
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From natural to synthetic hydrogels: how much biochemical complexity is required for mechanotransduction?†
Johnick F.
van Sprang
abc,
Imke P. M.
Smits
ac,
Jasper C. H.
Nooten
ab,
Peter-Paul K. H.
Fransen
ab,
Serge H. M.
Söntjens
 d,
Michel H. C. J.
van Houtem
d,
Henk M.
Janssen
d,
Martin G. T. A.
Rutten
ab,
Maaike J. G.
Schotman
ab and
P. Y. W.
Dankers
*abc
d,
Michel H. C. J.
van Houtem
d,
Henk M.
Janssen
d,
Martin G. T. A.
Rutten
ab,
Maaike J. G.
Schotman
ab and
P. Y. W.
Dankers
*abc
aInstitute for Complex Molecular Systems, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: p.y.w.dankers@tue.nl
bLaboratory of Chemical Biology, Department of Biomedical Engineering, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
cLaboratory of Cell and Tissue Engineering, Department of Biomedical Engineering, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
dSyMO-Chem B.V., Den Dolech 2, 5612 AZ Eindhoven, The Netherlands
First published on 27th November 2024
Abstract
The biochemical complexity of a material determines the biological response of cells triggered by a cell-material interaction. The degree in which this complexity influences basic cell-material interactions such as cell adhesion, spreading, and mechanotransduction is not entirely clear. To this end, we compared three different hydrogel systems, ranging from completely natural to synthetic, in their ability to induce mechanotransduction in kidney epithelial cells (HK-2). A natural hydrogel system was developed based on a decellularized kidney extracellular matrix (dECM). Supramolecular ureido-pyrimidinone (UPy)-glycinamide molecules, with self-associative behavior, were used for a hybrid and complete synthetic system. A hybrid system was engineered by co-assembling this monovalent UPy molecule with a hyaluronic acid, functionalized with ∼7 UPy-groups (UPy-HA), into a transient network. A similar approach was used for the synthetic hydrogel system, in which the multivalent UPy-HA was replaced with a bivalent UPy-PEG molecule with bioinert properties. Both hybrid and synthetic hydrogel systems were more mechanically tunable compared to the dECM hydrogel. The higher bulk stiffness in combination with the introduction of collagen type I mimicking UPy-additives allowed these materials to induce more nuclear yes-associated protein translocation in HK-2 cells compared to the biochemically complex dECM hydrogel. This demonstrated that minimal biochemical complexity is sufficient for inducing mechanotransduction.
1. Introduction
The holy grail for the biomaterial field is to recreate the complexity found in the native extracellular matrix, in terms of mechanical properties and biochemical composition, in a synthetic fashion.1,2 This feat would allow unprecedented control over cell-material interactions and thereby dramatically improve regenerative cell therapies. However, synthetic replication is currently not feasible due to the large amount of complex molecular species found in this natural material.3 The extracellular matrix demonstrates viscoelastic behavior in soft tissues and its main constituents are fibrillar proteins, glycoproteins, proteoglycans, and carbohydrates with various degrees of sulfated substitution.4–9 This large number of molecular species hints at complex function. Traditionally, these functions are subdivided into four categories: (1) a support structure for cellular adhesion and mechanotransduction, which allows cytoskeletal and chromatin remodeling, (2) compartmentalization of microstructures in the tissue (3) a local storage depot for biochemical molecules (i.e., growth factors and cytokines), and (4) a modulator for presentation of these biochemical molecules to cell surface receptors.8–10Repurposing animal tissue to generate biomaterials is one possible strategy to introduce a high degree of biological information into a material. These animal tissues are decellularized using surfactants. The resulting ‘naked’ matrix is processed into a hydrogel through milling and enzymatic treatment.11–14 Recently, these natural-derived hydrogels have been used for tissue engineering, in which they are used as a natural microenvironment for developing organoids.15–19 The unparalleled biochemical complexity makes these materials eminently suitable for such complex cultures. However, these natural hydrogels suffer from batch-to-batch variation, and poor control over hierarchical organization that is found in their native counterparts, which is critical for mechanical properties.13,20 Furthermore, the required amount of biochemical complexity is dependent on the desired biological response that the cell-material interaction needs to induce.21
Supramolecular hydrogels based on ureido-pyrimidinone (UPy) motifs have been used as artificial matrix that approximate certain mechanical and structural aspects of the native extracellular matrix.22–24 The UPy-moieties in these hydrogels dimerize through quadruple hydrogen bonding, and stack using π–π interactions and additional hydrogen bonds of a flanking urea group.25 The supramolecular stacks associate through hydrophobic interactions into fibrous superstructures reminiscent of the fibrillar architecture found in native extracellular matrix.22,26,27 Furthermore, the self-associative behavior of the UPy-moieties allows for different molecules containing complementary supramolecular moieties to co-assemble into one transient network.24,28 This modular feature of supramolecular materials enables a mix-and-match strategy in which the biochemical complexity and mechanical properties are tunable.24
Here, we investigated to which extent mechanotransduction in renal epithelial cells is dependent on biochemical complexity and mechanical properties using three types of hydrogels ranging from completely natural to synthetic (Fig. 1a). The natural hydrogel constituted a porcine processed, decellularized kidney extracellular matrix (dECM) derived from porcine. The hybrid and synthetic hydrogel systems incorporated a monovalent UPy-functionalized glycinamide (UPy-Gly) molecule, which required bi, -or multivalent UPy molecules to co-assemble into an entangled, hierarchical network.24 The hybrid hydrogel system consisted of a hyaluronic acid biopolymer, functionalized with ∼7 UPy moieties (UPy-HA), to cross-link the UPy-Gly assemblies (Fig. 1b). Hyaluronic acid is an important constituent of many tissues, interacts with hyaladherins (e.g., CD44, RHAMM, C1QBP, and HABP2) on the cell membrane surface, and modulates growth factor storage and presentation to cells.29–33 Additional biochemical complexity was incorporated into the hybrid system by introducing UPy-functionalized collagen type I mimetic polypeptides. One cell-adhesive peptide was based on a triple-helix forming polypeptide conjugated to a UPy-moiety (UPy-GFOGER),34 which binds to the α2β1 integrin receptor.35,36 The other additive was an RGD-sequence enriched, recombinant collagen type I peptide with on average two UPy-groups conjugated to lysine residues (UPy-RCP), which binds to the α5β1, -and αvβ3 integrin receptor.23,37,38
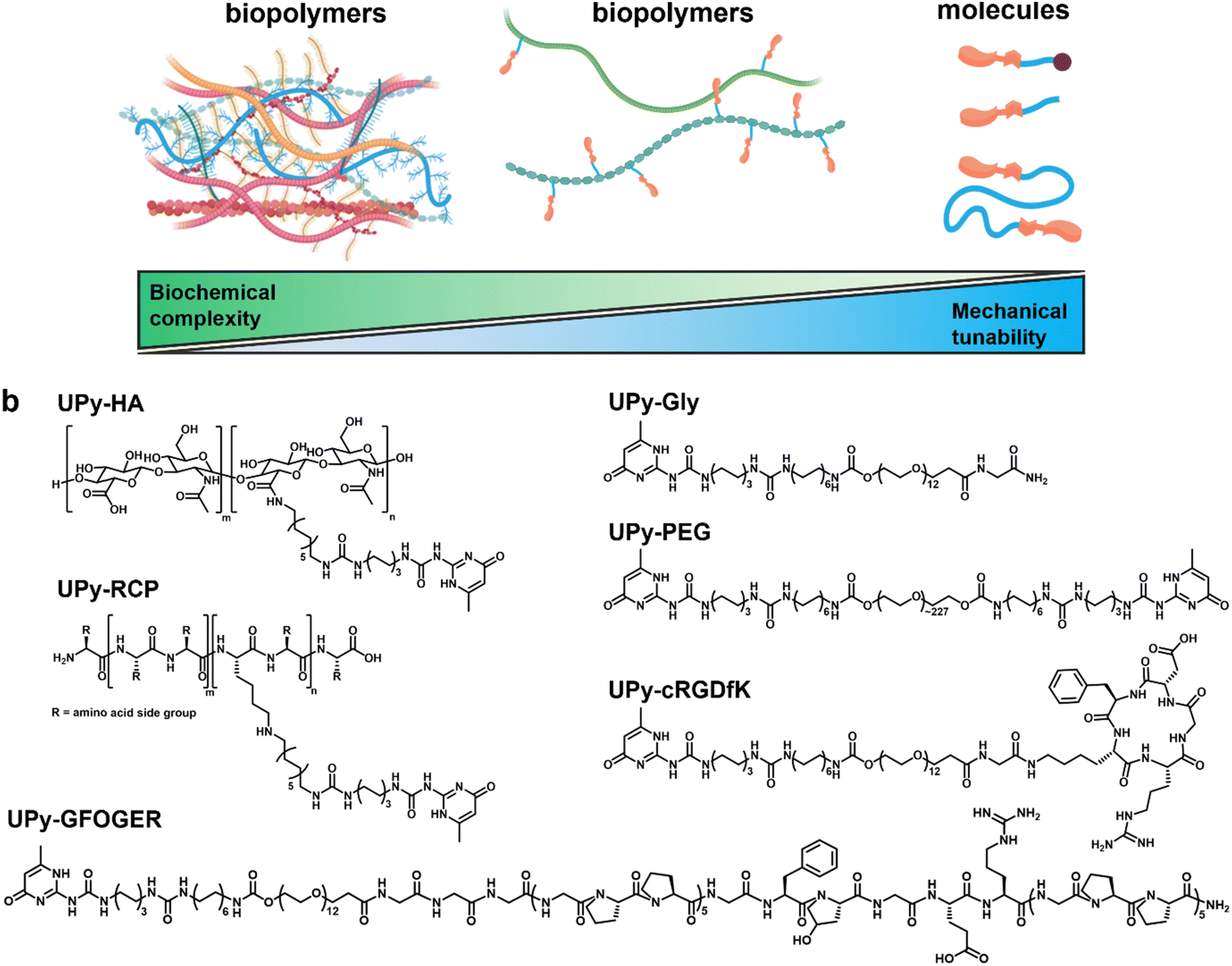 | ||
| Fig. 1 From natural to synthetic hydrogel systems. (a) Schematic overview illustrating the difference in biochemical complexity and mechanical tunability between different material classes. (b) Molecular structures of UPy-modified molecules used in both the hybrid (left side) and synthetic (right side) hydrogel systems. | ||
A poly(ethylene glycol) functionalized at both ends with UPy-groups (UPy-PEG) was used for the synthetic hydrogel system to reversibly cross-link the UPy-Gly assemblies (Fig. 1b).24 The hydrophilic PEG backbone of the bivalent UPy molecule is relatively bioinert.39 The synthetic hydrogel system incorporated a UPy-conjugated cRGDfK peptide (UPy-cRGDfK), which is considered the minimal peptide sequence required to induce cell adhesion (Fig. 1b).24 The mechanical tunability of these three hydrogel systems was investigated and their ability to induce cell adhesion, spreading, and mechanotransduction in immortalized proximal tubular epithelial cell line from human kidney (HK-2) was subsequently evaluated with respect to mechanical properties and biochemical complexity.
2. Experimental
2.1 Synthesis of UPy-RCP and UPy-Sulfo-Cy5
The synthesis of UPy-RCP, UPy-GFOGER, and UPy-Sulfo-Cy5 was described previously.23,24,342.2 Synthesis of tert-butyl (12-aminododecyl)carbamate (1)
A mixture of 1,12-diaminododecane (10 g, 50 mmol) and tert-butyl phenyl carbonate (9.8 g, 1 eq.) in ethanol (150 mL) was heated under reflux for 16 hours (Scheme 1). The resulting mixture was evaporated to dryness, affording a mixture of protected amines. This mixture was then separated by column chromatography (SiO2, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9:90 TEA/MeOH/CHCl3) to afford 6.8 g (91%) of the title compound. 1H-NMR (CDCl3) δ = 4.75–4.10 (br.m, 1H), 3.10 (q, 2H), 3.68 (t, 2H), 1.55–1.20 (br.m, 31H) ppm. 13C-NMR (CDCl3) δ = 156.12, 79.12, 42.41, 40.78, 33.99, 30.21, 29.75, 29.71, 29.68, 29.64, 29.43, 28.58, 27.03, 26.95 ppm. IR (ATR) ν = 3368, 2976, 2920, 2851, 1686, 1526, 1467, 1390, 1365, 1323, 1278, 1248, 1175, 870, 781, 722, 589 cm−1. MS (MALDI, positive reflector mode) Calculated: C17H36N2O2 = 300.28 g mol−1; m/z found: 301.28 [M + H]+, 323.26 [M + Na]+.
:9:90 TEA/MeOH/CHCl3) to afford 6.8 g (91%) of the title compound. 1H-NMR (CDCl3) δ = 4.75–4.10 (br.m, 1H), 3.10 (q, 2H), 3.68 (t, 2H), 1.55–1.20 (br.m, 31H) ppm. 13C-NMR (CDCl3) δ = 156.12, 79.12, 42.41, 40.78, 33.99, 30.21, 29.75, 29.71, 29.68, 29.64, 29.43, 28.58, 27.03, 26.95 ppm. IR (ATR) ν = 3368, 2976, 2920, 2851, 1686, 1526, 1467, 1390, 1365, 1323, 1278, 1248, 1175, 870, 781, 722, 589 cm−1. MS (MALDI, positive reflector mode) Calculated: C17H36N2O2 = 300.28 g mol−1; m/z found: 301.28 [M + H]+, 323.26 [M + Na]+.
 | ||
| Scheme 1 Synthetic procedure for the functionalization of a hyaluronic acid backbone with UPy-moieties. Reaction conditions: (i) CHCl3, 50 °C, 16 hours, 91% yield; (ii) TFA: DCM 1:1 v/v%, 2.5 hours, yield 101%; (iii) 0.08 eq. hyaluronic acid, 1.1 eq. PyBOP, 23.3 eq. TEA, DMSO, r.t., 16 hours, 53% yield. | ||
2.3 Synthesis of tert-butyl(12-(3-(6-(3-(6-methyl-4-oxo-1,4-dihydropyrimidin-2- yl)ureido)hexyl)ureido)dodecyl)carbamate (2)
A solution of 1-(6-isocyanatohexyl)-3-(6-methyl-4-oxo-1,4-dihydropyrimidin-2-yl)urea (1.98 g, 6.8 mmol) and mono-Boc-protected 1,12-diaminododecane 1 (2.18 g, 1.07 eq.) was stirred in chloroform at 50 °C for 16 hours, affording a white dispersion (Scheme 1). The solvent was evaporated, and the white solid was redissolved in 300 mL 1:1 chloroform/methanol under gentle heating. The slightly turbid solution was flushed over a silica plug, which was subsequently flushed with another 100 mL of the 1:1 solvent mixture. The combined solvent fractions were combined and evaporated to dryness. The material was again dissolved in 300 mL 1:1 chloroform/methanol and subjected again to the procedure above. After evaporation of the combined solvent fractions, this resulted in 3.7 g (91%) of the title compound as a white solid. 1H-NMR (DMSO, 50 °C) δ = 11.5–8.5 (br.s, 2H), 7.48 (br.s, 1H), 6.59 (br.s, 1H), 5.75 (s, 1H), 5.65 (q, 2H), 3.14 (q, 2H), 2.96 (m, 4H), 2.90 (q, 2H), 2.10 (s, 3H), 1.56–1.15 (br.m, 37H) ppm. 13C-NMR (DMSO, 50 °C) δ = 161.42, 157.95, 155.36, 154.62, 151.31, 104.27, 77.07, 39.46, 38.85, 38.60, 29.85, 29.78, 29.28, 28.90, 28.81, 28.76, 28.73, 28.67, 28.59, 28.49, 28.09, 26.19, 26.04, 25.83, 25.80, 22.85 ppm. IR (ATR) ν = 3335, 2921, 2851, 1699, 1668, 1625, 1576, 1520, 1480, 1464, 1444, 1414, 1390, 1364. 1309, 1254, 1172, 1137, 1041, 1017, 974, 942, 869, 844, 807, 784, 769, 742, 601, 564, 526, 465 cm−1. MS (MALDI, positive reflector mode) Calculated: C30H55N7O5 = 593.43 g mol−1; m/z found: 616.44 [M + Na]+, 632.42 [M + K]+.
2.4 1-(12-Aminododecyl)-3-(6-(3-(6-methyl-4-oxo-1,4-dihydropyrimidin-2-yl)ureido)hexyl)urea trifluoroacetate (3)
Boc-protected moiety 2 (3.0 g) was stirred in 25 mL DCM and 25 mL TFA for 2½ hours at room temperature (Scheme 1). The resulting mixture was partially evaporated at 25–30 °C on a rotary evaporator to remove most of the DCM, after which the product was precipitated with 200 mL diethyl ether. The supernatant was decanted and the precipitate was stirred with 50 mL for 20 minutes, decanted, dried, stirred with another 150 mL for 1 h, collected by filtration, and dried under vacuum to afford 3.1 g (101%) of the title compound as a white solid. 1H-NMR (DMSO) δ = 12.26–10.70 (br.s, 1H), 10.70–9.00 (br.s, 1H), 8.12–7.06 (br.m, 4H), 5.77 (br.s, 3H), 3.12 (q, 2H), 2.95 (m, 4H), 2.77 (m, 2H), 2.10 (s, 3H), 1.57–1.16 (br.m, 28H). 13C-NMR (DMSO) δ 164.66 (br), 161.25 (br), 158.19 (q, TFA), 158.13, 154.83 (br), 151.43 (br), 117.21 (q, TFA), 104.55, 39.21, 38.97, 38.83, 30.07, 30.00, 29.10, 29.04, 28.99, 28.90, 28.81, 28.50, 26.98, 26.39, 26.02, 26.00, 25.76, 23.17 (br) ppm. IR (ATR) ν = 3322, 2924, 2853, 1697, 1661, 1619, 1577, 1523, 1464, 1442, 1379, 1307, 1253, 1201, 1179, 1132, 1030, 943, 837, 799, 766, 740, 722, 646, 602, 565, 519 cm−1. MS (LC/MS, positive mode): Calculated: C25H47N7O3 = 493.37 g mol−1; m/z found: 494.42 [M + H]+, 987.33 [2M + H]+.2.5 Synthesis of UPy-HA (4)
Hyaluronic acid (0.5 g, 2.6 × 10−2 mmol), UPy-derivative 3 (193 mg, 12 eq.) and PyBOP (248 mg, 18 eq.) were dried under vacuum at room temperature for 1 hour. The solids were subsequently dispersed in 100 mL DMSO. After addition of TEA (1 mL, 280 eq.) the reaction was stirred for another 16 hours at room temperature (Scheme 1). The resulting reaction mixture was dialyzed using a Spectra/Por 3.5 kDa regenerated cellulose dialysis membrane against DMSO (1 L, 3 × 0.5 L) over 2 days. The solvent was subsequently exchanged from DMSO to water (0.5 L, 1 L) and subsequently to 1:1 water/THF (4 × 1 L) over about 7 hours. The resulting solution was partly evaporated on a rotary evaporator to remove most of the THF and lyophilized to afford an off-white solid. This solid was subsequently stirred for 90 minutes with 20 mL chloroform, collected by filtration, stirred with 30 mL 1:5 methanol/chloroform for 1 hour, collected by filtration, dried under vacuum, stirred again with 20 mL 1:3 methanol/chloroform for 140 minutes, collected by filtration, and finally dried under vacuum to afford 315 mg of the desired compound (53%) as an off-white solid. 1H-NMR (D2O/KOH) δ = 5.89 ppm (br.m, 0.14 H), 4.65–4.35 (br.m, 2H), 4.05–3.20 (br.m, 10.5 H), 3.16–3.01 (br.m, 0.8 H), 2.60 (q, 0.9 H), 2.23–1.84 (br.m, 3.4 H), 1.66–1.13 (br.m, 4.3 H), 1.04 (t, 1.3 H) ppm. Based on these NMR data, comparing the acetyl absorption of the hyaluronic acid (overlapping with the UPy Me-signal) with other signals, the polymer contained approximately 7 UPy units and 0.15 eq. TEA on average per polymer chain.
2.6 dECM hydrogel formulation
Porcine decellularized kidneys were received from Leiden University Medical Center. Kidneys were decellularized as previously reported.40 The kidneys were washed 2× with milliQ and incubated in milliQ for 24 hours at 4 °C. The ureter and calyx were removed and the remaining tissue was dissected. The tissue sections were lyophilized and formed into a powder using a Mikro-Dismembrator (Sartorius). The dECM powder was enzymatically digested for 72 h using 1 mg mL−1 gastric mucosa pepsin (Sigma-Aldrich, P7012-1G) and 20 mM HCl solution. This yielded a white, cloudy suspension with a pH ∼2, which was then neutralized with 0.1 M NaOH solution at 4 °C and lyophilized to obtain a dECM digested powder. The dECM hydrogels were prepared by stirring the dECM digested powder overnight in PBS solution at 4 °C. The resulting mixture was allowed to gelate at 37 °C to obtain a dECM hydrogel.2.7 UPy hydrogel formulation
Solutions of UPy-Gly (SyMO-Chem), UPy-PEG (SyMO-Chem), and UPy-HA molecules were prepared by dissolving the compounds in alkaline PBS solution at 50 °C for 1 hour. The alkaline PBS contained 160 mM NaOH. The alkaline UPy-solutions were neutralized to a pH ∼ 7.4 with 2 M HCl. For cell-adherent hydrogels the UPy-Gly solution also contained 2 mM UPy-cRGDfK (SyMO-Chem), 0.18 mM UPy-RCP, or 6 mM UPy-GFOGER. After neutralization, the UPy-Gly solution was mixed with either the UPy-PEG or UPy-HA solution at a 1:1 volume ratio, which induced hydrogel formation. Table S1, ESI† provides an overview of concentrations and mol ratios of the hydrogel compositions used within this work.
2.8 Cryogenic transmission electron microscopy
Samples were prepared by mixing the lyophilized dECM digest with PBS overnight at 4 °C at 0.1 wt%. Lacey carbon film grids (200 mesh, 50 μm hole size; Electron Microscopy Sciences) were surface plasma treated at 5 mA for 40 seconds using a Cressington 208 carbon coater, and each dispersion (3 μL) was applied onto each grid. Using an automated vitrification robot (FEI Vitrobot Mark III), excess sample was removed through blotting with filter paper for 3 seconds at −3 mm. Thin films of dispersions were vitrified by plunging the grids into liquid ethane just above its freezing point. Imaging was carried out on a FEI-Titan TEM equipped with a field emission gun operating at 300 kV. Samples were imaged using a post-column Gatan energy filter and a 2048 × 2048 Gatan CCD camera. Micrographs were recorded at low dose conditions, using a defocus setting of −10 μm at 25000× magnification.
2.9 Rheological characterization
Rheological measurements were carried out on a TA Instruments Dynamic Hybrid Rheometer 3. The dECM hydrogels were measured using a 20 mm cone-plate geometry (2.007°) with a truncation gap of 56 μm. Samples were loaded as a pre-gel solution at 10 °C. After an initial frequency measurement, samples were heated to 37 °C to induce gelation, and the complex modulus G* (γ = 0.01, ω = 1 rad s−1) was measured for 1 hour to ensure stable gel formation. A solvent trap was used to minimize sample drying.Mono- and bivalent or multivalent UPy solutions were mixed at desired ratios and were allowed to gelate in the fridge at 4 °C overnight. Rheological measurements of these samples were carried out using an 8 mm cross hatched steel plate-plate geometry which was lowered until it made full contact with the gel, resulting in a typical gap of 250–725 μm. Low viscosity silicon oil (47 V 100, RHODORSIL®) was used around the hydrogel to minimize sample drying. Samples were loaded at 37 °C after which the complex modulus G* (γ = 0.01, ω = 1 rad s−1) was measured for 8 hours to ensure that samples were at a stable plateau modulus and were and not altered or damaged during loading.
Frequency sweep measurements were performed at ω = 0.1 rad s−1 to 100 rad s−1, at a strain of γ = 0.01. Stress-relaxation was measured by applying a strain of γ = 0.075 with a strain rise time of 0.09 s and monitoring the stress for 1000 s. The data were normalized using the stress at 1 second as starting point.
2.10 Fluorescence recovery after photo-bleaching
FRAP measurements were performed on a Leica TCS SP5 X confocal microscope (Leica Microsystems). UPy hydrogels with 25 μM UPy-Sulfo-Cy5 were formulated in 8-well μ-slide with glass bottom (Ibidi) by mixing freshly prepared UPy-Gly solution, which contained UPy-Sulfo-Cy5, and bivalent UPy-PEG, -or multivalent UPy-HA solution. After mixing, the samples were allowed to gelate and equilibrate overnight in the fridge at 4 °C, while protected from light. Afterwards, 200 μL PBS solution was pipetted on top to prevent drying during the measurement. A circular area, with a diameter of 50 μm, was excited in each sample with a white laser at 646 nm wavelength and emission was detected between 660 and 760 nm wavelength using a hybrid detector. The fluorescence intensity in the bleached spot was normalized by the fluorescence intensity of the reference spot.2.11 Cell culture of HK-2 cells
HK-2 cells (ATCC) were routinely cultured in T75 culture flasks in 10 mL Dulbecco's modified Eagle Medium (DMEM; 41966, Gibco), supplemented with 10 v/v% fetal bovine serum (Greiner Bio-One), and 1 v/v% penicillin and streptomycin (Invitrogen) at 37 °C and 5% CO2. Cells were passaged at 70–90% confluency as single cells and seeded at a density of 100000 cells cm−2. The supplemented DMEM was replaced every 2–3 days. Cells were routinely checked for mycoplasma contamination.
2.12 Culture of HK-2 cells on hydrogels
Hydrogels were prepared in a 96-well plate one day prior to seeding of HK-2 cells. The hydrogels were UV-sterilized 3 × 5 minutes and washed 3 × 10 minutes with supplemented DMEM. Afterwards, hydrogels were placed in the incubator overnight at 37 °C and 5% CO2. HK-2 cells at a confluence of ∼70% were enzymatically removed from T75 flasks and seeded on the hydrogels at a density of 30000 cells cm−2. The cells were cultured for 3 days at 37 °C and 5% CO2, and medium was replaced only on day 1 to remove unattached cells.
2.13 Immunohistochemistry
On day 3 of culture, the HK-2 cells were washed 2× with sterile PBS solution and fixated in 4% formaldehyde solution for 20 minutes in the fridge at 4 °C. The samples were washed 2× with PBS solution after fixation and incubated in 10% donkey serum + 0.3% Triton X-100 in PBS solution for 2 hours at room temperature. After blocking and permeabilization, samples were incubated with primary anti-YAP1 antibody (1:100 dilution; Santa Cruz Biotechnology) overnight in the fridge at 4 °C and incubated with a donkey host-derived anti-mouse alexa568-conjugated antibody (1:400 dilution; Jackson ImmunoResearch) and Alexa Fluor™ 555 Phalloidin (Thermofisher Scientific) for 2 hours at room temperature. Afterwards, samples were stained with NucBlue™ Live ReadyProbes™ Reagent (Hoechst 33342) (Thermofisher Scientific) for 10 minutes and mounted on glass coverslips (24 × 60 mm). Mounted samples were imaged using a Leica TCS SP8 X confocal microscope (Leica Microsystems).
2.14 1,9-dimethylmethylene blue assay
The renal dECM powder was digested in papain digestion buffer, which was composed of 100 mM phosphate buffer (pH = 6.5), 5 mM L-cysteine (Sigma-Aldrich), 5 mM EDTA (Sigma-Aldrich), and 140 μg mL−1 papain (Sigma-Aldrich), at 60 °C for 16 h. The digested supernatants were centrifuged and used to determine the sGAG concentration, using a 1,9-dimethylmethylene blue (DMMB) assay with chondroitin sulfate from shark cartilage (Sigma-Aldrich) as a standard. Next, 40 μL of the digested sample was transferred to a 96-wells plate and 150 μL DMMB solution was added. The absorbance was measured at 540 and 595 nm using a spectrophotometer (SynergyHT).2.15 Bicinchoninic acid (BCA) assay
The dECM digest powder (0.05 wt%) was solubilized in PBS by 15 minute sonication at 4 °C. Protein levels of the digests were measured using a BCA assay (Pierce). Solubilized dECM samples (25 μL) were mixed with 200 μL BCA working reagent and incubated at 37 °C for 30 minutes. Absorbance was measured at 562 nm using a spectrophotometer (SynergyHT). Bovine serum albumin (Roche) was used to generate a standard curve.2.16 Statistics and reproducibility
Data are represented as mean ± standard error of mean (s.e.m.). Statistical differences between conditions were tested with ANOVA analysis of variance followed by Bonferroni's post hoc test. Data were statistically significant if P < 0.05. Sample size and P values are included in the figure legends. Different significance levels (P values) are indicated in each figure with asterisks: **P < 0.01, ***P < 0.001, and ****P < 0.0001. Graphad Prism was used as software for statistical analysis.3. Results and discussion
3.1 Natural hydrogel system based on decellularized kidney extracellular matrix
A natural-derived hydrogel was produced using porcine kidneys. These kidneys were decellularized using a combination of SDS, Triton X-100, heparin, and PBS solution as previously reported.40,41 The resulting matrix was processed into a powder through a combination of lyophilization and mechanical homogenization using a vibrating mill. Matrix digestion was continued with enzymatic treatment of a pepsin solution at pH ∼2 for 72 hours, followed by neutralization with aqueous NaOH solution (1 M) at 4 °C (Fig. 2a). Above the critical gelation concentration, the dECM pre-solution is able to gelate upon increasing the temperature to 37 °C. A dilution series (0.05–0.75 wt%) was prepared to investigate the gelation threshold in terms of dECM weight content, which was determined to be around 0.3 wt% based on vial-inversion tests (Fig. 2b). Cryogenic transmission electron microscopy was used to investigate the physical structure of the hydrogel network, which demonstrated characteristic collagen bundles (Fig. 2c).42 Rheological measurements supported the observations of the vial-inversion test, except for 0.1 and 0.2 wt% dECM solution that behaved as a liquid (G′ < G′′) during the vial-inversion test and measured as a hydrogel (G′ > G′′) in the rheological measurement. From 0.3 wt% dECM these two tests aligned in result, leading us to conclude that gelation was induced upon increasing the concentration to 0.3 wt%. The storage modulus of this hydrogel was 10 Pa, which is a soft gel. Increasing the dECM content in the hydrogel up to 2 wt% allowed an increase of the storage modulus to 200 Pa (Fig. 2d). However, further increase was not possible due to solubility limitations of the dECM powder. This may be caused by the high collagen content, as collagens are often insoluble.43 Another possibility is excess removal of sulfated carbohydrate compounds during the decellularization and washing steps. The charged groups on the sulfated carbohydrates improve solubility through electrostatic interactions with the aqueous surroundings. To this end, a 1,9-dimethylmethylene blue assay was used to investigate the sulfated glycosaminoglycan content in the dECM hydrogel, which was found to be 1.1 ± 0.1 wt% (Fig. S1, ESI†). Indeed, this may explain the low solubility of the digested dECM powder in water, which limited the mechanical tunability. Finally, the protein concentration was determined to be at 18.0 ± 1.9 wt% as measured using a BCA assay (Fig. S1, ESI†).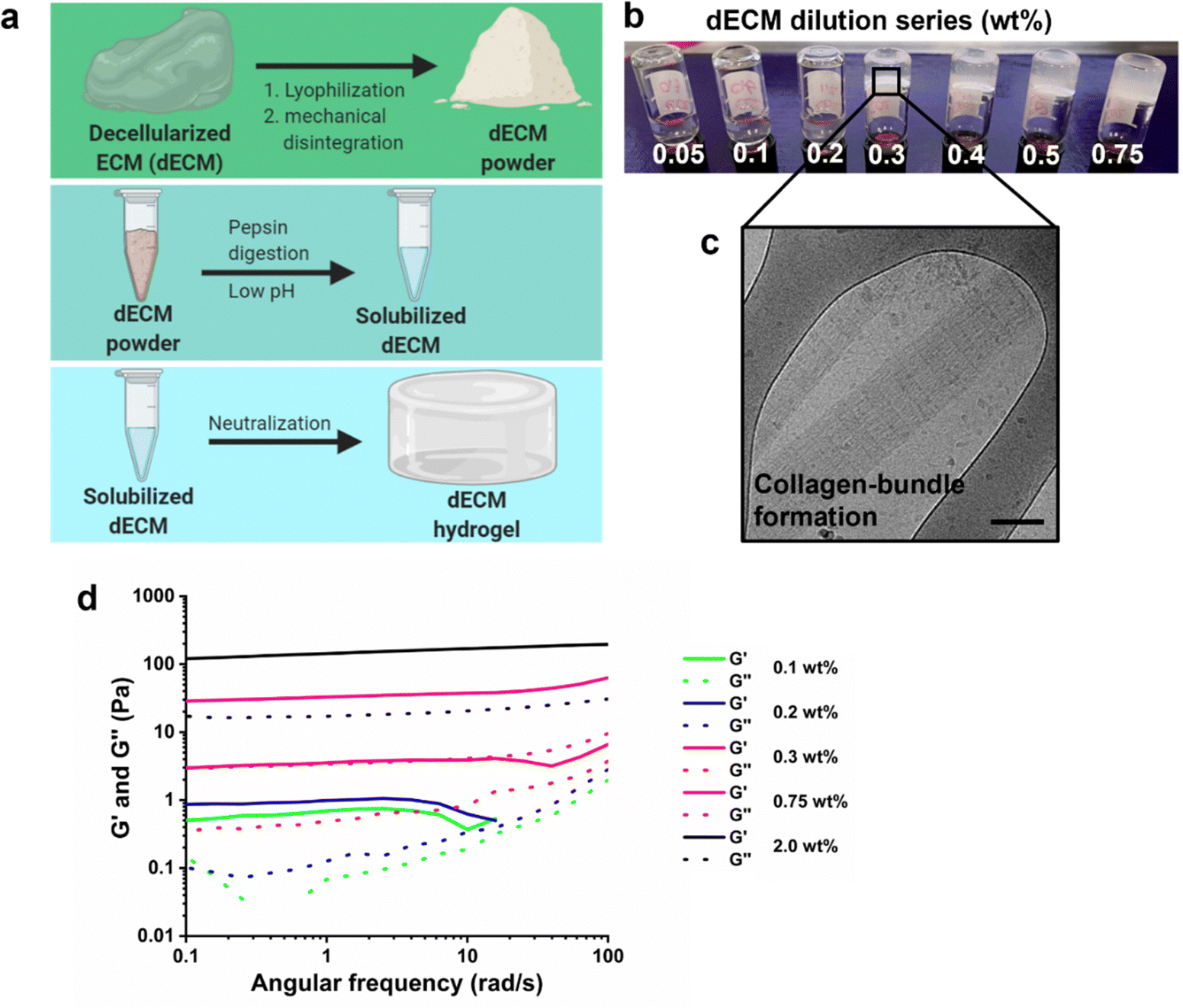 | ||
| Fig. 2 Preparation and rheological characterization of kidney dECM hydrogels. (a) Schematic overview illustrating the procedure to prepare a dECM hydrogel from a decellularized kidney. (b) A dilution series of dECM solutions (0.1–0.2–0.3–0.4–0.5–0.75–2.0 wt%) to determine the critical gelation concentration through a vial-inversion test. (c) A cryogenic transmission electron micrograph of the dECM solution demonstrating collagen-bundle formation. (d) Frequency dependence of storage (G′) and loss (G′′) moduli of dECM hydrogels at different concentrations. | ||
3.2 Development of a multivalent UPy-HA based hydrogel
We designed a multivalent UPy compound with a polycarbohydrate backbone (UPy-HA) (Fig. 3a). The UPy-HA molecule was composed of a hyaluronic acid (Mn ∼ 20 kg mol−1), with on average seven UPy-moieties that were reacted to the carboxylic acid group of the glucuronic acid in the repeating disaccharide unit. The UPy-moieties were flanked by a urea group, with a short hexyl spacer separating the two hydrogen bonding units. The urea group and the glucuronic acid were separated by a dodecyl spacer. The alkyl spacers were incorporated to provide a hydrophobic pocket that protects the self-associative hydrogen bonds from the aqueous environment and matches the UPy-urea-alkyl motif of UPy-Gly and UPy-PEG.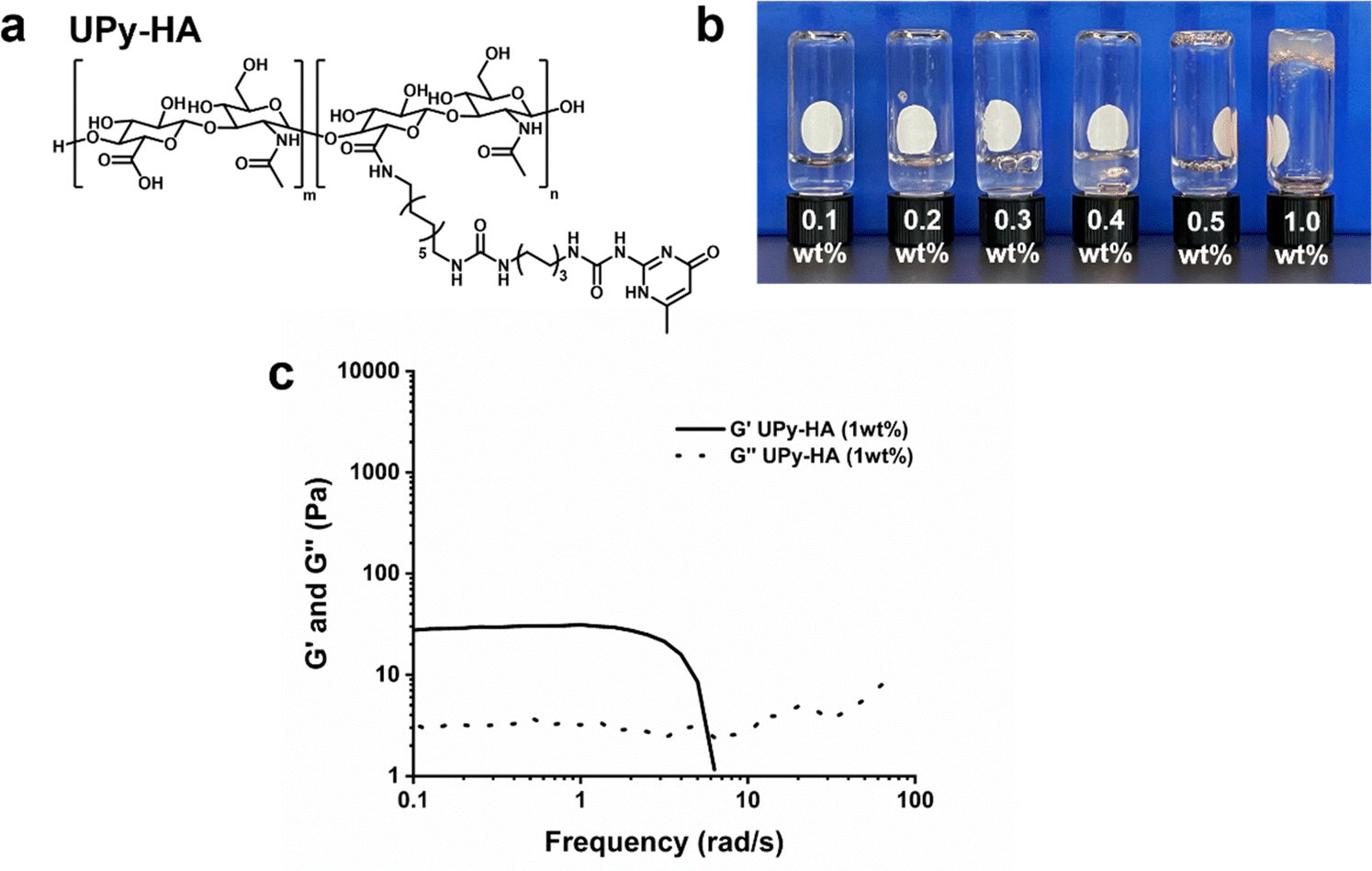 | ||
| Fig. 3 Hydrogel formation based on UPy-HA molecules. (a) Molecular structure of the multivalent UPy-HA molecule. (b) A dilution series of UPy-HA solution (0.1–1 wt%) used for vial-inversion test. (c) Frequency dependence of storage (G′) and loss (G′′) moduli of a 1 wt% UPy-HA hydrogel. | ||
The critical gelation concentration of the UPy-HA compound was investigated by preparing a dilution series of UPy-HA for a vial-inversion test (Fig. 3b). Samples were prepared by deprotonating UPy-HA in alkaline PBS solution and heating to 50 °C to molecularly dissolve the compound, after which it was neutralized using an HCl solution. This demonstrated that UPy-HA remained a solution up to 0.5 wt% and gelation was induced at a concentration of 1 wt%. The rheological behavior of the 1 wt% UPy-HA hydrogel was evaluated and demonstrated a soft bulk stiffness (∼30 Pa) based on a frequency sweep (Fig. 3c). After a frequency of ∼4 rad s−1, the measurement is no longer accurate due to the softness of the hydrogel, which causes an artifact in the measurement as seen by the steep drop in in G′.
3.3 Formulation of hybrid and synthetic hydrogels
Bivalent or multivalent UPy compounds are required to cross-link UPy-Gly assemblies into a transient viscoelastic network.24 By mixing monovalent UPy-Gly solutions and multivalent UPy-HA solutions it was investigated whether the resulting co-assemblies induced hydrogel formation (Fig. 4a and b) (Table S1, ESI†). Previous work by our group demonstrated that a co-assembly of UPy-Gly and UPy-PEG at a ratio of 84:1 mol resulted in a supramolecular hydrogel with exchange dynamics that allowed optimal cell adhesion.24 We rationalized that the number of UPy-groups in the material was a critical parameter in establishing the exchange dynamics. To this end, the incorporated amount of UPy-HA was normalized to the number of UPy-groups in the bivalent UPy-PEG, which resulted in a fixed ratio of 282:1 mol ratio between UPy-Gly and UPy-HA in the hybrid hydrogel system. Both compounds were separately dissolved in alkaline PBS solution at 70 °C, neutralized using HCl solution, and mixed to induce co-assembly.
 | ||
| Fig. 4 Supramolecular hydrogel formation based on UPy-HA and UPy-Gly co-assemblies. (a) Molecular structures of multivalent UPy-HA and monovalent UPy-Gly. (b) Vial-inversion test demonstrating the co-assembly of UPy-Gly and UPy-HA into a hydrogel at different concentrations. The left vial contains a 5 wt% UPy-Gly solution that does not gelate. The remaining vials contain a mixture of UPy-Gly and UPy-HA at a fixed 282:1 mol ratio. (c) Frequency dependence of storage (G′) and loss (G′′) moduli of hydrogels based on co-assembly of UPy-Gly and UPy-HA. (d) Stress-relaxation behavior of UPy-hydrogels measured by subjecting the hydrogels to 7.5% strain. | ||
The mixtures formed hydrogels at tested concentrations of 1.2, 2.4, and 4.8 wt%, indicating that the UPy-HA molecules behaved as effective inter-fiber cross-linkers in this system due to its multivalent nature (Fig. 4b). The mechanical bulk properties of these hybrid materials were characterized using rheological measurements at 37 °C, which confirmed formation of a viscoelastic solid (G′ > G′′). The bulk stiffness in the hybrid hydrogels increased as a function of hydrogelator content, with observed storage moduli of 300 Pa (1.2 wt%), 5 kPa (2.4 wt%), and 26.5 kPa (4.8 wt%). Measuring the storage and loss moduli over time demonstrated that it required ∼8 hours for these hydrogels to obtain stable bulk mechanical behavior, after being placed in the rheometer. Furthermore, the 1.2 wt% hybrid hydrogel demonstrated a frequency-dependent viscoelastic response with a cross over at ∼ 60 rad seconds−1 at which point the material showed liquid-like behavior (G′′ > G′) (Fig. 4c). This is most likely due to the weak physical interactions (i.e., hydrogen bonding) that is unable to withstand the rapid oscillatory deformation. The 2.4 and 4.8 wt% hybrid hydrogels did not demonstrate a limited frequency-dependent viscoelastic behavior (Fig. 4c). The viscoelastic behavior was further studied through stress relaxation experiments. A strain of 1% was applied, after which the stress decay was monitored for a period of 1000 seconds. Similar stress-relaxation behavior was observed for the hybrid hydrogels, irrespective of hydrogelator weight content. However, the hybrid hydrogels did not display sufficient stress relaxation within this time to determine the relaxation half time (Fig. 4d). The hybrid hydrogel system demonstrated slightly softer rheological properties compared to the synthetic hydrogel system, which was based on a co-assembly of UPy-Gly and UPy-PEG. The previously reported storage moduli for the synthetic UPy-hydrogel system were 1.8 kPa (1.25 wt%), 7.2 kPa (2.5 wt%), and 30 kPa (5 wt%).24
3.4 Comparing exchange dynamics between the synthetic and hybrid UPy systems
Fluorescence recovery after photo-bleaching (FRAP) experiments were performed to determine how the exchange dynamics compared between the hybrid and synthetic hydrogel system. This was done by incorporating 25 μM of a fluorescent UPy-Sulfo-Cy5 molecule into a 2.4 wt% hybrid hydrogel composed of UPy-Gly and UPy-HA at a 282:1 mol ratio and comparing it to a 2.5 wt% synthetic hydrogel composed of UPy-Gly and UPy-PEG at an 84:1 mol ratio. The FRAP studies demonstrated similar kinetic recovery profiles (Fig. 5a). However, a difference in the fraction of immobile molecules was observed. The hybrid hydrogel, formed from UPy-Gly and UPy-HA co-assemblies, had a lower fraction of immobile molecules (68 ± 1%) compared to the synthetic hydrogel composed of UPy-Gly and UPy-PEG molecules (80 ± 5%) (Fig. 5b). This dissimilarity suggested differences in underlying diffusion phenomenon of the UPy-additives between supramolecular assemblies, which may be caused due to differences in electrostatic charge and/or the molecular architecture of the supramolecular cross-linkers of each respective hydrogel system.
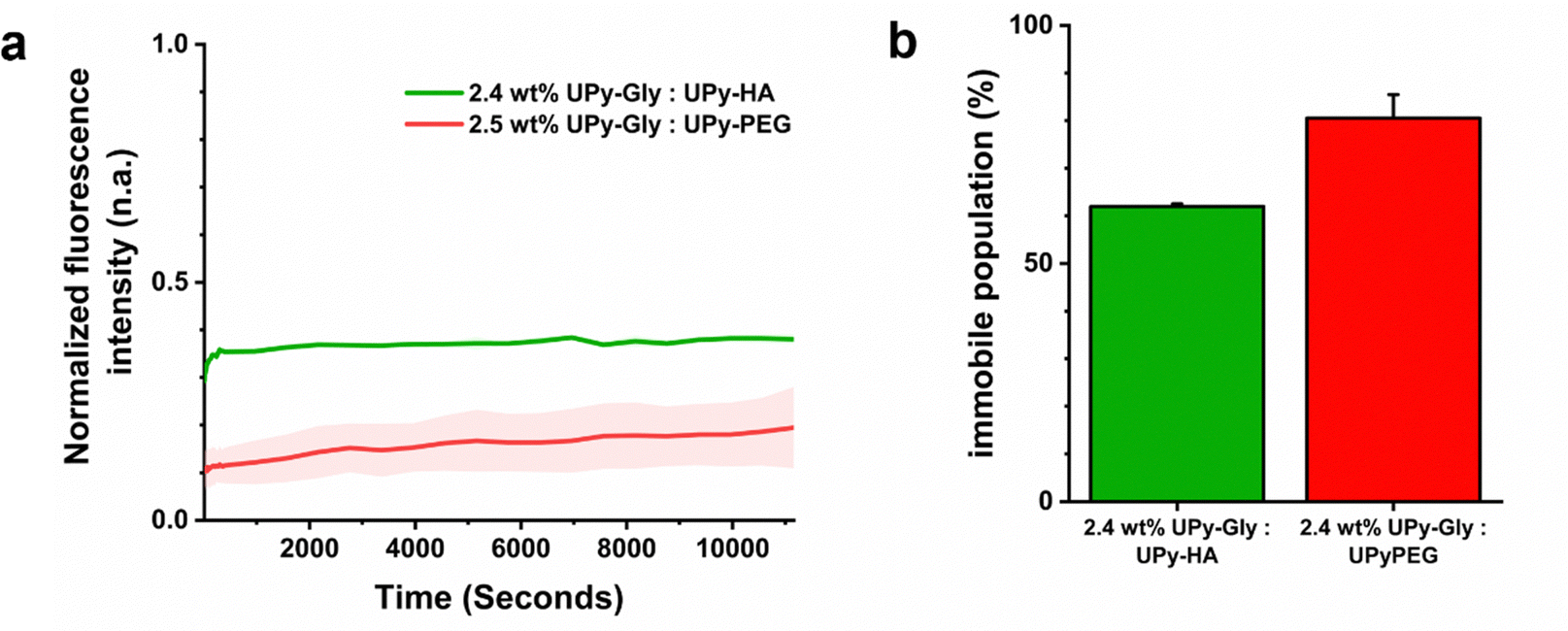 | ||
| Fig. 5 Fluorescence recovery after photobleaching of UPy-Gly hydrogels cross-linked with UPy-HA or UPy-PEG. (a) Graph shows fluorescence recovery after photo-bleaching (FRAP) results of UPy-Gly hydrogels cross-linked with UPy-HA or UPy-PEG. Each sample contained 25 μM of UPy-Sulfo-Cy5. Data are represented as mean ± s.e.m., N = 3 technical replicates. (b) Graph shows immobile populations of UPy-Sulfo-Cy5 molecules as determined by FRAP experiments. Data are represented as mean ± s.e.m., N = 3 technical replicates. | ||
3.5 The importance of biological activation for cell adhesion and spreading
Mechanotransduction is a process that is initiated by a cell-material interaction that allows cell-adhesion and spreading. To this end, a biomaterial requires a chemical motif to which cells are able to bind. The natural hydrogel system based on the renal dECM contained collagen-like bundles to which cells are able to adhere. However, the hybrid (i.e., UPy-HA + UPy-Gly) hydrogel and synthetic hydrogel (i.e., UPy-PEG + UPy-Gly) system are proposed to require additional bioactivation through additive incorporation. To this end, an integrin-binding UPy-cRGDfK additive was included in the synthetic hydrogel at a concentration of 1 mM. Furthermore, collagen type I mimicking additives UPy-RCP and UPy-GFOGER were included in the hybrid hydrogel, respectively. The number of integrin-binding sites of these two additives was normalized to the 1 mM of the UPy-cRGDfK in the synthetic hydrogel, to ensure that differences observed were not caused by ligand density. One UPy-RCP molecule contains eleven RGD sites, which meant that 0.09 mM UPy-RCP was incorporated the hybrid hydrogel. Integrin activation using the UPy-GFOGER additive is only possible in a triple-helix formation, which meant that 3 mM UPy-GFOGER was incorporated (Table S1, ESI†).HK-2 cells were seeded on the hydrogel surfaces to test which components induced cell adhesion and subsequent spreading. Both the natural dECM hydrogel and the synthetic hydrogel allowed HK-2 cells to adhere and spread (Fig. 6). However, cell adhesion to the hybrid hydrogel system depended on which UPy-additive was incorporated. No cells were observed if no UPy-additive was incorporated (Fig. 6). This indicated that there was no interaction between the carbohydrate backbone of UPy-HA with the CD44 receptor on the HK-2 cell membrane or the binding did not induce actin polymerization. HK-2 cells were observed upon incorporation of the UPy-GFOGER. However, these cells remained clustered together and showed a rounded appearance, lacking stress fibers (Fig. 6). Lack of cell spreading may be caused by low expression of the α2β1 integrin, which is the receptor for UPy-GFOGER. Another possibility is that triple-helix formation is inhibited by co-assembly with either the UPy-Gly or UPy-HA molecules. Nonetheless, the hybrid hydrogel system was able to allow proper HK-2 cell adhesion and spreading if the UPy-RCP additive was included (Fig. 6).
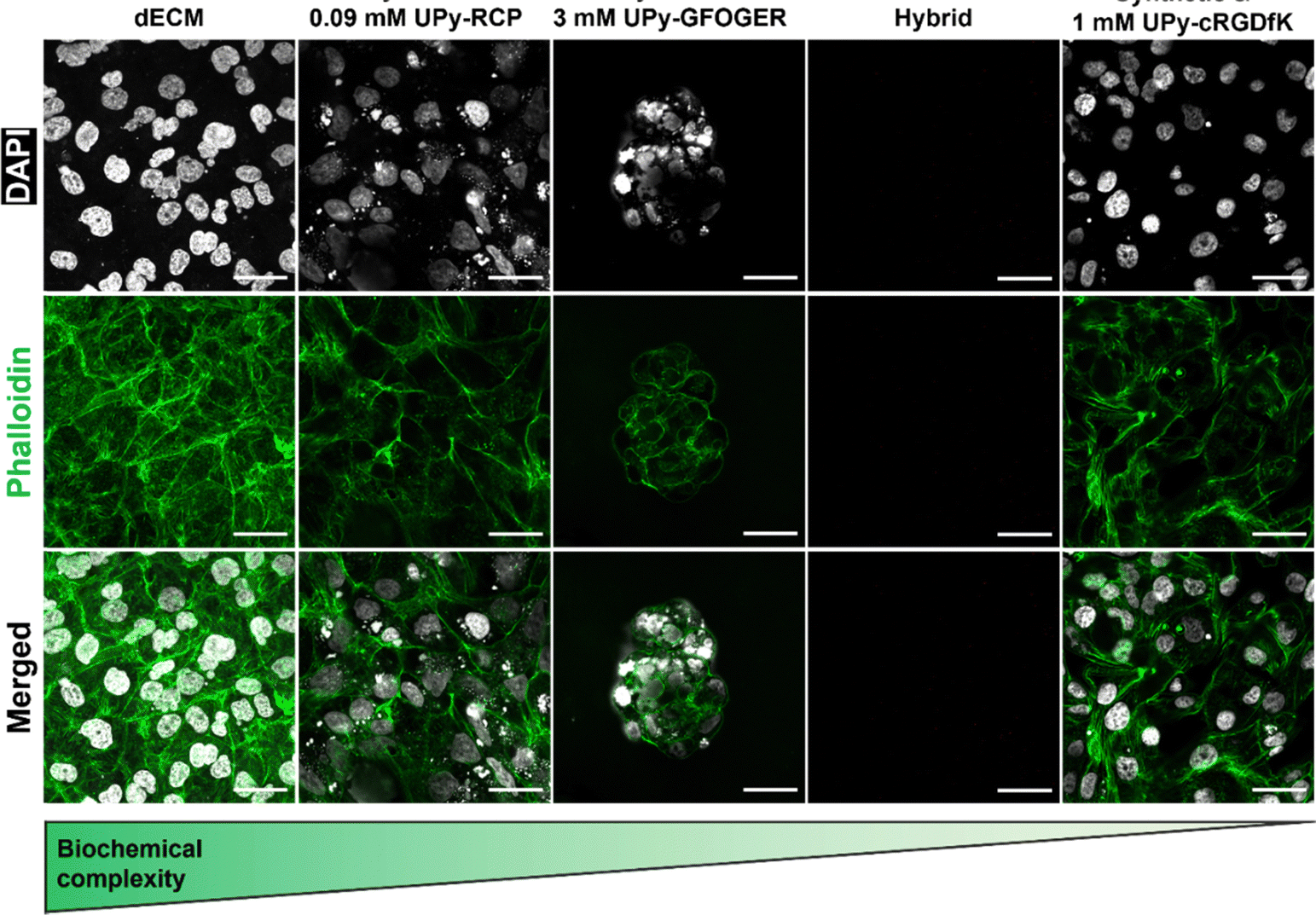 | ||
| Fig. 6 HK-2 cell adhesion and spreading on hydrogels with variation in biochemical complexity. Immunofluorescent confocal microscopy images of HK-2 cells on top of hydrogels with variation in biochemical complexity after 3 days of culture. Scale bars, 40 μm. | ||
3.6 Mechanotransduction on natural, hybrid, and synthetic hydrogels
HK-2 cells were seeded on the natural dECM, hybrid, -and synthetic hydrogel to elucidate the degree of biochemical complexity required to induce mechanotransduction (Fig. 7a and b). A 2 wt% dECM hydrogel was used with a storage modulus of ∼0.1 kPa. The hybrid hydrogel (1.2, 1.4, and 4.8 wt%) and synthetic hydrogels (1.25, 2.5, and 5 wt%) were varied in terms of hydrogelator content, which demonstrated their higher mechanical tunability in terms of bulk stiffness compared to the natural dECM hydrogel (Table S1, ESI†). Mechanotransduction in cells is strongly dependent on bulk stiffness of a material.44 The subcellular location of yes-associated protein (YAP) was used as a quantification parameter for mechanotransduction. This protein is considered a biological actuator of mechanical cues, due to actin stress fiber formation preventing YAP cytoplasmic degradation and allowing nuclear translocation.45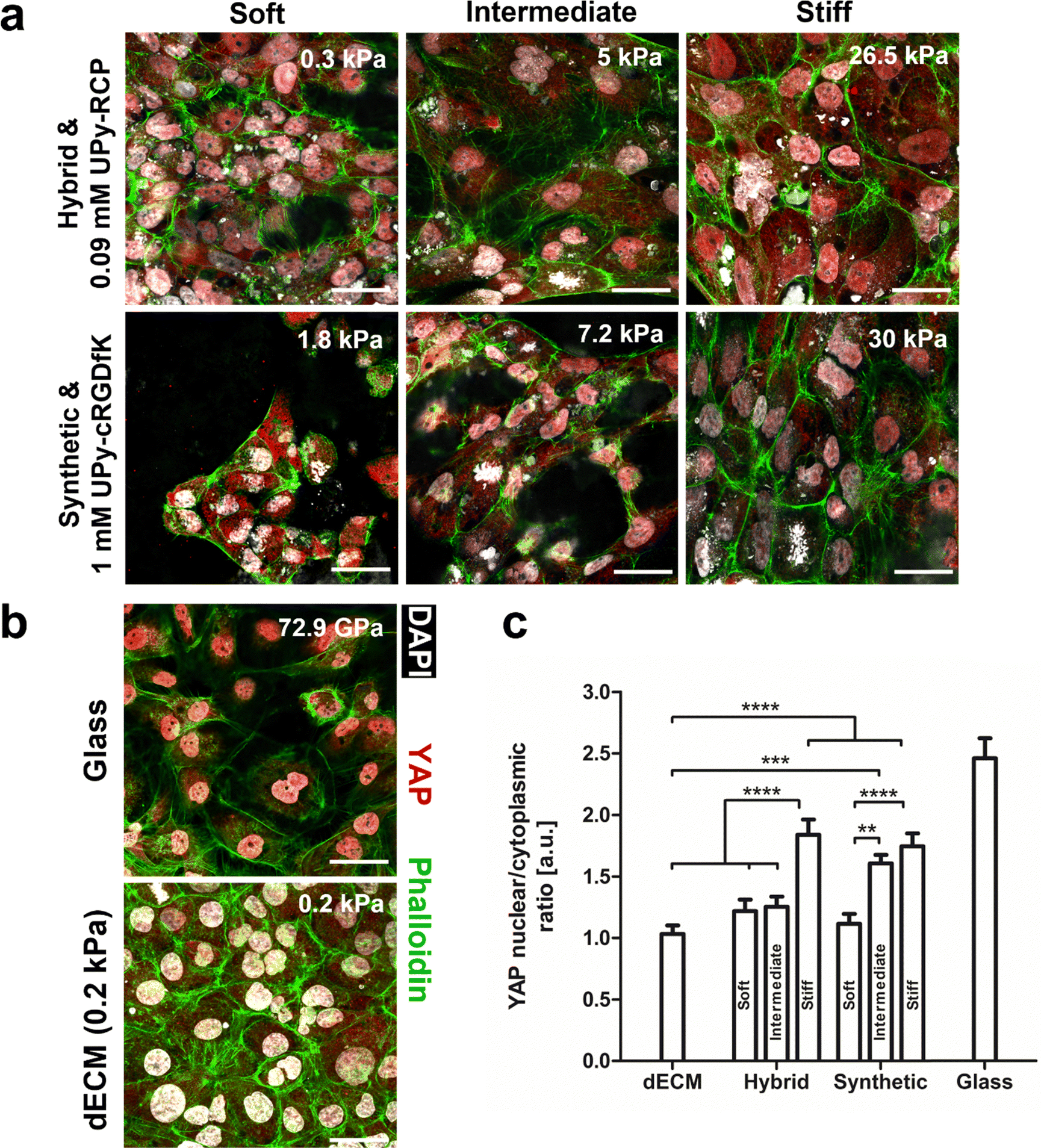 | ||
| Fig. 7 YAP subcellular localization in HK-2 cells on hydrogels with variations in biochemical complexity. (a) Immunofluorescence confocal microscopy images of YAP subcellular localization in HK-2 cells on hybrid and synthetic UPy-hydrogels after 3 days of culture. Hydrogelator content was varied for both systems to acquire hydrogels with different stiffnesses, which were denoted as soft, intermediate, and stiff. The corresponding storage modulus of each condition is also shown in each image. Scale bars, 40 μm. (b) Immunofluorescence confocal microscopy images of YAP subcellular localization in HK-2 cells on glass and dECM hydrogels after 3 days of culture. The corresponding elastic and storage modulus of each condition is shown in each image. Scale bars, 40 μm. (c) Graph shows the quantification of the ratio between nuclear and cytoplasmic YAP in HK-2 cells on day 3. Data are represented as a mean ± s.e.m., N = 20 cells, from three independent samples. Statistical significance was attributed to values of P < 0.05 as determined by two-way ANOVA with Bonferroni's post hoc test. **P < 0.01, ***P < 0.001*, and ****P < 0.001*. The HK-2 cells on glass were left out of the statistical comparison. | ||
HK-2 cells seeded on glass (GPa order of magnitude)46 were included in this study as a positive control for mechanotransduction, on which cells showed a nuclear to cytoplasmic ratio of 2.5 ± 0.1. Indeed, the stiffness was an important parameter for YAP nuclear localization in HK-2 cells for both the hybrid and synthetic hydrogel system. The highest nuclear to cytoplasmic YAP ratio was found to be similar between these systems for the most concentrated hydrogel samples and were significantly higher compared to all other tested hydrogels. Besides these hydrogel samples, only the 2.5 wt% synthetic hydrogel with intermediate stiffness (7 kPa) induced significantly more YAP nuclear translocation compared to the natural dECM hydrogel. The lowest nuclear to cytoplasmic YAP ratio was found in HK-2 cells grown on the dECM hydrogel (Fig. 7b and c). Together these results show that a high degree of biochemical complexity in a material is not required to induce mechanotransduction. Indeed, a minimal amount of bioactivity is required for cell adhesion. However, bulk stiffness appears to be a more important parameter in governing mechanotransduction as a biological response to a biomaterial in this cell line.
Conclusions
In this work, three different hydrogel systems, with a variation in biochemical complexity and mechanical properties, were designed and synthesized to investigate their importance with regard to mechanotransduction. The material with the highest degree of biochemical complexity was a natural hydrogel based on porcine renal dECM. At the same time, this material demonstrated the lowest degree of mechanical tunability, due to low solubility in aqueous environment, which limits practical application. A hybrid system introduced a multivalent UPy-HA molecule capable of co-assembling with monovalent UPy-Gly into a transient network. These two systems were compared to a synthetic supramolecular system composed of UPy-Gly and bivalent UPy-PEG. Both the hybrid and synthetic UPy-hydrogel systems demonstrated a higher degree of mechanical tunability compared to the dECM hydrogel system. This allowed these systems to obtain a higher stiffness through increasing the hydrogelator content in the materials. This proved to be critical in YAP nuclear translocation, which was taken as a quantifiable variable to indicate mechanotransduction. Together this indicated that a high degree of biochemical complexity is not required to induce mechanotransduction and may even prevent this biological response due to limited mechanical tunability combined with little control of molecular composition.Data availability
Our data is available from the first author Dr. Johnick van Sprang, or the corresponding author Prof. Patricia Dankers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge Franca M.R. Witjas and Marten A. Engelse for providing decellularized porcine kidneys, Laura Rijns for useful discussions regarding FRAP experiments, and part of the figures that were created with https://BioRender.com.This research was financially supported by the Ministry of Education, Culture and Science (Gravity Programs 024.003.013 and 024.005.020), by the partners of Regenerative Medicine Crossing Borders (RegMed XB) powered by HealthHolland, Top Sector Life Sciences & Health, and by the European Union's Horizon research and innovation program under grant agreement 101079482 (“SUPRALIFE”).
References
- O. J. G. M. Goor, S. I. S. Hendrikse, P. Y. W. Dankers and E. W. Meijer, Chem. Soc. Rev., 2017, 46, 6621–6637 RSC.
- T. J. Keane and S. F. Badylak, Semin. Pediatr. Surg., 2014, 23, 112–118 CrossRef PubMed.
- V. Vogel and G. Baneyx, Annu. Rev. Biomed. Eng., 2003, 5, 441–463 CrossRef CAS PubMed.
- J. E. Bischoff, E. M. Arruda and K. Grosh, Biomech. Model. Mechanobiol., 2004, 3, 56–65 CrossRef PubMed.
- H. Eskandari, S. E. Salcudean, R. Rohling and J. Ohayon, Phys. Med. Biol., 2008, 53, 6569–6590 CrossRef PubMed.
- A. Maccabi, A. Shin, N. K. Namiri, N. Bajwa, M. S. John, Z. D. Taylor, W. Grundfest and G. N. Saddik, PLoS One, 2018, 13, 1–18 CrossRef PubMed.
- J. K. Mouw, G. Ou and V. M. Weaver, Nat. Rev. Mol. Cell Biol., 2014, 15, 771–785 CrossRef CAS PubMed.
- K. Y. Tsang, M. C. H. Cheung, D. Chan and K. S. E. Cheah, Cell Tissue Res., 2010, 339, 93–110 CrossRef CAS PubMed.
- A. D. Theocharis, S. S. Skandalis, C. Gialeli and N. K. Karamanos, Adv. Drug Delivery Rev., 2016, 97, 4–27 CrossRef CAS PubMed.
- S. L. K. Bowers, I. Banerjee and T. A. Baudino, J. Mol. Cell. Cardiol., 2010, 48, 474–482 CrossRef CAS PubMed.
- W. Zhang, A. Du, S. Liu, M. Lv and S. Chen, Regener. Ther., 2021, 18, 88–96 CrossRef CAS PubMed.
- Y. Seo, S. Jeong, J. J. Chung, S. H. Kim, N. Choi and Y. Jung, ACS Biomater. Sci. Eng., 2020, 6, 610–620 CrossRef CAS PubMed.
- S. Sasikumar, S. Chameettachal, B. Cromer, F. Pati and P. Kingshott, Curr. Opin. Biomed. Eng., 2019, 10, 123–133 CrossRef.
- L. T. Saldin, M. C. Cramer, S. S. Velankar, L. J. White and S. F. Badylak, Acta Biomater., 2017, 49, 1–15 CrossRef CAS PubMed.
- S. A. Nam, E. Seo, J. W. Kim, H. W. Kim, H. L. Kim, K. Kim, T. M. Kim, J. H. Ju, I. G. Gomez, K. Uchimura, B. D. Humphreys, C. W. Yang, J. Y. Lee, J. Kim, D. W. Cho, B. S. Freedman and Y. K. Kim, Exp. Mol. Med., 2019, 51, 1–13 CrossRef PubMed.
- M. Saheli, M. Sepantafar, B. Pournasr, Z. Farzaneh, M. Vosough, A. Piryaei and H. Baharvand, J. Cell. Biochem., 2018, 119, 4320–4333 CrossRef CAS PubMed.
- G. G. Giobbe, C. Crowley, C. Luni, S. Campinoti, M. Khedr, K. Kretzschmar, M. M. De Santis, E. Zambaiti, F. Michielin, L. Meran, Q. Hu, G. van Son, L. Urbani, A. Manfredi, M. Giomo, S. Eaton, D. Cacchiarelli, V. S. W. Li, H. Clevers, P. Bonfanti, N. Elvassore and P. De Coppi, Nat. Commun., 2019, 10, 5658 CrossRef PubMed.
- R. Simsa, T. Rothenbücher, H. Gürbüz, N. Ghosheh, J. Emneus, L. Jenndahl, D. L. Kaplan, N. Bergh, A. M. Serrano and P. Fogelstrand, PLoS One, 2021, 16, 1–22 CrossRef PubMed.
- S. Kim, Y. S. Choi, J. S. Lee, S. H. Jo, Y. G. Kim and S. W. Cho, J. Ind. Eng. Chem., 2022, 107, 155–164 CrossRef CAS.
- R. H. J. De Hilster, P. K. Sharma, M. R. Jonker, E. S. White, E. A. Gercama, M. Roobeek, W. Timens, M. C. Harmsen, M. N. Hylkema and J. K. Burgess, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2020, 318, L698–L704 CrossRef CAS PubMed.
- S. E. Sakiyama-Elbert and J. A. Hubbel, Annu. Rev. Mater. Res., 2001, 31, 183–201 CrossRef CAS.
- A. C. H. Pape, M. M. C. Bastings, R. E. Kieltyka, H. M. Wyss, I. K. Voets, E. W. Meijer and P. Y. W. Dankers, Int. J. Mol. Sci., 2014, 15, 1096–1111 CrossRef CAS PubMed.
- S. Spaans, P. P. K. H. Fransen, M. J. G. Schotman, R. Van Der Wulp, R. P. M. Lafleur, S. G. J. M. Kluijtmans and P. Y. W. Dankers, Biomacromolecules, 2019, 20, 2360–2371 CrossRef CAS PubMed.
- M. Diba, S. Spaans, S. I. S. Hendrikse, M. M. C. Bastings, M. J. G. Schotman, J. F. van Sprang, D. J. Wu, F. J. M. Hoeben, H. M. Janssen and P. Y. W. Dankers, Adv. Mater., 2021, 33(37), 2008111 CrossRef CAS PubMed.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- M. M. C. Bastings, S. Koudstaal, R. E. Kieltyka, Y. Nakano, A. C. H. Pape, D. A. M. Feyen, F. J. van Slochteren, P. A. Doevendans, J. P. G. Sluijter, E. W. Meijer, S. A. J. Chamuleau and P. Y. W. Dankers, Adv. Healthcare Mater., 2014, 3, 70–78 CrossRef CAS PubMed.
- P. Y. W. Dankers, T. M. Hermans, T. W. Baughman, Y. Kamikawa, R. E. Kieltyka, M. M. C. Bastings, H. M. Janssen, N. A. J. M. Sommerdijk, A. Larsen, M. J. A. Van Luyn, A. W. Bosman, E. R. Popa, G. Fytas and E. W. Meijer, Adv. Mater., 2012, 24, 2703–2709 CrossRef CAS PubMed.
- M. M. C. Bastings, T. M. Hermans, A. J. H. Spiering, E. W. L. Kemps, L. Albertazzi, E. E. Kurisinkal and P. Y. W. Dankers, Macromol. Biosci., 2019, 19, 1–11 CrossRef PubMed.
- B. P. Toole, Curr. Opin. Cell Biol., 1990, 2, 839–844 CrossRef CAS PubMed.
- A. J. Day and G. D. Prestwich, J. Biol. Chem., 2002, 277, 4585–4588 CrossRef CAS PubMed.
- A. M. DeSimone, J. Leszyk, K. Wagner and C. P. Emerson, Sci. Adv., 2019, 5(12), eaaw7099 CrossRef CAS PubMed.
- J. I. Sumiya, S. Asakawa, T. Tobe, K. Hashimoto, K. I. Saguchi, N. H. Choi-Miura, Y. Shimizu, S. Minoshima, N. Shimizu and M. Tomita, J. Biochem., 1997, 122, 983–990 CrossRef CAS PubMed.
- Y. Xie, Z. Upton, S. Richards, S. C. Rizzi and D. I. Leavesley, J. Controlled Release, 2011, 153, 225–232 CrossRef CAS PubMed.
- A. F. Vrehen, J. F. Van Sprang, M. J. G. Schotman and P. Y. W. Dankers, Mater. Today Bio., 2024, 26, 101021 CrossRef CAS PubMed.
- J. Emsley, C. G. Knight, R. W. Farndale and M. J. Barnes, J. Mol. Biol., 2004, 335, 1019–1028 CrossRef CAS PubMed.
- C. G. Knight, L. F. Morton, A. R. Peachey, D. S. Tuckwell, R. W. Farndale and M. J. Barnes, J. Biol. Chem., 2000, 275, 35–40 CrossRef CAS PubMed.
- E. Koivunen, D. A. Gay and E. Ruoslahti, J. Biol. Chem., 1993, 268, 20205–20210 CrossRef CAS.
- G. Mondal, S. Barui and A. Chaudhuri, Biomaterials, 2013, 34, 6249–6260 CrossRef CAS PubMed.
- E. M. Moore and J. L. West, Regener. Eng. Transl. Med., 2019, 5, 167–179 CrossRef CAS.
- D. G. Leuning, F. M. R. Witjas, M. Maanaoui, A. M. A. de Graaf, E. Lievers, T. Geuens, C. M. Avramut, L. E. Wiersma, C. W. van den Berg, W. M. P. J. Sol, H. de Boer, G. Wang, V. L. S. LaPointe, J. van der Vlag, C. van Kooten, B. M. van den Berg, M. H. Little, M. A. Engelse and T. J. Rabelink, Am. J. Transplant., 2019, 19, 1328–1343 CrossRef CAS PubMed.
- J. P. Guyette, S. E. Gilpin, J. M. Charest, L. F. Tapias, X. Ren and H. C. Ott, Nat. Protoc., 2014, 9, 1451–1468 CrossRef CAS PubMed.
- B. D. Quan and E. D. Sone, Cryo-TEM analysis of collagen fibrillar structure, Elsevier Inc., 1st edn, 2013, vol. 532 Search PubMed.
- K. H. Stenzel, T. Miyata and A. L. Rubin, Annu. Rev. Biophys. Bioeng., 1974, 3, 231–253 CrossRef CAS PubMed.
- Q. Sun, Y. Hou, Z. Chu and Q. Wei, Bioact. Mater., 2022, 10, 397–404 CAS.
- S. Dupont, L. Morsut, M. Aragona, E. Enzo, S. Giulitti, M. Cordenonsi, F. Zanconato, J. Le Digabel, M. Forcato, S. Bicciato, N. Elvassore and S. Piccolo, Nature, 2011, 474, 179–184 CrossRef CAS PubMed.
- A. Seal, A. K. Dalui, M. Banerjee, A. K. Mukhopadhyay and K. K. Phani, Bull. Mater. Sci., 2001, 24, 151–155 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01774a |
| This journal is © The Royal Society of Chemistry 2025 |