
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotodegradable polyacrylamide tanglemers enable spatiotemporal control over chain lengthening in high-strength and low-hysteresis hydrogels†
Joshua S.
Lee‡
a,
Bruce E.
Kirkpatrick‡
 abc,
Abhishek P.
Dhand
d,
Lea Pearl
Hibbard
a,
Benjamin R.
Nelson
ab,
Nathaniel P.
Skillin
abc,
Makayla C.
Johnson
a,
Dilara
Batan
be,
Benjamin D.
Fairbanks
a,
Timothy J.
White
af,
Christopher N.
Bowman
af,
Jason A.
Burdick
abf and
Kristi S.
Anseth
*abf
abc,
Abhishek P.
Dhand
d,
Lea Pearl
Hibbard
a,
Benjamin R.
Nelson
ab,
Nathaniel P.
Skillin
abc,
Makayla C.
Johnson
a,
Dilara
Batan
be,
Benjamin D.
Fairbanks
a,
Timothy J.
White
af,
Christopher N.
Bowman
af,
Jason A.
Burdick
abf and
Kristi S.
Anseth
*abf
aDepartment of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, USA. E-mail: kristi.anseth@colorado.edu
bBioFrontiers Institute, University of Colorado Boulder, Boulder, USA
cMedical Scientist Training Program, University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA
dDepartment of Bioengineering, University of Pennsylvania, Pennsylvania, USA
eDepartment of Biochemistry, University of Colorado Boulder, Boulder, USA
fMaterials Science and Engineering Program, University of Colorado Boulder, Boulder, USA
First published on 6th December 2024
Abstract
Covalent hydrogel networks suffer from a stiffness-toughness conflict, where increased crosslinking density enhances the modulus of the material but also leads to embrittlement and diminished extensibility. Recently, strategies have been developed to form highly entangled hydrogels, colloquially referred to as tanglemers, by optimizing polymerization conditions to maximize the density and length of polymer chains and minimize the crosslinker concentration. It is challenging to assess entanglements in crosslinked networks beyond approximating their theoretical contribution to mechanical properties; thus, in this work, we synthesize and characterize polyacrylamide tanglemers using a photolabile crosslinker, which allows for direct assessment of covalent trapping of entanglements under tension. Further, this chemistry allows tuning of the modulus in situ by crosslink photocleavage (from tensile modulus (ET) = 100 kPa to <25 kPa). Beyond cleavage of crosslinks, we demonstrate that even non-degradable tanglemer formulations can be photo-softened and completely degraded through Fe3+-mediated oxidation of the polyacrylamide backbone. While both photodegradation methods are useful for spatial patterning and result in softer gels with reduced fracture strength, only crosslink photocleavage improves gel extensibility via light-induced chain lengthening (εF = 700% to >1500%). Crosslink photocleavage in tanglemers also affords significant control over localized swelling and diffusivity. In sum, we introduce a simple and user-directed approach for probing entanglements and asserting spatiotemporal control over stress–strain responses and small molecule diffusivity in polyacrylamide tanglemers, suggesting a multitude of potential soft matter applications including controlled release and tunable bioadhesive interfaces.
Introduction
Entanglements span an impressive range of length scales in biology.1 In subcellular processes, entanglements between telomeres of dividing yeast regulate cell survival in a thermally dependent manner,2 while entangled cytoskeletal proteins and internal membrane architectures provide eukaryotic cells with remarkable mechanical properties, including high toughness and rapid force dampening.3,4 At the tissue level, similar entangled structures—composed of extracellular matrix (ECM),5 cells,6 and, in some cases, entire organisms7—contribute to complex biological systems that are intrinsically strong, flexible, and capable of withstanding substantial deformation. Although entanglements store elastic energy on short timescales, these physical interactions between polymer chains cannot maintain long-term mechanical stability in the absence of constraining covalent crosslinks that stabilize the topology of an entangled network and prevent the disentanglement of strands.8 As such, the study of entanglement in synthetic polymeric networks has emerged as a promising strategy for preparing bioinspired materials with enhanced resistance to fatigue and fracture.9,10 For additional detailed discussion of polymer network structure and entanglements, we recommend several recent reviews.11–13Innovations spearheaded by Kim, Suo, and coworkers in fabricating entangled polymer networks (“tanglemers”) have demonstrated the potential of these systems to mimic biological resilience and improve the mechanical performance of synthetic materials under extreme conditions (e.g., cyclic loading, high strain, long-term interfacial wear). Initially, efforts were limited to single-network polyacryloyl hydrogels and elastomers, wherein essentially saturated solutions of monomer containing very low amounts of crosslinker and photoinitiator were polymerized to form long and densely entangled chains.14–16 Further, these authors developed a low-intensity mixing and annealing process of high-molecular weight polymer chains including poly(ethylene glycol), cellulose, and polybutadiene, which facilitated the preparation of entangled hydrogels and elastomers.17,18 Combining these methods with phase-separating interpenetrating networks,19,20 nanocomposite elements,21 sacrificial crosslinks,22,23 and double networks24 has resulted in hydrogels, rubbers, elastomers, and glasses with low hysteresis and unprecedented toughness and durability. Other groups introduced the concept of using redox-based initiation in place of photoinitiation for preparing entangled polyacrylamide hydrogels,25,26 and a recent report described a light-based additive manufacturing technique for preparing tanglemers with a combination of photoinitiator and redox-based initiator, affording tough and tissue-adhesive gels with complex 3D geometries.27 Beyond the aforementioned work, entangled hydrogels prepared by these methods have been applied as strain sensors,28,29 electrolytes,30 actuators,31,32 and for water harvesting and evaporative cooling technologies.33,34
While commonly used in many hydrogel formulations, including “tough gels” (e.g., double networks comprised of polyacrylamide and alginate35), photodegradable crosslinkers have yet to be incorporated in tanglemers.36–38 Such a system presents several interesting opportunities to tailor the material properties, as well as better understand structure–property relationships in these complex systems. For example, photodegradation provides a contact-free mechanism of crosslink cleavage, enabling direct assessment of the role of covalent crosslinks in the tanglemers. Although changes in entanglement density are detected in soluble polymeric systems via changes in viscosity,39,40 trapped entanglements are difficult to characterize in crosslinked networks beyond their relative contribution to mechanical and physical properties,41 which itself is challenging to precisely determine.12,13,42,43 In an entangled material, on-demand and near-instantaneous dissolution of crosslinks helps improve our understanding of how physical entanglements bear loads and become mobile in the absence of confining covalent bonds. Moreover, tanglemer hydrogels are prepared with a very low concentration of crosslinker, allowing for the use of highly absorptive species such as o-nitrobenzyl (oNB) moieties that would otherwise result in hydrogels of high optical density. Finally, photoresponsive chemistries permit spatiotemporal patterning via structured illumination for facile preparation of monolithic polymer networks with independent spatial variations in entanglement and crosslink density, swelling, and solute diffusivity.
Herein, we prepare and characterize highly entangled polyacrylamide hydrogels containing photodegradable oNB-containing crosslinks. To avoid premature photocleavage of the crosslinker, we use the redox-based initiator system of ammonium persulfate (APS) and tetramethylethylenediamine (TEMED).44 Due to the low concentration of the oNB chromophore, these materials attenuate <5% of incident 365 nm light through thicknesses >1 mm. By comparing crosslink photolysis in these materials to an alternative degradation approach involving photo-oxidative cleavage of the polyacrylamide backbone, we show that hydrogel extensibility depends on chain length while modulus and fracture strength are reduced by either approach. To conclude, we use spatial patterning of crosslink cleavage to generate single-network materials with programmable strain responses and small molecule release rates. This work reveals the interplay between entanglements and covalent crosslinks in entangled hydrogels and presents new strategies for preparing photoresponsive soft materials with highly tunable physical properties.
Results and discussion
We first sought to verify that the specific molecular structure of diacryloyl poly(ethylene glycol) (PEG) crosslinkers did not strongly influence the properties of polyacrylamide tanglemers. Two linear PEG-acryloyl crosslinkers (MW ∼ 3500 Da) were selected (Fig. 1A), with one bearing terminal acrylamide moieties (PEG-diAcm) and the other functionalized with oNB-acrylate endgroups (PEG-diPDA). While the diAcm crosslinker is not susceptible to cleavage by hydrolysis or photodegradation, the ester linkage of the diPDA crosslinker is cleaved by either light or water. As such, for the present study, all mechanical tests were performed within two weeks of gel fabrication to minimize softening as a result of hydrolysis. At longer timescales (on the order of several weeks), we observed uncontrolled softening of hydrogels, likely due to ester hydrolysis. However, alternative synthetic routes are available to prepare carbamate-linked oNB units that are more stable in water.45,46 | ||
| Fig. 1 Highly entangled polyacrylamide hydrogels have crosslinker-independent mechanical properties before light exposure. (A) Schematic representation of the formation of entangled polyacrylamide networks using PEG-diPDA (photodegradable diacrylate, red) and PEG-diAcm (non-degradable diacrylamide, blue) crosslinkers. (B) Representative tensile stress–strain curves of tanglemers prepared with each crosslinker, strained at 10 percent per second. (C) Mechanical properties of diPDA- and diAcm-crosslinked tanglemers including (i) Young's modulus, (ii) strain to failure, (iii) strength and (iv) work of fracture. (D) Hydrogels exhibit negligible hysteresis regardless of crosslinker identity. (E) Tensile stress–strain curves under active irradiation (shaded region) beginning at 250% strain show photoresponsiveness in diPDA-crosslinked hydrogels only. n = 3–4 gels per condition, with representative data shown in panels (B), (D), and (E). | ||
Tanglemers were prepared at a molar ratio of water to monomer of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which translates to roughly 66 wt% monomer. PEG crosslinkers were added at a molar ratio of monomer to crosslinker of 1:3.2 × 10−5. To solubilize this mixture fully, samples were placed on a hotplate set to 45 °C for five minutes before APS and TEMED were respectively added at final concentrations of 2.2 and 8.6 mM (equating to 0.05 and 0.1 wt%). After the addition of APS/TEMED, the tanglemer precursor solution was transferred by a pre-warmed glass pipette to hydrophobic glass slides separated by 500 μm plastic spacers and allowed to cool to room temperature on the bench as polymerization proceeded. Samples were left to react for one hour before removal from the glass slides and equilibration in water for 24 hours. An important consideration when preparing polyacryloyl tanglemers via free-radical polymerization of highly concentrated monomer solution is its propensity for auto-acceleration, limiting fabrication approaches to relatively thin or layer-by-layer systems where the exothermic reaction is mitigated by heat transfer; in thicker systems, runaway reactions are possible where the solvent boils and dangerous amounts of heat are generated.
:1, which translates to roughly 66 wt% monomer. PEG crosslinkers were added at a molar ratio of monomer to crosslinker of 1:3.2 × 10−5. To solubilize this mixture fully, samples were placed on a hotplate set to 45 °C for five minutes before APS and TEMED were respectively added at final concentrations of 2.2 and 8.6 mM (equating to 0.05 and 0.1 wt%). After the addition of APS/TEMED, the tanglemer precursor solution was transferred by a pre-warmed glass pipette to hydrophobic glass slides separated by 500 μm plastic spacers and allowed to cool to room temperature on the bench as polymerization proceeded. Samples were left to react for one hour before removal from the glass slides and equilibration in water for 24 hours. An important consideration when preparing polyacryloyl tanglemers via free-radical polymerization of highly concentrated monomer solution is its propensity for auto-acceleration, limiting fabrication approaches to relatively thin or layer-by-layer systems where the exothermic reaction is mitigated by heat transfer; in thicker systems, runaway reactions are possible where the solvent boils and dangerous amounts of heat are generated.
Equilibrated tanglemers had similar tensile stress–strain curves irrespective of crosslinker identity (Fig. 1B), with pronounced strain-stiffening after ε ∼ 250% (Fig. S1, ESI†), marking the accumulation of stress in fully extended entanglements trapped at covalent crosslinks. Both crosslinkers resulted in materials with elastic moduli (ET) on the order of 100 kPa, strain to failure (εF) exceeding 600%, fracture strength of 600–1000 kPa, and work of fracture >1000 kJ m−3 (Fig. 1C). As is characteristic for entangled networks, tanglemers prepared with both the diPDA and the diAcm crosslinkers showed negligible hysteresis (<5%) in cyclic loading and unloading to 300% strain (Fig. 1D). At high strains (500%), 10–20% hysteresis was observed with repeated loading, with the greatest hysteresis on the first cycle (Fig. S2, ESI†). Collectively, these results suggest that the specific molecular structure of the crosslinker (i.e., acrylate vs. acrylamide) is relatively unimportant for influencing tanglemer mechanical properties. Increasing the concentration of PEG in these hydrogels resulted in phase separation between polyacrylamide and PEG, rendering tanglemers prepared with 35 kDa PEG diacrylate crosslinker opaque and quite soft (ET ∼ 5 kPa) compared to homogenous tanglemers prepared from 3.5 kDa crosslinker (Fig. S3, ESI†).
With these formulations in hand, we next asked whether there were immediate changes in the mechanical properties of these materials under irradiation. To test this, we performed the same strain-to-failure tensile measurement as before but introduced 365 nm light exposure (100 mW cm−2) when the gels reached ∼250% strain. As expected, diAcm-crosslinked materials displayed similar stress–strain behavior independent of light exposure. However, stress in the diPDA-crosslinked tanglemers immediately plateaued upon exposure to light before strain-stiffening behavior eventually developed at ε ∼ 600% (Fig. 1E). Although fracture strength and toughness decreased in the diPDA-crosslinked tanglemers with active irradiation, the samples became significantly more extensible (Fig. S4, ESI†). These findings demonstrate that physical associations in tanglemers bear a significant proportion of the stress in the network while covalent crosslinks effectively shorten chains and trap entanglements, leading to increased extensibility but an amplified reduction in fracture mechanics as crosslinks are cleaved. The measured modulus of diPDA-crosslinked tanglemers is two orders of magnitude greater than predicted by rubberlike elasticity theory, highlighting the dominant role of trapped entanglements as the primary stress-bearing medium. During photodegradation, both covalent crosslinks and their associated entanglements are disrupted in a balanced manner, further supporting their combined contribution to the mechanics of these networks.
Next, we aimed to better understand how crosslink cleavage affects tanglemer properties, including chain mobility with varied light exposure in these materials (Fig. 2A). To ensure that tanglemers would degrade uniformly through the thickness of the gel, we calculated the attenuation of 365 nm light by the diPDA crosslinker at various concentrations (Fig. 2B). Hydrogels with 1–10 wt% diPDA content are optically thick (meaning <80% of the incident light reaches the bottom of the sample) within the first 200 μm of the material, which is comparable to diPDA-crosslinked PEG hydrogels reported by Tibbitt and colleagues.47,48 In comparison, our tanglemers contain only 0.1 wt% diPDA macromer; considering that these materials swell after crosslinking approximately 7-fold by mass, the final diPDA content in the photodegradable tanglemer formulation is approaching 0.01 wt%. As a result, these hydrogels have negligible attenuation through thicknesses even >1 mm.
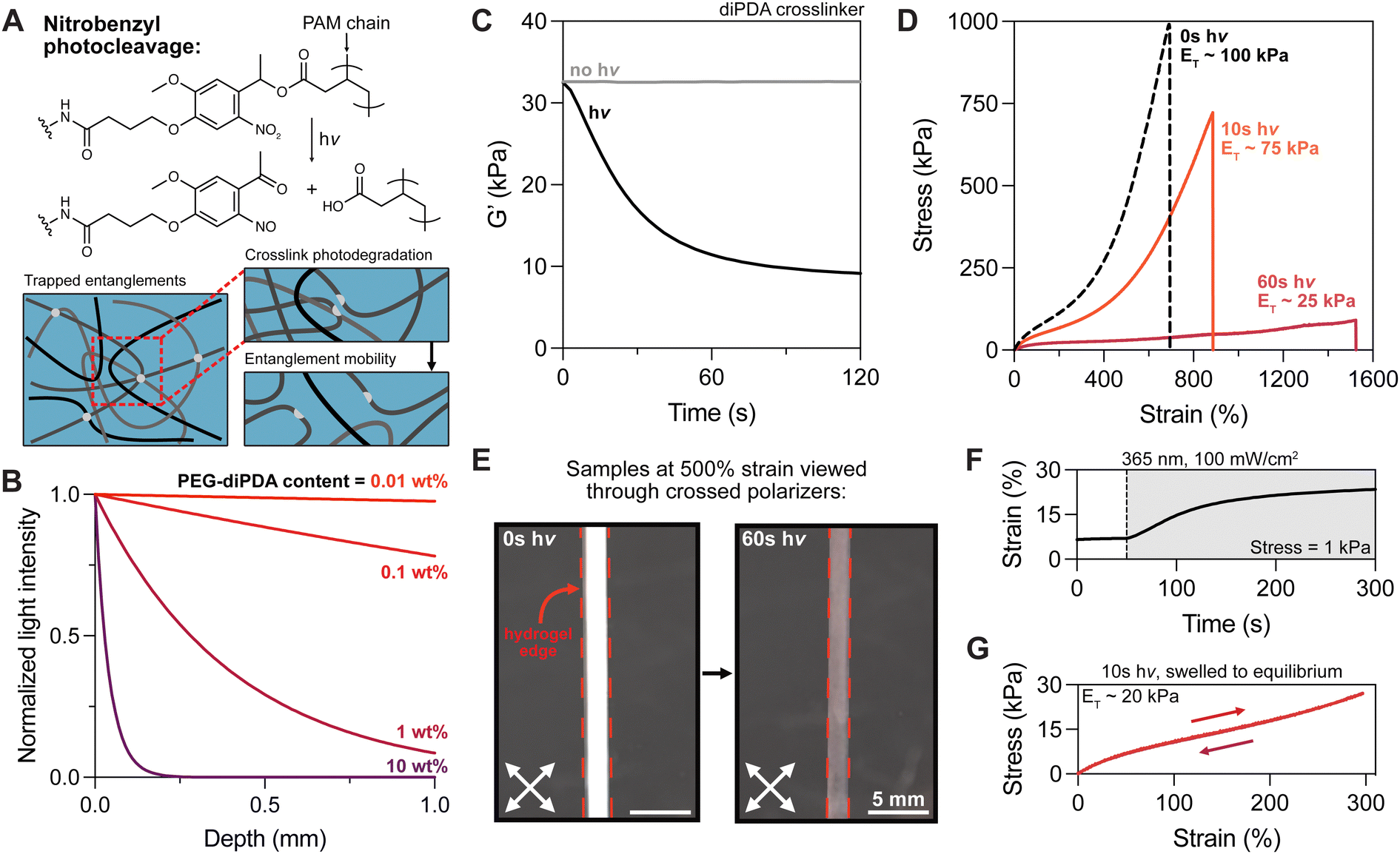 | ||
| Fig. 2 Photodegradation of diPDA crosslinks results in tanglemer chain lengthening. (A) Photocleavage of the oNB moiety enables greater polymer chain mobility and reptation of entangled strands. (B) Calculated attenuation of 365 nm light by PEG-diPDA crosslinker at various concentrations. (C) In situ oscillatory rheology of diPDA hydrogels ±hv displays a ∼3-fold decrease in storage modulus, mirroring changes in elastic modulus (D) during tensile testing with varied light exposure (0 s, 10 s, 60 s of 365 nm light at 100 mW cm−2). (E) Pre-strained tanglemers exhibit strong birefringence when viewed through crossed polarizers, which is lost as crosslink photodegradation reduces strain-induced chain alignment. (F) In situ oscillatory creep test held at 1 kPa shows plastic deformation during crosslink photocleavage. (G) Hydrogels re-equilibrated after partial degradation (10 s of 365 nm at 100 mW cm−2) retain negligible hysteresis despite decreased crosslink and entanglement density. n = 3 gels per condition, with representative data shown. | ||
To ascertain the dynamics of photodegradation in tanglemers, we turned to in situ oscillatory shear rheology, which was performed with and without light exposure (100 mW cm−2). Under irradiation, the storage modulus of diPDA tanglemers decreased 3-fold (from ∼33 to <10 kPa) within 60 seconds, after which it effectively plateaued (Fig. 2C), suggesting near-total cleavage of the oNB crosslinks. Modeling the conversion of the oNB group based on the known reaction order, absorbance, concentration, pathlength, and light intensity in this system, reaching 100% cleavage of this moiety in less than a minute requires quantum yields of 0.025–0.1 (Fig. S5, ESI†), which is consistent with other studies of this chemical species.49 With this in mind, tanglemers provide an exceptional platform for the inclusion of dilute but highly absorptive reactive groups for modifying network connectivity or functionality. Moreover, tanglemers enable a unique mode of amplified crosslinking density reduction, wherein photolysis of covalent crosslinks results in subsequent disentanglement events, further relaxing stresses and softening the network.
Tensile measurements were performed on diPDA tanglemers irradiated with 365 nm light at 100 mW cm−2 for 0 s (control), 10 s (partially degraded), and 60 s (fully degraded) immediately before testing (i.e., with no time to equilibrate in solvent following degradation). The gels showed a graded change in moduli, extensibility, and fracture strength (Fig. 2D). ET decreased from 100 kPa to <25 kPa while the respective strain and stress at break of 700% and 1 MPa (without light) changed to 1500% and 90 kPa, indicating an approximate doubling in extensibility but 10-fold reduction in strength. Appreciating the magnitude of this range, these materials have remarkably tunable properties. Similar to the decrease in modulus shown by oscillatory shear rheology during irradiation and the related estimation of oNB quantum yield, varied irradiation time before tensile testing also indicated rapid and relatively constant softening before reaching high conversion and a plateau value in tensile modulus by 60 seconds (Fig. S6, ESI†). Following degradation, these samples are out of equilibrium as a result of the high-MW polymer being confined to a relatively small gel volume, but un-trapped entanglements drive swelling and reptation over the course of hours to days. Although toughness measurements were beyond the scope of this study, conventional (non-entangled) polyacrylamide hydrogels containing the oNB crosslinker have been shown to undergo photodegradation-mediated crack tip softening in notched samples, dramatically increasing their toughness by more than 3-fold.50 This strategy could be deployed in these materials, potentially improving their already impressive resistance to failure.
Intriguingly, both diAcm and diPDA gels exhibited strong birefringence when viewed through crossed polarizers at ε = 500%, which disappeared in the diPDA samples after light exposure (Fig. 2E and Movie S1, ESI†). We hypothesize that this birefringence is caused by strain-induced chain alignment that diminishes as covalent crosslinks trapping the entanglements are cleaved, leaving chains in the network with increased mobility. Photodegraded tanglemers showed little birefringence even at fracture. Furthermore, an in situ oscillatory creep test (held at a stress of 1 kPa) was performed during exposure to 365 nm light, revealing plastic deformation of the material as entanglements were liberated (Fig. 2F). Notably, samples of the partially degraded tanglemers (10 s of light exposure) kept in water to re-equilibrate displayed weaker mechanics but retained their negligible hysteresis, suggesting that tanglemers with controlled degradation can be deliberately softened to specific extents without sacrificing their hyperelastic properties (Fig. 2G).
While the oNB crosslinker provides excellent control over photoresponsive tanglemer properties through controlled degradation, we also wondered if there were complementary approaches to induce photodegradation in tanglemers regardless of the specific crosslinking chemistry. Here, we turned to historical reports of oxidative photolysis of polyacrylamide, which is readily catalyzed by ferric salts.51–53 Serendipitously, as we were undertaking these studies, Boyer and coworkers reported a general strategy for FeCl3-catalyzed photo-oxidative degradation of a variety of polymers, including polyacryloyls.54 With this in mind, we theorized that irradiation of tanglemers equilibrated in FeCl3 would result in indiscriminate cleavage events throughout the network (Fig. 3A) that degrade both poly(ethylene glycol) and polyacrylamide, decreasing the molecular weight of the polymer chains and disrupting entanglements through an alternative mechanism to crosslink cleavage. Other polymers, hydrogels, and elastomers have been prepared with synthetic linkers (e.g., β-amino esters and acrylates, thioketals) that undergo photo-oxidative degradation,55–59 but these are more similar to the oNB group in facilitating selective cleavage of a specific bond compared to the uncontrolled degradation of the backbone that the Fe3+-polyacrylamide system offers.
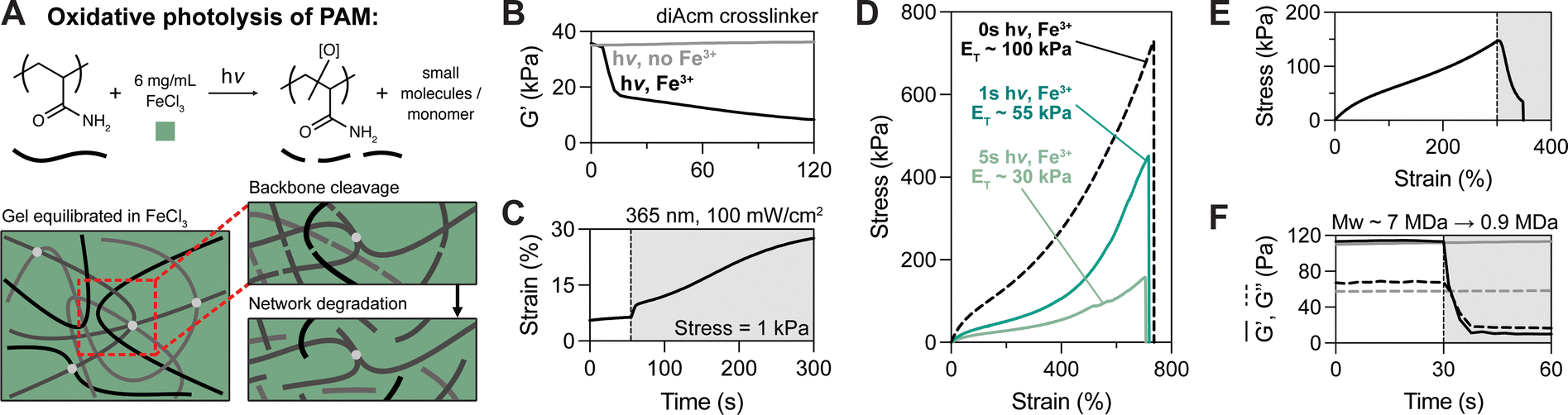 | ||
| Fig. 3 Fe3+-mediated photolysis of polyacrylamide backbone results in softening without increased extensibility. (A) FeCl3-mediated photo-oxidative degradation of diAcm-crosslinked polyacrylamide tanglemers reduces backbone molecular weight and eliminates entanglements through a distinct mechanism to oNB cleavage; PEG crosslinkers are also likely cleaved to some degree by photo-oxidation. (B) and (C) Shear rheology and creep tests illustrate that irradiation with 365 nm light in the presence of Fe3+ facilitates reduced storage modulus (B) and increased plasticity (C). (D) Tanglemers equilibrated in Fe3+ and exposed to varied light doses (0 s, 1 s, and 5 s at 100 mW cm−2) have decreased modulus and strength but comparable strain to failure. (E) Tensile response of Fe3+-containing tanglemer with active irradiation beginning at ε ≥ 250%, resulting in rapid stress relaxation and fracture. (F) Shear rheology of non-crosslinked “tanglemers” shows de-gelation upon light exposure in the presence of ferric salt; MALS analysis of these polymers suggests a decrease in molecular weight from 7 MDa to <1 MDa. n = 3 gels per condition, with representative data shown. | ||
The otherwise non-degradable diAcm-crosslinked tanglemers were equilibrated in FeCl3 (6 mg mL−1) and irradiated with 365 nm light at 100 mW cm−2. To confirm that tanglemers could be degraded by this method, shear photo-rheology was performed with and without Fe3+ (Fig. 3B). In the absence of ferric salt, diAcm-crosslinked tanglemers showed no response to irradiation (consistent with the previous results in Fig. 1E), but the storage modulus of Fe3+-containing tanglemers decreased from ∼36 kPa to <10 kPa, which was comparable to the extent of softening observed in the diPDA-crosslinked system. Similarly, a creep test (held at a stress of 1 kPa) revealed dramatic plastic deformation of the diAcm-linked gels upon irradiation in the presence of Fe3+ (Fig. 3C), suggesting that backbone cleavage enabled chain pull-out and reptation in a manner akin to the diPDA tanglemers. As Fe3+ is consumed during this process, post-degradation properties are controlled by both the light dose and the initial concentration of Fe3+ in the sample.
We next performed tensile tests on diAcm-crosslinked tanglemers equilibrated in Fe3+ and irradiated with varied doses of 365 nm light (1 s, 3 s, and 5 s at 100 mW cm−2). Notably, these exposure times are all less than that of the intermediate light dose tested with diPDA-crosslinked tanglemers. As photo-oxidation is a free radical process, propagation of radical species may result in multiple chain scission events per each absorbed photon, amplifying degradation in this mechanism compared to oNB photocleavage. In this same vein, when patterning through photomasks, we observed complete de-gelation (i.e., solubilization) of diAcm-crosslinked tanglemers with longer irradiation times (Movie S2, ESI†), which is not possible via oNB cleavage alone. Tensile measurements of the Fe3+-photodegraded gels showed some interesting contrasts to the diPDA-crosslinked tanglemers. Namely, the elastic modulus and fracture strength decreased following photo-oxidation while extensibility remained fixed (Fig. 3D). Where diPDA-linked samples showed yielding and increased extensibility when irradiated at ε ≥ 250% during tensile testing (as shown in Fig. 1E), Fe3+-mediated backbone cleavage resulted in rapid dissipation of stress, followed quickly by fracture (Fig. 3E), due to the disruption of entanglements caused by strand breaks throughout the polymer backbone.
Unlike diPDA photodegradation, which results in chain lengthening, the molecular weight of the polyacrylamide backbone sharply decreases during photo-oxidation, significantly weakening the material and leading to premature failure of the network. This chain-shortening effect was confirmed via shear photo-rheology and quantified using multi-angle light scattering (MALS). To acquire un-crosslinked polyacrylamide for further analysis, we prepared “gels” using our typical tanglemer formulation with the crosslinker removed. These polymers swelled dramatically (approximately 20-fold by mass after polymerization) and formed a highly viscous, jelly-like substance (Fig. S7, ESI†), demonstrating the important role of sparse crosslinks in topologically stabilizing entanglements to form robust hydrogel networks. We characterized this material by shear photo-rheology and, as with the diAcm-crosslinked tanglemers, found no response to irradiation without Fe3+ but complete solubilization (i.e., tan(δ) > 1) in the presence of ferric salts. By performing MALS on the polymers exposed to light with and without Fe3+, we found that the initial molecular weight of the polyacrylamide chains is on the order of 7 MDa (corresponding to a degree of polymerization of nearly 100000) and decreases following photo-oxidation to <1 MDa (Fig. 3F). Pre-softened diPDA-crosslinked tanglemers could also be fully solubilized by photo-oxidation (Movie S3, ESI†), offering sequential modes of network degradation for applications where initially robust soft materials must be precisely softened before total degradation to a sol.
One of the unique benefits of incorporating photolabile crosslinkers into hydrogels is spatiotemporal control over the network structure. While spatially patterned photodegradation in conventional hydrogels has been thoroughly investigated, spatial patterning of crosslinks within entangled hydrogels has yet to be explored. First, we assessed the performance of our two photodegradation modalities (i.e., selective crosslink cleavage versus oxidative chain scission) for spatial patterning using a confocal microscope equipped with a 405 nm laser. Both oNB and Fe3+-mediated degradation were compatible with patterning single-micron positive and negative features, although the Fe3+ system resulted in more swelling and deformation around the patterned regions (Fig. 4A). This is likely explained by the mobility of hydroxyl radicals and other reactive species that facilitate oxidative degradation, whereas oNB cleavage alone cannot fully erode the network nor are these moieties mobile or capable of propagating degradation events. To probe the effects of spatial patterning on macroscale material properties, we prepared patterned gels using photomasks (Fig. S8, ESI†) with fixed ratios of bright to dark fringes of 3:1, 1:1, and 1:3 mm (i.e., 75, 50, and 25% photodegraded). As expected from our previous results, photopatterned diPDA-crosslinked tanglemers held at 500% strain showed little birefringence under crossed polarizers in the bright fringes while strong birefringence was observed in the dark fringes (Fig. 4B). Although other “topoarchitected” materials have been prepared with block-like structures of varied mechanical properties,60 our system provides this capability from a single monolithic network.
 | ||
| Fig. 4 Multimodal approaches for spatial patterning yield tunable mechanics and diffusivity in highly entangled hydrogels. (A) Hydrogels patterned using the diPDA crosslinker show superior micron-scale resolution (i.e., reduced swelling) compared to Fe3+ photo-oxidation. (B) Photomasks with alternating stripes used for tanglemer photopatterning (top), creating alternating regions of birefringence in dark and bright fringes when viewed through crossed polarizers at 500% strain (bottom). (C) Varied photomask dimensions yield controlled modulation of tensile properties in diPDA-crosslinked tanglemers. Inset: Patterned photodegradation via Fe3+-mediated oxidation results in gel embrittlement and dramatically reduced fracture stress and strain. (D) and (E) Swelling ratio (D) and diffusivity (E) in photodegradable tanglemers. *** Indicates p ≤ 0.001 by one-way ANOVA. n = 3 gels per condition, with representative data shown in panel (C). | ||
As with varied light doses in the bulk tanglemers, tensile stress–strain curves of diPDA-crosslinked gels with varied degrees of spatial degradation showed predictable decreases in fracture strength and increases in extensibility, while Fe3+-mediated spatial softening caused dramatic embrittlement (Fig. 4C). We attribute the poor mechanics observed in tanglemers patterned via photo-oxidation to the presence of fracture-prone interfaces between bright and dark fringes (Fig. S9, ESI†), where differences in polymer molecular weight and crosslink density predispose the material to failure. After irradiation, swelling followed a predictable and relatively monotonic trend for diPDA-crosslinked tanglemers, with increasing exposure areas yielding higher equilibrium swelling at a rate of approximately 0.06 mg per mg per % irradiated (i.e., degrading 1% of the volume of the gel would increase swelling by 0.06 mg mg−1). In contrast, swelling in gels degraded via photo-oxidation showed little difference between 50% and 100% exposure areas (Fig. 4D). This observation is likely a product of two factors: network degradation in the dark fringes of the partially illuminated sample due to diffusion of the oxidative species and, more significantly, diffusion of degraded fragments out of the gels. Whereas the very high-MW and de-crosslinked chains in the diPDA gels are generally trapped and create a strong driving force for water uptake, the smaller oxidized fragments in the degraded diAcm gels are free to diffuse from the network and do not strongly influence swelling.
Finally, we conducted fluorescence recovery after photobleaching (FRAP) measurements on a soluble small molecule fluorophore (calcein, rH ∼ 0.7 nm) to determine diffusivity in a single diPDA-crosslinked tanglemer with spatial gradations in light exposure (0 s, 10 s, 60 s, Fig. S10, ESI†). We observed well-controlled modulation of small molecule diffusion (a proxy for release rate) by varied light exposure, with a more than 2-fold increase in the diffusion coefficient of a nanometer-scale probe in portions of the gel irradiated for 60 s and equilibrated for 24 hours (Fig. 4E). Taken together, these results demonstrate that the mechanics and other physical properties of our diPDA-crosslinked tanglemers can be tuned over a large range via both bulk irradiation under different light doses and by using a fixed light dose with varied spatial exposures.
Ample opportunities remain for this type of photoresponsive tanglemer system. As mentioned, a hydrolytically stable photodegradable crosslinker would enhance the performance of these hydrogels over long timescales. Similarly, other (or multiple) chromophores and functional groups could be used to provide redshifted, wavelength-dependent, or more amplified degradation responses.61–64 Additionally, alternative molecular design of the tanglemer precursors might enable incorporation of a photocleavable unit into the backbone to allow for more selective degradation than is offered by Fe3+-mediated photo-oxidation.
Some hydrogel formulations are much stiffer, stronger, and/or tougher than polyacrylamide tanglemers; these include amphiphilic double network gels,65 poly(vinyl alcohol) gels prepared by solvent exchange and wet annealing (although these have relatively high hysteresis),66,67 and protein-crosslinked hydrogels.68,69 Although our tanglemers already had relatively high moduli, extensibility, and work of fracture, incorporation of nanocomposite elements or dynamic crosslinks could further improve these properties.24,70,71 Moreover, direct tuning of polymer network topology (i.e., “molecular weaving”) could substantially increase extensibility and toughness,72,73 as has been shown in polyacrylamide gels prepared via nanoconfined polymerization that templates spatial relationships between growing chains.74 In this same regard, programming macroscale topology (i.e., tanglemer metamaterials or knitted materials prepared from tanglemer yarns) may offer benefits for controlling strain-stiffening responses and the Poisson ratio of the gel construct.75,76 Clearly, tanglemers present a rich platform for exploring the consequences of network and crosslinker structure and functionality on physical and mechanical properties, as well as unique opportunities for synthesizing tough, highly elastic, and stimuli-responsive hydrogels for a range of applications.
Conclusions
We presented two new strategies for photodegradation of highly entangled polyacrylamide networks; one uses an oNB-functional diacrylate for selective cleavage of crosslinks, while the other exploits a non-selective photo-oxidation reaction to degrade the polymer backbone. Using these approaches, in situ hydrogel mechanics (i.e., modulus, strain to failure, and fracture strength) were tuned across a broad range. Notably, the oNB crosslinker allows for chain lengthening, as photodegradation results in elimination of network junctions without disrupting the integrity of the polymer backbone. Leveraging this property, we prepared spatially patterned tanglemers with highly controlled variations in extensibility, swelling, and diffusivity. Photodegradable tanglemers open new avenues for the design of robust, resilient, and programmable hydrogels with precisely controlled mechanical and physical properties, ideal for next-generation soft materials.Methods
Materials
All reagents were purchased from Sigma-Aldrich and used as received unless otherwise noted. PEG-diAcm was purchased from Creative PEGWorks and PEG-diPDA was prepared according to a previously reported method44 with some modifications. Briefly, 2.2 eq. of hydroxyethyl photolinker (Advanced ChemTech) was dissolved in DMF with 1 eq. of PEG diamine (MW ∼ 3400), 4 eq. of HATU, and 8 eq. of DIPEA. This solution was protected from light and allowed to stir for 24 hours at room temperature before precipitation in ice-cold diethyl ether. Subsequently, the precipitated polymer was re-dissolved in DCM and mixed with TEA (4 eq.) on ice, followed by 4 eq. of acryloyl chloride (dropwise) and overnight stirring at room temperature. Finally, solvent was removed via rotary evaporation and the residual oil was dissolved in a minimal amount of chloroform and again precipitated in cold diethyl ether before dialysis against DI water through a regenerated cellulose membrane. Dry PEG-diPDA polymer was isolated by flash-freezing and lyophilization.Tanglemer fabrication
Several parameters control the composition of polyacrylamide tanglemers, including the molar ratios of water to monomer (W) and crosslinker to monomer (C). We selected a formulation with W = 2, which translates to roughly 66 wt% monomer, and C = 3.2 × 10−5 (∼1 crosslinker for every 30000 monomers). For a typical gel, 400 mg of acrylamide was combined with 200 μL of water containing PEG crosslinker and solubilized at 45 °C before the addition of initiator (final APS/TEMED concentration of 0.05/0.1 wt%). After transferring the warm macromer solution to room-temperature hydrophobic glass slides separated by 500 μm-thick spacers, the visual appearance of the gel precursor changed, where the edges of the liquid monomer blend took on polygonal geometries, suggesting partial crystallization that may have further templated propagation-driven chain growth and inhibited termination events.77 Gels were removed from the slides after one hour and equilibrated in DI water; samples used for tensile testing and shear rheology were cut or biopsy-punched to the appropriate dimensions.
Characterization of mechanical properties
Mechanical testing was performed using an RSA-G2 DMA and a DHR-3 rheometer equipped with a quartz photo-rheology stage (both TA Instruments). Strips of sandpaper were taped onto the tensile fixtures to prevent gel slippage at high strains; similarly, a sandblasted 8 mm parallel plate geometry was used for shear rheology. All tensile tests were performed at 10% strain per second. Tensile modulus was determined by calculating the slope of the stress–strain curve between 5–15%. Shear rheology was performed at 1% strain and 1 rad s−1, with samples compressed to an axial force of 0.5 N. Creep tests used a fixed stress of 1 kPa. All photodegradation was performed with a fixed intensity of 100 mW cm−2.Characterization of tanglemer degradation
For tensile measurement of photodegraded samples, tanglemers were exposed to light (after equilibration in 6 mg mL−1 FeCl3 solution for 10 minutes, in the case of the diAcm-crosslinked tanglemers) immediately before loading on the tensile stage unless otherwise specified (i.e., in situ degradation was performed during tensile testing while the sample was actively under strain). Photo-rheology of tanglemer degradation was performed using the same instrument settings as noted above. Swelling measurements were taken immediately after crosslinking, after equilibration, and after re-equilibration following exposure to light. For MALS analysis, “tanglemers” containing no crosslinker were prepared following the same method as for typical samples. These swelled considerably compared to gels containing crosslinker. Portions of swollen polymer corresponding to 1 mg of dry polymer ±Fe3+ were weighed, irradiated, and solubilized in 1 mL of DI water before filtering through 0.45 μm PTFE and subsequent analysis with a Treos II multiangle light scattering unit. The high molecular indicated by MALS is consistent with our qualitative observations from handling the sample irradiated without ferric salt; we initially attempted to pass the 1 mg mL−1 solution through a 0.22 μm filter, but this resulted in immediate clogging and rupture of the PTFE membrane. Micropatterning was performed using a Zeiss LSM 710 and custom .ovl files prepared in Matlab. Macroscopic photodegradation was performed using vinyl photomasks prepared with a Cricut printer.78 FRAP was performed according to a previously described method on a Nikon A1R.79Estimation of attenuation and quantum yield
Photophysical properties, including attenuation and consumption of the oNB group at different quantum yields, were calculated using a custom Python script implementing the Beer–Lambert law and first-order reaction kinetics. The script modeled light attenuation through the sample based on the initial concentration of the photoabsorber, the molar absorptivity (extinction coefficient of oNB at 365 nm of approximately 4300 M−1 cm−1 per ref. 47), and the depth of the sample. Quantum yield estimates were based on a PEG-diPDA concentration of 0.0125 wt% and tanglemer thickness of 1 mm, which corresponds to 8-fold volumetric swelling of the as-prepared tanglemer. Although gels containing Fe3+ were somewhat yellow-brown, the ferric salt is known to photobleach,80 meaning that optical thickness was less of a concern with this approach compared to the oNB crosslinker; this was evidenced by the reverse gelation shown in Movie S2 (ESI†).Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the ESI.† Raw data are available from the authors upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Connor Miksch and Dr Tayler Hebner for assistance with related preliminary studies, Dr Manuela Garay-Sarmiento and Arkodip Mandal for experimental assistance, Dr Joselle McCracken, Melvin Colorado Escobar, and Paula Pranda for assistance with equipment, and Drs Marianela Lemon and Monica Ohnsorg for helpful conversations. Funding for this study was generously provided by DARPA W911NF-19-2-0024 (KSA, CNB), NIH R01 DK120921 (KSA), as well as NSF-REU (2348856) and the CU YSSRP (JSL, MCJ).References
- T. C. Day, S. A. Zamani-Dahaj, G. O. Bozdag, A. J. Burnetti, E. P. Bingham, P. L. Conlin, W. C. Ratcliff and P. J. Yunker, bioRxiv, 2023, preprint DOI:10.1101/2023.06.13.544814.
- R. K. Nageshan, R. Ortega, N. Krogan and J. P. Cooper, Nat. Commun., 2024, 15, 4707 CrossRef CAS PubMed.
- S. Nageswaran, J. Haipeter, J. F. E. Bodenschatz, R. Meyer, S. Koster and C. Steinem, Biomacromolecules, 2023, 24, 2512–2521 CrossRef CAS PubMed.
- R. Chang and M. Prakash, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2303940120 CrossRef CAS.
- A. K. Schulz, M. Plotczyk, S. Sordilla, M. Boyle, K. Singal, J. S. Reidenberg, D. L. Hu and C. A. Higgins, bioRxiv, 2023, preprint DOI:10.1101/2023.08.11.553031.
- G. O. Bozdag, S. A. Zamani-Dahaj, T. C. Day, P. C. Kahn, A. J. Burnetti, D. T. Lac, K. Tong, P. L. Conlin, A. H. Balwani, E. L. Dyer, P. J. Yunker and W. C. Ratcliff, Nature, 2023, 617, 747–754 CrossRef CAS PubMed.
- V. P. Patil, H. Tuazon, E. Kaufman, T. Chakrabortty, D. Qin, J. Dunkel and M. S. Bhamla, Science, 2023, 380, 392–398 CrossRef CAS PubMed.
- F. Burla, Y. Mulla, B. E. Vos, A. Aufderhorst-Roberts and G. H. Koenderink, Nat. Rev. Phys., 2019, 1, 249–263 CrossRef.
- Y. S. Zhang and A. Khademhosseini, Science, 2017, 356, eaaf3627 CrossRef PubMed.
- X. Zhao, X. Chen, H. Yuk, S. Lin, X. Liu and G. Parada, Chem. Rev., 2021, 121, 4309–4372 CrossRef CAS.
- Y. Gu, J. Zhao and J. A. Johnson, Angew. Chem., Int. Ed., 2020, 59, 5022–5049 CrossRef CAS.
- Y. Gu, J. Zhao and J. A. Johnson, Trends Chem., 2019, 1, 318–334 CrossRef CAS.
- F.-S. Wang, L. M. Kosovsky, E. C. Krist, B. J. Kruse and A. V. Zhukhovitskiy, Trends Chem., 2024, 6, 447–458 CrossRef CAS.
- J. Kim, G. Zhang, M. Shi and Z. Suo, Science, 2021, 374, 212–216 CrossRef CAS PubMed.
- J. Kim, T. Yin and Z. Suo, J. Mech. Phys. Solids, 2022, 168, 105017 CrossRef CAS.
- Y. Wang, G. Nian, J. Kim and Z. Suo, J. Mech. Phys. Solids, 2023, 170, 105099 CrossRef CAS.
- G. Nian, J. Kim, X. Bao and Z. Suo, Adv. Mater., 2022, 34, e2206577 CrossRef.
- X. Bao, G. Nian, Y. Kutsovsky, J. Kim, Q. Jiao and Z. Suo, Soft Matter, 2023, 19, 5956–5966 RSC.
- G. Zhang, J. Kim, S. Hassan and Z. Suo, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2203962119 CrossRef CAS.
- G. Zhang, J. Steck, J. Kim, C. H. Ahn and Z. Suo, Sci. Adv., 2023, 9, eadh7742 CrossRef CAS.
- J. Steck, J. Kim, Y. Kutsovsky and Z. Suo, Nature, 2023, 624, 303–308 CrossRef CAS.
- M. Shi, J. Kim, G. Nian and Z. Suo, Extreme Mech. Lett., 2023, 59, 101953 CrossRef CAS.
- R. Zhu, Z. Zheng, D. Zhu and X. Wang, J. Colloid Interface Sci., 2024, 677, 687–696 CrossRef PubMed.
- R. Zhu, D. Zhu, Z. Zheng and X. Wang, Nat. Commun., 2024, 15, 1344 CrossRef CAS.
- C. Norioka, Y. Inamoto, C. Hajime, A. Kawamura and T. Miyata, NPG Asia Mater., 2021, 13, 34 CrossRef CAS.
- D. Zheng, S. Lin, J. Ni and X. Zhao, Extreme Mech. Lett., 2022, 51, 101608 CrossRef.
- A. P. Dhand, M. D. Davidson, H. M. Zlotnick, T. J. Kolibaba, J. P. Killgore and J. A. Burdick, Science, 2024, 385, 566–572 CrossRef CAS.
- J. Liu, X. Chen, B. Sun, H. Guo, Y. Guo, S. Zhang, R. Tao, Q. Yang and J. Tang, J. Mater. Chem. A, 2022, 10, 25564–25574 RSC.
- D. Ji, D. Y. Kim, Z. Fan, C. S. Lee and J. Kim, Small, 2024, 20, e2309217 CrossRef PubMed.
- Q. He, Z. Chang, Y. Zhong, S. Chai, C. Fu, S. Liang, G. Fang and A. Pan, ACS Energy Lett., 2023, 8, 5253–5263 CrossRef CAS.
- O. Skarsetz, R. Mathes, R. S. Schmidt, M. Simon, V. Slesarenko and A. Walther, ACS Appl. Mater. Interfaces, 2024, 16, 38511–38519 CrossRef CAS.
- Y. E. Cho, J. M. Park, W. J. Song, M. G. Lee and J. Y. Sun, Adv. Mater., 2024, e2406103, DOI:10.1002/adma.202406103.
- J. Sun, F. Ni, J. Gu, M. Si, D. Liu, C. Zhang, X. Shui, P. Xiao and T. Chen, Adv. Mater., 2024, 36, e2314175 CrossRef.
- L. Xu, D.-W. Sun, Y. Tian, L. Sun and Z. Zhu, Chem. Eng. J., 2024, 479, 147795 CrossRef CAS.
- J. Y. Sun, X. Zhao, W. R. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS.
- R. G. Fonseca, F. De Bon, P. Pereira, F. M. Carvalho, M. Freitas, M. Tavakoli, A. C. Serra, A. C. Fonseca and J. F. J. Coelho, Mater. Today Bio, 2022, 15, 100325 CrossRef CAS.
- R. G. Fonseca, A. Kuster, P. P. Fernandes, M. Tavakoli, P. Pereira, J. R. Fernandes, F. De Bon, A. C. Serra, A. C. Fonseca and J. F. J. Coelho, Adv. Healthcare Mater., 2023, 12, e2300918 CrossRef.
- R. G. Fonseca, A. Hajalilou, M. Freitas, A. Kuster, E. Parvini, A. C. Serra, J. F. J. Coelho, A. C. Fonseca and M. Tavakoli, Adv. Mater. Technol., 2023, 8, 2301007 CrossRef CAS.
- I. N. Haugan, M. J. Maher, A. B. Chang, T. P. Lin, R. H. Grubbs, M. A. Hillmyer and F. S. Bates, ACS Macro Lett., 2018, 7, 525–530 CrossRef CAS.
- Y. Ruan, Q. Zou, H. Zhang, Y. Zhang, H. Zhang, W. Wu, H. Liu, J. Yan and G. Liu, Macromolecules, 2024, 57, 6583–6592 CrossRef CAS.
- E. C. Krist, B. J. Kruse, J. C. Dickenson and A. V. Zhukhovitskiy, Macromolecules, 2024, 57(16), 7878–7883 CAS.
- S. P. O. Danielsen, H. K. Beech, S. Wang, B. M. El-Zaatari, X. Wang, L. Sapir, T. Ouchi, Z. Wang, P. N. Johnson, Y. Hu, D. J. Lundberg, G. Stoychev, S. L. Craig, J. A. Johnson, J. A. Kalow, B. D. Olsen and M. Rubinstein, Chem. Rev., 2021, 121, 5042–5092 CrossRef CAS.
- A. V. Dobrynin, Y. Tian, M. Jacobs, E. A. Nikitina, D. A. Ivanov, M. Maw, F. Vashahi and S. S. Sheiko, Nat. Mater., 2023, 22, 1394–1400 CrossRef CAS.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS PubMed.
- P. J. LeValley, R. Neelarapu, B. P. Sutherland, S. Dasgupta, C. J. Kloxin and A. M. Kloxin, J. Am. Chem. Soc., 2020, 142, 4671–4679 CrossRef CAS.
- T. Liu, B. Bao, Y. Li, Q. Lin and L. Zhu, Prog. Polym. Sci., 2023, 146, 101741 CrossRef CAS.
- M. W. Tibbitt, A. M. Kloxin, L. Sawicki and K. S. Anseth, Macromolecules, 2013, 46, 2785–2792 CrossRef CAS PubMed.
- M. W. Tibbitt, A. M. Kloxin and K. S. Anseth, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1899–1911 CrossRef CAS.
- L. T. B. Nguyen and M. Abe, Bull. Chem. Soc. Jpn., 2024, 97, uoae067 CrossRef.
- B. Liu, T. Yin, J. Zhu, D. Zhao, H. Yu, S. Qu and W. Yang, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2217781120 CrossRef.
- U. Gröllmann and W. Schnabel, Polym. Degrad. Stab., 1982, 4, 203–212 CrossRef.
- J. E. Woodrow, J. N. Seiber and G. C. Miller, J. Agric. Food Chem., 2008, 56, 2773–2779 CrossRef CAS PubMed.
- B. Xiong, R. D. Loss, D. Shields, T. Pawlik, R. Hochreiter, A. L. Zydney and M. Kumar, npj Clean Water, 2018, 1, 17 CrossRef CAS.
- M. Michelas, L. Wimberger and C. Boyer, Macromol. Rapid Commun., 2024, e2400358, DOI:10.1002/marc.202400358.
- C. T. B. Paula, P. Pereira, J. F. J. Coelho, A. C. Fonseca and A. C. Serra, Mater. Sci. Eng., C, 2021, 131, 112520 CrossRef CAS.
- C. T. B. Paula, A. B. Madeira, P. Pereira, R. Branco, P. V. Morais, J. F. J. Coelho, A. C. Fonseca and A. C. Serra, Eur. Polym. J., 2022, 177, 111447 CrossRef CAS.
- C. T. B. Paula, A. Leandro, P. Pereira, J. F. J. Coelho, A. C. Fonseca and A. C. Serra, Macromol. Biosci., 2024, 24, e2300289 CrossRef PubMed.
- C. T. B. Paula, S. Saraiva, P. Pereira, J. F. J. Coelho, A. C. Fonseca and A. C. Serra, Polymer, 2024, 294, 126697 CrossRef CAS.
- J. I. Bowman, J. B. Young, V. L. Thompson, K. A. Stewart, L. E. Diodati, K. C. Stevens, C. B. Eades and B. S. Sumerlin, Macromolecules, 2024, 57, 7547–7555 CrossRef CAS.
- X. Liu, J. Wu, K. Qiao, G. Liu, Z. Wang, T. Lu, Z. Suo and J. Hu, Nat. Commun., 2022, 13, 1622 CrossRef CAS PubMed.
- V. X. Truong, F. Li and J. S. Forsythe, ACS Appl. Mater. Interfaces, 2017, 9, 32441–32445 CrossRef CAS.
- J. L. Pelloth, P. A. Tran, A. Walther, A. S. Goldmann, H. Frisch, V. X. Truong and C. Barner-Kowollik, Adv. Mater., 2021, 33, e2102184 CrossRef PubMed.
- T. E. Brown, I. A. Marozas and K. S. Anseth, Adv. Mater., 2017, 29, 11 CrossRef.
- B. D. Fairbanks, S. P. Singh, C. N. Bowman and K. S. Anseth, Macromolecules, 2011, 44, 2444–2450 CrossRef PubMed.
- X. Hou, B. Huang, L. Zhou, S. Liu, J. Kong and C. He, Adv. Mater., 2023, 35, e2301532 CrossRef PubMed.
- Y. Wu, Y. Zhang, H. Wu, J. Wen, S. Zhang, W. Xing, H. Zhang, H. Xue, J. Gao and Y. Mai, Adv. Mater., 2023, 35, e2210624 CrossRef PubMed.
- H. Wu, Y. Wu, J. Yan, Y. Wang, H. Zhang, Z. Liu, H. Li, J. Wang and J. Gao, Polymer, 2024, 306, 127223 CrossRef CAS.
- L. Fu, L. Li, Q. Bian, B. Xue, J. Jin, J. Li, Y. Cao, Q. Jiang and H. Li, Nature, 2023, 618, 740–747 CrossRef CAS.
- K. Wang, Y. Zhang, T. Chen, L. Bai, H. Li, H. Tan, C. Liu and X. Qu, Composites, Part B, 2023, 266, 110991 CrossRef CAS.
- D. Sun, Y. Gao, Y. Zhou, M. Yang, J. Hu, T. Lu and T. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 49389–49397 CrossRef CAS PubMed.
- S. Pruksawan, J. W. R. Lim, Y. L. Lee, Z. Lin, H. L. Chee, Y. T. Chong, H. Chi and F. Wang, Commun. Mater., 2023, 4, 75 CrossRef CAS.
- G. Li, J. Zhao, Z. Zhang, X. Zhao, L. Cheng, Y. Liu, Z. Guo, W. Yu and X. Yan, Angew. Chem., Int. Ed., 2022, 61, e202210078 CrossRef CAS.
- Z. H. Zhang, B. J. Andreassen, D. P. August, D. A. Leigh and L. Zhang, Nat. Mater., 2022, 21, 275–283 CrossRef CAS PubMed.
- W. Li, X. Wang, Z. Liu, X. Zou, Z. Shen, D. Liu, L. Li, Y. Guo and F. Yan, Nat. Mater., 2024, 23, 131–138 CrossRef CAS.
- N. Munding, M. Fladung, Y. Chen, M. Hippler, A. D. Ho, M. Wegener, M. Bastmeyer and M. Tanaka, Adv. Funct. Mater., 2023, 34, 2301133 CrossRef.
- K. Singal, M. S. Dimitriyev, S. E. Gonzalez, A. P. Cachine, S. Quinn and E. A. Matsumoto, Nat. Commun., 2024, 15, 2622 CrossRef CAS.
- K. A. Berchtold, B. Hacioglu, J. Nie, N. B. Cramer, J. W. Stansbury and C. N. Bowman, Macromolecules, 2009, 42, 2433–2437 CrossRef CAS PubMed.
- L. M. Harrison, B. M. Brisard, K. D. Cashwell, A. L. Mulkey and C. A. Schmidt, bioRxiv, 2024, preprint DOI:10.1101/2024.04.29.591694.
- T. S. Hebner, B. E. Kirkpatrick, B. D. Fairbanks, C. N. Bowman, K. S. Anseth and D. S. W. Benoit, Adv. Sci., 2024, 11, e2402191 CrossRef PubMed.
- J. G. Schauberger, G. Riess and W. Kern, J. Appl. Polym. Sci., 2013, 129, 3623–3628 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb02149e |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2025 |