Catalysts for CO2/epoxide copolymerisation†
Michael R.
Kember
,
Antoine
Buchard
and
Charlotte K.
Williams
*
Department of Chemistry, Imperial College London, London, SW7 2AZ, UK. E-mail: c.k.williams@imperial.ac.uk; Fax: +44 (0)20 7594 5790
First published on 12th October 2010
Abstract
The article reviews recent developments (mostly since 2004 until June 2010) in catalysts for CO2/epoxide copolymerisation and in the properties of the polycarbonates.
 Michael R. Kember, Charlotte K. Williams and Antoine Buchard (left to right) | Michael Kember graduated from the University of Oxford in 2007, where he spent four years studying for an MChem at St. Edmund Hall. During his fourth year master's research project, he worked in Prof. Jon Dilworth's group on C–H activation with palladium catalysis. In 2007, Mike started a PhD at Imperial College London supervised by Charlotte Williams, where he has been investigating new catalysts for the copolymerisation of epoxides and CO2. Mike is currently writing his thesis before starting as a postdoctoral research associate in the Williams group. |
Antoine Buchard graduated in 2005 from the Ecole Polytechnique (Paris, France). He obtained an MSc in Organometallic Chemistry from the same university, in 2006, as part of this he spent 10 months working with Prof. K. Narasaka at Tokyo University. Antoine then completed a PhD in the Laboratoire Hétéroéléments et Coordination at the Ecole Polytechnique, in 2009, supervised by Prof. Pascal Le Floch and Dr Audrey Auffrant. His doctoral thesis concerned the coordination chemistry of iminophosphoranes and the development of new catalytic systems. During his PhD he collaborated with Charlotte Williams, working on lactide polymerisation, and was a visiting student at Imperial College London for 2 months in 2008. In November 2010, he returned to Imperial College London as a post-doctoral research associate, working with Charlotte Williams, on CO2/epoxide copolymerisation catalysis. |
Charlotte K Williams is a reader in catalysis and polymer chemistry at Imperial College London and an EPSRC advanced research fellow. Her research interests are in polymerisation catalysis, the activation and use of renewable resources, degradable polymer synthesis and the synthesis of electroactive polymers. Her research has been recognized by the RSC Energy, Environment and Sustainability Forum Early Career Award (2009), the RSC Meldola Medal (2005) and by the RSC Laurie Vergnano Award (2001). Charlotte studied for a PhD (1998–2001) at Imperial College London working with Professors Nick Long and Vernon Gibson FRS. She worked on the synthesis of new catalysts for ethylene polymerisation. She was a postdoctoral researcher at the University of Minnesota (2001–2002), where she worked with Professors Marc Hillmyer and Bill Tolman on the synthesis of Zn(II) initiators for lactide polymerisation. She returned to the UK, as a postdoctoral researcher working, with Professors Andrew Holmes FRS and Richard Friend FRS, at Cambridge University, on the synthesis of organometallic polymers for electronics. She was appointed to the chemistry department at Imperial College London in 2003. |
1. Introduction
1.1 Chemical syntheses using CO2
The development of methods to activate and use CO2 to prepare chemicals and materials is an attractive research goal. Carbon dioxide is abundant, renewable, of low toxicity and is emitted as a waste product from a myriad of industrial processes. A long-standing goal of synthetic chemistry has been to develop catalysts and processes which consume it, however, such reactions pose significant challenges.1–4 As the most highly oxidized state of carbon, CO2 is the lowest energy state of all carbon-containing binary neutral species: indeed, CO2 and water are the end-products of most energy releasing processes, including combustion and metabolic pathways. Table 1 illustrates the free energy of formation of C-1 molecules: the large energy required to reduce it is the most significant obstacle. This energy can either be directly input as physical energy or indirectly via the use of reactive chemical species as reagents; it is the latter strategy which powers the copolymerisation of epoxides and CO2.| Carbon compound | Carbon oxidation state | ΔGf (free energy of formation)/kJ mol−1 |
|---|---|---|
| CH4 (g) | −4 | −50.75 |
| CH3OH (l) | −2 | −166.1 |
| C | 0 | 0 |
| HCOOH (l) | +2 | −345.09 |
| CO (g) | +2 | −137.15 |
| CO2 (g) | +4 | −394.01 |
Nature is successful in transforming approximately 200 billion tonnes per year of CO2 into carbohydratesviaphotosynthesis. Synthetic chemistry has been less successful, so far there are only a limited range of reactions which can transform CO2 to useful products, those that yield materials with high market volumes and/or economics are even scarcer. Successful reactions include the synthesis of urea (146 Mt y−1, 2008), inorganic carbonates (45 Mt y−1, 2008, mostly Na2CO3via the Solvay process), methanol (6 Mt y−1), salicylic acid (60 kt y−1, 2003, via the Kolbe–Schmitt process), organic carbonates (100 kt y−1, 2009; the subject of recent reviews)6,7 and polycarbonates (a few kt y−1). Current production volumes for aliphatic polycarbonates produced from CO2 are small,8 however, the polycarbonates sector as a whole is large and growing. In Asia alone the sector is forecast to grow by 8–10%, resulting in the construction of new polycarbonate plants and opportunities for new technologies.6 Finally, it is important to note that CO2 consumption by chemical processes (approx. worldwide ∼100 Mt y−1) cannot impact global CO2 levels, nor are they a means to address climate change (UK CO2 emissions in 2008 from power stations exceeded 200 Mt y−1).9 However, they could be a means to add value to a portion of the CO2 from carbon sequestration and storage (CSS) processes.10
1.2 Copolymers from CO2
Various copolymers can be synthesised by the sequential copolymerisation of CO2 and strained heterocyclic molecules, including epoxides, aziridines, episulfides and oxetanes.11–13 The most widely studied is the copolymerisation with epoxides to produce polycarbonates (Fig. 1).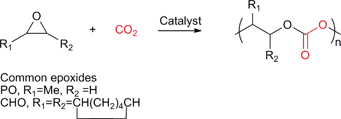 | ||
| Fig. 1 The sequential copolymerisation of epoxides and CO2. | ||
The manufacture of polymers is a growth industry, with worldwide production exceeding 150 Mt y−1.14 By far the most commonly used feedstocks are petrochemicals, derived from fossil fuels, with approximately 7% of worldwide oil and gas being consumed annually in polymer production. Such resources, although technically renewable, are estimated to be depleted and/or become uneconomical within the next century. In addition, issues with security of supply, cost and environmental impact drive the development of alternative polymer syntheses using renewable resources. CO2 represents an attractive co-monomer, not least because as a gas it would be compatible with many current polymerisation processes, and there may even be a future opportunity to couple carbon capture and storage (CSS) with CO2-to-polymer synthesis. So far, aliphatic polycarbonates are niche products, applied in lithographic and nano-technology applications as sacrificial binders.8 However, the safety and toxicity concerns associated with the production of conventional polycarbonate (from bis(phenol)A and phosgene), as well as consumer pressure for sustainable polymers, could enable more widespread opportunities for such materials. So far, polycarbonates produced from CO2 cannot match the properties of conventional polycarbonate; however, they have been proposed as alternatives for commodity applications, including packaging, engineering polymers and elastomers. The cost of production and the properties of the materials need to be improved to allow widespread impact. The catalysts for the copolymerisation are essential to control both the efficiency (cost) and the physical/chemical properties of polycarbonates: improvements to catalyst activity and selectivity offer tremendous potential for the production of copolymers from CO2.
1.3 CO2 copolymerisation catalysts
The copolymerisation of CO2 and epoxides is catalysed by various Lewis acidic metal halides, carboxylate or alk/aryloxide complexes, it is proposed to occur via a coordination–insertion mechanism, illustrated in Fig. 2.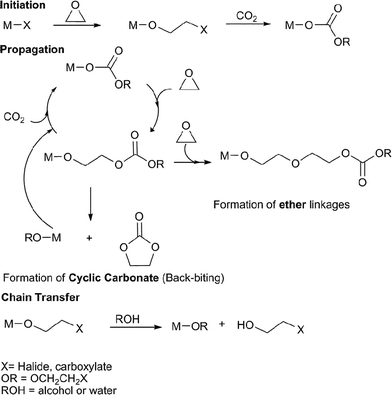 | ||
| Fig. 2 The proposed catalytic cycle for the copolymerisation of CO2 and epoxides. | ||
The metal complex initiates the copolymerisation by coordinating the epoxide and attacking it with the nucleophilic group, X (halide, carboxylate), leading to epoxide ring-opening and formation of a metal bound alkoxide. Certain metal alkoxides are able to undergo CO2 insertion to form a metal carbonate; it is this latter reaction which limits the range of catalysts. If the X group is an alkoxide/aryloxide, the initiation step is CO2 insertion.15 The cycle propagates by nucleophilic attack of the metal bound carbonate on a coordinated epoxide molecule, producing a new metal alkoxide, which inserts CO2; multiple repetitions of the series of reactions lead to copolymerisation. In principle this should lead to a copolymer with only carbonate linkages (100% carbonate), however, some catalysts can also homopolymerise epoxides or undergo decarboxylation reactions, and therefore form ether linkages in the copolymer backbone (thus, in the review the % carbonate incorporation is noted). In a controlled copolymerisation, the initiation reaction occurs more rapidly than propagation and leads to a degree of polymerisation (DP) dependent on the concentration of catalyst. The DP correlates with the number average molecular weight Mn (henceforth referred to as the molecular weight). The degree of control is judged by the fulfillment of the following criteria: linear increase in Mn with % conversion, linear increase in Mn with 1/[initiator]0, narrow polydispersity index (PDI = Mw/Mn), the ability to undergo sequential monomer addition (i.e. to enable block copolymer construction), high ki/kp (where ki = rate of initiation, kp = rate of propagation) and a high kp/ktr (where kp = rate of propagation, ktr = rate of intramolecular chain transfer). There are two possible chain transfer reactions: intramolecular backbiting and/or reaction with externally added alcohol/water/acid. The backbiting reaction occurs when the metal alkoxide chain end attacks a carbonate linkage on the copolymer chain forming a cyclic carbonate by-product and re-generating a metal alkoxide/X species. Five membered ring carbonates are thermodynamically stable and do not undergo any further ring-opening polymerisation; they are often produced as by-products. In the review, catalysts that are particularly selective for sequential copolymerisationvs. cyclic carbonate production (% selectivity) are highlighted. Chain transfer reactions can also occur if the reaction mixture is exposed to alcohols or water (this frequently occurs via contaminants), forming a hydroxyl terminated copolymer chain and a new metal alkoxide/hydroxide species, which can initiate/propagate. Chain transfer reactions result in a reduction in the number of repeat units and Mn and a so-called ‘immortal polymerisation’.16
1.4 Scope of the review
This review focuses on the catalysts for CO2–epoxide copolymerisation. The key criteria for assessing catalyst performance include the productivity (defined as turn-over-number, TON), activity (turn-over-frequency, TOF) and the quality of the copolymer produced (% carbonate, % selectivity, Mn) (Table 2). In this article, for all homogeneous catalysts the TONs are defined as moles of epoxide consumed per mole of metal in the catalyst (in order to compare mono-, di- and multi-metallic catalysts) and the TOF is TON h−1. For the heterogeneous catalysts the TONs are generally reported as g copolymer/g catalyst, and the TOF is TON h−1. Another important consideration are the conditions under which the catalyst will operate, including the pressure of CO2 (here, reported in the SI unit: atmospheres, atm. Where 1 atm = 0.1 Mpa, 1.01 bar, 14.7 psi, 760 mmHg), temperature (reported in °C) and any additives (co-catalysts or solvents). Conventionally the copolymerisation has been conducted using relatively forcing conditions, in particular high pressures of CO2. However, recently a number of groups, including our own, have reported catalysts capable of operating under mild conditions, including at 1 atm CO2 pressure and/or at room temperature. These advances could be significant because they reduce the energy input to the reaction, thereby enabling CO2 to be genuinely consumed.| Epoxide | Catalyst | T/°C | p[CO2]/atm | TONb | TOF c | % Carbonated | % Selectivityd | M n e | PDIe | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a All catalysts reported before Coates' 2004 review,17 with the exception of salen catalysts which were all reported before Darensbourg's salen review in 2007.18 b Moles epoxide consumed per metal. c Moles epoxide consumed per metal per hour. d Determined by 1H NMR spectroscopy. e Determined by gel permeation chromatography (GPC) using polystyrene standards. f Not specified. g Figures taken from similar catalyst run as actual figures not mentioned. | ||||||||||
| CHO | [(tpp)AlCl]/EtPh3PBr | 20 | 48 | 100 | 0.3 | >99 | >99 | 6200 | 1.06 | 24 |
| CHO | Zinc bis-phenoxide | 80 | 55 | 620 | 9.6 | >90 | —f | 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
3.8 | 26 |
| CHO | 2 | 50 | 7 | 380 | 2290 | 90 | >99 | 22900 |
1.09 | 34 |
| CHO | 18b/[PPN]N3 | 80 | 35 | 2686 | 1153 | >99 | >99 | 50000 |
1.13 | 36 |
| PO | 3 | 25 | 7 | 470 | 235 | >99 | 75 | 36700 |
1.13 | 35 |
| PO | 23b/[PPN]Cl | 22 | 14 | 620 | 620 | >98 | 99 | 26800 |
1.13 | 37 |
| PO | 19/[PPN]Cl | 60 | 34 | 770 | 192 | >99 | 93 | 26000g |
1.11g | 38 |
| PO | 26 | 60 | 14 | 602 | 602 | >99 | 90 | 7100 | 1.22 | 39 |
Recently there have been a number of interesting developments in the copolymerisation catalysis (Sections 2 and 3) and copolymer properties (Section 4) which will be highlighted in this feature article. The field has been comprehensively reviewed several times before;11,17–22 the most recent comprehensive review was published by Coates and Moore in 200417 and therefore this review focuses on developments since then (until June 2010) (Table 3). In particular, it was shown that highly active zinc β-diiminate catalysts exhibit a bimetallic active site,15 a finding that corroborates earlier mechanistic proposals (Fig. 3).11 The proposed bimetallic mechanism has led to the deliberate preparation of various new bimetallic catalysts some of which show improved productivity and activity. Metal salen complexes are a very promising class of catalyst and have been comprehensively reviewed by Darensbourg, in 2007.18 Recent developments, highlighted here, include the preparation of Cr(III) or Co(III) salen complexes and the single component catalysts where the co-catalyst is incorporated into the salen backbone via a saturated linking group. There have also been developments with more exotic ligands, metals and heterogeneous catalysts which will be briefly summarised. The preparation of catalysts able to operate under very low pressures of CO2 significantly improves the energy balance of the co-polymerisation and throughout the review we will draw particular attention to catalyst operating at 1 atmosphere pressure CO2. So far, only a narrow range of epoxides have been studied, with the majority of studies addressing copolymerisations of CO2 and either cyclohexene oxide (CHO) or propylene oxide (PO) which restricts the range of properties and potential applications for the resulting copolymers (Fig. 1). The copolymers produced from the commonly applied epoxides are poly(cyclohexene carbonate) (PCHC) and poly(propylene carbonate) (PPC). The final section focusses on new catalysts and epoxide monomers which can tailor the properties of the resulting copolymers, particular attention is paid to stereocontrol (for CHO and PO), regiocontrol (for PO) and the production of terpolymers and copolymers.
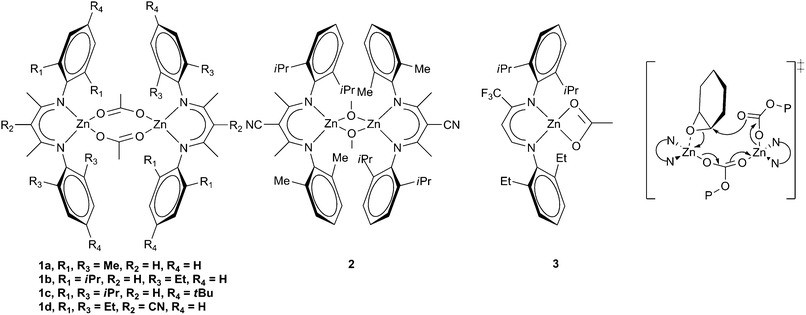 | ||
| Fig. 3 Zinc β-diiminate complexes and proposed bimetallic transition state. | ||
| Epoxide | Catalyst | T/°C | p[CO2]/atm | TONb | TOF c/h−1 | % Selectivityd | M n e | PDIe | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a All catalysts reported after Coates' 2004 review,17 with the exception of salen catalysts which are reported after Darensbourg's salen review in 2007.18 b Moles epoxide consumed per metal. c Moles epoxide consumed per metal per hour. d Calculated by 1H NMR spectroscopy. e Determined by GPC with broad polystyrene standards. f Not specified. g Bimodal weight distribution observed. | |||||||||
| CHO | 21e/[PPN]N3 | 60 | 34 | 1620 | 405 | —f | 19500 |
1.19 | 40 |
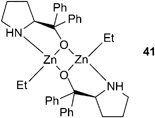
| CHO | 22/[PPN]Cl | 70 | 13 | 420 | 210 | >99 | 8800 | 1.14 | 41 |
| CHO | 5 | 80 | 14 | 9440 | 2860 | —f | 245000 |
1.20 | 42 |
| CHO | 11a | 100 | 10 | 1675 | 70 | 96 | 14000g |
1.03 | 43 |
| CHO | 17 | 100 | 10 | 186 | 1870 | >99 | 8800g | 1.04 | 44 |
| PO | 23c/[PPN]Cl | 45 | 20 | 980 | 1400 | 97 | 25900 |
1.08 | 45 |
| PO | 27 | 80 | 20 | 3300 | 3300 | 94 | 71000 |
1.25 | 46 |
| PO | 28e | 70 | 20 | 15000 |
15000 |
>99 | 300000 |
1.31 | 47 |
| PO | 32b | 100 | 25 | 10880 |
10880 |
97 | 60200 |
1.23 | 48 |
2. Homogeneous catalysts
2.1 Early discoveries
The copolymerisation of epoxides and CO2 was first discovered by Inoue et al. in the late 1960s, where a 1∶1 mixture of ZnEt2 and water copolymerised PO and CO2, producing low weight poly(propylene carbonate) with a TOF of 0.12 h−1 (mol. epoxide consumed per mol. cat per hour) at 20–50 atm CO2 and 80 °C.23 This discovery led to the development, during the 1970s and 1980s, of a variety of other heterogeneous catalysts, based upon a mixture of either ZnEt2 or ZnO and alcohols, with TOFs increased to 1 h−1; however the precise nature of the active sites was unclear. The difficulty in defining the active catalyst sites made structure/activity relationships, and therefore catalyst design, very tough. This, combined with the highly polydisperse copolymers produced, due to inequivalent active sites and a generally low CO2 incorporation, drove the development of well defined homogeneous catalysts and enabled significant developments in mechanistic understanding and catalyst design/activity.
The first homogeneous catalysts (aluminium tetraphenylporphyrin (tpp) complexes) were also reported by Takeda and Inoue, in 1978.24 These were active for the copolymerisation of both CHO and PO, with EtPh3PBr as a co-catalyst, producing copolymers with molecular weights between 3500 and 6000, with polydispersity indices (PDIs) below 1.10, although the reactions took 13 days to reach completion.
The development of discrete zinc bis-phenoxide complexes was an important breakthrough as the first highly active well-defined zinc complexes, however these catalysts produced polymers with very high polydispersity indices (probably due to catalyst aggregation phenomena) and as they were found to be active for epoxide homopolymerisation, leading to high proportions of ether linkages.25–31
The next major breakthrough came from Coates and co-workers, who reported a series of zinc β-diiminate complexes which showed much greater activity. A variety of substituents and initiating groups were investigated, and the influence correlated with activity.32,33 Depending on the steric bulk of the substituents R1 and R3 (Fig. 3), and the initiating groups, the complexes could form dimeric structures both in the solid state and in solution. Complexes with extremely bulky substituents (e.g.1c, Fig. 3) formed tightly held dimers and showed limited activity with CHO, whilst sterically unencumbered complexes (1a) gave a monomeric structure which showed similarly low activity.15 High activity with CHO was attained where the steric environment promoted loosely held dimeric structures such as with 1b, where a TOF of 729 h−1 was reported at 50 °C and 7 atm, producing a copolymer with 99% carbonate linkages, a Mn of 23300 and a PDI of 1.15, showing excellent control. The introduction of an electron withdrawing cyano-group on the backbone increased the activity further; 1d giving a TOF of 917 h−1 at 50 °C and 7 atm. A similar zinc methoxide complex, 2, gave an unprecedented TOF of 2290 h−1 under the same conditions, although only 90% carbonate linkages were observed.34 As with other zinc complexes, up to 5% cyclic carbonate was observed. The complexes were less active with PO, the best TOF achieved being 235 h−1 with complex 3 at 25 °C and 7 atm.35 A copolymer selectivity of only 75% was observed, but a reasonable molecular weight of 36700 was reported. It was suggested that the mechanism involved a bimetallic pathway (Fig. 3), with one metal pre-coordinating the epoxide, enabling a second metal to feed the growing copolymer chain to ring-open the epoxide. This mechanistic proposal has since led to the preparation of many bimetallic zinc complexes.
2.2 Bimetallic complexes
In 2005, Lee and co-workers reported a series of zinc anilido-aldimine complexes (Fig. 4, 4a–f) which showed extremely high TONs (700–3000) for the production of PCHC, as well as having high molecular weights (90000–280000).49 The PDIs (1.30–1.70) were somewhat broad, and the carbonate inclusion ranged from 85–96%, a common feature of zinc catalysts. The catalysts were the first discrete complexes to yield copolymers of such high molecular weights and TONs, because they could operate under lower loadings (down to 1∶16800, catalyst∶CHO) than previously reported catalysts. Structural variation revealed that bulky R groups on the phenyl rings linking with the ligand backbone reduced the activity (4f), whilst a bulkier R′ group on the terminal phenyl rings (4c and 4e) increased the activity. This latter effect reflected similar structure/activity studies carried out with the zinc BDI complexes. Interestingly, similar macrocyclic complexes showed no activity at all.
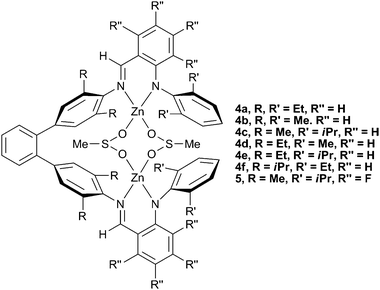 | ||
| Fig. 4 Bimetallic zinc anilido-aldimine complexes. | ||
A series of fluorinated analogues were later synthesised by the same group.42 The same structure/activity relationships were observed, with 5 being the most active. Under the same conditions (1∶5600, 80 °C, 14 atm) fluorinated 5 showed a TOF of 785 h−1, approximately 2.5 times greater than its unfluorinated analogue (4c). This increase in activity was attributed to two effects: firstly, the electron-withdrawing fluorine substituents could reduce the electron density at the metal centres, aiding CO2/epoxide binding, and secondly, the electron-withdrawing fluorines could decrease the basicity of the anilido nitrogen donor, making the complex less sensitive to protic impurities. This second influence was used to rationalise the increased stability of the complex under even lower catalyst loadings (1∶50000) which gave much higher TON/TOFs of 9440 and 2860 h−1, respectively, amongst the highest activities attained with CHO. The fluorinated complexes produced PCHC of similar molecular weights (>100000), however the PDIs were broader than their unfluorinated analogues (1.20–2.50) and the carbonate content was reduced to 60–80%. The complexes were later shown to be inactive with PO.46
In the same year, Xiao et al. reported a dizinc complex, coordinated with the Trost phenolate ligand, which was moderately active for CHO/CO2 copolymerisation, although the precise nature of the catalyst was not described as it was prepared in situ by the reaction between the ligand, ZnEt2 and ethanol (Fig. 5).50 The structure proposed was 6, and although a variety of alcohols were investigated, ethanol gave the best activity, producing PCHC with a TOF of 142 h−1 at 1∶500 loading, 20 atm and 80 °C. Most interestingly, the catalyst was active under just 1 atm CO2 pressure, albeit at a catalyst loading of 5 mol% (1∶20), giving a TOF of 3 h−1. Replacing the two zinc centres with magnesium (7) reduced the activity, under lower loadings, however the catalyst was more active at 1 atm CO2 pressure, producing a similar TOF at 60 °C instead of 80 °C.51 The copolymers produced had reasonable weights (20000–40000), although the PDIs were around 1.60.
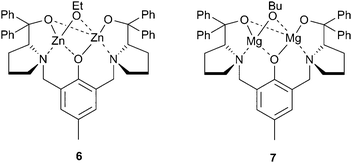 | ||
| Fig. 5 Bimetallic complexes of Trost's phenolate ligand. | ||
Various attempts to further alter the catalyst structure of zinc BDIs have been reported, with limited success. A series of complexes with both a cyano group and an ethoxy group on the backbone were prepared (Fig. 6). Changing the aryl substituents showed that 8 was the most active, producing PCHC with a TOF of 210 h−1, a result which correlates well with earlier detailed studies on BDI zinc complexes.34 In general the carbonate inclusion was low, between 60–80%, and the reactions were carried out under high pressures and temperatures (40 atm, 90–100 °C).52,53 A similar ligand formed clusters containing 12 zinc sites, and was significantly less active, giving a best TOF of 50 h−1, with only 34% carbonate linkages and an extremely broad PDI (20), which is not surprising given the aggregated structure.54
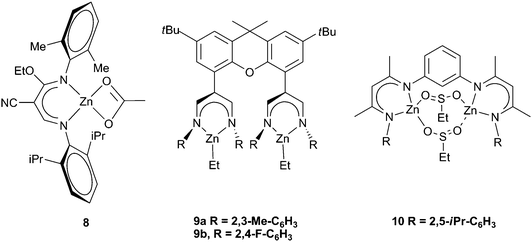 | ||
| Fig. 6 Various bimetallic zinc catalysts. | ||
A novel bimetallic BDI ligand structure (Fig. 6, 9a,b) which places two zinc sites parallel to each other, rather than face to face, was prepared with the aim of testing any cooperative bimetallic mechanism.55 The resulting dizinc complexes showed very low activity with CHO (max TOF 9 h−1), probably due to the catalyst steric bulk. The bridging of two BDI sites via para- and meta-substituted phenyl groups and m-pyridinyl gave substantially more active complexes (with CHO), of which the best was 10 (Fig. 6).56 It showed a TON of 1196 and a maximum TOF of 262 h−1, at 10 atm and 60 °C, with 99% carbonate linkages, quite high molecular weights (45000–100000) and PDIs of 1.20–1.40. Analogous calcium complexes were completely inactive.
Our research group have reported a series of bimetallic complexes coordinated by a novel ‘reduced Robson's type’ macrocyclic ligand, which were highly active with CHO under 1 atm CO2 pressure (see Table 5 for a comparison of catalysts active at this pressure). Complexes 11a–c (Fig. 7) showed good activity at 1 atm CO2, 11a giving a best TOF of 13 h−1 at 100 °C.43 Variation of the p-phenyl substituent showed that an electron-donating methoxy group (11c) significantly reduced the activity.57 The macrocyclic ligand environment and bimetallic structure were both proposed to be essential to the activity of the catalysts, as ‘open’ and monometallic analogues 13 and 14 (which are very active for lactide polymerisation) showed no activity.58,59 Trimetallic zinc complexes were easily formed in the presence of an excess of Zn(OAc)2. These trimetallic complexes (12a–c) showed reasonable activity at 1 atm pressure, but significantly less than their bimetallic analogues. This suggests that the external metal centre is inactive, or at least much less active, towards CHO, and possibly blocks one face of the complex. This reinforces the proposal that the macrocyclic bimetallic environment is key to the high activity.
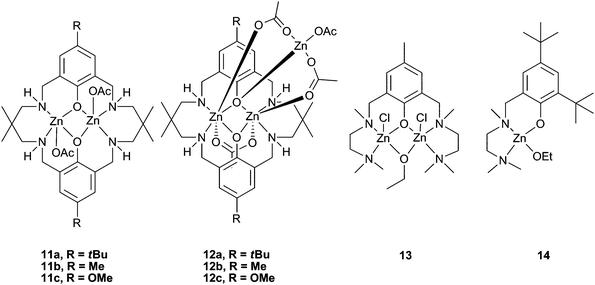 | ||
| Fig. 7 Highly active reduced Robson type zinc complexes (11 and 12) and inactive analogues (13 and 14). | ||
The copolymers produced were of low molecular weights (3000 to 9000), but had very narrow PDIs (<1.20). MALDI-ToF mass spectrometry of the PCHC showed acetate, hydroxyl and alcohol (cyclohexanol, cyclohexenol and cyclopentanol) end groups, the latter of which were proposed to derive from Meerwein–Pondorff–Verley–Oppenauer (MPVO) side reactions between the zinc catalyst and CHO. Thus, the alcohols and contaminating water were efficient chain transfer agents reducing the molecular weights.60Catalyst 11a successfully copolymerised unpurified (i.e. wet) CHO with a small loss in molecular weight, showing it was robust and tolerant, unlike many other zinc catalysts. This was also shown in the high TONs achieved (up to 1700 under 10 atm pressure and a 1∶10000 loading).
Substituting the zinc centres with cobalt led to the preparation of cobalt(II) analogues of 11a and 12a (15 and 16 respectively, Fig. 8), as well as a mixed valence cobalt(II)/cobalt(III) complex (17).44Catalyst 16 was less active than its zinc analogue, presumably as it is less robust, whilst 15 and 17 showed unprecedented activity for CHO copolymerisation under 1 atm pressure of CO2. At 100 °C, 17 produced PCHC with a TOF of 250 h−1, whilst 15 was slightly less active with a TOF of 200 h−1. Raising the pressure to 10 atm with 17, a maximum TOF of 1700 h−1 was achieved. The catalysts were much more selective than their dizinc analogues, producing PCHC with no observable cyclic carbonate. The copolymer molecular weights were similar to those produced with 11a, the main chain transfer agent in this case being trace amounts of water.
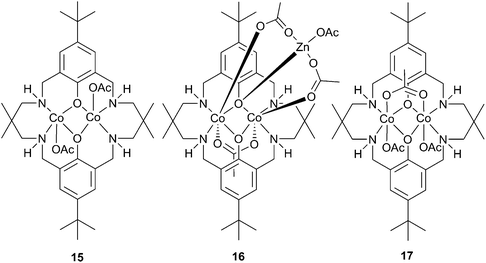 | ||
| Fig. 8 Cobalt acetate complexes of reduced Robson ligand. | ||
The high activity of these complexes at low pressures was attributed to the coordinative flexibility of the ligand, and the proximity of the two metal centres, both of which could facilitate a bidentate carboxylate chain binding mode, lowering the energy barrier to CO2 insertion.41 The significant increase in activity found by replacing zinc with cobalt could be due to the increased nucleophilicity of the cobalt-carbonate propagating species, and that the rate determining step is likely to be the epoxide ring opening by the nucleophilic carbonate chain. If the rate were dependent on epoxide binding, the more Lewis acidic 17 would be expected to show significantly increased activity.
2.3 Chromium salen complexes
The first example, in the academic literature, of the use of a chromium complex as a catalyst came from the Holmes research group, who reported a fluorinated chromium porphyrin complex, with a DMAP co-catalyst, which showed a TOF exceeding 150 h−1 for CHO, under harsh conditions (110 °C in scCO2 (222 atm)).61 The catalyst system yielded copolymers with a high degree of carbonate linkages and narrow PDIs. In the same year, Jacobsen and co-workers published a patent revealing that chiral chromium salen complexes were viable catalysts for the production of polycarbonates, with PPC being prepared under 1 atm CO2 pressure, however, no further details were provided.62 The Darensbourg group, inspired by the work of Jacobsen on asymmetric ring opening of epoxides63 (although, at that time, unaware of the patent), developed the chromium salen (salen = salicylaldimine, see Fig. 9) catalysts which showed high activity, stereoselectivity and stability and which have become (together with cobalt analogues) work-horses for this catalysis.18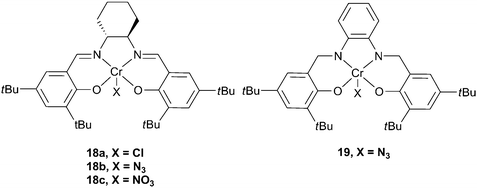 | ||
| Fig. 9 Chromium salen complexes. | ||
Catalyst
18a was active for CHO, but only with a nucleophilic co-catalyst, N-methylimidazole (N-MeIm).64 The co-catalyst function is generally proposed to be to bind at a site trans to the initiating group, weakening the axial bond and allowing for facile epoxide insertion, although recently both ionic and neutral co-catalysts have also been shown to be involved in the initiation step (vide infra).18 A wide variety of ligand substitutions, initiating groups and co-catalysts have been investigated,36,65–67 leading to increased activity, with complex 18b and [PPN]N3 (PPN: bis(triphenylphosphoranylidene)ammonium) giving a maximum TOF of 1153 h−1 at 80 °C and 35 atm. These complexes have also shown activity for PO copolymerisation,6819 being the most active of these, producing PPC with a TOF of 192 h−1 at 60 °C and 34 atm, with 93% copolymer selectivity and 99% carbonate linkages.38 The molecular weights are in the range 13000–26000, with PDIs of around 1.10. For a comprehensive review of earlier studies with metal salen catalysts, see Darensbourg's 2007 review.18 There have subsequently been several important developments which are outlined below.
More recently, detailed studies have been carried out in an attempt to understand the role of the co-catalyst and elucidate the reaction mechanism. One report found that several equivalents of ionic co-catalyst (including [PPN]Cl) reduced the activation barrier associated with the backbiting reactions, thereby giving an increase in cyclic carbonate content. They concluded that ionic co-catalysts should be restricted to 1 or 2 equivalents.69 Several anionic hexa-coordinated salen intermediates have been isolated and characterised by X-ray crystallography, including [salenCrX2]−[PPN]+, which was proposed to be the active catalyst.70 [salenCrCl] in the presence of 1 equivalent of [PPN]X (X ≠ Cl) forms a Schlenk equilibrium, with X-ray crystal structures reported for [salenCrCl2]−, [salenCr(N3)2]− and [salenCr(N3)Cl]−. The addition of epoxides to these octahedral complexes (bearing the co-catalyst trans to the initiating group) yielded [salenCr(N3)(epoxide)] at room temperature, however this reaction was an equilibrium that strongly favoured the anionic complexes. This coordination to yield the neutral species [salenCr(N3)(epoxide)] and the subsequent epoxide ring-opening were observed using in situATR FTIR spectroscopy, by following the different N3 stretching environments. It was suggested that the displaced anion initiated the copolymerisation by ring-opening the bound epoxide (Fig. 10).
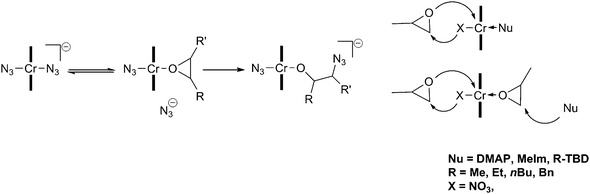 | ||
| Fig. 10 Proposed initiation mechanism with chromium salen complexes and ionic (left) and neutral (right) co-catalysts. | ||
The use of catalyst 18c with bulky nucleophiles, such as N-substituted 1,5,7-triazabicyclo-[4.4.0]-dec-5-enes (TBDs), increased the stereoselectivity of the copolymerisation when using rac-PO.71 Using N-methyl-TBD (MTBD) as the co-catalyst, ESI-Q-ToF mass spectrometry showed MTBD as a copolymer end-group, suggesting that instead of coordinating to the metal centre, it acted as a nucleophile in epoxide ring opening. As both MTBD and the anionic nucleophile (X = NO3−, in this case) can initiate, it was proposed that chains initiate and propagate on both sides of the catalyst (Fig. 10).
More recently, Rao et al. reported that [salanCrX] (salan = reduced salen) complexes (21a–d, Fig. 11), with DMAP as a co-catalyst, were up to 30 times more active for the copolymerisation of PO and CO2 than the salen counterparts (20a–d).72 Unlike the salen complexes, 21a–d did not show an induction time. The difference in the activities was proposed to be due to the sp3 hybridised amino donors in the salan ligands reducing the electrophilicity of the metal centre, thus facilitating reversible epoxide/DMAP binding. It was proposed that the dissociation and re-association of DMAP and growing chains was essential for high activity, and thus a less electrophilic metal centre produced a more active catalyst.72−74 ESI mass spectrometry of copolymers, at low conversions, showed DMAP as a chain end-group, again showing that bases can initiate the copolymerisation and suggesting reversible coordination to the metal centre.
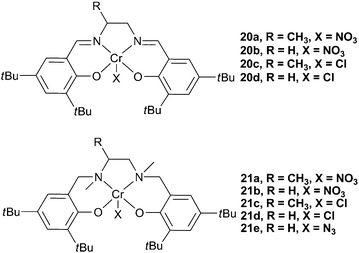 | ||
| Fig. 11 Chromium salen and salan complexes. | ||
The active species was proposed to be [(salan/salen)CrX(DMAP)] (C, Fig. 12), a species readily observed using in situESI-MS upon the combination of salan complexes and DMAP, a phenomenon that had also been reported using MeIm. It was observed, in the mass spectra, that salen complexes predominantly formed [salenCr(DMAP)2]+ (B), corroborating the suggestion that DMAP binding is much stronger with the more electrophilic salen complexes. The induction time with salen complexes was, therefore, proposed to be due to the slow loss of one DMAP to form the active [salenCrX(DMAP)] species. A TOF of 86 h−1 was reported with 21a, at 25 °C and 6 atm. 21a was also reported to be stereo- and regio-selective in the copolymerisation of rac-PO, producing highly isotactic PPC.73
![Proposed mechanism of copolymerisation with [(salen/salan)CrX] and DMAP.](/image/article/2011/CC/c0cc02207a/c0cc02207a-f12.gif) | ||
| Fig. 12 Proposed mechanism of copolymerisation with [(salen/salan)CrX] and DMAP. | ||
Complex 21e was subsequently reported to be active for the copolymerisation of CO2 and both CHO and PO, and for the terpolymerisation of both epoxides, in combination with [PPN]N3 co-catalysts. A maximum TOF of 405 h−1 was observed with CHO, at 60 °C and 34 atm, and 21 h−1 with PO, at 25 °C and 14 atm.40 The flexible salan ligand formed a pentacoordinate complex that, unlike salen complexes, has the vacant coordination site in an equatorial position of the coordination octahedra (see Fig. 13 for comparable structure with salalen complexes), something that was proposed by Rao et al. with complexes 21a–d and DMAP, but not verified.
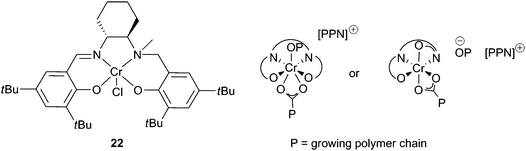 | ||
| Fig. 13 Salalen chromium complex and proposed carboxylate coordination modes. | ||
Half-reduced ‘salalen’ complexes were active with CHO, e.g. complex 22 and 1 equivalent of [PPN]Cl produced PCHC with a maximum TOF of 230 h−1, at 70 °C and 34 atm (Fig. 13).41 The copolymers had molecular weights around 8000, with PDIs 1.10–1.15. MALDI-ToF mass spectrometry revealed both chloride and hydroxyl end groups, suggesting chain transfer reactions with water were responsible for the low molecular weights. Catalyst 22 was remarkably active at reduced CO2 pressures, giving a TOF of 100 h−1 at 1 atm. This excellent activity, under mild conditions, was attributed to the flexibility of the ligand system, which like the salan complexes and unlike the salen complexes is not held in a rigid plane, and can bind in both axial and equatorial sites. This coordinative flexibility was proposed to facilitate bidentate binding of the growing carbonate chain, reducing the energy barrier to CO2 insertion and inhibiting decarboxylation, producing copolymers with 99% carbonate linkages.
A lysine based Cr salen catalyst, with a longer carbon backbone was considerably less active than conventional salen catalysts with two carbon diimine backbones, producing PCHC with a TOF of 76 h−1 under harsh conditions (80 °C, 45 atm).75 Ionic liquids, based upon dialkylimidazolium salts (such as 1-butyl-3-methylimidazolium (bmim) bromide), were used as co-catalysts with 7 for CHO copolymerisation.76 The length of the alkyl chains on the bmim cation affected the activity, with C12 substituents yielding equivalent activity to [Bu4N]Br, albeit at a higher CO2 pressure. A moderate TOF of 245 h−1 was reported, although this was at 55 atm CO2 pressure.
2.4 Two component cobalt salen catalysts
The substitution of chromium for cobalt in salen catalysts increased both the activity and selectivity for copolymer formation, particularly with PO. The formation of PPC can be complicated by the formation of cyclic carbonate (PC) by-product under thermodynamic conditions; it is notable that CHO has a much reduced tendency to form cyclic by-product due to the strained bicyclic rings. The first cobalt catalyst (23a, Fig. 14), reported by Coates and co-workers in 2003, was >99% selective for PPC formation, with 99% carbonate linkages and TOFs around 70 h−1, albeit at high catalyst loadings (200–500∶1, PO∶cat).77 With the addition of co-catalysts, the activities were increased significantly.78 The following year, Coates and co-workers published an excellent study using enantiopure (R,R) complexes 23a, b and e and comparing the influence of the initiating group and co-catalyst.37 In the absence of a co-catalyst the TOFs were dependent on the X group, with highest activities resulting from 23e (90 h−1, 500∶1, PO∶23e, 22 °C, 54 atm), this was due to a difference in the initiation period with the different complexes. Using [PPN]Cl and at lower loadings, catalyst 23b produced PPC selectivity from rac-PO with a TOF of 620 h−1, giving a copolymer with a molecular weight of 26800 and a PDI of 1.13.37 Using GPC analyses, it was concluded that when the complexes were used alone, one copolymer chain grew per cobalt centre, whereas when the co-catalysts were applied, two copolymer chains grew per cobalt centre (due to initiation from the co-catalyst). Interestingly, the same report studied the stereochemistry (and regiochemistry) using racemic and chiral catalysts/monomers. When S-PO was used instead of rac-PO with 23e, the TOF increased to 1100 h−1 (under the same conditions) and isotactic PPC was produced, highlighting the stereoselectivity of the catalyst. The discovery of cobalt salen catalysts was also covered extensively in the 2007 review.18
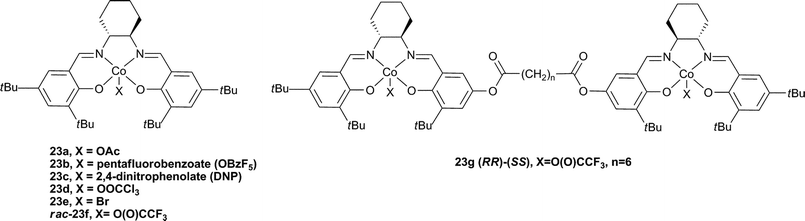 | ||
| Fig. 14 Cobalt salen complexes. | ||
In late 2006, Lu and co-workers reported a detailed study on salenCoX complexes and PO, investigating a number of different ligand substitutions, initiating groups (such as Cl, Br, NO3, ClO4), and a variety of different co-catalysts (R4NX, [PPN]X, MeIm, MTBD).45 Using 23c and ionic salts, it was ascertained that the ideal ionic co-catalyst in terms of activity and selectivity was a combination of a bulky cation ([PPN]+ > [nHept4N]+ > [nBu4N]+) and a nucleophilic anion with poor leaving-group ability (Cl− > Br− > I− > ClO4−). When neutral Lewis base co-catalysts were used, it was found that like the chromium analogues, bulky nucleophiles such as MTBD were highly effective co-catalysts, and also showed improved enantioselectivity. In contrast with the chromium catalysts, in the presence of non-sterically hindered bases such as MeIm the system was completely inactive, presumably because MeIm is bound too tightly to the metal centre and hinders epoxide binding. Even in the presence of one equivalent of active co-catalysts MTBD or [PPN]Cl, the addition of 1 or 2 equivalents of MeIm with 23c rendered the catalyst almost completely inactive. An increased loading of MTBD or [PPN]Cl in the absence of MeIm led to a significant increase in activity. For example, one equivalent of MTBD at 25 °C and 6 atm gave a TOF of 77 h−1 whilst four equivalents under the same conditions gave a TOF of 205 h−1. A higher co-catalyst loading also resulted in a decrease in copolymer molecular weight, and in the case of ionic co-catalysts, an increase in cyclic carbonate production; a result in agreement with the findings from chromium salen catalysts.69
A maximum TOF of 1400 h−1 was observed with one equivalent of [PPN]Cl at 45 °C and 20 atm, giving a copolymer with a Mn of 25900 and a PDI of 1.08, with 97% copolymer selectivity. As with 18c, ESI-Q-ToF mass spectrometry revealed MTBD as an initiating group, promoting a mechanism very similar to that subsequently proposed with MTBD and DMAP for chromium (Fig. 12) where the copolymer chain can dissociate and itself acts as a nucleophile, ring-opening the next metal-bound epoxide. In the case of both neutral and ionic co-catalysts, the molecular weight, and therefore the number of copolymer chains, was dependent on the concentration of the co-catalyst rather than 23c, leading to the suggestion that the two catalyst systems operate via a similar mechanism. It was noted that neither 23c, nor MTBD, showed any activity for the copolymerisation by themselves.
Enantiopure complex 24 (Fig. 15), in conjunction with DMAP copolymerised PO and CO2 giving a maximum TOF of 501 h−1, at 60 °C and 20 atm, producing a copolymer with a Mn of 5400 and a PDI of 1.10.79 Higher molecular weights were attained at longer reaction times and milder conditions, the highest being 70000, although this was achieved with a TOF of only 5 h−1. The catalyst was generally >99% selective for copolymer production. Using a longer but less bulky carbon backbone, 25 was less active, giving a TOF of 123 h−1, producing a copolymer with a molecular weight of 11500 and a PDI of 1.18, under similar conditions.80
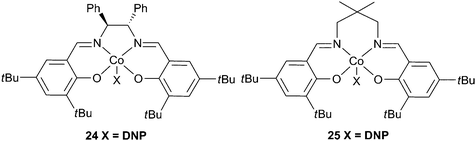 | ||
| Fig. 15 Cobalt salen complexes. | ||
Very recently Nozaki and co-workers have compared a mononuclear Co-salen complex, rac-23f , with a series of di-cobalt salen complexes of varying linker distance (Fig. 14, e.g.23g).81 The most active complex was 23g (n = 6) which showed a TOF of 180 h−1 (3000∶1, PO∶Co, 20 °C, 52 atm), 84% carbonate linkages and a molecular weight of 36700 (PDI = 1.07). This was markedly higher than the mononuclear analogue (cf.23f, TOF = 20 h−1 under the same conditions) and also higher than analogues with longer bridge lengths. This dependence of activity on a bimetallic complex, on the bridge length and increased activity at lower loadings indicated a bimetallic mechanism. Curiously, the iso-selectivity and regioselectivity (proportion of Head to Tail (HT) linkages, see Fig. 22) was decreased in the bimetallic complexes. On addition of [PPN]Cl (0.5 eq. to Co), 23f and 23g showed almost equivalent activities and copolymer properties, indicative of a monometallic active site, regardless of the nuclearity of the catalyst.
2.5 Single component salen catalysts
In 2006, Nakano et al. reported a novel cobalt salen catalyst with two ‘side arms’ bearing piperidine and piperidinium groups (26). The protonated piperidinium arm was proposed to prevent cyclic carbonate formation by protonating the copolymer chain upon its dissociation from the metal centre, preventing the backbiting reaction that leads to cyclic carbonate formation (Fig. 16). This allowed copolymerisation of PO to occur at 60 °C, with only 10% cyclic carbonate produced.39 This has since led to the development of several other single component cobalt salen catalysts with specially designed side arms which act as in-built co-catalysts, enabling further improvements in both activity and selectivity.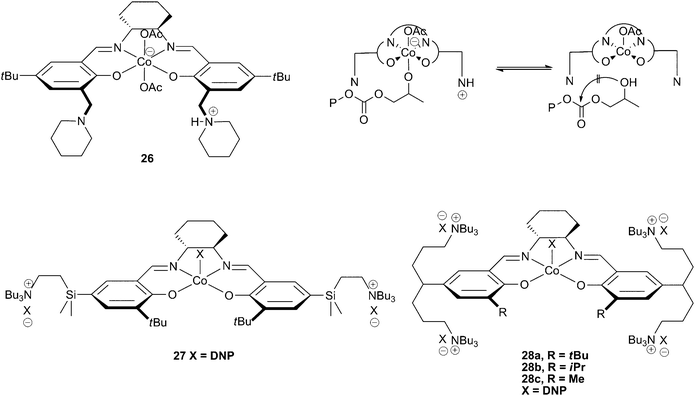 | ||
| Fig. 16 Cobalt salen complexes with cationic ‘arms’ and proposed mechanism for prevention of cyclic formation with 26 (top right). | ||
Complex 27, a derivative of catalyst 23c containing two tertiary amine cations on pendant arms, was designed to keep the dissociating anionic growing copolymer chains close to the metal centre.46 The concept is essentially an intramolecular combination of salenCoX and ionic co-catalyst (such as Bu4NX), allowing activity at lower loadings as well as at higher temperatures. The catalyst was active at loadings down to 1∶50000 (27∶PO), allowing very high TONs, TOFs and molecular weights as these are related to the catalyst loading. A TON of 14500 and a TOF of 3200 h−1 were reported at 1∶50000, at 80 °C and 20 atm, producing a copolymer with a high molecular weight of 53000 with a PDI of 1.35. The highest molecular weight attained was 95000, significantly greater than those produced by the two-component systems such as 23c/co-catalyst, which aren't able to function under such low loadings and generally produce copolymers with maximum molecular weights of approximately 30000. The intramolecular nature of the co-catalyst also allows activity under commercially relevant temperatures (80 °C) whilst maintaining a reasonable selectivity for copolymer formation (84–100%), indeed 27 was the first catalyst for PO to give good copolymer selectivity at these temperatures.
The replacement of the SiMe2 groups in the arms with CH2 groups decreased the activity significantly.82 However, changing the phenyl R substituent from a bulky tert-butyl group to a methyl group enhanced the activity significantly, giving similar activities to 27 under the same conditions. Catalysts 28a–c contain four tertiary amine cations in contrast to the two in catalyst 27. The same structure/activity relationship was observed, with 28c being the most active, producing PPC with a TOF of 26000 h−1 at 1∶25000 loading, 80 °C and 20 atm; by comparison, under the same conditions 27 gave a TOF of 3300 h−1. 28c also showed >99% selectivity for copolymer formation, even at a loading of 1∶100000, which produced a copolymer with a molecular weight of 285000 with a TON/TOF of 22000 in one hour. The catalyst was still active at a loading of 1∶150000 and the selectivity was an impressive 96%.
A method was proposed whereby the catalyst could be recovered from the copolymer. This has perhaps two main advantages: firstly, catalyst costs and waste streams could be significantly reduced. Secondly, metal residue in the copolymer is significantly decreased; residue which can cause discolouration and could raise concerns about potential toxicity. The coloured complex was caught on a short silica pad when the copolymerisation solution was passed through it, giving a colourless copolymer with roughly 1–2 ppm cobalt contamination. The tertiary amine arms were proposed to tether the complex to the silica, before the complex was removed from the silica with NaBF4. Addition of two equivalents of 2,4-dinitrophenol enabled regeneration of the catalyst, which showed little reduction in activity even after being recycled five times (the TON remained the same, although the TOF was reduced by 1/3 by the 5th recovery).
Further NMR, IR and DFT analysis led to the proposal that 28c in fact adopts an unusual structure, with the salen ligand only adopting a bidentate coordination mode and with the imine arms remaining uncoordinated (Fig. 17).47 In contrast to this, 28a–b, having bulkier aryl substituents, adopt the usual tetradentate coordination mode (Fig. 16). Structural variations of the imine backbone and the aryl substituents showed that bulky groups prevented the formation of the bidentate coordination mode. The ‘regular’ salen complexes were significantly less active for the copolymerisation of PO and CO2 than those which formed the bidentate structure. NMR studies showed that in coordinating solvents, such as THF or DMSO, two of the four anionic groups were easily dissociated from the metal centre and were replaced by coordinating solvent molecules. The two displaced anions were proposed to be held in the coordination sphere by the cationic side arms.
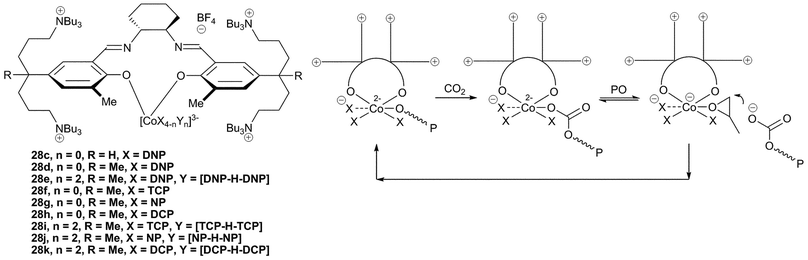 | ||
| Fig. 17 Salen Co complexes with unusual bidentate ligand coordination mode and proposed copolymerisation mechanism. | ||
It was proposed that the high activity observed with these bidentate species was due to ‘scrambling’ of the anionic ligands. The negatively charged cobalt centre was proposed to allow facile neutral/anionic ligand substitution reactions (as demonstrated by NMR). The proposed copolymerisation mechanism involves dissociation of the anionic chain whilst the epoxide coordinates, followed by epoxide ring-opening by the anionic copolymer chain, which is held close to the metal centre by the quaternary ammonium arms (Fig. 17). An induction period was observed with some of the catalysts; this was attributed to trace amounts of contaminating water. Replacement of [CoX4] with [CoX2Y2] (28e) where X is DNP and Y is the dinitrophenol–dinitrophenolate homoconjugation couple [DNP–H–DNP]− improved the TOF from 13000 h−1 to 16000 h−1, at 70 °C and 20 atm, and showed no induction time, suggesting it was less sensitive to water. The copolymer selectivity was also increased to >99%, giving PPC with an Mn of 300000 and a PDI of 1.31. The copolymers showed a bimodal distribution in the GPC traces, with one copolymer containing two hydroxyl end-groups, and one containing a DNP and a hydroxyl end-group. This clearly showed that water was responsible for some chain-transfer reactions.
A variety of different anions were also trialled to replace DNP, which is potentially explosive and is purchased in its hydrated form because of this.83 Substitution of DNP in 28d with 2,4,5-trichlorophenolate (TCP), 4-nitrophenolate (NP) and 2,4-dichlorophenolate (DCP) resulted in a slight reduction of TOF (10000, 8800 and 8300 h−1 respectively), with selectivities between 94–96% and similar PDIs. Use of the homoconjugated pairs [X–H–X]− with these same phenolates gave increased TOFs of 11000, 16000 and 13000, respectively. Analysis of the molecular weights suggested all four anions, as well as the alcohols present in the homocouple initiate the copolymerisation. As NP is not explosive or toxic (unlike the chlorinated phenols) and was the most active, this was deemed to be the best replacement for DNP.
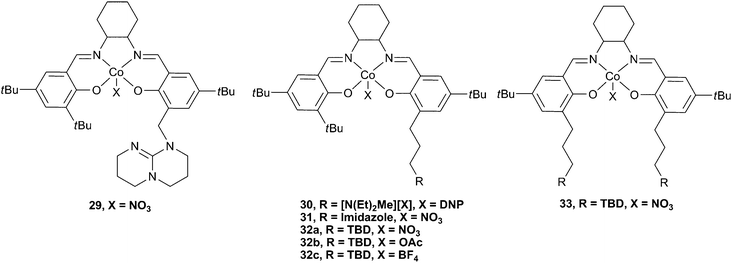
The use of Lewis base or tertiary amine co-catalysts attached via a pendant arm was also investigated (Fig. 18).48 The attachment of an anchored TBD group at the 2-position of one phenol donor (cf. substituents at the 4-position for 27 and 28) via a methylene (29) and propylene (32a–c) link was achieved, as well as the addition of two TBD substituents viapropylene linking groups (33). Complexes 32a and 32b were by far the most active of these complexes, with 32b converting PO to PPC with a TOF of 10880 h−1 at a loading of 1∶10000, at 100 °C and 25 atm. The copolymer selectivity was a remarkable 97%, even at 100 °C. This high selectivity, even at high temperatures, was proposed to be due to the ability of the pendant groups to stabilise the active Co(III) species against decomposition to an inactive Co(II) species. Unsubstituted cobalt salen complexes are known to have a tendency to be reduced and deactivated in this manner at high temperatures and low CO2 pressures. It is perhaps also of interest that Co(II) salen species have previously shown good activity catalysts for the coupling of epoxides and CO2, producing cyclic carbonates.8432b was also active under just 1 atm CO2 pressure, giving an excellent TOF of 265 h−1. In situESI MS analysis of the copolymerisation mixture showed that 29, with the methylene linker, was not as good at stabilising the Co(III) active species, which could explain why it showed very little activity. 31, which features an imidazole group rather than TBD, was also relatively inactive, although it did prevent decomposition to the Co(II) species. This result was perhaps not surprising as N-MeIm deactivated 23c, presumably because it coordinated strongly to the metal centre, inhibiting epoxide binding.4533, with two tethered TBD groups, also showed little activity, although it stabilised the Co(III) species, probably due to steric protection of the active site by the two large groups. 30, which features the tertiary ammonium cation showed approximately 2/3 the activity of 32b under the same conditions, gave a TOF of 3860 h−1, at 80 °C and 20 atm. However, it was later reported as an excellent catalyst for CHO copolymerisation under 1 atm pressure, giving a TOF of 263 h−1 at just 50 °C, and producing a perfectly alternating high molecular weight copolymer (Mn = 48200, PDI = 1.12).85
 | ||
| Fig. 18 Cobalt salen complexes with neutral ‘arms’. | ||
Elegant mechanistic studies using in situmass spectrometry suggested that the tethered TBD arm was capable of nucleophilic epoxide ring-opening in an analogous manner to the DMAP and MTBD co-catalysts, forming a tethered MTBD–epoxide adduct (I and II, Fig. 19).71,72In situFTIR spectroscopy showed two carbonate species: one assigned to the growing copolymer chain with the X end-group (IV, V) and the other assigned to a TBD–carbonate adduct (III) whereby CO2 inserted into species II. Addition of N2 into the ESI-MS experiment quickly removed this peak, suggesting the CO2 addition was reversible, explaining why the decarboxylated species (II) was observed in the MS studies instead of (III). It was this labile TBD-adduct which was proposed to stabilise the Co(III) species, associating and dissociating to allow the copolymer chain to grow in the trans position.
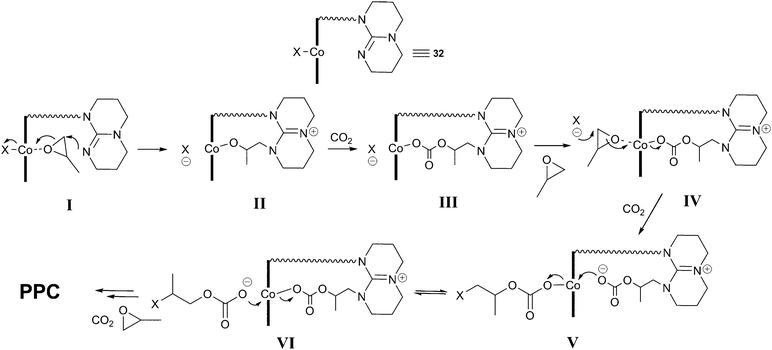 | ||
| Fig. 19 Proposed copolymerisation mechanism with 32a–c. | ||
A related mechanism of association and dissociation of labile groups, coordinated trans to one another at the cobalt centre, was also tentatively proposed for the binary salenCoX/ionic co-catalyst systems, although the intermolecular rather than intramolecular nature means that activity was significantly reduced at low catalyst loadings and higher temperatures, leading to decomposition to an inactive Co(II) species.
Finally, a series of salenCoX complexes with a neutral Lewis base substituent on the 4-position of the phenyl ring (neutral analogues of 27) were developed, with different co-ligands. The complexes showed poor activity, with X = NO3 being the most active, producing PPC with a TOF of 59 h−1 and 95% selectivity at 45 °C and 30 atm.
2.6 Miscellaneous ligand and metal systems
Fig. 20 and Table 4 outline the most active of these for CHO copolymerisation (since 2004).17–22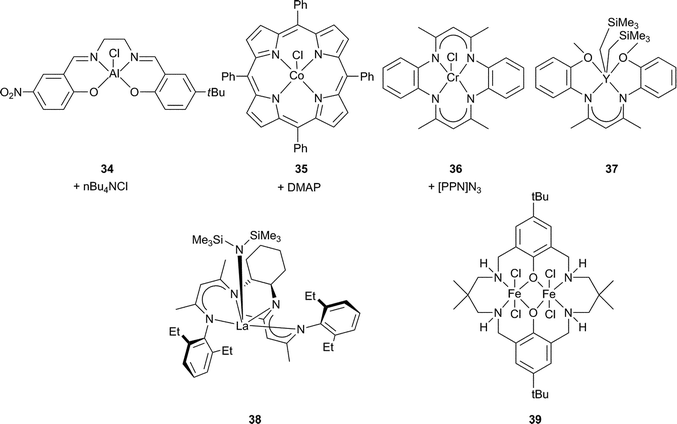 | ||
| Fig. 20 Selected homogeneous catalytic systems for CHO copolymerisation. | ||
| Catalyst (loading) | p(CO2)/atm | T/°C | t/h | TONa | TOF b | % Carbonatec | % Selectivityc | M n d | PDId | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a Moles epoxide consumed per mol. metal. b Moles epoxide consumed per mol. metal per hour. c Determined by 1H NMR spectroscopy. d Determined by GPC with polystyrene standards. | ||||||||||
| 34 + nBu4NCl (0.06 + 0.06 mol%) | 34 | 80 | 8 | 283 | 35 | 96 | 97 | N.A. | N.A. | 86 |
| 35 + DMAP (0.2 + 0.1 mol%) | 50 | 80 | 24 | 500 | 21 | >99 | 100 | 14500 |
1.13 | 87 |
| 36 + [PPN]N3 (0.06 + 0.03 mol%) | 34 | 80 | 1 | 1300 | 1300 | 95 | >99 | 27300 |
1.07 | 88 |
| 37 (0.17 mol%) | 15 | 130 | 10 | 474 | 47 | 99 | 96 | 19000 |
1.70 | 89 |
| 39 (0.1 mol%) | 10 | 80 | 24 | 694 | 29 | 99 | 99 | 11700 |
1.13 | 90 |
Building on the success of Cr(III) and Co(III) salen catalysts, other metal salens have been investigated. Aluminium copolymerisation catalysts were already well precedented, e.g. homoleptic aluminium alkoxides (with scCO2)91–93 and tetraphenylporphyrin (tpp) aluminium complexes.24 In addition, there were mechanistic parallels between the metal porphyrin and salen catalysts,16,74,94 and [(salen)AlCl] and [(tpp)AlCl] had shown similar affinities for PO.95 Darensbourg and Billodeaux thus reported a series of Al salen catalysts (including 34) for CHO, with tetrabutylammonium (nBu4NX, X = Cl, OAc, N3) or Lewis base (DMAP, N-MeIm or pyridine) co-catalysts.86 At 34 atm and 80 °C, the TOFs ranged from 5 to 35 h−1, considerably lower than the Cr(III)/Co(III) salen analogues. Sugimoto et al. also examined closely related Al(III) salen catalysts, where the co-ligand was acetate instead of chloride, obtaining low TOFs (8 h−1) and molecular weights (7100) with narrow PDIs (1.30).96Iron(III), zinc, gallium and manganese(III) salen complexes were either inactive or only active for cyclic carbonate production.74,97–99
Metal porphyrin catalysts have also been widely investigated.24 In 2003, 40 (Fig. 21) under 50 atm and at 80 °C, catalysed CHO/CO2 copolymerisation with a TOF of 16 h−1, producing PCHC with 99% carbonate linkages but with low molecular weight (6700, PDI = 1.3).100 At 80 °C and only 1 atm of CO2, the same catalyst exhibited a TOF of 3 h−1 (95% carbonate linkages, Mn = 3000, PDI = 1.6); a significant result as it was the first example of the copolymerisation at this low pressure.
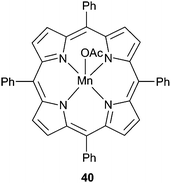 | ||
| Fig. 21 Manganese(III) porphyrin 39, first catalyst for the copolymerisation of CHO and CO2 under 1 atm (2003). | ||
The analogous cobalt porphyrin complex (35, Fig. 20) was slightly more active (TOF = 21 h−1), under the same high pressure and temperature conditions, and yielded PCHC of slightly higher molecular weight (carbonate linkages >99%, Mn = 14500, PDI = 1.13).87 It was also active under 1 atm pressure of CO2 and at room temperature (TON = 75, TOF = 3 h−1, >99% carbonate linkages, Mn = 1500, PDI = 1.11). All the PCHC samples showed bimodal molecular weight distributions. At low conversions, MALDI-ToF mass spectrometry showed chains with two hydroxyl end-groups (proposed to derive from water contamination) and chains with a chloride and hydroxyl end-group.16 Careful drying of both reagents and apparatus increased the molecular weight of the copolymer (29600), but the bimodality still remained. In contrast to 40, complex 35 catalysed PO copolymerisation, even under 1 atm CO2, to efficiently produce PPC, albeit of very low molecular weight (TON = 125, TOF = 5 h−1, 95% selectivity, 98% carbonate linkages, Mn = 1600, PDI = 1.22) (Table 5). Inspired by the success of the metal porphyrin catalysts, Darensbourg et al. prepared a conjugated macrocyclic ligand, tmtaa (tetramethyltetraazaannulene) ligand, which was more electron-donating than a porphyrin.101 The chromium(III) complex (36, Fig. 20) with an ionic co-catalyst ([PPN]N3), at 34 atm and 80 °C, displayed high TON (1300) and TOF (1300 h−1) with CHO, producing PCHC with a good molecular weight (27000) and narrow PDI (1.03).88,102 The PCHC molecular weight decreased on increasing the co-catalyst loading, but in contrast to related salen catalyst system 18b–[PPN]N3, cyclic carbonate production was not further enhanced.69 The increase in cyclic carbonate production in the presence of excess anionic co-catalyst is usually attributed to competition between the anion and the growing copolymer chain to coordinate at the metal centre. It was, therefore, concluded that 36 binds the copolymer chain with a greater affinity than its salen analogue. More recently a modified tmtaa ligand with a sterically hindered substituent was prepared which blocked one side of the Cr(III) complex and thereby showed copolymer chain growth from only one side of the catalyst.88
| Epoxide | Catalyst | Loading (mol%) | T/°C | TONa | TOF b/h−1 | % Carbonatec | % Selectivityc | M n d | PDId | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a Moles epoxide consumed per mol. metal. b Moles epoxide consumed per mol. metal per hour. c Determined by 1H NMR spectroscopy. d Determined by GPC in THF with polystyrene standards. e Not specified. | ||||||||||
| CHO | 1b | —e | — | — | — | — | 97 | — | — | 15 |
| CHO | 6 | 5 | 60 | 10 | 2 | — | — | 19200 |
1.56 | 50 |
| CHO | 7 | 0.67 | 60 | 43 | 22 | 94 | —e | 12900 |
1.29 | 51 |
| CHO | 11a | 0.1 | 80 | 219 | 9 | >99 | 96 | 6200 | 1.19 | 43 |
| CHO | 17 | 0.1 | 100 | 200 | 240 | >99 | >99 | 7300 | 1.03 | 44 |
| CHO | 22 | 0.1 | 70 | 200 | 100 | 98 | >99 | 8700 | 1.10 | 41 |
| CHO | 30 | 0.02 | 50 | 1315 | 263 | >99 | — | 48200 |
1.12 | 85 |
| PO | 32b | 0.02 | 25 | 1325 | 265 | >99 | >99 | 73100 |
1.10 | 48 |
| CHO | 35/DMAP | 0.2 | 25 | 75 | 3 | >99 | >99 | 1500 | 1.11 | 112 |
| PO | 35/DMAP | 0.2 | 25 | 125 | 5 | 98 | 95 | 1600 | 1.22 | 112 |
| CHO | 38 | 0.2 | 80 | 295 | 3 | 95 | — | 3000 | 1.60 | 100 |
| CHO | 40 | 0.2 | 75 | 304 | 13 | 88 | 95 | 13500 |
1.60 | 105 |
Rare earth metals (including Y and La) in combination with other metals (e.g.Zn) are well known to form active heterogeneous catalysts,103 for example [Nd(CO2CCl3)3], [ZnEt2], and glycerol copolymerise PO with an activity of 1.8 h−1 and a molecular weight of 62000.104 In 2005, the first examples of homogeneous rare-earth catalysts were reported simultaneously by the groups of Hultzsch105 and Hou.106 A cyclohexyl-bridged bis(β-diketiminato)(amido) lanthanum catalyst 38, at 1 atm and 75 °C, showed a TON of 304 and a TOF of 13 h−1 for CHO and produced PCHC of moderate molecular weight (13000) and broad PDI (1.6).105 The activity was, however, considerably higher than the well-known zinc BDI catalysts under similar conditions (which showed very low activity under 1 atm).15 A dialkyl lutetium complex, [(C5Me4SiMe3)Ln(CH2SiMe3)2(THF)], the most active of a series, produced PCHC at 70 °C and 12 atm of CO2 (Mn = 23000, PDI = 4.0, 92% carbonate linkages), with a TON of 306 and a TOF of 13 h−1. In contrast, under similar conditions, the Sc(III) analogue gave a high content of ether linkages (23% carbonate linkages). The stoichiometric reaction of those complexes with CO2 afforded quantitatively the bis(carboxylate) complexes, which were also moderately active. A range of other cyclopentadienyl lanthanide catalysts were then investigated.107,108 However, the most active species reported to date is 37 (Fig. 20), an o-anisol-substituted β-diketiminate yttrium dialkyl complex.8937 was active for CHO copolymerisation, under harsh conditions (15 atm, 130 °C), in dioxane solution, with a good TON (474) and TOF (48 h−1), producing PCHC with high carbonate content (99%), moderate molecular weight (19000) and broad PDI (1.7).
Finally, very recently, we have reported the use of a di-iron(III) complex 39, coordinated by a ‘reduced Robson's type’ macrocyclic ligand, for the copolymerisation of CHO and CO2.90 This catalyst showed a good activity at 10 atm CO2, giving a TOF of 107 h−1 at 80 °C, and producing a copolymer with a Mn of up to 17200 and narrow polydispersity (1.03). To the best of our knowledge, this is the first example of an iron catalyst for the homogeneous copolymerisation of CHO and CO2. Previously, some heterobimetallic tert-butoxides iron complexes ([(t-BuO)5FeLa] and [(t-BuO)4FeZn]) have been reported for the copolymerisation of PO and CO2.109 Also, several researchers have investigated model complexes for the double metal cyanide heterogeneous catalysts, some of which contain Fe(II) (vide infra).110,111
3. Heterogeneous catalysts
Zinc glutarate [Zn(O2C(CH2)3CO2)]n is the most widely applied heterogeneous catalyst for the copolymerisation of PO and CO2 due to its ease of preparation, good productivity and high molecular weight copolymer product. It was generally prepared by reaction between zinc oxide with glutaric acid, in warm toluene followed by azeotropic water removal and vacuum drying at elevated temperature.113 Various synthetic routes, alternative zinc precursors, stirring methods and additives have been investigated;113–120 the key factors controlling productivity were the degree of crystallinity and the size of the crystallites.117Zinc glutarate has very low solubility and this prevented growth of single crystals for a number of years, however, structural information from a range of spectroscopic and X-ray diffraction techniques indicated a layered structure with carboxylate groups bound at the surface sites.115,121 Recently single crystal X-ray diffraction confirmed this hypothesis, the structure showed that each zinc centre was tetrahedrally coordinated by four oxygen atoms from different glutarate groups leading to an alternating layered structure of zinc and glutarate ions.122Catalyst surface composition analysis indicated unsaturated zinc centres and reversible binding of CO2.115 The crystalline zinc glutarates were typically used at 40–50 atm CO2 pressure, 60 °C, with excess PO (300∶1, PO∶Zn glutarate) as the solvent. The copolymerisations were quite slow, with >90% conversion being achieved only after 40 h, however, such high conversions enable PPC to be prepared with high molecular weights. Typical productivities were approx. 70 g PPC/g Zn, although under optimised conditions this was increased somewhat.113,117,123 The molecular weights exceeded 100000 and the PDI values were relatively narrow (2–3), perhaps indicating a common surface initiating site. Furthermore, the quality of the copolymer was excellent with only minor contamination from ether linkages or propylene carbonate.113,117,123 Recently, Eberhardt et al. prepared zinc glutarate derivatives from diethyl zinc and glutaric acid, using sub-stoichiometric quantities of the diacid and subsequently reacting with SO2 to form ethylsulfinate groups.114 Although these complexes likely had lower crystallinity than conventional zinc glutarate they showed higher productivity (300 g PPC/g Zn) and equivalent copolymer properties.114 An excellent review of zinc glutarate catalysts and the materials properties of the PPC has recently been published by Luinstra (BASF).124
Double metal cyanides (DMCs), i.e.Zn3[M(CN)6]2, where M = Fe(III) or Co(III), are also a promising class of heterogeneous catalyst.125–134 Many of the reports on the synthesis and application of these species are in the patent literature,125–127,130,131 indeed it is notable that DMCs are also widely applied as epoxide homopolymerisation catalysts. In contrast to the findings for zinc glutarates, the best DMC catalysts were amorphous materials, in some cases highly crystalline materials were completely inactive.129 The catalyst activities were also highly dependent on the preparation method and additives; it was significant that the catalysts almost always include non-stoichiometric quantities of water, alcohols and/or metal halides.125–134 The productivities of the Fe–Zn DMCs tended to be lower for both PO and CHO copolymerisation than the Co–Zn analogues which showed productivities of 500–1000 g copolymer/g Zn.128,129,132 However, a drawback of these systems was the low CO2 incorporation, probably because the DMCs are such effective epoxide homopolymerisation catalysts. Thus, the % of carbonate linkages tended to be lower for PPC than PCHC (as PO is easier to homopolymerise), e.g. typical values for PPC are 20–40% whilst those for PCHC can reach 90%.129,132,133 The copolymerisation conditions were harsh, with temperatures in the range 80–130 °C and pressures from 50–100 atm.128,129,132,133 Finally, the copolymer molecular weights were only moderate (10000–30000) with wide polydispersity indices, as would be expected from a catalyst with multiple initiating sites.128,129,132,133 Some groups have attempted to shed light on the active species by preparing well defined analogues of DMCs. Thus, Darensbourg and co-workers prepared mixed Fe–Zn complexes, one of which, [CpFe(PPh3)(í-CN)2Zn(2,6-OC6H3(tert-butyl)2)(THF)]2, showed reasonable incorporation of CO2 (% carbonate linkages >85%) in PCHC, however, the productivities were significantly lower than for the DMCs.110,111 Also, Robertson et al. reported a two-dimensional Co[Ni(CN)4] catalyst which showed promising copolymerisation activity for PO and CO2.135 The catalyst was well characterised and the best results were obtained using anhydrous samples. Impressive TOFs of 1860 mole PO (mole Co)−1 h−1 were obtained and high molecular weight polycarbonate (74000) was produced, although with rather low CO2 incorporation. At lower temperatures the activity decreased but the loading of carbonate was increased.
4. Towards new materials
4.1 Properties of polycarbonates
With current concerns about the environmental impact of the plastics industry, in terms of resource utilisation and disposal, polycarbonates produced from the copolymerisation of carbon dioxide and epoxides are a promising alternative to petrochemicals.136 There are a very large range of commercially available and naturally occurring epoxides, from these it could be possible to produce various new polycarbonate materials that could provide suitable properties to address the growing demand for sustainably produced materials. So far, however, only a limited range of aliphatic polycarbonates have been produced, including polyethylene carbonate (PEC), PPC, polybutene carbonate (PBC) and PCHC. Furthermore compared to the widely applied poly(oxycarbonyloxy-1,4-phenylene isopropylidene-1,4-phenylene) (from bisphenol-A—usually known as Polycarbonate), which is partially crystalline, transparent, with a high impact strength (9.1 N cm−1),137 and a high glass transition temperature (Tg = 149 °C),137 their moderate thermal stability and facile thermal deformation prevent widespread applications.124,138–140Table 6 shows some selected thermal and mechanical properties of common polycarbonates produced by the copolymerisation of CO2 and epoxides, and those of polycarbonate.| Copolymer | T g a/°C |
T
d
50%
b/°C |
Tensile strength/MPa | Tensile modulus/MPa | Elongation at break (%) | Ref. |
|---|---|---|---|---|---|---|
| Polymers produced by the copolymerisation of CO2 and epoxides, except Polycarbonate* which is produced by condensation polymerisation.a Glass transition temperature determined by differential scanning calorimetry (DSC).b Temperature at which 50% mass loss is observed determined by thermogravimetric analysis (TGA). | ||||||
| Poly(ethylene)carbonate | 10 | 229 | — | — | — | 138 |
| Poly(propylene)carbonate | 42 | 252 | 7–30 | 700–1400 | 600–1200 | 85, 124 |
| Poly(butylene)carbonate | 9 | 241 | — | — | — | 141 |
| Poly(cyclohexene)carbonate | 118 | 310 | 40–44 | 3500–3700 | 1.1–2.3 | 85, 140 |
|
149 | 458 | 43–51 | 2000–2800 | 15–75 | 137, 142 |
The thermo-mechanical properties of polymers (including glass transition temperature, crystalline melting temperature, tensile, compressive, and flexural strengths, fatigue and impact resistance) are highly dependent on molecular weight, typically levelling off once chain entanglement becomes significant (which generally corresponds to a molecular weight exceeding 20000). Thus, the properties of aliphatic polycarbonates are highly dependent on molecular weight, a phenomenon which has caused some disparity in the literature values. For example, for PPC, a Tg of 27 °C was reported for a Mn of 26900 and a value of 42 °C at Mn of 114000.143 These variations were especially apparent at lower molecular weights where the chain end-groups exert a significant influence, thus PCHC with a Mn of 1400 had a Tg of 52 °C, whilst for a Mn of 3500, a Tg of 85 °C was reported.144 For the other copolymers, even at high molecular weight, discrepancies of as much as 30 °C are common (for PEC, values for Tg from 5–31 °C were reported).143,145Nevertheless, the low glass transition temperature of most aliphatic polycarbonates is disadvantageous, even if PPC is a potential synthetic elastomer. Despite having a higher glass transition temperature, the tensile properties of PCHC (Table 6) remain inferior to the corresponding properties of the polycarbonate. Three main strategies have therefore been used to try to modify the thermo-mechanical properties of aliphatic polycarbonates: the preparation of stereo- and regioregular copolymers, the use of new epoxide co-monomers and the preparation of terpolymers.
4.2 Asymmetric copolymerisation
Mono-substituted epoxides, e.g.PO, can be copolymerised in different regiochemistries (Fig. 22) including with the substituents on alternate carbon atoms (Head–Tail, HT) or with the substituents on consecutive carbon atoms (Head–Head, HH, or Tail–Tail, TT, linkages) (Fig. 22). In addition to this regioregularity, there are different stereochemistries, or tacticities, for the copolymer chain including all the substituents having the same stereochemistry (isotactic) or alternating stereochemistries (syndiotactic) or a random distribution of stereocentres (atactic) (Fig. 22).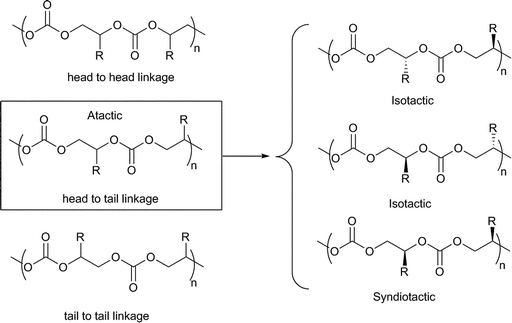 | ||
| Fig. 22 Regio- and stereochemistry of polycarbonates. | ||
Stereo- and regioregularity are desirable as they can increase crystallinity and have the potential to impact properties such as thermal resistance, toughness, stiffness, opacity and density. Significant research has been dedicated to the production of regioregular PPC. One report showed that increasing the degree of HT linkages from 70 to 77% led to an increase in the glass transition temperature from 37 to 42 °C.146
The desymmetrization of meso-molecules, e.g.CHO, with chiral catalysts or reagents is also regarded as a valuable strategy for the synthesis of enantiomerically enriched products. The ring opening of a meso-epoxide proceeds with inversion of one of the stereocentres, so asymmetric ring opening of the epoxide is expected to produce optically active polycarbonates, with either (R,R)- or (S,S)-trans-1,2-cyclohexylene repeat units. In 1999, Nozaki and co-workers were the first to report such a copolymerisation, catalysed by a chiral amino-alkoxide zinc complex 40 (Fig. 23), and producing optically active PCHC with 70% ee (measured by hydrolyzing the copolymer and analyzing the resulting diol using chiral GC) and a Tg of 117 °C (a value close to that of atactic PCHC).147,148 Coates and colleagues reported a well-defined, chiral Zn oxazoline complex, which showed similar enantioselectivity, but higher activity and excellent control of the molecular weight, producing a copolymer with a Tg and Tm of 120 and 220 °C, respectively.149
 | ||
| Fig. 23 Chiral amino-alkoxide zinc complex used for asymmetric copolymerisations. | ||
The use of chiral salen cobalt complexes, with various co-catalysts, enhanced both the regioselectivity and stereoselectivity of PPC. Coates and coworkers first reported that 23a, [(R,R)-(salen)CoOAc], catalyzed the alternating copolymerisation of CO2 (55 atm) and rac-PO or (S)-PO to afford alternating PPC with 80% head to tail linkages.77 Then, through the addition of quaternary ammonium salt co-catalysts, Lu and Wang showed that the [(R,R)-(salen)CoX] complexes (23a, c, d) were highly efficient catalysts for the copolymerisation of CO2 with PO, under mild conditions (25 °C, 2 atm) and also afforded perfectly alternating PPC but with >95% HT linkages.78 Simultaneously, Cohen et al. showed that the copolymerisation of rac-PO and CO2, catalysed by 23b, ([(R,R)-(salen)CoOBzF5]), and [PPN]Cl, at 14 atm pressure and 25 °C yielded iso-enriched PPC with 94% HT linkages. Interestingly, in the absence of co-catalyst, 23e produced regioregular atactic PPC from rac-PO, whilst rac-23e, [rac-(salen)CoBr], yielded syndio-enriched PPC with 84% HT linkages (based on 13C NMR assignments).37 The PPC syndiotacticity was proposed to result from a chain-end control mechanism, with alternate enchainment of (S)- and (R)-PO, depending on the stereochemistry of the catalyst, and through continuous chain-exchange reactions.
The 23c/[PPN]Cl system was active for the stereoselective alternating copolymerisation of CO2 and rac-PO (as well as working for CHO), giving polycarbonates with 54% ee (36% ee for CHO) and >99% carbonate linkages, at 25 °C and 14 atm.45 Shi et al. demonstrated that 22d/[PPN]Cl, at 25 °C and 6 atm, afforded isotactic-enriched PCHC (up to 38% ee), with a good molecular weight (Mn = 20600, PDI = 1.17). Increasing the CO2 pressure to 52 atm led to dramatic decreases in catalyst activity and enantioselectivity. In contrast, increasing the temperature from 25 to 80 °C, increased the catalytic activity with only a slight decrease in enantioselectivity (28% ee).150
4.3 Copolymerisation of CO2 and other epoxides
Fig. 24 and Table 7 highlight some of the other epoxides that have been tested and the properties of the resultant copolymers, including, where available, the thermal characterisation. The thermal values should be interpreted with some caution due to their dependence on the molecular weight and the proportion of carbonate linkages in the copolymer. The main limitation of these copolymerisations remains the lack of catalytic activity for epoxides other than PO, CHO, EO and BO. Inoue was one of the first researchers to explore the possibility of applying new monomers for the copolymerisation. The copolymerisation of carbon dioxide and trimethylsilyl glycidyl ether (A), using a diethyl zinc–water heterogeneous system, produced a readily degradable polycarbonate with a pendant hydroxyl group (obtained via the deprotection of the silyl ether during the work-up of the copolymer) (entry 1, Table 5).151Limonene oxide (B/B′), an epoxide derived from citrus fruits, was copolymerised using the BDI zinc catalysts at 25 °C and 7 atm to yield a copolymer with moderate molecular weight (10800) and a high Tg (111 °C).152 Another interesting copolymerisation involved end-capping a polycaprolactone chain with an epoxy group (L) and using the copolymerisation with CO2 to cross-link the copolymers, producing hyperbranched materials.153
 | ||
| Fig. 24 Selected other monomers used in CO2 copolymerisation reactions (see Table 7). | ||
| Monomer | Catalyst | p(CO2)/atm | T/°C | TONa | TOF b/h−1 | % Carbonatec | % Selectivityc | M n d | PDId | T g e/°C | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Moles epoxide consumed per mol. metal. b Moles epoxide consumed per mol. metal per hour. c Determined by 1H NMR. d Determined by GPC with polystyrene standards. e Glass transition temperature determined by DSC. | |||||||||||
| A (1000 equiv.) | [(salen)CrCl] + [PPN]Cl | 50 | 80 | — | — | — | — | — | — | — | 151 |
|
B∶B′ (300 equiv.∶250 equiv.) |
[(BDI)ZnOAc] | 7 | 25 | 74 | 37 | 100 | 100 (trans > 99) | 10800 |
1.12 | 111 | 152 |
| C | Y(CF3CO2)3 Zn(Et)2pyrogallol ternary catalyst | 28 | 60 | 11 | 1 | 98 | 100 | 132000 |
3.70 | −19 | 154 |
| D | DMC: K3Co(CN)6/ZnI2/PTME glycol | 10 | 80 | 266 | 22 | 33 | 100 | 2000 | 1.40 | — | 133 |
| E | DMC: K3Co(CN)6/ZnCl2//PTME glycol | 10 | 80 | 179 | 15 | 73 | 100 | 10100 |
2.20 | — | 132 |
| F | DMC: K3Co(CN)6/ZnCl2//PTME glycol | 10 | 50 | 250 | 21 | 7 | 100 | 2300 | 1.24 | — | 132 |
| G | DMC: K3Co(CN)6/ZnCl2//PTME glycol | 10 | 80 | 237 | 20 | 12 | 100 | 2800 | 1.44 | — | 132 |
| H | None | 220 | 100 | 35% conv. | In 24 h | 62 | 100 | 24700 |
6.15 | — | 12 |
| I (1300 equiv.) | [(salen)CrCl] + nBu4Cl | 35 | 110 | 309 | 41 | 97 | 97 | 10100 |
1.58 | T d 50% = 260 | 155, 156 |
| J (20 equiv.) | BMImCl | 10 | 80 | 13 | 2 | 99 | 100 | 1900 | 1.02 | — | 157 |
| K (320 equiv.) | [(EtBDI)ZnOEt] | 9 | 50 | 269 | 3 | 100 | 100 | 2300 | 4.40 | 53 | 144 |
| L (0.43 equiv.) | ZnO/glutaric acid | 103 | 60 | — | — | 100 | 100 | 7300 | 1.36 | — | 153 |
| M (33 equiv.) | [Y(CCl3COO)3]–[ZnEt2]–glycerine | 40 | 65 | 0.51 | 0.051 | 92.3 | 100 | 77000 |
2.27 | −6 | 158 |
4.4 Terpolymerisations
Another way to adjust copolymer properties is to copolymerise CO2 with two different epoxides, this has been accomplished either simultaneously (terpolymerisation) or sequentially (block copolymerisation) (Table 8). In contrast to the brittle behaviour of PCHC (elongation at break of 1–2%), PPC has good flexibility (elongation at break between 600 and 1200%), but its low Tg (35–45 °C) limits its use. To try to address this, terpolymerisations of PO, CHO, and CO2 have been undertaken, however, until 2006 only limited success was achieved mainly because of the different rates of polymerisation (leading to copolymers with high degrees of compositional drift). Bis(2,6-difluorophenoxide)zinc catalysts were tested, at 55 °C and 41–48 atm, with equimolar quantities of PO and CHO, but the resulting copolymer was mostly composed of cyclohexene carbonate linkages (85% PCHC, 12% PPC and 3% polypropylene ether) and the main side-product of the reaction was cyclic propylene carbonate.27 As a result of the low incorporation of PPC, the Tg was reduced from 115 °C for PCHC to 102 °C for the terpolymer. In addition, the formation of PC, which is a better ligand for zinc than epoxides, seriously hindered the catalytic activity. A ternary [Y(CCl3COO)3]–[ZnEt2]–glycerine catalyst was later tested, at 60–70 °C and 35–40 atm, producing terpolymers with variable compositions and Tg (from 37 to 126 °C) depending on PO/CHO ratios.159 These terpolymers typically showed two glass transition temperatures, indicating the formation of block copolymers (compositional drift) rather than an alternating terpolymer. An additional complicating factor was that aliphatic epoxides have an increased tendency, compared to alicyclic epoxides, to form cyclic carbonate by-products.| Monomers | Catalyst | p(CO2)/atm | T/°C | TOF a | % Selectivityb | % Alternating units | M n c | PDIc | T g d | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a Moles epoxide consumed per mol. metal per hour. b Determined by 1H NMR spectroscopy. c Determined by GPC with polystyrene standards. d Glass transition temperature determined by DSC. e 52% maleic linkages. | ||||||||||
|
PO/CHO (1∶1) |
28e | 20 | 75 | 6500 | >99 | Random | 134000 |
1.20 | 65 | 141 |
| 33% PC | (Td50% = 258) | |||||||||
|
PO/BO (1∶1) |
28e | 20 | 75 | 7700 | >99 | Random | 120000 |
1.13 | 19 | 141 |
| 32% PC | (Td50% = 237) | |||||||||
|
PO/HO (1∶1) |
28e | 20 | 75 | 9900 | >99 | Random | 154000 |
1.24 | 27 | 141 |
| 38% PC | (Td50% = 232) | |||||||||
|
CHO/PO (1∶1) |
30 | 25 | 90 | 3590 | >99 | Random | 50900 |
1.12 | 79 | 85 |
| 52% PCHC | (Td50% = 292) | |||||||||
|
CHO/EO (1∶1) |
30 | 25 | 90 | 4250 | >99 | Random | 48000 |
1.18 | 32 | 85 |
| 37% PCHC | (Td50% = 292) | |||||||||
|
CHO/BO (1∶1) |
30 | 25 | 90 | 2564 | >99 | Random | 42100 |
1.10 | 68 | 85 |
| 56% PCHC | (Td50% = 298) | |||||||||
|
CHO/HO (1∶1) |
30 | 25 | 90 | 1958 | >99 | Random | 39700 |
1.15 | 72 | 85 |
| 65% PCHC | (Td50% = 302) | |||||||||
|
PO/maleic anhydride (1∶1) |
DMC Zn/Fe | 40 | 60 | — | 19e | Random | 72200 (Mv) |
— | 52 | 160 |
| CHO/rac-lactide | BDI zinc acetate 33 | 40 | 90 | 43 | 15 | Random | 32300 |
1.22 | 58 | 161 |
|
CHO/diglycolic anhydride (5∶1) |
BDI zinc acetate | 7 | 50 | — | 70 | Blocks only (PE then PC) | 37000 |
1.20 | — | 162 |
|
PO/γ-butyrolactone (1∶1) |
DMC Zn/Fe | 40 | 60 | — | — | — | 123000 (Mv) |
— | 41 | 163 |
|
PO/ε-caprolactone (1∶1) |
Zinc glutarate | 28 | 60 | — | 70 | 4.6% then blocks | 234000 |
1.54 | 18 (Tm = 51) | 164 |
However, these limitations have been overcome through the development of higher activity catalysts. In 2006, [(salen)CoX] (23) and [PPN]Cl systems were efficient catalysts for the terpolymerisation of CHO, PO, and CO2.150 Using 23d, the terpolymerisation, at 25 °C and 15 atm, with equimolar quantities of CHO and PO, yielded polycarbonates (TOF = 129 h−1) with Mn = 24400, PDI = 1.24, and more than 99% carbonate linkages. The terpolymer also showed a single Tg and just one thermolysis peak which was attributed to an alternating copolymer microstructure. The Tg could also be adjusted between 50 and 100 °C by varying the epoxide ratios: increasing the cyclohexene carbonate content from 30–60 mol% thus increased the Tg from 60–81 °C. The unprecedented alternating nature of the terpolymer was attributed to: (1) the relatively high basicity and coordination ability of CHO inhibiting the reactivity of PO; (2) the relatively low reactivity and the steric hindrance of CHO retarding its homopolymerisation; (3) the dissociation of the propagating carboxylate from the metal center being much faster than propagation; and (4) the quaternary ammonium co-catalyst preventing backbiting and the formation of cyclic carbonate. Recently, a Co(III)-salen catalyst with a quaternary ammonium salt arm, 30,82 was tested with CHO and aliphatic epoxides (PO, BO, HO, EO, used in 1∶1 ratio with CHO), at 90 °C and 25 atm, and showed good TOFs (1958–3560 h−1) and high molecular weights (39700–50900, PDI ≈ 1.1). The terpolymers showed 50% CHC linkages and a single Tg (32–79 °C range, lower than that of PCHC).8528e was also used to terpolymerise CO2, PO, and 1-hexene oxide (HO) or 1-butene oxide (BO), without the formation of any cyclic carbonates and ether linkages.141 In addition, the PO mole fractions in the feed vs. the % of PC linkages in the terpolymers were in sufficient agreement to enable Fineman-Ross plots to be used to determine the monomer reactivity ratios. There was a linear dependency between the terpolymer Tg and the proportions of the third monomers used, thus enabling tuning of the Tg between −15 and 32 °C for CO2/PO/HO terpolymers, and between 9 and 33 °C for CO2/PO/BO terpolymers.
Finally, epoxides and CO2 have been coupled with monomers other than epoxides, including maleic anhydride,160ε-caprolactone,164,165γ-butyrolactone,163rac- and (S,S)-lactide,161 or diglycolic anhydride.162 So far, no catalyst system has been able to produce a perfectly alternating terpolymer, but block terpolymers have shown some interesting physical properties, e.g. degradability.
5. Conclusions
The copolymerisation of CO2 and epoxides is a promising route to prepare new sustainable copolymers and materials. The success and viability of the copolymerisaiton is critically dependent on the catalyst. Although the reaction has been known for more than 40 years, it has not yet received the intensive research efforts of related polymerisation catalyses (e.g.olefin polymerisation).Significant efforts have been devoted, by a number of academic groups, to the preparation and study of various homogeneous catalysts for the reaction. Recent kinetic studies have indicated that two leading ligand types, the salen (without co-catalyst) and β-diiminate, lead to catalysts which show bimetallic active sites. This has led to the deliberate preparation of various bimetallic complexes, some of which have shown improved productivities, activities and stabilities vs. monometallic analogues. However, our own work, and that of others, has shown that it is not only the bimetallic active site which is important but also the nature of the ancillary ligand. Metal salen complexes have an excellent precedent for this catalysis and recently a number of related derivatives, including reduced salalens and salans, have led to some improvements. The most active and selective metal salen complexes are Cr(III) and Co(III) species; recent developments and insights using these catalysts have been presented. Nozaki and co-workers reported the preparation of single component metal salen complexes, species where the co-catalyst is bound to the salen ligand via a saturated linking group. These species, and closely related reduced derivatives, have shown significant improvements in activity and stability vs. the two component systems. Finally, a number of new ligand types and metal centres, including lanthanides, have shown some activity. Also, a number of groups, including our own, have reported catalysts which are active under very mild conditions, including at just 1 atm pressure of CO2. Such catalysts are able to improve the energy balance (and hence the net CO2 consumption). There is significant scope for both more detailed kinetic and mechanistic studies of known catalysts and the development of new catalysts. In particular, the range of active metal centres is still surprisingly narrow and there is much scope for the development of new catalysts using inexpensive and abundant metal centres. In addition, the range of successful ancillary ligands is also narrow, with many reports focussing on derivatives of β-diiminate or salen ligands: there is much scope to move beyond these systems. The research field needs improved understanding of the copolymerisation kinetics and mechanism, which can only come from more detailed studies using homogeneous catalysts. Finally, the potential to develop new materials from more selective and active catalysts will open up the applications for this type of polycarbonate.
Heterogeneous catalysts have received significantly less attention, despite the excellent stabilities and good productivities they can show. The range of heterogeneous species could be widened and the development of methods to support homogeneous catalysts could facilitate catalyst recycling/removal.
Aliphatic polycarbonates are currently niche products, due in part to cost but also to the limited range of materials properties. The recent development of catalysts that enable the controlled synthesis of block and random terpolymers and control of stereo and regiochemistry is of great interest. This research area must continue to be developed in order to understand the structure–property relationships of aliphatic polycarbonates. In particular, the development of sustainable polymers, including polycarbonates, with improved thermal and mechanical properties could enable the replacement of engineering thermoplastics and open up large potential markets.
Acknowledgements
The Engineering and Physical Sciences Research Council (EP/C544846/1 and EPC544838/1) and the Leverhulme Foundation (F/07 058/BD) are thanked for research funding.References
- H. Arakawa, M. Aresta, J. N. Armour, M. A. Barteau, E. J. Beckman, A. T. Bell, J. E. Bercaw, C. Creutz, E. Dinjus, D. A. Dixon, K. Domen, D. L. DuBois, C. A. Eckert, E. Fujita, D. H. Gibson, W. A. Goddard, D. W. Goodman, J. Keller, G. J. Kubas, H. H. Kung, J. E. Lyons, L. E. Manzer, T. J. Marks, K. Morokuma, K. M. Nicholas, R. Periana, L. Que, J. Rostrup-Nielson, W. M. H. Sachtler, L. D. Schmidt, A. Sen, G. A. Somorjai, P. C. Stair, B. R. Stults and W. Tumas, Chem. Rev., 2001, 101, 953–996 CrossRef CAS.
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS.
- Carbon Dioxide as Chemical Feedstock, ed. M. Aresta, Wiley-VCH, Weinheim, 2010 Search PubMed.
- D. R. Lide, Handbook of Chemistry and Physics, Taylor and Francis, Boca Raton, FL, USA, 1993–1994 Search PubMed.
- T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312–1330 RSC.
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110(8), 4554–4581 CrossRef CAS.
- http://www.empowermaterials.com/ and http:// www.novomer.com/, accessed 20 June 2010.
- http://www.decc.gov.uk/en/content/cms/statistics/climate_change/gg_emissions/uk_emissions/2008_final/2008_final.aspx, accessed 20 June 2010.
- N. MacDowell, N. Florin, A. Buchard, J. Hallett, A. Galindo, G. Jackson, C. S. Adjiman, C. K. Williams, N. Shah and P. Fennell, Energy Environ. Sci., 2010 10.1039/c004106h.
- W. Kuran, Prog. Polym. Sci., 1998, 23, 919–992 CrossRef CAS.
- O. Ihata, Y. Kayaki and T. Ikariya, Macromolecules, 2005, 38, 6429–6434 CrossRef CAS.
- K. Soga, W. Chiang and S. Ikeda, J. Polym. Sci., Part A: Polym. Chem., 1974, 12, 121–131 Search PubMed.
- M. Okada, Prog. Polym. Sci., 2002, 27, 87–133 CrossRef CAS.
- D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911–11924 CrossRef CAS.
- S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2861–2871 CrossRef CAS.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS.
- D. J. Darensbourg and M. W. Holtcamp, Coord. Chem. Rev., 1996, 153, 155–174 CrossRef CAS.
- D. J. Darensbourg, R. M. Mackiewicz, A. L. Phelps and D. R. Billodeaux, Acc. Chem. Res., 2004, 37, 836–844 CrossRef CAS.
- K. Nozaki, Pure Appl. Chem., 2004, 76, 541–546 CrossRef CAS.
- H. Sugimoto and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5561–5573 CrossRef CAS.
- S. Inoue, H. Koinuma and T. Tsuruta, J. Polym. Sci., Part B: Polym. Lett., 1969, 7, 287–292 CrossRef CAS.
- N. Takeda and S. Inoue, Macromol. Chem. Phys., 1978, 179, 1377–1381 Search PubMed.
- D. J. Darensbourg and M. W. Holtcamp, Macromolecules, 1995, 28, 7577–7579 CrossRef CAS.
- D. J. Darensbourg, M. W. Holtcamp, G. E. Struck, M. S. Zimmer, S. A. Niezgoda, P. Rainey, J. B. Robertson, J. D. Draper and J. H. Reibenspies, J. Am. Chem. Soc., 1999, 121, 107–116 CrossRef CAS.
- D. J. Darensbourg, J. R. Wildeson, J. C. Yarbrough and J. H. Reibenspies, J. Am. Chem. Soc., 2000, 122, 12487–12496 CrossRef CAS.
- W. Kuran, Appl. Organomet. Chem., 1991, 5, 191–194 CAS.
- W. Kuran and T. Listos, Macromol. Chem. Phys., 1994, 195, 1011–1015 CrossRef CAS.
- W. Kuran and T. Listos, Macromol. Chem. Phys., 1992, 193, 945–956 Search PubMed.
- T. Listos, W. Kuran and R. Siwiec, J. Macromol. Sci., Part A: Pure Appl. Chem., 1995, 32, 393–403 Search PubMed.
- M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 1998, 120, 11018–11019 CrossRef CAS.
- M. Cheng, D. R. Moore, J. J. Reczek, B. M. Chamberlain, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 8738–8749 CrossRef CAS.
- D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, Angew. Chem., Int. Ed., 2002, 41, 2599–2602 CrossRef CAS.
- S. D. Allen, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 14284–14285 CrossRef CAS.
- D. J. Darensbourg, R. M. Mackiewicz and D. R. Billodeaux, Organometallics, 2005, 24, 144–148 CrossRef CAS.
- C. T. Cohen, T. Chu and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 10869–10878 CrossRef CAS.
- D. J. Darensbourg and A. L. Phelps, Inorg. Chem., 2005, 44, 4622–4629 CrossRef CAS.
- K. Nakano, T. Kamada and K. Nozaki, Angew. Chem., Int. Ed., 2006, 45, 7274–7277 CrossRef CAS.
- D. J. Darensbourg, M. Ulusoy, O. Karroonnirum, R. R. Poland, J. H. Reibenspies and B. Cetinkaya, Macromolecules, 2009, 42, 6992–6998 CrossRef CAS.
- K. Nakano, M. Nakamura and K. Nozaki, Macromolecules, 2009, 42, 6972–6980 CrossRef CAS.
- T. Bok, H. Yun and B. Y. Lee, Inorg. Chem., 2006, 45, 4228–4237 CrossRef CAS.
- M. R. Kember, P. D. Knight, P. T. R. Reung and C. K. Williams, Angew. Chem., Int. Ed., 2009, 48, 931–933 CrossRef CAS.
- M. R. Kember, A. J. P. White and C. K. Williams, Macromolecules, 2010, 43, 2291–2298 CrossRef CAS.
- X. B. Lu, L. Shi, Y. M. Wang, R. Zhang, Y. J. Zhang, X. J. Peng, Z. C. Zhang and B. Li, J. Am. Chem. Soc., 2006, 128, 1664–1674 CrossRef CAS.
- E. K. Noh, S. J. Na, S. Sujith, S. W. Kim and B. Y. Lee, J. Am. Chem. Soc., 2007, 129, 8082–8083 CrossRef CAS.
- S. J. Na, S. S, A. Cyriac, B. E. Kim, J. Yoo, Y. K. Kang, S. J. Han, C. Lee and B. Y. Lee, Inorg. Chem., 2009, 48, 10455–10465 CrossRef CAS.
- W.-M. Ren, Z.-W. Liu, Y.-Q. Wen, R. Zhang and X.-B. Lu, J. Am. Chem. Soc., 2009, 131, 11509–11518 CrossRef CAS.
- B. Y. Lee, H. Y. Kwon, S. Y. Lee, S. J. Na, S. I. Han, H. S. Yun, H. Lee and Y. W. Park, J. Am. Chem. Soc., 2005, 127, 3031–3037 CrossRef CAS.
- Y. L. Xiao, Z. Wang and K. L. Ding, Chem.–Eur. J., 2005, 11, 3668–3678 CrossRef CAS.
- Y. L. Xiao, Z. Wang and K. L. Ding, Macromolecules, 2006, 39, 128–137 CrossRef CAS.
- M. Kröger, C. Folli, O. Walter and M. Döring, Adv. Synth. Catal., 2005, 347, 1325–1328 CrossRef.
- M. Kröger and M. Döring, Catal. Today, 2006, 115, 146–150 CrossRef.
- M. Kröger, C. Folli, O. Walter and M. Döring, J. Organomet. Chem., 2006, 691, 3397–3402 CrossRef.
- M. F. Pilz, C. Limberg, B. B. Lazarov, K. C. Hultzsch and B. Ziemer, Organometallics, 2007, 26, 3668–3676 CrossRef CAS.
- D. F. J. Piesik, S. Range and S. Harder, Organometallics, 2008, 27, 6178–6187 CrossRef CAS.
- M. R. Kember, A. J. P. White and C. K. Williams, Inorg. Chem., 2009, 48, 9535–9542 CrossRef CAS.
- C. K. Williams, L. E. Breyfogle, S. K. Choi, W. W. Nam, V. G. Young Jr., M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2003, 125, 11350–11359 CrossRef CAS.
- C. K. Williams, N. R. Brooks, M. A. Hillmyer and W. B. Tolman, Chem. Commun., 2002, 2132–2133 RSC.
- W. J. van Meerendonk, R. Duchateau, C. E. Koning and G. J. M. Gruter, Macromolecules, 2005, 38, 7306–7313 CrossRef CAS.
- S. Mang, A. I. Cooper, M. E. Colclough, N. Chauhan and A. B. Holmes, Macromolecules, 2000, 33, 303–308 CrossRef CAS.
- E. N. Jacobsen, M. Tokunaga and J. F. Larrow, WO, 009463 2000, 1999.
- E. N. Jacobsen, Acc. Chem. Res., 2000, 33, 421–431 CrossRef CAS.
- D. J. Darensbourg and J. C. Yarbrough, J. Am. Chem. Soc., 2002, 124, 6335–6342 CrossRef CAS.
- D. J. Darensbourg and R. M. Mackiewicz, J. Am. Chem. Soc., 2005, 127, 14026–14038 CrossRef CAS.
- D. J. Darensbourg, R. M. Mackiewicz, J. L. Rodgers, C. C. Fang, D. R. Billodeaux and J. H. Reibenspies, Inorg. Chem., 2004, 43, 6024–6034 CrossRef CAS.
- D. J. Darensbourg, R. M. Mackiewicz, J. L. Rodgers and A. L. Phelps, Inorg. Chem., 2004, 43, 1831–1833 CAS.
- R. Eberhardt, M. Allmendinger and B. Rieger, Macromol. Rapid Commun., 2003, 24, 194–196 CrossRef CAS.
- D. J. Darensbourg, P. Bottarelli and J. R. Andreatta, Macromolecules, 2007, 40, 7727–7729 CrossRef CAS.
- D. J. Darensbourg and A. I. Moncada, Inorg. Chem., 2008, 47, 10000–10008 CrossRef CAS.
- B. Li, R. Zhang and X.-B. Lu, Macromolecules, 2007, 40, 2303–2307 CrossRef CAS.
- D.-Y. Rao, B. Li, R. Zhang, H. Wang and X.-B. Lu, Inorg. Chem., 2009, 48, 2830–2836 CrossRef CAS.
- B. Li, G.-P. Wu, W.-M. Ren, Y.-M. Wang, D.-Y. Rao and X.-B. Lu, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6102–6113 CrossRef CAS.
- G. A. Luinstra, G. R. Haas, F. Molnar, V. Bernhart, R. Eberhardt and B. Rieger, Chem.–Eur. J., 2005, 11, 6298–6314 CrossRef CAS.
- L. Guo, C. Wang, W. Zhao, H. Li, W. Sun and Z. Shen, Dalton Trans., 2009, 5406–5410 RSC.
- X. Xu, C. Wang, H. Li, Y. Wang, W. Sun and Z. Shen, Polymer, 2007, 48, 3921–3924 CrossRef CAS.
- Z. Qin, C. M. Thomas, S. Lee and G. W. Coates, Angew. Chem., Int. Ed., 2003, 42, 5484–5487 CrossRef CAS.
- X.-B. Lu and Y. Wang, Angew. Chem., Int. Ed., 2004, 43, 3574–3577 CrossRef CAS.
- Y. Niu, W. Zhang, X. Pang, X. Chen, X. Zhuang and X. Jing, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5050–5056 CrossRef CAS.
- Y. Niu, H. Li, X. Chen, W. Zhang, X. Zhuang and X. Jing, Macromol. Chem. Phys., 2009, 210, 1224–1229 CrossRef CAS.
- K. Nakano, S. Hashimoto and K. Nozaki, Chem. Sci., 2010, 1, 369–373 RSC.
- S. S, J. K. Min, J. E. Seong, S. J. Na and B. Y. Lee, Angew. Chem., Int. Ed., 2008, 47, 7306–7309 CrossRef.
- J. Yoo, S. J. Na, H. C. Park, A. Cyriac and B. Y. Lee, Dalton Trans., 2010, 39, 2622–2630 RSC.
- Y.-M. Shen, W.-L. Duan and M. Shi, J. Org. Chem., 2003, 68, 1559–1562 CrossRef CAS.
- W.-M. Ren, X. Zhang, Y. Liu, J.-F. Li, H. Wang and X.-B. Lu, Macromolecules, 2010, 43, 1396–1402 CrossRef CAS.
- D. J. Darensbourg and D. R. Billodeaux, Inorg. Chem., 2005, 44, 1433–1442 CrossRef CAS.
- H. Sugimoto and K. Kuroda, Macromolecules, 2008, 41, 312–317 CrossRef CAS.
- D. J. Darensbourg and S. B. Fitch, Inorg. Chem., 2007, 46, 5474–5476 CrossRef CAS.
- Z. Zhang, D. Cui and X. Liu, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6810–6818 CrossRef CAS.
- A. Buchard, M. R. Kember, K. Sandeman and C. K. Williams, Chem. Commun., 2010, 46 10.1039/c0cc02205e.
- T. Sarbu and E. J. Beckman, Macromolecules, 1999, 32, 6904–6912 CrossRef CAS.
- T. A. Zevaco, A. Janssen, J. Sypien and E. Dinjus, Green Chem., 2005, 7, 659–666 RSC.
- T. A. Zevaco, J. Sypien, A. Janssen, O. Walter and E. Dinjus, Catal. Today, 2006, 115, 151–161 CrossRef CAS.
- M. H. Chisholm and Z. Zhou, J. Mater. Chem., 2004, 14, 3081 RSC.
- P. Chen, M. H. Chisholm, J. C. Gallucci, X. Zhang and Z. Zhou, Inorg. Chem., 2005, 44, 2588–2595 CrossRef CAS.
- H. Sugimoto, H. Ohtsuka and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4172–4186 CrossRef CAS.
- D. J. Darensbourg, C. G. Ortiz and D. R. Billodeaux, Inorg. Chim. Acta, 2004, 357, 2143–2149 CrossRef CAS.
- D. J. Darensbourg and E. B. Frantz, Inorg. Chem., 2007, 46, 5967–5978 CrossRef CAS.
- A. Decortes, M. M. Belmonte, J. Benet-Buchholz and A. W. Kleij, Chem. Commun., 2010, 46, 4580–4582 RSC.
- H. Sugimoto, H. Ohshima and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3549–3555 CrossRef CAS.
- P. Mountford, Chem. Soc. Rev., 1998, 27, 105–116 RSC.
- D. J. Darensbourg and S. B. Fitch, Inorg. Chem., 2009, 48, 8668–8677 CrossRef CAS.
- X. Chen, Z. Shen and Y. Zhang, Macromolecules, 1991, 24, 5305–5308 CrossRef CAS.
- B. Liu, X. Zhao, X. Wang and F. Wang, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2751–2754 CrossRef CAS.
- D. V. Vitanova, F. Hampel and K. C. Hultzsch, J. Organomet. Chem., 2005, 690, 5182–5197 CrossRef CAS.
- D. Cui, M. Nishiura and Z. Hou, Macromolecules, 2005, 38, 4089–4095 CrossRef CAS.
- B. B. Lazarov, F. Hampel and K. C. Hultzsch, Z. Anorg. Allg. Chem., 2007, 633, 2367–2373 CrossRef CAS.
- D. Cui, M. Nishiura, O. Tardif and Z. Hou, Organometallics, 2008, 27, 2428–2435 CrossRef CAS.
- A. V. Nikitinskii, L. N. Bochkarev, S. Y. Khorshev and M. N. Bochkarev, Russ. J. Gen. Chem., 2004, 74, 1197–1200 CrossRef CAS.
- D. J. Darensbourg, M. J. Adams and J. C. Yarbrough, Inorg. Chem., 2001, 40, 6543–6544 CrossRef CAS.
- D. J. Darensbourg, M. J. Adams, J. C. Yarbrough and A. L. Phelps, Inorg. Chem., 2003, 42, 7809–7818 CrossRef CAS.
- H. Sugimoto and K. Kuroda, Macromolecules, 2008, 41, 312–317 CrossRef CAS.
- M. Ree, J. Y. Bae, J. H. Jung and T. J. Shin, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 1863–1876 CrossRef CAS.
- R. Eberhardt, M. Allmendinger, M. Zintl, C. Troll, G. A. Luinstra and B. Rieger, Macromol. Chem. Phys., 2004, 205, 42–47 CrossRef CAS.
- J. S. Kim, M. Ree, S. W. Lee, W. Oh, S. Baek, B. Lee, T. J. Shin, K. J. Kim, B. Kim and J. Luning, J. Catal., 2003, 218, 386–395 CrossRef CAS.
- Y. Z. Meng, L. C. Du, S. C. Tiong, Q. Zhu and A. S. Hay, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3579–3591 CrossRef CAS.
- M. Ree, Y. Hwang, J. S. Kim, H. Kim and G. Kim, Catal. Today, 2006, 115, 134–145 CrossRef CAS.
- S. J. Wang, L. C. Du, X. S. Zhao, Y. Z. Meng and S. C. Tjong, J. Appl. Polym. Sci., 2002, 85, 2327–2334 CrossRef CAS.
- Q. Zhu, Y. Z. Meng, S. C. Tjong, Y. M. Zhang and W. Wan, Polym. Int., 2003, 52, 799–804 CrossRef CAS.
- Q. Zhu, Y. Z. Meng, S. C. Tjong, X. S. Zhao and Y. L. Chen, Polym. Int., 2002, 51, 1079–1085 CrossRef CAS.
- J. S. Kim, M. Ree, T. J. Shin, O. H. Han, S. J. Cho, Y. T. Hwang, J. Y. Bae, J. M. Lee, R. Ryoo and H. Kim, J. Catal., 2003, 218, 209–219 CrossRef CAS.
- J. S. Kim, H. Kim and M. Ree, Chem. Mater., 2004, 16, 2981–2983 CrossRef CAS.
- Y. Hwang, H. Kim and M. Ree, Macromol. Symp., 2005, 224, 227–237 CrossRef CAS.
- G. A. Luinstra, Polym. Rev., 2008, 48, 192–219 Search PubMed.
- W. J. Kruper Jr. and D. J. Swart, The Dow Chemical Company, US,4,500,704, 1985.
- J. Kuyper, P. W. Lednor and G. A. Pogany, Shell Int. Research, NL, EP0222453, US4826887, US4826953, US4826952; 1989.
- W. Hinz, E. M. Dexheimer, E. Bohres and G. H. Grosch, BASF Corporation, US 6762278, 2004.
- S. Chen, Z. Hua, Z. Fang and G. Qi, Polymer, 2004, 45, 6519–6524 CrossRef CAS.
- S. Chen, G.-R. Qi, Z.-J. Hua and H.-Q. Yan, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5284–5291 CrossRef CAS.
- W. Hinz, J. Wildeson, E. M. Dexheimer and R. Neff, BASF Corporation, US, 6,713,599, 2004.
- W. Hinz, J. Wildeson and E. M. Dexheimer, BASF Corporation, US, 7268204, 2007.
- I. Kim, M. J. Yi, S. H. Byun, D. W. Park, B. U. Kim and C. S. Ha, Macromol. Symp., 2005, 224, 181–192 CrossRef CAS.
- I. Kim, M. J. Yi, K. J. Lee, D.-W. Park, B. U. Kim and C.-S. Ha, Catal. Today, 2006, 111, 292–296 CrossRef CAS.
- X. K. Sun, X. H. Zhang, F. Liu, S. Chen, B. Y. Du, Q. Wang, Z. Q. Fan and G. R. Qi, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3128–3139 CrossRef CAS.
- N. J. Robertson, Z. Q. Qin, G. C. Dallinger, E. B. Lobkovsky, S. Lee and G. W. Coates, Dalton Trans., 2006, 5390–5395 RSC.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, Jr., J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef.
- M. P. Stevens, Polymer Chemistry, an introduction, Oxford University Press, 1999 Search PubMed.
- S. D. Thorat, P. J. Phillips, V. Semenov and A. Gakh, J. Appl. Polym. Sci., 2003, 89, 1163–1176 CrossRef CAS.
- B. Liu, L. Chen, M. Zhang and A. Yu, Macromol. Rapid Commun., 2002, 23, 881–884 CrossRef CAS.
- C. Koning, J. Wildeson, R. Parton, B. Plum, P. Steeman and D. J. Darensbourg, Polymer, 2001, 42, 3995–4004 CrossRef CAS.
- J. E. Seong, S. J. Na, A. Cyriac, B.-W. Kim and B. Y. Lee, Macromolecules, 2010, 43, 903–908 CrossRef CAS.
- X.-G. Li and M.-R. Huang, Polym. Int., 1999, 48, 387–391 CrossRef CAS.
- X. H. Li, Y. Z. Meng, G. Q. Chen and R. K. Y. Li, J. Appl. Polym. Sci., 2004, 94, 711–716 CrossRef CAS.
- R. Duchateau, W. J. van Meerendonk, L. Yajjou, B. B. P. Staal, C. E. Koning and G.-J. M. Gruter, Macromolecules, 2006, 39, 7900–7908 CrossRef CAS.
- S. Inoue, T. Tsuruta, T. Takada, N. Miyazaki, M. Kambe and T. Takaoka, J. Appl. Polym. Sci.: Appl. Polym. Symp., 1975, 26, 257–267 Search PubMed.
- B. Liu, X. Zhao, X. Wang and F. Wang, Polymer, 2003, 44, 1803–1808 CrossRef CAS.
- K. Nozaki, K. Nakano and T. Hiyama, J. Am. Chem. Soc., 1999, 121, 11008–11009 CrossRef CAS.
- K. Nakano, T. Hiyama and K. Nozaki, Chem. Commun., 2005, 1871–1873 RSC.
- M. Cheng, N. A. Darling, E. B. Lobkovsky and G. W. Coates, Chem. Commun., 2000, 2007–2008 RSC.
- S. Gendler, S. Segal, I. Goldberg, Z. Goldschmidt and M. Kol, Inorg. Chem., 2006, 45, 4783 CrossRef CAS.
- S. Inoue, J. Macromol. Sci., Part A: Pure Appl. Chem., 1979, 13, 651–664 Search PubMed.
- C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 CrossRef CAS.
- J. Zhou, W. Wang, S. Villarroya, K. J. Thurecht and S. M. Howdle, Chem. Commun., 2008, 5806–5808 RSC.
- C.-S. Tan, C.-C. Juan and T.-W. Kuo, Polymer, 2004, 45, 1805–1814 CrossRef CAS.
- D. J. Darensbourg, P. Ganguly and W. Choi, Inorg. Chem., 2006, 45, 3831–3833 CrossRef CAS.
- D. J. Darensbourg, A. I. Moncada, W. Choi and J. H. Reibenspies, J. Am. Chem. Soc., 2008, 130, 6523–6533 CrossRef CAS.
- D.-W. Park, N.-Y. Mun, E.-H. Lee, Y. Choe and S.-W. Park, React. Kinet. Catal. Lett., 2006, 89, 149–156 CrossRef CAS.
- Y. Hu, L. Qiao, Y. Qin, X. Zhao, X. Chen, X. Wang and F. Wang, Macromolecules, 2009, 42, 9251–9254 CrossRef CAS.
- Z. Quan, J. Min, Q. Zhou, D. Xie, J. Liu, X. Wang, X. Zhao and F. Wang, Macromol. Symp., 2003, 195, 281–286 CrossRef CAS.
- Y. Liu, K. Huang, D. Peng and H. Wu, Polymer, 2006, 47, 8453–8461 CrossRef CAS.
- M. Kröger, C. Folli, O. Walter and M. Döring, Adv. Synth. Catal., 2006, 348, 1908–1918 CrossRef.
- R. C. Jeske, J. M. Rowley and G. W. Coates, Angew. Chem., Int. Ed., 2008, 47, 6041–6044 CrossRef CAS.
- L. Lu and K. Huang, Polym. Int., 2005, 54, 870–874 CrossRef CAS.
- Y. Hwang, J. Jung, M. Ree and H. Kim, Macromolecules, 2003, 36, 8210–8212 CrossRef CAS.
- S. Liu, H. Xiao, K. Huang, L. Lu and Q. Huang, Polym. Bull., 2006, 56, 53–62 CrossRef CAS.
Footnote |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| This journal is © The Royal Society of Chemistry 2011 |