
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing sodium structures and dynamics in hard carbon for Na-ion batteries using 23Na operando solid-state NMR spectroscopy†
Matej
Gabrijelčič
 ab,
Blaž
Tratnik
a,
Gregor
Kapun
a,
Elena
Tchernychova
a,
Nataša
Zabukovec Logar
ac,
Andraž
Krajnc
a,
Robert
Dominko
ade and
Alen
Vizintin
*a
ab,
Blaž
Tratnik
a,
Gregor
Kapun
a,
Elena
Tchernychova
a,
Nataša
Zabukovec Logar
ac,
Andraž
Krajnc
a,
Robert
Dominko
ade and
Alen
Vizintin
*a
aNational Institute of Chemistry, Hajdrihova 19, 1000, Ljubljana, Slovenia. E-mail: alen.vizintin@ki.si
bFaculty of Mathematics and Physics, University of Ljubljana, Jadranska 19, 1000 Ljubljana, Slovenia
cPostgraduate School, University of Nova Gorica, Vipavska 13, 5000 Nova Gorica, Slovenia
dFaculty of Chemistry and Chemical Technology, University of Ljubljana, Večna pot 113, 1000, Ljubljana, Slovenia
eALISTORE-European Research Institute, CNRS FR 3104, Hub de l'Energie, Rue Baudelocque, 80039, Amiens Cedex, France
First published on 25th November 2024
Abstract
Hard carbons are promising negative electrode materials for Na-ion batteries (SIBs), and the process of (de)insertion of Na+ ions into/from hard carbons has attracted much attention in recent research. Being a relatively new technology compared to lithium-ion batteries, the precise operational mechanism and degradation pathways of SIBs remain elusive. In this investigation, we focus on elucidating the physical and electrochemical attributes of corncob-derived hard carbon synthesized at 1400 °C using operando, in situ and ex situ solid-state NMR spectroscopy techniques, complemented by other advanced characterization methods, such as SEM-EDX, XPS and electrochemistry. Through the analysis of 23Na NMR spectra of hard carbon, we gained insights into sodium insertion within the active material, which is characterized by the shifting NMR peak during the (de)sodiation process. Furthermore, our findings revealed the formation of a solid electrolyte interphase (SEI) at the hard carbon electrode and pore surface, predominantly in the form of Na2CO3 in the bulk of the SEI, and NaF at the SEI surface, alongside the occurrence and differentiation of plating and metal sodium dendrites.
Introduction
Na-ion batteries (SIBs) have emerged as a promising alternative to Li-ion batteries (LIBs), particularly for use in power grids, due to their safety and the availability of sodium resources.1–4 However, the development of high-performance anode and cathode materials for SIBs remains a major challenge.5–7 Hard carbon has recently emerged as a promising anode material for SIBs.8–12Hard carbons are characterized by their disordered structure, comprising randomly oriented and curved graphite-like domains with defects and nanopores, and offer intriguing properties for various applications.13 The disordered structure with a certain percentage of sp3 hybridized carbon atoms and defects that are obtained at temperatures above 1000 °C facilitates the insertion of Na+ ions.14 However, it is noted that the specific capacity for Na+ insertion decreases as the carbonization temperature increases above 1700 °C. This decrease is attributed to the transition of a disordered to a more graphitic structure, which is less permeable to Na+ ion insertion.8,15
The electrochemical performance of hard carbons is consequently strongly influenced by their microstructure, which is determined by the choice of organic precursor and carbonization temperature.16,17 Typically, the galvanostatic curves of hard carbons display two distinct regions: a sloping region above 0.1 V and a plateau region below 0.1 V.18 While the exact sodium insertion mechanism into hard carbons is still under debate, the literature provides a general understanding of the sodiation (discharge) processes.5,18–21 These processes include the adsorption of Na+ ions at the hard carbon surface, defect sites, or heteroatoms, intercalation of Na+ ions between graphene layers in hard carbon, the formation of quasi-metallic Na clusters in pores, and the formation of the solid-electrolyte interphase (SEI).10,22 Additional processes may also occur, especially at the extreme limits of the potential, leading to the metallic nature of Na.23 During cell operation, polarization tends to increase over time or under high current densities, possibly pushing the hard carbon potential below 0 V and increasing the likelihood of Na atom deposition on the hard carbon electrode surface as Na metal plating.23,24 Since most research on hard carbons is conducted in half-cells, dendrite growth on the Na metal electrode occurs during Na+ ion extraction from the hard carbons.25
As already mentioned, the proximity of the plateau region's potential to the Na metal plating potential could easily result in the coexistence of Na in ionic, quasi-metallic and metallic states. Under severe cycling conditions, SIBs may encounter overcharging, causing excessive Na to deposit unevenly on the hard carbon electrode surface in the form of plated Na metal. Analogously, in the half-cell configuration, Na metal plating occurs at an underpotential below 0 V.23,24 During half-cell operation, the continuous dissolution/deposition of the Na metal electrode takes place. The dissolution and deposition behaviour can be explained as follows: during dissolution, a large resistance must be overcome to successfully dissolve Na from the electrode surface followed by continuous dissolution from pits. During deposition, a nucleation barrier must be overcome to spark the dendrite growth followed by continuous deposition.26,27 While this behaviour is similar to that of a Li metal electrode, the dendrite morphology differs; Na tends to form mossy dendrites, whereas Li forms needle-like structures.27 Both Na metal plating and dendrite growth present significant safety issues and seriously hamper the battery's performance.
The electrochemical performance of hard carbons is also influenced by the decomposition of the electrolyte at the hard carbon electrode and pore surface, leading to the formation of the solid electrolyte interface (SEI). The SEI acts as an electrically insulating layer that prevents direct contact between the electrolyte and the electrode, while its ionic conductivity facilitates the transport of Na+ ions to the surface of the negative electrode.28 This process of Na+ ion insertion is reversible, with the SEI layer serving as an ion conductor.29 The composition of the SEI layer depends on the electrolyte's composition and the state of sodiation, which vary due to the differing redox potentials of solvents.30–32 The formation of the SEI is critical for achieving optimal and stable cell performance, as it acts as a protective barrier, mitigating continuous electrolyte decomposition and enabling efficient Na+ ion transport.28 Compared to LIBs, the SEI in SIBs contains more inorganic species due to the different reactivity of alkali metal cations.33,34 However, the SEI in SIBs is less stable, leading to higher solubility, increased self-discharge, and the dissolution of the SEI during cycling, which exposes the carbon surface to further degradation and consumes more active material.28 Therefore, a thorough understanding and precise control of SEI formation are essential for developing high-performance and durable battery systems.
Nuclear magnetic resonance (NMR) spectroscopy has been proven to be a valuable tool for studying the dynamics and structural changes in SIBs.35,36 Originally, ex situ approaches were used in which the sample was subjected to rotation at a magic angle (MAS) with respect to the direction of the magnetic field, resulting in improved resolution of the NMR spectrum. This technique facilitated the analysis of morphologies, crystal structures, and electrochemical reaction products during the (de)sodiation of the batteries.37 However, the ex situ method has its limitations as it can perturb the battery's state and overlook transient phases formed during electrochemical reactions. To overcome these limitations, research has recently shifted to in situ and operando NMR measurements, which allow real-time observation of dynamic structural changes and processes during battery operation without disturbing the battery.38,39 Nevertheless, operational challenges such as sample manipulation in confined spaces and strong magnetic fields hinder the wide application of operando NMR. In addition, the static sample conditions during operando NMR measurements lead to broader signal peaks and lower resolution compared to ex situ MAS measurements.40,41
Despite these challenges, operando NMR measurements are the focus of recent research, since they provide valuable insights into battery operation and complement ex situ approaches. In particular, for paramagnetic materials such as metallic sodium, bulk magnetic susceptibility effects significantly influence NMR signals. This effect, resulting from dipolar couplings between nuclear spins and electron magnetic moments, leads to frequency shifts and line broadening in NMR spectra, especially when the shape or paramagnetic spin distribution of the sample is non-uniform.42–44
In this study, we aim to (i) provide an in-depth analysis of the formation and composition of the SEI, (ii) present NMR spectroscopy as a powerful tool to understand and differentiate the sodium storage processes occurring during the (de)sodiation of hard carbon, and (iii) distinguish between Na metal plating and dendrite growth. With ex situ NMR, energy-dispersive X-ray spectroscopy (EDX), and X-ray photoelectron spectroscopy (XPS), we determine the composition and the content of the main constituents of the SEI. Through operando/in situ NMR measurements, we separate the sodium storage processes occurring during different stages of sodiation and correlate them with SAXS measurements from a previous study.8 Further employing operando/in situ NMR spectroscopy, we differentiate between Na metal plating and dendrite formation due to their distinct chemical environments. Coupled with scanning electron microscopy (SEM) imaging, we could visualize different morphologies of both forms of metallic Na. Finally, correlations between chemical shifts and the electrochemical behavior were established, providing a comprehensive new insight into the hard carbon pore filling and degradation mechanisms of SIBs.
Experimental section
Materials
Hard carbon was synthesized through a two-step pyrolysis of corncob, as reported in our previous work.8 Briefly, the initial heat treatment was performed at 900 °C with a 1 h hold. The second heat treatment involved heating to 1400 °C and 1600 °C with another 1 h hold. After the pyrolysis, the obtained hard carbon was ground in a mixer mill (SPEX SamplePrep, Retsch) for 30 min. The obtained materials were denoted as CC@900 °C, CC@1400 °C and CC@1600 °C.Scanning electron microscopy
Scanning electron microscopy (SEM) analysis of the pristine and cycled CC@1400 °C electrodes was performed using a field-emission scanning electron microscope (FE-SEM) Apreo 2 S (Thermo Fisher Scientific, Netherlands), equipped with a Clean Connect vacuum transfer system (Thermo Fisher Scientific, Netherlands) and energy dispersive spectrometer (EDS) Ultim Max 100 (Oxford, UK).The samples were mounted into a Clean Connect transfer capsule in an argon filled glovebox and transferred directly to the FE-SEM instrument without washing and exposure to air. Morphological images of the sample were acquired using a low energy electron beam (2 kV @ 25 pA) by simultaneously collecting secondary electrons (SE) and backscattered electrons (BSE) with detectors placed at various positions. EDS elemental analysis and elemental mappings were performed under low voltage analytical conditions (7 kV @ 0.4 nA) using AZtec 6.1 SP1 processing software (Oxford, UK).
Electrode preparation, cell assembly and electrochemical measurements
Hard carbon electrodes were prepared with a composition of 90 wt% of active material (CC@1400 °C), 5 wt% conductive carbon Super C65 (Timcal) and 5 wt% polyvinylidene fluoride (PVdF, Aldrich), unless stated otherwise. The mixture was dissolved in N-methyl pyrrolidone (NMP, Aldrich) and ball milled for 30 min at 300 rpm to obtain a homogeneous slurry. The slurry was then cast on carbon coated Al foil (Armor, France) with a doctor blade applicator with a thickness of 100 μm. The coated slurry was dried at 80 °C overnight.Electrodes with a diameter of 15 mm were punched out the next day and transferred to an argon-filled glovebox with water and oxygen contents below 0.5 ppm. The loadings were maintained between 1.5 and 2 mg cm−2. CC@1400 °C electrodes were used as working electrodes whereas sodium metal (Aldrich) was used as the counter electrode. Both electrodes were separated by a glass fiber separator (Whatman GF-A), immersed in NP30 electrolyte (1 M NaPF6 in a solvent mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 vol%)). Each cell was filled with 100 μL of the electrolyte.
:1 vol%)). Each cell was filled with 100 μL of the electrolyte.
Electrochemical measurements were carried out within a potential window between 2 V and 0.005 V vs. Na/Na+ employing the following protocol: 5 formation cycles were conducted at a current of 30 mA g−1 (theoretical capacity was taken according to the model proposed by Bommier,5i.e., 301.6 mA h g−1), which roughly translates to a rate of C/10. After the formation cycles, the current was increased to 300 mA g−1 (corresponding to a rate of 1C) and the measurement continued for another 100 cycles. At the end of each sodiation a constant voltage step was applied at the lower cut-off, limited to 10 h or until the current rate was lower than C/100.
XPS
Ex situ X-ray photoelectron spectroscopy (XPS) of the sodiated electrode at 5 mV vs. Na/Na+ was performed using a Versa probe 3 AD (Physical Electronics, Chanhassen, U.S.A) with a monochromatic Al Kα X-ray source operating at 15 kV and 13.3 mA. The negative electrodes were washed with DMC to remove the electrolyte salt from the electrode surface in order to measure the penetration depth during XPS analysis. The sodiated electrode was put on a non-conductive double-sided scotch tape in a “floating” setup and placed at the center of the XPS holder inside an argon filled glove box. The samples were transferred inside into the XPS intro chamber with a transfer vessel without air exposure. For each sample, spectra were acquired by scanning a 1 × 1 mm area with a spot size of 200 μm. High-resolution spectra were measured at a pass energy of 27 eV with a step size of 0.05 eV, to obtain high quality data. Analysis of the peaks was performed with Multipak software using a weighted sum of Lorentzian and Gaussian component curves after performing Shirley background subtraction. The binding energies were referenced to the internal gold standard.Ex situ solid-state NMR
Ex situ NMR measurements were performed on hard carbon powder material, assembled in a Swagelok type cell. Cells were assembled in an argon-filled glovebox with water and oxygen contents below 0.5 ppm. About 20 mg of CC@1400 °C powder was used as the working electrode and sodium metal (Aldrich) was used as the counter electrode. The CC@1400 °C powder and sodium metal were separated by two glass fiber separators (GF-A), wetted with 150 μl of NP30. The cells were cycled in a voltage range between 2 V and 0.005 V vs. Na/Na+ with a current of 6 mA g−1 (C/50). The cells were then transferred to a glovebox and disassembled. The CC@1400 °C powder was recovered from the cells and put into NMR 1.6 mm rotors. During the process of disassembling the cells within the glovebox and obtaining the powder for the ex situ NMR study, the powder was not washed in any way.Solid-state magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectra were recorded on a 600 MHz VNMRS spectrometer (Agilent Technologies) using a 1.6 mm Triple Resonance HXY FastMAS Varian Probe. The shift axis in all the spectra was referenced using an external, secondary reference of adamantane (the 1H signal was set to 1.85 ppm). We can consider the reported shifts to be accurate within ±0.2 ppm. Larmor frequencies of 23Na, 1H and 19F nuclei were 158.539 MHz, 599.314 MHz and 563.919 MHz, respectively. The sample MAS frequency was 20 kHz. 23Na spectra were recorded using an excitation pulse with duration of 0.5 μs, 20000 scans and a 0.1 s delay between scans. The 1H spectra were recorded using a Hahn echo pulse sequence with π/2 and π pulses of 2.6 μs and 5.1 μs durations, respectively, with 200 scans and a 10 s delay between scans. 19F spectra were recorded using a Hahn echo pulse sequence with π/2 and π pulses of 2.8 μs and 5.6 μs durations, respectively, with 200 scans and a 10 s delay between scans.
23Na operando solid-state NMR
Hard carbon electrodes for operando NMR measurements were prepared at various pyrolysis temperatures (CC@900 °C, CC@1400 °C and CC@1600 °C) by mixing the active material, C65 and the polytetrafluoroethylene (PTFE, 60 wt% dispersion in water) binder in a mass ratio of 85:5:10. The composites were pressed on carbon coated Al mesh foil and dried at 80 °C overnight. Electrodes with dimensions of 5 mm × 15 mm were punched out the next day and transferred to an argon-filled glovebox. The loadings were maintained between 10 and 14 mg cm−2.
For operando NMR measurements, batteries were assembled in the form of in situ cells (Fig. S1†).40 The CC@900 °C, CC@1400 °C and CC@1600 °C electrodes, pressed onto the Al mesh, were placed into the cavities with the remaining mesh twisted into thin wires, which were then passed through the holes and extended outside the cells. A small drops of fast hardening epoxy and O-rings were applied to secure and seal the outgoing side of the Al wires. Sheets of glass fiber separators were cut into the appropriate size for the cavities and pressed onto the working electrodes. On the Na side, a Cu mesh twisted into thin wires are glued onto Viton O-rings. The O-rings seal the holes through which the Cu wires exit the chambers. The remaining Cu mesh is placed inside the cavities with thin Teflon sheets, cut to size depending on the desired pressure inside the cells. The Na metal electrodes (13 mm × 4 mm) were gently placed onto the Cu mesh. Subsequently, the glass fiber separators were soaked with approximately 100 μl of NP30. Finally, both cavities were pushed together and fixed using the capsules.
The in situ cells were cycled in a voltage range between 2 V and 0.005 V vs. Na/Na+ with a current of 15 mA g−1 (C/20). At the end of each sodiation, a constant voltage step was applied at the lower cut-off, limited to 10 h or until the current rate was lower than C/100.
In the plating test, the Na‖CC@1400 °C cell was sodiated at a current of 15 mA g−1 (C/20) till −1 V vs. Na/Na+ as the threshold condition.
For the Na‖Na symmetrical cell, two identical Na plates (13 mm × 4 mm, with a Na surface area of 0.5 cm−2) were used. The electrodes were stripped and deposited at a current density of 5.0 mA cm−2, with each half-cycle lasting for 2 h.
For the Na‖Cu plating test, a Na plate (15 mm × 5 mm) was used as the working electrode and Cu mesh was used as the counter electrode. The cell was cycled at a current density of 0.2 mA cm−2, with each half-cycle lasting for 2 h.
Operando solid-state NMR spectra were recorded on a Bruker AVANCE NEO 400 MHz NMR spectrometer using an ATMC in situ NMR 400 SB Probe (Fig. S2†). The shift axis in all the spectra was referenced using an external, secondary reference, NaCl (with the 23Na signal set to 7.2 ppm). The Larmor frequency of 23Na nuclei was 105.845 MHz and the sample remained static (without rotation) during the measurements. The 23Na spectra were recorded using an excitation pulse with a duration of 4.75 μs, collecting16384 scans, with a delay of 0.05 s between consecutive scans. The NMR spectra were analysed using ssNake and TopSpin software.
Results and discussion
Electrochemical testing of the corncob-derived negative electrode in a Na-ion cell
For the investigation of the sodium storage mechanism, we used hard carbon obtained from corncob (CC@1400 °C) as the negative electrode material. The selection was based on the favorable structural, textural, and morphological properties of the material, which led to improved electrochemical performance, as described elsewhere.8,45Fig. 1 shows the electrochemical characterization of the CC@1400 °C sample in a half-cell configuration with the Na metal electrode as the counter electrode. Fig. 1a illustrates the specific capacity evolution during cycling, highlighting the stable sodiation capacities over 100 cycles. The low initial coulombic efficiency (ICE) of 72% is primarily due to the initial loss of Na+ ions in the first cycle, associated with the decomposition of the electrolyte and the formation of a solid electrolyte interphase (SEI).46 The initial five formation cycles performed at a current density of C/10 result in a consistent sodiation capacity of approximately 300 mA h g−1. During the transition to a higher current density of 1C, a slight decrease in capacity is observed, which stabilizes at about 260 mA h g−1, maintaining stable cycling performance throughout 100 cycles.
 | ||
| Fig. 1 (a) Cycling stability over 100 cycles. The first five cycles were measured at C/10, with the rest cycles at 1C and (b) galvanostatic curve from the first cycle and the differential capacity (dQ/dV) curve is shown in the inset. | ||
For a detailed understanding of the electrochemical phenomena occurring in CC@1400 °C during (de)sodiation, we present the galvanostatic and differential capacity (dQ/dV) curves of the 1st cycle in Fig. 1b. CC@1400 °C exhibits a characteristic sodiation/desodiation galvanostatic curve, featuring two distinct regions, namely, a sloping region above 0.1 V and a plateau region below 0.1 V vs. Na/Na+.5 The sloping region highlights the interplay between Na+ adsorption and electrolyte decomposition at the hard carbon surface during sodiation. Both processes are evident in the dQ/dV curve between 0.8 V and 0.5 V vs. Na/Na+ (inset in Fig. 1b), with the first process attributed to SEI formation and the second to Na+ ion adsorption. At lower voltages, the processes in the plateau region exhibit more complex behavior. The (de)insertion of Na+ ions is observed in the differential capacity curve at voltages near the cut-off voltage (inset in Fig. 1b). However, several mechanistic studies suggest that micropore filling occurs simultaneously, leading to the formation of metallic Na clusters.5,18,20,47 Unfortunately, simple galvanostatic experiments cannot distinguish between insertion and micropore filling.
The galvanostatic and differential capacity curves (dQ/dV) provide an initial insight into the processes associated with sodium storage in hard carbon-based electrodes. Sodium storage involves the passivation of the hard carbon electrode and pore surface (formation of the SEI) as well as the formation of metastable quasi-metallic Na clusters in the bulk. A comprehensive understanding of these phenomena requires transition from a macroscopic perspective of electrochemical behavior to a microscopic viewpoint. This can be attained through employment of advanced non-invasive surface methods (SEM, XPS, and NMR) to reveal the details of SEI formation, and volumetric methods including operando measurements during battery cycling (NMR) to depict the sodium storage mechanisms inside the hard carbon electrode.
SEI study
As already mentioned, the electrolyte decomposes on the carbon surface during battery operation, which leads to the formation of the SEI. This process is a critical parameter for the performance of hard carbon materials as it determines the amount of Na+ ions consumed during the initial sodiation and controls/determines the subsequent cell performance. To visualize the formed SEI's morphology, CC@1400 °C was sodiated to 5 mV vs. Na/Na+, and ex situ SEM imaging was performed (Fig. S3†). The SEM micrographs (Fig. S3c and d†) reveal that the formed SEI creates a distinct surface film on the hard carbon particles. In addition, smaller spherical particles approximately 100 nm in size were observed on the surface. EDX elemental mapping (Fig. 2a) was performed to investigate the chemical composition of the SEI. The elemental mapping revealed a uniform coverage with sodium and oxygen prevalence at the surface.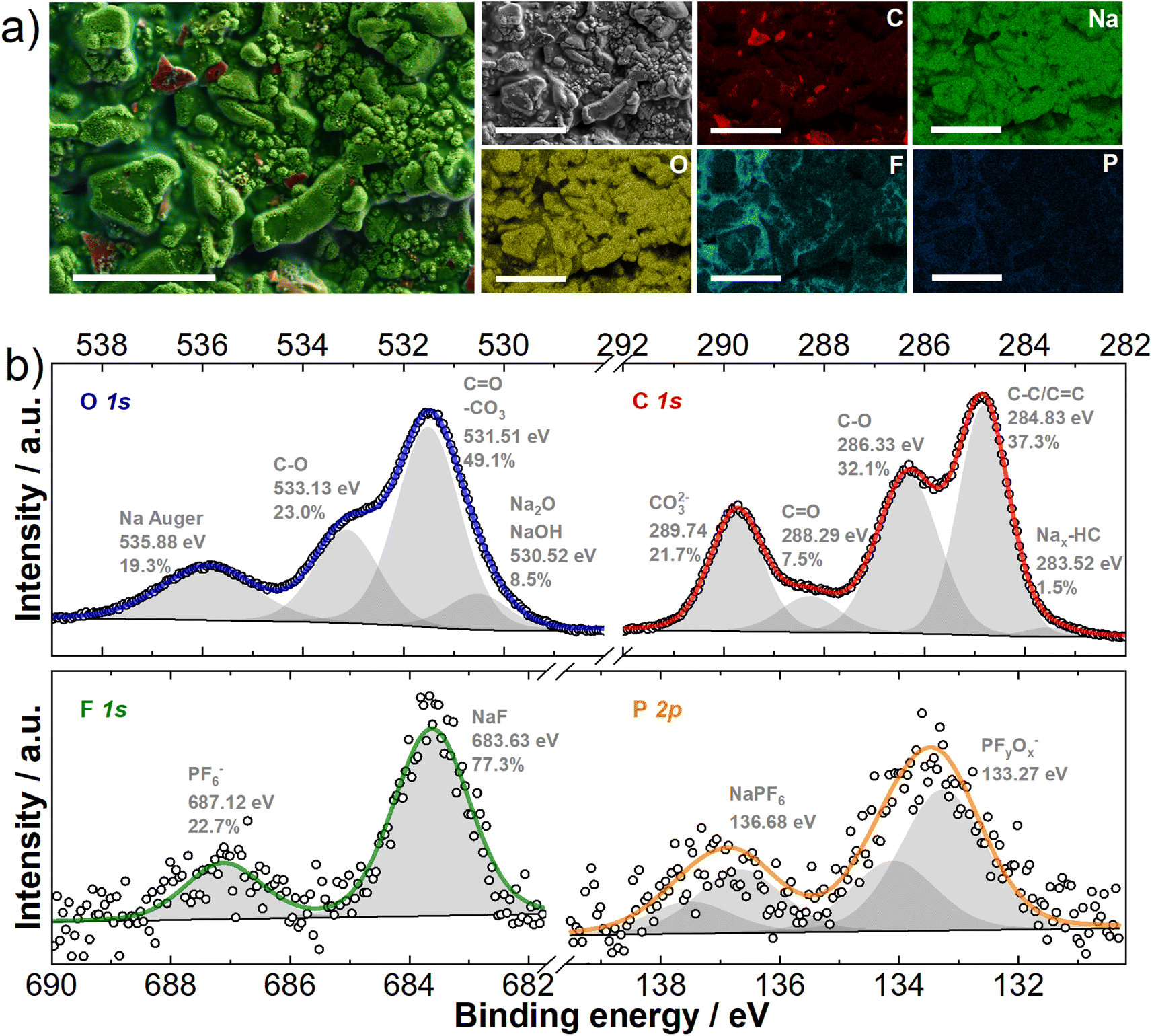 | ||
| Fig. 2 CC@1400 °C electrodes sodiated at 5 mV vs. Na/Na+ (a) EDX mapping of the electrode surface; scale bar 10 μm; (b) high resolution C 1s, O 1s, F 1s and P 2p XPS spectra. | ||
To further decipher the chemical composition, an XPS analysis was carried out. The results for the core contents of carbon (C), oxygen (O), fluorine (F), and phosphorus (P) are shown in Fig. 2b. The high-resolution C 1s XPS spectra presented in Fig. 2b, show several distinct peaks indicating different chemical states and compositions of the SEI. Peaks in the range of 288.29–289.74 eV correspond to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of sodium alkyl carbonate (NaCO3R) and sodium carbonate (Na2CO3), while a peak at approximately 286.33 eV is attributed to C–O compounds, found in sodium alkoxides and polyethylene glycol (PEG).47–52 The formation of sodium alkyl carbonate and sodium carbonate results from dimethyl carbonate (DMC) reduction, and PEG polymers are formed by both ethylene carbonate (EC) ring opening with further polymerization and DMC decomposition (Fig. S4†). Additionally, the most intense peak, representing sp3/sp2 carbon bonds is linked to CC@1400 °C, C65 carbon black, and hydrocarbons in the SEI. Finally, a less pronounced peak at 283.52 eV indicates the presence of inserted sodium within the hard carbon structure (Nax-HC).47–52 The O 1s peak fitting (Fig. 2b) reveals additional surface species: one at lower binding energies (530.52 eV) attributed to Na2O or NaOH, and another at higher binding energies (535.88 eV), indicative of the Na Auger signal. Interestingly, the F 1s XPS high-resolution spectra (Fig. 2b) show the presence of NaF (683.63 eV) in the SEI. The presence of NaF is attributed to the decomposition of DMC and NaPF6, as shown in Fig. S4.† In addition, the electrochemical reduction of PF6− at the hard carbon negative electrode results in the formation of PFyOx species (Fig. 2b). Analyzing the SEI chemical composition, the C 1s XPS curve shows high contents of CO and CO32− compounds, with the ratio of integrals of 32.1% and 21.7% respectively, while the contribution of the CO integral is only 7.5%. This suggests that the SEI is rich in Na2CO3, Na2O, or NaOH species and in sodium alkoxides/PEG.52–54
O groups of sodium alkyl carbonate (NaCO3R) and sodium carbonate (Na2CO3), while a peak at approximately 286.33 eV is attributed to C–O compounds, found in sodium alkoxides and polyethylene glycol (PEG).47–52 The formation of sodium alkyl carbonate and sodium carbonate results from dimethyl carbonate (DMC) reduction, and PEG polymers are formed by both ethylene carbonate (EC) ring opening with further polymerization and DMC decomposition (Fig. S4†). Additionally, the most intense peak, representing sp3/sp2 carbon bonds is linked to CC@1400 °C, C65 carbon black, and hydrocarbons in the SEI. Finally, a less pronounced peak at 283.52 eV indicates the presence of inserted sodium within the hard carbon structure (Nax-HC).47–52 The O 1s peak fitting (Fig. 2b) reveals additional surface species: one at lower binding energies (530.52 eV) attributed to Na2O or NaOH, and another at higher binding energies (535.88 eV), indicative of the Na Auger signal. Interestingly, the F 1s XPS high-resolution spectra (Fig. 2b) show the presence of NaF (683.63 eV) in the SEI. The presence of NaF is attributed to the decomposition of DMC and NaPF6, as shown in Fig. S4.† In addition, the electrochemical reduction of PF6− at the hard carbon negative electrode results in the formation of PFyOx species (Fig. 2b). Analyzing the SEI chemical composition, the C 1s XPS curve shows high contents of CO and CO32− compounds, with the ratio of integrals of 32.1% and 21.7% respectively, while the contribution of the CO integral is only 7.5%. This suggests that the SEI is rich in Na2CO3, Na2O, or NaOH species and in sodium alkoxides/PEG.52–54
23Na solid-state NMR on hard carbon
The movement of Na+ ions, surface reaction mechanisms, SEI formation, and dendrite growth patterns in the CC@1400 °C hard carbon sample were examined by using 23Na solid-state NMR. The technique also reveals the impact of sodium metal plating on battery performance.The ex situ powder samples were analysed at the initial state, at sodiation potentials of 0.5 V and 5 mV vs. Na/Na+, and at desodiation potentials of 0.5 V and 2 V vs. Na/Na+. The 23Na MAS NMR spectrum (Fig. S5†) of the cell on its initial state shows a single peak at −11.5 ppm, which corresponds to the solvated sodium of the salt in the electrolyte (NaPF6). Alongside this electrolyte contribution, an increase in the Na2CO3 signal at −6.5 ppm is observed at a sodiation potential of 0.5 V vs. Na/Na+. This Na2CO3 contribution further increases at a sodiation potential of 5 mV vs. Na/Na+ and then remains consistent throughout both desodiation measurements. This behavior is attributed to the growth and stabilization of the SEI after the first cycle, which is also confirmed by XPS. Additionally, 1H and 19F MAS NMR measurements were conducted. The contribution of electrolyte solvents EC and DMC are visible in the 1H MAS NMR measurements, appearing at 4.2 ppm and 3.4 ppm, respectively.55 Intense and narrower signals are present at the same chemical shifts at a desodiation potential of 2 V vs. Na/Na+, which is attributed to the additional electrolyte captured during rotor filling. At a sodiation potential of 5 mV vs. Na/Na+ and a desodiation potential of 0.5 V vs. Na/Na+, a trace contribution of NaOH is detected at 1.3 ppm56 in 1H MAS NMR, potentially indicating its presence in the SEI, as also confirmed by XPS. In 19F MAS NMR, the electrolyte contribution (PF6−) is evident from the onset of cycling at −75 ppm.57 Furthermore, a more intense and narrower signal at this chemical shift is observed at the end of the cycle at 2 V vs. Na/Na+, again indicating a greater amount of electrolyte captured during the rotor filling process. Notably, bulk-sensitive 19F MAS NMR does not reveal any NaF formation at −228 ppm (dashed line in Fig. S5†),58 in contrast to surface-sensitive XPS, which shows that the surface of SEI is rich in NaF.
To explore the impact of electrochemical reactions on both the Na metal electrode and the CC@1400 °C electrode, and to identify potential metastable phases undetectable through ex situ analysis, we performed 23Na operando solid-state NMR. The pseudo-2D spectrum shown in Fig. 3 represents a series of 1D spectra plotted versus time. The spectrum is plotted in a contour plot format with the chemical shift on the x-axis and time on the y-axis. The spectrum is segmented into three distinct regions. On the left side, it shows the features of metallic Na, spanning from 1150 ppm to 1110 ppm, which are characteristic of the paramagnetic part of the spectrum. The central section, which ranges from 1000 ppm to 400 ppm, shows quasi-metallic Na clusters which have a semi-paramagnetic nature. Finally, the right-hand segment corresponds to the electrolyte/SEI region, situated around 0 ppm, indicative of the diamagnetic component of the spectrum. At 50 mV vs. Na/Na+ during sodiation, an enhanced signal at 720 ppm is detected. As the potential decreases towards 5 mV vs. Na/Na+, the signal gradually shifts downfield to 850 ppm. According to the literature,59 this signal belongs to quasi-metallic Na clusters that form during the pore-filling phase with Na+ ions. While holding the voltage at 5 mV vs. Na/Na+, this signal continues to increase, indicating the progressive saturation of pores. Subsequently, as the current drops below C/100, initiating desodiation, the signal begins to decrease reversibly and shifts upfield. Above 50 mV vs. Na/Na+ during desodiation, the signal diminishes into the noise. Only minor peak broadening around the electrolyte region was observed during the cycle, which could be attributed to the detection of SEI growth.
 | ||
| Fig. 3 23Na operando solid-state NMR spectrum for the Na‖CC@1400 °C half cell at 90° orientation with respect to the external magnetic field of the spectrometer. First cycle electrochemistry is shown on the right. | ||
To more accurately map the changes in the quasi-metallic region, we have additionally aligned the baseline and used the logarithmic plot of NMR signal intensity levels versus time, as shown in Fig. 4a. Due to the logarithmic scale, we were able to observe changes before reaching 50 mV vs. Na/Na+, which marks the middle of the plateau, and detect them even at the beginning of the plateau at 100 mV vs. Na/Na+. This approach allowed us to detect a subtle additional signal (indicated by red arrows), alongside the existing signals at 1100 ppm and 850 ppm. Furthermore, at the end of the potential plateau at 100 mV vs. Na/Na+ in the desodiation process, the signal begins to shift upfield towards the electrolyte signal region. In the second sodiation, this signal begins to move downfield at the beginning of the second plateau at 100 mV vs. Na/Na+ and reaches the metallic Na region at a potential of 5 mV vs. Na/Na+. Once again, at the end of the plateau at 100 mV vs. Na/Na+ during the process of desodiation, it moves upfield and towards the electrolyte region. The weaker signal intensity during the first sodiation compared to the second is a result of Na+ ion loss during SEI formation in the initial cycle. To gain further insight, we conducted 23Na operando NMR measurements on hard carbon subjected to two extremely different carbonization temperatures, 900 °C and 1600 °C, designated as CC@900 °C and CC@1600 °C, respectively, as shown in Fig. S6.† No signal corresponding to quasi-metallic clusters was observed for CC@900 °C. However, an additional signal was detected at 100 mV vs. Na/Na+ during sodiation, similar to the potential observed in CC@1400 °C. Initially, this signal shifted downfield between the electrolyte and metallic Na signals; however, at 100 mV vs. Na/Na+, it shifted back upfield towards the electrolyte signal. For CC@1600 °C, an even more intense quasi-metallic cluster signal was detected with a peak at 900 ppm, indicating a downfield shift with respect to CC@1400 °C. Once again, an additional signal appeared at the same potential of 100 mV vs. Na/Na+ and shifted in the same manner as in CC@1400 °C, but it was significantly more intense and was already well detected in the first sodiation process.
 | ||
| Fig. 4 (a) 23Na operando solid-state NMR spectrum of the Na‖CC@1400 °C half cell at 90° orientation with respect to the external magnetic field of the spectrometer shown with a logarithmic plot of NMR signal intensity levels. Red arrows indicate the movement of an additional signal. Electrochemistry on the right-hand side represents the first two cycles. (b) Filling the surfaces of the pore walls with quasi-metallic Na atoms and (c) volumetric pore filling and formation of quasi-metallic Na clusters. | ||
In view of the additional measurements for CC@900 °C and CC@1600 °C, further conclusions can be drawn. In all three samples subjected to carbonization at different temperatures, a signal was detected that emerged at the beginning of the plateau, shifted downfield towards the metal Na signal at the end of sodiation, and then started to return upfield at the end of the plateau in the desodiation process. In terms of electrochemistry and the literature,60,61 this additional signal can be attributed to the Na+ ions inserted between the graphene layers. When the signal shifts to the region associated with metallic Na, it is indicative of quasi-metallic Na atoms filling the surfaces of the pore walls (Fig. 4b). At higher carbonization temperatures, where there are more insertion sites and larger pores, this signal is most intense in CC@1600 °C due to the greater number of contributing nuclei. The volume fraction (ϕpores [%]) and average width (Wp [nm]) of the nanometer sized pores, as measured by ex situ SAXS in ref. 8 are 6.1% and 0.33 nm for CC@900 °C, 6.7% and 0.45 nm for CC@1400 °C, and 8.6% and 0.52 nm for CC@1600 °C. Quasi-metallic Na clusters are not observed in CC@900 °C, where the pores are too small (ϕpores is 6.1% and Wp is 0.33 nm) to facilitate the formation of larger Na3 clusters.14,62 Instead, filling the surface of pore walls with quasi-metallic Na atoms is the only remaining process at the end of sodiation. However, additional volumetric pore filling (Fig. 4c) with quasi-metallic Na clusters is detected in CC@1400 °C. The intensity of the signal is even more pronounced in CC@1600 °C, where the pores are larger (ϕpores is 8.6% and Wp is 0.52 nm) and the nature of the quasi-metallic Na clusters is more metallic.14,62 This results in a downfield shift towards a more paramagnetic region compared to CC@1400 °C.
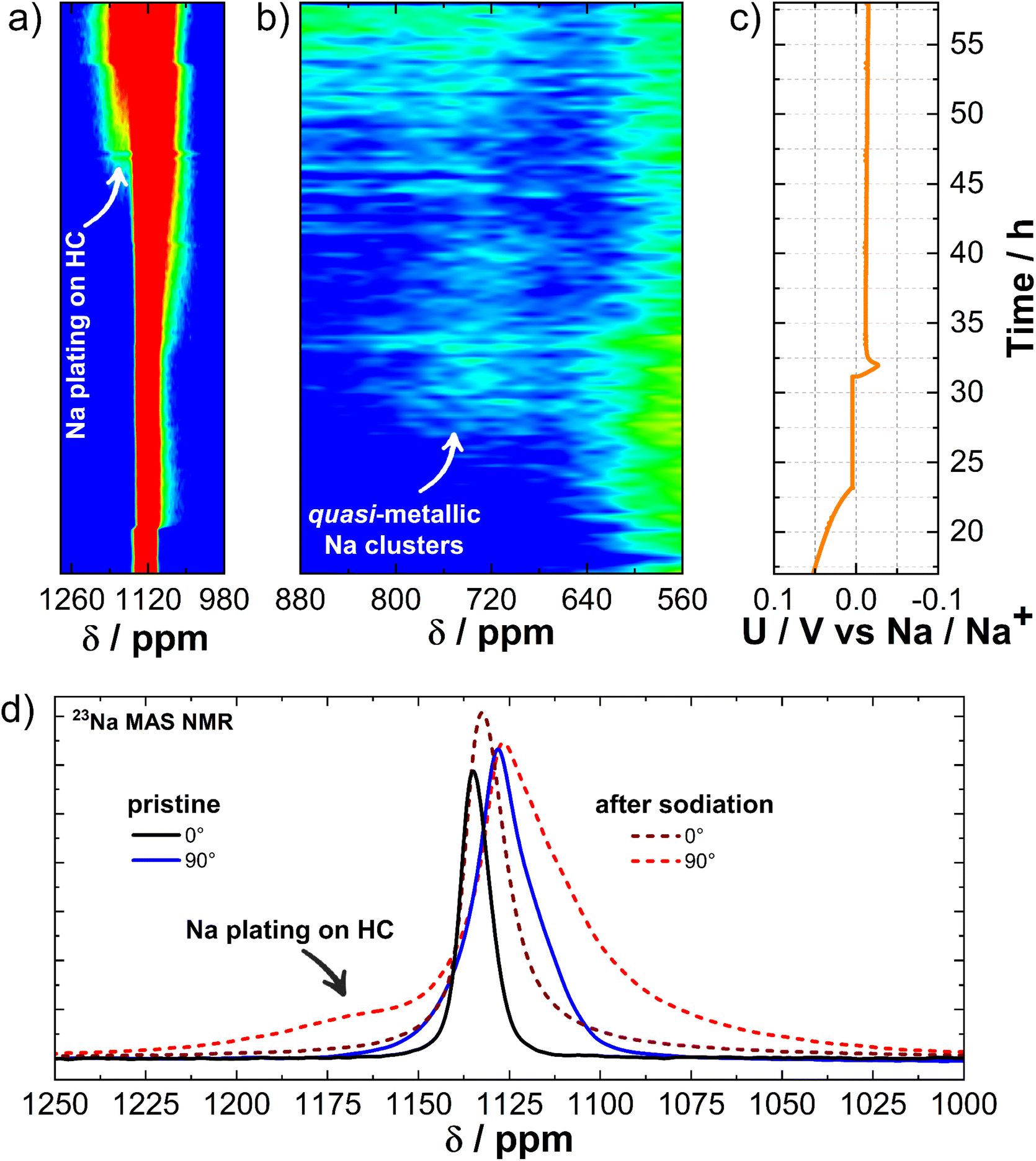
At a desodiation potential of 2 V vs. Na/Na+, an additional signal appears at 1133 ppm, close to the signal of the Na metal electrode resonating at 1120 ppm (Fig. 3). Fig. S7† shows this spectral region and its features when the Na metal is oriented at angles of 90° and 0° relative to the external magnetic field. The additional signal belongs to the Na dendrites growing on the surface of the metal Na electrode.41
Besides detecting inserted Na+ ions and quasi-metallic clusters in the hard carbon, NMR spectroscopy also proved useful for studying dendrites on the metallic Na electrode surface and plating on the hard carbon electrode surface. In both cases, metallic Na is present in different chemical environments that can be detected and identified by NMR.23 Using in situ/operando NMR at different orientations with respect to the external magnetic field, the magnetic susceptibility effect can be used to determine and distinguish between the morphology of different surface structures.
The study of dendrite formation and plating with 23Na solid-state NMR
To mimic the dendrite detection environment, we assembled symmetrical Na‖Na cells and cycled them at a current density of 5.0 mA cm−2. Analysis of the electrochemical curves depicting Na stripping/deposition in these symmetrical cells (Fig. 5a and b) demonstrates the successful acceleration of dendrite formation. Given the susceptibility of dendrite signal positioning to magnetic fields, we investigated how it is affected by the orientation of the cell in relation to the external magnetic field of the spectrometer. The layout diagram is presented in Fig. S8a.† To achieve this, we conducted in situ measurements at various angles both before and after the operando measurement (Fig. 5c). Additionally, two operando measurements were conducted at horizontal (90°) and vertical (0°) orientations with respect to the external magnetic field of the spectrometer to test dendrite growth dynamics over time (Fig. 5a and b).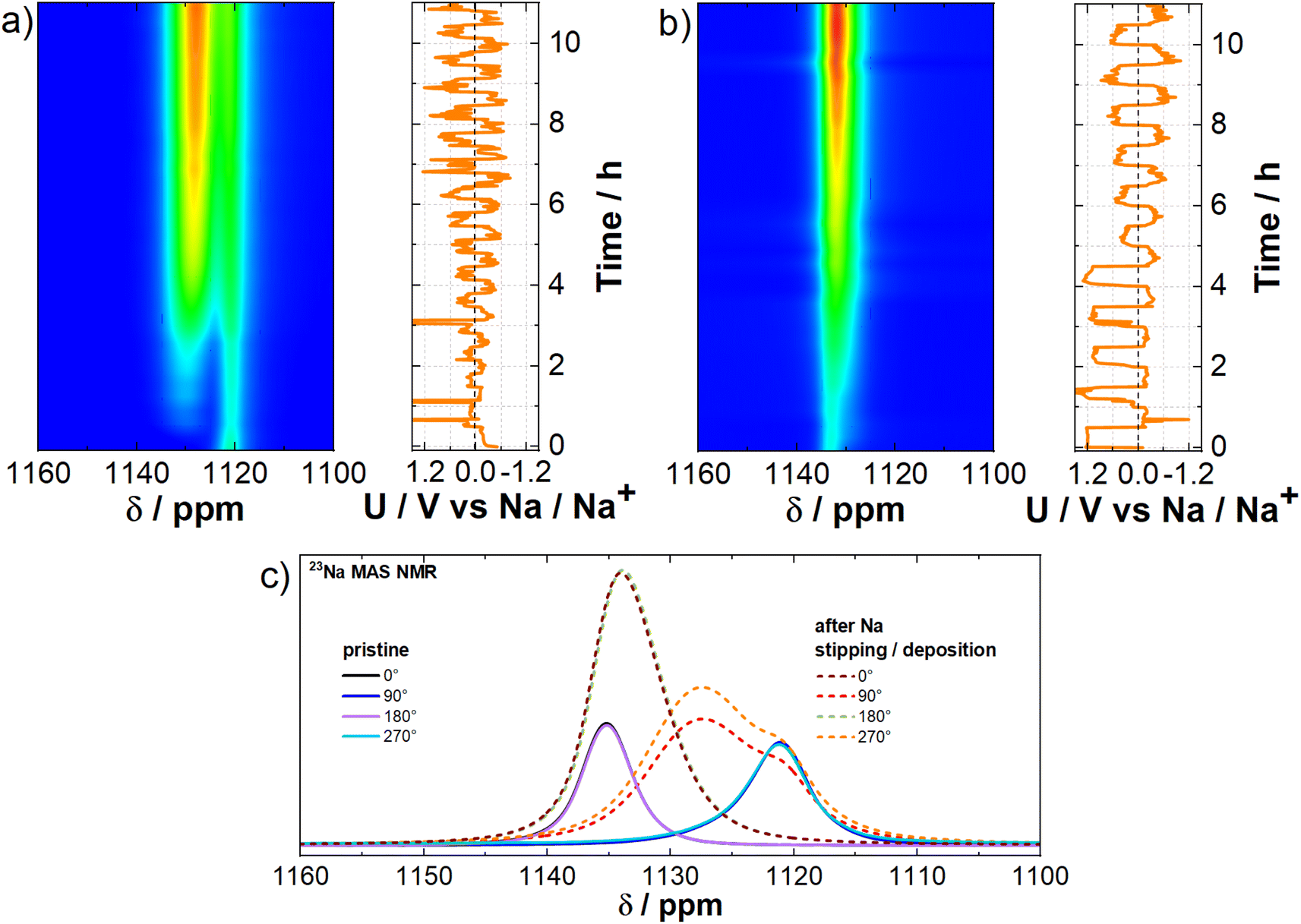 | ||
| Fig. 5 (a) 23Na operando solid-state NMR spectrum of the Na–Na symmetric cell at 90° orientation with respect to the external magnetic field of the spectrometer with related electrochemistry. (b) 23Na operando solid-state NMR spectrum of the Na–Na symmetric cell at 0° orientation with respect to the external magnetic field of the spectrometer with related electrochemistry. (c) 23Na in situ solid-state NMR spectrum before and after operando measurement at 4 different orientations of the Na–Na cell with respect to the external magnetic field. | ||
Magnetic susceptibility plays an important role42,43 in measurements involving metallic sodium, as it is paramagnetic. In our case, the chemical shift depends on the cross-section of the Na metal electrode along the external magnetic field. The Na metal signal shifts downfield to 1133 ppm at a 0° orientation and upfield to 1120 ppm at a 90° orientation. At intermediate angles, the Na metal signal shifts between these two chemical shifts (Fig. S8b and c†). Dendritic formations grow from the Na metal electrode surface at different angles (Fig. S8d†), resulting in a different chemical shift relative to the Na metal. This allows us to observe and track dendrites during real-time measurements. According to the measured chemical shift of the initial Na‖Na cell (Fig. S8b and c†) the orientation of the dendrites is on average between 45° and −45° relative to the normal on the plane of the Na metal electrode (Fig. 5c). The chemical shift is highly sensitive to the battery's orientation in the external magnetic field due to the geometry of dendrite growth and the associated bulk magnetic susceptibility effect.42,63 As shown in Fig. 5a, the 90° orientation is more advantageous than other angles, as the difference in chemical shifts between the Na metal electrode and the dendrites reaches 8 ppm, compared to only 2 ppm at the 0° orientation (Fig. 5b). This highlights that the 90° orientation significantly improves the distinction between NMR signals, allowing for better resolution of different components in the system. The NMR signal intensity of the dendrites even exceeds that of the Na metal electrode due to the distinct physical and chemical properties of these two forms of sodium.64 Dendrites, with their irregular structure and larger surface area, expose more sodium nuclei to the radio frequency field, enhancing signal detection. In contrast, the bulk Na metal's high conductivity limits radio frequency field penetration due to the skin effect, reducing the number of excited nuclei and thus weakening the signal. Additionally, dendrites contain more defects and impurities, creating diverse local environments that result in longer relaxation times and stronger NMR signals. In comparison, the ordered crystalline structure of bulk Na metal leads to faster relaxation and weaker NMR signals. In the SEM micrographs (Fig. 6a–c), we can observe the states where Na metal was stripped and subsequently redeposited on the sodium surface, leading to the formation of visible dendrites in a cubic-like form (deposits). A closer examination of these sodium deposits (Fig. 6b and c) reveals varying textures; some areas appear smoother, while others show rougher or more granular characteristics. Such observations align with the findings from the NMR measurements, which predict the cubic-like shape of the individual Na deposits at an average side angle of 45°.65
 | ||
| Fig. 6 SEM micrographs: (a) fresh Na, (b) Na electrode after stripping and deposition; (c) higher magnification of Na deposits, (d) fresh CC@1400 °C electrode, (e) CC@1400 °C electrode after the plating test, and (f) higher magnification of the plated CC@1400 °C electrode. | ||
In addition, a Na‖Cu cell was tested to detect dendrite stripping and deposition (Fig. S9†). The Na metal plate was used as the working electrode, and copper mesh was used as the counter electrode. The 23Na operando NMR measurement shows a comparable behavior to that of the Na‖Na cell. The difference lies in the growth of the signal downfield from the metal Na signal. In the case of the Na‖Na cell, the signal starts to grow at the final chemical shift (Fig. 5a), whereas in the case of the Na‖Cu cell the signal gradually shifts downfield from the position of the metal Na signal (Fig. S9b†). The results show that plating leads to a more uniform growth of micro-structures all over the surface of the Cu, gradually evolving into their final orientation (Fig. S10†). In contrast, the dendrites show a well-defined localized growth from the beginning of the cycling on the Na metal electrode.
Furthermore, the difference in the NMR spectra of the quasi-metallic states arising from Na plating on the HC electrode surface is still an open question.66,67 To confirm the Na metal deposition (plating) on the HC electrode surface, the cell was sodiated below the potential of sodium metal. The galvanostatic curve during sodiation at C/20 in Fig. S11,† shows a minimum close to −28 mV vs. Na/Na+, followed by an increase in voltage, although it remains negative. According to the literature,68,69 this minimum demonstrates the onset of sodium nucleation, followed by increased growth of sodium deposits at the surface of the hard carbon electrode. This is evident in Fig. 6d–f, where the pristine hard carbon electrode surface is compared to the surface of the electrode, which was sodiated below the potential of sodium metal. The pristine CC@1400 °C electrode has a relatively smooth surface with some degree of porosity (Fig. 6d). During the plating process in the negative voltage regime, the electrode surface exhibits an increase in roughness. The plated sodium forms clusters at the CC@1400 °C electrode surface (Fig. 6e and f). Furthermore, the electrode surface and sodium clusters are covered by the formed SEI as shown by XPS. To validate the differences in the quasi-metallic states resulting from Na plating on the hard carbon electrode, 23Na operando solid-state NMR was performed. An enlarged operando spectrum along with the galvanostatic curve sodiated under the potential of sodium is presented in Fig. 7. We aim to determine whether NMR can detect changes on the surface of the HC electrode in the region of the electrochemical plating. As already shown, the Na metal signal is detected at 1125 ppm (Fig. 7a). In the NMR spectra (Fig. 7b), spanning the region between 880 ppm and 560 ppm, the growth of quasi-metallic Na clusters after 25 hours at a potential of 5 mV vs. Na/Na+ is detected (Fig. 7c). The signal appears at 730 ppm and stabilizes around this chemical shift value during underpotential deposition. After 45 hours at a potential of −12 mV vs. Na/Na+, an additional signal starts to increase from the baseline at 1176 ppm, corresponding to Na plating on the hard carbon electrode surface.
 | ||
| Fig. 7 Na-HC plating test: 23Na operando solid-state NMR spectrum of the Na‖CC@C1400 °C half cell at 90° orientation with respect to the external magnetic field of the spectrometer around (a) Na metal region and (b) quasi-metallic Na clusters region. (c) Related galvanostatic curve for the measurement when sodiated to negative potentials. (d) 23Na in situ solid-state NMR spectrum before and after operando measurement at 2 different orientations of the Na‖CC@1400 °C cell with respect to the external magnetic field. | ||
The measurement shows that NMR spectroscopy can distinguish between the formation of quasi-metallic clusters and Na metal plating. Fig. 7d shows the 1D in situ NMR spectrum at the beginning and end of the operando measurement at 0° and 90° orientations relative to the magnetic field of the spectrometer. Due to the bulk magnetic susceptibility effect, NMR measurements again exhibit greater resolution of surface changes when the sample is oriented at 90° (horizontal) to the external magnetic field compared to 0° (vertical).42,63 This orientation-dependent resolution is crucial for accurately detecting and interpreting subtle changes in the magnetic properties of the sample. A change occurs in both cases. In the 90° orientation, an increase in the additional signal at 1176 ppm is detected downfield from the existing metal Na signal at 1125 ppm. However, in the 0° orientation, an additional signal increase at 1131 ppm is detected upfield from the existing metal Na signal at 1133 ppm.
The differences in the NMR chemical shift between dendrites and plating are attributed to their different structural and chemical properties (Fig. 8).70 Sodium deposits/dendrites (Fig. 8a), characterized by cubic-like morphology and a large surface area, exhibit a higher proportion of defects and impurities compared to the more uniform and homogeneous plating observed during continuous stripping and deposition.71,72 These structural differences lead to differences in the local chemical environments and interactions with the electrolyte, which in turn influence the electronic density and the local magnetic fields around the sodium nuclei. Consequently, these variations influence the shielding of sodium nuclei, resulting in different NMR chemical shifts for dendrites and plating.
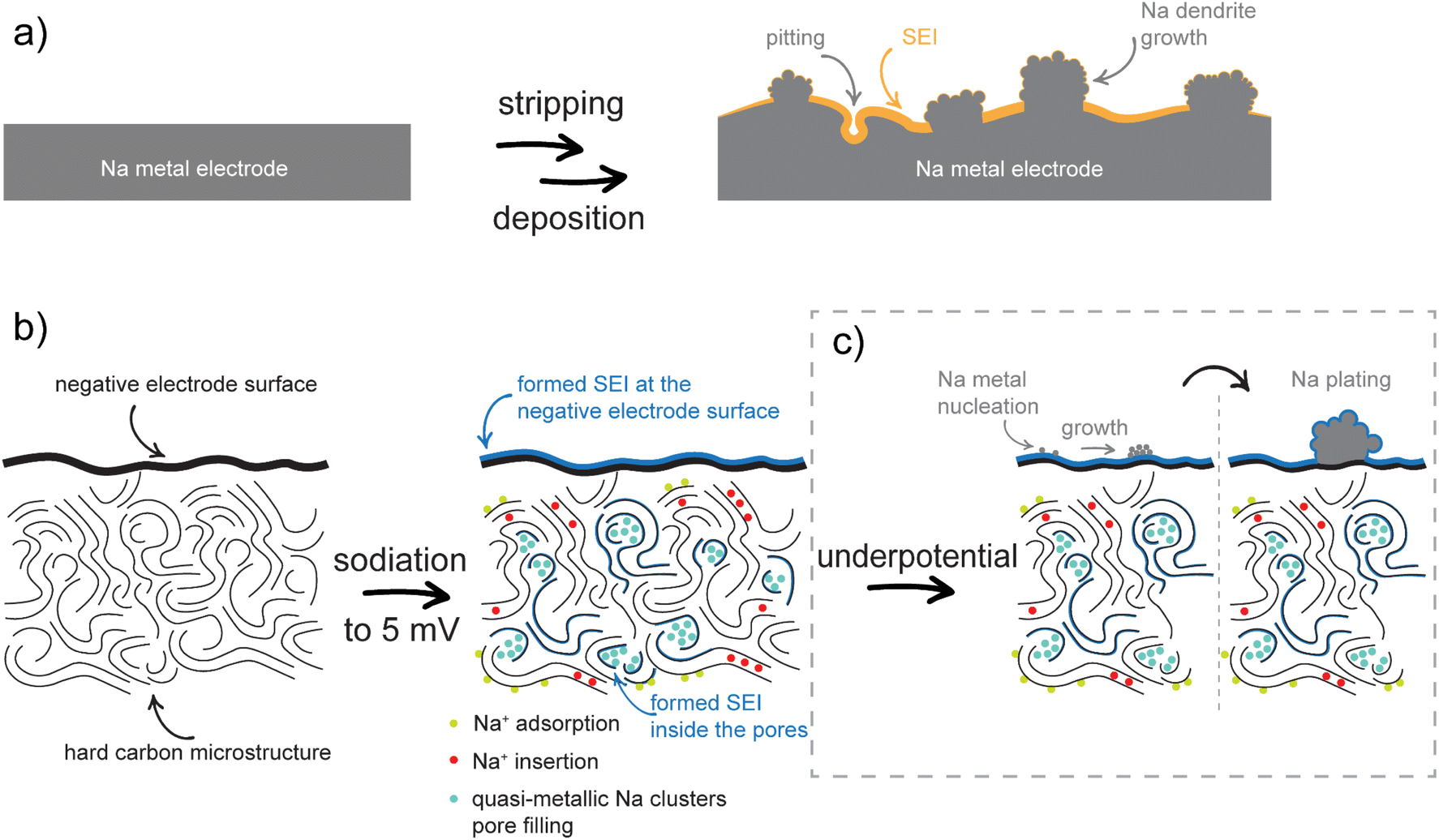 | ||
| Fig. 8 (a) Scheme of metal stripping and deposition on the Na metal electrode. (b) Scheme of SEI formation and sodiation mechanisms. (c) Na plating mechanisms on a hard carbon electrode. | ||
The proposed sodiation mechanism is shown in Fig. 8b. During the first sodiation, an SEI is formed and stabilized on the surface and inside the pores of the negative hard carbon electrode. The sequence of the next sodiation processes indicates that the adsorption of Na+ ions on the hard carbon surface and their insertion between the graphene layers occurs first, followed by the filling of the surface of the pore walls. If the pores are large enough to accommodate more than one layer of quasi-metallic Na atoms, the continuous filling results in the formation of quasi-metallic Na clusters (Na3). During desodiation, the process is reversed. In the first phase of the process, the quasi-metallic Na atoms, which are part of the quasi-metallic Na clusters, are extracted from the structure. This is followed by the extraction of the quasi-metallic Na atoms that are part of the pore wall surface and finally, the Na+ ions between the graphene sheets and on surface are removed. In the case of underpotential deposition, nucleation of Na metals occurs at the surface of the negative electrode, resulting in Na metal plating (Fig. 8c). The chemical environments of the Na plating and the Na dendrites are different due to their different structural and chemical properties and can be distinguished by solid-state NMR.
Conclusions
We demonstrated the versatility of operando, in situ and ex situ solid-state NMR spectroscopy in elucidating sodium storage mechanisms in Na-ion batteries. This technique, complemented by XPS and SEM, enhanced our understanding of the sodium environment in hard carbon. We explained SEI stabilization during the first cycle and monitored electrochemical mechanisms during sodiation/desodiation, distinguishing Na+ ion insertion between graphene layers from pore filling, and differentiating dendrite formation from underpotential Na plating.Hard carbon derived from corncobs (CC@1400 °C) showed stable sodiation capacities over 100 cycles. Analysis revealed interactions between Na+ ion adsorption and electrolyte decomposition, with ex situ SEM, EDX, and XPS confirming SEI formation and stabilization after the first cycle, predominantly NaF at the surface and Na2CO3 in the bulk. Ex situ NMR corroborated the XPS findings and provided additional insights into the electrolyte solvent contributions.
Operando/in situ NMR identified specific stages of Na+ ion insertion between graphene layers and pore filling during sodiation, with larger pores at higher carbonization temperatures supporting the formation of quasi-metallic Na clusters. With NMR, we were able to determine and differentiate the stages of the plateau where the processes of insertion and pore filling take place. Additionally, the volume fraction and average width of nanometre-sized pores, measured by ex situ SAXS in the previous study, increase with carbonization temperature and are consistent with the NMR findings. NMR also distinguished dendrite formation from Na metal plating, tracking the processes via chemical shifts. SEM images of Na metal deposits are consistent with NMR predictions.
A comprehensive mechanism for Na+ ion behavior in hard carbon was proposed: during initial sodiation, Na+ ions adsorb on the hard carbon surface and insert between graphene layers and then fill the pore walls and at the end quasi-metallic Na clusters are formed if the pores are large enough. During desodiation, this process reverses. Underpotential deposition leads to Na metal plating, with distinct shifts reflecting the different properties of plated Na and dendrites.
Data availability
Data for this article, including NMR, XPS and electrochemical data are available at ZENODO at https://doi.org/10.5281/zenodo.13896896.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are highly grateful to Prof. Gregor Mali for his generous long-time contribution to the development of instrumentation and theory in the field of solid-state NMR at the National Institute of Chemistry, as well as his scientific and personal guidance, on which this research was subsequently built. We would like to thank the Slovenian NMR Centre for the use of their spectrometer and Uroš Javornik for his advice on the NMR measurements. This research is part of the PhD project “Unravelling the electrochemical mechanisms of battery degradation by operando NMR and X-ray absorption spectroscopy” supported by CERIC-ERIC. This research also received financial support from the Slovenian Research and Innovation Agency (ARIS) under research programs P2-0423 and P1-0021 and project N2-0266. Additionally, this research was supported by the NATO Science for Peace and Security (SPS) Programme under grant G5836-SUPERCAR.References
- E. Irisarri, A. Ponrouch and M. R. Palacin, Review—Hard Carbon Negative Electrode Materials for Sodium-Ion Batteries, J. Electrochem. Soc., 2015, 162, A2476–A2482 CrossRef CAS.
- X. Chen, Y. Zheng, W. Liu, C. Zhang, S. Li and J. Li, High-performance sodium-ion batteries with a hard carbon anode: transition from the half-cell to full-cell perspective, Nanoscale, 2019, 11, 22196–22205 RSC.
- C. Matei Ghimbeu, A. Beda, B. Réty, H. El Marouazi, A. Vizintin, B. Tratnik, L. Simonin, J. Michel, J. Abou-Rjeily and R. Dominko, Review: Insights on Hard Carbon Materials for Sodium-Ion Batteries (SIBs): Synthesis – Properties – Performance Relationships, Adv. Energy Mater., 2024, 14, 2303833 CrossRef CAS.
- R. R. Gaddam and G. Zhao, Handbook of Sodium-Ion Batteries, Jenny Stanford Publishing, 2022, DOI:10.1201/9781003308744.
- C. Bommier, T. W. Surta, M. Dolgos and X. Ji, New Mechanistic Insights on Na-Ion Storage in Nongraphitizable Carbon, Nano Lett., 2015, 15, 5888–5892 CrossRef CAS PubMed.
- L. Wang, Y. Lu, J. Liu, M. Xu, J. Cheng, D. Zhang and J. B. Goodenough, A Superior Low-Cost Cathode for a Na-Ion Battery, Angew. Chem., Int. Ed., 2013, 52, 1964–1967 CrossRef CAS PubMed.
- H. Kim, J. Hong, G. Yoon, H. Kim, K.-Y. Park, M.-S. Park, W.-S. Yoon and K. Kang, Sodium intercalation chemistry in graphite, Energy Environ. Sci., 2015, 8, 2963–2969 RSC.
- B. Tratnik, N. Van de Velde, I. Jerman, G. Kapun, E. Tchernychova, M. Tomšič, A. Jamnik, B. Genorio, A. Vizintin and R. Dominko, Correlating Structural Properties with Electrochemical Behavior of Non-graphitizable Carbons in Na-Ion Batteries, ACS Appl. Energy Mater., 2022, 5, 10667–10679 CrossRef CAS PubMed.
- D. A. Stevens and J. R. Dahn, High Capacity Anode Materials for Rechargeable Sodium-Ion Batteries, J. Electrochem. Soc., 2000, 147, 1271 CrossRef CAS.
- M. Anji Reddy, M. Helen, A. Groß, M. Fichtner and H. Euchner, Insight into Sodium Insertion and the Storage Mechanism in Hard Carbon, ACS Energy Lett., 2018, 3, 2851–2857 CrossRef CAS.
- D. Chen, W. Zhang, K. Luo, Y. Song, Y. Zhong, Y. Liu, G. Wang, B. Zhong, Z. Wu and X. Guo, Hard carbon for sodium storage: mechanism and optimization strategies toward commercialization, Energy Environ. Sci., 2021, 14, 2244–2262 RSC.
- P. Yu, W. Tang, F.-F. Wu, C. Zhang, H.-Y. Luo, H. Liu and Z.-G. Wang, Recent progress in plant-derived hard carbon anode materials for sodium-ion batteries: a review, Rare Met., 2020, 39, 1019–1033 CrossRef CAS.
- F. Xie, Z. Xu, Z. Guo and M.-M. Titirici, Hard carbons for sodium-ion batteries and beyond, Prog. Energy, 2020, 2, 042002 CrossRef.
- A. Rajh, M. Gabrijelčič, B. Tratnik, K. Bučar, I. Arčon, M. Petric, R. Dominko, A. Vizintin and M. Kavčič, Structural and chemical analysis of hard carbon negative electrode for Na-ion battery with X-ray Raman scattering and solid-state NMR spectroscopy, Carbon, 2024, 228, 119398 CrossRef CAS.
- K. Gotoh, 23 Na Solid-State NMR Analyses for Na-Ion Batteries and Materials, Batteries Supercaps, 2021, 4, 1267–1278 CrossRef CAS.
- F. RosaLinp E, Crystallite growth in graphitizing and non-graphitizing carbons, Proc. R. Soc. London, Ser. A, 1951, 209, 196–218 Search PubMed.
- U. Mittal, L. Djuandhi, N. Sharma and H. L. Andersen, Structure and function of hard carbon negative electrodes for sodium-ion batteries, JPhys Energy, 2022, 4, 042001 CrossRef CAS.
- X. Chen, J. Tian, P. Li, Y. Fang, Y. Fang, X. Liang, J. Feng, J. Dong, X. Ai, H. Yang and Y. Cao, An Overall Understanding of Sodium Storage Behaviors in Hard Carbons by an “Adsorption-Intercalation/Filling” Hybrid Mechanism, Adv. Energy Mater., 2022, 12, 2200886 CrossRef CAS.
- N. Sun, Z. Guan, Y. Liu, Y. Cao, Q. Zhu, H. Liu, Z. Wang, P. Zhang and B. Xu, Extended “Adsorption–Insertion” Model: A New Insight into the Sodium Storage Mechanism of Hard Carbons, Adv. Energy Mater., 2019, 9, 1901351 CrossRef.
- Z. V Bobyleva, O. A. Drozhzhin, K. A. Dosaev, A. Kamiyama, S. V Ryazantsev, S. Komaba and E. V Antipov, Unveiling pseudocapacitive behavior of hard carbon anode materials for sodium-ion batteries, Electrochim. Acta, 2020, 354, 136647 CrossRef.
- H. Au, H. Alptekin, A. C. S. Jensen, E. Olsson, C. A. O'Keefe, T. Smith, M. Crespo-Ribadeneyra, T. F. Headen, C. P. Grey, Q. Cai, A. J. Drew and M. M. Titirici, A revised mechanistic model for sodium insertion in hard carbons, Energy Environ. Sci., 2020, 13, 3469–3479 RSC.
- P. C. Tsai, S. C. Chung, S. K. Lin and A. Yamada, Ab initio study of sodium intercalation into disordered carbon, J. Mater. Chem. A, 2015, 3, 9763–9768 RSC.
- K. Gotoh, T. Yamakami, I. Nishimura, H. Kometani, H. Ando, K. Hashi, T. Shimizu and H. Ishida, Mechanisms for overcharging of carbon electrodes in lithium-ion/sodium-ion batteries analysed by operando solid-state NMR, J. Mater. Chem. A, 2020, 8, 14472–14481 RSC.
- H. Zhou, Y. Song, B. Zhang, H. Sun, I. A. Khurshid, Y. Kong, L. Chen, L. Cui, D. Zhang, W. Wang, L. Yang and X. Du, Overview of electrochemical competing process of sodium storage and metal plating in hard carbon anode of sodium ion battery, Energy Storage Mater., 2024, 71, 103645 CrossRef.
- P. M. Bayley, N. M. Trease and C. P. Grey, Insights into Electrochemical Sodium Metal Deposition as Probed with in Situ23Na NMR, J. Am. Chem. Soc., 2016, 138, 1955–1961 CrossRef CAS.
- M. Mandl, J. Becherer, D. Kramer, R. Mönig, T. Diemant, R. J. Behm, M. Hahn, O. Böse and M. A. Danzer, Sodium metal anodes: Deposition and dissolution behaviour and SEI formation, Electrochim. Acta, 2020, 354, 136698 CrossRef CAS.
- Y. Deng, J. Zheng, A. Warren, J. Yin, S. Choudhury, P. Biswal, D. Zhang, L. A. Archer, Y. Deng, J. Zheng, A. Warren, S. Choudhury, P. Biswal, L. A. Archer Robert Frederick, J. Yin and D. Zhang, On the Reversibility and Fragility of Sodium Metal Electrodes, Adv. Energy Mater., 2019, 9, 1901651 CrossRef CAS.
- R. Mogensen, D. Brandell and R. Younesi, Solubility of the Solid Electrolyte Interphase (SEI) in Sodium Ion Batteries, ACS Energy Lett., 2016, 1, 1173–1178 CrossRef CAS.
- F. A. Soto, P. Yan, M. H. Engelhard, A. Marzouk, C. Wang, G. Xu, Z. Chen, K. Amine, J. Liu, V. L. Sprenkle, F. El-Mellouhi, P. B. Balbuena and X. Li, Tuning the Solid Electrolyte Interphase for Selective Li- and Na-Ion Storage in Hard Carbon, Adv. Mater., 2017, 29, 1606860 CrossRef PubMed.
- G. G. Eshetu, S. Grugeon, H. Kim, S. Jeong, L. Wu, G. Gachot, S. Laruelle, M. Armand and S. Passerini, Comprehensive Insights into the Reactivity of Electrolytes Based on Sodium Ions, ChemSusChem, 2016, 9, 462–471 CrossRef CAS PubMed.
- Y. Huang, L. Zhao, L. Li, M. Xie, F. Wu and R. Chen, Electrolytes and Electrolyte/Electrode Interfaces in Sodium-Ion Batteries: From Scientific Research to Practical Application, Adv. Mater., 2019, 31, 1808393 CrossRef PubMed.
- M. A. Muñoz-Márquez, M. Zarrabeitia, E. Castillo-Martínez, A. Eguía-Barrio, T. Rojo and M. Casas-Cabanas, Composition and evolution of the solid-electrolyte interphase in Na2Ti3O7 electrodes for Na-Ion batteries: XPS and auger parameter analysis, ACS Appl. Mater. Interfaces, 2015, 7, 7801–7808 CrossRef PubMed.
- C. Bommier, D. Leonard, Z. Jian, W. F. Stickle, P. A. Greaney and X. Ji, New Paradigms on the Nature of Solid Electrolyte Interphase Formation and Capacity Fading of Hard Carbon Anodes in Na-Ion Batteries, Adv. Mater. Interfaces, 2016, 3, 1600449 CrossRef.
- D. I. Iermakova, R. Dugas, M. R. Palacín and A. Ponrouch, On the Comparative Stability of Li and Na Metal Anode Interfaces in Conventional Alkyl Carbonate Electrolytes, J. Electrochem. Soc., 2015, 162, A7060–A7066 CrossRef CAS.
- Z. Shadike, E. Zhao, Y. Zhou, X. Yu, Y. Yang, E. Hu, S. Bak, L. Gu and X. Yang, Advanced Characterization Techniques for Sodium-Ion Battery Studies, Adv. Energy Mater., 2018, 8, 1702588 CrossRef.
- Y. Chen, Z. Dong, S. Lai, Y. Li, W. Lv, Y. He, F. Kang and M. Liu, Review for Advanced NMR Characterization of Carbon-Based and Metal Anodes in Sodium Batteries, Adv. Funct. Mater., 2024, 2408657 CrossRef CAS.
- K. Gotoh, T. Ishikawa, S. Shimadzu, N. Yabuuchi, S. Komaba, K. Takeda, A. Goto, K. Deguchi, S. Ohki, K. Hashi, T. Shimizu and H. Ishida, NMR study for electrochemically inserted Na in hard carbon electrode of sodium ion battery, J. Power Sources, 2013, 225, 137–140 CrossRef CAS.
- J. M. Stratford, P. K. Allan, O. Pecher, P. A. Chater and C. P. Grey, Mechanistic insights into sodium storage in hard carbon anodes using local structure probes, Chem. Commun., 2016, 52, 12430–12433 RSC.
- J. M. Bray, C. L. Doswell, G. E. Pavlovskaya, L. Chen, B. Kishore, H. Au, H. Alptekin, E. Kendrick, M.-M. Titirici, T. Meersmann and M. M. Britton, Operando visualisation of battery chemistry in a sodium-ion battery by 23Na magnetic resonance imaging, Nat. Commun., 2020, 11, 2083 CrossRef CAS.
- O. Pecher, J. Carretero-González, K. J. Griffith and C. P. Grey, Materials' Methods: NMR in Battery Research, Chem. Mater., 2017, 29, 213–242 CrossRef CAS.
- O. Pecher, P. M. Bayley, H. Liu, Z. Liu, N. M. Trease and C. P. Grey, Automatic Tuning Matching Cycler (ATMC) in situ NMR spectroscopy as a novel approach for real-time investigations of Li- and Na-ion batteries, J. Magn. Reson., 2016, 265, 200–209 CrossRef CAS PubMed.
- L. Zhou, M. Leskes, A. J. Ilott, N. M. Trease and C. P. Grey, Paramagnetic electrodes and bulk magnetic susceptibility effects in the in situ NMR studies of batteries: Application to Li1.08Mn1.92O4 spinels, J. Magn. Reson., 2013, 234, 44–57 CrossRef CAS PubMed.
- R. Pigliapochi, L. O'Brien, A. J. Pell, M. W. Gaultois, Y. Janssen, P. G. Khalifah and C. P. Grey, When Do Anisotropic Magnetic Susceptibilities Lead to Large NMR Shifts? Exploring Particle Shape Effects in the Battery Electrode Material LiFePO 4, J. Am. Chem. Soc., 2019, 141, 13089–13100 CrossRef CAS PubMed.
- A. Kubo, T. P. Spaniol and T. Terao, The Effect of Bulk Magnetic Susceptibility on Solid State NMR Spectra of Paramagnetic Compounds, J. Magn. Reson., 1998, 133, 330–340 CrossRef CAS PubMed.
- B. Tratnik, S. Aina, E. Tchernychova, M. Gabrijelčič, G. Mali, M. P. Lobera, M. Bernechea, M. Morcrette, A. Vizintin and R. Dominko, Exploring hybrid hard carbon/Bi 2 S 3 -based negative electrodes for Na-ion batteries, Green Chem., 2024, 26, 6089–6099 RSC.
- A. Beda, F. Rabuel, M. Morcrette, S. Knopf, P.-L. Taberna, P. Simon and C. Matei Ghimbeu, Hard carbon key properties allow for the achievement of high Coulombic efficiency and high volumetric capacity in Na-ion batteries, J. Mater. Chem. A, 2021, 9, 1743–1758 RSC.
- H. Kim, J. C. Hyun, D. Kim, J. H. Kwak, J. B. Lee, J. H. Moon, J. Choi, H. Lim, S. J. Yang, H. M. Jin, D. J. Ahn, K. Kang, H. Jin, H. Lim and Y. S. Yun, Revisiting Lithium- and Sodium-Ion Storage in Hard Carbon Anodes, Adv. Mater., 2023, 35, e2209128 CrossRef PubMed.
- G. G. Eshetu, T. Diemant, M. Hekmatfar, S. Grugeon, R. J. Behm, S. Laruelle, M. Armand and S. Passerini, Impact of the electrolyte salt anion on the solid electrolyte interphase formation in sodium ion batteries, Nano Energy, 2019, 55, 327–340 CrossRef CAS.
- M. Ma, H. Cai, C. Xu, R. Huang, S. Wang, H. Pan and Y.-S. Hu, Engineering Solid Electrolyte Interface at Nano-Scale for High-Performance Hard Carbon in Sodium-Ion Batteries, Adv. Funct. Mater., 2021, 31, 2100278 CrossRef CAS.
- A. Ponrouch, E. Marchante, M. Courty, J.-M. Tarascon and M. R. Palacín, In search of an optimized electrolyte for Na-ion batteries, Energy Environ. Sci., 2012, 5, 8572 RSC.
- A. Ponrouch, R. Dedryvère, D. Monti, A. E. Demet, J. M. Ateba Mba, L. Croguennec, C. Masquelier, P. Johansson and M. R. Palacín, Towards high energy density sodium ion batteries through electrolyte optimization, Energy Environ. Sci., 2013, 6, 2361 RSC.
- J. Fondard, E. Irisarri, C. Courrèges, M. R. Palacin, A. Ponrouch and R. Dedryvère, SEI Composition on Hard Carbon in Na-Ion Batteries After Long Cycling: Influence of Salts (NaPF 6 , NaTFSI) and Additives (FEC, DMCF), J. Electrochem. Soc., 2020, 167, 070526 CrossRef CAS.
- M. Dahbi, T. Nakano, N. Yabuuchi, S. Fujimura, K. Chihara, K. Kubota, J. Y. Son, Y. T. Cui, H. Oji and S. Komaba, Effect of Hexafluorophosphate and Fluoroethylene Carbonate on Electrochemical Performance and the Surface Layer of Hard Carbon for Sodium-Ion Batteries, ChemElectroChem, 2016, 3, 1856–1867 CrossRef CAS.
- M. Carboni, J. Manzi, A. R. Armstrong, J. Billaud, S. Brutti and R. Younesi, Analysis of the Solid Electrolyte Interphase on Hard Carbon Electrodes in Sodium-Ion Batteries, ChemElectroChem, 2019, 6, 1745–1753 CrossRef CAS.
- B. Ravdel, K. Abraham, R. Gitzendanner, J. DiCarlo, B. Lucht and C. Campion, Thermal stability of lithium-ion battery electrolytes, J. Power Sources, 2003, 119–121, 805–810 CrossRef CAS.
- J. M. Rimsza, E. G. Sorte and T. M. Alam, Hydration and Hydroxylation of MgO in Solution: NMR Identification of Proton-Containing Intermediate Phases, ACS Omega, 2019, 4, 1033–1044 CrossRef CAS.
- P. Barnes, K. Smith, R. Parrish, C. Jones, P. Skinner, E. Storch, Q. White, C. Deng, D. Karsann, M. L. Lau, J. J. Dumais, E. J. Dufek and H. Xiong, A non-aqueous sodium hexafluorophosphate-based electrolyte degradation study: Formation and mitigation of hydrofluoric acid, J. Power Sources, 2020, 447, 227363 CrossRef CAS.
- A. Sadoc, M. Body, C. Legein, M. Biswal, F. Fayon, X. Rocquefelte and F. Boucher, NMR parameters in alkali, alkaline earth and rare earth fluorides from first principle calculations, Phys. Chem. Chem. Phys., 2011, 13, 18539–18550 RSC.
- J. M. Stratford, P. K. Allan, O. Pecher, P. A. Chater and C. P. Grey, Mechanistic insights into sodium storage in hard carbon anodes using local structure probes, Chem. Commun., 2016, 52, 12430–12433 RSC.
- M. P. Mercer, M. Nagarathinam, E. M. Gavilán-Arriazu, A. Binjrajka, S. Panda, H. Au, M. Crespo-Ribadeneyra, M.-M. Titirici, E. P. M. Leiva and H. E. Hoster, Sodiation energetics in pore size controlled hard carbons determined via entropy profiling, J. Mater. Chem. A, 2023, 11, 6543–6555 RSC.
- Q. Li, X. Liu, Y. Tao, J. Huang, J. Zhang, C. Yang, Y. Zhang, S. Zhang, Y. Jia, Q. Lin, Y. Xiang, J. Cheng, W. Lv, F. Kang, Y. Yang and Q.-H. Yang, Sieving carbons promise practical anodes with extensible low-potential plateaus for sodium batteries, Natl. Sci. Rev., 2022, 9(8), nwac084 CrossRef CAS PubMed.
- R. Morita, K. Gotoh, M. Fukunishi, K. Kubota, S. Komaba, N. Nishimura, T. Yumura, K. Deguchi, S. Ohki, T. Shimizu and H. Ishida, Combination of solid state NMR and DFT calculation to elucidate the state of sodium in hard carbon electrodes, J. Mater. Chem. A, 2016, 4, 13183–13193 RSC.
- N. M. Trease, L. Zhou, H. J. Chang, B. Y. Zhu and C. P. Grey, In situ NMR of lithium ion batteries: Bulk susceptibility effects and practical considerations, Solid State Nucl. Magn. Reson., 2012, 42, 62–70 CrossRef CAS PubMed.
- R. Bhattacharyya, B. Key, H. Chen, A. S. Best, A. F. Hollenkamp and C. P. Grey, In situ NMR observation of the formation of metallic lithium microstructures in lithium batteries, Nat. Mater., 2010, 9, 504–510 CrossRef CAS PubMed.
- J. Lee, J. Kim, S. Kim, C. Jo and J. Lee, A review on recent approaches for designing the SEI layer on sodium metal anodes, Mater. Adv, 2020, 1, 3143 RSC.
- Y. Youn, B. Gao, A. Kamiyama, K. Kubota, S. Komaba and Y. Tateyama, Nanometer-size Na cluster formation in micropore of hard carbon as origin of higher-capacity Na-ion battery, npj Comput. Mater., 2021, 7, 48 CrossRef CAS.
- E. Šić, K. Schutjajew, U. Haagen, H. Breitzke, M. Oschatz, G. Buntkowsky and T. Gutmann, Electrochemical Sodium Storage in Hard Carbon Powder Electrodes Implemented in an Improved Cell Assembly: Insights from In Situ and Ex Situ Solid-State NMR, ChemSusChem, 2024, 17, e202301300 CrossRef PubMed.
- Z. Xu, Z. Guo, R. Madhu, F. Xie, R. Chen, J. Wang, M. Tebyetekerwa, Y.-S. Hu and M.-M. Titirici, Homogenous metallic deposition regulated by defect-rich skeletons for sodium metal batteries, Energy Environ. Sci., 2021, 14, 6381–6393 RSC.
- Y. Yui, M. Hayashi and J. Nakamura, In situ Microscopic Observation of Sodium Deposition/Dissolution on Sodium Electrode, Sci. Rep., 2016, 6, 22406 CrossRef CAS PubMed.
- Y. Zhao, K. R. Adair and X. Sun, Recent developments and insights into the understanding of Na metal anodes for Na-metal batteries, Energy Environ. Sci., 2018, 11, 2673–2695 RSC.
- B. Lee, E. Paek, D. Mitlin and S. W. Lee, Sodium Metal Anodes: Emerging Solutions to Dendrite Growth, Chem. Rev., 2019, 119, 5416–5460 CrossRef CAS PubMed.
- Z. W. Seh, J. Sun, Y. Sun and Y. Cui, A Highly Reversible Room-Temperature Sodium Metal Anode, ACS Cent. Sci., 2015, 1, 449–455 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta07135b |
| This journal is © The Royal Society of Chemistry 2025 |