
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pd-η3-C6H9 complexes of the Trost modular ligand: high nuclearity columnar aggregation controlled by concentration, solvent and counterion†
Daugirdas Tomas
Racys‡
 a,
Julian
Eastoe
a,
Per-Ola
Norrby
bc,
Isabelle
Grillo
d,
Sarah E.
Rogers
e and
Guy C.
Lloyd-Jones
*f
a,
Julian
Eastoe
a,
Per-Ola
Norrby
bc,
Isabelle
Grillo
d,
Sarah E.
Rogers
e and
Guy C.
Lloyd-Jones
*f
aSchool of Chemistry, University of Bristol, Office S314, Cantock's Close, Clifton, Bristol BS8 1TS, UK. E-mail: Julian.Eastoe@bristol.ac.uk
bAstraZeneca Pharmaceutical Development, Global Medicines Development, Pepparedsleden 1, SE-431 83 Mölndal, Sweden. E-mail: per-ola.norrby@astrazeneca.com
cDepartment of Chemistry and Molecular Biology, University of Gothenburg, Kemigården 4, #8076, SE-412 96 Göteborg, Sweden
dILL, CS20156, 38042 Grenoble Cedex 9, France. E-mail: grillo@ill.fr
eISIS-STFC, Rutherford Appleton Laboratory, Chilton, Oxon OX11 0QX, UK. E-mail: sarah.rogers@stfc.ac.uk
fSchool of Chemistry, University of Edinburgh, Joseph Black Building, West Mains Road, Edinburgh, Scotland EH9 3JJ, UK. E-mail: guy.lloyd-jones@ed.ac.uk
First published on 15th July 2015
Abstract
Under optimised conditions, the Trost modular ligand (TML) series induces high levels of asymmetric induction in an extraordinarily wide range of reactions involving palladium π-allyl intermediates. Prior mechanistic investigations into reactions involving Pd-η3-C6H9 intermediates have focussed on the monomeric 13-membered ring formed via P,P-chelation of the ligand to Pd. However, it is also recognised that ring-opening oligomerisation provides a pool of high nuclearity Pd-η3-C6H9 species that, by affording a low level, or even the opposite sense, of asymmetric induction relative to the mononuclear species, are responsible for a reduction in selectivity under non-optimised conditions. Herein we describe an investigation by NMR spectroscopy, molecular mechanics, molecular dynamics, and small-angle neutron scattering (SANS), of a Pd-η3-C6H9 cation bearing the 1,2-diaminocyclohexane TML ligand (2). Using both nondeuterated and perdeuterated (D47) isotopologues of the resulting complexes ([1]+), we show that a two-stage oligomerisation-aggregation process forms self assembled cylindrical aggregates of very high nuclearity (up to 56 Pd centres). We also investigate how concentration, solvent and counter-anion all modulate the extent of oligomerisation.
Introduction
The Trost modular ligand (TML) series1 has been applied to an extraordinarily wide range of allylic alkylation (Tsuji-Trost) reactions.2 Under carefully optimised conditions, these ligands frequently provide very high enantioselectivity in reactions that have proven challenging with other chiral ligands, particularly those involving cyclic allylic substrates.3–6 These features have led to broad use of the TML in the synthesis of natural products,7 as well as industrial application for the construction of high-enantiopurity chiral building blocks. However, reactions involving the TML can exhibit memory effects,8,9 and a high sensitivity of the enantioselectivity to reaction temperature, catalyst concentration, solvent and nucleophile counter-ion.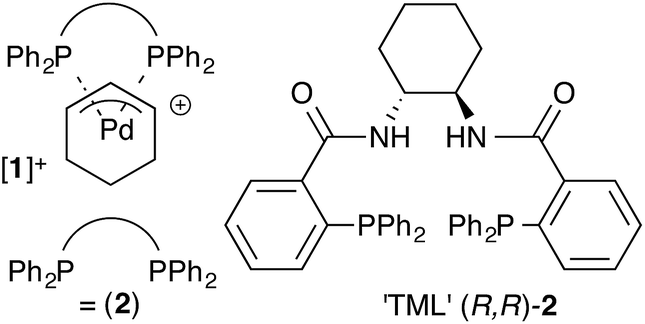
Our previous mechanistic studies of this system focussed on the monomeric cationic complex [1]+, in which a Pd(η3-C6H9) unit is chelated by the 1,2-diaminocyclohexane-derived TML ligand (2).10 The monomeric cation [1]+ was identified as an intermediate capable of leading to high asymmetric induction on attack of, for example, a malonate anion nucleophile, Scheme 1. Detailed NMR studies facilitated by isotopic labelling – in conjunction with MM-DFT simulations – led to a model10 in which the amide units in the catalyst facilitate enantioselective ligand-accelerated catalysis.11
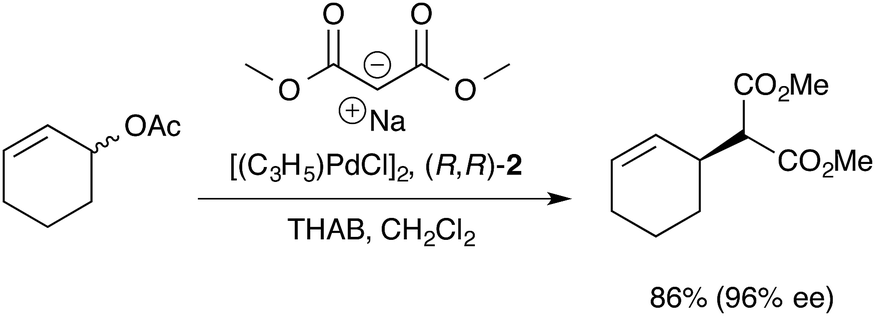 | ||
| Scheme 1 Asymmetric allylic alkylation of racemic 2-cyclohexenyl acetate; 2 = 1,2-diaminocyclohexane TML ligand, THAB = tetrahexylammonium bromide. | ||
For the ligand-accelerated catalysis to function efficiently, cation [1]+ requires a degree of flexibility. This flexibility is provided by the 13-membered chelate ring, but at a cost: complex [1]+ can readily undergo ring-opening oligomerisation to generate polynuclear species ([1]+)n, Scheme 2.12 Competing nucleophilic attack on the oligomer, rather than the monomer [1]+ is, in part, responsible for a reduction in overall enantioselectivity under non-optimised conditions.13,14
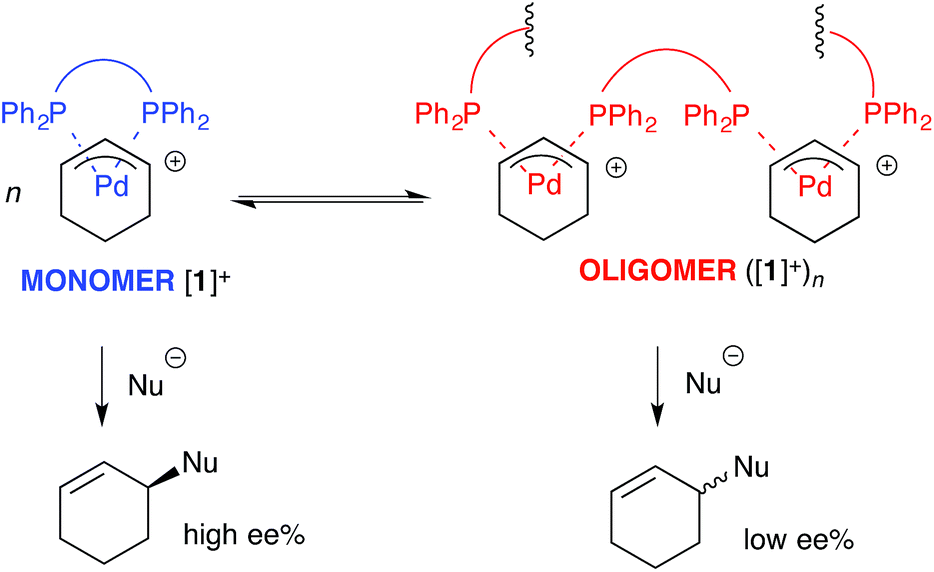 | ||
| Scheme 2 Oligomerisation of [1]+ erodes enantioselectivity during asymmetric Pd-catalysed allylic alkylation mediated by 2 (as Scheme 1). Nu = nucleophile. | ||
To date, the structure and origin of the formation of these oligomeric species has not been studied in detail. Herein, we describe an investigation of the oligomerisation of D0 and D47 isotopologues of [1]+, employing NMR spectroscopy, molecular mechanics (MM), molecular dynamics (MD), and contrast variation small-angle neutron scattering (SANS). The data obtained indicate that the impact of a first-stage of depletion of the monomeric species [1]+ from the catalyst pool, via cyclic oligimerisation, is amplified by a second-stage process involving columnar aggregation of the oligomers, leading to species with very high nuclearity (up to 56 Pd centres). The effects of solvent, ligand enantiopurity and counter-ion on the degree of aggregation are explored in detail, and it is concluded that a relatively small and restricted set of conditions facilitate dissolution of the complexes in a low aggregation state, consistent with the extensive optimisation frequently required for these catalyst systems.
Results and discussion
Preliminary NMR studies and synthesis of [D47]-[1][B(C6F5)4]
Despite extensive efforts,15 we have been unable to crystallise any Pd(η3-C6H9) complexes of 2, in either oligomeric or monomeric forms. Indeed, to date, the only the X-ray crystal structures of Pd-allyl complexes of ligand 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16 are η3-C3H5 complexes with triflate counter anions: one a racemic tetranuclear cyclo-oligomer,12 the other an acyclic dinuclear bis-P,O-chelate.17
16 are η3-C3H5 complexes with triflate counter anions: one a racemic tetranuclear cyclo-oligomer,12 the other an acyclic dinuclear bis-P,O-chelate.17
The extent of solution-phase oligomerisation of cationic complexes of type [1]+ can be conveniently estimated by 31P{1H} NMR spectroscopy.10,12 Analysis of [(R,R)-1][BAr4] complexes in CH2Cl2, where Ar = C6Cl5, 3,5-(CF3)2C6H3, or C6F5 (“BArF”), indicates a maximum monomer concentration ([1]+) of about 4 mM, Fig. 1. In THF, the monomer maximum is lower (approx. 1.6 mM) and decreases as [Pd]tot is raised above 10 mM. With smaller, less charge-diffuse, counter-anions such as chloride or triflate, the maximum monomer concentrations are lower still. We were unable to fit simple analytical solutions18 for monomer-oligomer distributions to any of the 31P NMR data, indicative that physicochemical effects dominate over simple solution-phase equilibria, even at low [Pd]tot.
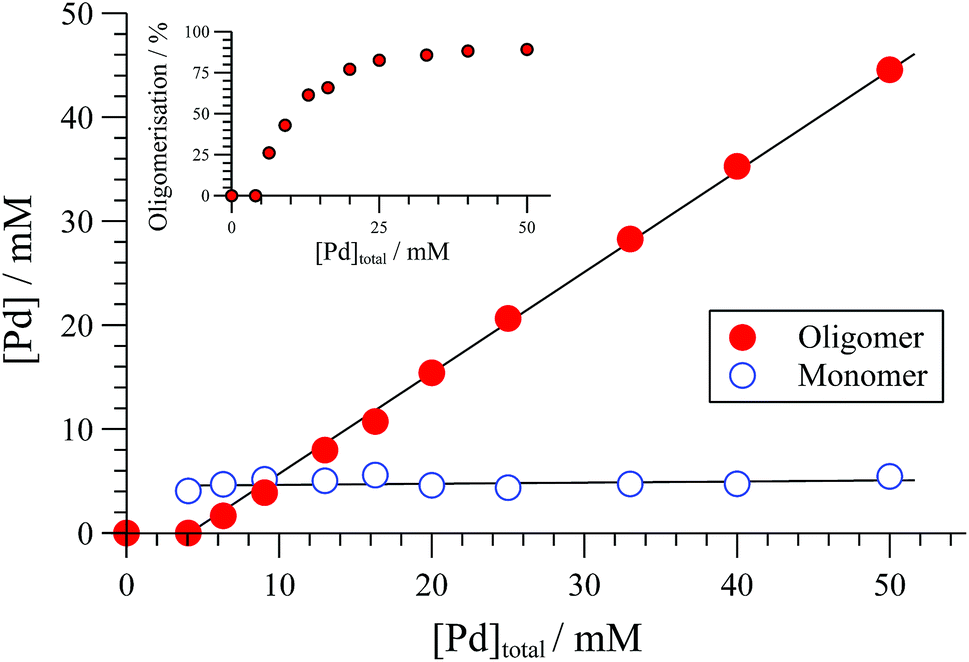 | ||
| Fig. 1 Speciation of [(R,R)-1][BArF] (BArF = B(C6F5)4) determined by 31P{1H} NMR (CD2Cl2, 25 °C). Solid lines through data are solely a guide to the eye. Inset shows proportion (%) of [Pd]tot that is in oligomeric form. | ||
We thus elected to study the oligomeric species by SANS – a technique that can be used for characterising the shape and dimensions of self-assembly structure and colloids.19 We began with [(R,R)-1][BArF],20 and, to aid the studies, also synthesised the perdeuterated enantiomeric complex [(S,S)-[D47]-1][BArF]. Not only does this facilitate SANS in a non-deuterated solvent, thus providing greater neutron scattering contrast,19 it also allows pseudo racemic and pseudo scalemic mixtures to be prepared by mixing [(S,S)-[D47]-1][BArF] with [(R,R)-1][BArF]. The perdeuterated complex was synthesised from benzoic acid ([D5]-3), chlorobenzene ([D5]-4) and cyclohexene ([D10]-5), Scheme 3. A major hurdle was the ortho-metallation of ester [D5]-6 with (TMP)2Mg·LiCl,21 which proceeded with an unexpectedly large net kinetic isotope effect (kH/kD ≈ 30).22 This required an excess of base to be employed, and interfered with a planned direct phosphination of the metallated intermediate. Instead, the intermediate was trapped with I2. The iodide [D4]-7 was then converted to a more conventional Grignard reagent,23 before reaction with chlorophosphine [D10]-810,24 to give phosphine [D14]-9, and thus acid [D14]-10.25
 | ||
| Scheme 3 Synthesis of pseudo enantiomer [(S,S)-[D47]-1][BArF]; BArF = B(C6F6)4. Conditions: (i) SOCl2, toluene, 100 °C; (ii) KOtBu, THF 0 °C; 84%; (iii) (TMP)2Mg·2LiCl, THF, 0 °C to 30 °C; (iv) I2, THF, -30 °C; 49%; (v) iPrMgCl, THF, -40 °C, 2 h; (vi) Mg, LiCl, THF, reflux; (vii) Et2NPCl2, THF; (viii) HCl, Et2O, -40 °C; 29%; (ix) [D10]-8 drop-wise addition, THF, -78 °C; 68%; (x) KOH, THF, r.t., 24 h; (xi) HCl; 58%; (xii) 5% Ru(PPh3)3Cl2, D2O, 10% SDS, microwave, 140 °C, 1 h; (xiii) mCPBA, CH2Cl2, NaHCO3; 51%; (xiv) (S)-1-phenylethylamine, MeCN, LiBr, 60 °C; 74%; (xv) DIAD, PPh3, THF, 0 °C to r.t.; 62%; (xvi) NaN3, CeCl3·7H2O, MeCN/H2O 9:1, reflux; (xvii) silica-gel chromatography; 61%; (xviii) 20% Pd(OH)2/C, MeOH, r.t., 1 atm H2; (xix) 20% Pd(OH)2/C, MeOH, 2 M HCl/Et2O, HCO2NH4, 65 °C; (xx) KOH, CH2Cl2, r.t.; (xxi) EDCI·HCl, HOBt·H2O, iPr2NEt, CH2Cl2, r.t.; (xxii) Et2O, HCl; 30%; (xxiii) NaCl, NaOAc, CuCl2, AcOH, Ac2O, 2 h, 95 °C; (xxiv) [D10]-5, 60 °C, 20 h; 13% (xxv) KBArF, MeCN, CH2Cl2, r.t.; (xxvi) CH2Cl2, r.t.; 99%. See ESI† for full details. | ||
Cyclohexene [D10]-5,26 was epoxidised, to give [D10]-11, and this ring-opened27 to give aminoalcohol [D10]-12.28 Aziridine [D10]-13, obtained under Mitsunobu conditions, was converted to azide [D10]-14.29 After diastereoisomer separation, hydrogenolysis30 gave (S,S)-diamine [D10]-15, which was coupled with acid [D14]-10 to afford Trost ligand [D38]-2. The chloro-bridged dimer [D18]-16, prepared31 from [D10]-5, was converted to cationic complex [D9]-17, and then reacted with [D38]-2 to generate [(S,S)-[D47]-1][BArF] in good yield.
SANS analysis of aggregation of [1][BArF] in THF
We began with SANS analysis32 of enantiopure [(R,R)-1][BArF] in THF-D8 at 25 °C, with [Pd]tot concentrations 16 to 64 mM, well above the oligomerisation threshold. The combined data sets,33 presented here as plots of scattered intensity (I(Q), y-axis) versus momentum transfer (Q, x-axis) were analysed with a range of different standard models for possible aggregate form factors, corresponding to various simple shapes,19Fig. 2.34 This clearly identified the particles as cylindrical or rod-like, and Guinier analyses (see ESI, Fig. S69†) established the radii (8-9 Å) and lengths (150-200 Å) as essentially invariant across the range of [Pd]tot explored. In other words, the number density of aggregates changes in response to [Pd]tot, but not their average dimensions,35 clearly indicative of a set of factors that tightly control the particle scale. | ||
| Fig. 2 SANS profiles of 1.6 to 64 mM (0.25-10.0 wt/vol%) {[(R,R)-1][BArF]}n in THF-D8, 25 °C; I(Q) = scattered intensity, Q/Å−1 = momentum transfer = (4π/λ)sin(θ/2); where scattering angle = θ and neutron wavelength = λ. The fitted curves correspond to a cylindrical particle form factor. The sample with 1.6 mM [Pd]tot has form factor fitted to radius ≤ 11 Å and length ≤ 50 Å. All other samples are fitted to radius 8-9 Å and length 150-200 Å. | ||
We have previously used 31P NMR spectroscopy to analyse the constitution of the solution-phase (i.e., lower-order) oligomers generated from various complexes of type [1][BAr4] in CD2Cl2.10,12 Using PPCOSY in combination with pairs of isotopically-differentiated ligands ([Dn]-2), we were able to determine that the oligomers are: i) non-chelated species (i.e. each of the ligands (2) in the oligomer are coordinated to two different Pd centres); ii) present in predominantly homochiral form (i.e. [(R,R)-1]+ and [(S,S)-1]+ oligomerise independently), and iii) contain no ‘free’ (i.e. not Pd-coordinated) P-centres in the ligand (2). Although a cyclic oligomer structure (Scheme 2) is fully consistent with these features, we were unable to determine the number (n) of ring-opened monomer units incorporated within the cyclo-oligomer ([1][BAr4])n.
As [Pd]tot in THF or CH2Cl2 solutions of complexes of type [1][BAr4] is increased, the 31P NMR bandshape of the signals arising from the cyclo-oligomer do not change in appearance, but the samples do become increasingly turbid. This behaviour suggests that in response to an increase in [Pd]tot, cyclo-oligomers ([1][BAr4])n do not incorporate more monomer (n), but instead aggregate to form larger particles, {([1][BAr4])n}m, containing ‘m’ cyclo-oligomers. It is these high nuclearity particles that are detected by SANS.12
In racemic or scalemic samples of [1][BArF], the homochiral cyclo-oligomers, ([1][BArF])n, could aggregate in three general forms: discrete homochiral, ordered heterochiral (e.g., alternating or co-block), or statistically distributed. SANS data of mixtures of [(R,R)-1][BArF] and [(S,S)-1][BArF] representing enantiopure, scalemic, and racemic samples, was uniform across the series, within experimental error, Fig. 3. The absence of a change in particle number density36 is consistent with a statistical cyclo-oligomer distribution in which there is no significant impact of homo- or hetero-chirality on the particle shape or size.
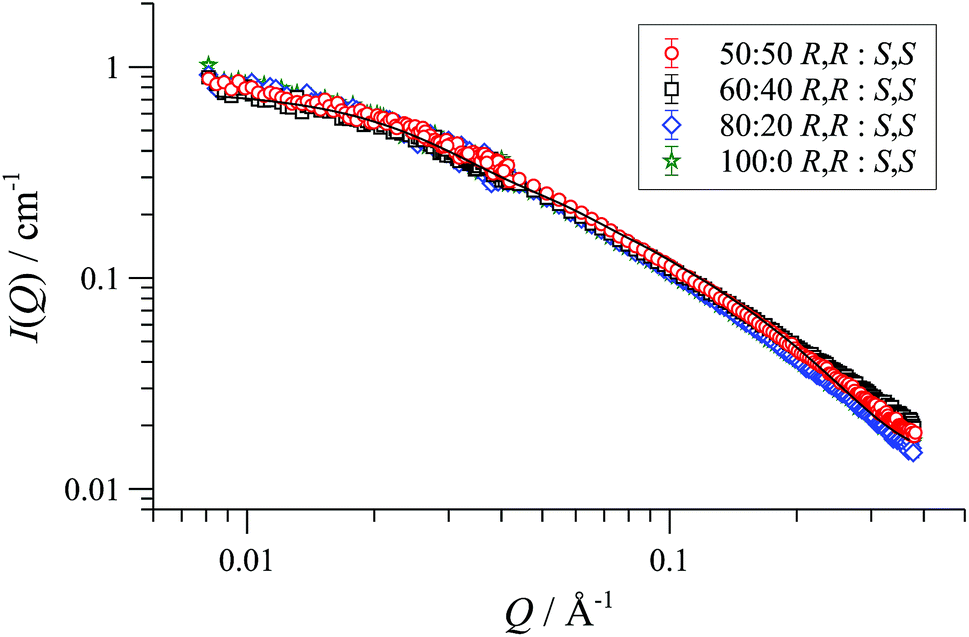 | ||
| Fig. 3 Fitted SANS profiles and cylinder form factor fits for 32 mM enantio-pure, racemic and scalemic samples of [1][BArF] in THF-D8, 25 °C. | ||
SANS data of the pseudo racemate [(S,S)-[D47]-1][BArF] + [(R,R)-[D0]-1][BArF]) in H8-THF, and in D8-THF, Fig. 4, and enantiomerically pure [(S,S)-[D47]-1][BArF] in D0-THF, show that the fully and partially deuterated systems retain the concentration-independent, cylindrical aggregate shape. A major difference is, however, observed in the dimensions: [D47]-[1][BArF] forms shorter (130 Å), slightly wider (10 Å radius) cylinders than [D0]-[1][BArF]. The pseudo racemic mixture measures as an average of its precursors (9-10 Å radius, 150 Å length), again consistent with a statistical distribution of cyclo-oligomers in the aggregate.37
 | ||
| Fig. 4 SANS data and cylinder form factor fits for pseudo racemic complex ([(S,S)-[D47]-1][BArF] + [(R,R)-[D0]-1][BArF]) in THF-D8 at 25 °C, at [Pd]tot ranging from 13 to 51 mM. | ||
Molecular mechanics (MM) and dynamics (MD)
Computational modelling was employed to probe the factors that control the aggregation phenomena, and to estimate the size (n) and number of cyclo-oligomers in the particle (m).38 The number of atoms in the aggregates (>2,000, vide infra) means that density functional theory (DFT) calculations of their structures would demand currently unattainable computational resources and inordinate simulation times. On the other hand, molecular mechanics (MM) can provide a good approximation in just a small fraction of the computational time required by DFT. Although MM in general calculates only a steric energy, not the full free energy, and ignores bond dissociation energies, accurate results can be obtained from MM3 calculations provided that comparisons are isodesmic and, more importantly, isoparametric.39 This requirement is fulfilled for all comparisons of ring oligomers and their non-covalent aggregates. We began by confirming that the structure of the 84-atom monomeric cationic P,P-chelate [1]+, optimised at MM3 level of theory with dielectric constant ε = 9.0, was almost identical to that obtained using DFT (B3LYP-D3) in a polarisable continuum model for dichloromethane.38 Further calculations involving ion-pairs, oligomers and aggregates were then performed using MM3 to provide analysis of the energies involved, albeit at a coarse-grained level of detail.Comparison of the MM3 optimised energies for monomeric P,P-chelate [1][BArF] with a series of homochiral cyclo- oligomers, ([1][BArF])n, normalised by the number of Pd atoms (n, the x-axis in Fig. 5) confirmed cyclo-oligomerisation to be exothermic, and probably also exergonic up to tetramers (n = 4). The cyclic dimer (n = 2) still suffers from ring strain, and a more substantial stability is afforded by trimerisation (n = 3) then tetramerisation40 (n = 4).
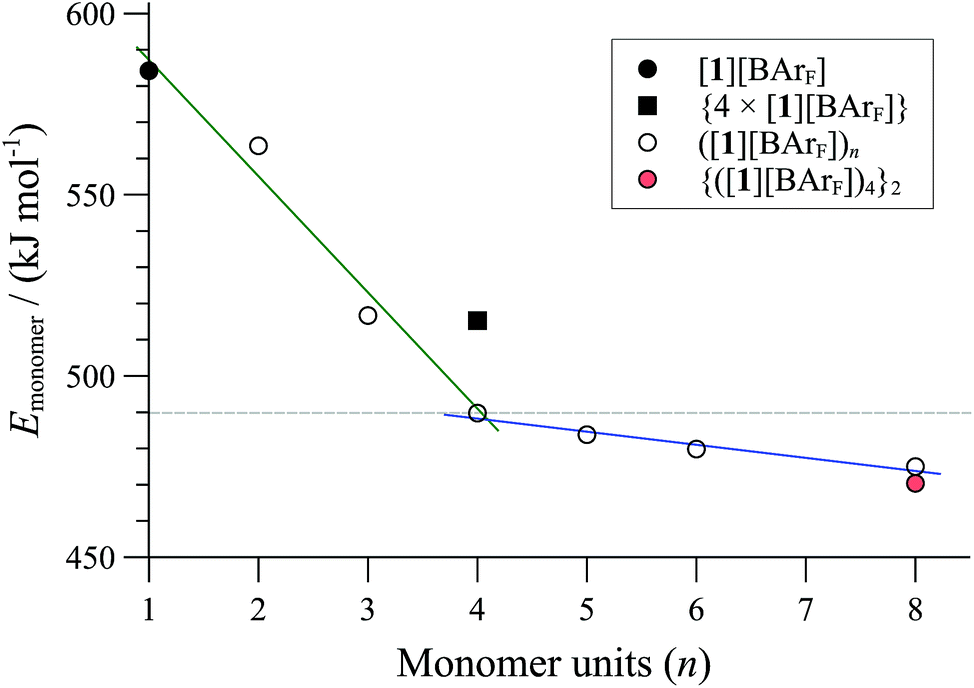 | ||
| Fig. 5 MM3 energies (ε = 9.0) of cyclo-oligomeric complexes ([1][BArF])n as a function of monomers incorporated (n, x-axis). {4 × ([1][BArF])} is a pre-orientated assembly of four monomeric P,P-chelates; {([1][BArF])4}2 is an aggregate formed from two tetrameric cyclo-oligomers (see m = 2 in Fig. 7). | ||
 | ||
| Fig. 6 MM3 structure of tetranuclear cyclo-oligomer ([1][BArF])4. For clarity, only two of the four BArF anions are shown. Colour coding: Pd, yellow; P, orange; O, red; N, dark blue; C, light grey; B, pink; F, green; η3-C6H9, light blue. | ||
 | ||
| Fig. 7 MM3 energies (ε = 9.0) of {([1][BArF])4}m (normalised per monomer), as a function of number (m) of cyclo-oligomeric tetramers aggregated. | ||
Further increase in oligomer ring size (n = 5, 6, 8) yields a modest reduction in the system energy but generates species with significantly higher radii than the 8-9 Å cylinder radius detected by SANS. In summary, the tetranuclear species (n = 4), Fig. 6, appears to be the dominant, if not exclusive, cyclo-oligomer, and aggregation of these cyclic tetramers ([1][BArF])4 was thus probed by MM3 as a process to generate the cylindrical particles, Fig. 7.
Positively charged rod-like structures with dissociated or removed anions were estimated by MM3 to be very much higher in energy than those where the anions were closely associated with the cationic cyclo-oligomeric building blocks. Anion interactions were thus explored more deeply, and although BArF is considered a weakly coordinating anion,41 the charge delocalisation over its surface reduces repulsive interactions with other BArF anions, and the presence of fluorine makes it significantly lipophilic. Indeed, the calculations indicated a favourable interleaving of the BArF anions in sandwich layers42 between cationic cyclo-oligomers. The estimated formation energies, (ΔE, Fig. 7) of such species {([1][BArF])4}m as a function of ‘m’ indicated that columnar aggregates are readily attainable, with the growing entropic cost (TΔS) placing limits on the aggregate length.34,40
These conclusions were further probed by molecular-dynamics (MD) simulations in which the MM3-minimised structures {([1][BArF])4}m were computationally excited (300-700 K) over short periods (300 ps) to test the relative structural integrity of the aggregate as a function of ‘m’. In the low dielectric constant medium used for the model, most systems (m = 4 to 16) did not undergo any significant changes in their tertiary structure at 300 K. As the energy input was increased the aggregate models exhibited varying degrees of structural deformation, undergoing rapid fragmentation at the highest energies. The most significant observations were made at intermediate energies (500-550 K): aggregates with m = 10-14 (e.g., Fig. 8, m = 12) retained a cylinder shape, albeit mildly distorted, over the full 300 ps simulation time, whereas higher or lower order aggregates significantly deformed, in some cases losing one or more BArF anions. The average dimensions of the MM3 aggregates with m = 10-14 (radius 8-9 Å and length 150-200 Å) are consistent with the particle dimensions determined by SANS, Fig. 2 and 3.
 | ||
| Fig. 8 MM3 structure of {([1][BArF])4}12. The monomer [1][BArF] (top left), with palladium coloured orange, is shown for scale. | ||
The effect of counter-ion and solvent on shape and extent of aggregation
The effect of solvent type on aggregation was probed by MM, 31P{1H} NMR (Fig. 9) and SANS, also comparing [1][BArF] with [1][OTf] to explore the impact of counter-ion. The aggregation mode for [1][BArF] determined by MM3, e.g., Fig. 8, involves multiple close-range electrostatic interactions that reduce the overall energy of the system. It is therefore not surprising that the solvent dielectric constant (εr) was found to modulate aggregation,43 and thus also solubility, with precipitation being the ultimate consequence of strong aggregation. | ||
| Fig. 9 Oligomerisation (31P{1H} NMR, %) of [1][BArF] and [1][OTf] at [Pd]tot = 15 mM in solvents of various dielectric constant (εr); overlaid areas in green (OTf) and blue (BArF) indicate regions where the complexes are insoluble. | ||
As indicated in Fig. 9, both [1][OTf] and [1][BArF] readily oligomerise and aggregate in all of the solvents that were explored, becoming essentially insoluble at the extremes of εr, (e.g., in alkanes, most ethers, chloroform, aromatic hydrocarbons, and at the opposite end of the scale, in water). The lipophilicity and charge-density of the anion also affects the solubility: [1][BArF] (but not [1][OTf]) readily dissolves in THF, and at the opposite end of the εr scale, [1][OTf] (but not [1][BArF]) is soluble in aqueous-organic mixtures.
SANS was employed to explore how the macromolecular composition of aggregates {([1][X])n}m is affected by solvation. Although [1][BArF] is not soluble in organic-aqueous mixtures, SANS data were attainable in polar aprotic solvents (e.g., MeCN, εr = 37.5; and DMSO, εr = 47). This confirmed that cylindrical aggregates were still formed, but were significantly shorter than those in THF, Fig. 10. Medium length cylinders were detected in a 50:50 mixture of THF and acetonitrile, consistent with the intermediate solvent polarity (εr ≈ 23). In all cases, the cylinders were of radius 8-10 Å, strongly suggesting the prevalence of the tetranuclear cyclo-oligomer building blocks, with the solvent modulating only the aggregation number ‘m’: {([1][BArF])4}m.
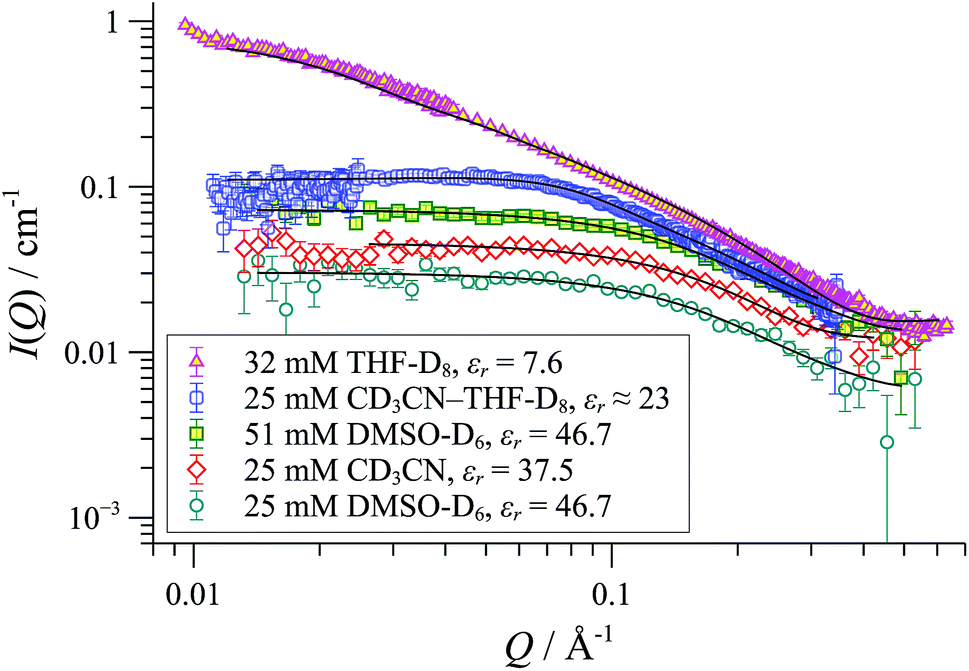 | ||
| Fig. 10 SANS data and cylinder form factor fits for [1][BArF] in various solvents at 25 °C. | ||
The [1][OTf] aggregates behaved differently. Although, cylinders of radius 8-10 Å were again detected in all cases, indicative of {([1][OTf])4}m aggregates, the flexibility, lengths and charge distribution in the particles were very different to those formed from [1][BArF].
In CD2Cl2, [1][OTf] forms cylindrical aggregates (up to 160 Å; Fig. 11) apparently with a degree of flexibility, a phenomenon that can be attributed to the small and interactive triflate anion being less able to rigidify the structures than the larger and more lipophilic BArF anion. The anion effect became even more pronounced in media of higher dielectric constant. In acetonitrile-based solvent mixtures (εr = 47-58) the SANS data indicated an additional minor structure factor contribution (S(Q)), consistent with weakly charged particles, Fig. 12. Weak repulsive interactions might arise from solvation-induced ion-pair separation of triflate from the cationic Pd(II) oligomeric cores. The increased cationic repulsion between the cyclo-oligomers appears to result in much shorter cylinders, just 30 Å in length, with misleadingly simple 31P NMR spectra.44 Similar conclusions were drawn from MD simulations with the medium set at εr = 35: only the shortest aggregates {([1][OTf])4}2-4 were structurally stable at elevated energies (500 K; 300 ps). All higher aggregates (m > 4) underwent rapid fragmentation.
 | ||
| Fig. 11 SANS data and flexible cylinder form factor fits for [1][OTf] in CD2Cl2 at 25 °C; fits are for radius 8-9 Å and lengths: 78 Å (13 mM); 156 Å (25 mM) and 332 Å (50 mM). | ||
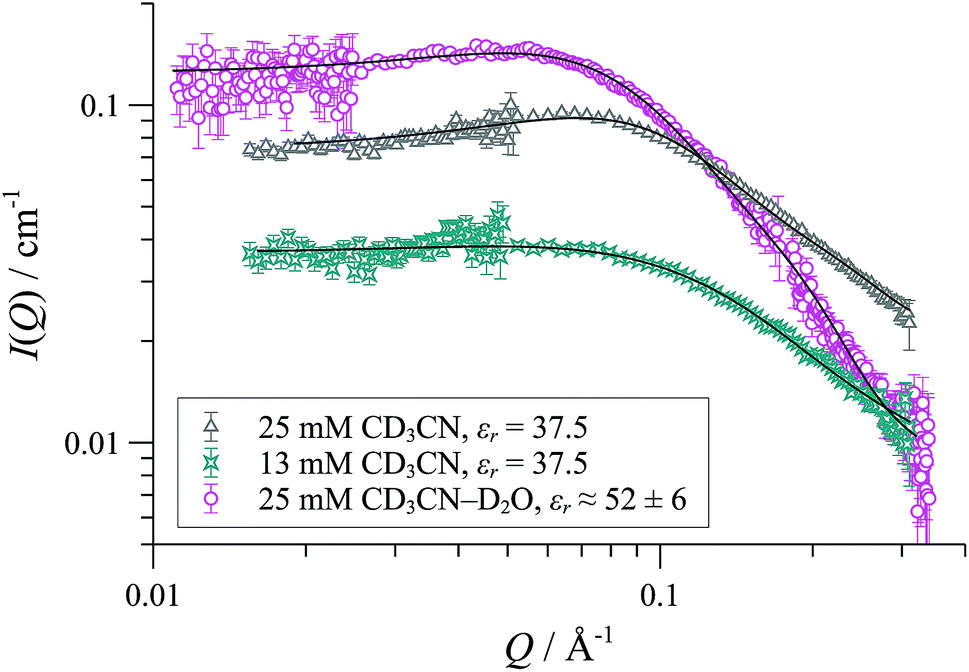 | ||
| Fig. 12 SANS data and cylinder form factor fits (including an effective Hayter-Penfold structure factor S(Q) to account for charged particles) for [1][OTf] in CD3CN at 25 °C. | ||
Finally, to probe the relevance of the higher aggregates to asymmetric alkylation (Scheme 1) SANS data were acquired on reaction mixtures in which [1][BARF] was employed as a pre-catalyst (10 mol%) for addition of tetrabutylamonium dimethylmalonate to cyclohexenylacetate in THF. While the effects of substrate background scattering, varying acquisition times and shorter Q-range slightly affected the data quality, it remained clear that the dominant structures in solution, for the whole duration of the catalytic process, were large cylinders. This result is consistent with previous conclusions that, in THF, the catalytic turnover proceeds via a small pool of highly-active monomeric catalyst species, in competition with cyclo-oligomers and aggregates.10,12
Conclusions
Since the initial report that [1]+ readily oligomerises,13 and that this is a largely undesirable property of an otherwise highly efficient catalyst, e.g., Scheme 1, there has been limited understanding of the oligomer structures.10,12 We have now identified, through NMR spectroscopy, SANS, and MM/MD simulations of [[Dn]-1][BArF] (n = 0, 47),45 that monomeric cations [1]+ undergo chelate-opening to form a tetranuclear cyclo-oligomer ([1][BArF])4; this being thermodynamically favoured over higher and lower nuclearity species. A second-stage process involving columnar-like interactions between the cyclo-oligomers then forms aggregates {([1][BArF])4}m. In THF these aggregates comprise alternating layers of cyclo-oligomer and interleaved BArF anions in the form of cylindrical particles (m = 10-14; radius 8-9 Å; length 150-200 Å) containing up to 56 Pd(η3-C6H9) centres, ligands (2) and BArF anions.The identity of the counter-anion has a pronounced effect on the proportion of oligomer generated from the monomer. Bulky, weakly-coordinating anions,46 reduce the extent of oligomerisation, particularly in low polarity solvents that cannot effectively stabilise charged particles. Here the role of the bulky and relatively lipophilic anions is to solvate the monomer [1]+. Smaller harder, less lipophilic anions are less able to solvate the monomer, and have the indirect effect of shifting the equilibrium towards the oligomer; an undesirable feature for catalysis. The diminutive size of the anion also results in greater flexibility of the resulting columnar aggregates, which are more ionic in nature, reducing their solubility in less polar solvents.
Overall, although the solvent polarity, counter-anion (X), and net concentration ([Pd]tot) all affect the degree of oligomerisation and aggregation (Fig. 13) of monomer [1][X], the solvent perhaps offers the greatest degree of scope for optimisation under the conditions of catalysis. In this regard, CH2Cl2 is favourable: solutions can be virtually free of oligomer at ambient temperature, provided [Pd]tot ≤ 4 mM. Intriguingly, SANS studies of ionic and non-ionic surfactants47 have revealed specific solvent combinations that can lead to “dead zones” where aggregation is suppressed, even for concentrated solutions. If such “dead zone” solvent combinations can be found for complexes of type [1][X], this may be highly advantageous for improving catalytic productivity whilst maintaining selectivity.
 | ||
| Fig. 13 Summary of upper-range aggregate cylinder lengths of [1][BArF] and [1][OTf] samples as determined by SANS experiments, in selected solvents., with [Pd]tot = 25 mM. | ||
Acknowledgements
We thank the ILL (France), ISIS (UK), and STFC (UK) for facilities (instruments D11, D16, D22 and D33 at ILL, and instruments SANS2d and LOQ at ISIS). The Bristol Chemical Synthesis Centre for Doctoral Training (EPSRC EP/G036764/1), AstraZeneca and the University of Bristol provided a PhD studentship to DTR.Notes and references
- B. M. Trost, M. R. Machacek and A. Aponick, Acc. Chem. Res., 2006, 39, 747–760 CrossRef CAS PubMed.
- Z. Lu and S. Ma, Angew. Chem., Int. Ed., 2007, 47, 258–297 CrossRef PubMed.
- B. M. Trost and R. C. Bunt, J. Am. Chem. Soc., 1994, 116, 4089–4090 CrossRef CAS.
- (a) S. Fuchs, V. Berl and J.-P. Lepoittevin, Eur. J. Org. Chem., 2007, 7, 1145–1152 CrossRef; (b) M. J. Moschitto, D. N. Vaccarello and C. A. Lewis, Angew. Chem., Int. Ed., 2015, 54, 2142–2145 CrossRef CAS PubMed.
- B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 2003, 125, 3090–3100 CrossRef CAS PubMed.
- B. M. Trost and G. Dong, J. Am. Chem. Soc., 2006, 128, 6054–6055 CrossRef CAS PubMed.
- B. M. Trost and N. Asakawa, Synthesis, 1999, 1491–1494 CrossRef CAS.
- B. M. Trost and R. C. Bunt, J. Am. Chem. Soc., 1996, 118, 235 CrossRef CAS.
- G. C. Lloyd-Jones and S. C. Stephen, Chem.–Eur. J., 1998, 4, 2539–2549 CrossRef CAS.
- C. P. Butts, E. Filali, G. C. Lloyd-Jones, P.-O. Norrby, D. A. Sale and Y. Schramm, J. Am. Chem. Soc., 2009, 131, 9945–9957 CrossRef CAS PubMed.
- D. J. Berrisford, C. Bolm and K. B. Sharpless, Angew. Chem., Int. Ed., 1995, 34, 1059–1070 CrossRef CAS.
- J. Eastoe, I. J. S. Fairlamb, J. M. Fernández-Hernández, E. Filali, J. C. Jeffery, G. C. Lloyd-Jones, A. Martorell, A. Meadowcroft, P.-O. Norrby, T. Riis-Johannessen, D. A. Sale and P. M. Tomlin, Faraday Discuss., 2010, 145, 27–47 RSC.
- I. J. S. Fairlamb and G. C. Lloyd-Jones, Chem. Commun., 2000, 2447–2448 RSC.
- Stoichiometric reaction of [(R,R)-[1][BAr4] (Ar = 3,5-(CF3)2C6H3) with tetrabutylammonium dimethylmalonate in THF at 21 °C: [Pd]tot = 1 mM affords alkylation product in 98% ee, whereas product of 6 % ee is obtained when [Pd]tot = 27 mM.10.
- Around 100 attempts were made to crystallise the following complexes ([(R,R)- and rac [1][X], X = BArF, B((3,5-(CF3)2C6H3)4, [Al(OC(CF3)3)4] and OTf) using diffusion, evaporation, cooling, seeding, co-crystallisation, and other methods, from a variety of binary and ternary solvent mixtures, including but not limited to chloroform, CH2Cl2/pentane, 1,2-dichloroethane/hexane, THF/heptane, 1,2-difluorobenzene/1,3-difluorobenzene, tetrachloroethane/hexafluorobenzene, MeCN/mesitylene, aqueous MeCN/MTBE, water, diethyl ether, etc. without success.
- For examples of X-ray structures of Pd complexes of 2 that are inactive as catalysts for allylic alkyation, see: (a) K. R. Campos, M. Journet, S. Lee, E. J. J. Grabowski and R. D. Tillyer, J. Org. Chem., 2005, 70, 268–274 CrossRef CAS PubMed; (b) C. Amatore, A. Jutand, L. Mensah and L. Ricard, J. Organomet. Chem., 2007, 692, 1457–1464 CrossRef CAS.
- C. P. Butts, J. Crosby, G. C. Lloyd-Jones and S. C. Stephen, Chem. Commun., 2000, 1707–1708 Search PubMed.
- D. C. Smith and G. M. Gray, J. Chem. Soc., Dalton Trans., 2000, 677 RSC.
- J. Eastoe, Surfactant Chemistry, Wuhan University Press, Wuhan, China, 2005 Search PubMed.
- Perfluoro tetraphenylborate [B(C6F5)4]− (“BArF”) was chosen to avoid SANS data being complicated by neutron scattering from protons or deuterons in the counter-ion, as this could then be used without perturbation in the perdeuterated complex.
- C. J. Rohbogner, A. J. Wagner, G. C. Clososki and P. Knochel, Org. Synth., 2009, 86, 374–384 CrossRef CAS.
- The large KIE (kH/kD ≈ 30, by comparison of approximate rates of metallation of the deuterated and non-deuterated substrates), may arise from multiple primary effects, involving the TMP-amide, and in situ generated [D]-TMP, or from tunnelling.
- It was found that slow addition of [D10]-8 to the Grignard drop-wise at -78 °C, reduced extensive by-product formation.
- F. M. Piller, P. Appukkuttan, A. Gavryushin, M. Helm and P. Knochel, Angew. Chem., Int. Ed., 2008, 47, 6802–6806 CrossRef CAS PubMed.
- E. Filali, G. C. Lloyd-Jones and D. A. Sale, Synlett, 2009, 205–208 CAS.
- K. Ishibashi and S. Matsubara, Chem. Lett., 2007, 36, 724–725 CrossRef CAS.
- M. Chandrasekhar, G. Sekar and V. K. Singh, Tetrahedron Lett., 2000, 41, 10079–10083 CrossRef CAS.
- A. K. Chakraborti, S. Rudrawar and A. Kondaskar, Eur. J. Org. Chem., 2004, 3597–3600 CrossRef CAS.
- V. M. Mastranzo, E. Santacruz, G. Huelgas, E. Paz, M. V. Sosa-Rivadeneyra, S. Bernès, E. Juaristi, L. Quintero and C. A. de Parrodi, Tetrahedron: Asymmetry, 2006, 17, 1663–1670 CrossRef CAS.
- In contrast to the report of Singh,27 hydrogenolysis with Pd(OH)2/C under acidic conditions in methanol failed to cleave the α-methylbenzyl auxiliary. Instead, after reduction of the azide, the hydrogen source was changed to ammonium formate: (a) A. D. Brown, M. E. Bunnage, P. A. Glossop, K. James, C. A. L. Lane, R. A. Lewthwaite and D. A. Price, WO 2005/092840 PCT/IB2005/000619, 2005; (b) T. Bieg and W. Szeja, Synthesis, 1985, 76–77 CrossRef CAS.
- The dimer was prepared using the literature procedure [see: (a) B. M. Trost, P. E. Strege, L. Weber, T. J. Fullerton and T. J. Dietsche, J. Am. Chem. Soc., 1978, 100, 3407–3415 CrossRef CAS; (b) S. Imaizumi, T. Matsuhisa and Y. Senda, J. Organomet. Chem., 1985, 280, 441–448 CrossRef CAS ]. However we identified degradation of the crude organometallic complex during the early work-up stages. Constant exposure of the crude material to air, avoiding vacuum operations until the complex has been purified, significantly increases the yield of 17.
- SANS experiments were conducted at the Institut Laue-Langevin (ILL) in Grenoble, France. Data was acquired in two detector movements on separate instrument: high-Q data on D16, and low-Q data on D22.
- Very similar results were obtained on SANS2D at ISIS, UK using a different batch of complex, indicative of the reproducibility of the data.
- Measurements were also conducted at higher and lower temperatures. Consistent with analysis by 31P{1H} NMR spectroscopy, aggregation is reversible, and favoured by lower temperatures. No significant change in particle dimensions was detected by SANS within the relatively narrow range of temperatures explored (10–40 °C).
- At higher concentrations the radius shrank slightly, possibly due to the accompanying increase in the ionic strength of the medium.12.
- It should be noted that most of the concentrations employed for the SANS experiments were, by necessity, well above the oligomerisation and aggregation threshold. Changes in particle number due to differential selectivity between homochiral and heterochiral aggregation of cyclo-oligomers may be small, and thus hard to distinguish.
- The resolution of the SANS instrument was insufficient to completely rule out the possibility that the scattering profile is the composite of two curves arising from segregated (homochiral) cylinders.
- (a) MM3 studies were conducted using Schrödinger MacroModel, version 9.9, Schrödinger, LLC, New York, NY, 2012employing a modified form of the force field reported in: (b) H. Hagelin, M. Svensson, B. Åkermark and P.-O. Norrby, Organometallics, 1999, 18, 4574–4583 CrossRef CAS; (c) DFT studies were conducted using Jaguar, version 7.9, Software, Schrödinger, LLC, New York, NY, 2012.
- See "Molecular Mechanics and Comparison of Force Fields", T. Liljefors, K. Gundertofte, P.-O. Norrby and I. Pettersson, in Computational Medicinal Chemistry for Drug Discovery, ed. P. Bultinck, J. P. Tollenaere, H. De Winter and W. Langenaeker, Marcel Dekker, New York, 2004, pp. 1–28 Search PubMed.
- Note that incorporation of each monomeric unit carries an entropic penalty of ca. 20–40 kJ/mol, not readily calculated by our current methods. To circumvent the entropy problem associated with molecularity, we compare assemblies with oligomers of the same size, and find that tetrameters are favored over monomers [([1][BArF])4versus {4([1][BArF])}, Figure 5] whereas an assembly of two tetramers is slightly favored over the octamer [{([1][BArF])4}2versus ([1][BArF])8, Figure 5]. The dimensions of the MM3-minimsed tetramer are similar to that of the Pd/ligand core of a cyclic tetranuclear complex [(η3-C3H5-Pd)4(2)4][OTf4], determined by single crystal X-ray diffraction.12.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS PubMed.
- A variety of other anion arrangements explored by MM3 modelling did not reveal any plausible alternative modes of aggregation, that would be compatible with the SANS data.
- The impact of solvent dielectric constant (εr) on ion-pair separation can be predicted by the Bjerrum length equation: λB=e2/(4πε0εrkBT) which indicates that the ion separation distance, λB, is inversely proportional to the dielectric constant (εr) of the media in which the solute is dissolved. The equation presumes point charges, although the general principles may still apply to more complex systems such as [1][X].
- These systems present an apparently simple pair of doublets (J = 38 Hz), almost identical in appearance, but not chemical shift, to the pair of doublets (2PdJPP) in the monomeric cationic chelate [1]+.12 However, it is very evident from the SANS data that the solutions are dominated by oligomers {([1][OTf])4}m, and from careful inspection of the 31P NMR spectrum that the monomer is indeed also present, but only in very low proportions (≤ 4%).
- The enantiomerically pure species [[D47]-1][BArF] is among the most synthetically complex perdeuterated species to have been prepared to date. For other examples see: (a) F. Aussenac, M. Laguerre, J.-M. Schmitter and E. J. Dufourc, Langmuir, 2003, 19, 10468–10479 CrossRef CAS; (b) R. G. Alken, US pat., US2005069276A1, 2005; (c) M. Schulz, J. Hirschmann, A. Draksharapu, G. Bindra, S. Soman, A. Paul, R. Groarke, M. T. Pryce, M. S. Rau, W. R. Browne and J. G. Vos, Dalton Trans., 2011, 40, 10545–10552 RSC; (d) T. Abe, A. Miyazawa, H. Konno and Y. Kawanishi, Chem. Phys. Lett., 2010, 491, 199–202 CrossRef CAS; (e) C. Lenges, P. White and M. Brookhart, J. Am. Chem. Soc., 1999, 121, 4385–4396 CrossRef CAS; (f) M. Ogasawara, K. Takizawa and T. Hayashi, Organometallics, 2002, 21, 4853–4861 CrossRef CAS; (g) R. Brainard, T. Miller and G. Whitesides, Organometallics, 1986, 5, 1481–1490 CrossRef CAS.
- All of the BAr4 anions tested, as well as Al(ORF)4 (“Krossing anions”: I. Krossing, Chem.–Eur. J., 2001, 7, 490–502 CrossRef CAS ), were effective in this regard.
- M. J. Hollamby, R. Tabor, K. J. Mutch, K. Trickett, K., J. Eastoe, R. K. Heenan and I. Grillo, Langmuir, 2008, 24, 12235–12240 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and Characterisation of compounds, and full details of aggregation studies by NMR, SANS, and MM3. See DOI: 10.1039/c5sc01181g |
| ‡ Current address: School of Chemistry, University of Glasgow, Joseph Black Building, University Avenue, Glasgow, Lanarkshire G12 8QQ, UK. E-mail: E-mail: daugirdas.racys@glasgow.ac.uk. |
| This journal is © The Royal Society of Chemistry 2015 |