |
| CoF2 | 295 mV@10 mA cm−2 | 83.9 | − | ||
| Co3O4 | 301 mV@10 mA cm−2 | 84.1 | − |
A heterocatalyst consisting of Fe2O3 and FeF2, referred to as an iron fluoride-oxide nanoporous film (IFONF), was prepared by anodization followed by fluorination using a piece of Fe foil, as depicted in Fig. 9a.20 After the anodization of the Fe foil, a porous Fe-oxide thin film (PTF) is formed. Subsequent fluorination results in the formation of IFONFs. Both XRD and TEM observations (Fig. 9b) confirm the formation of mixed Fe2O3 oxide and FeF2 fluoride phases. The IFONFs-45 catalyst, with 45 min of fluorination, showed an overpotential of 260 mV at a current density of 10 mA cm−2, which is higher than that of RuO2 (170 mV) but lower than that of Fe-oxide (480 mV). Its Tafel slope was 45 mV dec−1, which is lower than that of Fe-oxide (207 mV dec−1) and RuO2 (125 mV dec−1), implying different RDSs for the given pathway. The enhanced OER activities could be attributed to the increased active sites and reduced charge-transfer resistance in the heterocatalyst.
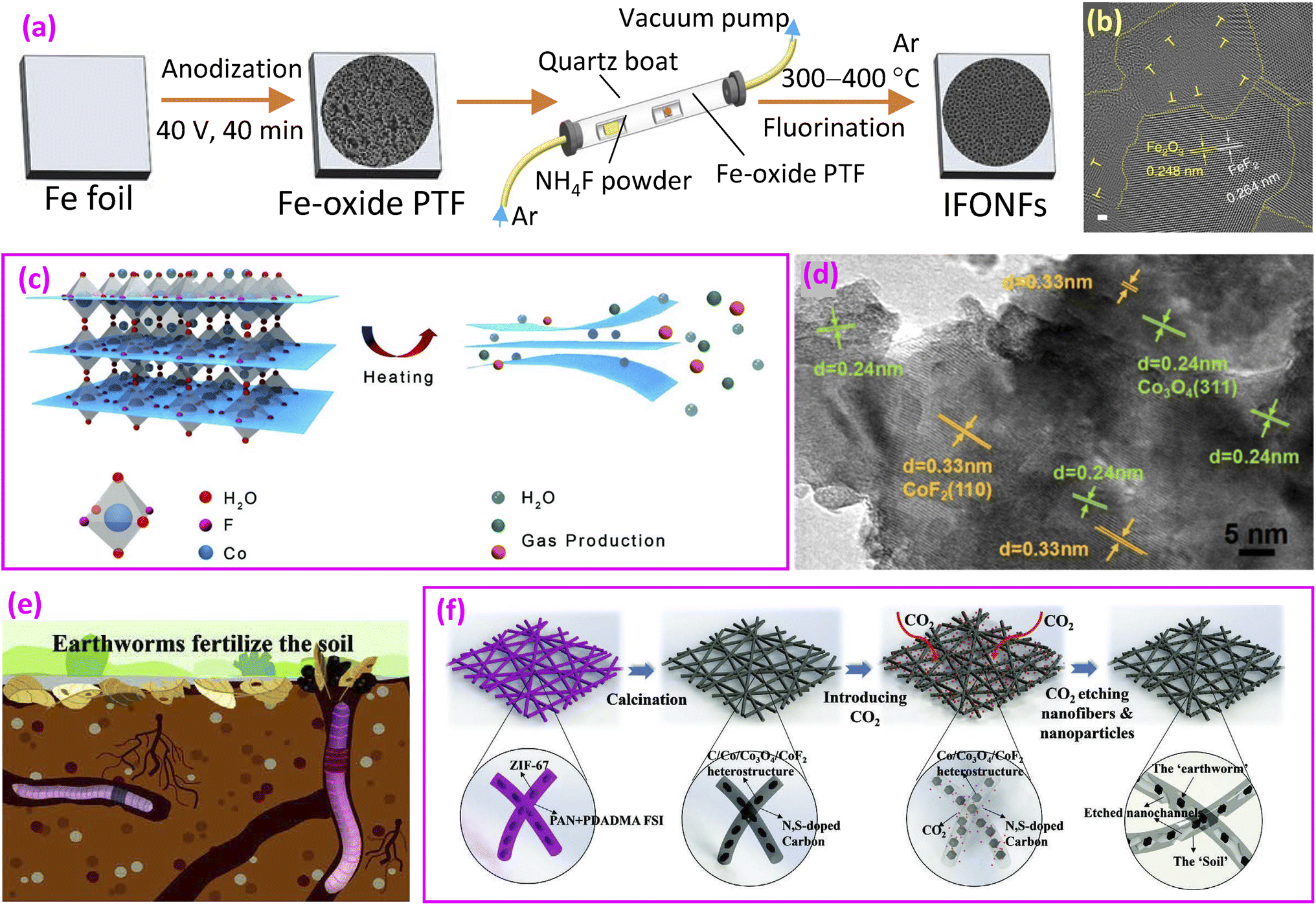 | ||
| Fig. 9 Fluoride/oxide heterocatalysts. (a) Synthesis of an iron fluoride-oxide nanoporous film (IFONF); (b) TEM image of the IFONF. Reproduced from ref. 20 with open access; (c) schematic illustration of the exfoliation of cobalt fluoride hydrate to ultrathin 2D CFO-RH400 nanosheets, driven by the rapid thermally generated and released water or other gaseous products; and (d) TEM image of CFO-RH400 nanosheets. Reproduced from ref. 148 with permission from American Chemical Society, copyright 2020. (e) Earthworms fertilizing the soil and (f) synthesis route of Co/Co3O4/CoF2@NSC-CC. Reproduced from ref. 149 with permission from John Wiley and Sons, copyright 2024. | ||
Since spinel NiCo2O4 has demonstrated effective electrochemical activity toward the OER,151 a heterocatalyst consisting of NiCo2O4, NiO and CoF2 was prepared.147 Starting from a bimetallic NiCo-MOF with an equal molar ratio of Ni and Co, fluorination using NH4F followed by high-temperature calcination produced a mixture of NiCo2O4/NiO/CoF2. The hybrid material delivers an overpotential of 240 mV at 10 mA cm−2 and a Tafel slope of 78 mV dec−1. The F doping in this hybrid material is thought to enhance charge transfer, improving both OER and HER activities.
Considering the unique structural features of 2D catalysts, such as large exposed surface areas with active sites,152,153 2D cobalt-fluoride-oxide (CFO) nanosheets were prepared. A method to obtain 2D Co fluoride-oxide was demonstrated through exfoliation by a rapid thermal annealing process (Fig. 9c).148 The material structure includes CoF2 fluoride and spinel Co3O4 oxide (Fig. 9d). The catalyst displayed a Tafel slope of 68.7 mV dec−1, lower than that of IrO2 (73.8 mV dec−1), indicating good OER kinetics and better catalytic activity. Nyquist electrochemical impedance spectroscopy revealed the smallest high-frequency interfacial charge-transfer resistance (Rct1), suggesting that the coexistence of M–O bonds and M–F bonds facilitates the charge transfer process and enhances the reaction kinetics.
Drawing inspiration from earthworms' soil fertilization process (Fig. 9e), researchers developed nanofibers consisting of Co/Co3O4/CoF2@N and sulfur-enriched carbon with restricted channels (NSC-CC).149 The fabrication method involved placing ZIF-67 nanoparticles on freshly spun nanofibers, followed by a calcination step. This process resulted in the formation of Co/Co3O4/CoF2 heterojunction particles embedded within the carbon framework (Fig. 9f). Carbon dioxide was subsequently introduced to create micro- and mesopores on the nanofiber surface. The N and S co-doped carbon matrix serves as “nano-soil”, while the Co/Co3O4/CoF2 heterostructure particles function as “nano-earthworms”. This heterogeneous catalyst demonstrated an overpotential of 310 mV at 10 mA cm−2, which is considerably lower than the overpotential of 430 mV observed in the catalyst lacking CoF2 and confined channels.
In a recent report, a cobalt oxide-fluoride heterojunction catalyst was synthesized on carbon cloth using a hydrothermal method.150 Subsequent heat treatment at 400 °C for 1 h in different atmospheres yielded varying results: an Ar environment produced a blend of Co3O4 and CoF2, air calcination led to Co3O4 formation, and an atmosphere with added NH4F resulted in CoF2. The research indicated that the heterocatalyst comprising mixed oxide and fluoride phases showed enhanced performance compared to individual oxide or fluoride components (Table 4). The interface between Co3O4 and CoF2 likely modifies the surface electronic configuration, leading to improved OER kinetics.
5. Fluorinated (oxy)hydroxides, carbonate hydroxides and their derived fluorides
A simple metal hydroxide is expressed as Mn+(OH)n, where metal M has a valence of n+. A meal oxyhydroxide is expressed as MOxH, where x = (n + 1)/2 for the metal Mn+.154 These compounds share a structural commonality in that they contain hydrogen in their structure. Transition-metal hydroxide and oxyhydroxide materials have been widely used for OER research due to several advantages, such as their distinct layered structure with large surface areas that favor the OER, excellent catalytic activity, high stability, and ease of preparation.155–160 Through fluorination, these materials have been reported to exhibit improved OER performance.5.1 Fluorinated hydroxides
(1) Fluorinated divalent M(OH)2 hydroxides. There are two important divalent M(OH)2 hydroxides, namely, β-Ni(OH)2 and α-Ni(OH)2·xH2O. β-Ni(OH)2 exists naturally as the mineral theophrastite, with a trigonal structure and a space group of P1m, containing edge-sharing hexagonally packed Ni(OH)6 octahedral layers (Fig. 10a).165,166 The spacing between the layers is 0.256 nm, which includes the hydrogen atoms on the distorted tetrahedral sites directed toward the adjacent layer. It is isostructural with other compounds when M = Mg (brucite), Ca (portlandite), Cd, and Co. α-Ni(OH)2·xH2O also has a trigonal structure with the same space group and similar Ni(OH)6 octahedral layers, as proposed by Bode et al.167 However, between the (001) planes, there are disordered H2O layers, resulting in an increase in the c-axis constant to c = 0.8 nm (Fig. 10b).168,169 The degree of hydration varies within the range x = 0.41–0.7, and the space between the octahedral layers reaches 0.596 nm.169 However, the trivalent structure of Fe(OH)3 is a 3D perovskite structure.170
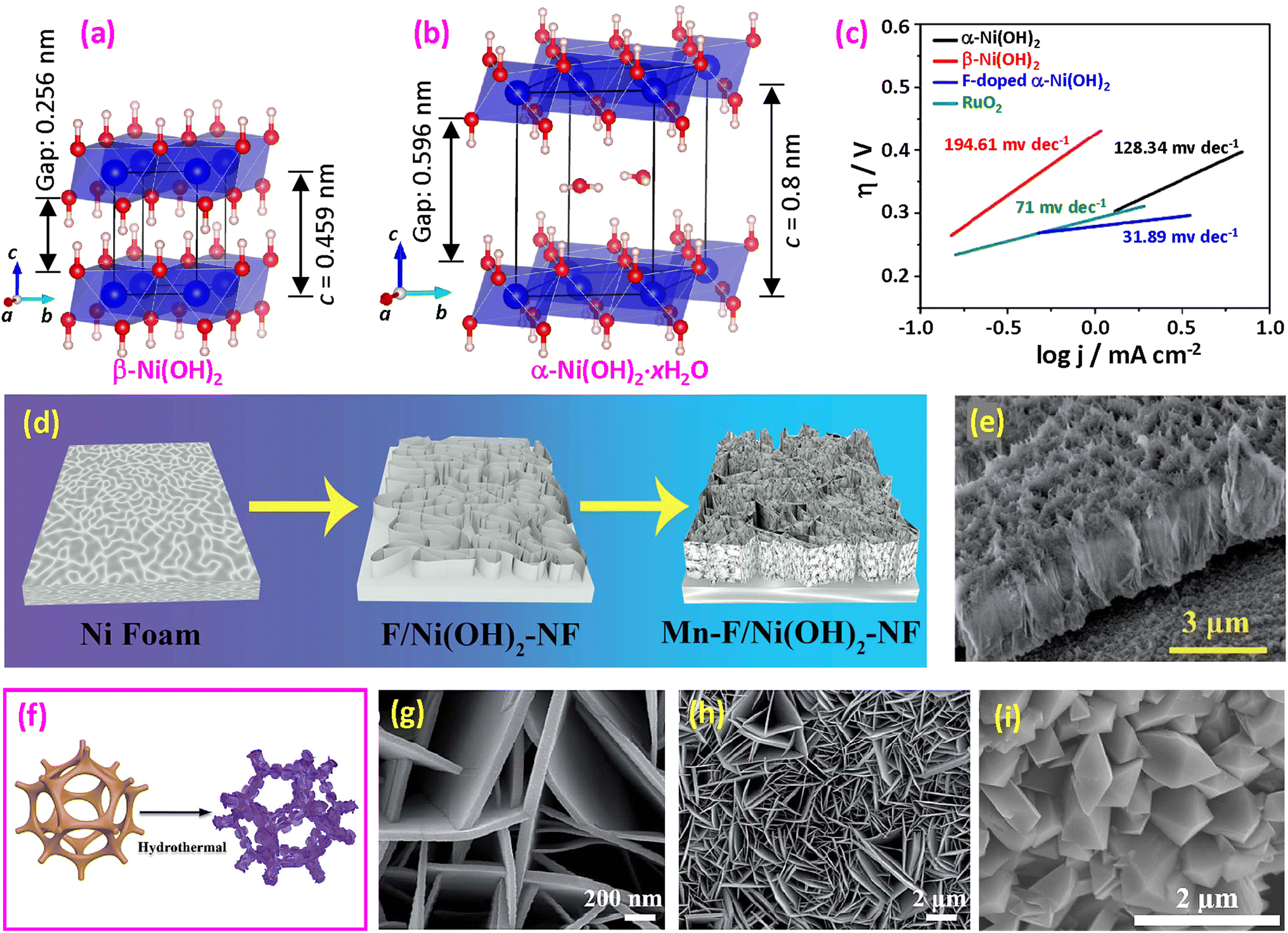 | ||
| Fig. 10 (a) Structural model of β-Ni(OH)2; (b) structural model of α-Ni(OH)2. (c) Tafel slope of OER catalysts. Reproduced from ref. 161 with permission from the Royal Society of Chemistry, copyright 2019. (d) Synthesis of Mn and F doped Ni(OH)2 on NF and (e) SEM side view of Mn-F/Ni(OH)2-NF. Reproduced from ref. 162 with open access. (f) Synthesis of CoxFey(OH)F via a one-step hydrothermal procedure; (g and h) SEM images. Reproduced from ref. 163 with permission from the Royal Society of Chemistry, copyright 2022. (i) SEM image of Co(OH)F. Reproduced from ref. 164 with permission from Elsevier, copyright 2022. | ||
The OER activities of fluorinated divalent hydroxides are listed in Table 5.
| Catalyst | Overpotential at specific current density | Tafel slope (mV dec−1) | Tested durability | Electrolyte | Source |
|---|---|---|---|---|---|
| a (*) Estimated in this study. | |||||
| NiFe-OH-F-SR | 181 mV@10 mA cm−2 | 22.6 | 165 h@50 mA cm−2 | 1 M KOH | 15 |
| NiFe–OH–F | 243 mV@10 mA cm−2 | 42.9 | — | ||
| F-doped α-Ni(OH)2 | 325 mV@10 mA cm−2 | 31.89 | 30 h@25 mA cm−2* | 1 M KOH | 161 |
| α-Ni(OH)2 | 430 mV@10 mA cm−2 | 128.34 | — | ||
| β-Ni(OH)2 | — | 194.61 | — | ||
| (Mn-F/Ni(OH)2-NF | 233 mV@20 mA cm−2 | 56.9 | 10 h@20 mA cm−2 | 1 M KOH | 162 |
| 2CoNiFe | 224 mV@10 mA cm−2 | 42 | 22 h@10 mA cm−2 | 1 M KOH | 171 |
| F-Ni(OH)2-16 h | 200 mV@10 mA cm−2 | 97.81 | 24 h@12 mA cm−2* | 1 M KOH | 172 |
| Ni(OH)2-16 h | 340 mV@10 mA cm−2 | 159.03 | — | ||
| Co(OH)F | 273 mV@10 mA cm−2 | 45 | 20 h@10 mA cm−2 | 1 M KOH | 164 |
| Co0.21Fe0.28(OH)F | 195 mV@10 mA cm−2 | 89.9 | 120 h@20 mA cm−2 | 1 M KOH | 163 |
| V–Co(OH)2 | 136 mV@10 mA cm−2 | 51.6 | 72 h@10 mA cm−2 | 1 M KOH | 173 |
| Fluorinated Ni/Fe hydroxide | 240 mV@10 mA cm−2 | 47 | 8.33 h@1.58 mA cm−2 | 0.1 M KOH | 174 |
Several approaches have been proposed for synthesizing fluorinated hydroxides. Hussain et al. synthesized ultrathin mesoporous Ni(OH)2 nanosheets by a hydrothermal method.161Fig. 10c demonstrates that α-Ni(OH)2 exhibits higher activity compared to β-Ni(OH)2, attributed to their structural variations. The introduction of F doping results in a significant decrease in the Tafel slope, from 128.34 mV dec−1 for α-Ni(OH)2 to a mere 31.89 mV dec−1, representing a 75.2% reduction. Additionally, the overpotential decreases from 430 mV for α-Ni(OH)2 to 325 mV at 10 mA cm−2. Computational studies using DFT indicate that the F-doped sample possesses lower adsorption energy, facilitating more rapid charge transfer. Fluorinated NiFe binary hydroxide nanosheets were also synthesized using the hydrothermal method with the addition of NH4F.15,175
As illustrated in Fig. 10d, F-doped α-Ni(OH)2 flakes were formed on a flat Ni foam surface through a hydrothermal reaction using Ni nitrate and urea precursors.162 Subsequently, a secondary reaction occurred where manganese nitrate was introduced to the precursor, resulting in the formation of Mn2+ and F− co-doped Ni(OH)2 flakes. These flakes were grown perpendicular to the Ni foam surface (Fig. 10e). The F doping could reduce the band gap and improve the conductivity and introduce oxygen vacancies to facilitate the OER. Patil et al. synthesized ultrathin 2D Ni(OH)2 nanosheets on 3D Ni foam through a hydrothermal route, where the Ni foam served as the Ni source and added NH4F was the F source.172 The F-doped Ni(OH)2 sample exhibited a well-defined nanosheet structure. The study revealed that F–Ni(OH)2 necessitates only 280 mV to achieve 50 mA cm−2, whereas unmodified Ni(OH)2 requires 320 mV. Additionally, the F-doped specimen exhibited a reduced Tafel slope of 97.81 mV dec−1, in contrast to the 126.64 mV dec−1 observed for the unmodified sample. According to DFT calculations, the introduction of fluorine doping resulted in a reduction of the bandgap from 2.19 eV to 1.25 eV. It was found that the Ni atoms located near the F dopants contribute to the improved electrical conductivity, while the F itself does not directly contribute. Instead of using flat NF, another study by Li et al. demonstrates the growth of CoFeOHF nanosheets on 3D NF via a one-step hydrothermal process, as shown in Fig. 10f.163 Regular nanosheets were formed on the NF, as imaged in Fig. 10g and h.
Rajesh et al. reported the synthesis of cobalt fluoride hydroxide, Co(OH)F, using nickel nitrate and NH4F as precursors in a hydrothermal reaction.164 The synthesized material displayed different crystallite shapes, ranging from 0.3 to 0.5 μm in size (Fig. 10i). Co(OH)F has an orthorhombic structure, which differs from that of Co(OH)2 and exhibits active OER performance (as shown in Table 5). Recently, a liquid phase deposition method was used to synthesize F-doped α-Ni(OH)2.176
(2) Fluorinated layered double hydroxides with mixed valence. In the structure of divalent α-Ni(OH)2·xH2O, the Ni(OH)6 octahedral layer is neutral and the interlamellar layer contains only neutral water molecules (Fig. 10b). In the structure of layered double hydroxides (LDHs), a portion of the M2+ divalent metal cations undergo isomorphous substitution with M3+ trivalent metal cations. This structural arrangement can be represented by a general formula [M1−x2+Mx3+(OH)2−]·[Ax/nn−·mH2O]. The expression contains two components: the initial bracket denotes the octahedral layer resembling brucite, which carries a positive charge of x+. The subsequent bracket signifies the intercalated layer, bearing a negative charge of x− to balance the positive charge from the octahedral layer. The value of x is the molar ratio of M2+/(M2+ + M3+), typically ranging from 0.2 to 0.33.177 An example of CoFe LDHs is shown in Fig. 11a, expressed as [Co182+Fe63+(OH)48−] [3(CO3)2− 12H2O] in a unit cell, where negatively charged (CO3)2− and neutral water molecules fill the space between the positively charged octahedral layers.181
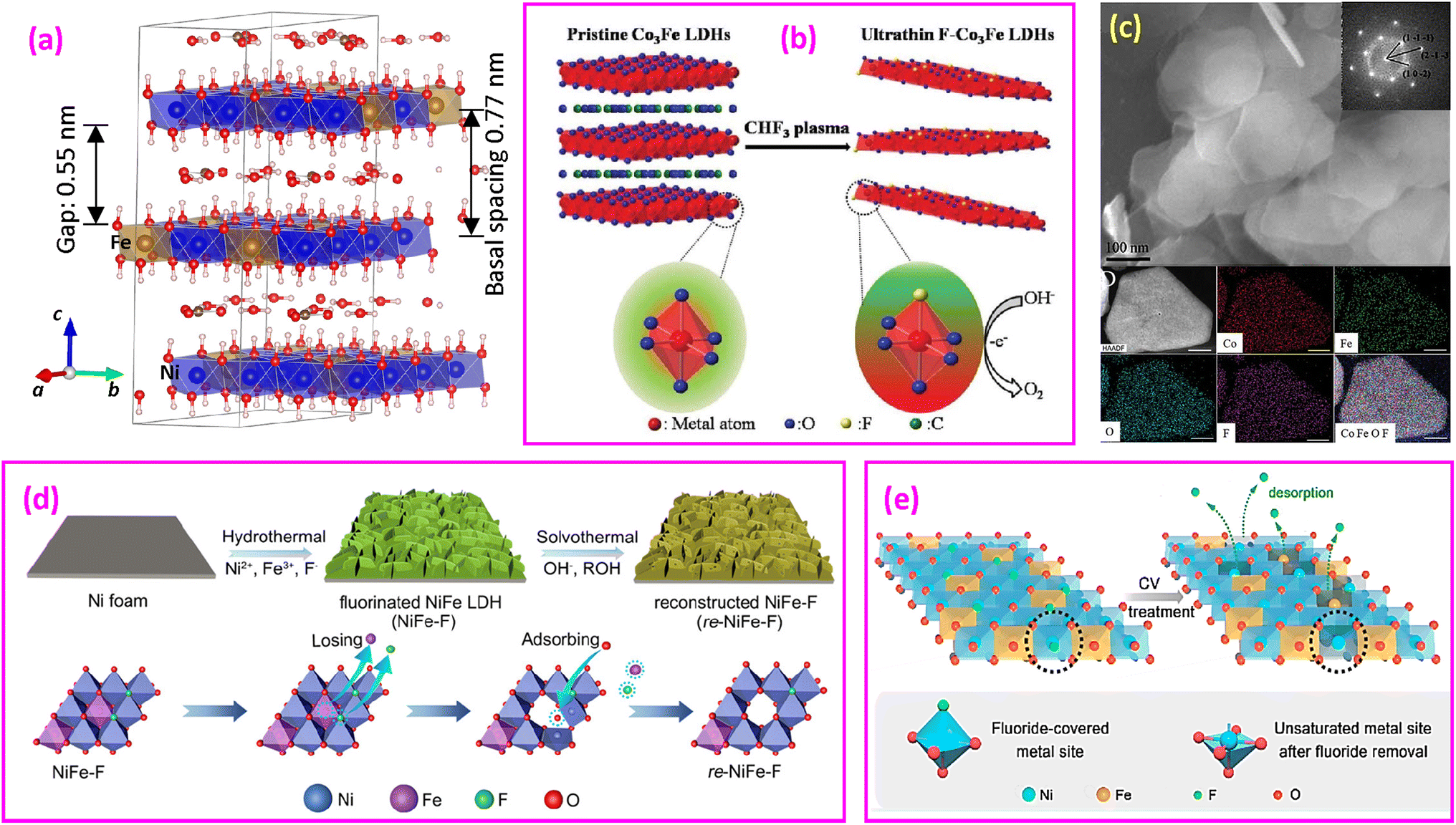 | ||
| Fig. 11 (a) Structural model of NiFe LDH. (b) Synthesis of F-doped Co3Fe LDHs and (c) TEM image (upper) and EDS maps (bottom) of Co3Fe LDHs. Reproduced from ref. 178 with permission from the Royal Society of Chemistry, copyright 2019. (d) Structural reconfiguration process to obtain reconstructed NiFe–F (re-NiFe–F). Reproduced from ref. 179 with permission from American Chemical Society, copyright 2023. (e) Fluorine pre-covered surface strategy, where F ions are removed by CV scans to improve the OER. Reproduced from ref. 180 with permission from American Chemical Society, copyright 2020. | ||
The OER activities of fluorinated LDHs are listed in Table 6.
| Catalyst | Overpotential at specific current density | Tafel slope (mV dec−1.) | Tested durability | Electrolyte | Source |
|---|---|---|---|---|---|
| a (*) Estimated in this study. | |||||
| F-Co3Fe LDH | 287 mV@10 mA cm−2 | 39.17 | — | 1 M KOH | 178 |
| Co3Fe LDH | 329 mV@10 mA cm−2 | 62.87 | — | 1 M KOH | |
| Fe-Ni-F-250 | 225 mV@10 mA cm−2 | 79 | 10 h@10 mA cm−2 | 1 M KOH | 182 |
| FeNi-LDH | 316 mV@10 mA cm−2 | 101 | — | 1 M KOH | |
| F-NiAl LDH-NF | 250 mV@10 mA cm−2 | 77 | 12 h@28.6 mA cm−2* | 1 M KOH | 31 |
| NiAl LDH-NF | 300 mV@10 mA cm−2 | 132 | — | 1 M KOH | |
| NiFe LDH-0.20 M | 243 mV@10 mA cm−2 | 50 | 10 h@1.580 V | 1 M KOH | 180 |
| NiFe LDH-0 M | 323 mV@10 mA cm−2 | 77* | — | 1 M KOH | |
| Fe-CoF2-300 | 230 mV@10 mA cm−2 | 41.9 | 10 h@10 mA cm−2 | 1 M KOH | 183 |
| CoFe LDH | 290 mV@10 mA cm−2 | 72.7 | — | 1 M KOH | |
| F-FeCoNi-Ov LDH/NF | 243.9 mV@50 mA cm−2 | 57.8 | 50 h@1.5 V | 1 M KOH | 184 |
| F-FeCoNi LDH/NF | 257.1 mV@50 mA cm−2 | 109.6 | — | 1 M KOH | |
| FeCoNi LDH/NF | 273.3 mV@50 mA cm−2 | 126.2 | — | 1 M KOH | |
| re-NiFe-F-CV | 152 mV@10 mA cm−2 | 92.1 | 100 h@10 mA cm−2 | 1 M KOH | 179 |
| NiFe-F-CV | 194 mV@10 mA cm−2 | 106.3 | — | 1 M KOH | |
| NiFe-F | 277 mV@10 mA cm−2 | 112.0 | — | 1 M KOH | |
| FeCoNi LDH/NF-3.0 mM | 196 mV@10 mA cm−2 | 22 | 120 h@100 mA cm−2 | 1 M KOH | 185 |
| FeCoNi LDH/NF-pristine | 243 mV@10 mA cm−2* | 39 | — | 1 M KOH | |
| 10F-NiCo | 240 mV@10 mA cm−2 | — | — | 1 M KOH | 186 |
| 0F-NiCo | 303 mV@10 mA cm−2 | — | — | 1 M KOH | |
Fig. 11b demonstrates the application of a CHF3-plasma etching method to incorporate F into ultrathin Co3Fe LDHs.178 The F-doped F-Co3Fe LDHs exhibit an overpotential of 287 mV to afford 10 mA cm−2, while the pristine Co3Fe LDHs exhibit a higher overpotential of 329 mV. Compared to the pristine sample with a Tafel slope of 62.87 mV dec−1, the fluorine-doped specimen exhibits a reduced Tafel slope of 39.17 mV dec−1. The layers along the c-axis exhibit nearly hexagonal shapes (Fig. 11c). It is believed that F ions filled oxygen vacancies to form metal–fluorine bonds, which can strongly modulate the electronic structure, facilitating the adsorption of OER intermediates and decreasing the reaction barriers. Tong et al. synthesized NiAl LDHs on NF and then fluorinated NiAl LDHs for comparison.31 The F-doped NiAl LDHs exhibited more active OER properties than the non-fluorinated ones. During the OER process, with the leaching of F, the metal–F bonds converted to form highly active Ni–OOH species, which enhances the efficiency of the oxygen evolution reaction (OER).31
As illustrated in Fig. 11d, a hydrothermal method was employed to cultivate F–NiFe LDHs on a nickel foam substrate.179 Subsequently, the precursor underwent a solvothermal process, resulting in the formation of reconstructed NiFe LDHs. The role of F was to facilitate surface reconstruction during the solvothermal processing, which involved the extraction of some metal ions (primarily Fe3+) and fluoride anions. This process generated voids and unsaturated coordination sites, facilitating the OER. The reconstructed product was then electrochemically activated by 30 CV scans at room temperature (denoted as re-NiFe LDH-F-CV). The reconstruction and CV activation significantly enhanced OER performance (Table 6). The formation of cavities following reconstruction enhances the adsorption process during the OER.
When NiFe LDHs were synthesized hydrothermally without the addition of NH4F, the resulting product exhibited a nearly spherical morphology and low degree of crystallinity,180 while with the addition of NH4F, [FeFn](n−3)− complex ions are formed, resulting in a product with an enhanced layered structure and increased crystallinity. In the case of fluorinated LDHs, F ions initially occupied the surface. To create highly active OER sites, an electrochemical process involving 2000 CV scans was employed to eliminate the surface F ions, exposing unsaturated metal sites (Fig. 11e). The sample synthesized with 0.2 M NH4F and treated by CV scans exhibits an overpotential of 243 mV@10 mA cm−2 and a Tafel slope of 50 mV dec−1, significantly lower than those of the sample without F, which had an overpotential of 323 mV and a Tafel slope of 77 mV dec−1. CV scan appears to be an effective way to boost the OER.
Yang et al. prepared LDHs with three metal cations Co2+, Ni2+ and Fe3+, with a Co/Ni/Fe ratio of about 1:1:2, and added different concentrations of NH4F in a solvothermal reaction.185 The research revealed that at a NH4F concentration of 3.0 mM, the catalyst demonstrated remarkable performance. It achieved a current density of 10 mA cm−2 with an exceptionally low overpotential of 196 mV, while exhibiting a Tafel slope as minimal as 22 mV dec−1. It was experimentally found that the F− ions leached gradually during the OER, promoting the formation of high-valent intermediates at the Co active site to enhance the catalyst activity. A recent DFT study reveals the interaction between the doped F in NiCo LDH and molecular orbitals.186 The fluorine atoms alter the active site by influencing the oxidation state of the metallic elements. A series of fluorinated NiCo LDHs were synthesized, and the lowest overpotential was recorded at 240 mV at 10 mA cm−2.
5.2 Fluorinated oxyhydroxides
Metal hydroxides and oxyhydroxides can transform under certain conditions. In the nickel metal system, there are various phases such as α-Ni(OH)2, β-Ni(OH)2, β-NiOOH, and γ-NiOOH.187 The Bode diagram167 of this system is shown in Fig. 12a, which outlines these phase transformations under different conditions.187,188 The OER activities of oxyhydroxides and carbonate hydroxides are listed in Table 7. | ||
| Fig. 12 (a) Bode diagram representing the transformation of various nickel (oxy)hydroxides. Reproduced from ref. 188 with open access. (b) Synthesis of F-doped NiOOH/Ni(OH)2/NF and (c) TEM image showing NiOOH on the surface. Reproduced from ref. 189 with permission from Elsevier, copyright 2023. (d) Synthetic process of Ni1−xFexO(OH)1−yFy/NiO/NF and (e) TEM image showing Fe, F–NiOOH. Reproduced from ref. 190 with permission from the Royal Society of Chemistry, copyright 2023. (f) Progressive reconstruction strategy to synthesize F-doped NiOOH and (g) TEM iamges showing NiOOH. Reproduced from ref. 191 with permission from the Royal Society of Chemistry, copyright 2024. | ||
| Catalyst | Overpotential at specific current density | Tafel slope (mV dec−1.) | Tested durability | Electrolyte | Source |
|---|---|---|---|---|---|
| a (*) Estimated in this study. | |||||
| Oxyhydroxides | |||||
| F modified β-FeOOH | 360 mV@10 mA cm−2 | 74.4 | 6 h@10–30 mA cm−2 | 1 M KOH | 192 |
| β-FeOOH | 410 mV@10 mA cm−2 | 81.5 | 6 h@10–30 mA cm−2 | 1 M KOH | |
| Fe1.9F4.75·0.95H2O | 320 mV@10 mA cm−2 | 44.6 | 6 h@10–30 mA cm−2 | 1 M KOH | |
| F-CoOOH/NF | 310 mV@50 mA cm−2 | 54 | 10 h@1.55 V | 1 M KOH | 13 |
| CoOOH/NF | 370 mV@50 mA cm−2 | 83 | — | ||
| F-NHO | 280 mV@10 mA cm−2 | 107 | 24 h@20 mA cm−2 (91.3% retention) | 1 M KOH | 193 |
| NHN | 340 mV@10 mA cm−2 | 118 | 24 h@20 mA cm−2 (59.3% retention) | 1 M KOH | |
| NiFe-PBAs-F | 190 mV@10 mA cm−2 | 57 | 50 h@200 mA cm−2 | 1 M KOH | 35 |
| NiFe-PBAs-Ar | 290 mV@10 mA cm−2 | 66.7 | — | 1 M KOH | |
| FeNiCo anodized 250 s | 260 mV@10 mA cm−2 | 42 | 200 h@100 mA cm−2 | 1 M KOH | 194 |
| As received | 342 mV@10 mA cm−2 | 40 | 81 h@100 mA cm−2* | 1 M KOH | |
| NiyFexOOH-20F | 259 mV@100 mA cm−2 | 60.3 | 24 h@50–200 mA cm−2 | 1 M KOH | 195 |
| NiyFexOOH-0F | 320 mV@100 mA cm−2 | 105.6 | — | 1 M KOH | |
| Ni1−xFexO(OH)1−yFy/NiO/NF | 186 mV@10 mA cm−2 | 32 | 100 h@500 mA cm−2 | 1 M KOH | 190 |
| Ni1−xFexOOH/NiO/NF | 225 mV@10 mA cm−2 | 55 | — | ||
| R′-NiF2 (NiOOH) | 228 mV@10 mA cm−2 | 54.3 | 15 days@10–100 mA cm−2 | 1 M KOH | 43 |
| Ni(OH)2·0.75H2O | 332 mV@10 mA cm−2 | 96.4 | — | 1 M KOH | |
| Fe–F–NiOxHy | 322 mV@10 mA cm−2 | — | — | 1 M KOH | 196 |
| Fe–NiOxHy | 337 mV@10 mA cm−2 | — | — | 1 M KOH | |
| F-NiOOH/Ni(OH)2/NF | 268 mV@10 mA cm−2 | 39 | 100 h@100 mA cm−2 | 1 M KOH | 189 |
| NiOOH/Ni(OH)2/NF | 300 mV@10 mA cm−2 | 89 | — | ||
| NH4NiF3/NF-AO | 240 mV@10 mA cm−2 | 59.5 | 400 h@20–200 mA cm−2 | 1 M KOH | 191 |
| Ni(OH)2/NF | 368 mV@10 mA cm−2 | 126.1 | — | 1 M KOH | |
|
|||||
| Carbonate hydroxides | |||||
| F-(MnCo)(CO3)0.5(OH)0.11·H2O | 240 mV@10 mA cm−2 | 78 | 24 h@10 mA cm−2 | 1 M KOH | 197 |
| (MnCo)(CO3)0.5(OH)0.11·H2O | 270 mV@10 mA cm−2 | 219 | — | 1 M KOH | |
Fluorinated oxyhydroxide has been reported to be synthesized via anodic electrooxidation. As shown in Fig. 12b, in the first step, Ni(OH)2 nanostructures on NF are synthesized through a hydrothermal route.189 Following this, the surface of Ni(OH)2 undergoes anodic electrooxidation in an alkaline solution containing fluorine. This process results in the formation of an F-doped Ni oxyhydroxide layer. As illustrated in the TEM image in Fig. 12c, this layer is typically amorphous and semicontinuous and has a thickness of less than 3 nm. Compared to the undoped sample, F-NiOOH/Ni(OH)2/NF showed a reduction in overpotential by 32 mV, a Tafel slope reduction by 50 mV dec−1, and significantly improved stability over 100 h. According to DFT calculations, F doping enhances the transfer of electrons from nickel to neighboring fluorine atoms. This leads to stronger OH adsorption and improves the rate-limiting step of deprotonation.
In another comparative study, F-doped Ni hydroxide was synthesized through an NH4F-mediated hydrothermal route, resulting in crystalline Ni3O2(OH)4.193 This sample requires only 230 mV to maintain 10 mA cm−2, with a Tafel slope of 107 mV dec−1, while the F-free sample, Ni3(OH)4(NO3)2, required 340 mV to maintain 10 mA cm−2, with a Tafel slope of 118 mV dec−1. The incorporation of F anions increased the Ni3+ content, hydrophilicity, and structural stabilities for the OER. F doping also improved the OER activity of NiOxHy.196
Starting from a Co-based precursor, Co(CO3)xOHy on NF, Chen et al. synthesized F-doped and undoped Co3O4 nanoparticles and F-doped crystalline CoOOH nanosheets for comparative studies.13 Materials doped with F demonstrated enhanced catalytic performance (Table 7), which was attributed to the distribution of F anions on the catalyst's surface and the resulting increase in hydrophilicity. DFT calculations reveal that F-doped crystals possess increased DOS at the conduction band edge compared to unmodified crystals, indicating that more carriers can be effectively transferred to the conduction band. An alternative synthesis method was reported using an anodized commercial FeNiCo alloy.194 The anodized layer containing (FeNiCo)F2 readily converts into a highly active porous oxyhydroxide (FeNiCo)OOH during anodic polarization in KOH. After only five CV cycles, the composition largely changed to F-doped oxyhydroxide.
An approach to co-dope Fe and F into nickel oxyhydroxide is illustrated in Fig. 12d.190 First, Ni(OH)2 is formed on NF. After calcination, NiO is formed on NF. Finally, anodic electrooxidation is conducted to produce Ni1−xFexO(OH)1−yFy, i.e., Fe and F doped NiOOH, in the form of an amorphous layer with 2 nm thickness on the surface, as shown in the TEM image in Fig. 12e. The optimized catalyst, Ni1−xFexO(OH)1−yFy/NiO/NF, exhibited excellent OER performance with a low overpotential of 186 mV to reach 10 mA cm−2, and a long stability over 100 h at 500 mA cm−2.
Recently, a progressive reconstruction method was proposed to synthesize F-doped NiOOH.191Fig. 12f illustrates the process of growing Ni(OH)2 nanosheets on NF through hydrothermal synthesis. Subsequently, Ni(OH)2/NF underwent heating at 300 °C for 30 min in molten ammonium fluoride, resulting in NH4NiF3/NF formation. The final step involved in situ activation via anodic oxidation, yielding the desired F-doped NiOOH/NF. TEM images in Fig. 12g confirm that the end product is crystalline γ-NiOOH. This fabricated catalyst showed favorable OER performance with a low overpotential of 240 mV to afford 10 mA cm−2 and maintained stability at 20–200 mA cm−2 for over 300 h.
Interestingly, phase transitions between hydroxides, fluorides, and oxyhydroxides were recently identified.43 Using hydrothermal synthesis, flower-like Ni(OH)2·0.75H2O was obtained. After fluorination at 450 °C for 1 h in the presence of NH4, the sheet-like hydroxide converted into cubic NiF2 nanoparticles. After CV testing the fresh NiF2 under HER and OER conditions, NiF2 transformed into Ni(OH)2 and NiOOH, respectively.
5.3 Fluorinated carbonate hydroxide
Shamloofard and Shahrokhian synthesized MnCo carbonate hydroxide (MnCo)(CO3)0.5(OH)0.11·H2O on graphite paper, using NH4F as the fluorine source to control the product's shape.197 The unaltered MnCo carbonate hydroxide displayed spherical formations made up of needle-like structures. However, when NH4F was introduced, the morphology transformed into cubic and pyramidal shapes. The F-doped material showed an overpotential of 240 mV to reach 10 mA cm−2 and a Tafel slope of 78 mV dec−1, which represented improvements compared to the pristine materials, which had an overpotential of 270 mV and the same Tafel slope of 78 mV dec−1. The sample's morphology, along with increased metal–F ionicity and chemical bond polarity, contributed to the improved the OER activities.1976. Fluorinated carbides, nitrides, phosphides and sulfides
Fluorinated carbides, nitrides, phosphides and sulfides have been reported for the OER, and their representative OER activities are listed in Table 8.| Catalyst | Overpotential at specific current density | Tafel slope (mV dec−1) | Tested durability | Electrolyte | Source |
|---|---|---|---|---|---|
| a (*) Estimated in this study. | |||||
| F-doped carbides | |||||
| TiTaFxC2 NP/rGO | 280 mV@10 mA cm−2 | 36 | 40 h@1.6 V | 1 M HClO4 | 198 |
| TiTaC2 NP/rGO | 420 mV@10 mA cm−2 | 106 | — | 1 M HClO4 | |
|
|||||
| F-doped nitrides | |||||
| PF/Ni1.5Co1.5N | 280 mV@10 mA cm−2 | 66.1 | 20 h@10 mA cm−2* | 1 M KOH | 199 |
| Ni1.5Co1.5N | 350 mV@10 mA cm−2 | 78.6 | — | 1 M KOH | |
|
|||||
| F-doped phosphides | |||||
| F-CoP NAs/CF | 231 mV@50 mA cm−2 | 73.19 | 100 h@50 mA cm−2 | 1 M KOH | 37 |
| CoP Nas/CF | 378 mV@50 mA cm−2 | 147.26 | — | ||
| Ni2P:5F | 230 mV@50 mA cm−2 | 28.06 | 24 h@1.55 V (95% retention) | 1 M KOH | 36 |
| Ni2P | 370 mV@50 mA cm−2 | 37.43 | 24 h@1.55 V (50% retention) | 1 M KOH | |
| F-Co2P/NF | 307 mV@50 mA cm−2 | 120 | 50 h@500–1000 mA cm−2 | 1 M KOH | 200 |
| Co2P/NF | 338 mV@50 mA cm−2 | 123 | — | ||
| F-NC2AL@ZFAL/NF | 177 mV@10 mA cm−2 | 67 | 20 h@10 mA cm−2 | 1 M KOH | 201 |
| Zn/F-NiCoP/NF | 285 mV@50 mA cm−2 | 78.33 | 40 h@10 mA cm−2 | 1 M KOH | 202 |
| Zn-NiCoP/NF | 331 mV@50 mA cm−2 | 102.84 | — | 1 M KOH | |
| F-FeCoPV@IF | 370 mV@1000 mA cm−2 | 118 | 20 h@100 mA cm−2 | 1 M KOH + sea water | 203 |
| Ni2P-F3 | 261 mV@50 mA cm−2 | 52.1 | 24 h@50 mA cm−2 | 1 M KOH | 204 |
| Ni2P | 412 mV@100 mA cm−2 | 168.8 | |||
|
|||||
| F-doped sulfides | |||||
| Ni(OH)2/F-Ni3S2/NF (FN-20) | 360 mV@100 mA cm−2 | 126 | 20 h@1.7 V (94.9% retention) | 1 M KOH | 205 |
| Pristine Ni3S2 | 580 mV@100 mA cm−2 | 211 | 20 h@1.7 V (20.9% retention) | ||
| F-Ni3S2/NF | 358 mV@10 mA cm−2 | — | 30 h@1.6 V | 1 M KOH | 206 |
| Fe–CoS2–F | 298 mV@10 mA cm−2 | 46.0 | 100 h@1.53 V | 1 M KOH | 207 |
| Fe–CoS2 | 345 mV@10 mA cm−2 | 64.2 | — | 1 M KOH | |
| CoS2–F | 350 mV@10 mA cm−2 | 107.6 | — | 1 M KOH | |
| CoS2 | 363 mV@10 mA cm−2 | 110.9 | — | 1 M KOH | |
| F-NiPx/Ni3S2-NF | 370 mV@100 mA cm−2 | 92 | 24 h@1.55 V | 1 M KOH | 208 |
| NiPx/Ni3S2-NF | 420 mV@100 mA cm−2 | 108 | — | 1 M KOH | |
6.1 F-doped carbides
Transition-metal carbides have demonstrated appealing properties, including high conductivity, chemical stability, thermal stability, and mechanical strength, making them potential candidates for catalyst applications.209–213 However, their OER catalytic activities are generally considered low, even in alkaline media.214–216Since monometallic carbides exhibit negligible OER activities in the acidic medium, Feng et al. reported bimetallic carbides (TiTaC2) doped with F.198 As illustrated in Fig. 13a, starting from precursors K2TaF7 and K2TiF6, a hydrothermal treatment yields fluoride K2MFx on reduced graphene oxide (rGO). Further annealing at an intermediate temperature of 1100 °C produces oxyfluorides TaO2F and TiOF on rGO, whereas at a higher temperature of 1200 °C, F-doped carbide TiTaFxC2 is formed on rGO. The TiTaFxC2 nanoparticles range in size from 20 to 50 nm. The TiTaFxC2 NP/rGO catalyst exhibits an overpotential of 280 mV at 10 mA cm−2, significantly lower than non-doped TiTaC2 NP/rGO, which has an overpotential of 420 mV. The Tafel slope is also significantly reduced from 106 to 36 mV dec−1 (Table 8). DFT studies indicate that the F dopant lowers the activation energy in the RDS by functioning as an electron acceptor in TiTaFxC2. This process involves charge transfer from the metal and adjacent carbon atoms to fluorine (Fig. 13b). The resulting charge redistribution enhances Ti's adsorption properties, strengthening the bonding of crucial intermediates on TiTaFxC2 and promoting the oxygen evolution reaction (OER) under acidic conditions.
 | ||
| Fig. 13 (a) Synthesis of fluorinated metal carbide and (b) DFT-calculated charge density distribution. Reproduced from ref. 198 with permission from American Chemical Society, copyright 2022. (c) Synthesis of Co-based nitride catalysts and (d) DFT calculation of Gibbs free energy, with insets of P and F doped Co1.5Ni1.5N (111) structural models. Reproduced from ref. 199 with permission from John Wiley and Sons, copyright 2017. | ||
Additionally, tantalum carbide doped with F and partially oxidized to TaCxFyOz was prepared on graphitized carbon.217 The synthesis process involving K2TaF7 as a precursor initially yielded TaO2F, which subsequently transformed into TaCxFyOz following high-temperature treatment in a nitrogen atmosphere. This resulting material exhibited remarkable electrocatalytic properties for oxidizing methanol in an acidic environment.
6.2 F-doped nitrides
Compounds formed by combining transition metals with nitrogen, known as transition-metal nitrides (TMNs), are characterized by their remarkably high melting temperatures, superior mechanical attributes, exceptional electrical conduction properties, and robust structural integrity.218 In fact, TMNs exhibit surface and adsorption properties similar to those of noble metals because nitrogen atoms can modulate their electronic and geometric structures.219,220 TMNs have shown promising electrocatalytic activities in the OER.221,222Bimetallic Ni–Co nitride is an effective OER catalyst.223 Bai et al. codoped P and F into bimetallic nitride Ni1.5Co1.5N mesoporous nanorods by using an ionic liquid method (Fig. 13c).199 The doped material exhibited better OER activity than the undoped version (Table 8). Gibbs free energy calculations using DFT, shown in Fig. 13d, reveal that the largest free energy difference occurs in the second step of the OER process, which has the highest ΔG2 for both doped and undoped materials. However, the doped material, with ΔG2 = 2.25 eV, has a lower energy barrier compared to the undoped material, with ΔG2 = 3.04 eV, indicating that less overpotential is required for the doped materials.
Initially, it was believed that single-phase graphitic carbon nitride (g-C3N4) lacked the ability to perform photocatalytic overall water splitting, primarily due to its inadequate oxygen evolution reaction (OER) capabilities.224 Nevertheless, in situ observation revealed that when using a fluorinated carbon nitride (Fx-CN) catalyst synthesized through hydrothermal treatment, the fluorination process inhibits the buildup of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O by creating CF bonds on the surface.225 In single-phased g-C3N4, the robust binding of intermediate CO presents a significant challenge for overall water splitting. DFT calculations indicate an enhanced OER pathway on adjacent nitrogen atoms through C–F interactions, which reduce the energy barriers associated with the OER.
O by creating CF bonds on the surface.225 In single-phased g-C3N4, the robust binding of intermediate CO presents a significant challenge for overall water splitting. DFT calculations indicate an enhanced OER pathway on adjacent nitrogen atoms through C–F interactions, which reduce the energy barriers associated with the OER.
6.3 F-doped phosphides
Due to their distinctive electronic structure and high catalytic performance, transition-metal phosphides (TMPs) have emerged as promising catalysts for the OER.226,227 However, the intrinsic activity and durability of pristine TMPs are limited. Research on F-doped phosphide CoP has demonstrated significantly improved OER performance, reducing the overpotential of undoped CoP from 378 to 285 mV at 10 mA cm−2 and lowering the Tafel slop from 147.26 to 73.19 mV dec−1.37 Fluorination creates phosphorus vacancies (PV), leading to increased active sites and regulation of charge distribution, which enhances the OER. A molten salt and gas-phase phosphorization method was used to synthesize F-doped FeCoP nanosheets with PV.203 As shown in Fig. 14a, a piece of 3D iron foam (IF) is used as the substrate, and FeCoO@IF nanosheets are first grown in situ using a molten salt mixture of cobalt nitrate and NH4F. Afterward, phosphorization is conducted in an inert atmosphere to obtain the final product, F-FeCoPv@IF. A SEM image in Fig. 14b shows the layered structure of the material. The synthesized catalyst was used for seawater splitting (Fig. 14c), and the obtained H2/O2 volume ratio was 2:1, indicating a faradaic efficiency of 100% in the 1.0 M KOH with seawater solution (Fig. 14d).
 | ||
| Fig. 14 (a) Synthesis procedure for F-FeCoPv@IF; (b) SEM image of F-FeCoPv@IF; (c) photographs of a homemade two-electrode cell; and (d) photographs of gases produced at the anode and cathode at different times. Reproduced from ref. 203 with permission from Elsevier, copyright 2023. (e) Synthesis route for NiCoP/NF and (f and g) SEM images of NiCoP. Reproduced from ref. 201 with permission from Elsevier, copyright 2022. (h) Synthesis of Zn/F-NiCoP/NF and (i) SEM of Zn/F-NiCoP/NF. Reproduced from ref. 202 with permission from Elsevier, copyright 2023. | ||
A two-step synthesis of cross-linked NiCoP nanosheets has been reported, as depicted in Fig. 14e.201 The process began with the production of F-NiCo2Al-LDH/NF, which was subsequently phosphatized to yield NiCoAlP/NF. This was followed by an acid wash to eliminate Al species. As depicted in Fig. 14f and g, the resulting nanosheets grow perpendicular to the NF surface, creating an interconnected porous network structure. Such a structure favors charge/electron transfer for the OER. The overpotential reached as low as 177 mV for a current density of 10 mA cm−2.
Recently, Zn/F-doped NiCo-MOFs were synthesized on NF via a hydrothermal reaction, followed by phosphorization to obtain Zn/F-doped NiCoP (Fig. 14h).202 The NiCo-MOF template appears as nanoprisms (Fig. 14i). The Zn/F co-doped NiCoP exhibited better activity than single-doped or RuO2 on NF. DFT calculations reveal that Zn and F codoping optimizes the adsorption energy of active sites for reactants and intermediates, enhancing the catalytic performance. Similarly prepared F-doped CoP/Ni2P nanowires were also studied for the HER.228
6.4 Fluorinated sulfides
Transition-metal sulfides (TMSs) have emerged as promising electrocatalysts for the OER, owing to their superior electrical conductivity and inherent catalytic activity.229 F-doped TMSs have been reported for the OER. Using Ni foam, and Na2S and NH4F solutions in a hydrothermal reaction, structural evolution was observed in the synthesis of heteronanorods, as shown in Fig. 15a.205 This process involved the formation of Ni(OH)2 nanosheets after 1 h, the formation of F–Ni3S2 nanorods on Ni(OH)2 nanosheets after 5 h, and finally the development of Ni(OH)2 nanosheets/F–Ni3S2 heteronanorods on nickel foam. The catalyst required a low overpotential of 360 mV to reach 100 mA cm−2. DFT calculations revealed that the incorporation of F modulated the electron density at the Fermi level of Ni3S2, contributing to elevated electrical conductivity and charge transfer efficiency.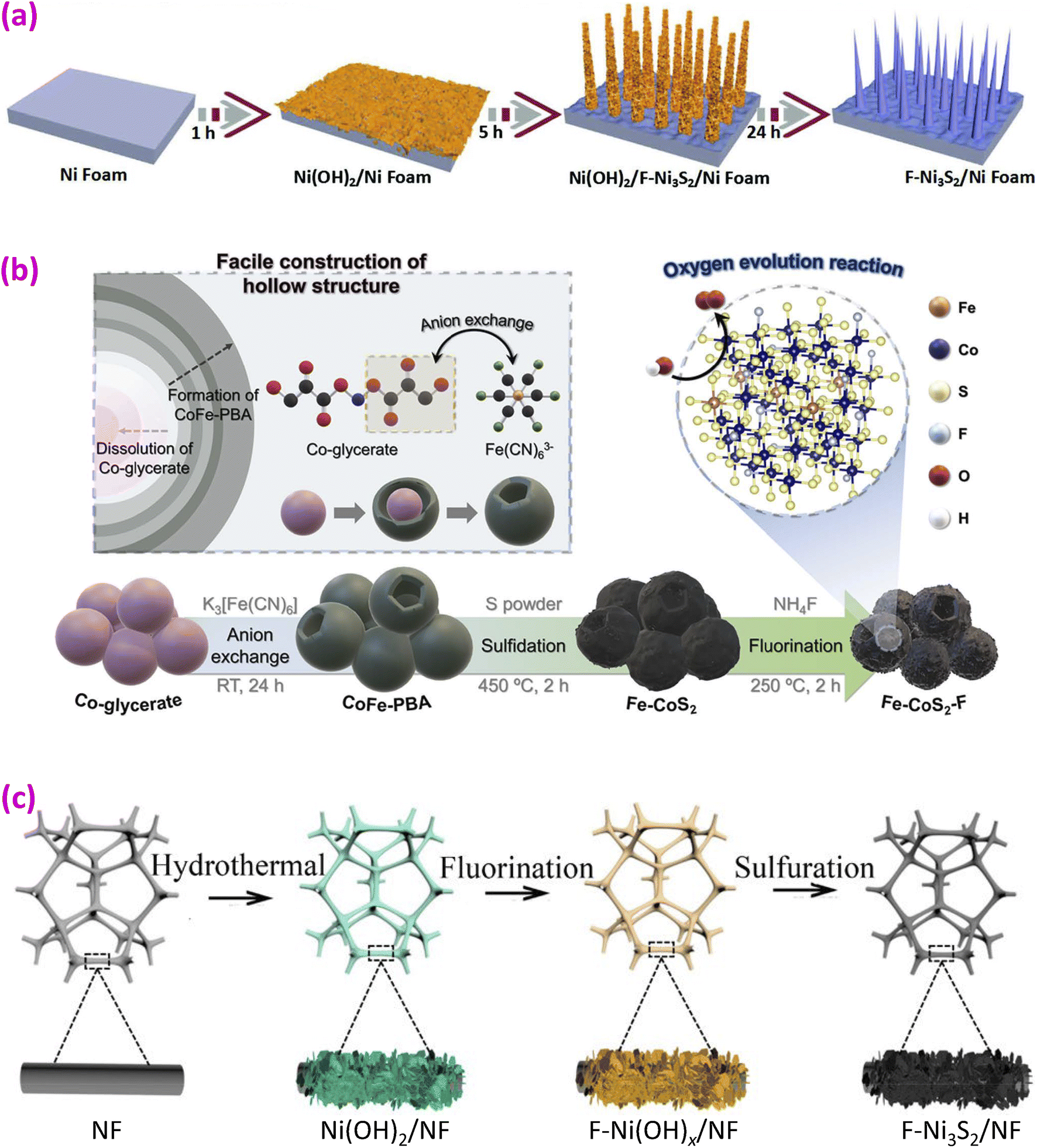 | ||
| Fig. 15 (a) Synthesis of the fabrication process of Ni(OH)2 nanosheets/F-doped Ni3S2 heteronanorods. Reproduced from ref. 205 with permission from the Royal Society of Chemistry, copyright 2018. (b) Synthesis of Fe and F dual-doped CoS2 hollow spheres using self-sacrificial templates. Reproduced from ref. 207 with permission from Elsevier, copyright 2024. (c) Synthesis of F–Ni3S2/NF. Reproduced from ref. 206 with permission from American Chemical Society, copyright 2019. | ||
A method to prepare Fe and F dual-doped CoS2 nanospheres (Fe–CoS2–F) was also reported.207Fig. 15b illustrates the process of creating Fe and F dual-doped hollow spheres. Initially, Co-glycerate nanospheres are synthesized. These solid spheres undergo a transformation into bimetallic CoFe-PBA hollow spheres through a room-temperature wet–chemical reaction lasting 24 h, utilizing K3[Fe(CN)6] and sodium citrate. Subsequently, CoFe-PBA is heat-treated with sulfur powder in an Ar environment, resulting in Fe-doped CoS2 hollow spheres (Fe–CoS2). The final step involves fluorination using NH4F, yielding Fe and F dual-doped hollow spheres (Fe–CoS2–F). The sample with dual doping demonstrated the most excellent hydrophilicity and surface wettability, as evidenced by the smallest contact angle (10.8°) of a water droplet, which is beneficial for the OER. Compared to single-doped or undoped samples, the dual-doped sample displayed the most favorable overpotential and Tafel values (Table 8). According to DFT calculations, the presence of dual dopants mitigates the excessively strong adsorption energy of reaction intermediates in the rate-determining steps, thereby improving OER performance.
In addition, F-doped Ni3S2 nanosheets have been reported, and the synthesis is depicted in Fig. 15c.206 The synthesis of F–Ni3S2 nanosheets on NF involved a multi-step process. Initially, Ni(OH)2 nanosheets were created on NF through hydrothermal synthesis. This was followed by a fluorination step to produce F–Ni(OH)x. The final stage involved hydrothermal sulfuration, resulting in F–Ni3S2 nanosheets on NF. Although F–Ni3S2/NF demonstrated superior HER performance, its OER capabilities were found to be suboptimal.
7. F-doped carbon-based catalysts
Nanocarbon materials possess exceptional properties, including superior electrical conductance, enhanced surface area, adjustable structural organization, extremely thin graphite-like layers, and characteristics associated with low dimensionality.230 These unique features make nanocarbons highly versatile for use in catalysts. However, carbon itself is generally inert for most electrochemical reactions, so surface or subsurface modifications are required to increase the binding energies of reactants and reaction intermediates on carbon.231 Carbon-based catalysts have been used for the OER, including both carbon catalysts and carbon hybrid catalysts.230–2327.1 F-doped carbon catalysts
The representative OER performances of F-doped carbon catalysts are listed in Table 9. Doped nanocarbon sheets have been used for the OER. Zhang and Dai doped graphene with N, P and F, as shown in Fig. 16a.233 The process began with the polymerization of graphene oxide (GO) to create GO coated with polyaniline (PANi), resulting in GO-PANi. Subsequently, this compound underwent pyrolysis in the presence of NH4FPF6, yielding a tri-doped material known as GO-PANI-FP, which incorporated fluorine, phosphorus, and nitrogen. SEM observations display a highly porous layered structure, as shown in Fig. 16b. This catalyst outperforms the RuO2 catalyst. Kim et al. found that F-doped carbon catalysts could enhance OER activity.235,237 Sim et al. synthesized N and F codoped graphene quantum dots (GQDs) (Fig. 16c and d), which showed an overpotential of 408 mV@10 mA cm−2, while a single N-doped GQD sample exhibited 425 mV and an undoped GQD sample showed 447 mV overpotentials.238 The F–C bonding enhances the OER performance of the GQDs. In another study, N and F codoped porous graphene nanosheets (NFPGNSs) demonstrated improved OER compared to N single-doped nano-graphene (NNG),234 as listed in Table 9.| Catalyst | Overpotential at specific current density | Tafel slope (mV dec−1) | Tested durability | Electrolyte | Source |
|---|---|---|---|---|---|
| a (*) Estimated in this study. | |||||
| GO-PANi31-FP | 460 mV@3 mA cm−2* | 136 | 10 h@10 mA cm−2 | 0.1 M KOH | 233 |
| GO-PANi-950 | 630 mV@3 mA cm−2* | 221 | — | ||
| NFPGNS | 340 mV@3 mA cm−2* | 78 | — | 1 M KOH | 234 |
| NNG | 490 mV@3 mA cm−2* | 109 | — | 1 M KOH | |
| F-doped carbon black | 150 mV@0.5 mA cm−2* | — | — | 0.1 M KOH | 235 |
| Carbon black | 270 mV@0.5 mA cm−2* | — | — | 0.1 M KOH | |
| p-FGDY/CC | 460 mV@10 mA cm−2 | 128 | 9 h@10 mA cm−2 | 1.0 M KOH | 236 |
| F-doped carbon | — | 183 | — | 1 M KOH (pH = 14) | 237 |
| N,F-GQDs | 408 mV@10 mA cm−2 | — | 22 h@1.6 V | 1 M KOH | 238 |
| N-DQDs | 425 mV@10 mA cm−2* | — | — | 1 M KOH | |
| GQDs | 447 mV@10 mA cm−2* | — | — | 1 M KOH | |
| F-doped CNTs | 280 mV@10 mA cm−2 | 76 | 10 h@15 mA cm−2 | 1 M KOH | 239 |
| Pristine CNTs | 340 mV@10 mA cm−2 | 75 | — | 1 M KOH | |
| C-NBF-G | 333 mV@10 mA cm−2 | 114 | — | 0.1 M KOH | 240 |
| FCl-CQDs/VG | 393 mV@10 mA cm−2 | 296 | 10 h@1.5 V | 1 M KOH | 241 |
| N, B, and F doped PCNFs | 280 mV@10 mA cm−2 | 83 | 50 h@10 mA cm−2 | 1 M KOH | 242 |
| PCFN-800 | 280 mV@10 mA cm−2 | 43 | — | 1 M KOH | 243 |
 | ||
| Fig. 16 F-doped carbon catalysts for the OER. (a) Preparation process of a GO-PANi-FP tri-functional catalyst and (b) SEM image of GO-PANi-FP. Reproduced from ref. 233 with permission from John Wiley and Sons, copyright 2016. (c) Synthesis of N and F codoped GQDs and (d) TEM image of a GQD. Reproduced from ref. 238 with permission from Elsevier, copyright 2020. (e) Photograph of porous fluorographdiyne networks on a carbon cloth p-FGDY/CC material. Reproduced from ref. 236 with permission from John Wiley and Sons, copyright 2019. (f) LSV plots of CNTs. Reproduced from ref. 239 with permission from Springer Nature, copyright 2021. (g) SEM of chain-like porous nanofibers. Reproduced from ref. 242 with permission from the Royal Society of Chemistry, copyright 2024. | ||
Another form of carbon, graphdiyne (GDY), a new carbon allotrope containing both sp- and sp2-hybridized carbon atoms, has been fluorinated for the OER.236 Its uneven surface charge distribution results in various active sites. The synthesis of 3D hierarchical porous fluorographdiyne networks on carbon cloth (p-FGDY/CC) was accomplished (Fig. 16e). The catalyst demonstrated exceptional performance in catalyzing the OER, the HER, and overall water splitting (OWS) reactions, showing remarkable durability across a broad spectrum of pH conditions, ranging from acidic to basic environments.
Carbon nanotubes (CNTs) can be functionalized with several dopants,244 and F doping has also been found to enhance the OER performance of CNTs.239 F-doped CNTs required only 280 mV to maintain 10 mA cm−2 current, while pristine CNTs required 340 mV (Fig. 16f). The F-doped CNTs also showed a lower Tafel slope. Surface modification using the more electronegative fluorine atoms created semi-ionic C–F and covalent C–F active sites, which improved OER efficiency.
Recently, Muthurasu et al. synthesized N, B and F tri-doped chain-like porous carbon nanofibers (PCNFs) using an electrospinning method, followed by stabilization (280 °C) and carbonization (1200 °C) in a furnace, as shown in Fig. 16g.242 The codoping enhanced the OER performance, surpassing that of the RuO2 catalyst.
7.2 F-doped carbon hybrid catalysts
Integrating TMs or TM compounds with carbon as hybrid catalysts can significantly enhance OER performance.245–247 The representative OER performance of F-doped TM/carbon hybrid catalysts are listed in Table 10. Fluorinated TM compounds combined with porous carbon forms, such as nanofibers,248 carbon encapsulated layers,249–251,255 graphene sheets,252,254,257,259 graphene quantum dots,256 carbon nanotubes,253 and nanocellulose-cornstalk aerogels,258 have been used for the OER.| Catalyst | Overpotential at specific current density | Tafel slope (mV dec−1) | Tested durability | Electrolyte | Source |
|---|---|---|---|---|---|
| a (*) Estimated in this study. | |||||
| FeNiF/NCF | 260 mV@10 mA cm−2 | 67 | 10 h@10 mA cm−2 | 1 M KOH | 248 |
| FeNiO/NCF | 330 mV@10 mA cm−2 | 90 | — | ||
| CoF2/NC | 294 mV@10 mA cm−2 | 70.0 | 10 h@1.52 V | 1 M KOH | 249 |
| FeNi@NC-1-8-F | 242 mV@10 mA cm−2 | 45.24 | 12 h@10–40 mA cm−2 | 1 M KOH | 250 |
| FeNi@NC-1-8 | 275 mV@10 mA cm−2 | 55.2 | — | ||
| C/O-FeNi/FeF2 | 253 mV@10 mA cm−2 | 52 | 16 h@10–20 mA cm−2 | 1 M KOH | 251 |
| WO3@F0.1-GS | 298 mV@10 mA cm−2 | 77.6 | 24 h@10 mA cm−2 | 1 M KOH | 252 |
| WO3@GS | 393 mV@10 mA cm−2 | 116.6 | Unstable | 1 M KOH | |
| CoFe@NCNTs-700-F-300 | 231 mV@10 mA cm−2 | 45.9 | 20 h@15 mA cm−2* | 1 M KOH | 253 |
| CoFe@NCNTs-700 | 290 mV@10 mA cm−2 | 56.0 | — | ||
| N,F-Co(OH)2/GO | 228 mV@10 mA cm−2 | 52.6 | 30 h@10 mA cm−2 | 1 M KOH | 254 |
| Co(OH)2/GO | 325 mV@10 mA cm−2 | 72.1 | — | 1 M KOH | |
| F-N/FeCoNC900 | 273 mV@10 mA cm−2 | 52.8 | — | 1.0 M KOH | 255 |
| GQD/F-NiF PBA | 318 mV@50 mA cm−2 | 34.7 | 30 h@100 mA cm−2 | 1.0 M KOH | 256 |
| GQD/NiF PBA | 339 mV@50 mA cm−2 | 41.79 | — | ||
| CoFeF-rGO | 245 mV@10 mA cm−2 | 90 | 10 h@1.475 V | 1 M KOH | 257 |
| CoFeO-rGO | 430 mV@10 mA cm−2* | 113 | — | 1 M KOH | |
| N,B,F@Co-CNF | 368 mV@10 mA cm−2 | 94.88 | — | 1 M KOH | 258 |
| CoFeNiF-rGAs | 238 mV@10 mA cm−2 | 78.8 | 20 h@10 mA cm−2 | 1 M KOH | 259 |
| Fe2Ni@NC-C-F | 247 mV@10 mA cm−2 | 44.81 | 60 h@10 mA cm−2 | 1 M KOH | 260 |
| Fe2Ni@NC-C | 314 mV@10 mA cm−2 | 71.33 | — | 1 M KOH | |
| Fe2Ni–C–F | 287 mV@10 mA cm−2 | 61.83 | — | 1 M KOH | |
| Fe2Ni–C | 337 mV@10 mA cm−2 | 70.82 | — | 1 M KOH | |
| AlF3@HPCNFs-3 | 310 mV@10 mA cm−2 | 121 | — | 1 M KOH | 45 |
| PCNFs | 520 mV@10 mA cm−2 | 405 | — | ||
| (MnNiCuCoZn)F2-PCNFs | 310 mV@10 mA cm−2 | 88.2 | 5.56 h with 94% retention | 1 M KOH | 261 |
Fluorination of N-doped porous nanofibers synthesized by electrospinning resulted in FeF2, NiF2, Ni3Fe and possibly oxides encapsulated with nanocarbon layers.248 Such hybrid catalysts exhibit high OER performances, due to the large surface area and roughness of the nanofibers. As illustrated in Fig. 17a, the selective fluorination of FeNi3 alloy oxides (NiO and Fe3O4) within N-doped porous carbon nanofibers (NiFeO/NCF) resulted in the formation of fluorides (NiF2 and FeF2) embedded in N-doped porous carbon nanofibers (FeNiF/NCF).248 The oxide catalyst exhibited an overpotential of 330 mV for 10 mA cm−2, which was reduced to 260 mV after fluorination. The Tafel slope was also reduced from 90 to 67 mV dec−1.
 | ||
| Fig. 17 F-doped carbon hybrid catalysts for the OER. (a) TEM image of FeNiF/NCF. Reproduced from ref. 248 with permission from Elsevier, copyright 2020. (b) TEM image (left) and elemental maps (right) of C/O–FeNi/FeF2. Reproduced from ref. 251 with permission from Elsevier, copyright 2022. (c) SEM image of F-N/FeCoNC900 derived from ZIF-67. Reproduced from ref. 255 with permission from Elsevier, copyright 2023. (d) Synthesis of Co-Fe-F/CNT with an inset of the SEM image of CoFe@NCNT. Reproduced from ref. 253 with permission from Elsevier, copyright 2023. (e) Gibbs free energy calculation of WO3 nanoparticles on F-doped graphite sheets. Reproduced from ref. 252 with permission from Elsevier, copyright 2023. (f) Synthesis of CoFeNiF-rGAs and (g) TEM image. Reproduced from ref. 259 with permission from Elsevier, copyright 2024. (h) TEM image of (MnNiCuCoZn)F2 nanoparticles in porous carbon nanofibers. Reproduced from ref. 261 with permission from the Royal Society of Chemistry, copyright 2024. | ||
A catalyst of carbon-confined iron–nickel alloy/iron fluoride doped with oxygen (C/O–FeNi/FeF2) is presented in Fig. 17b.251 The nanoparticles are O-doped FeNi and FeF2, embedded in carbon nanolayers, as revealed by elemental mapping on the right side of Fig. 17b. This catalyst exhibited a low overpotential of 253 mV to reach 10 mA cm−2 which is even lower than that of IrO2. ZIF-67 was used as a template to develop an Fe, N and F co-doped porous carbon catalyst, as shown in Fig. 17c.255 The catalyst was evaluated for both the ORR and OER, showing promise as a ZIF-derived bifunctional non-precious metal catalyst. N-doped CNTs were used to connect the CoFe binary alloy and fluoride nanoparticles, as shown in Fig. 17d.253 This setup achieved a low overpotential of 231 mV to drive 10 mA cm−2, while RuO2 required an overpotential of 325 mV to reach the same current density.
In WO3-decorated F-doped graphite sheets, F doping reduced the overpotential largely by 95 mV and Tafel slope by 39 mV dec−1.252 DFT calculations revealed that the determining step is the deprotonation of surface-adsorbed OH, and the F-GS sample required the lowest overpotential, as shown in Fig. 17e. Functionalized fluorographene262 could improve the OER performance of CoN4.263 F-doped graphene oxide was also used to enhance the OER.254 N, F-doped Co(OH)2/GO showed an overpotential of 228 mV for 10 mA cm−2 and a Tafel slope of 52.6 mV dec−1, whereas the undoped version had an potential of 370 mV and a Tafel slope 75.9 mV dec−1. The presence of highly electronegative fluorine in graphene oxide stabilized the Co2+ active site, enhancing the transfer of charge and adsorption processes. This resulted in improved performance of the oxygen evolution reaction (OER).254
Yu and coworkers added graphene oxide in a hydrothermal synthesis and obtained Co–Fe fluorides on graphene.257 The fluorides, composed of FeF2 and CoF2, were in nanosheet shape. This catalyst exhibited an overpotential of 245 mV for 10 mA cm−2. The same group also reported tri-metallic fluorides Co–Fe–Ni–F on reduced graphene architecture (rGA), as illustrated in Fig. 17f.259 The fluorides were identified as CoF2 and NiF2 in nanosheet form, as shown in Fig. 17g. This catalyst showed an overpotential of 238 mV for 10 mA cm−2. DFT analysis showed that the Co–Fe–Ni–F-rGA trimetallic compound exhibits enhanced electronic states close to the Fermi level, leading to superior conductivity and an elevated d-band center εd, which promotes better adsorption.
Recently, high-entropy fluoride (MnNiCuCoZn)F2 nanoparticles were synthesized in porous carbon nanofibers (PCNFs), as shown in the TEM image in Fig. 17h.261 The approach leveraged the active sites provided by the HEF and exploited the Zn component's ability to convert HEF nanoparticles from single-crystals to polycrystals, effectively enhancing electrocatalytically active sites. The (MnNiCuCoZn)F2-PCNFs catalyst exhibited exceptional performance in both OER and ORR processes.
8. Summary and concluding remarks
In this review article, we summarize the synthesis, structure, and OER performance of state-of-the-art fluorinated catalysts for the OER, including (1) transition-metal fluorides with binary, ternary, quaternary, and high-entropy systems; (2) fluoride-oxide catalysts of oxyfluorides, fluorinated oxides, and fluoride/oxide heterocatalysts; (3) fluorinated hydroxides, oxyhydroxides and carbonate hydroxides and their derived fluorides; (4) fluorinated carbides, nitrides, phosphides and sulfides; and (5) fluorinated carbon and carbon hybrid catalysts. Fluorine's exceptional electronegativity, the highest among all elements, leads to metal–fluorine bonds that are highly ionic. This characteristic allows these bonds to be readily broken down in electrolyte solutions, providing an inherent advantage for catalysts containing fluorine in the OER.This review details the OER performances of various fluorinated catalysts, emphasizing the unique role of fluorine in enhancing OER activity. The application of pure transition-metal fluorides in the OER is constrained by their poor electrical conductivity. However, incorporating heteroatomic dopants can enhance OER performance by improving electrical conductivity and structural stability. This doping approach has been successfully implemented in both binary and ternary systems. Furthermore, researchers have developed high-entropy fluoride catalysts that exhibit increased structural stability. Oxides, known for their prolonged stability, can benefit from fluorination, which may improve conductivity and lower energy barriers for electron transfer. Hydroxides and related catalysts exhibit superior OER performance due to their unique open surface structures, where fluorine can further facilitate surface reconstruction, enhancing OER activity. Additionally, fluorinated carbides, nitrides, phosphides, and sulfides have shown improved electrical conductivity, contributing to OER enhancement. Carbon-based catalysts hold high potential for OER applications owing to their large surface areas and efficient electron mobility, and their hybrid materials offer synergistic improvements in OER performance.
The fluorination of existing OER catalysts has been explored across a wide range of materials, with various fluorination methods introduced for each type of catalyst. In catalyst synthesis for the OER, creating highly porous nanostructures with accessible surface areas and active sites is essential. Fluorinated catalysts offer a notable advantage through their capacity for surface reconstruction, as evidenced by the examples in this study. As a result, fluorination emerges as a promising approach to enhance catalyst efficiency, and its potential for further improvements should be explored in OER catalyst development. Based on existing research data, fluorination has been shown to decrease the overpotential by an average of 21.6% and reduce the Tafel slope by 29.6% across various catalysts. Enhancing their OER performance through fluorination might be worth exploring for newly developed OER catalysts, provided this approach has not been previously attempted.
In contrast to the well-researched oxide-based catalysts for the OER, studies on fluorides or fluorinated catalysts are scarce and demand an extensive investigation. Creating materials with multiple elements doping to boost OER performance is complex, requiring optimization and theoretical evaluation. The review presents numerous instances demonstrating that developing heterocatalysts, rather than single catalysts, is an effective approach to enhancing the OER. Improving structural stability and durability is essential for fluorides to produce highly stable and corrosion-resistant fluorinated catalysts for OER applications. Although most reported materials have been synthesized using wet chemistry techniques, scaling up for practical applications necessitates industrial-grade precursors. Prototype development in laboratory research for these applications is crucial and requires collaboration across disciplines.
Data availability
This review article is based entirely on publicly available data sourced from published literature. All the data analyzed in this paper are derived from previously published studies and can be accessed through the original publications. No new experimental data were generated or analyzed for this review. All relevant sources and references are provided in the article's reference section, and the full-text articles can be accessed through the respective journals or databases. Readers interested in specific datasets or studies referenced in this review can access them directly through the provided citations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. NSF PREM project, DMR-1827731. Some of the research facilities at the author's laboratory were funded by NSF grants ECCS-1900837 and EES-2106181. The author expresses gratitude to previous team members, Drs Gibin George and Sivasankara Rao Ede, for their valuable input and engaging discussions, as well as their assistance with technical matters.References
- S. Y. Tee, K. Y. Win, W. S. Teo, L.-D. Koh, S. Liu, C. P. Teng and M.-Y. Han, Adv. Sci., 2017, 4, 1600337 CrossRef.
- A. Hayat, M. Sohail, H. Ali, T. A. Taha, H. I. A. Qazi, N. Ur Rahman, Z. Ajmal, A. Kalam, A. G. Al-Sehemi, S. Wageh, M. A. Amin, A. Palamanit, W. I. Nawawi, E. F. Newair and Y. Orooji, Chem. Rec., 2023, 23, e202200149 CrossRef CAS PubMed.
- N. Mlilo, J. Brown and T. Ahfock, Technol. Econ. Smart Grids Sustain. Energy, 2021, 6, 25 CrossRef.
- B. M. Hunter, H. B. Gray and A. M. Müller, Chem. Rev., 2016, 116, 14120–14136 CrossRef CAS PubMed.
- S. R. Ede and Z. Luo, J. Mater. Chem. A, 2021, 9, 20131–20163 RSC.
- S. Anantharaj, S. Kundu and S. Noda, Nano Energy, 2021, 80, 105514 CrossRef CAS.
- F. Zeng, C. Mebrahtu, L. Liao, A. K. Beine and R. Palkovits, J. Energy Chem., 2022, 69, 301–329 CrossRef CAS.
- G. Dong, L. Yan and Y. Bi, J. Mater. Chem. A, 2023, 11, 3888–3903 RSC.
- Z. Liu, H. Liu, X. Gu and L. Feng, Chem. Eng. J., 2020, 397, 125500 CrossRef CAS.
- K. Lemoine, J. Lhoste, A. Hémon-Ribaud, N. Heidary, V. Maisonneuve, A. Guiet and N. Kornienko, Chem. Sci., 2019, 10, 9209–9218 RSC.
- M. Li, H. Liu and L. Feng, Electrochem. Commun., 2021, 122, 106901 CrossRef CAS.
- Z. Zhang, Y. Zhu, Y. Zhong, W. Zhou and Z. Shao, Adv. Energy Mater., 2017, 7, 1700242 CrossRef.
- P. Chen, T. Zhou, S. Wang, N. Zhang, Y. Tong, H. Ju, W. Chu, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2018, 57, 15471–15475 CrossRef CAS PubMed.
- W. Wang, Y. Yang, D. Huan, L. Wang, N. Shi, Y. Xie, C. Xia, R. Peng and Y. Lu, J. Mater. Chem. A, 2019, 7, 12538–12546 RSC.
- B. Zhang, K. Jiang, H. Wang and S. Hu, Nano Lett., 2019, 19, 530–537 CrossRef CAS.
- J. Zhang, Y. Ye, Z. Wang, Y. Xu, L. Gui, B. He and L. Zhao, Adv. Sci., 2022, 9, 2201916 CrossRef CAS PubMed.
- S. Guddehalli Chandrappa, P. Moni, D. Chen, G. Karkera, K. R. Prakasha, R. A. Caruso and A. S. Prakash, ACS Appl. Energy Mater., 2021, 4, 13425–13430 CrossRef CAS.
- Q. Dong, T. Su, W. Ge, Y. Ren, Y. Liu, W. Wang, Q. Wang and X. Dong, Adv. Mater. Interfaces, 2020, 7, 1901939 CrossRef CAS.
- W. Li, A. Murisana, Q. Zhang, S. Wang and G. De, Electrochem. Commun., 2022, 141, 107363 CrossRef CAS.
- X. Fan, Y. Liu, S. Chen, J. Shi, J. Wang, A. Fan, W. Zan, S. Li, W. A. Goddard and X.-M. Zhang, Nat. Commun., 2018, 9, 1809 CrossRef PubMed.
- E. A. Dawi, M. Padervand, S. Ghasemi, S. Hajiahmadi, K. Kakaei, Z. Shahsavari, S. Karima, M. Baghernejad, M. Signoretto, Z. H. Ibupoto, A. Tahira and C. Wang, J. Water Process Eng., 2023, 54, 103979 CrossRef.
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- Y. Peng, H. Hajiyani and R. Pentcheva, ACS Catal., 2021, 11, 5601–5613 CrossRef CAS.
- Q. Liang, G. Brocks and A. Bieberle-Hütter, J. Phys.: Energy, 2021, 3, 026001 CAS.
- Q. Ma and S. Mu, Interdiscipl. Mater., 2023, 2, 53–90 CrossRef.
- X. Liu, Z. He, M. Ajmal, C. Shi, R. Gao, L. Pan, Z.-F. Huang, X. Zhang and J.-J. Zou, Trans. Tianjin Univ., 2023, 29, 247–253 CrossRef CAS.
- J. Li, W. Tian, Q. Li and S. Zhao, ChemSusChem, 2024, 17, e202400239 CrossRef CAS PubMed.
- X. Ren, Y. Zhai, N. Yang, B. Wang and S. Frank) Liu, Adv. Funct. Mater., 2024, 34, 2401610 CrossRef CAS.
- Z. Li, P. Wei and G. Wang, Energy Fuels, 2022, 36, 11724–11744 CrossRef CAS.
- Y. Liu, X. Liang, H. Chen, R. Gao, L. Shi, L. Yang and X. Zou, Chin. J. Catal., 2021, 42, 1054–1077 CrossRef CAS.
- Y. Tong, H. Mao, P. Chen, Q. Sun, F. Yan and F. Xi, Chem. Commun., 2020, 56, 4196–4199 RSC.
- C. Wang, S. Wei, F. Li, X. Long, T. Wang, P. Wang, S. Li, J. Ma and J. Jin, Nanoscale, 2020, 12, 3259–3266 RSC.
- Q. Xu, M. Chu, M. Liu, J. Zhang, H. Jiang and C. Li, Chem. Eng. J., 2021, 411, 128488 CrossRef CAS.
- G. Li, Y. Gou, R. Ren, C. Xu, J. Qiao, W. Sun, Z. Wang and K. Sun, Energy Mater. Adv., 2023, 4, 0029 CrossRef CAS.
- F. Ma, Q. Wu, M. Liu, L. Zheng, F. Tong, Z. Wang, P. Wang, Y. Liu, H. Cheng, Y. Dai, Z. Zheng, Y. Fan and B. Huang, ACS Appl. Mater. Interfaces, 2021, 13, 5142–5152 CrossRef CAS PubMed.
- S. D. Ghadge, O. I. Velikokhatnyi, M. K. Datta, K. Damodaran, P. M. Shanthi and P. N. Kumta, J. Electrochem. Soc., 2021, 168, 064512 CrossRef CAS.
- Y. Sun, W. Sun, L. Chen, A. Meng, G. Li, L. Wang, J. Huang, A. Song, Z. Zhang and Z. Li, Nano Res., 2023, 16, 228–238 CrossRef CAS.
- L. Gao, X. Cui, C. D. Sewell, J. Li and Z. Lin, Chem. Soc. Rev., 2021, 50, 8428–8469 RSC.
- Y. Zeng, M. Zhao, Z. Huang, W. Zhu, J. Zheng, Q. Jiang, Z. Wang and H. Liang, Adv. Energy Mater., 2022, 12, 2201713 CrossRef CAS.
- H. Chai, P. Wang, T. Wang, L. Gao, F. Li and J. Jin, ACS Appl. Mater. Interfaces, 2021, 13, 47572–47580 CrossRef CAS.
- Y.-N. Zhou, M.-X. Li, S.-Y. Dou, H.-Y. Wang, B. Dong, H.-J. Liu, H.-Y. Zhao, F.-L. Wang, J.-F. Yu and Y.-M. Chai, ACS Appl. Mater. Interfaces, 2021, 13, 34438–34446 CrossRef CAS.
- Q. Xu, H. Jiang, X. Duan, Z. Jiang, Y. Hu, S. W. Boettcher, W. Zhang, S. Guo and C. Li, Nano Lett., 2021, 21, 492–499 CrossRef CAS.
- P. Ji, D. Zheng, H. Jin, D. Chen, X. Luo, J. Yang, Z. Wang and S. Mu, Small Struct., 2023, 4, 2300013 CrossRef CAS.
- J. Qian, Q. Shu, B. He, Y. Hua, X. Ge, D. Sun, W. Liu, Q. Zhang and L. Zhou, Int. J. Hydrogen Energy, 2024, 68, 777–787 CrossRef CAS.
- Q. Zeng, N. Deng, G. Wang, Y. Feng, W. Kang and B. Cheng, J. Colloid Interface Sci., 2024, 654, 1063–1079 CrossRef CAS.
- Z. Xu, W. Zuo, Y. Yu, J. Liu, G. Cheng and P. Zhao, Adv. Sci., 2024, 11, 2306758 CrossRef CAS.
- J. Xie, R. Ding, Y. Li, J. Guo, Y. Zhang, Q. Fang, M. Yan, Y. He, Z. Yan, Z. Chen, X. Guo, Q. Yang, J. Luo, Y. Zhang, X. Sun and E. Liu, Nano Energy, 2024, 126, 109669 CrossRef CAS.
- W. Liu, Q. Chen, Y. Shang, F. Liu, R. He, J. Zhang, Q. Li, H. Chai, Y. Tan and S.-J. Bao, Adv. Funct. Mater., 2024, 2410325 CrossRef CAS.
- K. Zhang and R. Zou, Small, 2021, 17, 2100129 CrossRef CAS PubMed.
- Y. Xue, Y. Wang, H. Liu, X. Yu, H. Xue and L. Feng, Chem. Commun., 2018, 54, 6204–6207 RSC.
- H. Liu, M. Zha, Z. Liu, J. Tian, G. Hu and L. Feng, Chem. Commun., 2020, 56, 7889–7892 RSC.
- X. Gu, Y.-G. Ji, J. Tian, X. Wu and L. Feng, Chem. Eng. J., 2022, 427, 131576 CrossRef CAS.
- J. Xu, M. Li, B. Dong and L. Feng, Chin. Chem. Lett., 2023, 108798 Search PubMed.
- X. Han, L. Lin, C. Pei and X. Yu, Energy Fuels, 2022, 36, 2123–2129 CrossRef CAS.
- W. Du, Y. Li, L. Zhang, J. Jiang, T. Zhao, T. Xie, Y. Yao, P. Li, Y. Feng and G. Xu, ACS Appl. Nano Mater., 2023, 6, 12754–12763 CrossRef CAS.
- Y. Zhang, B. Wang, C. Hu, M. Humayun, Y. Huang, Y. Cao, M. Negem, Y. Ding and C. Wang, Chin. J. Struct. Chem., 2024, 43, 100243 CrossRef CAS.
- C. Pei, H. Chen, B. Dong, X. Yu and L. Feng, J. Power Sources, 2019, 424, 131–137 CrossRef CAS.
- Y. Li, G. Xu, J. Yang, J. Jiang, C. Wang and L. Zhang, Appl. Surf. Sci., 2021, 566, 150696 CrossRef CAS.
- R. Gao, B. Hu, Z. Fang, M. Deng, Y. Wu, Q. Yan, W. Yuan, D. Chen, W. Han and Z. Chen, Catal. Commun., 2022, 169, 106482 CrossRef CAS.
- S. E. Balaghi, S. Heidari, M. Benamara, H. Beyzavi and G. R. Patzke, Int. J. Hydrogen Energy, 2022, 47, 1613–1623 CrossRef CAS.
- X. Gu, Z. Liu, H. Liu, C. Pei and L. Feng, Chem. Eng. J., 2021, 403, 126371 CrossRef CAS.
- Y. Du, G. Hao, T. Zhao, D. Li, G. Liu, D. Zhong, J. Li and Q. Zhao, Chem. Commun., 2024, 60, 4182–4185 RSC.
- Y. Li, J. Li, X. Zhai, Y. Liu, G. Wang, X. Yang and G. Ge, ACS Appl. Energy Mater., 2022, 5, 13981–13989 CrossRef CAS.
- H. Yao, Y. Zheng, S. Yue, S. Hu, W. Yuan and X. Guo, Inorg. Chem. Front., 2023, 10, 804–814 RSC.
- T. Wang, H. Chen, Z. Yang, J. Liang and S. Dai, J. Am. Chem. Soc., 2020, 142, 4550–4554 CrossRef CAS PubMed.
- P. A. Sukkurji, Y. Cui, S. Lee, K. Wang, R. Azmi, A. Sarkar, S. Indris, S. S. Bhattacharya, R. Kruk, H. Hahn, Q. Wang, M. Botros and B. Breitung, J. Mater. Chem. A, 2021, 9, 8998–9009 RSC.
- P. Yang, Y. An, C. Feng, Y. Liu, S. Liu, L. Gao, Y. Zhou, X. Li, P. Li and F. Zeng, Int. J. Hydrogen Energy, 2024, 51, 1218–1228 CrossRef CAS.
- Z. Hao, Z. Du, T. Deng, D. Wang, Y. Zeng, S. Yu, Z. Meng, X. Hu, X. Hao and H. Tian, J. Mater. Chem. A, 2023, 11, 22884–22890 RSC.
- W. H. Baur and A. A. Khan, Acta Crystallogr. B, 1971, 27, 2133–2139 CrossRef CAS.
- M. A. Hepworth, K. H. Jack, R. D. Peacock and G. J. Westland, Acta Crystallogr., 1957, 10, 63–69 CrossRef CAS.
- S. Thundiyil, S. Kurungot and R. N. Devi, ACS Omega, 2019, 4, 31–38 CrossRef CAS.
- D. Kowalski, H. Kiuchi, T. Motohashi, Y. Aoki and H. Habazaki, ACS Appl. Mater. Interfaces, 2019, 11, 28823–28829 CrossRef CAS.
- I. Yamada, M. Kinoshita, S. Oda, H. Tsukasaki, S. Kawaguchi, K. Oka, S. Mori, H. Ikeno and S. Yagi, Chem. Mater., 2020, 32, 3893–3903 CrossRef CAS.
- S. R. Ede, C. N. Collins, C. D. Posada, G. George, H. Wu, W. D. Ratcliff, Y. Lin, J. Wen, S. Han and Z. Luo, ACS Catal., 2021, 11, 4327–4337 CrossRef CAS.
- M. Leblanc, G. Ferey and R. de Pape, Mater. Res. Bull., 1984, 19, 1581–1590 CrossRef CAS.
- E. V. Peresypkina and V. A. Blatov, Acta Crystallogr. Sect. B, 2003, 59, 361–377 CrossRef CAS PubMed.
- J. Kohl, D. Wiedemann, S. Nakhal, P. Bottke, N. Ferro, T. Bredow, E. Kemnitz, M. Wilkening, P. Heitjans and M. Lerch, J. Mater. Chem., 2012, 22, 15819–15827 RSC.
- J. Kohl, D. Wiedemann, S. I. Troyanov, E. Palamidis and M. Lerch, Dalton Trans., 2015, 44, 13272–13281 RSC.
- M. Leblanc, V. Maisonneuve and A. Tressaud, Chem. Rev., 2015, 115, 1191–1254 CrossRef CAS PubMed.
- D. H. McTaggart, J. D. Sundberg, L. M. McRae and S. C. Warren, Sci. Data, 2023, 10, 90 CrossRef CAS PubMed.
- G. George, S. R. Ede and Z. Luo, Fundamentals of Perovskite Oxides: Synthesis, Structure, Properties and Applications, CRC Press, 2020 Search PubMed.
- D. Liu, P. Zhou, H. Bai, H. Ai, X. Du, M. Chen, D. Liu, W. F. Ip, K. H. Lo, C. T. Kwok, S. Chen, S. Wang, G. Xing, X. Wang and H. Pan, Small, 2021, 17, 2101605 CrossRef CAS.
- H. J. Song, H. Yoon, B. Ju and D.-W. Kim, Adv. Energy Mater., 2021, 11, 2002428 CrossRef CAS.
- Y. Zhu, W. Zhou, Y. Zhong, Y. Bu, X. Chen, Q. Zhong, M. Liu and Z. Shao, Adv. Energy Mater., 2017, 7, 1602122 CrossRef.
- G. George, S. L. Jackson, C. Q. Luo, D. Fang, D. Luo, D. Hu, J. Wen and Z. Luo, Ceram. Int., 2018, 44, 21982–21992 CrossRef CAS.
- W. Wang, M. Xu, X. Xu, W. Zhou and Z. Shao, Angew. Chem., Int. Ed., 2020, 59, 136–152 CrossRef CAS.
- G. George, S. R. Ede and Z. Luo, Fundamentals of Perovskite Oxides: Synthesis, Structure, Properties and Applications, CRC Press, Boca Raton, Florida, 2020 Search PubMed.
- N. Han, M. Race, W. Zhang, R. Marotta, C. Zhang, A. Bokhari and J. J. Klemeš, J. Clean. Prod., 2021, 318, 128544 CrossRef CAS.
- F. Zhang, Y. Mao, T.-J. Park and S. S. Wong, Adv. Funct. Mater., 2008, 18, 103–112 CrossRef CAS.
- W. Mi, C. Dai, S. Zhou, J. Yang, Q. Li and Q. Xu, Mater. Lett., 2018, 227, 66–69 CrossRef CAS.
- W. Rüdorff, G. Lincke and D. Babel, Z. Anorg. Allg. Chem., 1963, 320, 150–170 CrossRef.
- L. Zhang, W. Cai and N. Bao, Adv. Mater., 2021, 33, 2100745 CrossRef CAS.
- Y. Sun and S. Dai, Sci. Adv., 2021, 7, eabg1600 CrossRef CAS PubMed.
- Z. Wang, J. You, Y. Zhao, R. Yao, G. Liu, J. Lu and S. Zhao, J. Environ. Chem. Eng., 2023, 11, 109080 CrossRef CAS.
- H. Han, J. Woo, Y.-R. Hong, Y.-C. Chung and S. Mhin, ACS Appl. Energy Mater., 2019, 2, 3999–4007 CrossRef CAS.
- H. Zhou, R. E. Ruther, J. Adcock, W. Zhou, S. Dai and J. Nanda, ACS Nano, 2015, 9, 2530–2539 CrossRef CAS PubMed.
- S.-W. Kim, N. Pereira, N. A. Chernova, F. Omenya, P. Gao, M. S. Whittingham, G. G. Amatucci, D. Su and F. Wang, ACS Nano, 2015, 9, 10076–10084 CrossRef CAS PubMed.
- K. Liang, L. Guo, K. Marcus, S. Zhang, Z. Yang, D. E. Perea, L. Zhou, Y. Du and Y. Yang, ACS Catal., 2017, 7, 8406–8412 CrossRef CAS.
- X. Yue, Y. Jin and P. K. Shen, J. Mater. Chem. A, 2017, 5, 8287–8291 RSC.
- N. Yamada, S. Kitano, Y. Yato, D. Kowalski, Y. Aoki and H. Habazaki, ACS Appl. Energy Mater., 2020, 3, 12316–12326 CrossRef CAS.
- K. Lemoine, Z. Gohari-Bajestani, R. Moury, A. Terry, A. Guiet, J.-M. Grenèche, A. Hémon-Ribaud, N. Heidary, V. Maisonneuve, N. Kornienko and J. Lhoste, ACS Appl. Energy Mater., 2021, 4, 1173–1181 CrossRef CAS.
- Z. Gohari-Bajestani, X. Wang, A. Guiet, R. Moury, J.-M. Grenèche, A. Hémon-Ribaud, Y. Zhang, D. Chartrand, V. Maisonneuve, A. Seifitokaldani, N. Kornienko and J. Lhoste, Chem Catal., 2022, 2, 1114–1127 CrossRef CAS.
- R. Mizuochi, K. Oka, Y. Inaguma and K. Maeda, Sustain. Energy Fuels, 2022, 6, 2423–2427 RSC.
- Y. Zhu, W. Dai, X. Zhong, T. Lu and Y. Pan, J. Colloid Interface Sci., 2021, 602, 55–63 CrossRef CAS PubMed.
- S. A. Patil, A. C. Khot, V. D. Chavan, I. Rabani, D. Kim, J. Jung, H. Im and N. K. Shrestha, Chem. Eng. J., 2024, 480, 146545 CrossRef CAS.
- S. Wang, C.-Z. Yuan, Y. Zheng, Y. Kang, K. S. Hui, K. Wang, H. Gao, D. A. Dinh, Y.-R. Cho and K. N. Hui, ACS Catal., 2024, 14, 3616–3626 CrossRef CAS.
- S. Zhang, Y. Xiao, Q. Feng and Z. Lei, ACS Appl. Nano Mater., 2024, 7, 8978–8987 CrossRef CAS.
- A. Terry, S. Mathiot, A. Guiet, E. Boivin, Z. Gohari-bajestani, V. Maisonneuve, A. Hémon-Ribaud, R. Moury, N. Kornienko and J. Lhoste, ACS Appl. Energy Mater., 2024 DOI:10.1021/acsaem.4c00259.
- M. Jain, D. Gill, S. Monga and S. Bhattacharya, J. Phys. Chem. C, 2023, 127, 15620–15629 CrossRef CAS.
- H. Wang, K. H. L. Zhang, J. P. Hofmann, V. A. de la, P. O'Shea and F. E. Oropeza, J. Mater. Chem. A, 2021, 9, 19465–19488 RSC.
- M. Chen, N. Kitiphatpiboon, C. Feng, A. Abudula, Y. Ma and G. Guan, eScience, 2023, 3, 100111 CrossRef.
- H. Sun, X. Xu, G. Chen and Z. Shao, Carbon Energy, 2024, e595 CrossRef CAS.
- C. Zhong, Z. Han, T. Wang, Q. Wang, Z. Shen, Q. Zhou, J. Wang, S. Zhang, X. Jin, S. Li, P. Wang, D. Gao, Y. Zhou and H. Zhang, J. Mater. Chem. A, 2020, 8, 10831–10838 RSC.
- C. Lyu, Y. Li, J. Cheng, Y. Yang, K. Wu, J. Wu, H. Wang, W.-M. Lau, Z. Tian, N. Wang and J. Zheng, Small, 2023, 19, 2302055 CrossRef CAS.
- K. Kadakia, M. K. Datta, O. I. Velikokhatnyi, P. H. Jampani and P. N. Kumta, Int. J. Hydrogen Energy, 2014, 39, 664–674 CrossRef CAS.
- S. D. Ghadge, P. P. Patel, M. K. Datta, O. I. Velikokhatnyi, R. Kuruba, P. M. Shanthi and P. N. Kumta, RSC Adv., 2017, 7, 17311–17324 RSC.
- S. D. Ghadge, O. I. Velikokhatnyi, M. K. Datta, P. M. Shanthi, S. Tan, K. Damodaran and P. N. Kumta, ACS Catal., 2019, 9, 2134–2157 CrossRef CAS.
- S. D. Ghadge, O. I. Velikokhatnyi, M. K. Datta, P. M. Shanthi, S. Tan and P. N. Kumta, ACS Appl. Energy Mater., 2020, 3, 541–557 CrossRef CAS.
- G. Li, X. Xu, H. Liu, X. Yang and M.-C. Lin, ChemCatChem, 2022, 14, e202201039 CrossRef CAS.
- H. Jia, X. Yang, X. Meng, G. Zhang and G. Li, New J. Chem., 2023, 47, 3658–3662 RSC.
- F. Shamsi and M. Rezaei, Colloids Surf. A Physicochem. Eng. Asp., 2023, 670, 131608 CrossRef CAS.
- L. Zhang, Q. Xu, S. Wen, H. Zhang, L. Chen, H. Jiang and C. Li, ACS Nano, 2024, 18, 22454–22464 CrossRef CAS PubMed.
- W. Xie, J. Huang, L. Huang, S. Geng, S. Song, P. Tsiakaras and Y. Wang, Appl. Catal., B, 2022, 303, 120871 CrossRef CAS.
- P. Wang, Q. Cheng, C. Mao, W. Su, L. Yang, G. Wang, L. Zou, Y. Shi, C. Yan, Z. Zou and H. Yang, J. Power Sources, 2021, 502, 229903 CrossRef CAS.
- H. Zeng, M. Oubla, X. Zhong, N. Alonso-Vante, F. Du, Y. Xie, Y. Huang and J. Ma, Appl. Catal., B, 2021, 281, 119535 CrossRef CAS.
- J.-Y. Xie, R.-Y. Fan, J.-Y. Fu, Y.-N. Zhen, M.-X. Li, H.-J. Liu, Y. Ma, F.-L. Wang, Y.-M. Chai and B. Dong, Int. J. Hydrogen Energy, 2021, 46, 19962–19970 CrossRef CAS.
- K. Xiao, Y. Wang, P. Wu, L. Hou and Z.-Q. Liu, Angew. Chem., Int. Ed., 2023, 62, e202301408 CrossRef CAS.
- B. Xiong, L. Ge, X. Lei, Y. Wang, J. Yang, W. Li, X. Li, Z. Cheng, Z. Fu and Y. Lu, Sci. China Mater., 2023, 66, 1793–1800 CrossRef CAS.
- Y.-G. Ji, J. Wu, H. Wen, S. Wang and L. Feng, Chem. Eng. J., 2024, 496, 154211 CrossRef CAS.
- X. Hong, Y. Gao, M. Ji, J. Li, L. Ding, Z. Yu and K. Chang, J. Alloys Compd., 2024, 1007, 176500 CrossRef CAS.
- J. Xiong, H. Zhong, J. Li, X. Zhang, J. Shi, W. Cai, K. Qu, C. Zhu, Z. Yang, S. P. Beckman and H. Cheng, Appl. Catal., B, 2019, 256, 117817 CrossRef CAS.
- T. Wang, J. Fan, C.-L. Do-Thanh, X. Suo, Z. Yang, H. Chen, Y. Yuan, H. Lyu, S. Yang and S. Dai, Angew. Chem., Int. Ed., 2021, 60, 9953–9958 CrossRef CAS.
- J. Ran, L. Wang, M. Si, X. Liang and D. Gao, Small, 2023, 19, 2206367 CrossRef CAS.
- K. Iwase, M. Ohtaka and I. Honma, Chem. Mater., 2023, 35, 2773–2781 CrossRef CAS.
- J. Kim, J. Lee, S. Kim and W. Jung, Electron. Mater. Lett., 2024, 20, 450–458 CrossRef CAS.
- X. Cao, Y. Hao, J. Zheng, H. Wang, Z. Lin, Y. Zhao, J. Liu, M. Zhang and Z. Shen, Energy Fuels, 2024, 38, 8095–8102 CrossRef CAS.
- I. A. Cordova, Q. Peng, I. L. Ferrall, A. J. Rieth, P. G. Hoertz and J. T. Glass, Nanoscale, 2015, 7, 8584–8592 RSC.
- X. Lang, L. Chen, Y. Zhu, X. Liu, H. Guo, T. You and H. Zhang, J. Phys. Chem. C, 2023, 127, 11932–11939 CrossRef CAS.
- P. Gayen, S. Saha and V. Ramani, Acc. Chem. Res., 2022, 55, 2191–2200 CrossRef CAS PubMed.
- G. A. Kaptagay, T. M. Inerbaev, Yu. A. Mastrikov, E. A. Kotomin and A. T. Akilbekov, Solid State Ionics, 2015, 277, 77–82 CrossRef CAS.
- B. Modak and S. K. Ghosh, J. Phys. Chem. C, 2015, 119, 7215–7224 CrossRef CAS.
- E. Fabbri, M. Nachtegaal, X. Cheng and T. J. Schmidt, Adv. Energy Mater., 2015, 5, 1402033 CrossRef.
- X. Xu, Y. Pan, W. Zhou, Y. Chen, Z. Zhang and Z. Shao, Electrochim. Acta, 2016, 219, 553–559 CrossRef CAS.
- T.-H. Shen, L. Spillane, J. Vavra, T. H. M. Pham, J. Peng, Y. Shao-Horn and V. Tileli, J. Am. Chem. Soc., 2020, 142, 15876–15883 CrossRef CAS PubMed.
- X. Xu, C. Su and Z. Shao, Energy Fuels, 2021, 35, 13585–13609 CrossRef CAS.
- X. Han, P. Liu, R. Ran, W. Wang, W. Zhou and Z. Shao, Mater. Today Energy, 2022, 23, 100896 CrossRef CAS.
- S. Wu, J. Liu, B. Cui, Y. Li, Y. Liu, B. Hu, L. He, M. Wang, Z. Zhang, K. Tian and Y. Song, Electrochim. Acta, 2019, 299, 231–244 CrossRef CAS.
- K. Huang, Z. Zhao, H. Du, P. Du, H. Wang, R. Wang, S. Lin, H. Wei, Y. Long, M. Lei, W. Guo and H. Wu, ACS Sustain. Chem. Eng., 2020, 8, 6905–6913 CrossRef CAS.
- H. Li, G. Yan, H. Zhao, P. C. Howlett, X. Wang and J. Fang, Adv. Mater., 2024, 36, 2311272 CrossRef CAS.
- P. Ji, D. Zheng, H. Jin and Y. Liao, Int. J. Hydrogen Energy, 2024, 57, 473–480 CrossRef CAS.
- S. V. Devaguptapu, S. Hwang, S. Karakalos, S. Zhao, S. Gupta, D. Su, H. Xu and G. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 44567–44578 CrossRef CAS PubMed.
- H. Yu, J. Ke and Q. Shao, Small, 2023, 19, 2304307 CrossRef CAS.
- A. Raza, J. Z. Hassan, U. Qumar, A. Zaheer, Z. U. D. Babar, V. Iannotti and A. Cassinese, Mater. Today Adv., 2024, 22, 100488 CrossRef CAS.
- T.-W. Chen, S.-M. Chen, G. Anushya, R. Kannan, P. Veerakumar, M. M. Alam, S. Alargarsamy and R. Ramachandran, Nanomaterials, 2023, 13, 2012 CrossRef CAS PubMed.
- S. Anantharaj, K. Karthick and S. Kundu, Mater. Today Energy, 2017, 6, 1–26 CrossRef.
- Y. Chen, K. Rui, J. Zhu, S. X. Dou and W. Sun, Chem.–Eur. J., 2019, 25, 703–713 CrossRef CAS PubMed.
- L. Yang, Z. Liu, S. Zhu, L. Feng and W. Xing, Mater. Today Phys., 2021, 16, 100292 CrossRef CAS.
- Z. Yu, Y. Bai, G. Tsekouras and Z. Cheng, Nano Sel., 2022, 3, 766–791 CrossRef CAS.
- B. Guo, H. Huo, Q. Zhuang, X. Ren, X. Wen, B. Yang, X. Huang, Q. Chang and S. Li, Adv. Funct. Mater., 2023, 33, 2300557 CrossRef CAS.
- S. Nagappan, A. Karmakar, R. Madhu, H. N. Dhandapani, S. S. Roy and S. Kundu, Catal. Sci. Technol., 2023, 13, 6377–6391 RSC.
- N. Hussain, W. Yang, J. Dou, Y. Chen, Y. Qian and L. Xu, J. Mater. Chem. A, 2019, 7, 9656–9664 RSC.
- J. Lv, X. Yang, K. Li, X. Chen, S. Sun, H.-Y. Zang, Y.-F. Chang, Y.-H. Wang and Y.-G. Li, Nanoscale Adv., 2019, 1, 4099–4108 RSC.
- Y. Li, X. Zhai, C. Fan, X. Chen, Y. Liu, J. Yang, L. Chen, G. Ge and J. Zhang, J. Mater. Chem. A, 2022, 10, 11774–11783 RSC.
- J. Anthuvan Rajesh, S.-H. Kang and K.-S. Ahn, Mater. Lett., 2022, 308, 131207 CrossRef CAS.
- C. Greaves and M. A. Thomas, Acta Crystallogr. Sect. B, 1986, 42, 51–55 CrossRef.
- K. Lawson, S. P. Wallbridge, A. E. Catling, C. A. Kirk and S. E. Dann, J. Mater. Chem. A, 2023, 11, 789–799 RSC.
- H. Bode, K. Dehmelt and J. Witte, Electrochim. Acta, 1966, 11, 1079–1087 CrossRef CAS.
- K. I. Pandya, W. E. O'Grady, D. A. Corrigan, J. McBreen and R. W. Hoffman, J. Phys. Chem., 1990, 94, 21–26 CrossRef CAS.
- D. S. Hall, D. J. Lockwood, C. Bock and B. R. MacDougall, Proc. R. Soc. A, 2015, 471, 20140792 CrossRef PubMed.
- W. D. Birch, A. Pring, A. Reller and H. W. Schmalle, Am. Mineral., 1993, 78, 827–834 CAS.
- K. Cysewska, M. Zając, M. Łapiński, J. Karczewski, M. K. Rybarczyk, B. Kamecki, P. Jasiński and S. Molin, Energy Technol., 2021, 9, 2100688 CrossRef CAS.
- S. J. Patil, N. R. Chodankar, S.-K. Hwang, G. S. Rama Raju, Y.-S. Huh and Y.-K. Han, Small, 2022, 18, 2103326 CrossRef CAS.
- P. Muthukumar, P. Nantheeswaran, M. Mariappan, M. Pannipara, A. G. Al-Sehemi and S. P. Anthony, Dalton Trans., 2023, 52, 4606–4615 RSC.
- L. Li, J. Wu, L. Huang, G. Lan, N. Wang, H. Zhang, X. Chen and X. Ge, New J. Chem., 2022, 46, 20490–20496 RSC.
- J. Yu, J. Li, R.-T. Gao, Y. Yang and L. Wang, Small, 2024, 20, 2310642 CrossRef CAS PubMed.
- T. Watanabe, K. Tsuchimoto, T. Fukushima, K. Murakoshi, M. Mizuhata and H. Minamimoto, Sustain. Energy Fuels, 2024, 8, 4813–4819 RSC.
- G. Fan, F. Li, D. G. Evans and X. Duan, Chem. Soc. Rev., 2014, 43, 7040–7066 RSC.
- Z. Liu, C.-L. Dong, Y.-C. Huang, J. Cen, H. Yang, X. Chen, X. Tong, D. Su, Y. Wang and S. Wang, J. Mater. Chem. A, 2019, 7, 14483–14488 RSC.
- S. Cai, H. Liu, H. Cheng, B. Sun, W. Xia, H. Hu and S. Zhou, ACS Appl. Nano Mater., 2023, 6, 7864–7872 CrossRef CAS.
- C. Wu, H. Li, Z. Xia, X. Zhang, R. Deng, S. Wang and G. Sun, ACS Catal., 2020, 10, 11127–11135 CrossRef CAS.
- F. Dionigi, Z. Zeng, I. Sinev, T. Merzdorf, S. Deshpande, M. B. Lopez, S. Kunze, I. Zegkinoglou, H. Sarodnik, D. Fan, A. Bergmann, J. Drnec, J. F. de Araujo, M. Gliech, D. Teschner, J. Zhu, W.-X. Li, J. Greeley, B. R. Cuenya and P. Strasser, Nat. Commun., 2020, 11, 2522 CrossRef.
- C. Pei, Y. Gu, Z. Liu, X. Yu and L. Feng, ChemSusChem, 2019, 12, 3849–3855 CrossRef CAS PubMed.
- M. Li, Y. Gu, Y. Chang, X. Gu, J. Tian, X. Wu and L. Feng, Chem. Eng. J., 2021, 425, 130686 CrossRef CAS.
- Z. Zhai, W. Yan and J. Zhang, Nanoscale, 2022, 14, 4156–4169 RSC.
- Y. Yang, Y. Wu, D. Guo and L. Liu, Appl. Surf. Sci., 2024, 644, 158810 CrossRef CAS.
- M.-J. Pei, Y.-K. Shuai, X. Gao, J.-C. Chen, Y. Liu, W. Yan and J. Zhang, Small, 2024, 20, 2400139 CrossRef CAS PubMed.
- A. V. der Ven, D. Morgan, Y. S. Meng and G. Ceder, J. Electrochem. Soc., 2005, 153, A210 CrossRef.
- P. W. Menezes, S. Yao, R. Beltrán-Suito, J. N. Hausmann, P. V. Menezes and M. Driess, Angew. Chem., 2021, 133, 4690–4697 CrossRef.
- J. Wang, L. A. Zhang, Y. Ren and P. Wang, Electrochim. Acta, 2023, 437, 141475 CrossRef CAS.
- J. Wang, Y. Ren and P. Wang, J. Mater. Chem. A, 2023, 11, 4619–4626 RSC.
- K. Guo, J. Jia, X. Lu, S. Wang, H. Wang, H. Wu and C. Xu, Inorg. Chem. Front., 2024, 11, 1479–1491 RSC.
- G.-F. Chen, Y. Luo, L.-X. Ding and H. Wang, ACS Catal., 2018, 8, 526–530 CrossRef CAS.
- L. Chen, J. Chang, Y. Zhang, Z. Gao, D. Wu, F. Xu, Y. Guo and K. Jiang, Chem. Commun., 2019, 55, 3406–3409 RSC.
- M. Nishimoto, S. Kitano, D. Kowalski, Y. Aoki and H. Habazaki, ACS Sustain. Chem. Eng., 2021, 9, 9465–9473 CrossRef CAS.
- B. M. Kathale, H. Xiao, S. Yang, H. Yin, T. Yu, X. Zhou, L. Qian, J. Xiao, P. Lei and X. Li, Electrochim. Acta, 2022, 406, 139831 CrossRef CAS.
- Y. Jang, S. Ha, H.-T. Lim and S. Lee, Chem. Commun., 2023, 59, 8298–8301 RSC.
- M. Shamloofard and S. Shahrokhian, Inorg. Chem., 2023, 62, 1178–1191 CrossRef CAS PubMed.
- M. Feng, J. Huang, Y. Peng, C. Huang, X. Yue and S. Huang, ACS Nano, 2022, 16, 13834–13844 CrossRef CAS.
- X. Bai, Q. Wang, G. Xu, Y. Ning, K. Huang, F. He, Z. Wu and J. Zhang, Chem.–Eur. J., 2017, 23, 16862–16870 CrossRef CAS PubMed.
- X. Chen, M. Wei and J. Zhou, J. Mater. Chem. A, 2021, 9, 22626–22634 RSC.
- Z. Hou, Y. Zhu, W. Du, X. Jia, T. Huang and K.-J. Huang, J. Ind. Eng. Chem., 2022, 112, 85–95 CrossRef CAS.
- J. Zhu, X. Zheng, C. Liu, Y. Lu, Y. Liu, D. Li and D. Jiang, J. Colloid Interface Sci., 2023, 630, 559–569 CrossRef CAS PubMed.
- J. Zhu, J. Chi, T. Cui, L. Guo, S. Wu, B. Li, J. Lai and L. Wang, Appl. Catal., B, 2023, 328, 122487 CrossRef CAS.
- X. Sun, S. Song, G. Yan, Y. Liu, H. Ding, X. Zhang and Y. Feng, Dalton Trans., 2024, 53, 8843–8849 RSC.
- P. Hao, W. Zhu, F. Lei, X. Ma, J. Xie, H. Tan, L. Li, H. Liu and B. Tang, Nanoscale, 2018, 10, 20384–20392 RSC.
- W. He, L. Han, Q. Hao, X. Zheng, Y. Li, J. Zhang, C. Liu, H. Liu and H. L. Xin, ACS Energy Lett., 2019, 4, 2905–2912 CrossRef CAS.
- H. Kim, K. Min, G. Song, J. Kim, H. C. Ham and S.-H. Baeck, J. Colloid Interface Sci., 2024, 665, 922–933 CrossRef CAS PubMed.
- K. Li, Y. Tong, D. Feng and P. Chen, J. Colloid Interface Sci., 2022, 625, 576–584 CrossRef CAS PubMed.
- S. T. Hunt, T. Nimmanwudipong and Y. Román-Leshkov, Angew. Chem., Int. Ed., 2014, 53, 5131–5136 CrossRef CAS PubMed.
- Y. Xiao, J.-Y. Hwang and Y.-K. Sun, J. Mater. Chem. A, 2016, 4, 10379–10393 RSC.
- Q. Gao, W. Zhang, Z. Shi, L. Yang and Y. Tang, Adv. Mater., 2019, 31, 1802880 CrossRef.
- X. Wu, S. Zhou, Z. Wang, J. Liu, W. Pei, P. Yang, J. Zhao and J. Qiu, Adv. Energy Mater., 2019, 9, 1901333 CrossRef.
- H. Wang, S. Zhu, J. Deng, W. Zhang, Y. Feng and J. Ma, Chin. Chem. Lett., 2021, 32, 291–298 CrossRef CAS.
- Y. Yu, J. Zhou and Z. Sun, Adv. Funct. Mater., 2020, 30, 2000570 CrossRef CAS.
- J. Chen, B. Ren, H. Cui and C. Wang, Small, 2020, 16, 1907556 CrossRef CAS PubMed.
- Y. Wang, Q. Wu, B. Zhang, L. Tian, K. Li and X. Zhang, Catalysts, 2020, 10, 1164 CrossRef CAS.
- X. Yue, C. He, C. Zhong, Y. Chen, S. P. Jiang and P. K. Shen, Adv. Mater., 2016, 28, 2163–2169 CrossRef CAS PubMed.
- R. S. Ningthoujam and N. S. Gajbhiye, Prog. Mater. Sci., 2015, 70, 50–154 CrossRef CAS.
- Y. Zhang, B. Ouyang, J. Xu, G. Jia, S. Chen, R. S. Rawat and H. J. Fan, Angew. Chem., 2016, 128, 8812–8816 CrossRef.
- Z. Meng, S. Zheng, R. Luo, H. Tang, R. Wang, R. Zhang, T. Tian and H. Tang, Nanomaterials, 2022, 12, 2660 CrossRef CAS PubMed.
- Y. Lu, Z. Li, Y. Xu, L. Tang, S. Xu, D. Li, J. Zhu and D. Jiang, Chem. Eng. J., 2021, 411, 128433 CrossRef CAS.
- L. Yang, L. Zhang, Y. Li, B.-H. Lee, J. Kim, H. S. Lee, J. Bok, Y. Ma, W. Zhou, D. Yuan, A.-L. Wang, M. S. Bootharaju, H. Zhang, T. Hyeon and J. Chen, J. Am. Chem. Soc., 2024, 146, 12556–12564 CrossRef CAS PubMed.
- X. Chen, X. Yu, G. Zhang, S. Wei, Y. Huang, H. Wang, J. Jiang, Z. Ma and Q. Li, J. Electroanal. Chem., 2024, 958, 118162 CrossRef CAS.
- G. Zhang, Z.-A. Lan and X. Wang, Chem. Sci., 2017, 8, 5261–5274 RSC.
- J. Wu, Z. Liu, X. Lin, E. Jiang, S. Zhang, P. Huo, Y. Yan, P. Zhou and Y. Yan, Nat. Commun., 2022, 13, 6999 CrossRef CAS PubMed.
- K. Liu, C. Zhang, Y. Sun, G. Zhang, X. Shen, F. Zou, H. Zhang, Z. Wu, E. C. Wegener, C. J. Taubert, J. T. Miller, Z. Peng and Y. Zhu, ACS Nano, 2018, 12, 158–167 CrossRef CAS PubMed.
- W. Wu, S. Luo, Y. Huang, H. He, P. K. Shen and J. Zhu, Mater. Chem. Front., 2024, 8, 1064–1083 RSC.
- K. Chen, Y. Li, G. Wu, Q. Wang, C. Fan, L. Zhang and S. Han, Int. J. Hydrogen Energy, 2024, 51, 1421–1428 CrossRef CAS.
- Y. Guo, T. Park, J. W. Yi, J. Henzie, J. Kim, Z. Wang, B. Jiang, Y. Bando, Y. Sugahara, J. Tang and Y. Yamauchi, Adv. Mater., 2019, 31, 1807134 CrossRef.
- P. Stelmachowski, J. Duch, D. Sebastián, M. J. Lázaro and A. Kotarba, Materials, 2021, 14, 4984 CrossRef CAS PubMed.
- L. Zhang, J. Xiao, H. Wang and M. Shao, ACS Catal., 2017, 7, 7855–7865 CrossRef CAS.
- L. Li, H. Yang, J. Miao, L. Zhang, H.-Y. Wang, Z. Zeng, W. Huang, X. Dong and B. Liu, ACS Energy Lett., 2017, 2, 294–300 CrossRef CAS.
- J. Zhang and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 13296–13300 CrossRef CAS.
- X. Yue, S. Huang, J. Cai, Y. Jin and P. K. Shen, J. Mater. Chem. A, 2017, 5, 7784–7790 RSC.
- J. Kim, R. Zhou, K. Murakoshi and S. Yasuda, RSC Adv., 2018, 8, 14152–14156 RSC.
- C. Xing, Y. Xue, B. Huang, H. Yu, L. Hui, Y. Fang, Y. Liu, Y. Zhao, Z. Li and Y. Li, Angew. Chem., Int. Ed., 2019, 58, 13897–13903 CrossRef CAS PubMed.
- J. Kim, T. Fukushima, R. Zhou and K. Murakoshi, Materials, 2019, 12, 211 CrossRef CAS PubMed.
- Y. Sim, S. J. Kim, G. Janani, Y. Chae, S. Surendran, H. Kim, S. Yoo, D. C. Seok, Y. H. Jung, C. Jeon, J. Moon and U. Sim, Appl. Surf. Sci., 2020, 507, 145157 CrossRef CAS.
- Z. Ali, M. Mehmood, J. Ahmad, A. Fatima and M. Ali, J. Appl. Electrochem., 2021, 51, 1573–1581 CrossRef CAS.
- M. Tan, Q. Wang, S. Wang, W. Liu, D. Wang, S. Luo, P. Hou, M. Zhou, Y. Zhang, S. Yan and X. Liu, J. Electrochem. Soc., 2022, 169, 096517 CrossRef CAS.
- Y. An, Z. Ren, Y. Kong, Y. Tian, B. Jiang and F. Shaik, Int. J. Hydrogen Energy, 2024, 58, 633–645 CrossRef CAS.
- A. Muthurasu, I. Pathak, D. Acharya, Y. R. Rosyara and H. Y. Kim, J. Mater. Chem. A, 2024, 12, 1826–1839 RSC.
- M. Nadeem, A. Ulfat, A. Kumar, M. H. Bhatti, F. Rabani, U. Yunus, M. Aamir, M. Sher, K. M. Alotaibi and G. Yasin, Int. J. Hydrogen Energy, 2024, 53, 457–467 CrossRef CAS.
- Z. Luo, A. Oki, L. Carson, L. Adams, G. Neelgund, N. Soboyejo, G. Regisford, M. Stewart, K. Hibbert, G. Beharie, C. Kelly-Brown and P. Traisawatwong, Chem. Phys. Lett., 2011, 513, 88–93 CrossRef CAS PubMed.
- Q. Hu, G. Li, Z. Han, Z. Wang, X. Huang, H. Yang, Q. Zhang, J. Liu and C. He, J. Mater. Chem. A, 2019, 7, 14380–14390 RSC.
- A. I. Douka, H. Yang, L. Huang, S. Zaman, T. Yue, W. Guo, B. You and B. Y. Xia, EcoMat, 2021, 3, e12067 CrossRef CAS.
- W. Li, C. Wang and X. Lu, J. Mater. Chem. A, 2021, 9, 3786–3827 RSC.
- M. Zha, C. Pei, Q. Wang, G. Hu and L. Feng, J. Energy Chem., 2020, 47, 166–171 CrossRef.
- X. Gu, C. Wu, S. Wang and L. Feng, Catal. Commun., 2022, 162, 106394 CrossRef CAS.
- Y. Zhou, H. Liu, X. Gu, X. Wu and L. Feng, Carbon Energy, 2022, 4, 924–938 CrossRef CAS.
- M. Li, S. Wang, X. Wang, X. Tian, X. Wu, Y. Zhou, G. Hu and L. Feng, Chem. Eng. J., 2022, 442, 136165 CrossRef CAS.
- K. Lu, Z. Wang, Y. Wu, X. Zhai, C. Wang, J. Li, Z. Wang, X. Li, Y. He, T. An, K. Yang, D. Yang, F. Yu and B. Dai, Chem. Eng. J., 2023, 451, 138590 CrossRef CAS.
- Y. Kuang, R. He, X. Gu, F. Yang, X. Tian and L. Feng, Chem. Eng. J., 2023, 456, 141055 CrossRef CAS.
- P. Muthukumar, P. Nantheeswaran, M. Mariappan, M. Pannipara, A. G. Al-Sehemi and S. P. Anthony, Dalton Trans., 2023, 52, 3877–3883 RSC.
- X. He, L. Chang, H. Wu, G. Liu, Y. Zhang and A. Zhou, J. Alloys Compd., 2023, 967, 171709 CrossRef CAS.
- A. M. Tarigan, S. Aulia, M. Rinawati, L.-Y. Chang, Y.-S. Cheng, C.-C. Chang, W.-H. Huang, J.-L. Chen, H. Setyawan and M.-H. Yeh, Chem. Eng. J., 2023, 476, 146754 CrossRef CAS.
- Y. Lu, X. Han, Y. Zhang and X. Yu, Nanomaterials, 2024, 14, 16 CrossRef CAS.
- D. Liu, Z. Qian, Y. Li, Y. Luo, C. Liu and J. Cui, Adv. Sustainable Syst., 2024, 8, 2300594 CrossRef CAS.
- Y. Lu, C. Pei, X. Han, Y. Li, H. S. Park, J. K. Kim and X. Yu, J. Electroanal. Chem., 2024, 957, 118108 CrossRef CAS.
- Z. Ma, J. Wu, F. Yang, S. Wang, H. Wen and L. Feng, Chem. Eng. J., 2024, 491, 152113 CrossRef CAS.
- G. Wang, H. Chi, Y. Feng, J. Fan, N. Deng, W. Kang and B. Cheng, J. Mater. Chem. A, 2024, 12, 19109–19122 RSC.
- R. A. Borse, M. B. Kale, S. H. Sonawane and Y. Wang, Adv. Funct. Mater., 2022, 32, 2202570 CrossRef CAS.
- B. B. Xiao, L. Yang, H. Y. Liu, X. B. Jiang, B. Aleksandr, E. H. Song and Q. Jiang, Appl. Surf. Sci., 2021, 537, 147846 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |