Computational design of orthogonal nucleoside kinases†‡
Lingfeng
Liu
a,
Paul
Murphy
b,
David
Baker
bc and
Stefan
Lutz
*a
aDepartment of Chemistry, Emory University, Atlanta, GA 30322, USA. E-mail: sal2@emory.edu; Fax: +1 404-727-6586; Tel: +1 404-712-2170
bDepartment of Biochemistry, University of Washington, Seattle, WA 98195, USA
cHoward Hughes Medical Institute, Seattle, WA 98195, USA
First published on 19th October 2010
Abstract
We report the computational enzyme design of an orthogonal nucleoside analog kinase for 3′-deoxythymidine. The best kinase variant shows an 8500-fold change in substrate specificity, resulting from a 4.6-fold gain in catalytic efficiency for the nucleoside analog and a 2000-fold decline for the native substrate thymidine.
Nucleoside analogs are prominent and potent small-molecule prodrugs for the treatment of viral infections and cancer. Following cellular uptake, the prodrugs are activated to their corresponding triphosphate anabolites by cellular kinases of the nucleoside salvage pathway, turning them into chain terminators for low-fidelity polymerases and reverse transcriptases.1 However, the high substrate specificity of the endogenous kinases reduces the effectiveness of many prodrugs.2 One strategy to enhance the potency of nucleoside analogs has been the introduction of exogenous, promiscuous kinases by suicide gene therapy.3,4 The broad-specificity kinases from Herpes Simplex virus and Drosophila melanogaster were shown to boost the cytotoxicity of ganciclovir by up to 80-fold.4,5 Nevertheless, the catalytic performance of these kinases is compromised by the competition of native substrates and prodrug for the active site. In addition, broad-specificity kinases can perturb the tightly regulated natural nucleotide metabolism.6
To avoid these problems, we have pursued the engineering of orthogonal nucleoside analog kinases with changed rather than broader substrate specificity.7 Previously, directed evolution of the 2′-deoxynucleoside kinase from D. melanogaster (DmdNK) in combination with FACS-based screening led to the identification of an orthogonal kinase for 3′-deoxythymidine (ddT, 2, Fig. 1A), a representative of the large category of nucleoside analogs whose biological function is compromised due to lack of phosphorylation.8 The laboratory-evolved ddT kinase had two active site mutations, E172V and Y179F, which resulted in a 6-fold higher activity for 2 compared to DmdNK, as well as a 20-fold kcat/KM preference for 2 over thymidine (1), an overall 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold change in substrate specificity.
000-fold change in substrate specificity.
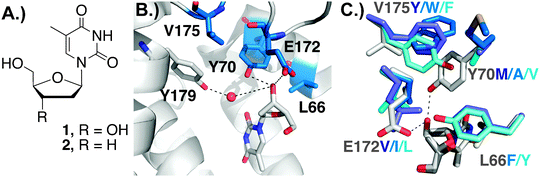 | ||
| Fig. 1 (A) Structures of thymidine (1) and 3′-deoxythymidine (2). (B) DmdNK co-crystallized with Thy (PDB: 1OT3).10 Active site residues that were found critical to switching the enzyme's substrate specificity from Thy to ddT by directed evolution (E172/Y179) and computational modeling (L66/Y70/E172/V175) are shown. (C) Overlay of key active site residues in the native structure and Rosetta model highlights the necessity for coevolution at positions 66, 70, and 175 due to steric constraints. | ||
Recent success in enzyme design by computational methods raised the question whether an alternate strategy using enzyme design by computational methods could recapitulate these findings or identify alternative kinase variants.9 The in silico approach enables a faster and far more thorough search of sequence space. In addition, it can accelerate the enzyme discovery process by reducing the number of required evolutionary iterations and by providing a quantitative predictive framework for protein engineers to explore questions of biocatalyst stability and substrate specificity.
Using crystallographic information for DmdNK in the presence of 1 (PDB: 1OT3),10 we applied an extension of the Rosetta suite of molecular modeling tools to redesign the active site of the kinase.11 Fixed-backbone design to optimize the specificity of DmdNK for 2 relative to 1 identified a set of four positions (L66, Y70, E172, and V175) in the vicinity of the substrate binding pocket and designs were made with altered amino acid identities for these residues (Fig. 1B).12 Individual designs were ranked based on the predicted energy of interaction of 2 (ΔGddT) for higher activity, as well as ΔGddT − ΔGThy for maximum specificity. Among the top performers in the computational model, the predictions for position 66 clearly favored a benzyl side chain to sterically block the proper orientation of the native substrate's 3′-hydroxyl group. The model was less conclusive about the substitutions of residues Y70 and V175. While the latter position favors large hydrophobic side chains (F, Y, W), predictions for substitutions in position 70 were nonconvergent and seemed to be largely compensatory in nature, accommodating the new bulky neighboring groups in positions 66 and 175 (Fig. 1C). Finally, Rosetta suggested substitutions at E172, one of the previously identified mutation hotspots.7 Predictions favored hydrophobic residues with β-branched side chains, eliminating hydrogen-bonding interactions with 1 and allowing for tighter protein packing.
The suggested amino acid substitutions in positions 66, 70, or 175 were of particular interest as they had not been observed in previous directed evolution experiments. We attributed their absence to the fact that all three substitutions require at least two or three nucleotide changes per codon, a highly improbable event in a whole-gene random mutagenesis library with a total of 2–4 nucleotide changes per 700-bases sequence.13 In addition, mutagenesis in one of the three positions likely requires compensatory changes of the neighboring amino acid(s) to preserve the structural and functional integrity of the enzyme, further reducing the prospects for such variants to exist in our experimental libraries. Nevertheless, the suggested Rosetta designs seemed sensible and hence were built and tested for their stability, as well as for catalytic performance with native substrate (1) and the targeted nucleoside analog (2).
Guided by the Rosetta predictions, we initially decided to lock in the most frequent substitution (L66F) and chose V175Y from among the suggested substitutions (F, Y, W). Within this framework, we tested two variants carrying either Y70V (RosD3) or Y70M (RosD4) which, based on the model, fit well in the newly created cavity between F66 and Y175. Both enzyme variants were assembled by site-directed mutagenesis, expressed in Escherichia coli host, and, after purification, characterized by steady-state kinetics (Table 1). Consistent with our model, the catalytic efficiency for 2 was preserved in RosD3 and increased ∼2.4-fold in RosD4 due to a drop in the Michaelis–Menten constant. At the same time, the kcat/KM values for 1 decreased by 20 and 58-fold for RosD3 and RosD4, respectively. The declines were largely due to higher KM values. The stability of both variants dropped from 70% residual activity after 10 minutes at 37 °C for DmdNK to 58% and 39% for RosD3 and RosD4, respectively.
| Enzyme | Thymidine | ddT | RS | TS (%) | ||||
|---|---|---|---|---|---|---|---|---|
| k cat/s−1 | K M/μM | k cat/KM (103 × s−1 M−1) | k cat/s−1 | K M/μM | k cat/KM (103 × s−1 M−1) | |||
| a Previously reported data;7 numbers in parentheses are fold changes in catalytic performance of the variant over DmdNK for the particular substrate. RS: relative specificity [kcat/KM (ddT)/kcat/KM (T)]. TS: thermostability expressed (in % residual activity) with a standard error of ±4%. | ||||||||
| DmdNK | 12.9 ± 0.9 | 2.7 ± 0.5 | 4813 | 0.53 ± 0.03 | 115 ± 22 | 4.6 | 0.001 | 70 |
| R4.V3-[85] | 0.13 ± 0.01 | 92 ± 14 | 1.4 | 1.36 ± 0.01 | 49 ± 3 | 28 | 20 | 34 |
| (E172V, Y179F, H193Y) | (−100) | (−34) | (−3438) | (+2.6) | (+2.3) | (+6) | ||
| RosD3 | 6.3 ± 0.2 | 27 ± 3 | 234 | 0.29 ± 0.01 | 79 ± 14 | 3.7 | 0.016 | 58 |
| (L66F, Y70V, V175Y) | (−2) | (−10) | (−20) | (−1.8) | (+1.5) | (−1.2) | ||
| RosD4 | 4.6 ± 0.1 | 56 ± 2 | 83 | 0.4 ± 0.01 | 36 ± 2 | 11 | 0.13 | 39 |
| (L66F, Y70M, V175Y) | (−3) | (−20) | (−58) | (−1.3) | (+3) | (+2.4) | ||
| RosD5 | 0.08 ± 0.01 | 96 ± 15 | 0.84 | 0.19 ± 0.01 | 35 ± 4 | 5.4 | 6.4 | 28 |
| (L66F, Y70M, E172V, V175Y) | (−160) | (−36) | (−5730) | (−2.8) | (+3.3) | (+1.1) | ||
| RosD6 | 0.21 ± 0.01 | 66 ± 7 | 3.2 | 0.41 ± 0.01 | 35 ± 4 | 12 | 3.7 | 50 |
| (L66F, Y70M, E172I, V175Y) | (−60) | (−24) | (−1500) | (−1.3) | (+3.3) | (+2.6) | ||
| RosD7 | 0.42 ± 0.02 | 173 ± 32 | 2.4 | 0.65 ± 0.02 | 32 ± 4 | 21 | 8.5 | 50 |
| (L66F, Y70M, E172I, V175W) | (−31) | (−64) | (−2000) | (+1.2) | (+3.6) | (+4.6) | ||
Next, we created a small site-directed mutagenesis library at position 172 skewed towards hydrophobic residues (Table S1, ESI‡). RosD4 was selected as the template for these experiments, based on its promising ddT activity and more favorable relative specificity. Interestingly, the kinetic properties of all eleven second-generation variants show 2-fold or less variation in their kinetics for 2 compared to RosD4. For 1, mutations at E172 affected mostly the enzymes' turnover rates. The observed 20 to 50-fold declines translated into comparable gains in relative substrate specificity. Among the tested variants, substitution of E172 to either V (RosD5), T, L, or I (RosD6) showed significant functional improvements. Although the most notable change in substrate specificity was observed for E172V/T, thermostability studies indicated that these two variants had significant lower residual activity compared to RosD4 (Table 1). In contrast, RosD6, despite being slightly less specific, retained higher residual activity than its parental enzyme. A possible explanation for the differences in stability of these variants can be derived from computational models (Fig. 2). Both, E172V and E172I, in conjunction with F66, remodel the enzyme active site to disfavor binding of substrates with 2′-deoxyribosyl moieties by eliminating the potential hydrogen-bonding partner and increasing steric constraint for the substrate's 3′-OH group. However, E172I shows noticeable tighter packing of the sec-butyl side chain compared to the isopropyl group of E172V, an observation consistent with the detected increases in protein thermostability.
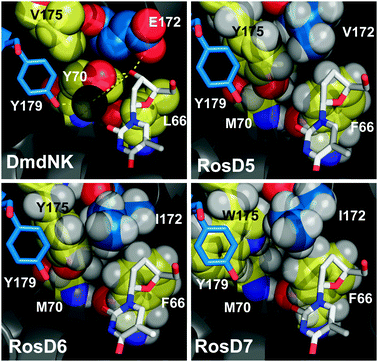 | ||
| Fig. 2 In silico remodeling of DmdNK focused on L66, Y70, E172, and V175 that form a hydrogen-bonding network with the substrate. In RosD5 and RosD6, substitutions of L66F, Y70M, and V175Y were explored in combination with E172V and E172I, respectively. The model suggests tighter packing for I172 in comparison to V172, consistent with the higher thermostability of the former. The conformational reorganization of Y175W in RosD7 further improves the catalytic performance. | ||
Finally, the ambiguity of the original Rosetta design regarding substitutions of residue 175 led us to revisit our initial choice and explore additional amino acid substitutions in that position. Working with RosD6 as the template, we prepared five mutations, replacing Y175 with I, L, M, F, or W. These variants were again characterized for their kinetic properties with 1 and 2 (Table S2, ESI‡). Among the substitutions, only Y175W (RosD7) and Y175F showed improvements in their relative specificity which is consistent with the predictions by Rosetta. In Y175F, the gain in specificity results from a combination of lower turnover rates and higher KM values, slightly more favorable for 2 than 1. More importantly, the elimination of the 4-hydroxyl group caused a drop in the protein's thermostability to 23% residual activity in our stability assay, the lowest value for any designer kinase in this study. In contrast, the replacement of the tyrosine side chain with an indole moiety in RosD7 translated into higher catalytic efficiency for 2 by raising substrate turnover. At the same time, RosD7 lowers the catalyst's performance for 1 by increasing its apparent binding constant. In addition to these very favorable functional changes, the protein stability remained unchanged at 50%. While the observed changes in catalytic function remain difficult to rationalize based on our current models, the computational structure predictions for these new variants can provide valuable guidance. For RosD7, the energy minimization by Rosetta causes the indole side chain of W175 to rotate 90° relative to Y175 (Fig. 2). The conformational reorganization positions the aromatic side chain in such a way that it can now stack against the benzyl portion of the neighboring Y179 while tightening the substrate binding pocket by slightly pushing I172 towards the bound nucleoside analog.
In summary, computational redesign of DmdNK by Rosetta in combination with site-directed mutagenesis has yielded a new, orthogonal designer kinase, RosD7, whose catalytic performance matches our previously evolved ddT kinase R4.V3-[T85].7 The designed enzyme exhibits 4.6-fold higher specific activity for 2 compared to the parental DmdNK and favors the nucleoside analog 8.5-fold over 1 (based on kcat/KM), an 8500-fold change in substrate specificity. Although the relative specificity of RosD7 is approximately 2-fold lower than the laboratory-evolved variant, our new in silico design possesses several superior properties. The lower KM of RosD7 for 2 compared to R4.V3-[T85] and a more favorable KM ratio of 5.4 for 2 over 1 compared to 1.9 for R4.V3-[T85] are critically important for in vivo applications as they minimize interference with nucleoside analog activation by native nucleosides (Liu and Lutz, unpublished results). Furthermore, the designer kinase is significantly more stable than our previously evolved ddT kinase. Our results demonstrate Rosetta's ability to successfully identify four positions in the active site of DmdNK critical for recognizing the sugar moiety of a nucleosidic substrate. While amino acid substitutions of E172 have previously been reported, mutations of L66, Y70 and V175 have to our knowledge never been observed, possibly due to their functional codependency. The latter positions' impact on substrate specificity clearly validates their relevance and supports our argument for the potential benefits of more extensive searches of protein sequence space made possible by computational methods. Our results also demonstrate some of the current limitations of in silico methods, accurately predicting suitable variations for some positions such as L66F while being ambiguous for others including Y70, E172 and V175. Nevertheless, local variability in predictions can easily be addressed experimentally by site-directed or site-saturation mutagenesis and, for DmdNK, proved highly effective in fine-tuning substrate specificity. Overall, the computational predictions can offer a powerful tool to complement experiments at the bench, guiding and accelerating the engineering process. Future structural studies of these engineered kinases will not only examine the accuracy of these models but also allow for refinements of the predictive framework. For the laboratory evolution of nucleoside analog kinases in general, the in silico approach presents a promising strategy to obtain lead enzymes for novel nucleoside analog prodrugs, especially for analogs showing little to no detectable activity with wild type kinases.
Financial support in part by the National Institutes of Health [GM69958 to SL] and the Howard Hughes Medical Institute (HHMI) is gratefully acknowledged.
Notes and references
- Y. Mehellou and E. De Clercq, J. Med. Chem., 2010, 53, 521–538 CrossRef CAS.
- E. S. Arner and S. Eriksson, Pharmacol. Ther., 1995, 67, 155–186 CrossRef CAS; S. Eriksson, B. Munch-Petersen, K. Johansson and H. Eklund, Cell. Mol. Life Sci., 2002, 59, 1327–1346 CrossRef CAS.
- K. W. Culver, Z. Ram, S. Wallbridge, H. Ishii, E. H. Oldfield and R. M. Blaese, Science, 1992, 256, 1550–1552 CrossRef CAS.
- F. L. Moolten, Cancer Res., 1986, 46, 5276–5281 CAS.
- N. Solaroli, M. Johansson, J. Balzarini and A. Karlsson, Gene Ther., 2007, 14, 86–92 CrossRef CAS.
- S. Song, Z. F. Pursell, W. C. Copeland, M. J. Longley, T. A. Kunkel and C. K. Mathews, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4990–4995 CrossRef CAS.
- L. Liu, Y. Li, D. Liotta and S. Lutz, Nucleic Acids Res., 2009, 37, 4472–4481 CrossRef CAS.
- M. A. Waqar, M. J. Evans, K. F. Manly, R. G. Hughes and J. A. Huberman, J. Cell. Physiol., 1984, 121, 402–408 CrossRef CAS.
- A. Korkegian, M. E. Black, D. Baker and B. L. Stoddard, Science, 2005, 308, 857–860 CrossRef CAS; P. M. Murphy, J. M. Bolduc, J. L. Gallaher, B. L. Stoddard and D. Baker, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 9215–9220 CrossRef CAS.
- N. E. Mikkelsen, K. Johansson, A. Karlsson, W. Knecht, G. Andersen, J. Piskur, B. Munch-Petersen and H. Eklund, Biochemistry, 2003, 42, 5706–5712 CrossRef CAS.
- R. Das and D. Baker, Annu. Rev. Biochem., 2008, 77, 363–382 CrossRef CAS.
- B. I. Dahiyat and S. L. Mayo, Science, 1997, 278, 82–87 CrossRef CAS; B. Kuhlman and D. Baker, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 10383–10388 CrossRef CAS; T. Kortemme, L. A. Joachimiak, A. N. Bullock, A. D. Schuler, B. L. Stoddard and D. Baker, Nat. Struct. Mol. Biol., 2004, 11, 371–379 CrossRef CAS.
- T. S. Wong, D. Roccatano, M. Zacharias and U. Schwaneberg, J. Mol. Biol., 2006, 355, 858–871 CrossRef CAS.
Footnotes |
| † This article is part of the ‘Enzymes and Proteins’ web-theme issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Methods and results for computational design, as well as preparation and characterization of enzyme variants. See DOI: 10.1039/c0cc02961k |
| This journal is © The Royal Society of Chemistry 2010 |