
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIodide-mediated intermediate regulation strategy enables high-capacity and ultra-stable zinc–iodine batteries†
Zhijie Xua,
Jiaqi Yanga,
Peng Sunb,
Yaoyu Chena,
Zhengxiao Jia,
Xusheng Wang c,
Min Xu*a,
Jinliang Li*b and
Likun Pan*a
c,
Min Xu*a,
Jinliang Li*b and
Likun Pan*a
aShanghai Key Laboratory of Magnetic Resonance, School of Physics and Electronic Science, Institute of Magnetic Resonance and Molecular Imaging in Medicine, East China Normal University, Shanghai 200241, China. E-mail: xumin@phy.ecnu.edu.cn; lkpan@phy.ecnu.edu.cn
bSiyuan Laboratory, Guangdong Provincial Engineering Technology Research Center of Vacuum Coating Technologies and New Energy Materials, College of Physics & Optoelectronic Engineering, Department of Physics, Jinan University, Guangzhou 510632, China. E-mail: lijinliang@email.jnu.edu.cn
cState Key Laboratory of Bio-based Fiber Materials, School of Materials Science and Engineering, Zhejiang Sci-Tech University, Hangzhou 310018, P. R. China
First published on 18th June 2025
Abstract
The practical implementation of aqueous zinc–iodine (Zn–I2) batteries is hindered by the limited cathode capacity, rampant Zn dendrite formation, and anode corrosion issues. In this work, we propose a novel iodide-mediated intermediate regulation strategy achieved through a rationally designed combination of zinc iodide (ZnI2) and high-loading cathodes. Mechanistic studies reveal that iodide ions (I−) generate abundant iodine active sites on the elemental iodine-embedded porous carbon cathode (I2@PAC), which facilitates the conversion of under-oxidized triiodide (I3−) to pentaiodide (I5−), thereby significantly enhancing cathode capacity. Concurrently, the I− coordinate with Zn2+ to suppress the decomposition of coordinated water molecules, effectively mitigating side reactions and enabling dendrite-free Zn deposition morphology. These mechanisms collectively contribute to exceptional Coulombic efficiency (>99.7%) and outstanding cycling stability. The optimized Zn–I2 full cell achieves a remarkable specific capacity of 250.2 mAh g−1 at 0.2 A g−1, along with ultralong cycling durability exceeding 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles while maintaining 85% capacity retention. This iodide-mediated intermediate regulation strategy provides a viable pathway for developing high-capacity and ultra-stable aqueous Zn–I2 batteries.
000 cycles while maintaining 85% capacity retention. This iodide-mediated intermediate regulation strategy provides a viable pathway for developing high-capacity and ultra-stable aqueous Zn–I2 batteries.
Introduction
Aqueous rechargeable batteries have emerged as a promising alternative to conventional lithium-ion systems, primarily owing to their intrinsic safety and environmental benignity.1–3 Among these, aqueous zinc–ion batteries (AZIBs) have attracted substantial research interest, with significant efforts dedicated to enhancing their anode reversibility.4,5 Nevertheless, the energy density of AZIBs remains constrained by the limited operating potential and specific capacity of cathode materials when compared to the zinc anode (−0.76 V vs. the standard hydrogen electrode, 820 mAh g−1).6,7 While conventional Zn2+ intercalation cathodes (e.g., vanadium-based oxides,8,9 manganese-based compounds,10,11 and Prussian blue analogues12,13) dominate current research, iodine-based conversion-type cathodes are gaining momentum due to their inherent advantages: abundant natural reserves (particularly from seawater with 50–60 μg L−1 iodine concentration), high theoretical capacity (211 mAh g−1 based on iodine mass), and relatively high discharge plateau (1.3 V vs. Zn2+/Zn).14In previous studies on aqueous Zn–I2 batteries, extensive research efforts have been dedicated to improving Zn anode reversibility, with electrolyte additives emerging as the predominant strategy.15,16 A representative work by Xiao et al.6 demonstrated that the 1-ethyl-3-methylimidazolium acetate (EMIM[OAc]) ionic liquid additive facilitates uniform Zn deposition via steric hindrance from surface-adsorbed EMIM+ cations while concurrently suppressing polyiodide shuttling through chemical binding. Additionally, N-containing heterocyclic compounds have been reported to act as pH buffers, inhibiting hydrogen evolution reactions (HER) and anode corrosion while facilitating non-dendritic Zn plating/stripping.17,18 Although these additives effectively enhance zinc anode stability and extend battery cycle life, they offer minimal improvement to the cathode's electrochemical performance. Furthermore, increasing the mass loading of iodine at the cathode represents an essential requirement for practical zinc–iodine batteries.14 Wang et al.19 utilized a novel binder, polyacrylonitrile copolymer, at the cathode to enhance iodine loading by bonding with polyiodides. However, these strategies still ignore the fundamental challenge of low iodine utilization rate. The core limitation originates from the intrinsic characteristics of the (I−/I0) redox system: upon oxidation of I− to I2, spontaneous reaction with excess I− generates triiodide species, leading to irreversible consumption of electrochemically active iodine.20 Further conversion of polyiodides and confining them to cathode materials is the key to improve iodine utilization and specific cathode capacity in advanced zinc–iodine battery systems.
In this work, we propose a novel iodide-mediated intermediate regulation strategy based on I− additive, enabling the development of high mass loading zinc–iodine batteries with ultra-low capacity decay. This strategy leverages iodine active sites from I2@PAC and I− in electrolyte, facilitating the conversion of under-oxidized I3− to I5− in electrolyte. This process significantly enhances iodide utilization efficiency and boosts cathode specific capacity. Density functional theory (DFT) calculations reveal that iodine sites on I2@PAC thermodynamically promote I5− formation. In situ Raman spectroscopy confirms the reversible generation of I5− at cathode interface. Distribution of relaxation times (DRT) analysis demonstrates new charge transfer processes (presumably I3− to I5−) induced by iodide additives. Meanwhile, I− replace coordinated water molecules in Zn2+ solvation sheath, promoting Zn (002) crystal plane growth while suppressing zinc dendrite formation and side reactions on anode. Consequently, the Zn∥Zn symmetric cell achieves stable cycling over 3000 h at 1 mA cm−2/1 mAh cm−2. The Zn∥I2@PAC full cell delivers a high capacity of 240.8 mAh g−1 (based on cathode iodine mass) at 1 A g−1, maintaining 85% capacity retention after 10000 cycles.
Results and discussion
Aiming to study the role of I− in zinc–iodine batteries, ZnI2 was selected as the I− source, and the 2 M ZnSO4 electrolyte (ZSO) was the contrast electrolyte. As shown in Fig. S1a,† ZSO containing x M ZnI2 additive was denoted as ZSO-x. To assess the electrochemical compatibility between the electrolyte and cathode material, a seven-day immersion stability test was conducted (Fig. S1b and c†). Scanning electron microscopy (SEM) images (Fig. S1d†) confirm that the electrode surface morphology remained intact throughout the immersion period. Furthermore, energy-dispersive X-ray spectroscopy (EDS) analysis (Table S1†) revealed a consistent iodine weight ratio of 40–47%, demonstrating robust interfacial stability and compatibility between the electrolyte and cathode material. The conductivity profiles and pH characteristics of ZSO-x are presented in Fig. S2.† A positive correlation was observed between iodine ion concentration and electrical conductivity, while all samples maintained weakly acidic pH values in the range of 4.8–5.1.In conventional zinc–iodine battery systems, Zn2+ deposit onto the zinc electrode during charging while I− are oxidized to I2, which will spontaneously form more soluble I3− with the I− that have not been oxidized. During discharging, these processes are reversed.21,22 This redox chemistry based on the I−/I3− couple enables a theoretical capacity of 141 mAh g−1, calculated based on the mass of iodide element. The corresponding electrochemical reactions are summarized in eqn (1)–(4):23
Anode:
| Zn2+ + 2e− ↔ Zn E = −0.763 V vs. SHE | (1) |
Cathode:
| I2 + 2e− ↔ 2I− E = 0.535 V vs. SHE | (2) |
| I− + I2 ↔ I3− | (3) |
| I3− + 2e− ↔ 3I− E = 0.536 V vs. SHE | (4) |
The iodide-mediated intermediate regulation strategy was formed by adding I− additive to the electrolyte and I2 on the porous active carbon of the cathode. As shown in Fig. 1a, during charging, the additional I− are oxidized on the porous carbon cathode to generate more I2@PAC sites, where I3− further react to form I5−, as described by eqn (5):24–26
| I3− + I2 ↔ I5− | (5) |
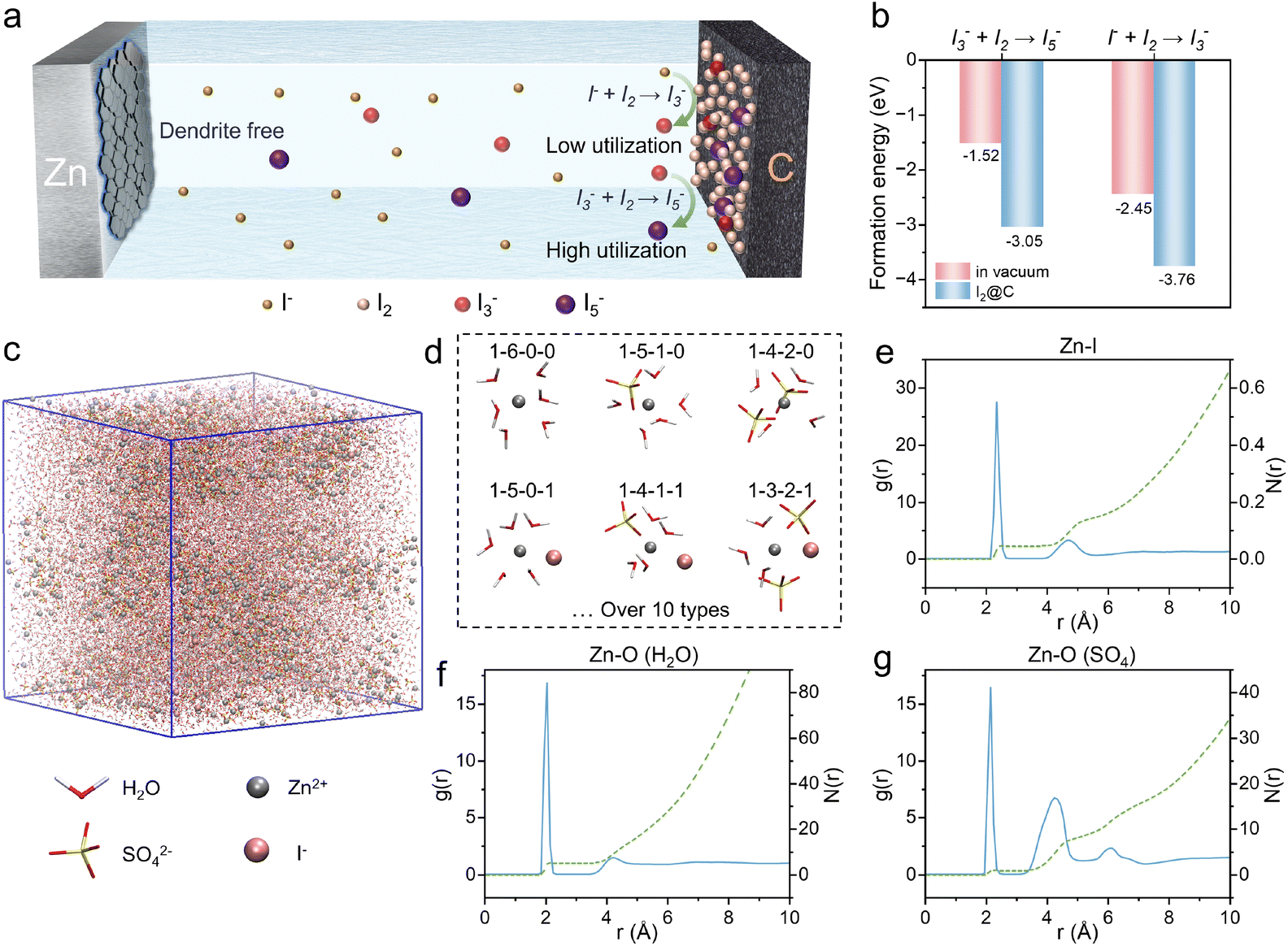 | ||
| Fig. 1 Theoretical calculations of iodide-mediated intermediate regulation strategy. (a) The schematic diagram of iodide-mediated intermediate regulation strategy mechanism. (b) The formation energy of I5− and I3− ions in vacuum and at the iodine sites of I2@PAC from DFT calculation. (c) MD simulation for the ZSO-0.1 electrolyte. (d) 3D snapshots of the representative solvation structures of Zn2+. (e–g) RDFs and the coordination number of Zn–I, Zn–O (H2O) and Zn–O (SO4) in ZSO-0.1 electrolyte respectively. | ||
For individual iodide ions, the transformation pathway from I− to I5− demonstrates superior redox chemical utilization compared to the I− to I3− conversion pathway. To quantitatively assess the thermodynamic feasibility of these transformations, we conducted DFT calculations to determine the formation energies of I5− and I3− species under two conditions: in vacuum (Fig. S3†) and at iodine sites on the PAC substrate (Fig. S4†). The computational results (Fig. 1b) reveal a significant energy difference, with formation energy decreasing from −1.52 eV to −3.05 eV for I5− and from −2.45 eV to −3.76 eV for I3− when reactions occurred on I2@PAC. This energetic preference indicates that I2@PAC substrate thermodynamically favors the evolution of I3− intermediates into I5− species with higher iodine utilization. Importantly, electrolyte enrichment with I− enhances the surface density of active iodine sites on PAC, thereby promoting the solution-phase conversion of I3− to I5−. This iodide-mediated intermediate regulation strategy ultimately elevates the overall conversion efficiency of the I−/I0 redox couple through stabilized polyiodide (I5−) formation.
The introduction of I− will also modulate the solvation structure of Zn2+. To elucidate this mechanism, molecular dynamics (MD) simulations were systematically conducted for both pristine ZSO (Fig. S5†) and iodide-containing ZSO-0.1 (Fig. 1c) electrolyte systems. The detailed molecular numbers are listed in Table S2.† Structural analysis reveals that Zn2+ coordinates with I−, sulfate anions, and water molecules through distinct Zn–I, Zn–O (SO4), and Zn–O (H2O) interactions, forming [Zn(H2O)x(SO42−)y(I−)z] (denoted as 1–x–y–z) coordination clusters (Fig. 1d). To quantitatively characterize the solvation environment, radial distribution functions (RDFs) and coordination number (CN) analyses were performed. Notably, the RDF profile exhibits a prominent Zn–I peak at 2.3 Å (Fig. 1e), demonstrating that I− can enter the primary solvation sheath of Zn2+. Comparative CN analysis further reveals structural reorganization: the CN of Zn–O (H2O) in ZSO-0.1 (Fig. 1f) registers 5.07 compared to 5.12 in ZSO (Fig. S6a†), while the CN of sulfate decreases from 1.01 (Fig. S6b†) to 0.88 (Fig. 1g). These findings collectively indicate that I− induce structural rearrangement in the Zn2+ solvation sheath. Specifically, I− competitively displace both water molecules and sulfate anions from the first coordination sheath, thereby reducing the hydration degree of Zn2+.
To systematically investigate the role of iodide-mediated intermediate regulation strategy on the electrochemical performance of Zn∥I2@PAC full cells, we first prepared the cathode material I2@PAC via a solution adsorption method.27,28 EDS mapping images (Fig. S7†) confirm homogeneous iodine distribution throughout the porous active carbon substrate. Thermogravimetric (TG) analysis confirmed an iodine content of 53.4 wt% in the composite (Fig. S8†), demonstrating effective iodine loading through this synthesis approach. The cells were assembled by varying I2 mass loadings (1.6, 2.4, and 3.2 mg I2 cm−2) with the electrolytes containing different I− concentrations at 1 A g−1 (Fig. 2a–c). The results reveal a dual enhancement mechanism: both increased I− concentration in the electrolyte and higher I2 loading in the cathode significantly improve specific capacity. This synergistic behavior suggests that optimizing iodine speciation across the electrolyte–cathode interface is critical for maximizing battery capacity. Notably, although the ZSO-0.2 electrolyte with the highest I− concentration demonstrated superior capacity values, it displayed significant capacity degradation under high cathode loading conditions (3.2 mg cm−2), failing to maintain stable cycling performance beyond 200 cycles as evidenced in Fig. 2c. This instability was further reflected in its low Coulombic efficiency (CE) of ∼99.0% (Fig. S9†), substantially lower than the >99.5% CE values consistently observed for other electrolytes. Through comprehensive evaluation of both specific capacity and cycling stability, ZSO-0.1 emerged as the optimal candidate, exhibiting balanced electrochemical performance. Consequently, ZSO-0.1 was selected for subsequent comparative experiments against the ZSO electrolyte. Additionally, we conducted cycling performance tests of Zn∥pure carbon cells in each electrolyte (Fig. S10a†). As clearly observed from the 100th cycle galvanostatic charge–discharge (GCD) curves (Fig. S10b†), with increasing I− concentration, the charge–discharge profile of the pure carbon cathode undergoes a gradual transition from typical capacitive behavior to battery-type characteristics. A distinct voltage plateau emerges between 1.1–1.3 V, corresponding to the I−/I0 redox couple. The specific capacity at the 100th cycle (when all cells were in the stable cycling stage) was compared and linearly fitted with the I− concentration in electrolytes, with the results shown in Fig. 2d. The slope k represents the rate at which the specific capacity of the cathode active material increases per mole of I− additive. For the pure carbon cathode, the slope is 218.9 mAh g−1 mol−1, indicating the efficiency of I− in boosting cathode specific capacity as a redox active species. In contrast, the I2@PAC cathode (loaded with 1.6 mg cm−2 I2) exhibits a significantly higher slope of 330.2 mAh g−1 mol−1 compared to pure carbon. This suggests that in the battery system with I2@PAC cathode, I− not only act as electrolyte additives but also activate additional faradaic reactions, thereby contributing extra capacity. Combined with the DFT calculation results in Fig. 1b, we speculate that increased I− concentration elevates the number of iodine sites on the porous carbon during charging, facilitating the transformation of I3− intermediates into I5− species with higher iodine utilization. Moreover, as the cathodic iodine mass loading increases, the corresponding slope demonstrates a gradual diminishing trend. This phenomenon occurs because the amount of I− additive in the electrolyte contributing to capacity is fixed, and its specific capacity contribution proportionally decreases with the elevation of cathode loading. As shown in Fig. 2e, the specific capacity of ZSO-0.1 presents a capacity of 250.2 mAh g−1 at 0.2 A g−1, ∼20% higher than ZSO's 208.8 mAh g−1. Even at a high current density of 5 A g−1, ZSO-0.1 also demonstrates even greater superiority with a specific capacity of 197.1 mAh g−1, outperforming ZSO's 143.9 mAh g−1 by ∼35%. The GCD profiles (Fig. S11†) reveal significantly smaller polarization for ZSO-0.1 compared to ZSO under high-current conditions. These results collectively indicate that ZSO-0.1 exhibits comprehensively superior rate performance to ZSO.
 | ||
| Fig. 2 Electrochemical performance of Zn∥I2@PAC full cells. Cycling performance of Zn∥I2@PAC cells with a mass loading of (a) 1.6 mg cm−2, (b) 2.4 mg cm−2 and (c) 3.6 mg cm−2 at 1 A g−1 in different electrolytes. (d) Comparison of the specific capacity at 1 A g−1 after 100 cycles. (e) Rate performance of Zn∥I2@PAC cells at different current densities. (f) Long cycling performance at 1 A g−1 of Zn∥I2@PAC cells. | ||
Moreover, the ZSO-0.1 exhibits exceptional long-term cycling stability (Fig. 2f), retaining 85% of its initial capacity after 10000 cycles with an average CE of 99.7%. The capacity decay of the cathode is multifaceted, potentially arising from side reactions, electrolyte decomposition-induced instability at the electrode–electrolyte interface, and dissolution of cathode iodine species.29 Notably, the GCD profiles in Fig. S12† remain highly stable throughout the cycling tests, indicating robust interfacial stability between the electrode and electrolyte. Previous studies have demonstrated that N-doped porous carbon exhibits superior iodine species adsorption capability compared to undoped porous carbon, significantly enhancing long-term capacity retention.30 However, the cathode material consists of undoped porous carbon in this work, as confirmed by the absence of N in the EDS analysis (Table S1†). We attribute the primary cause of cathode capacity decay to the weak adsorption of iodine species by the undoped carbon, leading to their dissolution during cycling. For a comprehensive evaluation, we compare the main parameters and cycling performance of this work with recently reported strategies in the literature, including electrolyte modification and cathode design approaches based on I−/I0 redox chemistry (Table S3†). The results demonstrate that our facile iodide-mediated intermediate regulation strategy achieves a high specific capacity of 250.2 mAh g−1 under low current densities while maintaining 198 mAh g−1 after cycling.
In situ electrochemical impedance spectroscopy (EIS) test at different state-of-charge (SoC) was conducted to gain insights into the charge transfer process on the I2@PAC cathode.31,32 As shown in Fig. 3a and b, the both EIS curves show the shape of two semicircles and a diagonal line. However, such a rough judgment would miss the coupled charge transfer process.33 To gain deeper insight into the cathode reaction mechanisms, we performed DRT analysis based on in situ EIS measurements. This approach allows for the quantification of interfacial resistance evolution by deconvoluting contributions from processes with distinct time constants.34 As shown in Fig. 3c, the DRT profiles exhibit three well-resolved peaks, each corresponding to a specific time constant, namely τ1, τ3, and τ4. Similarly, Fig. 3d reveals four discernible peaks associated with τ1, τ2, τ3, and τ4. Generally, the τ1 peak at 10−5–10−4 s can be assigned to the contact resistance.35 To confirm the attribution of τ2, τ3, and τ4, a set of EIS experiments were conducted at the open circuit voltage (OCV), OCV ± 0.05 V and OCV ± 0.1 V. When a larger overpotential was applied, the Rct would reduce. Otherwise, it would be assigned to the ion transport progress across the SEI layer.36 From the results in Fig. S13,† it is found that all time constants (τ2, τ3, and τ4) are assigned to Rct (the specific impedance values are listed in Tables S4 and S5†). We speculate that the extra time constant τ2 in ZSO-0.1 is due to the charge transfer process in eqn (5). To clarify the reaction mechanism in ZSO-0.1, in situ Raman spectroscopy was conducted to monitor the evolution of electrochemical products during charging/discharging cycles (Fig. 3e and f). When discharged to 1.25 V, a distinct I5− peak at 168 cm−1 and a weaker I3− peak (108 cm−1) emerged.17,24 Notably, both peaks gradually diminished as the discharge progressed. Upon subsequent charging to 1.1 V, the I5− peak reappeared and persisted until fully charged, whereas the I3− signal temporarily re-emerged before completely disappearing upon reaching full charge. These findings indicate that I3− solely as an intermediate with low concentration in ZSO-0.1, while I5− exhibits remarkable stability upon full SoC and undergoes complete conversion to I− during discharge, confirming its superior electrochemical reversibility.
 | ||
| Fig. 3 Redox chemistry and kinetic analysis of Zn∥I2@PAC cells. In situ Nyquist plots of I2@PAC cathode in (a) ZSO and (b) ZSO-0.1 electrolyte at different SoC. DRT curves from in situ EIS measurements at different SoC in (c) ZSO and (d) ZSO-0.1 electrolyte. (e) Current and cell voltage as a function of time and (f) the corresponding 2D in situ Raman spectra of the Zn∥I2@PAC cells in ZSO-0.1 electrolyte during the initial discharging–charging processes. GITT and the related diffusion coefficient curves in (g) ZSO and (h) ZSO-0.1 electrolyte. (i) CV curves and (j) Tafel plots of the Zn∥I2@PAC cells in ZSO and ZSO-0.1 electrolyte. | ||
To investigate the reaction kinetics, galvanostatic intermittent titration technique (GITT) analysis was performed on Zn∥I2@PAC cells with ZSO (Fig. 3g) and ZSO-0.1 (Fig. 3h) electrolytes. The calculated zinc ion diffusion coefficients demonstrate significant enhancement in ZSO-0.1 (10−9–10−8 cm2 s−1) compared to ZSO (10−10–10−9 cm2 s−1), representing nearly an order of magnitude improvement. This accelerated zinc ion transport in ZSO-0.1 directly correlates with its superior fast charge–discharge performance. The addition of additives increases the conductivity of the electrolyte (Fig. S2†), which also explains the increase in diffusion kinetics of Zn2+.37 Further kinetic evaluation through cyclic voltammetry (CV) at 0.1 mV s−1 revealed distinct advantages in ZSO-0.1 for the I0/I− redox reaction (Fig. 3i). The I− → I0 oxidation peak appears at 1.25 V with a corresponding reduction peak at 1.16 V, showing higher current density in ZSO-0.1 compared to ZSO. As the scan rate escalates, a gradual increase in overpotential is observed (Fig. S14†), a phenomenon attributed to polarization effects occurring during electrochemical cycling.38 The b-value analysis indicates that while both ZSO and ZSO-0.1 demonstrate surface capacitance-dominated charge storage mechanisms, ZSO-0.1 exhibits a greater contribution from diffusion-controlled processes compared to ZSO. Tafel slope analysis derived from CV curves provides additional kinetic insights (Fig. 3j). The significantly smaller Tafel slope for ZSO-0.1 during reduction processes (vs. ZSO) confirms faster reaction kinetics, while comparable oxidation slopes suggest similar anodic behavior. Furthermore, we conducted Tafel curve measurements on Zn∥Zn symmetric cells to evaluate the exchange current densities.39 Remarkably, ZSO-0.1 demonstrated a significantly higher exchange current density of 4.51 mA cm−2 compared to the 0.449 mA cm−2 observed for ZSO (Fig. S15†), highlighting its superior charge transfer kinetics. These comprehensive kinetic analyses collectively demonstrate that ZSO-0.1 achieves enhanced reaction kinetics through improved ion transport and charge transfer characteristics, ultimately explaining its superior rate performance shown in Fig. 2e.
Beyond the cathode performance, the overall functionality of Zn∥I2@PAC cells are critically dependent on the compatibility between the electrolyte and Zn anode. To assess the applicability of ZSO-0.1 electrolyte for Zn anode, linear sweep voltammetry (LSV) measurements using Zn∥Ti asymmetric cells were employed to evaluate the electrochemical stability windows of both pristine ZSO and modified ZSO-0.1 electrolytes (Fig. S16†). ZSO-0.1 exhibited a characteristic oxidation peak at 2.2 V corresponding to the I−/I0 redox conversion on the Ti foil, while both electrolyte systems demonstrated oxygen evolution reactions (OER) initiating at approximately 2.7 V. This comparative assessment indicates that while the modified ZSO-0.1 electrolyte maintains the inherent OER threshold, it introduces specific iodine-related redox reactions within the operational voltage range. To investigate the influence of I− on the reversibility of zinc anodes, Zn∥Cu asymmetric cells were assembled and tested. As shown in Fig. 4a, ZSO fails rapidly after 115 cycles with an average CE of 96.9%, whereas ZSO-0.1 demonstrated exceptional stability over 1000 cycles with a significantly higher average CE of 99.6%. To evaluate zinc plating/stripping reversibility, Fig. 4b compares the initial CE (ICE) and voltage–capacity profiles of both electrolyte systems. Notably, ZSO-0.1 achieved a superior ICE of 94.9% compared to 91.9% for ZSO, suggesting enhanced electrochemical reversibility through I− additive. The rate performance of Zn∥Zn symmetric cells was conducted with a cutoff capacity of 1 mAh cm−2. The comparative analysis in Fig. 4c reveals that ZSO-0.1 exhibits a reduced overpotential compared to pristine ZSO. To further investigate the zinc deposition behavior, Zn∥Ti asymmetric cells were assembled to monitor the zinc nucleation and deposition processes on titanium substrates. Electrochemical measurements demonstrate that the zinc deposition overpotential decreases from 45 mV in ZSO to 37 mV in ZSO-0.1 (Fig. 4d). Complementary CV analysis (Fig. S17†) reveals distinct nucleation characteristics: the cross potential (point C) observed during positive scanning represents the typical nucleation signature, while points A and B where Zn2+ begins to be reduced on the electrode correspond to the nucleation overpotential.17 Obviously, the introduction of I− increased the nucleation overpotential by 17 mV relative to pristine ZSO. This phenomenon can be explained by the established principle that elevated nucleation overpotentials create enhanced driving forces during initial nucleation phases, which typically promote more uniform zinc deposition with refined grain size.40 To evaluate the stability of Zn2+ plating/stripping processes in electrolytes of various I− concentrations, Zn∥Zn symmetric cells were conducted as shown in Fig. 4e. Notably, ZSO-0.1 can maintain stable cycling for 3000 h under 1 mA cm−2/1 mAh cm−2, while the other electrolytes failed within less than 250 h. As illustrated in the inset of Fig. 4e, the ZSO-0.2 displayed distinct soft shorts behavior during charge/discharge cycles after cycling for 244 h.41 The Nyquist plots in Fig. 4f further reveal that ZSO-0.1 exhibits a significantly lower charge-transfer resistance compared to ZSO. These results collectively demonstrate the critical role of I− additives in optimizing nucleation dynamics, enhancing ion transport kinetics, and stabilizing zinc anode performance.
 | ||
| Fig. 4 Stability of Zn anodes by electrolyte regulation. (a) CE responses of Zn∥Cu asymmetric cells with ZSO and ZSO-0.1 electrolyte at 1 mA cm−2/1 mAh cm−2 and (b) the corresponding voltage profiles at selected cycles. (c) Rate performance of Zn∥Zn symmetric cells at different current densities. (d) Profiles displaying voltage–time variations of Zn∥Ti cells at a current density of 1 mA cm−2. (e) Voltage responses of Zn∥Zn symmetric cells in ZSO and ZSO-0.1 electrolytes at 1 mA cm−2/1 mAh cm−2 for 3000 h (insets are magnified voltage profiles). (f) EIS tests of the Zn∥Zn symmetric cells at OCV. | ||
To investigate Zn deposition behavior and side reactions occurring on the Zn anode, the cycled Zn anodes and the pristine Zn anode were investigated by employing X-ray diffraction (XRD). As shown in Fig. 5a, the cycled ZSO anode exhibited additional diffraction peaks corresponding to Zn4SO4(OH)6·5H2O (PDF#39-0688), indicating by-product formation during cycling.42 In contrast, no impurity phase was detected in ZSO-0.1 (Fig. 5b), and all the diffraction peaks were assigned to the Zn metal (PDF#04-0831). The evolution of preferred crystallographic orientation was quantified through the intensity ratio of (002) to (100) diffraction peaks I(002)/I(100). Particularly, ZSO-0.1 demonstrated a progressive increase in this ratio during cycling, reaching 0.41 after 50 cycles, while ZSO showed a declining trend that culminated at 0.10 (Fig. 5c). This divergence suggests distinct crystal growth patterns: the iodide-containing electrolyte promotes preferential development of (002)-oriented crystallites, whereas ZSO favors (100) crystal plane growth. Complementary analysis through relative texture coefficient (RTC) calculations revealed that ZSO-0.1 achieved a 10.5% RTC value for the Zn (002) plane after 50 cycles (Fig. 5d). These observations confirm that I− effectively regulate zinc crystallographic orientation. The higher (002) crystal plane index contributes to suppressing dendrite growth.43 The Zn electrodes after plating in Zn∥Zn symmetric cells were conducted to observe the surface morphology of zinc deposition. The captured SEM images show random lamellar dendrite clusters (Fig. 5e) and disordered by-products (Fig. 5f) in ZSO, while regular hexagonal zinc deposition (Fig. 5g) and flat zinc surface without by-product protrusions (Fig. 5h) can be observed in ZSO-0.1. Complementary in situ optical monitoring further elucidated the temporal evolution of zinc deposition behavior. As illustrated in Fig. 5i, substantial dendritic deposits and bulk by-products formed on the bare ZSO electrolyte's zinc electrode within 10 min of deposition, with progressive thickening of these protrusions over time that ultimately created a highly irregular surface topography. By contrast, zinc electrodes immersed in the ZSO-0.1 electrolyte exhibited only minor deposition thickening, maintaining a strikingly smooth surface morphology throughout the observation period (Fig. 5j). This pronounced contrast in deposition behavior can be reasonably attributed to the role of iodide additives. In conventional ZSO electrolyte, the side reactions and inherent tip-enhanced polarization facilitate the sequential formation of dendritic structures through three key processes: (1) competitive decomposition of coordinated water molecules generates OH− and by-product; (2) local pH elevation promotes the precipitation of Zn4SO4(OH)6·5H2O; (3) preferential zinc deposition at protrusion tips establishes self-accelerating dendritic growth.44,45 However, in ZSO-0.1, I− can reshape the Zn2+ solvation structure and suppress water activity, thereby significantly reducing the generation of by-products. Additionally, I− can induce the growth of zinc (002) crystal planes and mitigate zinc dendrite formation, thus achieving more uniform zinc deposition.
 | ||
| Fig. 5 Regulation of Zn deposition and suppression of dendritic growth. (a and b) XRD patterns of Zn electrodes before and after cycling in electrolytes. (c) Plots of the I(002)/I(100) ratio and (d) the RTC value. (e–h) SEM images of Zn anode after plating for 2 h at 1 mA cm−2. (i and j) In situ optical microscopy investigations of the Zn deposition in ZSO and ZSO-0.1 electrolytes at 1 mA cm−2. | ||
Conclusions
In this work, we propose an iodide-mediated intermediate regulation strategy by introducing ZnI2 to simultaneously address the challenges of low iodine utilization at the cathode and dendritic growth at the anode in zinc–iodine batteries. We demonstrate that the added I− act as a solvation sheath modifier, where I− coordinates with Zn2+ to suppress water decomposition in the coordination sphere, thereby mitigating side reactions and enabling dendrite-free Zn deposition. This optimization extends the lifespan of Zn∥Zn symmetric cells to 3000 h at 1 mA cm−2/1 mAh cm−2. Moreover, the introduced I− enhance the diffusion coefficient to 10−9–10−8 cm2 s−1 and reduce the overpotential of the I−/I0 redox couple, significantly improving electrochemical kinetics. As a cathode activator, I− facilitates the formation of abundant I2@PAC active sites, promoting the I3− → I5− conversion and boosting iodine utilization. The optimized Zn∥I2@PAC full cell delivers a high specific capacity of 250.2 mAh g−1 under stringent conditions (iodine loading: 3.2 mg cm−2, 0.2 A g−1) and retains 199.5 mAh g−1 (85% capacity retention) after 10000 cycles at 1 A g−1. We believe that our work will provides fundamental insights into the I−/I0 redox chemistry and offers a feasible strategy for developing high-capacity, ultra-stable zinc–iodine batteries.
Data availability
All the data used to support the findings of this study can be made available by the corresponding authors upon request.Author contributions
Z. Xu: writing – original draft, investigation, data curation, conceptualization, formal analysis; J. Yang: formal analysis, data curation, conceptualization; P. Sun: investigation, data curation; Y. Chen, investigation, data curation; Z. Ji, investigation, data curation; X. Wang, investigation, data curation; M. Xu, writing – review & editing, visualization, supervision, funding acquisition, conceptualization; J. Li, writing – review & editing, visualization, supervision, funding acquisition, conceptualization; L. Pan: writing – review & editing, visualization, supervision, funding acquisition, conceptualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support from the Guangxi Science and Technology Program (2024AB08156), International Partnership Program of Chinese Academy of Sciences (124GJHZ2023031MI), National Natural Science Foundation of China (21875068), and Fundamental Research Funds for the Central Universities (2020ECNU-GXJC003).References
- J. Zheng, Q. Zhao, T. Tang, J. Yin, C. D. Quilty, G. D. Renderos, X. Liu, Y. Deng, L. Wang, D. C. Bock, C. Jaye, D. Zhang, E. S. Takeuchi, K. J. Takeuchi, A. C. Marschilok and L. A. Archer, Science, 2019, 366, 645–648 CrossRef CAS PubMed.
- M. Zhao, J. Rong, F. Huo, Y. Lv, B. Yue, Y. Xiao, Y. Chen, G. Hou, J. Qiu and S. Chen, Adv. Mater., 2022, 34, 2203153 CrossRef CAS PubMed.
- Y. Su, Y. Zhang, W. Feng, G. Zhang, Y. Sun, C. Yin, G. Yuan, Y. Tang, W. Zhou, H.-C. Chen and H. Pang, Angew. Chem., Int. Ed., 2025, 64, e202502752 CrossRef CAS PubMed.
- M. Zhao, Y. Lv, Y. Xu, H. Yang, Z. Bo and J. Lu, Nat. Commun., 2025, 16, 2843 CrossRef CAS PubMed.
- W. Deng, Z. Xu and X. Wang, Energy Storage Mater., 2022, 52, 52–60 CrossRef.
- T. Xiao, J.-L. Yang, B. Zhang, J. Wu, J. Li, W. Mai and H. J. Fan, Angew. Chem., Int. Ed., 2024, 63, e202318470 CrossRef CAS PubMed.
- S. Back, L. Xu, J. Moon, J. Kim, Y. Liu, S. Y. Yi, D. Choi and J. Lee, Small, 2024, 20, 2405487 CrossRef CAS PubMed.
- Y. Dai, C. Zhang, J. Li, X. Gao, P. Hu, C. Ye, H. He, J. Zhu, W. Zhang, R. Chen, W. Zong, F. Guo, I. P. Parkin, D. J. L. Brett, P. R. Shearing, L. Mai and G. He, Adv. Mater., 2024, 36, 2310645 CrossRef CAS PubMed.
- J. Gao, C. Cheng, L. Ding, G. Liu, T. Yan and L. Zhang, Chem. Eng. J., 2022, 450, 138367 CrossRef CAS.
- W. Qu, Y. Cai, B. Chen and M. Zhang, Energy Environ. Mater., 2024, 7, e12645 CrossRef CAS.
- Z. Zhou, Z. Chen, X. Luo, L. Wang, J. Liang, W. Peng, Y. Li, F. Zhang and X. Fan, ACS Appl. Mater. Interfaces, 2022, 14, 24386–24395 CrossRef CAS PubMed.
- S. Liu, Z. Sun, B. Li, X. Liu and C. Xue, J. Energy Storage, 2025, 111, 115310 CrossRef.
- L. Ye, H. Fu, R. Cao and J. Yang, J. Colloid Interface Sci., 2024, 664, 423–432 CrossRef CAS PubMed.
- H. Wu, J. Hao, S. Zhang, Y. Jiang, Y. Zhu, J. Liu, K. Davey and S.-Z. Qiao, J. Am. Chem. Soc., 2024, 146, 16601–16608 CrossRef CAS PubMed.
- K. Qiu, G. Ma, Y. Wang, M. Liu, M. Zhang, X. Li, X. Qu, W. Yuan, X. Nie and N. Zhang, Adv. Funct. Mater., 2024, 34, 2313358 CrossRef CAS.
- F. Wang, W. Liang, X. Liu, T. Yin, Z. Chen, Z. Yan, F. Li, W. Liu, J. Lu, C. Yang and Q.-H. Yang, Adv. Energy Mater., 2024, 14, 2400110 CrossRef CAS.
- Y. Lyu, J. A. Yuwono, P. Wang, Y. Wang, F. Yang, S. Liu, S. Zhang, B. Wang, K. Davey, J. Mao and Z. Guo, Angew. Chem., Int. Ed., 2023, 62, e202303011 CrossRef CAS PubMed.
- L. Su, H. Wu, S. Zhang, C. Cui, S. Zhou and H. Pang, Adv. Mater., 2025, 37, 2414628 CrossRef CAS PubMed.
- K. Wang, H. Li, Z. Xu, Y. Liu, M. Ge, H. Wang, H. Zhang, Y. Lu, J. Liu, Y. Zhang, Y. Tang and S. Chen, Adv. Energy Mater., 2024, 14, 2304110 CrossRef CAS.
- C. Chen, Z. Li, Y. Xu, Y. An, L. Wu, Y. Sun, H. Liao, K. Zheng and X. Zhang, ACS Sustain. Chem. Eng., 2021, 9, 13268–13276 CrossRef CAS.
- H. Yang, Y. Qiao, Z. Chang, H. Deng, P. He and H. Zhou, Adv. Mater., 2020, 32, 2004240 CrossRef CAS PubMed.
- W. Shang, J. Zhu, Y. Liu, L. Kang, S. Liu, B. Huang, J. Song, X. Li, F. Jiang, W. Du, Y. Gao and H. Luo, ACS Appl. Mater. Interfaces, 2021, 13, 24756–24764 CrossRef CAS PubMed.
- Z. Pei, Z. Zhu, D. Sun, J. Cai, A. Mosallanezhad, M. Chen and G. Wang, Mater. Res. Bull., 2021, 141, 111347 CrossRef CAS.
- S.-J. Zhang, J. Hao, H. Li, P.-F. Zhang, Z.-W. Yin, Y.-Y. Li, B. Zhang, Z. Lin and S.-Z. Qiao, Adv. Mater., 2022, 34, 2201716 CrossRef CAS PubMed.
- M. Liu, Q. Chen, X. Cao, D. Tan, J. Ma and J. Zhang, J. Am. Chem. Soc., 2022, 144, 21683–21691 CrossRef CAS PubMed.
- C. Prehal, H. Fitzek, G. Kothleitner, V. Presser, B. Gollas, S. A. Freunberger and Q. Abbas, Nat. Commun., 2020, 11, 4838 CrossRef CAS PubMed.
- Y. Zou, T. Liu, Q. Du, Y. Li, H. Yi, X. Zhou, Z. Li, L. Gao, L. Zhang and X. Liang, Nat. Commun., 2021, 12, 170 CrossRef CAS PubMed.
- W. Zong, J. Li, C. Zhang, Y. Dai, Y. Ouyang, L. Zhang, J. Li, W. Zhang, R. Chen, H. Dong, X. Gao, J. Zhu, I. P. Parkin, P. R. Shearing, F. Lai, K. Amine, T. Liu and G. He, J. Am. Chem. Soc., 2024, 146, 21377–21388 CrossRef PubMed.
- D. Lin and Y. Li, Adv. Mater., 2022, 34, 2108856 CrossRef CAS PubMed.
- Y. Li, X. Guo, S. Wang, W. Sun, D. Yu, N. Li, H. Zhou, X. Zhang and H. Pang, Adv. Sci., 2025, 2502563 CrossRef PubMed.
- W. Hu, Y. Peng, Y. Wei and Y. Yang, J. Phys. Chem. C, 2023, 127, 4465–4495 CrossRef CAS.
- P. Chen, Z. Wang, Q. Zhang, Y. Zeng, T. Liu, S. Bao, D. Li and L. Wang, Chem. Eng. J., 2025, 508, 160944 CrossRef CAS.
- Y. Lu, C.-Z. Zhao, J.-Q. Huang and Q. Zhang, Joule, 2022, 6, 1172–1198 CrossRef CAS.
- T. H. Wan, M. Saccoccio, C. Chen and F. Ciucci, Electrochim. Acta, 2015, 184, 483–499 CrossRef CAS.
- J. Wang, P. Yang, Y. Wang and S. Wang, Carbon, 2024, 228, 119432 CrossRef CAS.
- J. Chen, E. Quattrocchi, F. Ciucci and Y. Chen, Chem, 2023, 9, 2267–2281 CAS.
- S. Rashidi, N. Karimi, B. Sunden, K. C. Kim, A. G. Olabi and O. Mahian, Prog. Energy Combust. Sci., 2022, 88, 100966 CrossRef.
- R. Yi, X. Shi, Y. Tang, Y. Yang, P. Zhou, B. Lu and J. Zhou, Small Struct., 2023, 4, 2300020 CrossRef CAS.
- Q. Zhang, Y. Ma, Y. Lu, Y. Ni, L. Lin, Z. Hao, Z. Yan, Q. Zhao and J. Chen, J. Am. Chem. Soc., 2022, 144, 18435–18443 CrossRef CAS PubMed.
- T. C. Li, Y. Lim, X. L. Li, S. Luo, C. Lin, D. Fang, S. Xia, Y. Wang and H. Y. Yang, Adv. Energy Mater., 2022, 12, 2103231 CrossRef CAS.
- Q. Li, A. Chen, D. Wang, Z. Pei and C. Zhi, Joule, 2022, 6, 273–279 CrossRef.
- S. Wang, Y. Zhao, H. Lv, X. Hu, J. He, C. Zhi and H. Li, Small, 2024, 20, 2207664 CrossRef CAS PubMed.
- J. Xu, Z. Huang, H. Zhou, G. He, Y. Zhao and H. Li, Energy Storage Mater., 2024, 72, 103596 CrossRef.
- L. E. Blanc, D. Kundu and L. F. Nazar, Joule, 2020, 4, 771–799 CrossRef CAS.
- K. Fic, A. Płatek, J. Piwek, J. Menzel, A. Ślesiński, P. Bujewska, P. Galek and E. Frąckowiak, Energy Storage Mater., 2019, 22, 1–14 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc03114a |
| This journal is © The Royal Society of Chemistry 2025 |