
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural control of dynamic covalent cages: kinetic vs. thermodynamic assembly and PFAS removal from water†
Tobias Pausch a,
Pablo Martínez Mestreb,
Fabiola Zapatab,
Andreas Mixc and
Bernd M. Schmidt*a
a,
Pablo Martínez Mestreb,
Fabiola Zapatab,
Andreas Mixc and
Bernd M. Schmidt*a
aInstitut für Organische Chemie und Makromolekulare Chemie, Heinrich-Heine-Universität Düsseldorf, Universitätsstraße 1, Düsseldorf 40225, Germany. E-mail: bernd.schmidt@hhu.de
bDepartamento de Química Orgánica, Universidad de Murcia, Edificio 19, Murcia, 30100, Spain
cInstitut für Anorganische Chemie und Strukturchemie, Universität, Bielefeld, Universitätsstr. 25, Bielefeld 33615, Germany
First published on 18th June 2025
Abstract
Dynamic covalent chemistry is a powerful tool to synthesise complex structures from simple building blocks. However, even minor variations in the numerous parameters governing self-assembly can drastically influence the size and structure of the resulting assemblies. Herein, we report the selective formation of three cages belonging to the low-symmetry Tri22Tri2 cage topology for the first time, using highly symmetric tritopic building blocks, confirmed by single-crystal X-ray (SC-XRD) analysis. Fluorinated and non-fluorinated aldehydes were combined with two amines differing in their degree of structural flexibility. Applying either kinetic or thermodynamic control through solvent selection allowed for the selective synthesis of either the low-symmetry Tri22Tri2 or the larger, highly symmetric Tri4Tri4 assemblies. While the fluorinated linker strongly preferred the formation of the Tri22Tri2 cage topology under thermodynamic control, the non-fluorinated linker selectively formed the Tri4Tri4 species. Kinetic control, using methanol as a poor solvent, allowed for the selective precipitation of the Tri22Tri2 intermediate. Reduction of the Janus-like fluorinated Tri22Tri2 cages yielded the cages Et2F2red and TREN2F2red, which showed high potential for removing perfluorooctanoic acid (PFOA) from water, with Et2F2red exhibiting structural rearrangements in organic solvents to accommodate PFOA, as observed by 1H and 19F NMR titrations in combination with 19F DOSY measurements.
Introduction
Molecular self-assembly using organic building blocks, in combination with metals or in their absence, gives access to a variety of nanostructures through the organisation of tailor-made molecular building blocks that exhibit suitable intra- and intermolecular interactions.1 Organic chemistry-rooted dynamic covalent chemistry (DCC) is therefore an efficient synthetic strategy that employs multitopic precursors to form reversible covalent bonds, combining the advantages of error correction during self-assembly with stability, allowing for in-solution analysis and application of the assembled molecule.2 It has enabled the synthesis of a variety of supramolecular architectures, from macrocycles3 to cages,4 polymers,5 and covalent organic frameworks (COFs),6 with potential energy surfaces being the central concept in understanding the assembly outcome.4a,7Building blocks of similar topology, through changes in geometry,1c,8 size,1,4,8 rigidity of linkers,1,9 or even changes in substituents10 can build a multitude of accessible cage topologies.4,11 Recently, Jelfs and co-workers rationalised expected topologies (connection patterns) by analysing the geometry and topology of building blocks through calculations combined with experimentally observed structures.11b They also introduced a systematic nomenclature for describing cage topologies, denoted as XmpYn, providing clarity and avoiding confusion with bracket notation, such as [4 + 2], which is more commonly associated with pericyclic reactions in organic chemistry.
By combining ditopic (Di), tritopic (Tri), or tetratopic (Tet) building blocks, a diverse library of cage geometries can be envisioned. Although several gaps have been filled in recent years, some predicted geometries remain unobserved, with no corresponding crystal structures reported. Within the TrinTrin family (Fig. 1), Tri4Tri4 is the only geometry for which crystal structures have been commonly obtained.12 In contrast, Tri1Tri1 has mostly only been observed in solution,13 while the lower-symmetry Tri22Tri2 topology has yet to be reported. The formation of such complex, lower-symmetry structures is relatively rare compared to highly symmetric Platonic or Archimedean solid-based topologies, such as cubes14 or tetrahedra.15 Computational studies have shown that the higher symmetry of such assemblies is generally preferred from an entropic point of view,16a however, it cannot be fully disregarded that the formation of multiple smaller structures, disregarding their symmetry, should be overall more beneficial for the entropic term.11b,16b
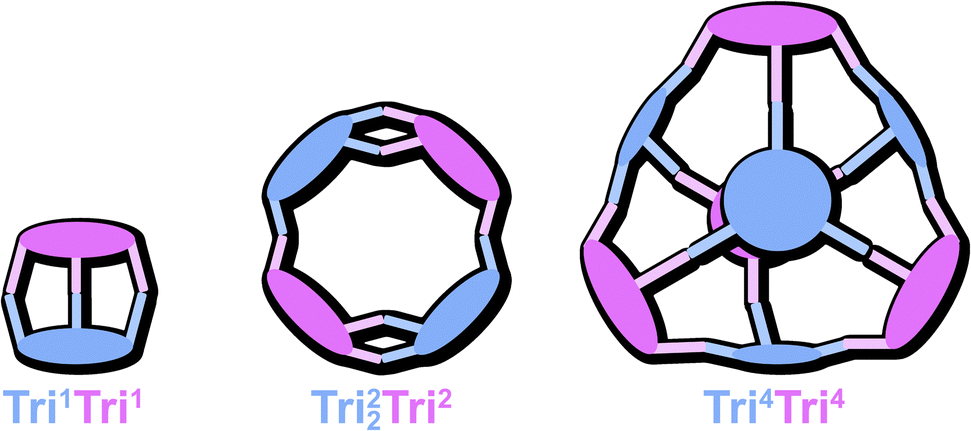 | ||
| Fig. 1 Schematic representation of the cage geometries obtained when reacting two tritopic building blocks with each other, giving different topologies. | ||
Especially in recent years, the interest in these intricate assemblies has grown due to their unique and selective host–guest chemistry.17,18 For metal–organic cages (MOCs), various strategies have been explored to achieve complex assemblies.17 One common approach involves employing multiple linkers to form heteroleptic cages, effectively disrupting the symmetry of the final structure.19 In DCC-based systems this approach is of great interest, however, the desired social self-sorting4c,20,21 is rare, and narcissistic self-sorting10,22 or statistical mixtures23 are dominating this space. Thus, lower-symmetry assemblies are typically achieved by employing less symmetric building blocks, often exhibiting an inherent chirality.18,22a,b,24 For example, He and Zhang et al. demonstrated that the use of C2 and C2v building blocks leads to the formation of a C2-symmetric imine cage of the unusual Tet44Di8 topology.24b
Beyond linker design, reaction conditions can significantly influence the assembly process and resulting topology. Even if using the same starting materials, solvent choice25 and/or the concentration26 used can drastically shift the equilibrium towards different topologies by either enhancing or suppressing inter-/intramolecular interactions, respectively.26a
These examples, however, predominantly focus on the formation of the thermodynamic product. While many examples support the thermodynamically controlled formation of imine cages, some observations suggest that they may instead be kinetically controlled products, especially when precipitating from the reaction mixture.12d,25b,27,28 This can be rationalised by considering the potential energy surface of the system, where the cage structure may correspond to a kinetically trapped state rather than the global thermodynamic minimum, and cage formation can be driven by precipitation, preventing further equilibration towards more stable assemblies, highlighting the complexity of controlling self-assembly.
Results and discussion
Synthesis and characterisation of the cages
Herein, we report the synthesis of three novel cages of the unique Tri22Tri2 geometry, including topological assembly control and the complexation of per- and polyfluoroalkyl substances (PFAS) in organic media and their removal from aqueous solutions. For that purpose, we chose the flexible aldehyde F, offering the combination of an electron-rich core and electron-deficient fluorinated panels, aiming for a Janus-type nanocavity to facilitate the intermolecular host–guest interactions.12c Stirring (2,4,6-triethylbenzene-1,3,5-triyl)trimethanamine (Et) with the flexible F in an equimolar ratio for 3 days in methanol at room temperature resulted in the precipitation of a colourless solid. To our surprise, both the 1H and 19F NMR of a redissolved sample showed a complex set of signals (Fig. 2c), whereas, on the contrary, MALDI-MS only showed a singular signal belonging to a condensation of two F with two Et building blocks. The lower C2h symmetry of the obtained cage Et2F2 results in a splitting of the observed resonance signals, showing a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio for all signals, which is in good accordance with the ratio expected for a cage of Tri22Tri2 geometry. This unrepresented cage topology distinguishes itself through its lowered symmetry as a result of the two double connections found within its structure (Fig. 2a).11b The resulting inherent strain and rigidity of the assembly cause a diastereotopic splitting of the protons HE and HF belonging to the double-connected amine motif (Fig. 2c).
:1 ratio for all signals, which is in good accordance with the ratio expected for a cage of Tri22Tri2 geometry. This unrepresented cage topology distinguishes itself through its lowered symmetry as a result of the two double connections found within its structure (Fig. 2a).11b The resulting inherent strain and rigidity of the assembly cause a diastereotopic splitting of the protons HE and HF belonging to the double-connected amine motif (Fig. 2c).
 | ||
| Fig. 2 (a) Synthetic route towards imine cage Et2F2 and the respective amine cage Et2F2red; (b) the SC-XRD structure of Et2F2red with thermal ellipsoids set at 50% probability, hydrogens and solvents were omitted for clarity; (c) 1H and 19F NMR (600 MHz/282 MHz, CDCl3, 298 K) spectra of the imine cage Et2F2 showing the splitting of all signals due to the reduced symmetry. | ||
Intrigued by these results, we investigated different conditions for the cage formation, aiming for either thermodynamic or kinetic control over the assemblies. For that purpose, we chose chloroform as solvent, heating to 60 °C to allow all intermediates to remain in solution, over time reaching the thermodynamic equilibrium. For experiments under kinetic conditions, we investigated the two most common “poor solvents” (lower solubility of intermediates and products), acetonitrile and methanol, to induce precipitation of possible intermediate structures. The respective mixtures were stirred at room temperature upon dropwise addition of a solution of the respective amine.27
Using acetonitrile as the solvent, stirring at room temperature for 3 days again selectively led to the formation of the Tri22Tri2 species Et2F2 precipitating from the reaction solution. Stirring Et and F in chloroform at 60 °C for three days resulted in the formation of a second, more symmetric species, without any precipitation occurring. MALDI-MS of the obtained mixture revealed the formation of the highly symmetric (Td) Tri4Tri4 topology alongside Et2F2 (see Fig. S1 and S2†). In an attempt to isolate the observed different cage topologies, the dynamic covalent imines were reduced by in situ reaction with sodium borohydride.29 The respective amine cage Et2F2red could be isolated in 37% yield from the one-pot two-step reaction of Et and F or in 94% yield from Et2F2. Again, HRMS (ESI†) and NMR analysis confirmed the formation of Et2F2red, whereas the larger Et4F4red structure could not be isolated. Et2F2red readily crystallised from a chloroform solution, and the obtained crystals were subjected to single-crystal X-ray analysis (SC-XRD), unambiguously confirming the anticipated Tri22Tri2 structure (Fig. 2b). As a result of the two double connections between two singular F and Et motifs, the amine cage is flattened overall, resembling a double-walled macrocycle of C2h symmetry, with two different angles for Ar–O–ArF bonds to accommodate the inherent strain expected with less flexible building blocks.11b One ethyl group of the Et motif is located between the fluorinated panels of each double connection. The same is true for the analogous hydrogen of the phloroglucinol motif, explaining the strong upfield shifts observed in 1H NMR (Fig. S123,† and 4b, Hf, Hg). Meanwhile, all free electron pairs of the amine nitrogen are pointing inwards, forming two main cavities where residual chloroform solvent molecules are located. Overall, even with the increased flexibility of the amine bonds compared to the more rigid imine bonds, the structure still appears potentially strained.
Thus, in addition to Et, the flexible tris(2-aminoethyl)amine (TREN) was investigated in combination with aldehyde F, anticipating that under thermodynamic control, enrichment of the geometry encoded in the linker would be observed, as the rigidity is reduced.
Interestingly, again the Tri22Tri2 topology was favoured over the larger Tri4Tri4 species (see Table S1†). Heating the reaction mixture in chloroform led to the almost exclusive formation of TREN2F2 over TREN4F4 (96:4). In contrast, precipitation from methanol or acetonitrile reaction mixtures favoured the formation of the Tri4Tri4 species, likely due to the increased solubility of the cages and their intermediates compared to the Et-based counterparts.27 This is supported by the observation that in acetonitrile solution, only TREN4F4 was found in the precipitate, whereas the filtrate contains a mixture of cage topologies similar to those observed in chloroform (acetonitrile filtrate TREN2F2:TREN4F4 ratio 94:6). Overall, these findings indicate that aldehyde F preferentially directs the formation of the Tri22Tri2 cage topology. Under thermodynamic control, this is less pronounced when employing the more rigid amine Et in comparison to TREN, with kinetic conditions exclusively leading to the formation of Et2F2 alongside insoluble oligomeric species typical for these conditions.12d The inverse behaviour of the TREN-based cages is likely a result of two factors; (a) their increased solubility, allowing for a higher proportion of Tri4Tri4 species to be formed before precipitation occurs, and (b) the distance between the fluorobenzene motifs, which is influenced by the rigidity of the amine building block.
We rationalised that the Tri22Tri2 topology is directed through small intramolecular interactions between the fluorinated benzene motifs leading to a preorganisation,30 where two panels are near to each other in solution leading to a preferred formation of a double connection between one Et molecule and two aldehyde groups of a singular F molecule. This is in line with previous observations where we found that using fluorinated aldehydes can result in the formation of the unusual Tri6Di9 topology alongside the otherwise strongly favoured, well-known Tri4Di6 topology.31 Additionally, the preferred formation of TREN2F2 over TREN4F4 suggests that these interactions are (a) lessened with the more preorganised and less flexible amine Et and (b) play a significant role in the stabilisation of the newly obtained Tri22Tri2 cage topology.
Therefore, we also investigated the non-fluorinated aldehyde derivative H, expecting no significant interactions between its panels. Aldehyde H was readily prepared in a two-step procedure starting from phloroglucinol (see the ESI†). To our delight, the initial screenings under either thermodynamic (in chloroform at 60 °C) or kinetic control (acetonitrile or methanol at room temperature) revealed that the highly soluble and flexible amine TREN exclusively formed the Tri4Tri4 geometry (Fig. 3). The respective imine cage TREN4H4 could be isolated from chloroform in quantitative yield (Fig. 3a). As expected for reactions under kinetic control, TREN4H4 precipitated from the reaction mixture along with insoluble by-products. Similarly, the Et-based cage (Et4H4) formed quantitatively under thermodynamic control. In contrast, NMR and MALDI-MS analyses of the precipitate from methanol revealed the clean formation of the lower symmetry cage Et2H2 of Tri22Tri2 topology (Fig. 3c). Upon extraction from the insoluble by-products, the lower symmetry cage could be isolated in 38% yield.
 | ||
| Fig. 3 (a) Synthesis of the non-fluorinated cages under either thermodynamic or kinetic control with a schematic representation of the Tri22Tri2 and Tri4Tri4 cage geometries and the respective 1H NMR (600 MHz, CDCl3, 298 K) spectra of (b) Et4H4, (c) Et2H2, and (d) TREN4H4. The signal assignments are shown in (a). | ||
Monitoring a redissolved sample of Et2H2 in CDCl3 (2.3 mM, consistent with the concentration used for cage synthesis) at either 60 °C or at room temperature resulted in the appearance of a new set of signals. After one day, noticeable amounts of Et4H4 had formed, whereas at room temperature only marginal amounts of the Tri4Tri4 species Et4H4 were observed. Over the course of seven days, the Tri4Tri4 species became increasingly enriched, and after 24 days, the complete conversion to Et4H4 was observed under both conditions. At 60 °C, this initial transformation proceeded notably quicker, reaching a 1:1 cage ratio already after five days, while at room temperature this was reached only after about nine days (Fig. S14–S16†). Similarly, when stirring Et2H2 in CDCl3 for 3 days at 60 °C, an almost complete cage-to-cage transformation towards Et4H4 was observed. At room temperature, only small amounts (∼34%) of Et4H4 were formed. Suspending Et4H4 in methanol and stirring for three days, even at 60 °C, did not result in any observable interconversion (Fig. S13†).
This underlines the bias towards the larger, highly symmetric, and less internally strained Tri4Tri4 topology over the low-symmetry Tri22Tri2 topology in solution,11b strongly suggesting that Et2H2 is an intermediate structure formed on the pathway towards Et4H4. To rule out that reaction temperature plays a role in this observation, we reacted Et with H in chloroform at room temperature, again selectively forming Et4H4, while stirring in methanol, even at 60 °C, leading to Et2H2 as the singular species, strongly suggesting that the solubility of the intermediates is the discriminating factor.
To test this, during cage formation studies, the chloroform content was fixed at 10% to fully dissolve the starting materials, while the acetonitrile concentration was varied. For a methanol/chloroform mixture (90:10), Et2H2 almost exclusively precipitated from the solution. As the acetonitrile content increased, the proportion of Et4H4 in the precipitate also increased, until at 40% acetonitrile content, only Et4H4 precipitated from the reaction solution (see Fig. S10 and S11†). This trend aligns with the observed solubility differences of aldehyde H, which is poorly soluble in methanol but highly soluble in acetonitrile. Additionally, the faster precipitation observed in mixtures with lower acetonitrile content (Fig. S12†) suggests that (a) the solubility of intermediates in the respective solvents can be roughly estimated from the solubility of the employed building blocks and (b) the primary factor driving the formation of Et2H2 is precipitation.25b These findings highlight that, under kinetic control, a mixture of species might precipitate, but adjusting the solvent composition can dramatically shift the equilibrium. Thus, to achieve a desired outcome, the solubility of the building blocks should be carefully considered when selecting the solvent for self-assembly reactions.
PFOA removal
With the understanding of the dynamic covalent chemistry behind the formation of the Tri22Tri2 cage topology, we shifted our focus back to the fluorinated Tri22Tri2 cages. As previously shown, the imine cages could be easily reduced in situ to the respective amine cages Et2F2red and TREN2F2red with sodium borohydride. Additionally, Et2F2red with the electron-rich cores with fluorinated, electron-deficient panels, featuring exposed amine groups, was investigated for the uptake of perfluorinated octanoic acid (PFOA) and trifluoroacetic acid (TFA). Per- and polyfluoroalkyl substances (PFAS), or “forever chemicals,” are synthetic compounds used in industry for their thermal stability and resistance to degradation. Found in coatings, foams, and textiles, they persist in the environment, accumulate in organisms, and pose serious health risks.32 Despite restrictions, PFAS contamination remains a major global challenge due to their removal complexity.33 Supramolecular chemistry approaches have been employed to tackle the complex challenge of sieving PFAS molecules, which exhibit both hydrophilic and lipophilic behaviour, such as leveraging the rich host–guest chemistry of supramolecular compounds34 including cyclodextrin derivatives,35d water-soluble iron-35e or palladium-based,35a and insoluble Zr-based35b MOCs. Recently, Sessler and Chi et al. used a water-insoluble fluorinated amine-based cage feasible for the removal of PFOA from aqueous solutions through the reduction of the respective dynamic covalent imine cage.35c Contrary to the binding of neutral compounds, e.g., perfluorocarbons (PFCs), which is primarily governed by hydrophobic effects and π-stacking within nonpolar, shape-complementary cavities,36 the binding of PFAS typically relies on an interplay of electrostatic and fluorophilic interactions by using the polar headgroup as an anchor.35c–eFirst, we performed NMR titrations in organic media (chloroform/methanol mixture 95:5) at a concentration of 2.5 mM, investigating the cage's ability to capture PFOA and to study present interactions. Additionally, the symmetric “open-cavity” model compound Bn3F1red was prepared for the comparison with the “closed-cavity” cages. Bn3F1red was synthesised by condensation of aldehyde F with benzylamine, followed by in situ reduction with sodium borohydride.
Upon addition of the cages to a PFOA solution, shifts for all PFOA signals could be observed in 19F NMR spectra. The CF2-group (Fa) neighbouring the carboxylic acid exhibited the most significant shift of 1.6–1.8 ppm, while the other signals, even including the terminal CF3-group (Fg), were roughly shifted by −0.2 ppm (see ESI†). For the symmetric model compound Bn3F1red, however, only marginal shifts of (<0.02 ppm) for all signals besides Fa were noticeable. The addition of PFOA to the respective hosts led to a shifting of all host signals in 1H and 19F NMR, with the CH2–NH–CH2 motif expressing the strongest shifts in 1H NMR, while other signals were only slightly shifted (<0.05 ppm) in the case of TREN2F2red and Bn3F1red. Et2F2red showed noticeable downfield shifts for almost all signals. Most interestingly, the protons Hf and Hg were strongly downfield shifted by 0.78 ppm and 0.48 ppm, respectively, until almost overlaying with the other ethyl groups (HF and HG) of the Et motif. Simultaneously, both phloroglucinol protons HD and Hd shifted upfield, indicating that Et2F2red undergoes a structural rearrangement. Upon addition of PFOA, the singular ethyl group of the Et located between the double-connected fluorinated benzenes of F is supposedly pushed outwards, away from the fluorinated panels, being deshielded in the process. This assumption is supported by 1H–1H NOESY NMR data, which reveal that upon the addition of PFOA, the previously isolated ethyl group exhibits a new correlation pattern similar to that observed for the other ethyl groups (Fig. S75†). A similar structural change can be observed in the solid-state. When comparing the crystal structures of PFOA@Et2F2red (grown from a dichloromethane/acetonitrile solution) and Et2F2red (Fig. S17–S19†). PFOA@Et2F2red shows a more elongated cage structure, where the ethyl groups are all pointing away from the fluorobenzenes and two PFOA molecules are located outside of the cage (Fig. 4a). The addition of a strong acid like trifluoroacetic acid did not result in a significant shifting of these signals, instead, a broadening of the signals is observed (Fig. S50 and S51†). When titrating octanoic acid, of comparable structure and with a pKa of 3.8 ± 0.1 (vs. 2.2 ± 0.2)37 no shifting of the cage signals can be observed (Fig. S45 and S46†), rendering the observed structural rearrangement of Et2F2red to be selective towards PFOA, not being a result of simple protonation. 19F DOSY experiments unambiguously confirmed the formation of a complex between PFOA and Et2F2red in organic media, showing almost identical diffusion coefficients (3.68 × 10−10 m2 s−1 for PFOA and 3.81 × 10−10 m2 s−1 for Et2F2red) corresponding well to the one observed for pure cage (3.64 × 10−10 m2 s−1, see Table S3†).
 | ||
| Fig. 4 (a) Schematic illustration of the PFOA-induced structural rearrangement in Et2F2red, where one ethyl group (pink square) of the Et motif is pushed away from the two neighbouring fluorobenzenes of the F motif; (b) stacked 1H NMR spectra (300 MHz, CDCl3/MeOD, 95:5, 2.5 mM, 298 K), highlighting the chemical shifts in Et2F2red upon the addition of PFOA, signal assignment analogous to Fig. 2a; (c) stacked 19F NMR spectra (282 MHz, CDCl3/MeOD, 95:5, 2.5 mM, 298 K), highlighting the chemical shifts of PFOA upon the addition of Et2F2red; (d) stacked 19F NMR spectra (565 MHz, 128 scans, D2O, 298 K) showing the removal of PFOA from an aqueous solution using Et2F2red as a heterogenous adsorbent. | ||
The observed 19F NMR shifts suggest that PFOA binding in organic media arises from a combination of electrostatic and fluorophilic interactions. Binding is primarily driven by electrostatic attraction between the carboxylic acid group of PFOA and the protonated, quaternary amines of the hosts. However, only the cages show additional interactions, as indicated by pronounced shifts of the PFOA signals, particularly of the terminal CF3 group. This points to a binding mode in which the perfluoroalkyl chain remains largely outside the cage, while the CF3 group partially inserts into the cage windows and interacts with the fluorinated panels. This interpretation is in good accordance with the preliminary SC-XRD data. Overall, this fluorophilic interaction appears to be weak and of dynamic nature, with the apparent encapsulation resulting mainly from spatial proximity rather than strong inclusion (see the ESI† for a detailed discussion). These findings are consistent with studies on COFs,38 porous polymers39 and macrocycle-based hydrogels,40 where quaternisation of amine groups enhances PFAS uptake from aqueous solutions through an interplay of electrostatic and hydrophobic effects.
Encouraged by these promising observations, we investigated the ability of these highly hydrophobic cages to remove PFOA from aqueous solution. A 1 mg mL−1 solution of PFOA in deionised water was prepared, to which 1 equivalent of the completely insoluble cages was added. After stirring the colourless suspension for one hour, the mixture was filtered through a syringe filter, and the clear filtrate was analysed by 19F NMR. To our delight, 1 equivalent of the cages already removed approximately 80% of the initial PFOA amount. Upon addition of 5 equivalents, no clear signals corresponding to PFOA or the cage were detected, indicating an almost complete removal (Fig. 4d). These results highlight the potential of these cages as heterogeneous, low-molecular-weight adsorbent materials for PFOA removal.
Conclusions
In conclusion, we were able to gain access to an unrepresented cage geometry of the Tri22Tri2 topology using only highly symmetric building blocks. This was accomplished by leveraging either thermodynamic or kinetic control over the self-assembly process. As building blocks, a fluorinated and a non-fluorinated aldehyde alongside one of two amines with differing degrees of preorganisation, flexibility, and solubility were used.Investigations revealed that the fluorinated linker favoured the Tri22Tri2 cage topology under thermodynamic control, whereas the non-fluorinated linker H exclusively formed the larger symmetric Tri4Tri4 derivatives under these conditions. Applying kinetic control allowed for the selective formation of the low-symmetry Tri22Tri2 cages that precipitated from the reaction mixtures. Studies conducted strongly suggest that the flexibility of the building blocks plays a crucial role, enabling the formation of the Tri22Tri2 species as an intermediate towards larger structures. This understanding allowed us to use solvent selection to direct the assembly pathway, enabling the formation of either the intermediate low-symmetry Tri22Tri2 cages under kinetic control or the larger high-symmetry Tri4Tri4 structures under thermodynamic control.
Additionally, the reduction of the fluorinated Tri22Tri2 cages led to the low-symmetry Janus-like cages Et2F2red and TREN2F2red, which demonstrated promising potential for the removal of PFOA from aqueous solutions. Even in organic solvents, Et2F2red indicated interactions selectively with PFOA, undergoing a structural rearrangement to accommodate PFOA.
Our findings highlight the delicate aspects of self-assembly pathways in directing cage assembly and provide new insights into the use of fluorinated and non-fluorinated linkers to tailor structural outcomes. This approach expands the toolbox of supramolecular chemists, offering a new route to design cage-like compounds with enhanced structural complexity, paving the way for enzyme-like complex host–guest structures and advanced lightweight functional materials.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for Et2F2red (2430764) has been deposited in the joint Cambridge Crystallographic Data Centre (CCDC) and Fachinformations-zentrum Karlsruhe Access Structures service, available free of charge.Author contributions
The study was conceptualised by T. P. and B. M. S. The majority of experiments were conducted by T. P., including the synthesis and characterisation, assisted by P. M. M. P. M. M. and F. Z. performed the titration experiments. A. M. was responsible for 19F DOSY NMR measurements and data analysis, B. M. S. conducted SC-XRD data acquisition and refinement. The first draft of the manuscript, review, editing, and data presentation was executed by T. P. and B. M. S. All authors approved the final submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Jona Voss, MSc, for help preparing 19F DOSY NMR samples. T. P. is grateful for a PhD fellowship from the Evangelisches Studienwerk Villigst. B. M. S. acknowledges the support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) SCHM 3101/6. F. Z. and P. M. M. are thankful towards the Ministerio de Ciencia e Innovación of Spain, FEDER project PID2020-113483GB-I00 and the Fundación Séneca Región de Murcia (CARM) Project 21956/PI/22. P. M. M. is very thankful for a mobility aid for beneficiaries of the pre-doctoral contract programme of the Plan de Fomento de la Investigación, according to resolution R.-606/2021, financed by the University of Murcia.Notes and references
- (a) C. J. T. Cox, J. Hale, P. Molinska and J. E. M. Lewis, Chem. Soc. Rev., 2024, 53, 10380–10408 RSC; (b) F. B. L. Cougnon, A. R. Stefankiewicz and S. Ulrich, Chem. Sci., 2024, 15, 879–895 RSC; (c) S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853–908 CrossRef CAS PubMed.
- M. E. Belowicha and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
- J. Yu, D. Qi and J. Li, Commun. Chem., 2020, 3, 189 CrossRef CAS PubMed.
- (a) S. Ivanova and F. Beuerle, Isr. J. Chem., 2024, 64, e202400025 CrossRef; (b) T. Kunde, T. Pausch and B. M. Schmidt, Eur. J. Org Chem., 2021, 27, 5844–5856 CrossRef; (c) K. Acharyya and P. S. Mukherjee, Angew. Chem., Int. Ed., 2019, 58, 8640–8653 CrossRef CAS PubMed; (d) T. Hasell and A. I. Cooper, Nat. Rev. Mater., 2016, 1, 16053 CrossRef CAS; (e) M. Mastalerz, Angew. Chem., Int. Ed., 2010, 49, 5042–5053 CrossRef CAS PubMed.
- N. Zheng, Y. Xu, Q. Zhao and T. Xie, Chem. Rev., 2021, 121, 1716–1745 CrossRef CAS PubMed.
- F. Beuerle and B. Gole, Angew. Chem., Int. Ed., 2018, 57, 4850–4878 CrossRef CAS PubMed.
- (a) A. J. Greenlee, C. I. Wendell, M. M. Cencer, S. D. Laffoon and J. S. Moore, Chem, 2020, 2, 1043–1051 CAS; (b) Q. Ji, R. C. Lirag and O. Š. Miljanić, Chem. Soc. Rev., 2014, 43, 1873–1884 RSC.
- B. Olenyuk, A. Fechtenkötter and P. J. Stang, J. Chem. Soc., Dalton Trans., 1998, 1707–1728 RSC.
- E. M. King, M. A. Gebbie and N. A. Melosh, Langmuir, 2019, 35, 16062–16069 CrossRef CAS PubMed.
- Q. Chen, Z. Li, Y. Lei, Y. Chen, H. Tang, G. Wu, B. Sun, Y. Wei, T. Jiao, S. Zhang, F. Huang, L. Wang and H. Li, Nat. Commun., 2023, 14, 4627 CrossRef CAS PubMed.
- (a) A. Tarzia, E. H. Wolpert, K. E. Jelfs and G. M. Pavan, Chem. Sci., 2023, 14, 12506–12517 RSC; (b) V. Santolini, M. Miklitz, E. Berardo and K. E. Jelfs, Nanoscale, 2017, 9, 5280–5298 RSC.
- (a) T. David, R. Oestreich, T. Pausch, Y. Wada, T. Fleck-Kunde, M. Kawano, C. Janiak and B. M. Schmidt, Chem. Commun., 2024, 60, 14762–14765 RSC; (b) K. Tian, X. Wang, M. P. Schuldt, S. M. Elbert, F. Rominger and M. Mastalerz, Org. Mater., 2023, 5, 91–97 CrossRef CAS; (c) T. Kunde, E. Nieland, H. V. Schröder, C. A. Schalley and B. M. Schmidt, Chem. Commun., 2020, 56, 4761–4764 RSC; (d) J. C. Lauer, W.-S. Zhang, F. Rominger, R. R. Schröder and M. Mastalerz, Chem.–Eur. J., 2018, 24, 1816–1820 CrossRef CAS PubMed; (e) S. M. Elbert, F. Rominger and M. Mastalerz, Chem.–Eur. J., 2014, 20, 16707–16720 CrossRef CAS PubMed.
- (a) V. W. L. Gunawardana, T. J. Finnegan, C. E. Ward, C. E. Moore and J. D. Badjić, Angew. Chem., Int. Ed., 2022, 61, e202207418 CrossRef PubMed; (b) Y. Wu, C. Zhang, S. Fang, D. Zhu, Y. Chen, C. Ge, H. Tang and H. Li, Angew. Chem., Int. Ed., 2022, 61, e202209078 CrossRef CAS PubMed; (c) H. Wang, S. Fang, G. Wu, Y. Lei, Q. Chen, H. Wang, Y. Wu, C. Lin, X. Hong, S. K. Kim, J. L. Sessler and H. Li, J. Am. Chem. Soc., 2020, 142, 20182–20190 CrossRef CAS PubMed; (d) K. Kataoka, T. D. James and Y. Kubo, J. Am. Chem. Soc., 2007, 129, 15126–15127 CrossRef CAS PubMed.
- (a) S. Ivanova, E. Köster, J. J. Holstein, N. Keller, G. H. Clever, T. Bein and F. Beuerle, Angew. Chem., Int. Ed., 2021, 60, 17455–17463 CrossRef CAS PubMed; (b) S. Klotzbach, T. Scherpf and F. Beuerle, Chem. Commun., 2014, 50, 12454–12457 RSC; (c) P. Wagner, F. Rominger, W.-S. Zhang, J. H. Gross, S. M. Elbert, R. R. Schröder and M. Mastalerz, Angew. Chem., Int. Ed., 2021, 60, 8896–8904 CrossRef CAS PubMed; (d) P. H. Kirchner, L. Schramm, S. Ivanova, K. Shoyama, F. Würthner and F. Beuerle, J. Am. Chem. Soc., 2024, 146, 5305–5315 CrossRef CAS PubMed.
- (a) S. Klotzbach and F. Beuerle, Angew. Chem., Int. Ed., 2015, 54, 10356–10360 CrossRef CAS PubMed; (b) M. Mastalerz, M. W. Schneider, I. M. Oppel and O. Presly, Angew. Chem., Int. Ed., 2011, 50, 1046–1051 CrossRef CAS PubMed; (c) T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS PubMed.
- (a) P. Skowronek, B. Warzajtis, U. Rychlewska and J. Gawroński, Chem. Commun., 2013, 49, 2524–2526 RSC; (b) R. R. Julian, S. Myung and D. E. Clemmer, J. Phys. Chem., 2005, 109, 440–444 CrossRef CAS PubMed.
- (a) C. T. McTernan, J. A. Davies and J. R. Nitschke, Chem. Rev., 2022, 122, 10393–10437 CrossRef CAS PubMed; (b) S. Pullen, J. Tessarolo and G. H. Clever, Chem. Sci., 2021, 12, 7269–7293 RSC.
- (a) C. Chen and S. Zhang, Acc. Chem. Res., 2025, 58, 583–598 CrossRef CAS PubMed; (b) A.-D. Manick, J.-P. Dutasta and A. Martinez, ChemPlusChem, 2023, 88, e202300291 CrossRef CAS PubMed.
- (a) E. Benchimol, A. Rivoli, M. Kabiri, B. Zhang, J. J. Holstein, P. Ballester and G. H. Clever, J. Am. Chem. Soc., 2025, 147, 3823–3829 CrossRef CAS PubMed; (b) K. E. Ebbert, F. Sendzik, L. Neukirch, L. Eberlein, A. Platzek, P. Kibies, S. M. Kast and G. H. Clever, Angew. Chem., Int. Ed., 2025, 64, e202416076 CrossRef CAS PubMed; (c) B. Zhang, H. Lee, J. J. Holstein and G. H. Clever, Angew. Chem., Int. Ed., 2024, 63, e202404682 CrossRef CAS PubMed; (d) K. Wu, E. B. A. Baksi and G. H. Clever, Nat. Chem., 2024, 16, 584–591 CrossRef CAS PubMed.
- F. Beuerle, S. Klotzbach and A. Dhara, Synlett, 2016, 27, 1133–1138 CrossRef CAS.
- (a) S. Klotzbach and F. Beuerle, Angew. Chem., Int. Ed., 2015, 54, 10356–10360 CrossRef CAS PubMed; (b) V. Abet, F. T. Szczypiński, M. A. Little, V. Santolini, C. D. Jones, R. Evans, C. Wilson, X. Wu, M. F. Thorne, M. J. Bennison, P. Cui, A. I. Cooper, K. E. Jelfs and A. G. Slater, Angew. Chem., Int. Ed., 2020, 59, 16755–16763 CrossRef CAS PubMed.
- (a) N. Schäfer, L. Glanz, A. Lützen and F. Beuerle, Org. Chem. Front., 2025, 12, 1763–1771 RSC; (b) D. Beaudoin, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2017, 56, 1244–1248 CrossRef CAS PubMed; (c) K. Acharyya and P. S. Mukherjee, Chem.–Eur. J., 2014, 20, 1646–1657 CrossRef CAS PubMed; (d) K. Acharyya, S. Mukherjee and P. S. Mukherjee, J. Am. Chem. Soc., 2013, 135, 554–557 CrossRef CAS PubMed.
- (a) T. Kunde, T. Pausch and B. M. Schmidt, Chem.–Eur. J., 2021, 27, 8457–8460 CrossRef CAS PubMed; (b) S. Jiang, J. Jones, T. Hasell, C. E. Blythe, D. J. Adams, A. Trewin and A. I. Cooper, Nat. Commun., 2011, 2, 207 CrossRef PubMed.
- (a) F. Qiu, X. Zhang, W. Wang, K. Su and D. Yuan, J. Am. Chem. Soc., 2025, 147, 8500–8512 CrossRef CAS PubMed; (b) L. Zhang, Y. Jin, G.-H. Tao, Y. Gong, Y. Hu, L. He and W. Zhang, Angew. Chem., Int. Ed., 2020, 59, 20846–20851 CrossRef CAS PubMed; (c) X. Wang, Y. Wang, H. Yang, H. Fang, R. Chen, Y. Sun, N. Zheng, K. Tan, X. Lu, Z. Tian and X. Cao, Nat. Commun., 2016, 7, 12469 CrossRef PubMed; (d) H. Qu, X. Tang, X. Wang, Z. Li, Z. Huang, H. Zhang, Z. Tian and X. Cao, Chem. Sci., 2018, 9, 8814–8818 RSC.
- (a) F. Wang, X. Shi, Y. Zhang, W. Zhou, A. Li, Y. Liu, J. L. Sessler and Q. He, J. Am. Chem. Soc., 2023, 145, 10943–10947 CrossRef CAS PubMed; (b) G.-F. Feng, J. Geng, F.-D. Feng and W. Huang, Sci. Rep., 2020, 10, 4712 CrossRef CAS PubMed; (c) X. Liu and R. Warmuth, J. Am. Chem. Soc., 2006, 128, 14120–14127 CrossRef CAS PubMed.
- (a) C. Ge, Z. Cao, T. Feng, Y. Wu, M. Xiao, H. Tang, K. Wang, L. Wang and H. Li, Angew. Chem., Int. Ed., 2024, 63, e202411401 CrossRef CAS PubMed; (b) N. Cao, Y. Wang, X. Zheng, T. Jiao and H. Li, Org. Lett., 2018, 20, 7447–7450 CrossRef CAS PubMed.
- T. H. G. Schick, F. Rominger and M. Mastalerz, J. Org. Chem., 2020, 85, 13757–13771 CrossRef CAS PubMed.
- S. Fisher, H.-H. Huang, L. Sokoliuk, A. Prescimone, O. Fuhr and T. Šolomek, J. Org. Chem., 2025, 90, 4158–4166 CrossRef CAS PubMed.
- T. Pausch, T. David, T. Fleck-Kunde, H. Pols, J. Gurke and B. M. Schmidt, Angew. Chem., Int. Ed., 2024, 63, e202318362 CrossRef CAS PubMed.
- (a) Z. Zhang and O. Š. Miljanić, Org. Mat., 2019, 1, 19–29 CAS; (b) M. I. Hashim, H. T. M. Le, T.-H. Chen, Y.-S. Chen, O. Daugulis, C.-W. Hsu, A. J. Jacobson, W. Kaveevivitchai, X. Liang, T. Makarenko, O. Š. Miljanić, I. Popovs, H. V. Tran, X. Wang, C.-H. Wu and J. I. Wu, J. Am. Chem. Soc., 2018, 140, 6014–6026 CrossRef CAS PubMed; (c) T.-H. Chen, I. Popov, W. Kaveevivitchai, Y.-C. Chuang, Y.-S. Chen, O. Daugulis, A. J. Jacobson and O. Š. Miljanić, Nat. Commun., 2014, 5, 5131 CrossRef CAS PubMed.
- T. Fleck-Kunde, E. H. Wolpert, L. zur Horst, R. Oestreich, C. Janiak, K. E. Jelfs and B. M. Schmidt, Org. Mater., 2022, 4, 255–260 CrossRef CAS.
- (a) N. Sharma, V. Kumar, V. Sugumar, M. Umesh, S. Sondhi, P. Chakraborty, K. Kaur, J. Thomas, C. Kamaraj and S. S. Maitra, Case Stud. Chem. Environ. Eng., 2024, 9, 100623 CrossRef CAS; (b) A. Leeson, T. Thompson, H. F. Stroo, R. H. Anderson, J. Speicher, M. A. Mills, J. Willey, C. Coyle, R. Ghosh, C. Lebrón and C. Patton, Environ. Toxicol. Chem., 2021, 40, 24–36 CrossRef CAS PubMed.
- A. M. Calafat, L.-Y. Wong, Z. Kuklenyik, J. A. Reidy and L. L. Needham, Environ. Health Perspect., 2007, 115, 1596–1602 CrossRef CAS PubMed.
- (a) G. Zhang, W. Lin, F. Huang, J. Sessler and N. M. Khashab, J. Am. Chem. Soc., 2023, 145, 19143–19163 CrossRef CAS PubMed; (b) S. Ohtani, K. Kato, S. Fa and T. Ogoshi, Coord. Chem. Rev., 2022, 462, 214503 CrossRef CAS.
- (a) M. D. Bairagya, P. S. Ntipouna, N. K. Stewart and N. Elgrishi, Chem. Commun., 2024, 60, 11084–11087 RSC; (b) D. Camdzic, H. K. Welgama, M. R. Crawley, A. Avasthi, T. R. Cook and D. S. Aga, ACS Appl. Eng. Mater., 2024, 2, 87–95 CrossRef CAS; (c) Y. He, J. Zhou, Y. Li, Y.-D. Yang, J. L. Sessler and X. Chi, J. Am. Chem. Soc., 2024, 146, 6225–6230 CrossRef CAS PubMed; (d) Z. Chen, Y.-L. Lu, L. Wang, J. Xu, J. Zhang, X. Xu, P. Cheng, S. Yang and W. Shi, J. Am. Chem. Soc., 2023, 145, 260–267 CrossRef CAS PubMed; (e) C. R. P. Fulong, M. G. E. Guardian, D. S. Aga and T. R. Cook, Inorg. Chem., 2020, 59, 6697–6708 CrossRef CAS PubMed.
- U. Kai, R. Sumida, Y. Tanaka and M. Yoshizawa, J. Am. Chem. Soc., 2025, 147, 10640–10646 CrossRef CAS PubMed.
- L. J. Musegades, O. P. Curtin and J. D. Cyran, J. Phys. Chem. C, 2024, 128, 1946–1951 CrossRef CAS PubMed.
- (a) W. Wang, Z. Zhou, H. Shao, S. Zhou, G. Yu and S. Deng, Chem. Eng. J., 2021, 412, 127509 CrossRef CAS; (b) W. Ji, L. Xiao, Y. Ling, C. Ching, M. Matsumoto, R. P. Bisbey, D. E. Helbling and W. R. Dichtel, J. Am. Chem. Soc., 2018, 140, 12677–12681 CrossRef CAS PubMed.
- M. E. Tighe, M. D. Thum, N. K. Weise and G. C. Daniels, Chem. Eng. J., 2024, 499, 156280 CrossRef CAS.
- A. Chaix, C. Gomri, B. T. Benkhaled, M. Habib, R. Dupuis, E. Petit, J. Richard, A. Segala, L. Lichon, C. Nguyen, M. Gary-Bobo, S. Blanquer and M. Semsarilar, Adv. Mater., 2024, 37, 2410720 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2430764. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc02247a |
| This journal is © The Royal Society of Chemistry 2025 |