
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComposite solid-state electrolytes for all solid-state lithium batteries: progress, challenges and outlook
Senhao Wang ,
Andrea La Monaca and
George P. Demopoulos*
,
Andrea La Monaca and
George P. Demopoulos*
Materials Engineering, McGill University, Montreal, QC H3A0C5, Canada. E-mail: george.demopoulos@mcgill.ca
First published on 17th December 2024
Abstract
Composite solid-state electrolytes (CSEs) with multiple phases offer greater flexibility to customize and combine the advantages of single-phase electrolytes, making them promising candidates for commercial all-solid-state batteries (ASSBs). Based on existing investigations, this review provides a comprehensive overview of the recent progress in CSEs. First, we introduce the historical development of solid-state ionic conductors, and then summarize the fundamentals including key materials and mechanisms of lithium-ion transport. Three main types of advanced structures for CSEs are classified and highlighted according to the recent progress, namely composite solid electrolytes with passive fillers, composite solid electrolytes with active fillers, and 3D framework composite solid electrolytes. Finally, the challenges and perspectives of the composite solid-state electrolytes are discussed.
1. Introduction
Energy is the constant driving force for the sustainable advancement of society. Electrochemical batteries are among the most crucial devices for efficiently storing and delivering energy enabling higher degree of mobility, electrification of transportation, and renewable energy development. The commercialization of rechargeable lithium-ion batteries (LIBs) in the early 1990s marked a significant milestone in the evolution of electrochemical energy storage devices. This innovation has revolutionized modern lifestyles with the widespread adoption of LIBs in portable electronics and electric vehicles. As the demand for energy storage continues to surge, the focus of future battery development will be on enhancing energy density and ensuring safety.1 One key approach to achieving a high energy density cell is to explore advanced electrode materials with high capacity and a broad voltage range. Li metal stands out as a promising candidate due to its ultra-high theoretical specific capacity (3860 mA h g−1, 10× times greater than that of graphite anodes in LIBs (Fig. 1a)) and most negative electrochemical potential (3.040 V compared to SHE),2 which enables larger capacity and operating voltage, ultimately leading to greater energy density in batteries. Consequently, Li-metal batteries (LMBs) have the potential to power electronic devices for extended periods while remaining lightweight.3 In traditional LIBs, non-aqueous solutions are commonly used to move Li ions between the positive and negative electrodes.4 The flammable and prone-to-leak nature of non-aqueous solutions poses a significant safety risk. Substituting the liquid organic solution with a robust solid-state electrolyte (SSE) offers a viable approach to enhancing LIB safety (Fig. 1b).5 Also, SSE allows for reducing thickness, potentially boosting volumetric energy density and enabling use in flexible and wearable devices.6 Thus, the combination of Li-metal anode with SSE in solid-state LMBs (SSLMBs) holds promise for high energy density and robust safety features.7,8 Despite the promising aspects, the path to commercializing SSLMBs encounters substantial hurdles.9 Li-metal anodes were initially suggested in the 1960s but have been largely disregarded in commercial products due to dendrite growth, extensive volume fluctuations, and the strong chemical and electrochemical reactivity of Li metal. These challenges lead to significant side reactions, safety issues, and short lifespan of LMBs as depicted in Fig. 1c.10–12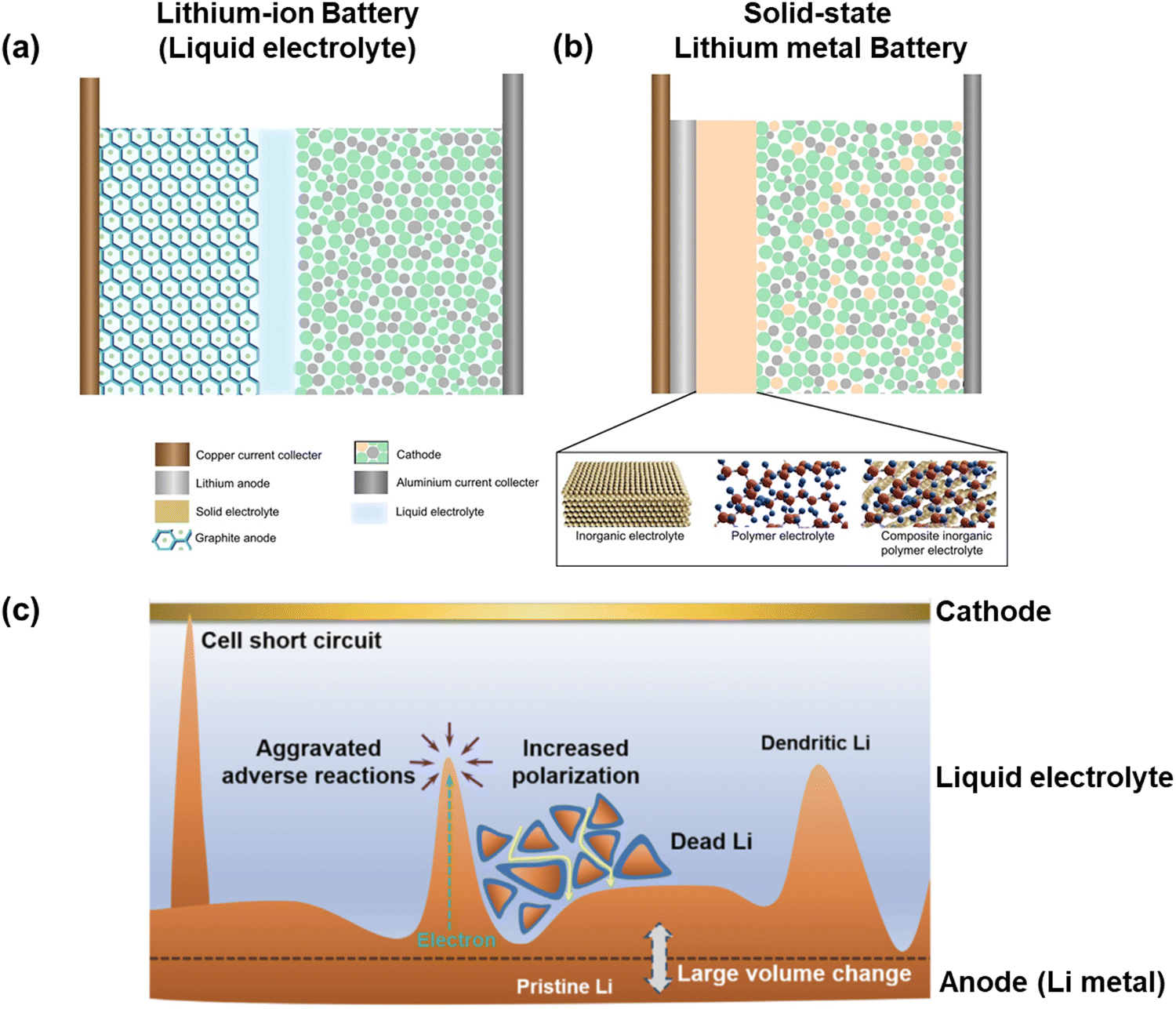 | ||
| Fig. 1 Schematics of (a) conventional Li-ion battery (LIB) and (b) solid-state Li metal battery (SSLMB);5 and (c) safety issues with Li-metal anodes in conventional LIB.10 | ||
SSEs offer an attractive opportunity to achieve high-energy-density and safe battery systems. These materials are in general non-flammable and some of them may prevent the growth of Li dendrites.13,14 There are two main categories of SSEs proposed for application in Li metal batteries: polymer solid-state electrolytes (PSEs)15 and inorganic solid-state electrolytes (ISEs).16 While both types have been extensively researched, each has its own strengths and weaknesses. ISEs exhibit relatively high Li-ion conductivity, electrochemical and thermal stability, as well as dendrite suppression capabilities. However, their brittleness and poor interfacial compatibility with electrodes pose significant challenges.17–19 On the other hand, PSEs offer good mechanical flexibility and interfacial compatibility with electrodes. This makes them suitable for flexible battery applications. Nevertheless, polymer electrolytes have lower ionic conductivity at room temperature, which is a major drawback for their application.20,21 Composite solid-state electrolytes (CSEs) comprised of a polymer SE and a ceramic SE can combine the benefits of both materials while mitigating their drawbacks.22–25 By carefully controlling the composition and employing an appropriate manufacturing process, these polymer–ceramic composites have the potential to exhibit the desired properties and performance characteristics. These may include adequate Li-ion conductivity at room temperature, optimal mechanical strength, an expanded electrochemical stability window, enhanced Li-ion transference efficiency, good interface interaction with electrodes, and the potential to suppress dendrite formation.26 This article provides an overview of composite solid-state electrolytes commonly currently considered for utilization in all-solid-state batteries (ASSBs). The evolution of solid-state electrolytes is discussed initially, followed by an examination of the characteristics and drawbacks of individual solid-state electrolytes. Their limitations are then addressed by switching to composite solid-state electrolytes. Subsequent sections focus on cutting-edge strategies designed to enhance the properties of CSEs in order to attain high ionic conductivity, low interfacial resistance, and enhanced stability towards electrodes for future applications in ASSBs.
2. Historical progress
Solid-state ionics (SSI) is a research area that covers a wide range of disciplines, focusing on the fast movement of mobile ions, and mixed ions within solid materials, and examining their physical and chemical characteristics. The term SSI was coined by Takahashi in the 1970s;27 however, this phenomenon has been recognized since the early 1800s when Michael Faraday uncovered mass movement in Ag2S and PbF2.28 In the investigation of ionic conductors, the achievement of enhanced Na+ ion mobility in a glass by Warburg and the initial Na+ ion transference number assessments by Warburg29 and Tegetmeier30 are noteworthy contributions. In 1897, Walther Nernst made impactful advances by formulating the Nernst equation and identifying ionic conduction in aliovalent-doped zirconia.31 Utilizing his discoveries, Nernst created the Nernst lamp employing yttria-stabilized zirconia (YSZ) as the filament.31,32 Subsequently, more effective tungsten filament lamps superseded these. While Nernst identified the mass transport in zirconia, he could not have comprehended the structural particulars of the material and the mode of transport until Wagner elucidated the fundamental mechanism of oxygen ion conduction in doped ZrO2 through oxygen concentration EMF measurements in 1943.33,34 Another significant milestone in SSI was the revelation of the extraordinary characteristics of α-AgI by Tubandt and Lorenz at Halle in 1914.35 In the 1930s, Tubandt conducted experiments to analyze the ionic conductivity of various metal halides utilizing novel techniques to assess both ionic and electronic contributions to conductivity. Tubandt employed specialized electrodes that permitted the passage of mobile ions in and out of the sample, determining the overall current flow through the sample through basic DC measurements, thereby approximating the electronic component of total conductivity. The transfer of ions in AgI was quantified by monitoring changes in electrode masses, thereby elucidating the ionic element of conductivity. Frenkel proposed two distinct diffusion mechanisms through interstitials and vacancies in 1925, postulating the existence of point defects.36 The concept of point defects was solidified in the 1920s and 1930s by Frenkel, Walter Schottky, and Carl Wagner,32,37 accompanied by the development of point-defect thermodynamics by Schottky and Wagner, aiding in the comprehension of ionic and electronic transport mechanisms in solids. In the 1950s, Wagner made significant contributions, including the Hebb–Wagner direct-current polarization method, which employed a blocking electrode to differentiate between ionic and electronic current carriers in predominantly ionically conducting solids.38,39 This technique continues to be utilized for assessing the ionic and electronic conductivity of diverse solid electrolytes.40–43 Kiukkola and Wagner employed solid electrolyte-based electrochemical sensors in 1957, conducting extensive potentiometric measurements.44 In the 1960s, solid silver ion conducting materials such as Ag3SI11 and RbAg4I5 were introduced and demonstrated for use in electrochemical cells by Takahashi and Yamamoto, and by Argue and Owens, respectively.45,46 Takahashi et al. synthesized a Cu+ ion superconductor, Rb4Cu16I7Cl13, exhibiting the highest room temperature ionic conductivity (0.34 S cm−1) ever recorded among solid electrolytes.47 In 1967, Yao and Kummer made a ground-breaking discovery regarding the high ion mobility present in alkali metal substituted β-alumina.48 Following this, Kummer and Weber successfully utilized Na-β-alumina in Na–S batteries.49 This initial discovery of β-alumina paved the way for the creation of newer superionic conductors, such as gallates (where Al is replaced by Ga) and ferrites (where Al is replaced by Fe).50,51 Then, in 1976, Goodenough and Hong introduced a sodium superionic conductor, now commonly referred to as NASICON.52 They produced a compound with the formula Na1+xZr2P3−xSixO12, with 0 < x < 3. Among these compounds, Na3Zr2PSi2O12 displayed the highest conductivity, reaching 0.2 S cm−1 at 300 °C, comparable to Na-β-alumina. Hong's research also exemplified how ionic substitutions in NASICON, integrating elements like Li, Ag, and K, led to the synthesis of a wide array of compounds.53With a keen focus on lithium-ion conductors, significant attention has been placed on their development since the 1970s due to the low ionic radii and weight of the Li+ ion. Breakthroughs in the ionic conductivity of both single and polycrystalline lithium nitride were achieved in the late 1970s.54 Notable advancements included the conductivity improvement observed by integrating Al2O3 into LiI, resulting in a conductivity 50 times higher than that of pure LiI.55 In 1980, Alan et al. showcased the application of LiI as an electrolyte in cardiac pacemakers, setting a new standard in medical technology.56 Following this, Hong57 introduced another variant of lithium superionic conductor known as LISICON, characterized by the general formula Li16−2xMx(TO4)4, where M represents a divalent cation (such as Mg2+, Zn2+) and T signifies a tetravalent cation (such as Si4+, Ge4+), with x ranging from 0 to 4. In 2000, Kanno et al. introduced thio-LISICON as a new lithium-ion conductor, with Li3.25Ge0.25P0.75S4 displaying the highest conductivity within this series.58 Thangadurai et al.59 further expanded on these findings in 2003, reporting the lithium-ion conductivity of garnet-type Li5La3M2O12 (where M stands for Ta or Nb). Of note is the inherent advantage of garnet-type electrolytes, as they can directly interface with Li metal without causing any damage. A different category of solid electrolytes known as Li-rich anti-perovskite was introduced by Zhao and colleagues in 2012.60 Within this category, Li3OCl0.5Br0.5 demonstrated a room temperature conductivity of 1.94 × 10−3 S cm−1. Over the past few decades, significant strides have been made in identifying and developing innovative solid electrolytes, especially for advanced solid-state battery systems, fuel cells, and sensors, as detailed in Fig. 2.
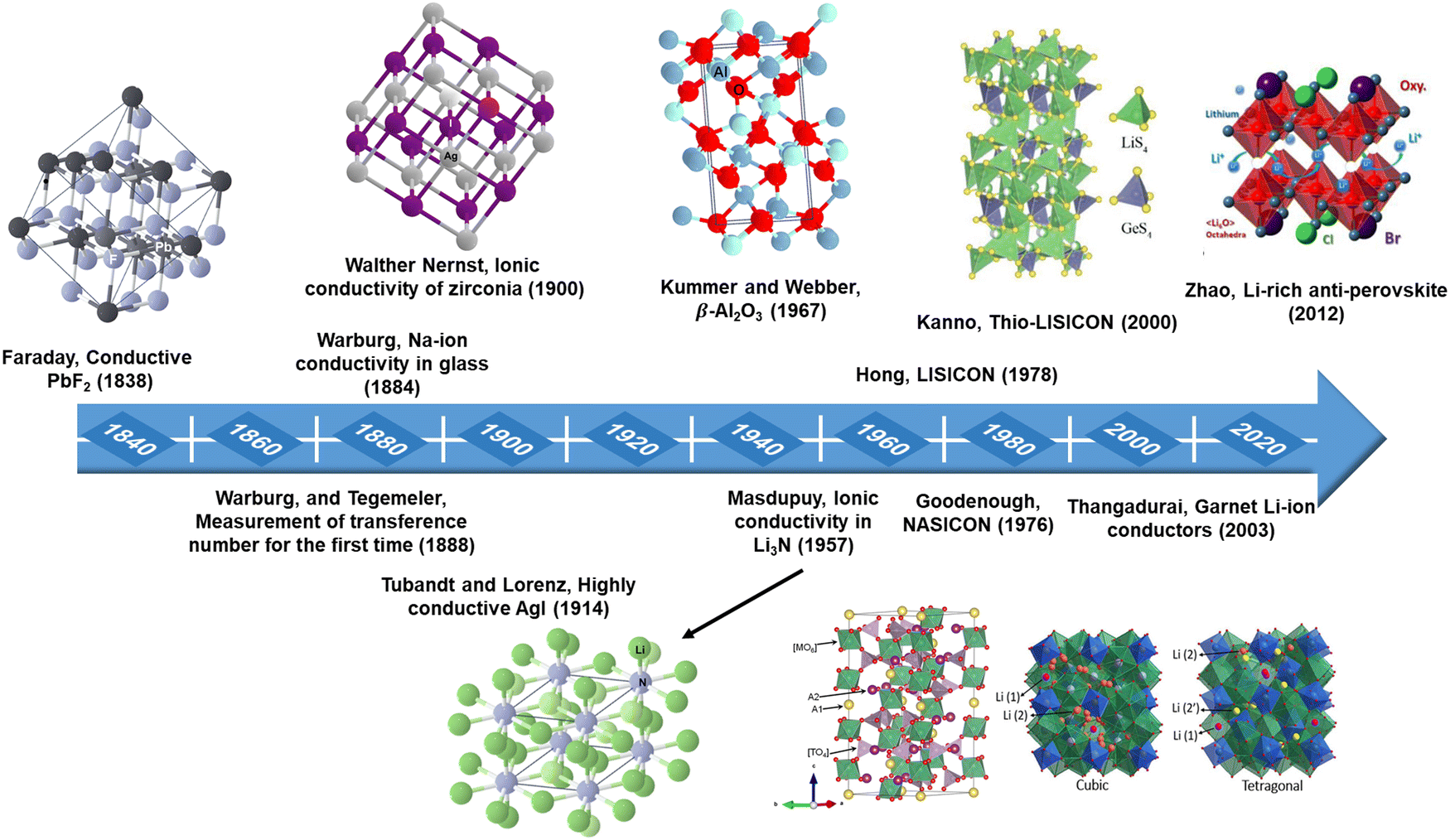 | ||
| Fig. 2 Historical development of solid electrolytes. | ||
3. Single inorganic solid electrolytes and polymer solid electrolytes
3.1. Inorganic solid electrolytes
Inorganic solid electrolytes include oxide, sulfide, and halogen types, specifically, oxide-type including garnet solid electrolytes, Li1+xAlxTi2−x(PO4)3 (0 ≤ x ≤ 0.5) (NASICON) solid electrolytes, perovskite solid electrolytes, anti-perovskite solid electrolytes, and sulfide-type including thio-LISICON solid electrolytes, Li10GeP2S12 (LGPS) solid electrolytes, Li9.54SiP1.44S11.7Cl0.3 solid electrolytes and argyrodite solid electrolytes. Owing to their high ionic conductivity, excellent mechanical properties, non-flammability and non-explosive safety, inorganic solid electrolytes have attracted wide attention from researchers. This part will introduce their structures and electrochemical properties, as well as the advantages and disadvantages of these electrolytes.Garnet-type solid electrolyte Li7La3Zr2O12 (LLZO) and its derivatives is considered as the most promising oxide solid electrolyte61,62 due to its high lithium-ion conductivity (10−3–10−4 S cm−1) at room temperature, wide electrochemical stability window, and good chemical stability with lithium metal. LLZO has two crystalline structures, the cubic and tetragonal phases (Fig. 3a),63 of which the desired one is the cubic structure as its ionic conductivity is about two orders of magnitude higher than that of the tetragonal phase. To obtain the cubic phase, extreme high sintering temperature is required in order to transition from tetragonal phase to cubic phase.64,65 The sintering temperature of cubic LLZO prepared by the traditional solid-state reaction method needs to reach 1150–1230 °C.66 Although the sintering temperature can be reduced by using spark plasma sintering,67 hot pressing sintering,68 and other methods, it still needs a high temperature above 1000 °C. Recently though a breakthrough low-temperature crystallization approach was reported by the authors enabling the synthesis of nanoscale cubic LLZO at only 600 °C via a hydrothermal enabled method exhibiting good ionic conductivity, electrochemical stability as well as improved relative density.69 In general, to obtain a stable LLZO cubic phase structure, elemental doping (Ta, Al, Ga, Nb, and Mg, etc.) is recommended.70,71 Element doping can also improve the relative density of electrolyte.72 There are though some disadvantages of LLZO that cannot be ignored, such as its chemical stability in water. At room temperature, LLZO reacts with water to form LiOH, which eventually reacts with CO2 to form Li2CO3. Other than causing contamination, this reaction process may also cause the transformation of cubic phase to tetragonal phase, reducing the total ionic conductivity.73,74 In addition, there are some interfacial side reactions between LLZO and electrode materials. For example, researchers seeking to obtain a tight interfacing between LLZO and cathode applied high temperature co-sintering and pulsed laser deposition giving rise to the formation of an interfacial phase.75–78 Thus, when LiCoO2 thin films were deposited on LLZO pellet by pulse deposition, a La2CoO4 intermediate phase of about 50 nm thick was formed at the interface between LLZO and LiCoO2 resulting in high interfacial resistance, hindering interfacial Li-ion transport.
 | ||
| Fig. 3 (a) Cubic and tetragonal crystal structure of garnet solid electrolyte.65 (Reproduced with permission from The Royal Society of Chemistry, UK.) (b) Schematic diagram of the crystal structure of NASICON-type solid electrolyte.79 (Reproduced with permission from the American Chemical Society.) (c) LixLa0.557TiO3 (0.303 ≤ x ≤ 0.370) cubic phase and tetragonal phase crystal structure.80 (Reproduced with permission from Elsevier.) (d) Crystal structures of β-Li3PS4.81 (Reproduced with permission from Elsevier.) (e) Crystal structure of LGPS, one-dimensional view of LGPS and Lit transport pathway.82 (Reproduced with permission from Nature Publishing Group.) (f) Structures and lithium-ion migration pathways of Li3MCl6 (where M = Er, Y, Ho, Yb) based on hexagonal close packed (hcp) anion framework.83 (Reproduced with permission from Nature Publishing Group.) | ||
The general formula of NASICON structure can be written as LiM2(PO4)3, where M can be Ti, Ge, Zr, and other elements. The NASICON skeleton includes PO4 tetrahedra and MO6 (M = Ti, Ge, Zr) octahedra, forming a three-dimensional (3D) network structure (Fig. 3b).84,85 Li1+xAlxTi(Ge)2−x(PO4)3 prepared by partial substitution of Ti4+ or Ge4+ with Al3+ exhibits an enhanced conductivity of 10−2–10−3 S cm−1 at room temperature, which approaches that of liquid electrolyte.86 However, the tetravalent (Ti4+, Hf4+) metal elements in the NASICON-type solid electrolyte is reduced by lithium metal leading to the distortion of the crystal structure, thus limiting its use in high energy density lithium metal batteries.87,88
The perovskite solid electrolyte with formula Li3xLa2/3−xTiO3, (0 < x < 0.16), it has the ABO3 structure, where position A represents Li and La with a total atomic occupancy less than 1 resulting in the existence of vacancies. In perovskite solid electrolytes, the Li-ion transport mechanism is the ion-vacancy transition type, based on which the bulk ionic conductivity at room temperature could reach as high as 10−3 S cm−1.89 However, the resistance of grain boundaries is much higher than the bulk (grain), leading to very low total conductivity. There are two-types of perovskite crystal structures, cubic phase and tetragonal phase (Fig. 3c), among which the ionic conductivity of the cubic phase is the higher one.90,91 Moreover, due to Ti4+ undergoing reduction reaction with lithium metal, resulting in Ti3+, their application in advanced lithium metal batteries is severely limited.92
Sulfide-type solid electrolytes exhibit higher ionic conductivity compared to oxide-type solid electrolytes comparable to that of liquid electrolytes, or even higher because of the larger ionic radius and lower electronegativity of S2− compared to O2−, which means less binding force for Li-ions and higher concentration of free-moving Li-ions.93 Moreover, sulfide-type solid electrolytes exhibit other advantages of good flexibility, processability, and interfacial contact-ability with electrodes.94 Generally, the crystal structures of sulfide-type solid electrolytes can be classified into glassy, glass-ceramic, and crystalline-ceramic. Specifically, there is no grain boundary impedance in glassy-type, and the structure is isotropic, thereby Li-ion transport is easier exhibiting higher ionic conductivity.95 Glass-ceramic type is obtained from glassy type via high-temperature partial-crystallization, and it is fundamentally a mixture of crystalline and amorphous phases. The glass-ceramic type exhibits a higher ionic conductivity compared to glassy type due to the Li-ion transport channels of ceramic crystalline phase. There are three main types of crystalline-ceramic sulfide solid electrolytes, namely Thio-LISICON type,96 sulfur–silver–germanium ore type,97 and Li10GeP2S12 type.98 Although sulfide-type solid electrolytes exhibit high room temperature Li-ion conductivity, they are highly hygroscopic and sensitive to air components making the synthesis process and preservation conditions challenging. Notably, solid-state batteries enabled by sulfide-type solid electrolytes produce H2S gas during the cycle process, causing their expansion, although additives could be used to inhibit the production of H2S gas without solving the fundamental problem.99,100 Moreover, sulfide solid electrolytes are not stable with lithium metal and traditional oxide cathode materials. Thus, the sulfide electrolyte Li3PS4 (Fig. 3d) may be reduced by lithium metal into low lithium-ion conductivity products, such as Li3P or Li2S, that increase the interface resistance, until a stable solid electrolyte interface (SEI) layer forms.101,102 The SEI layer formed by the reaction of Li10GeP2S12 (crystal structure shown in Fig. 3e) with lithium metal, contains not only Li3P and Li2S but also Li–Ge alloy, which can act as electron conductor. As a consequence, the interface characterized by ion and electron conductivities is unstable, leading to SEI thickening and interface impedance increase with increased of cycling.103 When a sulfide electrolyte and an oxide cathode (LiCoO2) are assembled into a battery, compared with sulfide, oxide has stronger binding ability to lithium ions, and lithium ions in sulfide electrolyte are more likely to enter oxide cathode materials, which leads to the decrease of lithium-ion concentration in electrolyte, and ultimately leads to the decrease of battery capacity.104 Theoretical calculations and experiments evaluated the electrochemical stability windows of different types of sulfide electrolytes, and the results showed that sulfide electrolytes have a narrow electrochemical stability window.105,106
Recently, a novel inorganic solid electrolyte material called lithium-rich anti-perovskite electrolyte has gained attention in the scientific community. This material was initially identified by Zhao et al.,107 who noted that its conductivity can be as high as 10−3 S cm−1 at room temperature. Comparatively, lithium-rich anti-perovskite shares a similar crystal structure to the traditional perovskite material ABO3, except for the reversal of cation and anion positions. The crystal structure of the anti-perovskite form is denoted as Li3AB, where the A site is occupied by an oxygen atom, and the B site is filled by a halogen atom such as chlorine or bromine.108 Specifically, the anti-perovskite compound Li3OCl features a cubic system arrangement, with oxygen atoms situated centrally within octahedral assemblies and lithium ions positioned at the reserved vertices of these octahedra, culminating in a lithium-rich configuration. This unique structure, characterized by a significant number of vacancies, not only facilitates the transportation of lithium ions but also lowers the activation energy required for such processes (see Fig. 3f).109
Deng et al.110 found that the use of Br to partially substitute Cl in Li3OCl for the synthesis of Li3OCl1−xBrx demonstrated that the resulting anti-perovskite was easier to create and led to an increase in ionic conductivity. In a similar study, Hood et al.111 discovered that the substitution of Li2OHX (X = Cl, Br) also resulted in an anti-perovskite structure. This compound could be easily obtained by heating a mixture of LiOH and LiX below 400 °C. The produced Li2OHX exhibited noticeable lattice imperfections, along with high ionic conductivity and a low activation energy. Additionally, Li2OHX exhibited stability towards lithium metal, even at temperatures surpassing the melting point of lithium metal.112 Despite previous research on Li3OX and Li2OHX, Song et al.113 demonstrated through experimental and theoretical investigations, such as Bonn–Oppenheimer molecular dynamics simulations, that most of the previously reported Li3OCl and Li3OBr compounds may actually be Li2OHCl and Li2OHBr. It was revealed that the short OH− groups in this anti-perovskite electrolyte rapidly formed Frenkel defects, creating additional space for the swift movement of lithium ions. The presence of H was shown to enhance lithium-ion conductivity.
The production process of inorganic solid electrolytes is intricate, requiring high sintering temperatures and substantial energy consumption. Furthermore, these electrolytes face challenges such as difficult powder compression, high quality standards, and the brittleness of the sintered electrolyte sheet. During cycling, the battery volume expands due to the repeated plating/stripping of lithium metal. The rigid structure of the electrolyte sheet fails to alleviate this stress adequately, leading to electrolyte ruptures and battery malfunctions. However, a drawback of inorganic solid electrolytes is the considerable interface impedance between the electrode and electrolyte, hindering their widespread utilization.
3.2. Mechanism of Li-ion transport in inorganic solid electrolytes
Ionic conductivity in crystalline solid materials is heavily influenced by the defects present in the crystal structure, including point defects, line defects, planar defects, volume defects, and electron defects.114 Among these, point defects are particularly important in the context of lithium-ion diffusion mechanisms. A diagram illustrating some common point defects is provided in Fig. 4a.115 The two most notable point defects are the Frenkel defect, which consists of an anion vacancy accompanied by a cation interstitial, and the Schottky defect, which involves a cation vacancy accompanied by an anion vacancy. Point defect-based mechanisms can be categorized into vacancy (defect) mechanisms and non-vacancy (non-defect) mechanisms. The former includes simple vacancy mechanisms, while the latter encompasses interstitial mechanisms, collective mechanisms, and interstitial-substitutional exchange mechanisms.116 The vacancy mechanism, illustrated in Fig. 4b, allows lithium ions to move from their original position to an adjacent vacancy for diffusion, involving minimal lattice strain and lower activation energy barriers.117 The concentration of vacancies within the lattice plays a crucial role in transport kinetics, impacting the pathways and energy barriers for lithium migration.118 Additionally, several other elements, such as the type of ions close to the diffusion pathway and the arrangement and proximity of the nearby vacancies or doped cations—can influence the energy barrier for lithium ion diffusion.119 For non-vacancy mechanisms, an important example is the interstitial mechanism, which encompasses both direct interstitial diffusion and interstitial knock-off diffusion, illustrated in Fig. 4c and d, respectively. In the direct interstitial mechanism, the interstitial ion moves directly to a neighboring interstitial site. Typically, interstitial atoms are much smaller than the matrix atoms, leading to significant lattice strain during their migration. Conversely, indirect interstitial diffusion, often observed in lithium-ion batteries, especially at high lithium ion concentrations, involves the interstitial atom striking a matrix atom. The displaced matrix atom then migrates to another nearby interstitial site, as depicted in Fig. 4d. Here, the interstitial atom may be the same size or similar to the matrix atom. This indirect interstitial mechanism, also known as the knock-off mechanism or collective mechanism, involves the simultaneous movement of at least two atoms.116 Another interstitial mechanism is the interstitial substitutional exchange, also classified as a collective mechanism, which can be further divided into direct exchange and ring diffusion, as shown in Fig. 4e. In the former case, two atoms simultaneously shift and exchange positions on the lattice, while in the latter scenario, a cluster of atoms (consisting of three or more) move collectively in a circular motion, shifting by one atom's length to new locations. Contrary to the vacancy mechanism, the non-vacancy mechanism, also known as non-defect diffusion, poses a greater challenge due to the elevated energy barrier for migration. Like the diffusion processes observed in inorganic crystalline electrolytes, the movement of lithium ions in inorganic amorphous materials (commonly found in glasses) involves transitioning from one local site to adjacent sites.115,120 In addition to the framework and arrangement of ions, the interaction between the structural framework and charge carriers plays a significant role in facilitating the diffusion process. Nonetheless, some inorganic solid electrolyte materials lack clear classification as ceramics or glasses, necessitating further investigation into their lithium-ion diffusion mechanisms.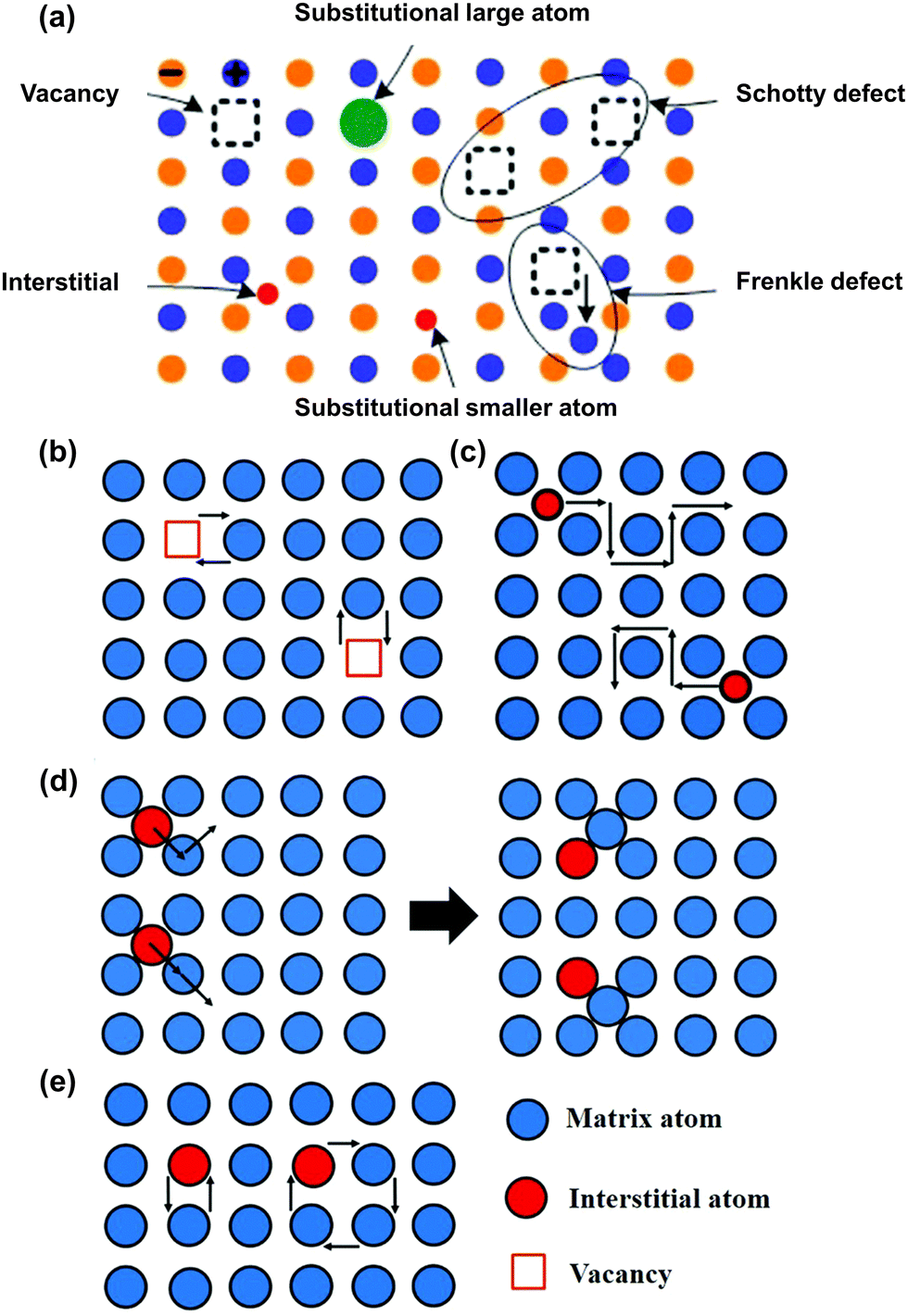 | ||
| Fig. 4 Schematic diagram for mechanisms of Li ionic transport in active inorganic region. (a) Some typical point defects in the inorganic solid electrolyte.114 (Reproduced with permission from Elsevier.) (b) Vacancy diffusion mechanism, (c) direct interstitial mechanism, (d) interstitial knock-off mechanism, and (e) direct exchange and ring mechanism.121 (Reproduced with permission from IOP Publishing Ltd.) | ||
3.3. Polymer solid electrolytes
In the 1980s, Wright and colleagues made a pivotal discovery by demonstrating that Li+ ions could be transported through polyethylene oxide (PEO)-based electrolytes combined with lithium salts.122 This transport involves a continual process of complexation and uncomplexation with the oxygen bonds in the PEO chains, leading to the material becoming ion-conductive. This breakthrough brought the concept of polymer electrolytes into focus, offering a novel direction for developing solid electrolytes. The mechanism through which lithium ions move in polymer electrolytes involves the lithium ions hopping between coordination sites and the segmented movements of the polymer chains. Consequently, the ion conductivity in these systems largely depends on the quantity of free lithium ions and the mobility of the polymer chains. To achieve polymer electrolytes with high ionic conductivity, selecting appropriate polymers and lithium salts is crucial. Polymers that possess high dielectric constants and polar functional groups (such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, –O–, –N–, –P–, and –C
O, –O–, –N–, –P–, and –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) are particularly favorable because they facilitate the dissociation of lithium salts, promoting their interaction with polar groups, forming complexes that repeatedly complex and de-complex along the polymer chains. Consequently, low lattice energy lithium salts, which ionize readily, are chosen to maximize the concentration of free lithium ions within the polymer matrix.123 The capability of polymer chains to accommodate lithium ion migration remarkably influences the ion transport within the electrolyte, thereby impacting its overall electrochemical performance. Factors such as the glass transition temperature and the crystallinity of the polymer are critical in determining the migratory ability of the polymer chains.124 These factors fundamentally shape the efficiency of lithium-ion movement and, thus, the performance of the polymer electrolyte. Li+ is unable to move freely below the Tg threshold in the polymer system (Tg refers to the temperature at which chain segments become mobile while the molecular chain remains static). In recent years, various polymers have been explored as electrolytes alongside PEO, such as polyacrylonitrile (PAN),125 poly(vinylidene fluoride) (PVDF),126 polymethylmethacrylate,127 polyvinylchloride,128 and polycaprolactone.129 PEO electrolytes have been extensively studied and are currently the most popular choice. PEO offers benefits of eco-friendliness, affordability, flexible chain segments, and enhanced lithium-ion transport capability. However, PEO's drawbacks include high room temperature crystallinity, low ionic conductivity, and high viscosity impairing film formation, resulting in weak mechanical strength and restrictions on its usage. PAN contains the cyano (–CN) group, an electron-withdrawing component that readily interacts with lithium ions, facilitating their movement by continually replacing active sites. This property contributes to PAN's high electrochemical stability, enhancing lithium-ion conductivity, thermal resilience, and electrochemical stability.130 PAN-based electrolytes exhibit exceptional high-voltage stability, making them particularly well-suited for pairing with high-voltage cathodes. Nevertheless, the compatibility of PAN with lithium metal is limited due to instability issues. PVDF boasts superior mechanical strength, dielectric properties, film-forming characteristics, and resistance to high pressures, rendering it a valuable polymer electrolyte option. The typical structures include α, β, and γ.131
N) are particularly favorable because they facilitate the dissociation of lithium salts, promoting their interaction with polar groups, forming complexes that repeatedly complex and de-complex along the polymer chains. Consequently, low lattice energy lithium salts, which ionize readily, are chosen to maximize the concentration of free lithium ions within the polymer matrix.123 The capability of polymer chains to accommodate lithium ion migration remarkably influences the ion transport within the electrolyte, thereby impacting its overall electrochemical performance. Factors such as the glass transition temperature and the crystallinity of the polymer are critical in determining the migratory ability of the polymer chains.124 These factors fundamentally shape the efficiency of lithium-ion movement and, thus, the performance of the polymer electrolyte. Li+ is unable to move freely below the Tg threshold in the polymer system (Tg refers to the temperature at which chain segments become mobile while the molecular chain remains static). In recent years, various polymers have been explored as electrolytes alongside PEO, such as polyacrylonitrile (PAN),125 poly(vinylidene fluoride) (PVDF),126 polymethylmethacrylate,127 polyvinylchloride,128 and polycaprolactone.129 PEO electrolytes have been extensively studied and are currently the most popular choice. PEO offers benefits of eco-friendliness, affordability, flexible chain segments, and enhanced lithium-ion transport capability. However, PEO's drawbacks include high room temperature crystallinity, low ionic conductivity, and high viscosity impairing film formation, resulting in weak mechanical strength and restrictions on its usage. PAN contains the cyano (–CN) group, an electron-withdrawing component that readily interacts with lithium ions, facilitating their movement by continually replacing active sites. This property contributes to PAN's high electrochemical stability, enhancing lithium-ion conductivity, thermal resilience, and electrochemical stability.130 PAN-based electrolytes exhibit exceptional high-voltage stability, making them particularly well-suited for pairing with high-voltage cathodes. Nevertheless, the compatibility of PAN with lithium metal is limited due to instability issues. PVDF boasts superior mechanical strength, dielectric properties, film-forming characteristics, and resistance to high pressures, rendering it a valuable polymer electrolyte option. The typical structures include α, β, and γ.131
Compared to inorganic solid electrolytes, polymer electrolytes offer greater flexibility and lighter weight, leading to decreased interface impedance between the electrode and electrolyte. This enhances battery energy density and commercial viability. Nonetheless, polymer electrolytes suffer from drawbacks such as low lithium-ion conductivity (10−5–10−8 S cm−1) at room temperature, subpar mechanical properties, inability to curb lithium dendrite formation and expansion, inadequate thermal stability, narrow electrochemical stability range, and incapacity to accommodate high-voltage cathode materials, constraining their usability.132
In essence, inorganic solid and polymer electrolytes present distinct advantages but face limitations that impede their real-world application. Proposing a novel approach to create composite electrolytes through a mix of different electrolyte types offers potential solutions. Composite electrolytes leverage the strengths of diverse electrolyte components and compensate for individual shortcomings. Incorporating fillers in the composite electrolyte can enhance mechanical properties, ionic conductivity, and lithium-ion mobility; increase the electrochemical stability range; and fortify electrolyte thermal stability. Furthermore, composite electrolytes address interface challenges of inorganic solid electrolytes, boosting electrolyte-to-electrode interaction. Additionally, the presence of inorganic solid electrolyte components in composite electrolytes as quick ion conductors can aid ion migration and facilitate even ion dispersion at the electrode–electrolyte interface, deterring lithium dendrite growth, and ultimately boosting overall solid electrolyte performance. Composite electrolytes with low weight have the potential to create batteries with increased energy density, opening exciting opportunities for the advancement of portable electronic devices, electric cars, and off-grid renewable energy autonomy.
3.4. Mechanism of Li-ion transport in polymer solid electrolytes
Organic (polymer) electrolytes are typically the matrix utilized in CSSEs in most instances. Within the realm of polymer electrolyte materials for lithium batteries, there are two primary categories: gel polymer and dry polymer. The latter is specifically suitable for ASSLBs and will be the primary focus of this discussion. Generally, lithium salts (inclusive of Li+ cations and anions) are added into solid polymers containing sequential polar functional groups (such as –O–,O, –S–, –N–, –P–, –CO, and –CN) to enhance the lithium ionic conductivities.133 The subsequent section will delve into the mechanisms of Li ion transport within solid polymer electrolytes that are impregnated with lithium salts, with a particular emphasis on the free-volume model and ion conduction model.134
The primary locations for ion transport within solid polymer electrolytes are the amorphous regions that lie above their respective glass transition temperatures (Tg).135,136 The free-volume model and its associated theoretical extensions are widely acknowledged means of elucidating the ion transport mechanisms within these amorphous regions. Specifically, the lithium ions find suitable coordination sites (such as –O– in polyethylene oxide, –CN in polyacrylonitrile, and –NR in polyamide) along the polymer's segmental chains. Additionally, the polymer chains exhibit localized segmental movements akin to a quasi-liquid state due to the presence of free volume around them. This molecular framework allows for the Li+ ion to transition from one coordination site to another via the available free volume within a single chain or among disparate chains under the influence of an electric field.133,137
In addition to the ion transport mechanism predominantly observed in amorphous regions, the model of ionic conduction is applied in solid polymer crystalline phases. The Li+ ion transport in these phases is characterized by a lower dependence on segmental motion and can be primarily described using the Arrhenius equation. While it was previously believed widely that ion transport kinetics in amorphous regions with activated chain segments were significantly faster compared to the crystalline phase,138,139 it has been proven that ion transport in crystalline phases can be significant and is sometimes even faster than in amorphous regions in an increasing number of instances. Z. Gadjourova and colleagues suggested that the ionic conductivity of the crystalline phase in a static, ordered environment might exceed that of the equivalent amorphous phase above the Tg.136 As illustrated in Fig. 5, in the crystalline phase of PEO6:LiXF6 (X = P, As, and Sb), polymer (PEO) chains pair up to create cylindrical tunnels where lithium ions occupy coordination sites (–O– in PEO), while XF6− anions are located externally and are not coordinated. Within this arrangement, Li+ ions can traverse along the tunnels through neighboring coordination sites without relying on the segmental motion of the polymer chains.133,140 Building on this, Z. Stoeva and co-workers, among others in the same research group, theorized that the ionic conductivity could witness a boost by 1.5 to 2 orders of magnitude through further modifying these stoichiometric crystalline complexes by replacing the XF6− ions.140–143 In summary, the mechanisms of ion transport in polymer electrolyte materials are influenced by various factors like temperature, polymer type, molecular weight, polymer structure, dissociation capability, and concentration of Li salt in the polymers.134
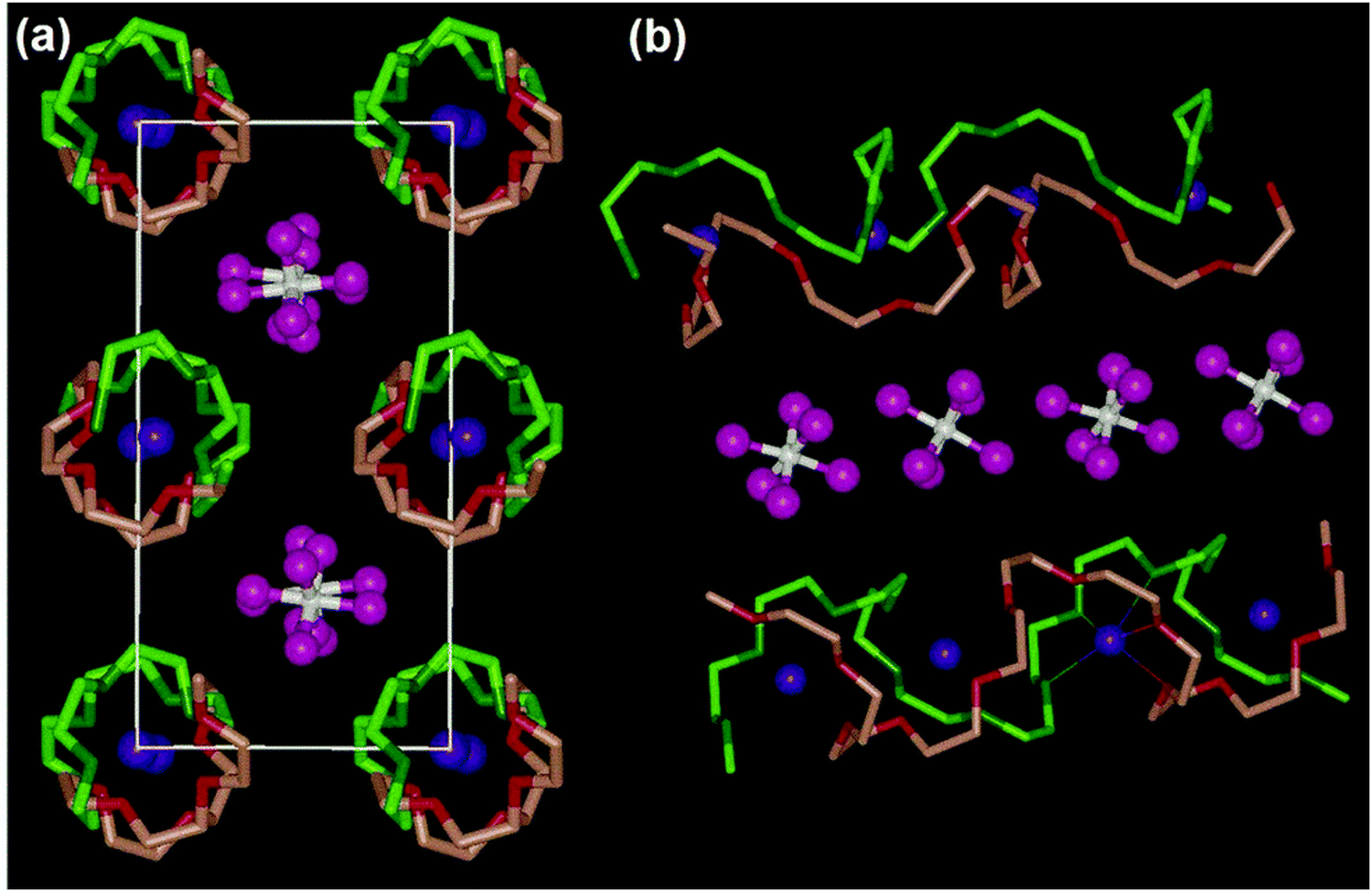 | ||
| Fig. 5 Structure of a typical solid polymer electrolyte material (poly(ethylene oxide)6:LiAsF6). (a) View along the chain axis for the Li+ transport pathway and (b) view of the relative position of the chains and their conformation. (Blue, Li; white, As; pink, F; light and dark green are for C and O in chain 1; and light and dark red are for C and O in chain 2, respectively.)136,144 (Reproduced with permission from Nature Publishing Group.) | ||
The assessed properties of individual single SPEs and ISEs reveal that they cannot independently provide adequate ionic conductivity, mechanical strength, suitable electrode compatibility, and efficient production (Fig. 6). Therefore, to reach the performance requirements of ideal SSEs for use in ASSBs, the most apparent solution is to combine two or more types of electrolyte materials with complementary advantages to construct composite solid-state electrolytes. Coupled electrolytes with appropriate rigid and flexible components can ensure good electrode wettability for low interfacial impedance, adequate Li-ion conductivity, and good mechanical strength to enable robust ASSLB fabrication.
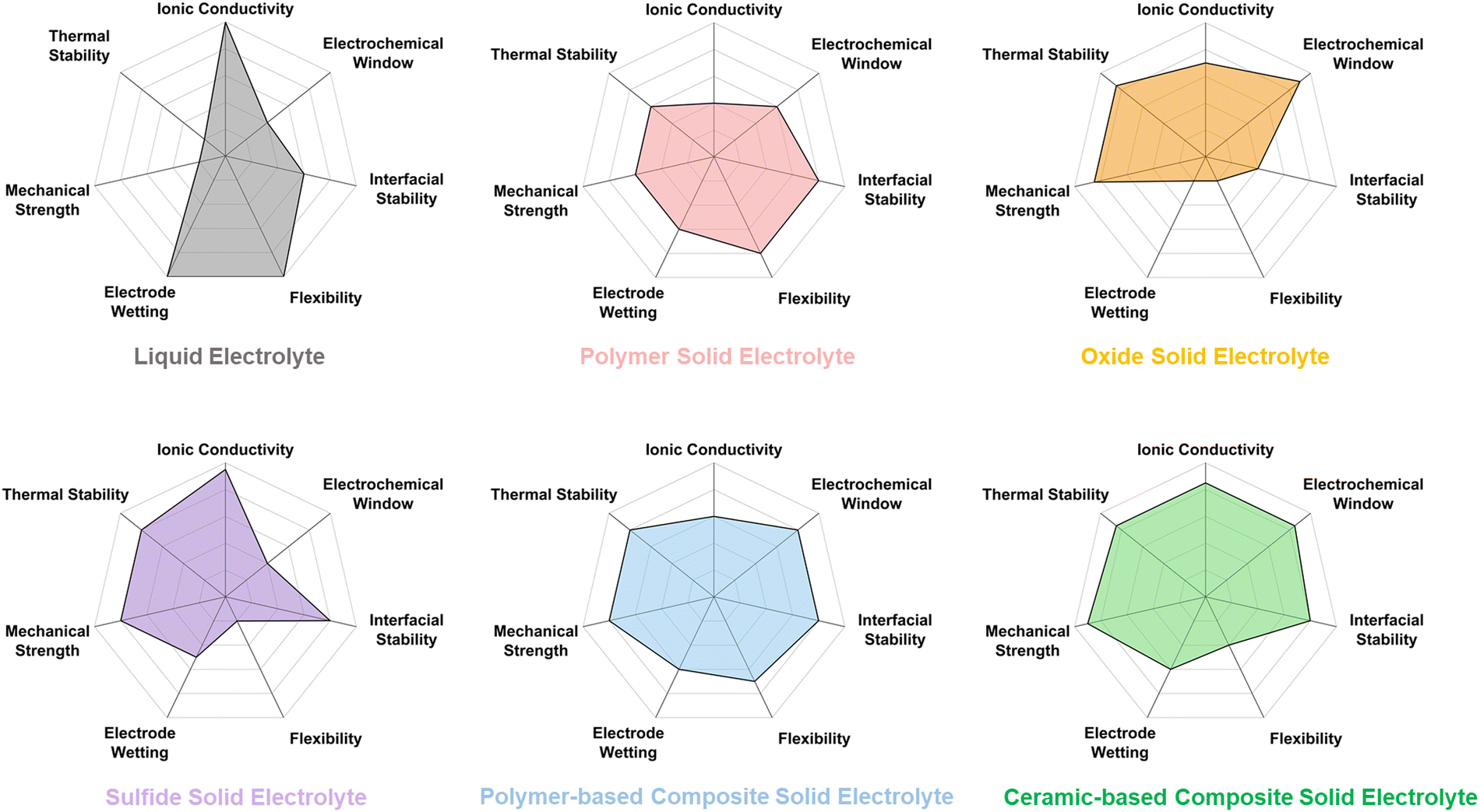 | ||
| Fig. 6 Comparison of the main features of the different categories of solid-state electrolytes compared to liquid electrolytes. | ||
4. Composite solid-state electrolytes
Generally, in an ASSLB with a composite solid-state solid electrolyte (CSE), the CSE is sandwiched between the cathode and anode, playing a crucial role in the electrochemical performance and stability of the battery during long-term operation. The performance requirements include high ionic conductivity, appreciable Li+ transference number, good electrochemical stability, excellent chemical and thermal stability, and desirable mechanical strength and flexibility. Three main types of CSEs and their performance are discussed below.4.1. Composite solid electrolytes with passive fillers
Extensive exploration into polymer electrolytes has revealed drawbacks such as limited practicality due to low lithium-ion conductivity, subpar mechanical properties, and a narrow electrochemical stability window. The addition of a specified quantity of inert inorganic filler to a polymer matrix has shown to enhance the amorphous phase ratio of the polymer, reduce the melting point (Tm) and glass transition temperature (Tg) of the polymer matrix, facilitate lithium-ion migration, improve lithium-ion conductivity, and bolster the mechanical properties and heat resistance of the electrolyte. In the 1980s, Weston et al.145 introduced α-Al2O3 filler into the polymer matrix to produce composite solid electrolytes. The incorporation of α-Al2O3 particles into the PEO/LiClO4 electrolyte matrix has proven to significantly enhance the mechanical strength and lithium-ion conductivity (10−5 S cm−1) of the solid composite electrolyte. This enhancement can be attributed to the Lewis acid–base interaction between the embedded inorganic filler and the polymer, which diminishes the polymer's crystallinity and augments the lithium ion migration within the polymer chain. While the introduction of inorganic fillers notably elevates ionic conductivity, it important to note that both anions and cations are present in the composite electrolyte. The migration of cations and anions between the positive and negative electrodes under the influence of an electric field is crucial for conductivity, with the lithium-ion transference number serving as a key metric for evaluating lithium-ion transport in the electrolyte. A higher lithium-ion transference number correlates with a reduced concentration polarization on the electrode surface, thus promoting interface stability between the electrode and electrolyte. Moreover, a higher lithium-ion transference number enables rapid charging. Xia et al.146 utilized single-layered nano-sheet of layered double hydroxide (SLN) in conjunction with a composite of poly(vinylidene fluoride)-hexafluoropropylene and lithium bis(trifluoromethanesulfonyl)imide (PVDF–HFP/LiTFSI). The SLN, bearing a positive charge is a Lewis acid that shows strong interaction with TFSI− anions, thereby enhancing the release of Li+. Calculations based on density functional theory demonstrate that the addition of SLN can effectively boost the binding energy of Li and TFSI ions, facilitating the break-up of LiTFSI. In comparison to LiTFSI, SLN and TFSI exhibit lower binding energy, which aids in the sequestration of TFSI−, resulting in an improved lithium-ion migration within the polymer. Incorporating 1 wt% SLN was found to yield the highest ionic conductivity of 2.2 × 10−4 S cm−1 at 25 °C, along with a lithium-ion transference number of 0.78 and a wide electrochemical stability window of approximately 4.9 V.In a separate study, Chen et al.147 employed calcium-doped cerium oxide (Ca–CeO2) to fabricate hollow nanotubes featuring a high specific surface area. The composite of PEO/LiTFSI was found to enhance the contact area between the polymer and Ca–CeO2, diminish the crystallinity of PEO, and bolster the mechanical properties of the electrolyte. Ca–CeO2 was revealed to elevate the concentration of oxygen vacancies, establish a positive potential region, and exhibit strong attraction towards the anions in LiTFSI. The hollow structure and expansive specific surface area provided ample reaction sites between the inorganic components and lithium salts, thereby augmenting the lithium-ion concentration. An ionic conductivity of 1.3 × 10−4 S cm−1 and a lithium-ion transference number of 0.453 were achieved at 60 °C. Notably, the size of the inert filler significantly impacts the conductivity of solid composite electrolytes. Smaller particle sizes translate to larger specific surface areas, facilitating enhanced contact with the polymer and lithium salt, ultimately leading to improved ionic conductivity and lithium-ion transference number. Croce et al.148 incorporated nanoscale TiO2 and Al2O3 inert fillers (ranging from 5.8 to 13 nm) into the PEO/LiClO4 matrix electrolyte, leading to an improvement in lithium-ion conductivity of the solid composite electrolyte by three orders of magnitude and achieving a high average lithium-ion transference number of 0.6 across various temperatures. However, due to the thinness (and softness of the polymer matrix) of the composite solid electrolyte membrane, it is prone to being punctured by lithium dendrites, which presents a risk of short circuiting within the battery. Additionally, uncontrolled growth of lithium dendrites will continue consuming active electrode materials, which results in irreversible capacity loss. The uniform dispersion of fillers can evenly distribute stress, thus hampering the growth of dendrites. Moreover, the fillers form a stable interface layer with the electrode, enhancing the interface contact between the electrode and electrolyte, inhibiting dendrite formation, and ensuring stable battery operation.
To achieve a more uniform filler distribution within solid composite electrolytes, advanced preparation technologies have also been employed in the manufacturing of these composite electrolytes. Bao et al.149 utilized vapor deposition technology to fabricate a ZnO/PEO/LiTFSI composite electrolyte with ZnO uniformly dispersed throughout by depositing ZnO quantum dots of an average size of 4 nm within the PEO/LiTFSI matrix. The chemical interaction between ZnO and PEO in the prepared electrolyte results in ZnO binding with –CH2CH2O– to create a Zn–O–C bond (refer to Fig. 7a), leading to a more effective reduction in the Tg and Tm of the polymer, ultimately enhancing the ionic conductivity of the composite electrolyte. The presence of ZnO and lithium metal on the surface of the composite electrolyte leads to the formation of a Li–Zn alloy, which significantly enhances the stability of the interface between the electrolyte and the electrode while decreasing the impedance at the interface. In comparison to electrolytes prepared using physical mixing and vapor deposition, the NCM811/SPEs/Li battery assembled with this electrolyte demonstrates greater initial discharge capacity and cycle stability (Fig. 7b). The in situ formation of the Li–Zn alloy at the interface also helps in suppressing the formation of lithium dendrites. Wang et al.150 further elucidated the mechanism by which lithium-rich alloys inhibit lithium dendrites, employing both theoretical calculations and experimental methods. The creation of a flexible protective lithium-rich alloy layer on the lithium metal surface decreases the diffusion barrier for lithium atoms, facilitating rapid movement of lithium atoms across the alloy surface and subsequent diffusion through the alloy layer. This process, in turn, reduces the occurrence of detrimental interface side reactions. It is important to highlight that not all lithium-rich alloys exhibit beneficial effects, as alloys that simultaneously transport ions and electrons can impede battery performance.
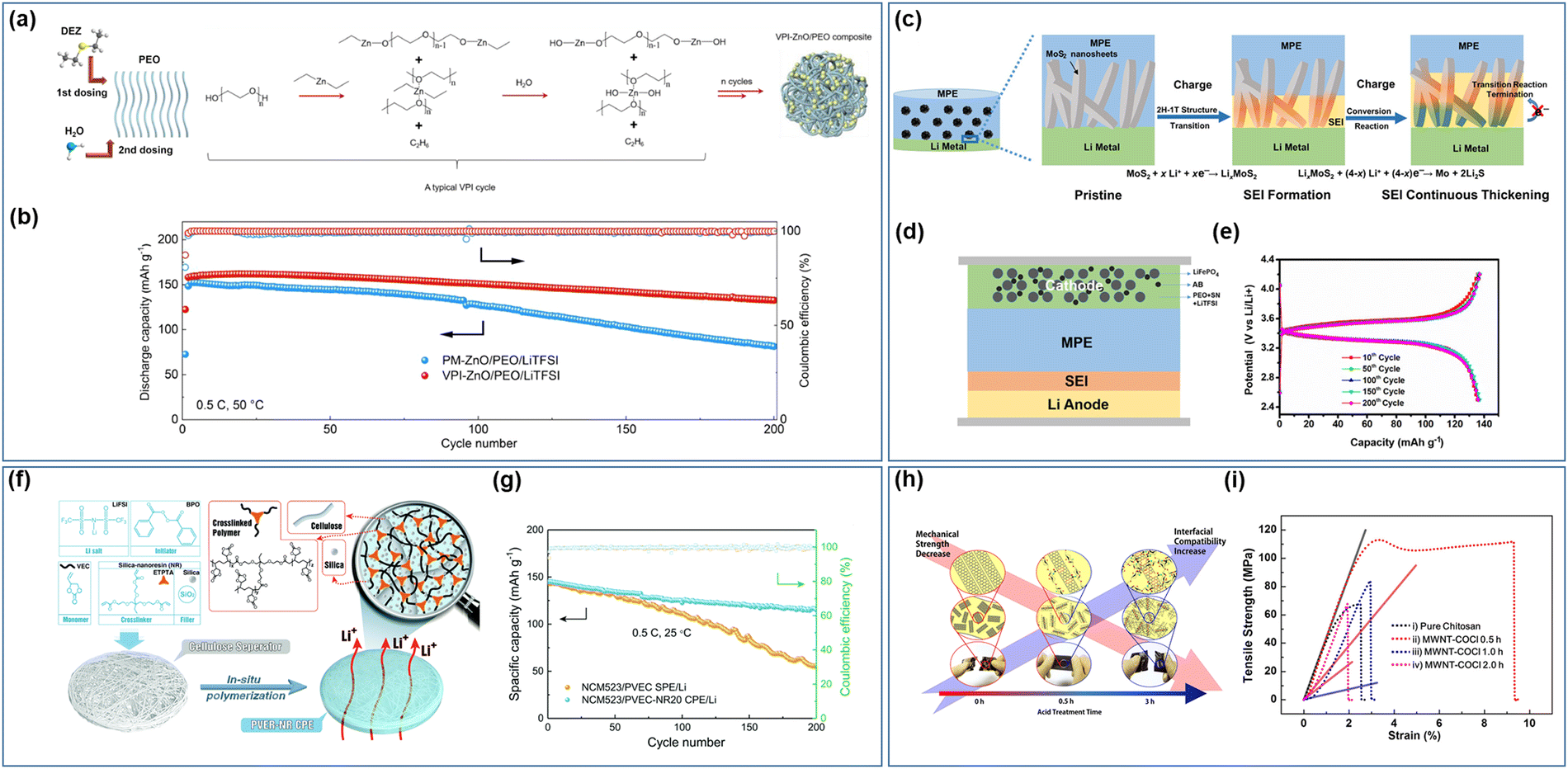 | ||
| Fig. 7 (a) Schematic illustration of the vapor phase infiltration (VPI) process and (b) cycle performance of the batteries with different electrolytes (PM-ZnO/PEO/LiTFSI and VPI ZnO/PEO/LiTFSI).149 (Reproduced with permission from Elsevier.) (c) Schematic of the SEI layer formed on the Li anode by the in situ conversion reaction of MoS2, (d) schematic illustration of the assembly of the battery with MoS2 nanosheets into PVDF–HFP electrolyte (MPE) after cycling, (e) and charge–discharge profiles of the solid batteries.151 (Reproduced with permission from the American Chemical Society.) (f) Schematic illustration of the fabrication process of the poly(vinyl ethylene carbonate)-silica-nanoresin (PVEC-NR) hybrid solid electrolyte and (g) cycling stability of NCM523/PVEC-NR20/Li and NCM523/PVEC/Li batteries.152 (Reproduced with permission from The Royal Society of Chemistry, UK.) (h) Schematic of the relationship between the mechanical strength of carbon nanotubes and (i) interfacial compatibility with the host polymer matrix and representative strength–strain curves of pure chitosan SPEs.153 (Reproduced with permission from Elsevier.) | ||
The introduction of an inorganic filler into the interfacial reaction promotes the formation of an SEI film in situ between the electrolyte and electrode interface. This SEI film plays a crucial role in inhibiting the formation of lithium dendrites and enhancing interfacial stability, contributing significantly to the overall performance of the battery system. Sun et al.151 published a study focusing on enhancing the ionic conductivity and manipulating interface properties of electrolytes through the incorporation of inorganic fillers for conversion reactions. A composite solid electrolyte, MoS2/PVDF–HFP/LiFSI (MPE), was developed by integrating petal-shaped nano sheets of MoS2 into PVDF–HFP (Fig. 7c). The unique morphology of the MoS2 nano sheets, with a substantial specific surface area, significantly enhances the interaction between the filler and polymer electrolyte. By reducing the crystallinity of the polymer, the migration of lithium ions within the chain is facilitated. Additionally, the Sulfur atoms in MoS2 facilitate the dissociation of lithium ions, ultimately leading to an enhanced ionic conductivity of the composite electrolyte (achieving a conductivity of 2.8 × 10−4 S cm−1 at ambient temperature). Through a two-step in situ reaction between MoS2 and lithium metal during the cyclic process, a gradient solid electrolyte interface (SEI) film was observed to form, effectively suppressing the growth of lithium dendrites and ensuring a stable electrode–electrolyte interface (refer to Fig. 7d and e). Furthermore, the interaction between the filler and polymer matrix improves the electrolyte's ability to inhibit lithium dendrite formation. An et al.154 successfully synthesized polymer network electrolytes through the chemical grafting of 2D boron nitride nano sheets with poly(ethylene glycol)diacrylate (PEGDA) utilizing a silane coupling agent. The grafting process significantly improved the mechanical properties of the electrolytes on a molecular level, resulting in an impressive tensile strength of 26.2 MPa, effectively suppressing lithium dendrites. Furthermore, the composite electrolyte exhibited enhanced ionic conductivity and lithium-ion transference number, along with an expanded electrochemical stability window, facilitating the compatibility with high voltage cathode materials and ultimately increasing the energy density of the battery. In a study by Kuai et al.,152 SiO2 nano-resin (NR) was utilized as both an inert filler and cross-linking agent in the production of composite polymer electrolyte (PVEC-NR CPE) based on PVEC crosslinking (Fig. 7f). Notably, the composite electrolyte demonstrated optimal performance when the NR content was 20 wt%. A one-step heat treatment of the entire assembled battery resulted in complete contact between the electrode and electrolyte through in situ free radical polymerization, leading to a further reduction in internal resistance (Fig. 7g).
Aside from traditional inorganic inert materials, the incorporation of novel fillers has gained considerable attention in recent research. Materials like covalent organic frameworks (COFs) or metal organic frameworks (MOFs) possess a high specific surface area, a uniformly distributed micro-pore structure, and easily modifiable organic functional groups. They can be physically mixed with a polymer matrix or chemically grafted to prepare composite solid electrolytes. Within the electrolyte, unsaturated coordination metal ions have the ability to coordinate with anions, restricting their movement and potentially increasing lithium-ion concentration. Alternatively, they can directly construct a negatively charged framework to enhance lithium-ion conduction.155 Furthermore, the derivative of organic ligand has the potential to enhance the bond at the interface of filler and polymer matrix, resulting in a reduction of impedance at the interface.156–158 It is important to highlight that the composite electrolyte, containing polymer with crystalline properties, tends to exhibit suboptimal performance at low temperatures, necessitating pre-testing heating for enhanced results. Nevertheless, the heat treatment of the electrolyte poses limitations in practical usage. Utilization of MOF as a filler in composite electrolytes offers a broader temperature range for operation and serves as a valuable reference for investigating the low-temperature behavior of electrolytes.159 In a study by He et al.,160 a composite electrolyte membrane was developed using halogen (Cl−, F−) modified Ce-MOF material and PVDF–HFP/LiTFSI composite. The presence of halogens (Cl−, F−) within the pores of the composite electrolyte helps to control the distribution of electrons and enhance the movement of lithium ions. Additionally, the inclusion of halogens assists in the creation of a uniform SEI layer, enhancing ionic conductivity and stabilizing the interface between the electrode and electrolyte, thus reducing lithium dendrite formation. The Li|SE-4Cl-Li+|LFP battery demonstrated outstanding discharge capacity and rate performance, with specific capacities at various rates, including 163, 160, 156, 148, and 124 mA h g−1. Even at −20 °C after a 0.1C rate cycle, a discharge capacity of 107 mA h g−1 was achieved, highlighting the potential for improved low-temperature battery performance.
Carbon materials have been extensively explored in various fields including optics, mechanics, and electricity. They are increasingly being integrated as fillers in composite solid electrolytes, such as zero-dimensional fullerenes (C60),161 one-dimensional carbon nanotubes (CNT),162 and two-dimensional graphene and its derivatives. CNTs and graphene offer numerous advantages, such as high aspect ratio, large specific surface area, and lightweight properties. Utilizing them as fillers can help to transfer stress within the polymer to the carbon material, thereby enhancing the mechanical strength of the electrolyte.163 To effectively distribute stress within the polymer into the carbon material, Kim et al.153 modified the multi-walled carbon nanotubes (MWNTs) by treating them with varying concentrations of acid acyl chloride, resulting in a multifunctional filler that exhibits excellent mechanical properties and good interface compatibility with the polymer. The functional filler's surface is enriched with acyl chloride groups, which can form amide bonds with the chitosan polymer matrix and facilitate stress transfer to the functional MWNTs (Fig. 7h). The study indicates that the electrolyte produced using MWNT-COCl for 0.5 hours shows the most promising mechanical properties (refer to Fig. 7i). Jia et al.164 utilized graphene oxide (GO) as the filler in PAN/LiClO4. The presence of Lewis acid–base interactions between GO and the numerous oxygen-containing functional groups on the surface facilitate lithium-ion migration within the electrolytes, enhancing lithium-ion conductivity. Moreover, the incorporation of GO leads to an improvement in the electrolyte's tensile modulus, and the Lewis acid–base interplay between GO and PAN reduces the polarity of ‘–CN’, resulting in a softer electrolyte. The highest ionic conductivity (4 × 10−4 S cm−1) was achieved when the GO content was 1.0 wt%. The solid-state battery comprising a GO composite electrolyte demonstrates increased capacity (165 mA h g−1 after 50 cycles) and Coulombic efficiency (99.4%). Interestingly, the combined use of CNTs and GO as fillers proves to have a synergistic effect, enhancing mechanical properties.165 Wu et al.166 introduced both carboxylated one-dimensional carbon nanotubes (CNTs) and two-dimensional graphene oxide (GO) into a polyelectrolyte complex (PEC) simultaneously. Through π–π interactions, these components formed a multifunctional 3D structure, where the strong interaction between their electrostatic attraction and the PEC matrix played a synergistic role. A PEC film incorporating 3 wt% GO-CNT (in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio) exhibited superior mechanical properties (tensile strength of 155.4 MPa and elongation at break of 9.0%) compared to films with either GO or CNT alone.
:1 weight ratio) exhibited superior mechanical properties (tensile strength of 155.4 MPa and elongation at break of 9.0%) compared to films with either GO or CNT alone.
Addressing the need for large-scale production, there is growing concern over the environmental impact and costs associated with manufacturing electrolytes. The abundant natural clay within the earth's crust offers plentiful mineral resources suitable for use as inert fillers in composite solid electrolytes. Halloy-site nanotubes (HNTs) are distinctive one-dimensional natural nano-fillers, characterized by their hollow tubular structure, high aspect ratio, excellent mechanical strength, and outstanding thermal stability. HNTs can consolidate naturally under pressure and heat flow, making them an affordable and widely available clay material.167 Lin and collaborators168 directly combined natural HNTs with PEO/LiTFSI to fabricate composite electrolytes. Due to the different chemical structures present on the inner and outer surfaces of HNTs, the internal surface holds positive charges to restrict anion movement, whereas the external surface carries negative charges to adsorb lithium ions. This unique configuration boosts lithium salt dissociation and significantly enhances lithium-ion conductivity. Montmorillonite (MMT) also displays a high aspect ratio and specific surface area. In summary, the dimensions, morphology, and concentration of inorganic inert fillers significantly affect the ionic conductivity and electrochemical performance of composite electrolytes. Incorporating inert fillers can impede the formation of lithium dendrites and ensure the stabilization of the electrode–electrolyte interface.169 However, one notable drawback of inert fillers is their incapacity to facilitate the transfer of lithium ions within the electrolyte.
4.2. Composite solid electrolytes filled with active fillers
In recent years, research has advanced, and the utilization of novel solid-state composite electrolytes integrating inorganic solid electrolytes as active fillers in solid polymer electrolyte matrix has garnered significant interest. Differing from inactive inorganic fillers, active components can supply extra lithium-ion transmission routes, exhibiting elevated lithium-ion conductivity and enhanced electrochemical efficacy. The active filler can also notably increase the electrochemical stability span, lithium-ion transference count, interface connection, and the capacity to suppress lithium dendrite formation by engaging with the polymer matrix via ionic dipole interactions, hydrogen bonding, p–p bonding, Lewis acid–base forces, and other mechanisms. In the case of ceramics in polymer, it is generally believed that the optimal proportion of ceramic filler ranges from 10% to 30% by weight. Excessive filler content can result in filler clustering, detrimentally impacting stress distribution and electrolyte performance. Conversely, ceramic nanoparticles with low loading levels are unable to establish a continuous network for lithium-ion transmission within the polymer matrix, as there exists an obstructive energy barrier between particles that impedes ion mobility.169 Employing 1D nanofibers in place of ceramic particles for constructing a continuous lithium-ion transmission path can alleviate the energy barrier hindering ion transport.The fabrication of ceramic nanofibers via electrospinning presents a fresh approach for fabricating composite electrolytes. While the initial studies and patents related to electrospinning date back to the 1900s, only in the late 1990s, in concomitance with the spread of novel nanotechnologies, this technique started drawing attention. More recently, electrospinning has been employed to fabricate highly ion-conductive materials in the form of nanofibers and nanowires170 The first works experimenting with this approach have been published in 2015.171–173 Among them, Liu et al.171 successfully synthesized Li0.33La0.557TiO3 (LLTO) nanowires and used them to enhance the performance of a PAN:LiClO4 electrolyte. Subsequently, they also explored the effect of well-aligned LLTO nanowires on the performance of the same composite electrolyte,174 reporting a ten-time higher conductivity when compared to randomly dispersed ones. Wan et al.175 designed and tested an integrated all-solid-state battery featuring a PEO:LiTFSI electrolyte with LLZO nanowires, which delivered a specific capacity of 158.7 mA h g−1 after 80 cycles at 0.1C and 45 °C. The composite electrolyte can reportedly suppress the lithium dendrite growth as it sustained a 1000 h cycling test in a Li symmetric cell. Similar studies have been conducted on NASICON materials, such as Li1.3Al0.3Ti1.7(PO4)3 (LATP) and Li1.5Al0.5Ge1.5(PO4)3 (LAGP), which have been fabricated in the form of ceramic nanowires by coupling sol–gel and electrospinning, and successfully used to enhance PEO-based electrolytes.176–179 Guo et al.180 developed a novel superfine spinning fiber composite SEI with an interwoven shell/core structure using coaxial co-spinning (Fig. 8a–c). The resulting film structure exhibits outstanding mechanical properties. Within the composite electrolyte, the spun fibers demonstrate anisotropic lithium-ion conduction vertically and horizontally. Additionally, the lithium-ion transmission rate is significantly higher along the superfine fiber compared to through it. This feature contributes to a more uniform electric field distribution within the electrolyte during circulation, reducing the occurrence of tip electric fields (Fig. 8d).
 | ||
| Fig. 8 (a) Schematic illustration of the design principle of electrospun-composite solid electrolyte (ES-CSE), (b) top-view SEM image of ES-CSE, (c) TEM image of ES-CSE, and (d) Li-ion transfer pathway in ES-CSE.180 (Reproduced with permission from Wiley-VCH.) (e) Schematic of vertically aligned and connected ceramic channels (randomly dispersed ceramic particles and vertically aligned ceramic structure) and (f) and (g) SEM images of the ice-template LATP channels.181 (Reproduced with permission from American Chemical Society.) (h) Complex structure in the PVDF/LLZTO CPE, (i) Raman spectra and (j) photographs of PVDF PSE and PVDF/LLZTO CPE.182 (Reproduced with permission from the American Chemical Society.) (k) Illustration of the Li salt state in the PVDF and PVBL electrolyte, (l) SEM image of the coupled BTO-LLTO nanowires, (m) high-resolution TEM image of the coupled BTO-LLTO nanowires and (n) SEM images of the cross-section & surface of the PVBL electrolyte.183 (Reproduced with permission from Nature Publishing Group.) | ||
The combination of enhanced mechanical properties and uniform electric field distribution assists in partially preventing the formation of lithium dendrites. Furthermore, the composite electrolyte displays high lithium-ion conductivity levels (1.5 × 10−4 S cm−1 at 35 °C and 1.5 × 10−3 S cm−1 at 55 °C) and impressive cycling stability. To optimize the utilization of the active filler in creating a continuous lithium-ion transmission pathway perpendicular to the electrode direction, Zhai et al.181 prepared a continuous nanoparticle transport channel of Li1+xAlxTi2−x(PO4)3 using the ice template method (Fig. 8e). By utilizing polyethylene glycol (PEG) as a plasticizer, the electrolyte achieved an ionic conductivity of 0.52 × 10−4 S cm−1 at room temperature, surpassing that of inorganic ceramic particles with equivalent composition by 3.6 times. It is important to highlight that the vertically aligned connection structure distinguishes itself from both the widely dispersed ceramic particle arrangement and the nearly parallel spun fiber structure, enabling enhanced ionic conductivity, minimized unnecessary filler incorporation, reduced electrolyte mass, and increased battery energy density (Fig. 8f and g). The inorganic constituent within the vertical structure promotes ionic conductivity and employing fillers with high intrinsic ionic conductivity (e.g., sulfide and halide electrolytes) can yield even better electrochemical performance. Conversely, in polymer–ceramic electrolytes, the polymer matrix acts as the principal component and exhibits high crystallinity at ambient temperature.
In the polymer electrolyte, lithium-ion transportation predominantly occurs in the amorphous phase, thus excessive crystallization of the polymer must be avoided as it would impact negatively the electrochemical performance of the electrolyte. Therefore, various techniques have been devised to decrease crystallinity alongside active filler incorporation. Similar to inert fillers, active fillers can partake in Lewis acid–base reactions with the polymer matrix to lower crystallinity and enhance performance. Zhang et al.182 utilized Li6.75La3Zr1.75Ta0.25O12 (LLZTO) to blend with a PVDF/LiClO4 polymer matrix (Fig. 8h). The introduction of LLZTO particles into PVDF enabled the La atoms in LLZTO to interact with the N atoms and CO groups in the solvent, as illustrated in Fig. 8i. This interaction resulted in the partial defluoridation of PVDF, leading to a decrease in polymer crystallinity (Fig. 8j). This crystallinity reduction promoted the complexation between LLZTO particles and lithium salt, facilitating the effective dissociation of lithium salt and ultimately achieving a solid composite electrolyte with lithium-ion conductivity of up to 5 × 10−4 S cm−1. In addition to the reaction between the inorganic filler and polymer matrix, various external techniques (e.g., hot pressing, UV curing, liquid nitrogen quenching, etc.) were applied to decrease the crystallinity of composite solid electrolytes. Li et al.184 optimized the polymer matrix by incorporating Li6.2Ga0.1La3Zr0.5Bi0.5O12 (LLZO) ceramic filler. When the weight ratio of PEO/PVDF was adjusted to 7:3, the PEO/PVDF blend matrix exhibited a lower melting point and increased thermal stability. With a LLZO content of 10 wt%, the highest ionic conductivity reached was 4.2 × 10−5 S cm−1 at 30 °C. Meanwhile, Siyal et al.185 demonstrated that composite electrolyte membranes consisting of Li1.3Al0.3Ti1.7(PO4)3 (LATP) with double-filled PVDF filled with Li0.33La0.557TiO3 (LLTO) achieved even higher ionic conductivity, 10−3 S cm−1 at room temperature.
Other than boosting conductivity, the utilization of double-filled composite electrolytes of LATP and LLTO nanoparticles effectively hindered the formation of lithium dendrites. Furthermore, research indicated that the inclusion of plasticizers could effectively diminish the crystallinity of the polymer matrix and increase the amorphous region. Depending on their physical state, plasticizers can be categorized as either solid or liquid. Solid plasticizers, in contrast to traditional liquid ones, are more beneficial in maintaining the mechanical strength of electrolytes. Rakumar et al.186 incorporated solid succinonitrile (SN) as a plasticizer within the LLZO–PEO–LiTFSI system. Notably, with an SN content of 30 wt%, the system's conductivity surged to 4.23 × 10−4 S cm−1 at 25 °C, yielding an impressive cycle rate performance when deployed in NCM811/Li battery systems.
The inclusion of a liquid plasticizer can markedly enhance conductivity, albeit at the expense of the electrolyte's mechanical integrity. Introducing an organic solvent as a plasticizer not only improves interface contact but also raises the electrolytes' flammability. Importantly, it is widely acknowledged that ionic liquids, consisting entirely of cations and anions, exhibit non-flammability. Consequently, the integration of ionic liquids into the electrolyte serves a similar function to flame retardants.187 Furthermore, ionic liquids possess a certain viscosity, which facilitates the creation of a viscoelastic interface between the electrode and electrolyte. This interface can interact with the electrode to form a solid electrolyte interface film, thereby exhibiting some capability to suppress lithium dendrite formation. Nevertheless, it should be recognized that the viscosity of ionic liquids can adversely impact conductivity. Therefore, attention must be paid to the types, structures, and temperatures of ionic liquids used in studies such as those by Yang et al.,188 Yang et al.,189 and Guo et al.190
Additionally, unlike the stability observed with LLZO and lithium metal, the Ti4+ ions in LATP and LLTO can undergo redox reactions with lithium metal, leading to its instability. Replacing Ti4+ with other tetravalent elements does not fully rectify this instability, which can promote electron transport and the formation of lithium dendrites within the electrolyte, ultimately causing a battery short circuit.191–193 By incorporating LATP and LLTO as fillers within polymer electrolytes, the polymer matrix can effectively encapsulate LATP and LLTO particles. This reduces their direct interaction with lithium metal significantly, thereby enhancing the stability of the interface between the electrode and electrolyte. Jia et al.194 developed a composite electrolyte (PVDF:LLTO@PDA) using LLTO particles coated with biodegradable polydopamine (PDA) and PVDF. This composite solid electrolyte significantly minimizes direct contact between LLTO and lithium metal, demonstrating outstanding stability with lithium metal. A Li/Li symmetrical cell assembled with PVDF:LLTO@PDA exhibited stable cycling for 800 hours at a current density of 0.1 mA cm−2 at 60 °C, whereas the PVDF:LLTO membrane experienced failure after cycling for just 25 hours under identical conditions. Shi et al.183 developed a robust method aimed at creating high-throughput pathways for Li-ion transport by coupling ceramic dielectrics with electrolytes to address the low ionic conductivity of hybrid solid-state electrolytes. A highly conductive and dielectric hybrid solid-state electrolyte was achieved by combining a poly(vinylidene difluoride) (PVDF) matrix with BaTiO3–Li0.33La0.56TiO3−x nanowires in a side-by-side heterojunction configuration (PVBL) (Fig. 8k). The polarized BaTiO3 dielectric significantly enhances the dissociation of lithium salt, thereby producing more mobile Li-ions which spontaneously and locally transfer to the coupled Li0.33La0.56TiO3−x, facilitating highly efficient transport (Fig. 8l–n). These synergistic effects lead to an impressive ionic conductivity of 8.2 × 10−4 S cm−1 and a lithium transference number of 0.57 at 25 °C in the PVBL. The composite solid electrolyte was also prepared using a perovskite electrolyte filler following the same method. Shin et al.195 utilized Li0.29La0.57TiO3 and PEO to fabricate a flexible, biphasic solid electrolyte (SBE) with a thickness of 30 μm, exhibiting an ionic conductivity of 1.2 × 10−4 S cm−1. To enhance the battery's volumetric energy density and minimize unnecessary components, the SBE was assembled into a three-unit cell configuration. The resulting battery pack operated effectively within a voltage range of 9.2–12.0 V and demonstrated a reversible capacity of 125 mA h g−1. To summarize, active fillers have the capacity to create uninterrupted lithium-ion transmission channels and significantly diminish the crystallinity within the polymer matrix. Nevertheless, an overloading of active fillers causes agglomeration, which severely impacts the mechanical and electrochemical properties of composite solid electrolytes.
4.3. 3D framework composite solid electrolytes
As the amount of filler increases, the composite electrolyte transitions from a ceramic-in-polymer structure to a polymer-in-ceramic structure, imparting certain ceramic characteristics to the electrolyte. This transition effectively enhances the mechanical properties of the electrolyte and increases its capability to resist lithium dendrite formation. However, increased filler content still encounters the issue of agglomeration. By casting the inorganic filler into a 3D framework with a porous structure and subsequently infusing it with a polymer matrix, it is possible to prevent agglomeration while also enhancing continuous ion transport channels. Concurrently, incorporating a specified amount of polymer electrolyte as a filler within the ceramic framework grants the composite electrolyte a degree of flexibility, facilitating close contact with the electrode interface and reducing interface impedance.Leveraging the advantage that inorganic fillers can create a 3D interconnected network within the polymer matrix has become a popular area of research, focusing on developing 3D inorganic frameworks with uniform porosity through straightforward methods. Lin et al.196 developed a three-dimensional porous silica aerogel structure and integrated it with PEO–LiTFSI PSE. This newly synthesized composite solid electrolyte possesses a significant elastic modulus of 0.43 GPa and a hardness close to 170 MPa, which is effective in suppressing dendrite formation (Fig. 9a). Due to the high specific surface area of the porous structure, it can engage thoroughly with the polymer matrix, leading to a Lewis acid–base reaction that enhances the dissociation of lithium salt. Additionally, the interaction between the incorporated plasticizer and SiO2 enhances movable Li-ion concentration, endowing the electrolyte with exceptional ionic conductivity of 6 × 10−4 S cm−1 at 30 °C. The 3D porous framework significantly diminishes the crystallinity of the polymer while also boosting the mechanical strength of the composite electrolyte. However, such an inert filler-based 3D framework does not facilitate lithium-ion transport. In contrast, when the 3D framework incorporates an active filler, it establishes continuous channels for lithium-ion transmission, thereby not only improving Li-ion conductivity but also promoting even deposition of lithium ions at the electrode–electrolyte interface, which is essential in mitigating lithium dendrite formation. Bae et al.197 incorporated LLTO precursor with PVA and utilized glutaraldehyde (GA) crosslinking agent for gelation to create LLTO hydrogel. Subsequently, the 3D structured LLTO framework was generated through calcination, followed by the injection of PEO/LiTFSI polymer matrix into the LLTO 3D framework to produce a composite solid electrolyte. The interconnected 3D structure maintains the integrity of the LLTO phase within the composite solid electrolyte, preventing particle aggregation and establishing a rapid conduction pathway within the polymer matrix, resulting in a Li-ion conductivity of 8.8 × 10−5 S cm−1.
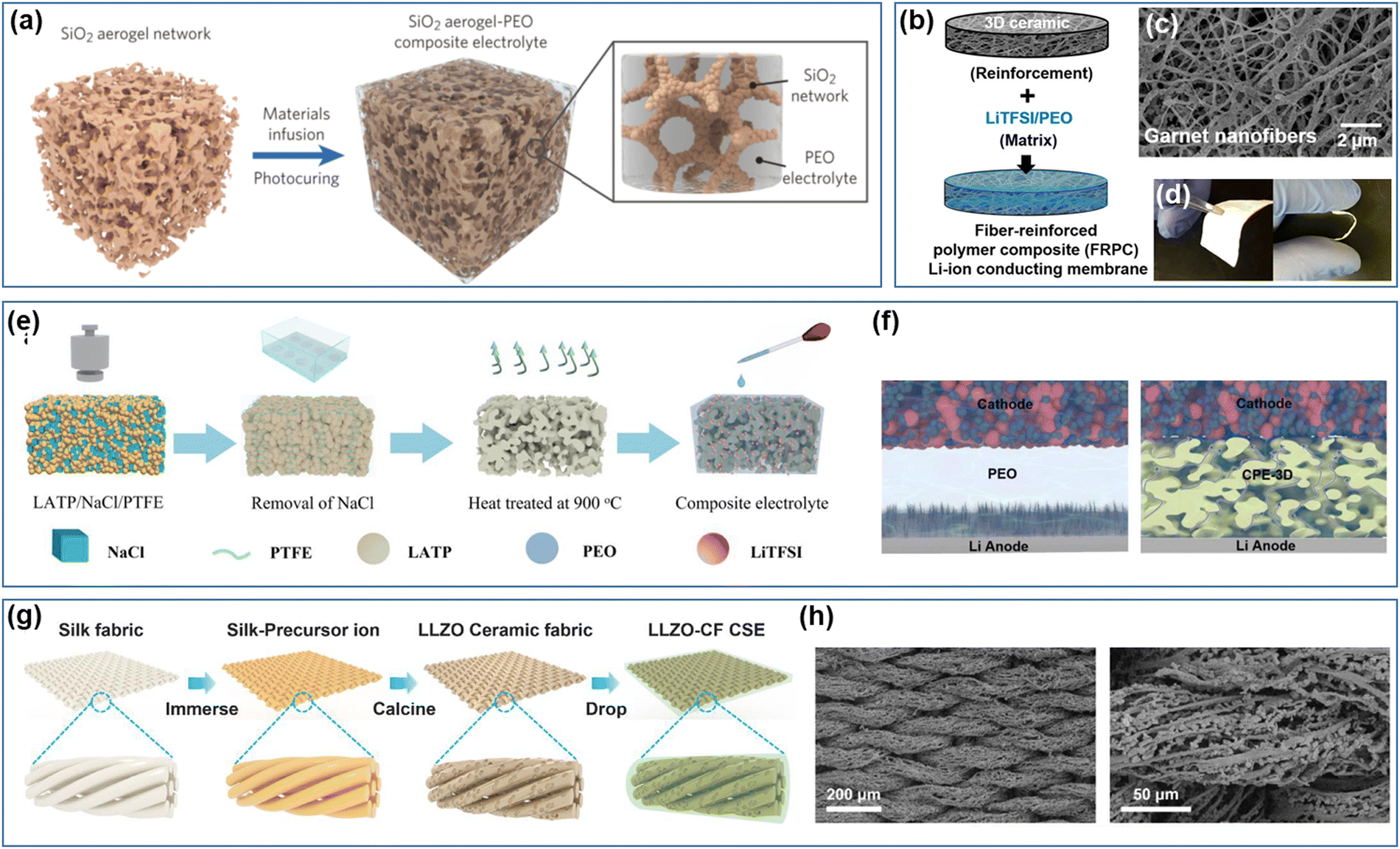 | ||
| Fig. 9 (a) Schematic showing the synthetic procedures of the SiO2–aerogel-reinforced composite solid electrolyte.196 (Reproduced with permission from Wiley-VCH.) (b) Schematic procedure to fabricate the fiber-reinforced polymer composite solid electrolyte, (c) SEM image of the garnet nanofiber network, and (d) digital image to show the flexible and bendable membrane.198 (Reproduced with permission from Proceedings of the National Academy of Science of the USA.) (e) Schematic diagram of the fabrication of LATP-PEO composite solid electrolyte and (f) structure schematic of LFP/PEO/Li&LFP/LATP-PEO/Li cells.199 (Reproduced with permission from Elsevier.) (g) Schematic of the preparation of the LLZO ceramic fiber composite solid electrolyte and (h) SEM images of LLZO ceramic fibers.200 (Reproduced with permission from Elsevier.) | ||
The electrospinning technique was also employed to fabricate a 3D framework, wherein continuous spinning, fiber deposition, film formation, and layer-by-layer stacking were employed. Fu et al.198 innovatively transformed one-dimensional nanofiber filler into a 3D interconnected fiber network, leading to the formation of a LLZO framework via a combination of electrospinning and calcination. Subsequently, the 3D framework was immersed in a lithium salt-polymer matrix to develop a composite solid electrolyte (Fig. 9b). The SEM image of the LLZO 3D framework is presented in Fig. 9c and the digital image of the composite solid electrolyte is shown in Fig. 9d. The solid electrolyte membrane exhibited exceptional flexibility and a high lithium-ion conductivity of 2.5 × 10−4 S cm−1 at 25 °C. Zhang et al.201 used a method in which PVDF–PEO/LiTFSI and LLZO nanoparticles were co-spun to create a continuous interconnected 3D network structure. This structure was then placed in a mixed solution of PEO and LiTFSI to produce a composite electrolyte. The resulting electrolyte membrane was incorporated into a Li/Li symmetrical battery, which exhibited excellent cycle stability with no short circuit occurrence over a period of 1000 hours. Additionally, the LiFePO4/Li battery that was created displayed outstanding rate performance, maintaining 99.2% of its initial capacity after 180 cycles. A flexible battery utilizing the composite electrolyte was able to power an LED to emit light normally even when bent or folded, showcasing the impressive flexibility of the electrolyte membrane. Yang et al.202 developed a three-dimensional network made of aluminum-doped Li0.33La0.557TiO3 (LLATO) nanofibers which has been then embedded in a PVDF–HFP matrix. They unveiled a strong chemical interaction upon introduction of LLATO nanofibers into the polymer electrolyte, which caused dehydrofluorination of PVDF chains, deprotonation of –CH2 groups and amorphization of the polymer matrix. This led to an improved ionic conductivity as high as 5.1 × 10−4 S cm−1 at 25 °C as well as a better cycling stability compared to the monolithic polymer electrolyte. Electrospinning has consistently proved to be a facile and low-cost technique for lab scale explorative studies, considering both the initial setup and the long-term use. Researchers have used it widely to fabricate ceramic nanowires and 3D frameworks for solid state electrolytes. Despite this, scaling up the process for potential industrial applications will be challenging, especially because of the low production rate and the high applied voltages. Moreover, since the electrospinning is based on the deposition of randomly distributed fibers on a grounded collector, it could yield inhomogeneous products when scaled up. It is also often reported to be very sensitive towards environmental parameters, such as temperature and relative humidity which can largely affect the reproducibility of the process.
To address these challenges and expedite the production of the 3D framework while minimizing the impact of environmental factors, the template method has gained attention. This method is favored for its simplicity, scalability, high content of inorganic fillers, and uniform pore distribution, making it a suitable approach for creating 3D frameworks of composite solid electrolytes. Wang et al.199 developed a 3D connected porous framework of LATP using NaCl as a template through a simple technique. They then incorporated a blend of polymer PEO and LiTFSI into the conductive frame to create a composite solid electrolyte (Fig. 9e). By adjusting the NaCl template content, it is possible to manage the amount of inorganic ceramics and the porosity of the 3D framework, which can be eliminated with water. The LATP content in the fabricated 3D structure reaches approximately 70 wt%, which imparts significant mechanical strength to prevent the formation of lithium dendrites and ensure the structural stability of the 3D framework at high temperatures (Fig. 9f). When subjected to a current density of 0.2 mA cm−2, a Li/Li symmetrical battery assembled with this composite electrolyte demonstrated stable operation for 1000 hours without experiencing any short circuits, which is double the performance of similar electrolytes containing LATP particles.
Furthermore, various natural porous materials like silk, cotton, hemp, and other fibers have been explored as templates for constructing 3D frameworks.203 These templates, derived from natural porous materials, maintain the original shape and dimensions, allowing polymer electrolytes to efficiently fill the voids through permeation and capillary action. This process facilitates intimate physical contact between polymers and 3D frameworks.204 Silk, a natural animal protein fiber with a high molecular weight, comprises predominantly of silk fibroin (70–80%) and sericin (20–30%). Proteins are composed of multiple peptide chains containing various amino acid residues (carboxyl, amino, hydroxyl, etc.) that can interact with metal ions. Pan et al.200 utilized silk and a LLZO precursor solution to create a complex via a coordination reaction, subsequently calcining it to obtain a LLZO template. This template was then combined with PEO/LiTFSI to form a sandwich composite solid electrolyte (Fig. 9g) and the SEM images of LLZO fibers are presented in Fig. 9h. When the LLZO content in the resultant electrolyte reached 70 wt%, it maintained good flexibility and exhibited good ionic conductivity (8.89 × 10−5 S cm−1 at 30 °C and 5.1 × 10−4 S cm−1 at 50 °C). The electrochemical stability window extended to 5.1 V, making it compatible with high-voltage cathode materials. In cathode material preparation, PEO/LiTFSI was utilized instead of a binder, achieving close interface contact between electrolyte and electrode through a melting treatment. The composite electrolyte's fusion-connected structure and various rapid lithium-ion transmission channels facilitated the electrolyte-assembled LiFePO4/Li full battery's stable cycling performance, even under high rates of 1C, with a reversible capacity of 107.2 mA h g−1 after 500 cycles. The natural fibers’ surface functional groups enhance compatibility between fillers and the polymer matrix, and the nano/micro pores in the framework fully fill the polymer matrix.
Whether through electrospinning fibers or the template method, maintaining porosity uniformity in 3D frameworks is crucial. To ensure uniform pore distribution in the 3D framework, new fabrication technologies have proven effective. 3D printing technology, a novel manufacturing technique based on layer-by-layer stacking, can produce precise framework structures that traditional methods cannot achieve, accurately control the template's shape and pore structure, and enhance raw material utilization. Zekoll et al.205 utilized 3D printing template technology to create various Li1.4Al0.4Ge1.6(PO4)3 (LAGP) 3D frame structures. These structures were then integrated with epoxy resin to form a 3D framework characterized by a uniform pore distribution and continuous ceramic channels (Fig. 10a). Results indicated that the electrolyte exhibited optimal conductivity (Fig. 10b) and mechanical properties when the 3D framework microstructure was configured as a rotator. Furthermore, 3D printing technology introduces innovative approaches for developing electrode materials with consistent voids. However, the compressive strength of the composite electrolyte was compromised due to the presence of polymer within the ceramic framework. Therefore, enhancing the mechanical properties of electrolytes remains a crucial aspect of electrolyte design. Kou et al.206 crafted an asymmetric LLTO framework featuring both a porous layer and a dense layer, which was then impregnated with a PEO/LiTFSI polymer solution (Fig. 10c) and the SEM images of asymmetric LLTO framework are presented in Fig. 10d. This process imparted good flexibility and processability to the composite electrolyte. The resultant composite electrolyte contained approximately 72.5 wt% LLTO ceramic and retained sufficient flexibility to bend without damage. At 30 °C, the ionic conductivity reached was 1.49 × 10−4 S cm−1, which is 40 times higher than that of PEO (Fig. 10e and f). The key advantage of this structure design lies in the asymmetric dense layer's ability to enhance the compressive strength of electrolytes and inhibit lithium dendrite formation. The assembled all-solid-state Li–S battery demonstrated stable cycle capacity and excellent rate performance. Additionally, 3D porous ceramic framework fabricated via an integrated sintering method could maximize the concentration of the active ionic conductor (over 90 wt%) leading to enhanced Li-ion conductivity, mechanical strength, and electrochemical stability of the ceramic-based composite solid electrolyte.
 | ||
| Fig. 10 (a) Schematic of the templating procedure used for the synthesis of structured composite solid electrolyte (CSE) and (b) the Arrhenius plot of the as-prepared CSE.205 (Reproduced with permission from The Royal Society of Chemistry.) (c) Schematic illustration of the synthesis of asymmetric LLTO framework (ALF) and PEO–LiTFSI/ALF CSE, (d) SEM images of ALF, and (e and f) lithium-ion conduction properties.206 (Reproduced with permission from Elsevier.) (g) Schematic synthesis illustration of the ceramic-based CSE, (surface & cross-section) SEM images of the 3D porous LLZO framework, and (h) SEM images of the ceramic-based CSE.207 (Reproduced with permission from Elsevier.) (i) Schematic synthesis illustration, (j) SEM images, and (k) Li-ion transport capability of the thin ceramic-based CSE, (l) schematic of the ASSB and (m) ASSB performance from 3 to 4.8 V.208 (Reproduced with permission from Cell Reports.) | ||
Wang et al.207 reported a unique 3D porous design of Li6.1Al0.3La3Zr2O12 (LLZO) scaffold via simple integrated sintering process demonstrated high ionic conductivity, porosity (30.56%), and mechanical strength. By introducing PEO–LiTFSI PSE into the as-designed 3D porous framework (the weight percentage of LLZO is over 97%), the mechanical properties are significantly enhanced (Fig. 10g and h). This enhancement allows the ceramic-based composite solid electrolyte to effectively inhibit the growth of Li dendrites. Moreover, the high porosity promotes widespread occupancy of PSE throughout the composite, forming metal-nitrogen bonding between the TFSI− groups in PSE and the La atoms in c-LLZO. The molecular bond of the polymer to the garnet scaffold surface establishes a strong interfacial connection between PSE/garnet, resulting in low interfacial impedance and mitigating the impact of grain boundaries on ionic conductivity. The ceramic-based composite solid electrolyte exhibits favorable electrochemical characteristics, including a room-temperature ionic conductivity of 0.547 mS cm−1 and 2 mS cm−1 at 80 °C. The Li/Li symmetric cells demonstrate exceptional long cycling stability against Li metal (over 500 h), and the assembled ASSB Li|ceramic-based composite solid electrolyte|LiFePO4 also display stable cycling and rate performance. From the same group, Wang et al.208 modified the synthesis method to design a 3D porous LLZO ultra-thin framework (Li6.1Al0.3La3Zr2O12) (Fig. 10i) exhibiting high Li+ conductivity, high porosity (45.74%) (Fig. 10j), and a wide electrochemical window (5.08 V) suitable for pairing with high-voltage cathode materials. The ceramic-based composite solid electrolyte (CSE) enabled by this thin 3D framework has a high ceramic-mass content (∼93%), which helps prevent the growth of Li dendrites. The porous structure of the LLZO framework allows for a high-volume occupancy of PVDF–LiTFSI, with metal-atom bonding between La from LLZO and N from TFSI− groups, as well as F from the PVDF polymer chains. The LLZO–TFSI− coupling promotes the dissociation of Li salts, enhances Li+ transport, and reduces interfacial impedance, leading to high ionic conductivity and Li transfer number of the ceramic-based CSE at 25 °C (0.437 mS cm−1 and 0.72) (Fig. 10k). The ceramic-based CSE interfaces well with TiO2-coated LiNi0.6Co0.2Mn0.2O2 (TiO2@NCM622) and Li metal, enabling uniform Li+ transport electric field for dendrite-free Li stripping and plating. The ASSB Li/ceramic-based CSE/TiO2@NCM622 (Fig. 10l) cycled stably from 3 to 4.8 V over demonstrates the excellent potential of this porous ceramic scaffold-based CSE architecture for practical applications (Fig. 10m).
Overall, the composite solid-state electrolytes with 3D framework structure allow to create a connected pathway for lithium-ion movement. This helps to evenly disperse lithium ions at the electrode and electrolyte interfacial region, preventing the formation of lithium dendrites. Additionally, the 3D framework enhances the mechanical properties and adds flexibility due to the polymer component. Despite these benefits, the weight increases with the inclusion of the 3D framework structure, negatively impacting its energy density. In this regard, how to achieve a composite solid-state electrolyte with both lightweight and excellent performance requires to further investigation.
4.4. Composite solid electrolytes with sulfide-type ceramic fillers
Research on polymer–ceramic electrolytes that incorporate sulfide-type fillers remains limited. The information available from several studies suggests that polymer–sulfide composites can achieve ionic conductivity levels comparable to other composite types, including polymer–NASICON, polymer–garnet, and polymer–perovskite systems. Zhao et al. documented a PEO/Li10GeP2S12/LiTFSI composite electrolyte capable of delivering an ionic conductivity of 1.21 × 10−3 S cm−1 at 80 °C.209 In addition, Villaluenga et al. created a non-flammable composite electrolyte composed of 77 wt% sulfide ceramic (25P2S5·75Li2S) and 23 wt% PFPE (perfluoropolyether), which demonstrated a conductivity of 10−4 S cm−1 at room temperature.210 The limited exploration of composites with sulfide fillers may be attributed to the susceptibility of sulfide materials to degradation when exposed to ambient air. Thus, this review is focusing on the oxide-based composite solid electrolytes.4.5. Interfacial issues
The polymeric phase present in ceramic composite electrolytes can enhance the contact surfaces between the electrodes and the electrolyte (both at the anode and the cathode). However, the interfacial challenges associated with polymer–ceramic composite electrolytes remain intricate and problematic. These challenges manifest at the junctions between the electrolyte and the lithium anode, as well as between the electrolyte and the cathode, in addition to the boundaries between the polymer matrices and the ceramic fillers. It is crucial to develop strategies that facilitate the establishment of low-resistance, stable interfaces, particularly on the anode side. The introduction of a super-thin layer of ionic liquid or ionic gel may aid the polymeric phase of the composite electrolyte in preserving a continuous ionic pathway at the interface between the lithium metal and the electrolyte. Within the polymer–ceramic composite, unanticipated voids within the structure of ceramic fillers may lead to inadequate contact between the matrix and the filler or potentially result in an incompatible interface. Thus, effective surface treatment techniques for ceramic fillers are vital. The interfacial issues encountered at the cathode side—whether with insertion or conversion cathodes—are even more complex. High-resistance and unstable solid interfaces may develop between cathode particles and the composite electrolyte. Such interphases could be unstable during prolonged cycling of batteries due to the volume fluctuations of the cathode particles. Therefore, exploring the construction of a stable and highly efficient ion conduction network at the interface between the cathode and the composite electrolyte is of significant importance.5. Manufacturability potential of ASSBs with various types of solid electrolytes
Among the different types of composite solid electrolytes, the manufacturing of polymer matrix based ASSBs is expected to be the most straightforward as it is building on the already commercial production of lithium metal–polymer batteries by Blue Solutions in France featuring PEO solid electrolyte.211 The production process differs from that for conventional liquid electrolyte LIBs as the wettability of PEO-based electrolytes on electrodes does not match that of polymer gels and liquid organic media. To achieve a closer interfacial contact between the PEO-based electrolyte and the electrode, it is essential to apply suitable hot-pressing techniques (such as calendaring, rolling, or hot isostatic pressing) or to create electrode composites (where the electrode layer and electrolyte layer are combined). This necessitates an upgrade and transformation of the existing production lines. By following this approach, it should become possible to fabricate winding cells and stacking cells using polymer electrolyte materials. Two examples of manufacturing process chains that have been developed,212 are illustrated in Fig. 11. In Fig. 11a, the creation of a cathode-supported half-cell is demonstrated, where the application of the solid electrolyte layer follows the processes of slurry mixing, tape casting, low-temperature sintering, and the shaping of the cathode composite layer. An aerosol deposition method is utilized to fabricate the solid-state electrolyte (SSE) layer, as this approach allows for the achievement of high quality and density without requiring a high-temperature co-sintering phase, thus minimizing undesirable side reactions. A thermal curing process at approximately 600 °C is subsequently implemented.213 The lithium metal anode and the current collector(s) are integrated prior to the assembly and packaging of the cell. This processing method is claimed to be advantageous because it eliminates high-temperature sintering phases and enables the integration of various solid electrolyte materials, such as LATP for the cathode composite and LLZO for the solid electrolyte layer.213 The overall readiness level of this technology chain is primarily influenced by the relatively immature state of the aerosol deposition process. | ||
| Fig. 11 Examples of manufacturing technology chains for (a) cathode-supported cells and (b) tri-layer cell.212 (Reproduced with permission from The Royal Society of Chemistry.) | ||
In Fig. 11b, the process sequence for a tri-layer SSE matrix is illustrated, featuring three consecutive tape casting operations designed to create the scaffolding, as noted in a recent study by Hitz et al.214 These layers can be made as separate green sheets, which are then laminated consecutively.214 Alternatively, it is possible to layer the casts directly on one another following each solvent evaporation step. The porosities of the outer layers can be adjusted through suitable slurry mixtures, including the addition of pore formers. This adjustment is critical for the subsequent infiltration of active materials215,216 and for enhancing surface area, thus allowing larger current densities.214 The green tapes are then trimmed to the appropriate dimensions before undergoing the high-temperature sintering process, during which the pore formers in the outer layers are eliminated. By precisely controlling the particle size and sintering conditions,217,218 a dense (over 99%) SSE layer can be formed between the two porous layers, effectively suppressing lithium dendrite formation during battery operation.214 To prevent the degradation of LLZO caused by humidity and Li2CO3 formation, it is advisable to conduct further processing following sintering in a dry or inert environment.219
The cathode slurry is introduced into the upper porous structure via a screen-printing technique. It is important to note that this technique might need to be executed multiple times to adequately fill the voids that persist after the evaporation of the solvent.215 Smaller cathode particles appear to promote a denser arrangement of particles and serve to mitigate volume fluctuations of the active materials throughout the battery's operation.220 Adjusting the viscosity of the slurry and the content of solids will be necessary to ensure proper filling of all the pores. Following the infiltration of the cathode, a sintering process is conducted at a reduced temperature to guarantee mechanical and ionic connectivity between the solid-state electrolyte (SSE) matrix and the cathode particles. Incorporating a low temperature melting glass, such as Li3BO3, into the cathode slurry can help to lower the temperatures required for sintering.221,222 Subsequently, the anode is infiltrated into the alternate porous layer of the scaffolding, potentially through melt processing.216 Given the high reactivity of molten lithium, it is crucial to implement safety measures and maintain an inert atmosphere during processing. A surface treatment may be employed to improve the wettability of lithium223 and create a mixed conducting network.224 During the infiltration process, it is essential to exercise caution to avoid creating an external short circuit. Ultimately, the current collector(s) are connected, and the cell is assembled. While the tri-layer configuration214 offers greater mechanical strength compared to free-standing layers, managing the delicate ceramic layers during large-scale production with fully automated machinery presents a significant challenge. For both design approaches illustrated (cathode-supported or tri-layer), precise adjustments to the process parameters during the fabrication and sintering of layers are essential for the successful functioning of all-solid-state batteries (ASSBs).
In contrast to the flexibility and adaptability of polymer materials, inorganic solid electrolytes, like garnet oxides, are considerably more rigid and fragile. Consequently, the only feasible method for producing garnet-based cells is through sheet stacking. A research study has systematically explored the possible production technologies and associated manufacturing costs for a specific type of garnet-based all-solid-state battery (ASSB). They selected a battery that uses LLZO as the electrolyte material and LiNi0.5Mn1.5O4 (LNMO) as the cathode material for detailed discussion and analysis.211 Theoretically, this battery type could achieve an energy density of 530 Wh kg−1 if optimally designed. As noted earlier, creating composite electrodes and electrolytes is the most practical approach for hard oxide ceramics; thus, co-sintering is a crucial process in the fabrication of all-solid-state cells. However, numerous studies indicate that high-performance LLZO electrolytes need to be sintered at temperatures exceeding 1000 °C to develop a dense structure, while LNMO cannot be sintered at temperatures above approximately 600 °C to avoid the formation of undesired by-products, which predominantly adversely affect ASSBs.225,226
The tri-layer cell technology chain comprising three distinct electrolyte layers is relatively advanced.215 The sandwich structure of the electrolyte consists of an intermediate dense layer flanked by two identical layers that include pore formers created through tape casting and evaporation techniques. This is followed by alternating processes like application of cathode slurry through screen printing and high-temperature sintering. However, the high-temperature processes entail significant energy usage; thus, an alternative approach is suggested—producing cathode-supported cells. This method involves tape casting a composite slurry for the cathode, followed by laser cutting, low-temperature sintering, laser shaping, aerosol deposition of the solid electrolyte, and a final low-temperature sintering phase. Regarding both strategies, the authors212 implicitly support the use of metallic lithium foil as the anode. Furthermore, the authors estimate that the cost of garnet-based ASSBs could drop below $150 per kWh, allowing them to compete effectively with traditional lithium-ion batteries. On the other hand, they argue that utilizing carbon materials, such as modified graphite or silicon–carbon composites, in place of lithium metal could significantly lower manufacturing cost and enhance safety. A critical requirement in sulfide-based ASSB manufacturing is the production environment; sulfides require processing in an inert atmosphere to prevent exposure to moisture and oxygen. This poses a production challenge and necessitates additional investment in manufacturing. Moreover, sulfide materials lack sufficient stability when in contact with metallic lithium as anode or high-voltage cathodes, which can lead to shorter cycle life and the emission of toxic gases. Thus pursuing composite electrolyte development that takes into account the manufacturability aspects from an industrial scale perspective is a critical consideration in moving forward.
6. Summary, challenges and perspectives
6.1. Summary of this review paper
To promote the advancement of composite solid-state electrolytes (CSEs) for all-solid-state lithium batteries (ASSBs), this paper provides a detailed overview of recent developments in advanced materials and structures. Initially, a brief history of solid-state ionic conductors is reviewed, followed by a summary of the fundamental aspects such as the key materials, mechanisms of Li ionic transport, and performance requirements for CSEs. The key materials and advanced structures of CSEs are then classified and summarized, including inorganic (inert & active) fillers in the polymer matrix, and 3D inorganic continuous frameworks with filled polymers composite electrolytes.6.2. Challenges and perspectives
Despite the rapid advances and increasing research efforts, this field is still in its infancy. It appears that the technical maturity of CSEs utilized in ASSBs for energy conversion and utilization has yet to meet the necessary requirements for practical implementation and commercialization. This is due to several significant challenges, such as the not fully understood mechanisms for ionic transport in CSEs, the sluggish ionic conductivities, stability issues (chemical, electrochemical, mechanical, and thermal) of ASSBs with CSEs, and insufficient economic and technological feasibility. Further details on these challenges and perspectives are presented below.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding received from McGill Sustainability Systems Initiative Innovation Fund and Natural Sciences & Engineering Research Council of Canada Strategic Projects Programme (NSERC STPGP 521217-18) is greatly acknowledged.References
- P. Albertus, S. Babinec, S. Litzelman and A. Newman, Nat. Energy, 2018, 3, 16–21 CrossRef CAS.
- X.-B. Cheng, J.-Q. Huang and Q. Zhang, J. Electrochem. Soc., 2018, 165, A6058–A6072 CrossRef CAS.
- G. Li, Z. Chen and J. Lu, Chem, 2018, 4, 3–7 CAS.
- X. Ren, S. Chen, H. Lee, D. Mei, M. H. Engelhard, S. D. Burton, W. Zhao, J. Zheng, Q. Li, M. S. Ding, M. Schroeder, J. Alvarado, K. Xu, Y. S. Meng, J. Liu, J.-G. Zhang and W. Xu, Chem, 2018, 4, 1877–1892 CAS.
- V. Raj, N. P. B. Aetukuri and J. Nanda, Curr. Opin. Solid State Mater. Sci., 2022, 26(4), 100999 CrossRef CAS.
- H. Gao, S. Xin, L. Xue and J. B. Goodenough, Chem, 2018, 4, 833–844 CAS.
- Y. Shen, Y. Zhang, S. Han, J. Wang, Z. Peng and L. Chen, Joule, 2018, 2, 1674–1689 CrossRef CAS.
- X.-Q. Zhang, X.-B. Cheng and Q. Zhang, Adv. Mater. Interfaces, 2018, 5, 1701097 CrossRef.
- A. Mauger, M. Armand, C. M. Julien and K. Zaghib, J. Power Sources, 2017, 353, 333–342 CrossRef CAS.
- X.-B. Cheng, R. Zhang, C.-Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- X.-B. Cheng, C.-Z. Zhao, Y.-X. Yao, H. Liu and Q. Zhang, Chem. Rev., 2019, 5, 74–96 CAS.
- A. Manthiram, X. W. Yu and S. F. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS.
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 16141 CrossRef.
- V. Thangadurai, S. Narayanan and D. Pinzaru, Chem. Soc. Rev., 2014, 43, 4714–4727 RSC; R. J. Chen, W. J. Qu, X. Guo, L. Li and F. Wu, Mater. Horiz., 2016, 3, 487–516 RSC.
- S. Choudhury, S. Stalin, D. Vu, A. Warren, Y. Deng, P. Biswal and L. A. Archer, Nat. Commun., 2019, 10, 4398 CrossRef PubMed.
- J. C. Bachman, S. Muy, A. Grimaud, H. H. Chang, N. Pour, S. F. Lux, O. Paschos, F. Maglia, S. Lupart, P. Lamp, L. Giordano and Y. Shao-Horn, Chem. Rev., 2016, 116, 140–162 CrossRef CAS PubMed.
- X. Wang, H. Zhai, B. Qie, Q. Cheng, A. Li, J. Borovilas., B. Xu, C. Shi, T. Jin, X. Liao, Y. Li, X. He, S. Du, Y. Fu, M. Dontigny, K. Zaghib and Y. Yang, Nano Energy, 2019, 60, 205–212 CrossRef.
- R. Chen, Q. Li, X. Yu, L. Chen and H. Li, Chem. Rev., 2019, 120, 6820–6877 CrossRef PubMed.
- S. Wang, H. Xu, W. Li, A. Dolocan and A. Manthiram, J. Am. Chem. Soc., 2018, 140, 250–257 CrossRef CAS PubMed.
- S. Xia, X. Wu, Z. Zhang, Y. Cui and W. Liu, Chem, 2019, 5, 753–785 CAS.
- Y. Jiang, X. Yan, Z. Ma, P. Mei, W. Xiao, Q. You and Y. Zhang, Polymers, 2018, 10, 1237 CrossRef PubMed.
- Y. Li, B. Xu, H. Xu, H. Duan, X. Lu, S. Xin, W. Zhou, L. Xue, G. Fu, A. Manthiram and J. B. Goodenough, Angew. Chem., 2017, 56, 753–756 CrossRef CAS PubMed.
- Y. T. Li, B. Y. Xu, H. H. Xu, H. N. Duan, X. J. Lu, S. Xin, W. D. Zhou, L. G. Xue, G. T. Fu, A. Manthiram and J. B. Goodenough, Angew. Chem., Int. Ed., 2017, 56, 753–756 CrossRef CAS.
- A. Li, X. Liao, H. Zhang, L. Shi, P. Wang, Q. Cheng, J. Borovilas, Z. Li, W. Huang, Z. Fu, M. Dontigny, K. Zaghib, K. Myers, X. Chuan, X. Chen and Y. Yang, Adv. Mater., 2019, 32, 1905517 CrossRef PubMed.
- G. Tan, F. Wu, C. Zhan, J. Wang, D. Mu, J. Lu and K. Amine, Nano Lett., 2016, 16, 1960–1968 CrossRef CAS PubMed.
- S. Li, S. Q. Zhang, L. Shen, Q. Liu, J. B. Ma, W. Lv, Y. B. He and Q. H. Yang, Adv. Sci., 2020, 7, 1903088 CrossRef CAS.
- O. Yamamoto, Sci. Technol. Adv. Mater., 2017, 18, 504–527 CrossRef PubMed.
- K. Funke, Sci. Technol. Adv. Mater., 2013, 14, 43502 CrossRef CAS.
- E. Warburg, Ann. Phys. Chem., 1994, 21, 662 Search PubMed.
- E. Warburg and F. Tegetmeier, Ann. Phys., 1888, 32, 455 CrossRef.
- W. Nernst and W. Z. Wild, Z. Elektrochem. Angew. Phys. Chem., 1900, 7, 373–376 CAS.
- C. Z. Wagner, Phys. Chem. B Chem., 1933, 22, 181 Search PubMed.
- K. Funke, Sci. Technol. Adv. Mater., 2013, 14, 43502 CrossRef CAS PubMed.
- C. Wagner, Naturwissenschaften, 1943, 31, 265–268 CrossRef CAS.
- C. Tubandt and E. Z. Lorenz, Phys. Chem., 1914, 87, 513–542 CAS.
- J. Frenkel, Eur. Phys. J. A, 1926, 35, 652–660 Search PubMed.
- C. Wagner and W. Z. Schottky, Phys. Chem. B Chem., 1930, 11, 163 CAS.
- M. H. Hebb, J. Chem. Phys., 1952, 20, 185–190 CrossRef CAS.
- J. B. Wagner and C. J. Wagner, Chem. Phys., 1957, 26, 1597–1601 CAS.
- C. Wagner, J. Chem. Phys., 1953, 21, 1819–1827 CrossRef CAS.
- V. Thangadurai and W. Weppner, Adv. Funct. Mater., 2005, 15, 107–112 CrossRef CAS.
- P. Hartmann, T. Leichtweiss, M. R. Busche, M. Schneider, M. Reich, J. Sann, P. Adelhelm and J. Janek, J. Phys. Chem. C, 2013, 117, 21064–21074 CrossRef CAS.
- G. M. Rupp, M. Glowacki and J. Fleig, J. Electrochem. Soc., 2016, 163, 1189–1197 CrossRef.
- K. Kiukkola and C. Wagner, J. Electrochem. Soc., 1957, 104, 379–387 CrossRef.
- B. B. Owens and G. R. Argue, Science, 1967, 157, 308–310 CrossRef CAS.
- J. N. Bradley and P. D. Greene, Trans. Faraday Soc., 1967, 63, 424–430 RSC.
- T. Takahashi, O. Yamamoto, S. Yamada and S. Hayashi, J. Electrochem. Soc., 1979, 126, 1654–1658 CrossRef CAS.
- Y. Y. Yao and J. T. Kummer, J. Inorg. Nucl. Chem., 1967, 29, 2453–2475 CrossRef CAS.
- J. T. Kummer and N. Weber, SAE Tech. Pap. Ser., 1967, 76, 88–89 Search PubMed.
- K. Kuwabara and T. Takahashi, J. Solid State Chem., 1976, 19, 147–153 CrossRef CAS.
- J. P. Boilot, J. Théry and R. Collongues, Mater. Res. Bull., 1973, 8, 1143–1151 CrossRef CAS.
- J. B. Goodenough, H. Y.-P. Hong and J. A. Kafalas, Mater. Res. Bull., 1976, 11, 203–220 CrossRef CAS.
- H. Y.-P. Hong, Mater. Res. Bull., 1976, 11, 173–182 CrossRef CAS.
- B. A. Boukamp and R. A. Huggins, Mater. Res. Bull., 1978, 13, 23–32 CrossRef CAS.
- C. C. Liang, J. Electrochem. Soc., 1973, 120, 1289–1292 CrossRef CAS.
- A. A. Schneider, D. E. Harney and M. J. Harney, J. Power Sources, 1980, 5, 15–23 CrossRef CAS.
- H. Y.-P. Hong, Mater. Res. Bull., 1978, 13, 117–124 CrossRef CAS.
- R. Kanno, T. Hata, Y. Kawamoto and M. Irie, Solid State Ionics, 2000, 130, 97–104 CrossRef CAS.
- V. Thangadurai, H. Kaack and W. J. F. Weppner, J. Am. Ceram. Soc., 2003, 86, 437–440 CrossRef CAS.
- Y. Zhao and L. L. Daemen, J. Am. Chem. Soc., 2012, 134, 15042–15047 CrossRef CAS.
- Z. Qin, X. Meng, Y. Xie, D. Qian, H. Deng, D. Mao, L. Wan and Y. Huang, Energy Storage Mater., 2021, 43, 190–201 CrossRef.
- Y. Pang, J. Pan, J. Yang, S. Zheng and C. Wang, Electrochem. Energy Rev., 2021, 4, 169–193 CrossRef CAS.
- Q. Liu, Z. Geng, C. Han, Y. Fu, S. Li, Y.-B. He, F. Kang and B. Li, J. Power Sources, 2018, 389, 120–134 CrossRef CAS.
- N. Zhao, W. Khokhar, Z. Bi, C. Shi, X. Guo, L.-Z. Fan and C.-W. Nan, Joule, 2019, 3, 1190–1199 CrossRef CAS.
- C. Wang, K. Fu, S. P. Kammampata, D. W. McOwen, A. J. Samson, L. Zhang, G. T. Hitz, A. M. Nolan, E. D. Wachsman, Y. Mo, V. Thangadurai and L. Hu, Chem. Rev., 2020, 120, 4257–4300 CrossRef CAS PubMed.
- R. Murugan, V. Thangadurai and W. Weppner, Angew. Chem., Int. Ed., 2007, 46, 7778–7781 CrossRef CAS PubMed.
- S.-W. Baek, J.-M. Lee, T. Y. Kim, M.-S. Song and Y. Park, J. Power Sources, 2014, 249, 197–206 CrossRef CAS.
- J. Sakamoto, E. Rangasamy, H. Kim, Y. Kim and J. Wolfenstine, Nanotechnology, 2013, 24, 424005 CrossRef PubMed.
- S. Wang, H.-c Chiu and G. P. Demopoulos, J. Power Sources, 2024, 595, 234061 CrossRef CAS.
- A. Kim, J. H. Kang, K. Song and B. Kang, ACS Appl. Mater. Interfaces, 2022, 14, 12331–12339 CrossRef CAS PubMed.
- L. Shen, L. Wang, Z. Wang, C. Jin, L. Peng, X. Pan, J. Sun and R. Yang, Solid State Ionics, 2019, 339, 114992 CrossRef CAS.
- Y. Luo, Q. Zhang, A. Shen, M. Shen, D. Xie and Y. Yan, Solid State Ionics, 2022, 374, 115812 CrossRef CAS.
- S. Rajendran, N. K. Thangavel, K. Mahankali and L. M. R. Arava, ACS Appl. Energy Mater., 2020, 3, 6775–6784 CrossRef CAS.
- X. Wang, J. Wang, F. Li, F. Zhu and C. Ma, Ceram. Int., 2020, 46, 18544–18550 CrossRef CAS.
- L. J. Miara, W. D. Richards, Y. E. Wang and G. Ceder, Chem. Mater., 2015, 27, 4040–4047 CrossRef CAS.
- C. Roitzheim, Y. J. Sohn, L.-Y. Kuo, G. H€auschen, M. Mann, D. Sebold, M. Finsterbusch, P. Kaghazchi, O. Guillon and D. Fattakhova-Rohlfing, ACS Appl. Energy Mater., 2022, 5, 6913–6926 CrossRef CAS.
- S. Hong, S. H. Song, M. Cho, S. Kim, S. H. Yu, D. Lee and H. Kim, Small, 2021, 17, e2103306 CrossRef.
- K. H. Kim, Y. Iriyama, K. Yamamoto, S. Kumazaki, T. Asaka, K. Tanabe, C. A. J. Fisher, T. Hirayama, R. Murugan and Z. Ogumi, J. Power Sources, 2011, 196, 764–767 CrossRef CAS.
- M. Avdeev, Chem. Mater., 2021, 33, 7620–7632 CrossRef CAS.
- X. Hu, X. Cheng, S. Qin, G. Yan, J. Malzbender, W. Qiang and B. Huang, Ceram. Int., 2018, 44, 1902–1908 CrossRef CAS.
- K. Homma, M. Yonemura, T. Kobayashi, M. Nagao, M. Hirayama and R. Kanno, Solid State Ionics, 2011, 182, 53–58 CrossRef CAS.
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto and A. Mitsui, Nat. Mater., 2011, 10, 682–686 CrossRef CAS PubMed.
- B. He, F. Zhang, Y. Xin, C. Xu, X. Hu, X. Wu, Y. Yang and H. Tian, Nat. Rev. Chem., 2023, 7, 826–842 Search PubMed.
- S. F. Cherif, M. F. Zid and A. Driss, Arabian J. Chem., 2019, 12, 3309–3315 CrossRef CAS.
- I. d'Anciaes Almeida Silva, A. M. Nieto-Munoz, A. C. M. Rodrigues and H. Eckert, J. Am. Ceram. Soc., 2020, 103, 4002–4012 CrossRef.
- J. K. Feng, L. Lu and M. O. Lai, J. Alloys Compd., 2010, 501, 255–258 CrossRef CAS.
- L. Li, Z. Zhang, L. Luo, R. You, J. Jiao, W. Huang, J. Wang, C. Li, X. Han and S. Chen, Ionics, 2020, 26, 3815–3821 CrossRef CAS.
- S. Wang, S. Li, B. Wei and X. Lu, J. Electrochem. Soc., 2020, 167, 100528 CrossRef CAS.
- Z. Jiang, H. Xie, S. Wang, X. Song, X. Yao and H. Wang, Adv. Energy Mater., 2018, 8, 1801433 CrossRef.
- K. P. Abhilash, P. Christopher Selvin, B. Nalini, K. Somasundaram, P. Sivaraj and A. Chandra Bose, J. Phys. Chem. Solids, 2016, 91, 114–121 CrossRef CAS.
- L. Xu, L. Zhang, Y. Hu and L. Luo, Nano Energy, 2022, 92, 106758 CrossRef CAS.
- M. J. Wang, J. B. Wolfenstine and J. Sakamoto, Adv. Funct. Mater., 2020, 30, 1909140 CrossRef CAS.
- S. Chen, D. Xie, G. Liu, J. P. Mwizerwa, Q. Zhang, Y. Zhao, X. Xu and X. Yao, Energy Storage Mater., 2018, 14, 58–74 CrossRef.
- J. A. Gonzalez-Leon, M. H. Acar, S. W. Ryu, A. V. Ruzette and A. M. Mayes, Nature, 2003, 426, 424–428 CrossRef CAS.
- H. Tsukasaki, S. Mori, H. Morimoto, A. Hayashi and M. Tatsumisago, Sci. Rep., 2017, 7, 4142 CrossRef PubMed.
- F. Sun, K. Dong, M. Osenberg, A. Hilger, S. Risse, Y. Lu, P. H. Kamm, M. Klaus, H. Mark€otter, F. García-Moreno, T. Arlt and I. Manke, J. Mater. Chem., 2018, 6, 22489–22496 RSC.
- R. P. Rao, N. Sharma, V. K. Peterson and S. Adams, Solid State Ionics, 2013, 230, 72–76 CrossRef CAS.
- T. Yajima, Y. Hinuma, S. Hori, R. Iwasaki, R. Kanno, T. Ohhara, A. Nakao, K. Munakata and Z. Hiroi, J. Mater. Chem., 2021, 9, 11278–11284 RSC.
- C. Singer, H.-C. Topper, T. Kutsch, R. Schuster, R. Koerver and R. Daub, ACS Appl. Mater. Interfaces, 2022, 14, 24245–24254 CrossRef CAS.
- M. Calpa, N. C. Rosero-Navarro, A. Miura, R. Jalem, Y. Tateyama and K. Tadanaga, Appl. Mater. Today, 2021, 22, 100918 CrossRef.
- D. H. S. Tan, E. A. Wu, H. Nguyen, Z. Chen, M. A. T. Marple, J.-M. Doux, X. Wang, H. Yang, A. Banerjee and Y. S. Meng, ACS Energy Lett., 2019, 4, 2418–2427 CrossRef CAS.
- N. Zhang, L. Wang, Q. Diao, K. Zhu, H. Li, C. Li, X. Liu and Q. Xu, J. Electrochem. Soc., 2022, 169, 020544 CrossRef CAS.
- S. Wenzel, S. Randau, T. Leichtweiß, D. A. Weber, J. Sann, W. G. Zeier and J. Janek, Chem. Mater., 2016, 28, 2400–2407 CrossRef CAS.
- J. H. Woo, J. E. Trevey, A. S. Cavanagh, Y. S. Choi, S. C. Kim, S. M. George, K. H. Oh and S.-H. Lee, J. Electrochem. Soc., 2012, 159, A1120–A1124 CrossRef CAS.
- J. Wu, S. Liu, F. Han, X. Yao and C. Wang, Adv. Mater., 2021, 33, e2000751 CrossRef.
- R. Xu, F. Han, X. Ji, X. Fan, J. Tu and C. Wang, Nano Energy, 2018, 53, 958–966 CrossRef CAS.
- Y. Zhao and L. L. Daemen, J. Am. Chem. Soc., 2012, 134, 15042–15047 CrossRef CAS.
- J. Zhang, J. Han, J. Zhu, Z. Lin, M. H. Braga, L. L. Daemen, L. Wang and Y. Zhao, Inorg. Chem. Commun., 2014, 48, 140–143 CrossRef CAS.
- Y. Tian, F. Ding, H. Zhong, C. Liu, Y.-B. He, J. Liu, X. Liu and Q. Xu, Energy Storage Mater., 2018, 14, 49–57 CrossRef.
- Z. Deng, B. Radhakrishnan and S. P. Ong, Chem. Mater., 2015, 27, 3749–3755 CrossRef CAS.
- Z. D. Hood, H. Wang, A. Samuthira Pandian, J. K. Keum and C. Liang, J. Am. Chem. Soc., 2016, 138, 1768–1771 CrossRef CAS PubMed.
- Y. Li, W. Zhou, S. Xin, S. Li, J. Zhu, X. Lu, Z. Cui, Q. Jia, J. Zhou, Y. Zhao and J. B. Goodenough, Angew. Chem., Int. Ed., 2016, 55, 9965–9968 CrossRef CAS PubMed.
- A. Y. Song, Y. Xiao, K. Turcheniuk, P. Upadhya, A. Ramanujapuram, J. Benson, A. Magasinski, M. Olguin, L. Meda, O. Borodin and G. Yushin, Adv. Energy Mater., 2018, 8, 1700971 CrossRef.
- B. Zhang, R. Tan, L. Yang, J. Zheng, K. Zhang, S. Mo, Z. Lin and F. Pan, Energy Storage Mater., 2018, 10, 139–159 CrossRef.
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 1–16 Search PubMed.
- M. Park, X. Zhang, M. Chung, G. B. Less and A. M. Sastry, J. Power Sources, 2010, 195, 7904–7929 CrossRef CAS.
- M. S. Islam, D. J. Driscoll, C. A. J. Fisher and P. R. Slater, Chem. Mater., 2005, 17, 5085–5092 CrossRef CAS.
- A. Van Der Ven and G. Ceder, Electrochem. Solid-State Lett., 2000, 3, 301–304 CrossRef CAS.
- A. Chroneos, B. Yildiz, A. Tarancon, D. Parfitt and J. A. Kilner, Energy Environ. Sci., 2011, 4, 2774–2789 RSC.
- C. Angell, Annu. Rev. Phys. Chem., 1992, 43, 693–717 CrossRef CAS.
- M. Wu, B. Xu and C. Ouyang, Chin. Phys. B, 2015, 25, 1–10 Search PubMed.
- D. E. Fenton, J. M. Parker and P. V. Wright, Polymer, 1973, 14, 589 CrossRef CAS.
- L. Liu, D. Zhang, X. Xu, Z. Liu and J. Liu, Chem. Res. Chin. Univ., 2021, 37, 210–231 CrossRef CAS.
- J. Park, S. Han, H. Park, J. Lee, S. Cho, M. Seo, B. J. Kim and S. Q. Choi, Macromolecules, 2021, 54, 9532–9541 CrossRef CAS.
- G. X. Dong, H. J. Li, Y. Wang, W. J. Jiang and Z. S. Ma, Ionics, 2021, 27, 2955–2965 CrossRef CAS.
- X. Shi, N. Ma, Y. Wu, Y. Lu, Q. Xiao, Z. Li and G. Lei, Solid State Ionics, 2018, 325, 112–119 CrossRef CAS.
- M. V. Leena Chandra, S. Karthikeyan, S. Selvasekarapandian, M. Premalatha and S. Monisha, J. Polym. Eng., 2017, 37, 617–631 CrossRef CAS.
- T. Janaki Rami Reddy, V. B. S. Achari, A. K. Sharma and V. V. R. Narasimha Rao, Ionics, 2007, 13, 435–439 CrossRef.
- W. Ye, M. Zaheer, L. Li, J. Wang, H. Xu, C. Wang and Y. Deng, J. Electrochem. Soc., 2020, 167, 110532 CrossRef CAS.
- P. Hu, J. Chai, Y. Duan, Z. Liu, G. Cui and L. Chen, J. Mater. Chem., 2016, 4, 10070–10083 RSC.
- P. Martins, A. C. Lopes and S. Lanceros-Mendez, Prog. Polym. Sci., 2014, 39, 683–706 CrossRef CAS.
- H. Wang, L. Sheng, G. Yasin, L. Wang, H. Xu and X. He, Energy Storage Mater., 2020, 33, 188–215 CrossRef.
- L. Long, S. Wang, M. Xiao and Y. Meng, J. Mater. Chem. A, 2016, 4, 10038–10039 RSC.
- J. Mindemark, M. J. Lacey, T. Bowden and D. Brandell, Prog. Polym. Sci., 2018, 81, 114–143 CrossRef CAS.
- S. K. Fullerton-Shirey and J. K. Maranas, Macromolecules, 2009, 42, 2142–2156 CrossRef CAS.
- D. P. Tunstall, P. G. Brue, Z. Gadjourova and Y. G. Andreev, Nature, 2001, 412, 1–4 CrossRef.
- C. Berthier, W. Gorecki, M. Minier, M. B. Armand, J. M. Chabagno and P. Rigaud, Solid State Ionics, 1983, 11, 91–95 CrossRef CAS.
- W. A. Henderson, N. R. Brooks and V. G. Young, J. Am. Chem. Soc., 2003, 125, 12098–12099 CrossRef CAS PubMed.
- W. A. Henderson and S. Passerini, Electrochem. Commun., 2003, 5, 575–578 CrossRef CAS.
- Z. Stoeva, I. Martin-Litas, E. Staunton, Y. G. Andreev and P. G. Bruce, J. Am. Chem. Soc., 2003, 125, 4619–4626 CrossRef CAS PubMed.
- A. M. Christie, S. J. Lilley, E. Staunton, Y. G. Andreev and P. G. Bruce, Nature, 2005, 433, 50–53 CrossRef CAS PubMed.
- C. Zhang, E. Staunton, Y. G. Andreev and P. G. Bruce, J. Am. Chem. Soc., 2005, 127, 18305–18308 CrossRef CAS.
- S. J. Lilley, Y. G. Andreev and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 12036–12037 CrossRef CAS PubMed.
- G. S. MacGlashan and Y. G. Andreev, Nature, 1999, 398, 792–794 CrossRef CAS.
- J. E. Weston and B. C. H. Steele, Solid State Ion., 1982, 7, 75–79 CrossRef CAS.
- S. Xia, B. Yang, H. Zhang, J. Yang, W. Liu and S. Zheng, Adv. Funct. Mater., 2021, 31, 2101168 CrossRef CAS.
- H. Chen, D. Adekoya, L. Hencz, J. Ma, S. Chen, C. Yan, H. Zhao, G. Cui and S. Zhang, Adv. Energy Mater., 2020, 10, 2000049 CrossRef CAS.
- F. Croce, G. B. Appetecchi, L. Persi and B. Scrosati, Nature, 1998, 394, 456–458 CrossRef CAS.
- W. Bao, L. Zhao, H. Zhao, L. Su, X. Cai, B. Yi, Y. Zhang and J. Xie, Energy Storage Mater., 2021, 43, 258–265 CrossRef.
- R. Wang, J. Yu, J. Tang, R. Meng, L. F. Nazar, L. Huang and X. Liang, Energy Storage Mater., 2020, 32, 178–184 CrossRef.
- Y. Sun, J. Wang, D. Fu, F. Zhang, Z. Wang, X. Chen, J. Xu, J. Hu and X. Wu, ACS Sustainable Chem. Eng., 2021, 9, 2237–2245 CrossRef CAS.
- Y. Kuai, F. Wang, J. Yang, H. Lu, Z. Xu, X. Xu, Y. NuLi and J. Wang, Mater. Chem. Front., 2021, 5, 6502–6511 RSC.
- J. S. Kim and J. K. Lim, React. Funct. Polym., 2021, 166, 105013 CrossRef CAS.
- H. An, Q. Liu, J. An, S. Liang, X. Wang, Z. Xu, Y. Tong, H. Huo, N. Sun, Y. Wang, Y. Shi and J. Wang, Energy Storage Mater., 2021, 43, 358–364 CrossRef.
- W. Xu, X. Pei, C. S. Diercks, H. Lyu, Z. Ji and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 17522–17526 CrossRef CAS.
- S. Bai, Y. Sun, J. Yi, Y. He, Y. Qiao and H. Zhou, Joule, 2018, 2, 2117–2132 CrossRef CAS.
- W.-H. Huang, X.-M. Li, X.-F. Yang, X.-X. Zhang, H.-H. Wang and H. Wang, Mater. Chem. Front., 2021, 5, 3593–3613 RSC.
- G. Wang, P. He and L. Z. Fan, Adv. Funct. Mater., 2020, 31, 2007198 CrossRef.
- Z. Wang, R. Tan, H. Wang, L. Yang, J. Hu, H. Chen and F. Pan, Adv. Mater., 2018, 30, 1704436 CrossRef PubMed.
- W. He, D. Li, S. Guo, Y. Xiao, W. Gong, Q. Zeng, Y. Ouyang, X. Li, H. Deng, C. Tan, Q. Zhang and S. Huang, Energy Storage Mater., 2022, 47, 271–278 CrossRef.
- A. Wang, H. Xu, X. Liu, S. Wang, Q. Zhou, J. Chen and L. Zhang, Compos. Sci. Technol., 2017, 152, 68–75 CrossRef CAS.
- T. Wu and J. N. Wang, RSC Adv., 2016, 6, 38187–38191 RSC.
- M. Ahmad and S. R. P. Silva, Carbon, 2020, 158, 24–44 CrossRef CAS.
- W. Jia, Z. Li, Z. Wu, L. Wang, B. Wu, Y. Wang, Y. Cao and J. Li, Solid State Ionics, 2018, 315, 7–13 CrossRef CAS.
- Y. Pan, H. Bao and L. Li, ACS Appl. Mater. Interfaces, 2011, 3, 4819–4830 CrossRef CAS PubMed.
- J.-K. Wu, C.-C. Ye, T. Liu, Q.-F. An, Y.-H. Song, K.-R. Lee, W.-S. Hung and C.-J. Gao, Mater. Des., 2017, 119, 38–46 CrossRef CAS.
- C. I. Idumah, A. Hassan, J. Ogbu, J. U. Ndem and I. C. Nwuzor, Compos. Interfaces, 2018, 26, 751–824 CrossRef.
- Y. Lin, X. Wang, J. Liu and J. D. Miller, Nano Energy, 2017, 31, 478–485 CrossRef CAS.
- M. Wu, D. Liu, D. Qu, Z. Xie, J. Li, J. Lei and H. Tang, ACS Appl. Mater. Interfaces, 2020, 12, 52652–52659 CrossRef CAS.
- A. L. Monaca, A. Paolella, A. Guerfi, F. Rosei and K. Zaghib, Electrochem. Commun., 2019, 104, 106483 CrossRef.
- W. Liu, N. Liu, J. Sun, P.-C. Hsu, Y. Li, H.-W. Lee and Y. Cui, Nano Lett., 2015, 15, 2740–2745 CrossRef CAS PubMed.
- T. Yang, Y. Li and C. K. Chan, J. Power Sources, 2015, 287, 164–169 CrossRef CAS.
- T. Yang, Z. D. Gordon, Y. Li and C. K. Chan, J. Phys. Chem. C, 2015, 119, 14947–14953 CrossRef CAS.
- W. Liu, S. W. Lee, D. Lin, F. Shi, S. Wang, A. D. Sendek and Y. Cui, Nat. Energy, 2017, 2, 17035 CrossRef CAS.
- Z. Wan, D. Lei, W. Yang, C. Liu, K. Shi, X. Hao, L. Shen, W. Lv, B. Li, Q. H. Yang, F. Kang and Y.-B. He, Adv. Funct. Mater., 2019, 29, 1805301 CrossRef.
- G. Lancel, P. Stevens, G. Toussaint, M. Marechal, N. Krins, D. Bregiroux and C. Laberty-Robert, Langmuir, 2017, 33, 9288–9297 CrossRef CAS PubMed.
- A. L. Monaca, G. Girard, S. Savoie, H. Demers, G. Bertoni, S. Krachkovskiy, S. Marras, E. Mugnaioli, M. Gemmi, D. Benetti, A. Vijh, F. Rosei and A. Paolella, J. Mater. Chem. A, 2021, 9, 13688–13696 RSC.
- A. L. Monaca, G. Girard, S. Savoie, G. Bertoni, S. Krachkovoskiy, A. Vijh, F. Pierini, F. Rosei and A. Paolella, J. Electrochem. Soc., 2021, 168, 110512 CrossRef.
- A. L. Monaca, G. Girard, S. Savoie, R. Veillette, S. Krachkovskiy, F. Pierini, A. Vijh, F. Rosei and A. Paolella, Nanoscale, 2022, 14, 5094 RSC.
- Z. Guo, Y. Pang, S. Xia, F. Xu, J. Yang, L. Sun and S. Zheng, Adv. Sci., 2021, 8, e2100899 CrossRef.
- H. Zhai, P. Xu, M. Ning, Q. Cheng, J. Mandal and Y. Yang, Nano Lett., 2017, 17, 3182–3187 CrossRef CAS.
- X. Zhang, T. Liu, S. Zhang, X. Huang, B. Xu, Y. Lin, B. Xu, L. Li, C. W. Nan and Y. Shen, J. Am. Chem. Soc., 2017, 139, 13779–13785 CrossRef CAS.
- P. Shi, J. Ma, M. Liu, S. Guo, Y. Huang, S. Wang, L. Zhang, L. Chen, K. Yang, X. Liu, Y. Li, X. An, D. Zhang, X. Cheng, Q. Li, W. Lv, G. Zhong, Y.-B. He and F. Kang, Nat. Nanotechnol., 2023, 18, 602–610 CrossRef CAS.
- J. Li, K. Zhu, Z. Yao, G. Qian, J. Zhang, K. Yan and J. Wang, Ionics, 2019, 26, 1101–1108 CrossRef.
- S. H. Siyal, S. S. A. Shah, T. Najam, M. S. Javed, M. Imran and J.-L. Lan, ACS Appl. Energy Mater., 2021, 4, 8604–8614 CrossRef CAS.
- R. Balasubramaniam, C.-W. Nam, V. Aravindan, J.-C. Seol, K. V. Ajeya, H.-Y. Jung and Y.-S. Lee, ChemElectroChem, 2022, 9, e202200317 CrossRef CAS.
- Z. Dai, J. Yu, J. Liu, R. Liu, Q. Sun, D. Chen and F. Ciucci, J. Power Sources, 2020, 464, 228182 CrossRef CAS.
- Y. Yang, Q. Wu, D. Wang, C. Ma, Z. Chen, Q. Su, C. Zhu and C. Li, J. Membr. Sci., 2020, 612, 118424 CrossRef CAS.
- G. Yang, M. L. Lehmann, S. Zhao, B. Li, S. Ge, P.-F. Cao, F. M. Delnick, A. P. Sokolov, T. Saito and J. Nanda, Energy Storage Mater., 2021, 35, 431–442 CrossRef.
- Q. Guo, Y. Han, H. Wang, S. Xiong, Y. Li, S. Liu and K. Xie, ACS Appl. Mater. Interfaces, 2017, 9, 41837–41844 CrossRef CAS.
- L. Yang, Y. Song, H. Liu, Z. Wang, K. Yang, Q. Zhao, Y. Cui, J. Wen, W. Luo and F. Pan, Small Methods, 2020, 4, 1900751 CrossRef CAS.
- K. Liu, R. Zhang, J. Sun, M. Wu and T. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 46930–46937 CrossRef CAS.
- T.-Q. Yang, C. Wang, W.-K. Zhang, Y. Xia, Y.-P. Gan, H. Huang, X.-P. He and J. Zhang, Rare Met., 2022, 41, 1870–1879 CrossRef CAS.
- M. Jia, Z. Bi, C. Shi, N. Zhao and X. Guo, ACS Appl. Mater. Interfaces, 2020, 12, 46231–46238 CrossRef CAS PubMed.
- H. S. Shin, W. G. Ryu, M. S. Park, K. N. Jung, H. Kim and J. W. Lee, ChemSusChem, 2018, 11, 3184–3190 CrossRef CAS PubMed.
- D. Lin, P. Y. Yuen, Y. Liu, W. Liu, N. Liu, R. H. Dauskardt and Y. Cui, Adv. Mater., 2018, 30, e1802661 CrossRef.
- J. Bae, Y. Li, J. Zhang, X. Zhou, F. Zhao, Y. Shi, J. B. Goodenough and G. Yu, Angew. Chem., Int. Ed., 2018, 57, 2096–2100 CrossRef CAS.
- K. K. Fu, Y. Gong, J. Dai, A. Gong, X. Han, Y. Yao, C. Wang, Y. Wang, Y. Chen, C. Yan, Y. Li, E. D. Wachsman and L. Hu, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7094–7099 CrossRef CAS PubMed.
- G. Wang, H. Liu, Y. Liang, C. Wang and L.-Z. Fan, Energy Storage Mater., 2022, 45, 1212–1219 CrossRef.
- P. Pan, M. Zhang, Z. Cheng, L. Jiang, J. Mao, C. Ni, Q. Chen, Y. Zeng, Y. Hu and K. Fu, Energy Storage Mater., 2022, 47, 279–287 CrossRef.
- M. Zhang, P. Pan, Z. Cheng, J. Mao, L. Jiang, C. Ni, S. Park, K. Deng, Y. Hu and K. K. Fu, Nano Lett., 2021, 21, 7070–7078 CrossRef CAS.
- H. Yang, J. Bright, B. Chen, P. Zheng, X. Gao, B. Liu, S. Kasani, X. Zhang and N. Wu, J. Mater. Chem. A, 2020, 8, 7261 RSC.
- Y. Gong, K. Fu, S. Xu, J. Dai, T. R. Hamann, L. Zhang, G. T. Hitz, Z. Fu, Z. Ma, D. W. McOwen, X. Han, L. Hu and E. D. Wachsman, Mater. Today, 2018, 21, 594–601 CrossRef CAS.
- R. Li, S. Guo, L. Yu, L. Wang, D. Wu, Y. Li and X. Hu, Adv. Mater. Interfaces, 2019, 6, 1900200 CrossRef.
- S. Zekoll, C. Marriner-Edwards, A. K. O. Hekselman, J. Kasemchainan, C. Kuss, D. E. J. Armstrong, D. Cai, R. J. Wallace, F. H. Richter, J. H. J. Thijssen and P. G. Bruce, Energy Environ. Sci., 2018, 11, 185–201 RSC.
- W. Kou, J. Wang, W. Li, R. Lv, N. Peng, W. Wu and J. Wang, J. Membr. Sci., 2021, 634, 119432 CrossRef CAS.
- S. Wang and G. P. Demopoulos, Energy Storage Mater., 2024, 71, 103604 CrossRef.
- S. Wang, S. Bessette, R. Gauvin and G. P. Demopoulos, Cell Rep. Phys. Sci., 2024, 5, 102213 CrossRef CAS.
- Y. R. Zhao, C. Wu, G. Peng, X. T. Chen, X. Y. Yao, Y. Bai, F. Wu, S. J. Chen and X. X. Xu, J. Power Sources, 2016, 301, 47–53 CrossRef CAS.
- I. Villaluenga, K. H. Wujcik, W. Tong, D. Devaux, D. H. C. Wong, J. M. DeSimone and N. P. Balsara, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 52–57 CrossRef CAS.
- Blue solutions: advantages of solid-state batteries, https://www.blue-solutions.com/en/battery-technology/#advantages-of-solid-state-batteries.
- J. Schnell, F. Tietz, C. Singer, A. Hofer, N. Billot and G. Reinhart, Energy Environ. Sci., 2019, 12, 1818–1833 RSC.
- D. Hanft, J. Exner and R. Moos, J. Power Sources, 2017, 361, 61 CrossRef CAS.
- T. Kato, S. Iwasaki, Y. Ishii, M. Motoyama, W. C. West, Y. Yamamoto and Y. Iriyama, J. Power Sources, 2016, 303, 65 CrossRef CAS.
- G. T. Hitz, D. W. McOwen, L. Zhang, Z. Ma, Z. Fu, Y. Wen, Y. Gong, J. Dai, T. R. Hamann, L. Hu and E. D. Wachsman, Mater. Today, 2019, 22, 50 CrossRef CAS.
- Y. Ren, T. Liu, Y. Shen, Y. Lin and C. W. Nan, Ionics, 2017, 23(9), 2521 CrossRef CAS.
- S. Xu, D. W. McOwen, L. Zhang, G. T. Hitz, C. Wang, Z. Ma, C. Chen, W. Luo, J. Dai, Y. Kuang, E. M. Hitz, K. Fu, Y. Gong, E. D. Wachsman and L. Hu, Energy Storage Mater., 2018, 15, 458 CrossRef.
- E. Yi, W. Wang, J. Kieffer and R. M. Laine, J. Power Sources, 2017, 352, 156 CrossRef CAS.
- J. Schnell and G. Reinhart, Proc. CIRP, 2016, 57, 568 CrossRef.
- W. Xia, B. Xu, H. Duan, X. Tang, Y. Guo, H. Kang, H. Li and H. Liu, J. Am. Ceram. Soc., 2017, 100(7), 2832 CrossRef CAS.
- D. Wang, Q. Sun, J. Luo, J. Liang, Y. Sun, R. Li, K. Adair, L. Zhang, R. Yang, S. Lu, H. Huang and X. Sun, ACS Appl. Mater. Interfaces, 2019, 11(5), 4954 CrossRef CAS PubMed.
- S. Ohta, J. Seki, Y. Yagi, Y. Kihira, T. Tani and T. Asaoka, J. Power Sources, 2014, 265, 40 CrossRef CAS.
- K. Fu, Y. Gong, G. T. Hitz, D. W. McOwen, Y. Li, S. Xu, Y. Wen, L. Zhang, C. Wang, G. Pastel, J. Dai, B. Liu, H. Xie, Y. Yao, E. D. Wachsman and L. Hu, Energy Environ. Sci., 2017, 10(7), 1568 RSC.
- S. Xu, D. W. McOwen, C. Wang, L. Zhang, W. Luo, C. Chen, Y. Li, Y. Gong, J. Dai, Y. Kuang, C. Yang, T. R. Hamann, E. D. Wachsman and L. Hu, Nano Lett., 2018, 18(6), 3926 CrossRef CAS PubMed.
- A. V. Potapenko, S. I. Chernukhin, I. V. Romanova, K. S. Rabadanov, M. M. Gafurov and S. A. Kirillov, Electrochim. Acta, 2014, 134, 442–449 CrossRef CAS.
- S. Yang, J. Chen, Y. Liu and B. Yi, J. Mater. Chem. A, 2014, 2, 9322–9330 RSC.
| This journal is © The Royal Society of Chemistry 2025 |