
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular tweezers for lysine and arginine – powerful inhibitors of pathologic protein aggregation
Thomas
Schrader
*a,
Gal
Bitan
*b and
Frank-Gerrit
Klärner
*a
aFaculty of Chemistry, University of Duisburg-Essen, Essen, Germany. E-mail: thomas.schrader@uni-due.de; frank.klaerner@uni-duisburg-essen.de
bDepartment of Neurology, David Geffen School of Medicine, Brain Research Institute, and Molecular Biology Institute, University of California at Los Angeles, Los Angeles, CA, USA. E-mail: gbitan@mednet.ucla.edu
First published on 29th July 2016
Abstract
Molecular tweezers represent the first class of artificial receptor molecules that have made the way from a supramolecular host to a drug candidate with promising results in animal tests. Due to their unique structure, only lysine and arginine are well complexed with exquisite selectivity by a threading mechanism, which unites electrostatic, hydrophobic and dispersive attraction. However, tweezer design must avoid self-dimerization, self-inclusion and external guest binding. Moderate affinities of molecular tweezers towards sterically well accessible basic amino acids with fast on and off rates protect normal proteins from potential interference with their biological function. However, the early stages of abnormal Aβ, α-synuclein, and TTR assembly are redirected upon tweezer binding towards the generation of amorphous non-toxic materials that can be degraded by the intracellular and extracellular clearance mechanisms. Thus, specific host–guest chemistry between aggregation-prone proteins and lysine/arginine binders rescues cell viability and restores animal health in models of AD, PD, and TTR amyloidosis.
 From right to left Thomas Schrader, Gal Bitan and Frank-Gerrit Klärner | Thomas Schrader received his PhD in 1988 with W. Steglich at Bonn University, Germany. After a postdoctorate with E. C. Taylor at Princeton university (USA), he moved to Düsseldorf university to begin his own research. Schrader was subsequently called to Marburg University (2000), where he served as Associate Professor. In 2006, he moved to Essen, where he now holds a chair in Organic Chemistry. His research aims at gaining control over biological functions using rationally designed synthetic receptor molecules. |
Gal Bitan received his PhD in organic chemistry in 1996 from the Hebrew University of Jerusalem, Israel. In his postdoctoral work, Dr Bitan introduced novel photochemical cross-linking techniques for investigation of amyloid β-protein oligomerization and discovered a key oligomer, the paranucleus. Currently, Dr Bitan is an Associate Professor of Neurology at UCLA, where he studies novel therapeutic and diagnostic tools for disorders caused by protein misfolding and aggregation, such as Alzheimer's and Parkinson's diseases. |
Frank-Gerrit Klärner received his PhD in 1968 with Emanuel Vogel at Köln University, Germany. In 1974 he finished his “Habilitation” and was Associate Professor at Bochum Ruhr-University from 1980–1992. Since 1992, he has been a full professor at the University of Duisburg-Essen. From 1998–2005 he chaired the collaborative DFG center of supramolecular research. He retired in 2006. His research interests include supramolecular chemistry (molecular tweezers and clips), pericyclic reactions and chemistry at high-pressure. |
1. Introduction
In the 1990s, several groups around the world pursued the construction of a new class of supramolecular tools, which they called “molecular tweezers”. It was reasoned that a U-shaped molecule with aromatic side walls would form a rigid, highly preorganized concave cavity, which in principle could host a guest molecule with an appropriate size and topology and exploit aromatic interactions. Various elegant solutions to this challenge were presented by Whitlock,1 Zimmerman,2 Harmata,3 Rebek,4 Nolte,5 Fuzakawa6 and others. However, their applications in biology remained rare, mainly because these systems operated only in organic solutions.In 1996, Klärner et al. introduced a highly preorganized hydrocarbon system with convergent aromatic π-systems and discovered the efficient inclusion of cationic guests inside its cavity.7 This binding mode was retained when anionic groups were later attached to the tweezer skeleton, rendering the new hosts water-soluble. With this change, molecular recognition became possible in buffered aqueous solution, opening the door for biological applications. In addition, affinities towards appropriate cationic guests increased substantially. The new host compounds were screened against a large number of small biomolecules – and furnished only two hits: lysine and arginine, which were complexed with low micromolar affinity.8 This marked the beginning of an exciting series of discoveries which culminated in the development of drug candidates against neurological disorders for which there is no cure to date.
Non-covalent inter- and intramolecular bonds involving aromatic rings (π–π and CH–π interactions) are generally important for the formation of highly organized chemical and biological systems.9–11 If a synthetic host molecule can be designed which exploits aromatic interactions for the specific recognition of single amino acid residues in peptides and proteins, it may be able to interfere in a predictable way with protein folding, aggregation or enzyme catalysis. Although a plethora of artificial receptor molecules have been developed for amino acids (e.g., crown ethers,12–14 calixarenes,15–19 cyclophanes,20 polyaza-arenes,21 galactose derivatives,22 molecular tweezers with substituted phosphonate groups,23 peptide units,24 or porphyrin rings25 as side walls), most of them provide little selectivity for a single residue. In this respect, molecular benzene tweezers 1 are remarkable, since they reject all other amino acids and only bind to lysine and arginine. Their structures are shown in Scheme 1.7,26–28 The alternating order of benzene and norbornadiene rings in 1 produces a belt-like arrangement with a horseshoe-shaped cavity, which preferentially accommodates extended alkane chains. However, if R1/R2 are anionic, only guests with a terminal ammonium or guanidinium cation are inserted because in addition to the electrostatic and dispersive attraction resulting from side-chain threading the cation efficiently forms an ion pair with the pendent tweezer anion.29–31 Various anionic groups also confer water solubility to the hydrocarbon skeleton; they are conveniently introduced into the central bridge as phosphates, phosphonates, sulphates or carboxylates leading to symmetrically disubstituted derivatives 1c–f.8,32,33 Monophosphate-substituted tweezers 1h–n turned out to be water-soluble, too, so that in principle, a second binding site at the central benzene bridge can be used to attach an additional recognition site for ditopic peptide and protein recognition.34,35 A systematic study of the influence of different anions and linkers on mono- and disubstituted tweezers has spurred an investigation of their potential application as new tools for peptide and protein recognition. In the past decade, we have unraveled the potential of molecular tweezers with their unique binding mode to counteract and revert pathological aggregation of amyloidogenic peptides and proteins, rendering them promising candidates for disease-modifying therapy of major neurological disorders.36–40
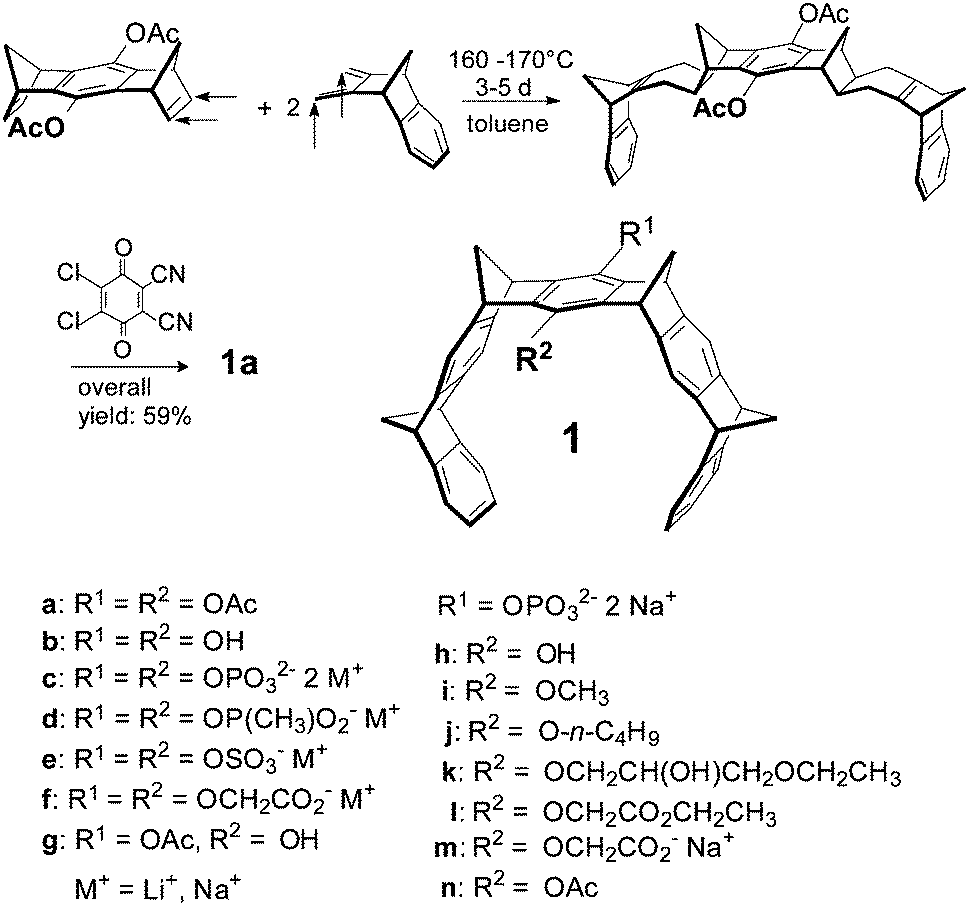 | ||
| Scheme 1 Synthesis of molecular tweezers of type 1. | ||
In this account, we discuss first the unique supramolecular properties of our new molecular tweezers and their recognition profile towards amino acids and peptides, and then proceed to their ability to inhibit peptide and protein aggregation. We introduce the concept of “process-specificity,” which explains why the tweezers are non-toxic and at the same time powerfully rescue cells and animals from life-threatening aberrant protein aggregation.
2. Molecular tweezers recognize amino acids and peptides
2.1 Synthesis of molecular tweezers 1a–n
The tweezer skeleton is accessible by repetitive Diels–Alder reactions of a 1,4,5,8-bismethanotetrahydroanthracene derivative as a bisdienophile with 2,3-bismethylene-5,6-benzonorbornene as a diene. Scheme 1 shows the synthesis of tweezers 1a substituted by two acetoxy groups in the central benzene bridge as a representative example.26 The key step in this synthesis is the Diels–Alder reaction which selectively proceeds on the exo face of the bisdienophile and the endo face of the diene leading to the bisadduct with all four methylene bridges syn to one another. Oxidative dehydrogenation of the cyclohexene moieties with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) produces 1a in an overall yield of 59%. Reduction of the acetoxy functions in 1a with LiAlH4 leads to hydroquinone tweezers 1b in 98% yield. 1b is the starting material for the preparation of water-soluble tweezers 1c–f each symmetrically substituted either by two phosphate, phosphonate, sulphate or O-methylenecarboxylate groups.8,32,33 Hydrolysis of 1a with one mole equivalent of NaOH leads to tweezers 1g (substituted by one hydroxy and one acetoxy function in the central benzene bridge) in 98% yield. 1g in turn is the starting material for the unsymmetrically substituted tweezers 1h–n which are also water-soluble.342.2 Molecular recognition of amino acids and peptides by tweezers 1c–n
The analysis of molecular recognition events inside the tweezer cavity is facilitated by very characteristic changes in the NMR and fluorescence spectra of their complexes. The 1H NMR spectra of the phosphate- and sulphate-substituted tweezers 1c and 1e are concentration-dependent in aqueous buffer. In particular, the 1H NMR signals assigned to the protons attached to the tips of the terminal benzene rings are shifted in aqueous solution at high tweezer concentration by Δδmax = 2.2 ppm (1c) and 2.0 ppm (1d), respectively, compared to the data measured in CD3OD. This finding indicates the formation of the self-assembled dimers (1c)2 and (1e)2 in aqueous solution, a fact that could severely compromise their ability to carry guest molecules. Fortunately, the respective dimerization constants KDim determined by NMR titration are very small (KDim = 60 and 370 M−1).33 Dimer formation is dependent on the size of the hydrophobic hydrocarbon units, and can be explained as a result of the non-classical hydrophobic effect. The 1H NMR spectra of the less polar tweezers 1d, 1f and 1h–n are not significantly concentration-dependent, so all these benzene tweezers exist as monomers in dilute aqueous solution. In the 1H NMR spectra of 1k and 1l, the signals assigned to the methyl group of the side chain (R2 = OCH2CH(OH)CH2OCH2CH3 or OCH2CH2CO2CH2CH3) display an upfield shift of Δδ ≈ 0.5 or 2.0 ppm34 indicating that the methyl groups point inside the cavity, comparable to the original tweezer 1 (R1 = R2 = OCH2CH2CO2CH2CH3).7,26,27 Such self-inclusion phenomena may also hinder guest binding.Finally, in buffer at an almost neutral pH value the phosphate-substituted tweezers 1c and 1h–n are partially protonated and carry ∼1.5 charges on each hydrogen phosphate. Tweezers 1 show strong emission bands at λem ≈ 330 nm in their fluorescence spectra on excitation at λexc = 285 nm. Comparison with the fluorescence spectrum of 1,4-dimethoxybenzene (λem = 320 nm) allows the assignment of the tweezers' emission band to the substituted central hydroquinone bridge as a chromophore.33 Binding of guest molecules by these hosts leads to a partial quenching of their emission bands. Thus, complex formation can also be monitored by fluorescence spectroscopy and the respective binding constants Ka and, hence, the dissociation constants Kd (Kd = 1/Ka) can be determined by fluorimetric titration experiments (Fig. 1).
 | ||
| Fig. 1 Dependence of the emission bands at λem = 336 nm of phosphate tweezers 1c (λexc = 285 nm) on the [Ac-Lys-OMe] or [Ac-Arg-OMe] concentration in aqueous phosphate buffer (10 mM, pH = 7.6). Reprinted with permission from J. Org. Chem. 2013, 78, 6721–6734. Copyright 2016 American Chemical Society. | ||
| Guest | K d [μM] | |||
|---|---|---|---|---|
| 1c | 1d | 1e | 1f | |
| Phosphate buffer.a 200 mM, pH = 7.6.b 10 mM, pH = 7.6.c 10 mM, pH = 7.2.d No binding. | ||||
| Ac-Lys-OMe | 17a | 68a | 28a | 226a |
| 9b | 19c | 643c | ||
| H-Lys-OH | 21a | 874a | 227b | 1170c |
| KAA | 30a | 905a | 303b | 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 333c 333c |
| KLVFF | 20a | 38b | ||
| KKLVFF | 4a | 71a | ||
| KKLVFFAK | 7a | |||
| KKKK | 10a | |||
| Ac-Arg-OMe | 60a | 178a | 882a | |
| 20b | 77c | 281c | ||
| H-Arg-OH | 699b | 609c | ||
| H-Arg-OMe | 160b | |||
| RGD | 86a | |||
| cRGDfV | 59b | |||
| cGRGDfL | 26b | |||
| IAPP1–7 | 9 | |||
| IAPP2–14 | 104 | |||
| IAPP2–7 | —d | |||
| Guest | K d [μM] | ||||||
|---|---|---|---|---|---|---|---|
| 1h | 1i | 1j | 1k | 1l | 1m | 1n | |
| Ac-Lys-OMe | 260 | 40 | >1000 | 370 | 45 | 70 | 35 |
| Ac-Arg-OMe | 110 | 120 | >1000 | 620 | 90 | 100 | 45 |
The accumulated data in Table 1 allow making the following conclusions: tweezers 1c, 1d and 1e are highly selective for lysine and arginine. Thus, 1c does not bind to peptide IAPP2–7 (lacking basic amino acids), but does bind to closely related fragments with a single lysine (K-1) or a single arginine residue (R-11). This confirms the earlier results obtained with phosphonate tweezers 1d, which also complexes lysine more strongly than arginine and much stronger than histidine. Other amino acids (e.g., Asp, Ser, Phe, Leu, Ala, or Gly) are not bound at all.8
The anion dependence of the tweezers' affinities decreases from phosphate (1c) over sulfate (1e) and phosphonate (1d) down to O-methylenecarboxylate (1f). This finding can be explained by the larger negative charge at the phosphate groups in 1c compared to those of the phosphonate, sulphate, or carboxylate groups in 1d–f. The electrostatic interaction between the positively charged ammonium or guanidinium end groups of the lysine or arginine side chains, respectively, and the anionic substituents on the tweezers clearly provides a significant contribution to the overall strength of the host–guest binding. Importantly, complexes with lysine derivatives are generally more stable than those with the corresponding arginine derivatives due to their delocalized guanidinium ion. This is impressively demonstrated by 1c whose affinity towards IAPP1–7 (containing only K1) is one order of magnitude superior to IAPP2–14 (containing only R11). Peptides containing two adjacent K units at their N terminus (KKKK, KKLVFF, and KKLVFFAK) form even more stable complexes with 1c. The host–guest interaction also depends on the competing phosphate buffer: affinities decrease two- to threefold from 10 mM to 200 mM.
| Host–guest complex | Δδmaxa [ppm] | ||
|---|---|---|---|
| ε-H | δ-H | γ-H | |
| a Δδmax = δ0 − δC; δ0, δC – chemical 1H NMR shifts of the free and complexed guest, respectively. b Diastereotopic H atoms. | |||
| Exp.: 1c·Ac-Lys-OMe | 3.91 | — | — |
| Calc.: 1c′·Ac-Lys-OMe′ | 3.62 | 5.51 | 4.62 |
| Exp.: 1c·KAA | 5.92 | 3.22 | 2.28 |
| Calc.: 1c′·KAA′ | 5.71 | 5.08 | 2.55 |
| Exp.: 1d·Ac-Lys-OMe | >4 | 1.57, 1.45b | — |
| Calc.: 1d′·Ac-Lys-OMe′ | 3.46 | 3.42, 3.21b | 1.72 |
| Exp.: 1e·Ac-Lys-OMe | 3.75 | 4.41 | 2.64 |
| Calc.: 1e′·Ac-Lys-OMe′ | 4.39 | 3.19 | 1.10 |
| Exp.: 1f·Ac-Lys-OMe | 0.94 | 0.54 | 0.40 |
| Calc.: (1f′·Ac-Lys-OMe′)in | 5.44 | 3.05 | 1.69 |
| Calc.: (1f′·Ac-Lys-OMe′)out | 0.03 | 0.72 | 0.41 |
| Exp.: 1c·Ac-Arg-OMe | 3.75 | 2.54 | |
| Calc.: 1c′·Ac-Arg-OMe′ | 5.46 | 2.46 | |
| Exp.: 1d·Ts-Arg-OMe | 3.90 | 4.09, 3.29b | |
| Calc.: 1d′·Ts-Arg-OMe′ | 4.30 | 2.51, 1.67b | |
| Exp.: 1e·Ac-Arg-OMe | 3.86 | 2.51 | |
| Calc.: 1e′·Ac-Arg-OMe′ | 3.86 | 0.63 | |
| Exp.: 1f·Ac-Arg-OMe | 0.96 | 0.62, 0.48a | |
| Calc.: (1f′·Ac-Arg-OMe′)in | 3.36 | 1.39, 1.04a | |
| Calc.: (1f′·Ac-Arg-OMe′)out | 0.26 | 0.38, 0.36a | |
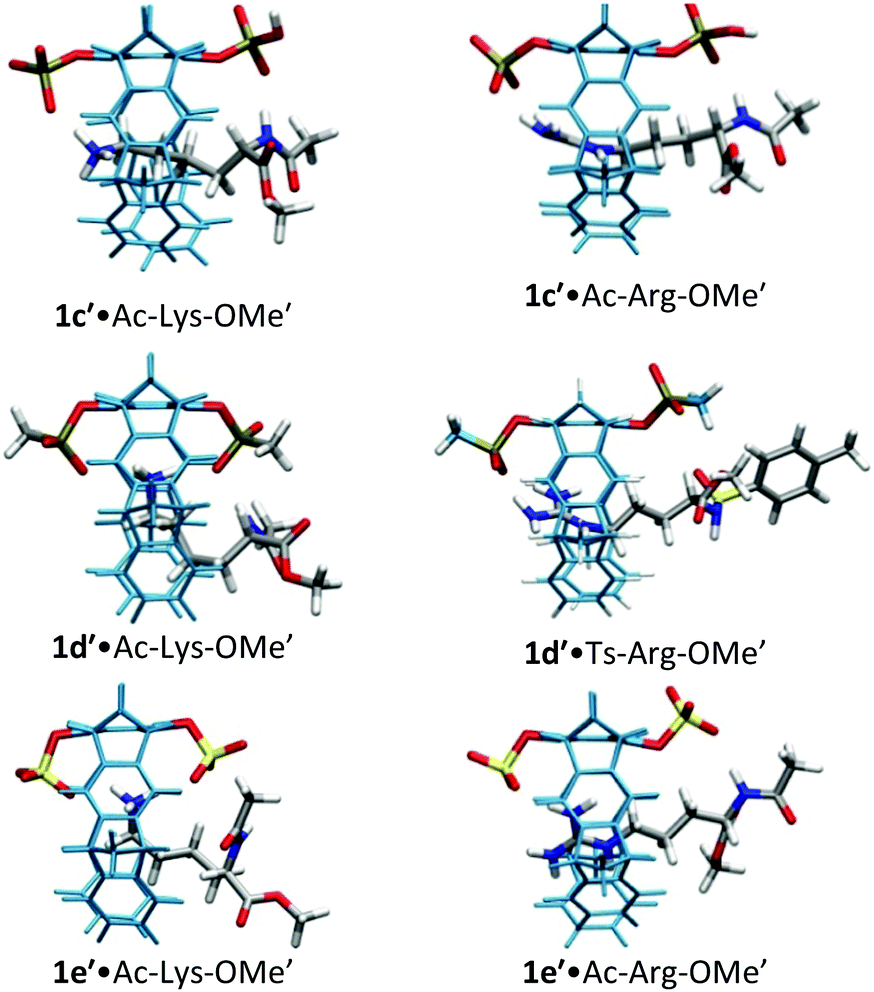 | ||
| Fig. 2 Host–guest complex structures of phosphate, phosphonate, and sulfate tweezers 1c′, 1d′ and 1e′ with lysine and arginine derivatives, optimized by QM/MM calculations without counter-ions. Each structure contains a 60 Å water layer (not shown). Reprinted with permission from J. Org. Chem., 2013, 78, 6721–6734. Copyright 2016 American Chemical Society. | ||
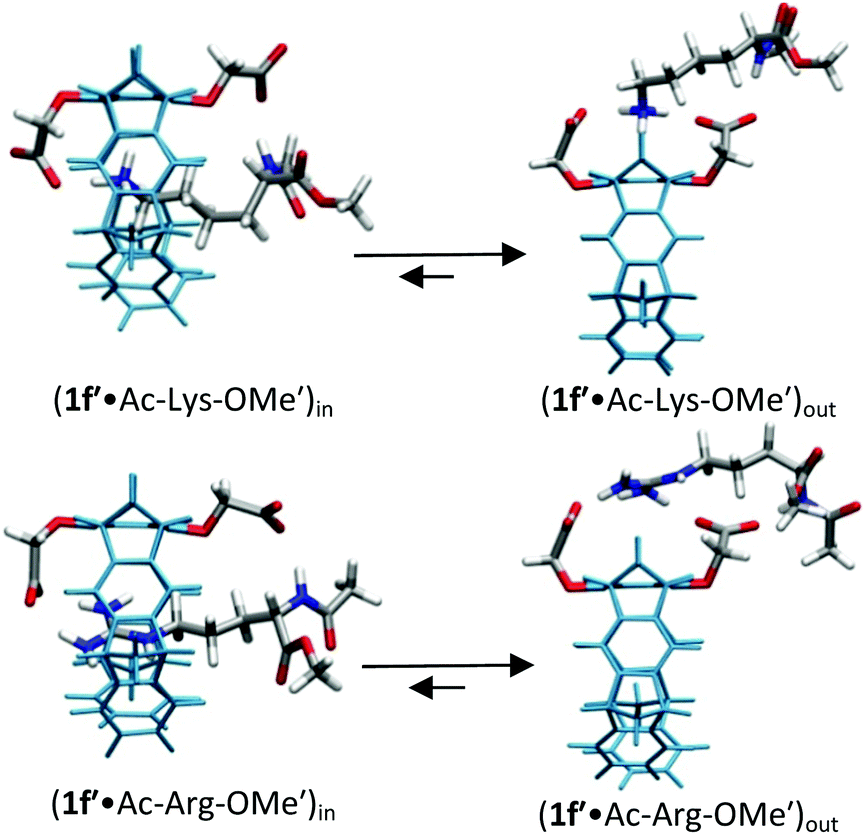 | ||
| Fig. 3 Host–guest complex structures of OCH2CO2−-substituted tweezers 1f′ with lysine and arginine derivatives optimized by QM/MM calculations without counter-ions. Each structure contains a 60 Å water layer (not shown). Reprinted with permission from J. Org. Chem., 2013, 78, 6721–6734. Copyright 2016 American Chemical Society. | ||
For lysine and arginine complexes with carboxylate-tweezers 1f, theoretical Δδmax values were independently calculated with the guest side chain positioned either inside or outside the cavity (Fig. 2). Comparison with experimental Δδmax values indicates that both complexes exist as rapid equilibria between the structures (1f′·Ac-Lys-OMe′)in and (1f′·Ac-Lys-OMe′)out or (1f′·Ac-Arg-OMe′)in and (1f′·Ac-Arg-OMe′)out, with a strong preference of the outside structures. Apparently, the extended OCH2CO2− groups in 1f block the tweezers' cavity and direct the guest molecule to a position outside the cavity where the major host–guest binding force is electrostatic attraction. QM/MM calculations produce chelate arrangements between both carboxylates in 1f and the complexed amino acid cation outside the tweezers' cavity, which are only possible because of the additional methylene group in the OCH2CO2− side chain which is absent in 1c–d (Fig. 2 and 3).33 The loss of CH–π and hydrophobic interactions in this geometry explains why the complexes of 1f are significantly less stable than those of phosphate or sulphate tweezers 1c or 1e. Evidently, dispersive interactions inside the tweezers' cavities and hydrophobic forces contribute substantially to the stability of the inclusion complexes.
Some of our ditopic tweezer derivatives of type 1 were tailored for RGD loops in peptides and proteins.35 To this end, the arginine-binding monophosphate tweezers 1 were connected through different linkers with a guanidiniocarbonylpyrrole unit which binds to aspartate. These hybrid receptors recognize RGD peptides, but unfortunately, the ether linkage prevents inclusion of the arginine side chain inside the tweezer cavity, so that affinities remain modest (>20 μM).
In summary, the unique threading binding mode for lysine and arginine side chains inside the cavity leads to an exceptional selectivity of most tweezer derivatives for basic amino acids. Hydrophobic forces and electrostatic attraction both contribute to the tweezers' affinity which is typically in the micromolar regime. Care must be taken to prevent self-dimerization of larger tweezers and self-inclusion of pendant arms for ditopic recognition. Likewise, a chelate-type guest binding outside the tweezers produces weak binders. The best candidate for lysine and arginine-containing peptides is 1c, the phosphate tweezers. In unstructured peptides, each basic amino acid seems to be well accessible and binds to a tweezers molecule. If such supramolecular interactions disturb peptide misfolding or hinder the formation of amyloid, 1c may become a candidate for the deliberate prevention of aberrant peptide aggregation.
3. Molecular tweezers modulate abnormal protein aggregation
Aberrant self-assembly of peptides and proteins into toxic oligomers and aggregates is a pathological phenomenon underlying over fifty diseases called amyloidoses or proteinopathies.47 Prominent examples include Alzheimer's disease (AD)48 and Parkinson's disease (PD).49 In each proteinopathy, certain proteins misfold and self-associate into abnormal, toxic oligomers and aggregates.In many of the rare proteinopathies, mutations in the cognate gene lead to deletions, amino-acid substitutions, or sequence expansion in the corresponding proteins, which normally are stable, well-structured proteins, e.g., Cu/Zn-superoxide dismutase 1 (SOD1) in familial amyotrophic lateral sclerosis,50 transthyretin (TTR) in familial amyloidotic polyneuropathy (FAP),51 and huntingtin poly-Q expansion in Huntington's disease.52 These sequence alterations destabilize the protein structure resulting in misfolding and aggregation. In contrast, the proteins forming the toxic oligomers and aggregates in the more common diseases, such as AD, PD, and type-2 diabetes, belong to the category of natively unstructured proteins.53 Thus, two natively unstructured proteins—amyloid β-protein (Aβ) and the microtubule-associated protein tau form the pathologic hallmarks of AD, amyloid plaques54 and neurofibrillary tangles,55,56 respectively; the natively unstructured α-synuclein is the main component of the pathologic hallmarks of PD—Lewy bodies and Lewy neurites;57 and the natively unstructured hormone IAPP (amylin) forms pancreatic amyloid in type-2 diabetes.58
3.1 Conceptual considerations
With the exception of tau, in which hyperphosphorylation and other pathological posttranslational modifications lead to aggregation, for the other proteins a simple increase in concentration in vivo is sufficient to initiate abnormal aggregation. Thus, it appears as if certain natively unstructured proteins have a high proclivity for abnormal aggregation. This may raise important questions. Why are such potentially harmful proteins part of our normal physiology? Why were they not eliminated during our evolution? The answer to these questions may be, at least partially, the fact that amyloidoses typically are diseases of old age and their onset in most cases occurs past reproductive age. Thus, evolutionary pressure to eliminate these proteins from human physiology is minimal or non-existent. However, because of the large increase in lifespan in recent generations, these diseases have become, or are becoming, epidemics.The structure of stably folded proteins has been optimized through millions of years of evolution. Nonetheless, in the late 1990s and early 2000s, a number of papers showed that even stable proteins can be “coerced” into forming amyloid under appropriate conditions,59–62 leading to the hypothesis that the amyloid structure, which is highly stable, might have been a common primordial protein structure against which evolution has selected the well-folded proteins we know today as life's building blocks.63,64 The stability of the structure is achieved in each case through the carefully optimized sum of the forces holding it together, hydrogen bonds, salt bridges, hydrophobic interactions, van der Waals interactions, and in some cases covalent bonds, e.g., disulfide and lactam bridges. The same forces mediate the abnormal self-assembly of amyloidogenic proteins, yet because the aberrant assemblies, particularly the most toxic species, the oligomers, were not optimized by evolution, the forces holding them together are much weaker, and hence the oligomers are metastable structures that exist in constantly changing dynamic mixtures.65 This distinction between normal, stable proteins and abnormal oligomers of amyloidogenic proteins is the key to the unique activity of molecular tweezers as selective inhibitors of amyloid proteins' toxicity.
As discussed above, molecular tweezers bind with high selectivity to Lys residues. The Lys residue is unique among the twenty proteinaceous amino acids in its ability to form both hydrophobic and electrostatic interactions. Both types of interactions are prominent mediators of the formation of the abnormal protein oligomers and Lys residues have been reported to be important players in these assembly processes in many cases.66–73 Thus, we hypothesized that disruption of these interactions could interfere with the process of abnormal self-assembly. However, for such disruption to be specific only for the loosely bound abnormal proteins, the disrupting compounds must bind to Lys with relatively weak affinity and the binding must be highly labile. Otherwise, binding of these compounds to normal proteins would disrupt their function, and possibly their structure. Yet if labile binding with moderate affinity could be achieved, it would lead to a novel kind of inhibitors that are “process-specific,” i.e., their specificity is to the process of aberrant protein self-assembly rather than to a particular protein.
Indeed, molecular tweezers were found to be ideal candidates for process-specific inhibition. They bind to Lys residues with affinity in the low μM range (Table 1)8,32–34 and their binding is highly labile, as evidenced by surface-plasmon resonance experiments.46 Moreover, due to their rigid structure, they bind only to exposed Lys residues, where steric interference is minimal. Thus, crystallographic experiments and molecular modeling showed that out of seventeen Lys residues in the small adaptor protein 14-3-3, only five bound to the molecular tweezers CLR01, whereas the other twelve were shielded from binding.46 In contrast, in the misfolded proteins comprising abnormal oligomers, most Lys residues are readily accessible to the molecular tweezers. Thanks to these unique characteristics, molecular tweezers effectively modulate protein self-assembly and prevent the formation of toxic oligomers and aggregates even in the complex environment of a cell or a whole organism.74
In the following pages, we report the exciting discoveries we made when we began exploring the interference of abnormal protein aggregation employing our supramolecular ligands. For clarity we selected four critical proteins, whose misfolding and subsequent aggregation trigger prominent neurological and other disorders: Aβ (Alzheimer's disease, inclusion-body myositis), tau (Alzheimer's disease and other tauopathies); α-synuclein (Parkinson's disease and other synucleinopathies); and transthyretin (TTR, familial amyloidotic polyneuropathy, familial amyloidotic cardiomyopathy, and senile systemic amyloidosis). In all cases, we characterized first the direct interaction between the isolated compounds (host–guest chemistry), moved on to cell-culture experiments, and finally progressed to animal experiments. Here is the story, which is focused on the effect of the phosphate-substituted tweezers 1c on these amyloid proteins. In most publications dealing with this effect, compound 1c is called CLR01 (Fig. 4A) and the phosphate-substituted bridge (lacking the tweezers' side arms), which has been used for control experiments, CLR03 (Fig. 4B).
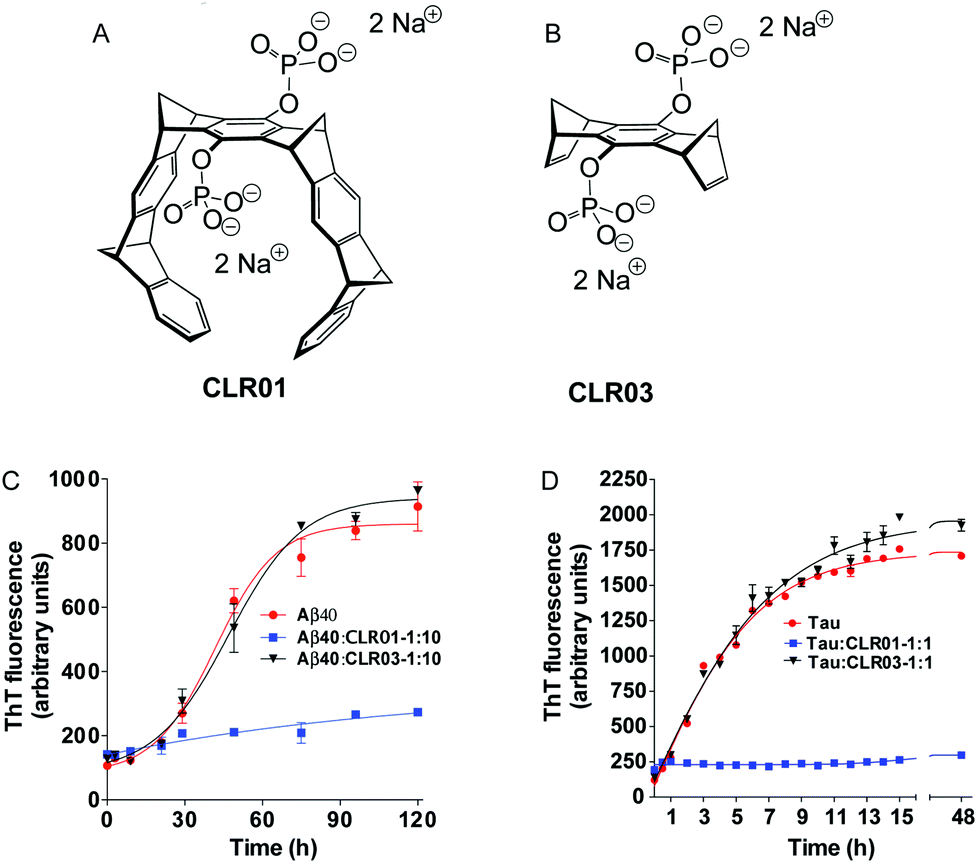 | ||
Fig. 4 The active compound, CLR01 versus the negative control, CLR03. (A) Structure of tweezer 1c ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CLR01. (B) Structure of bridge CLR03 (each compound contains two disodium phosphate groups). (C and D) The effect of CLR01 and CLR03 on β-sheet formation by (C) Aβ40 or (D) the embryonic isoform of tau was assessed by measuring Thioflavin T fluorescence. Reprinted with permission from J. Am. Chem. Soc., 2011, 133, 16958–16969. Copyright 2011 American Chemical Society. CLR01. (B) Structure of bridge CLR03 (each compound contains two disodium phosphate groups). (C and D) The effect of CLR01 and CLR03 on β-sheet formation by (C) Aβ40 or (D) the embryonic isoform of tau was assessed by measuring Thioflavin T fluorescence. Reprinted with permission from J. Am. Chem. Soc., 2011, 133, 16958–16969. Copyright 2011 American Chemical Society. | ||
3.2 Inhibition of oligomerization and aggregation of proteins involved in Alzheimer's disease (AD) – Aβ and tau
In the etiology of AD, a modified version75 of the so-called “amyloid cascade hypothesis”76 is nowadays generally accepted. The original hypothesis suggested that the aggregation of Aβ into β-sheet-rich amyloid was the trigger for a long chain of subsequent events, including tau hyperphosphorylation and aggregation, inflammation, and neuronal death resulting in the complex clinical picture of AD,76 whereas the currently accepted view is that the real culprits are the elusive Aβ oligomers rather than the amyloid fibrils. This creates a challenge for therapy development because the oligomers are metastable species that lack a stable structure, which typically is the starting point for drug discovery. However, as explained above, supramolecular agents, such as molecular tweezers, have favorable characteristics allowing them to disrupt selectively the metastable structure of the oligomers and thus interfere with the primary cause of the disease at a very early stage.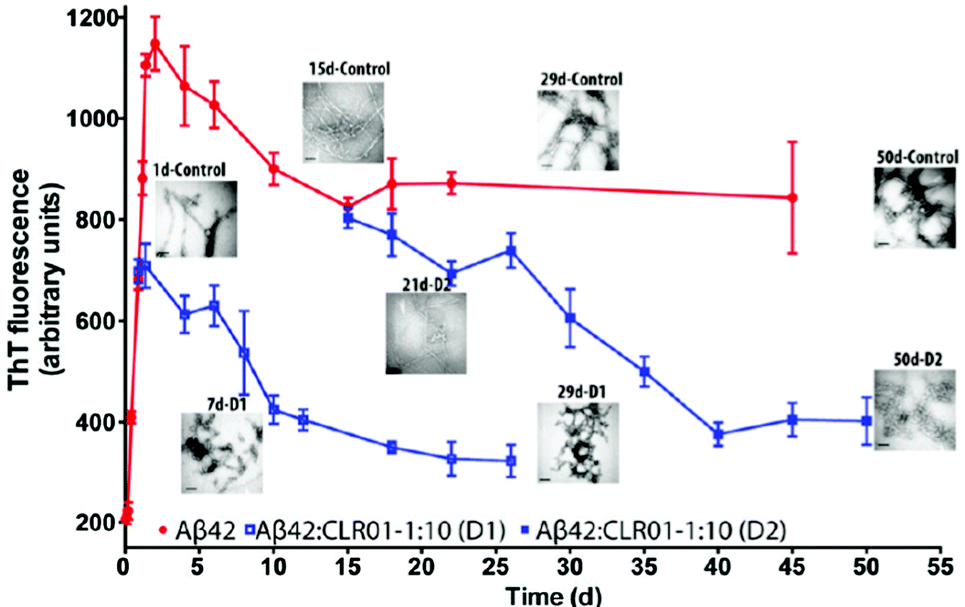 | ||
| Fig. 5 CLR01 disaggregates Aβ fibrils. Disaggregation of preformed Aβ42 fibrils by CLR01 was studied by adding a 10-fold excess of CLR01 to aggregating solutions of 10 μM Aβ42 at 21 h (disaggregation reaction D1) or 15 days (D2) after initiation of aggregation. The reactions were monitored using ThT fluorescence and TEM. Reprinted with permission from J. Am. Chem. Soc., 2011, 133, 16958–16969. Copyright 2011 American Chemical Society. | ||
Similarly, CLR01 was found to be an efficient inhibitor of the aggregation of several other disease-related amyloidogenic proteins, including islet amyloid polypeptide (IAPP, amylin),36,41 calcitonin,36 insulin,36 β2-microglobulin,36 mutant p53,82 and the HIV-infection-enhancing semen protein fragments PAP(248–286), PAP(85–120), and SEM1(45–107).40 In most cases, the protein:CLR01 ratio needed for complete inhibition was in the range 1:0.1–1:3. In one case studied so far, the amyloidogenic peptide PrP(106–126), CLR01 was unable to inhibit the aggregation (measured using turbidity), although the morphology of the aggregates in the presence of CLR01 was amorphous rather than fibrillar.36
Subsequently, dynamic light scattering (DLS) was employed to investigate the impact of CLR01 on the oligomer size distribution of Aβ directly and non-invasively. Previously, Aβ40 and Aβ42 had been shown to form particle distributions with hydrodynamic radii (RH) of 2–6 nm and 8–60 nm, respectively, which grew in size over several days as each peptide aggregated.84 At a 1:1 ratio, CLR01 produced oligomers of similar size, but they did not grow into larger aggregates. Importantly, the newly formed oligomeric species were not toxic anymore, as evidenced by the following cell culture experiments.
In all the experiments, Aβ42 was treated with 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) to dissociate any pre-existing aggregates.87 Changes in dendritic spine density were measured in rat primary hippocampal neurons whose dendritic spines were visualized by staining with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) at 2000× magnification. As it has been documented before,88,89 the treatment of neurons with 3 μM Aβ42 for 72 h led to a dramatic depletion of the dendritic spines to ∼20% of the baseline level and to the appearance of abundant “varicosities” due to the arrest of cargo transport along the dendrites. In contrast, when neurons were incubated with Aβ42 in the presence of a 10-fold excess of CLR01, the density of dendritic spines was recovered to ∼80% of the baseline level and varicosities were not observed, demonstrating potent inhibition of Aβ42 synaptotoxicity by CLR01.38 As expected, the negative control compound, CLR03, had no effect on Aβ42 synaptotoxicity.
CLR01 and CLR03 were tested next in electrophysiological assays to examine their effect on the synapse function in autaptic microcultures of mouse hippocampal neurons. The amplitude and frequency of both evoked excitatory postsynaptic currents and spontaneous miniature excitatory postsynaptic currents were reduced to 50–70% of their baseline level following treatment with 200 nM Aβ42 and recovered back to the baseline level with a 10-fold excess of CLR01.38
Another early characteristic of Alzheimer's disease is the loss of synaptic plasticity. Long-term potentiation (LTP) is a cellular correlate of synaptic plasticity, which is the basis for learning and memory.90 Thus, the measurement of LTP allows an assessment of the loss or gain of synaptic plasticity in cell culture, and has been used commonly to assess the synaptotoxic effect of Aβ oligomers.91,92 To assess the capability of CLR01 to protect against the deleterious effect of Aβ42 on synaptic plasticity, field excitatory postsynaptic potentials (field EPSP) evoked by Schaffer collateral stimulation were recorded from the CA1 subfield of the hippocampus in hippocampal slices obtained from 8-week-old wild-type mice. In these experiments, 200 nM Aβ42 reduced the LTP to 46% of its baseline level. Co-application of 200 nM Aβ42 and 2 μM CLR01 significantly ameliorated the LTP inhibition to 65% of the baseline level. Though this rescuing effect was statistically significant, its magnitude was small relative to the protective effects of CLR01 in the primary neuronal cultures used for the measurement of baseline synaptic activity as described above. A potential explanation is differences in diffusion to the cellular targets between Aβ42 oligomers and CLR01, which might have diminished the effectiveness of CLR01 in brain slices relative to cultured neurons. To test this hypothesis, we examined whether a 1 h incubation of Aβ42 with CLR01 before application to hippocampal slices would produce stronger protection. Indeed, 1 h pre-incubation of Aβ42 with CLR01 provided a stronger protective effect, raising the field EPSP amplitude potentiation to 82% of the baseline level.38 In contrast, CLR03 did not show any protective effect. Overall, the morphological and electrophysiological experiments showed that CLR01 was an effective inhibitor of Aβ42 synaptotoxicity, supporting its development as a drug lead for Alzheimer's disease.
Immunohistochemical analysis of brain sections of vehicle-treated mice using monoclonal antibody (mAb) 6E10, which is specific for residues 3–8 in Aβ (674–679 in APP), showed extracellular amyloid plaques deposited predominantly in the subiculum and CA1 regions of the hippocampus, as reported previously by La Ferla and co-workers who developed this 3 × Tg mouse model.93 In addition, the mice showed neurofibrillary tangles detected by mAb AT8 predominantly in the CA1 and CA3 regions. mAb AT8 recognizes phosphorylated S202 and T205 in hyperphosphorylated tau (p-tau) (Fig. 6).
 | ||
| Fig. 6 CLR01 decreases amyloid-β protein and p-tau deposition. Triple-transgenic mice were treated with 0.04 mg kg−1 per day CLR01 or the vehicle. (A and C) Vehicle-treated transgenic mouse hippocampus. (B and D) CLR01-treated transgenic mouse hippocampus. (A and B) transgenic mouse brain stained with mAb 6E10 showing amyloid plaque deposition. (C and D) transgenic mouse brain showing AT8-positive neurofibrillary tangles in the CA1 region. Reprinted with permission from Brain, 2012, 135, 3735–3748. Copyright 2012 Oxford Journals. | ||
Mice treated with CLR01 showed a significant decrease in Aβ deposition in multiple brain areas, including those affected the most in AD—the hippocampus (35% reduction) and cortex (50% reduction). Similarly, reduction in AT8-positive p-tau was observed in the CA1 (33%) and CA3 (46%) regions, respectively, in mice treated with CLR01. In contrast, there was no effect on normal tau.38 As tau hyperphosphorylation and aggregation are believed to be downstream of Aβ-induced toxicity, the data may reflect both direct and indirect effects of CLR01 treatment on neurofibrillary tangles. The fact that the reduction in neurofibrillary tangles was observed in relatively old mice (15 months of age) suggests that the effect was at least partially a direct effect of CLR01 on tau, because previously, La Ferla's group showed that Aβ immunotherapy led to clearance of early, but not late p-tau aggregates.94 Compared to vehicle-treated mice, the CLR01-treated mice showed significant reduction in the number of microglia per hippocampal area.38 In contrast, there was essentially no difference between vehicle- and CLR01-treated wild-type mice in the level of microgliosis, suggesting that the compound itself did not cause any inflammatory response.
In contrast to many older transgenic models of AD, which had only amyloid but not tau pathology, the 3 × Tg model has both lesions and therefore is considered to be fairly advanced. Nonetheless, no animal model faithfully recapitulates all the aspects of the human disease,95,96 and it is therefore important to examine whether drug effects can be observed in more than one model. To that end, CLR01 was evaluated in a transgenic rat model of AD, which expresses familial AD-linked mutant forms of human APP (K670N/M671L/V717I) and PS1 (M146V).97 CLR01 was administered in a manner similar to the experiment using the 3 × Tg mouse model at a dose of 0.1 or 0.3 mg kg−1 per day. The animals were mixed-gender and were treated at 9-months of age, an age in which they are expected to have moderate plaque pathology.98 The amyloid plaque burden was evaluated in this study using the Aβ-specific mAb MOAB-2.99 The treatment led to 45% and 52% reduction in plaque burden in the 0.1 and 0.3 mg kg−1 treatment groups, respectively (whole brain analysis),100 demonstrating that the effect of the compound was not limited to one model or one species.
In summary, the specific host–guest interaction between the 3 basic amino acids of Aβ and the molecular tweezers CLR01 enabled the compound to protect cells from Aβ toxicity and counteract synaptotoxicity and AD pathology in animal brains; this also implies that CLR01 penetrated through the blood brain barrier (BBB). It might be argued that CLR01 acted peripherally, similarly to the “peripheral sink” hypothesis proposed for anti-Aβ immunotherapy.101 However, for the micromolar affinity and highly labile binding mode of CLR01, this possibility is entirely implausible.
3.3 Inhibition of α-synuclein toxicity
Misfolding and aggregation of the natively unstructured protein, α-synuclein, which is thought to be important for synaptic vesicle release, lead to several diseases called synucleinopathies, of which the most prevalent is PD. Similarly to AD, only symptomatic treatment is available for PD, but no disease-modifying therapy.CLR01 inhibits α-synuclein fibrillogenesis, disassembles mature fibrils and redirects protein aggregation towards non-toxic oligomers. Initially, experiments similar to the case of Aβ were carried out with the lysine-rich (15 K, ∼11% of the sequence) α-synuclein. α-Synuclein aggregation, which usually occurs after an extended lag phase of 4 days and reaches a plateau after 10 days, could be completely suppressed with equimolar CLR01, and even with 10 mol% it was still strongly inhibited. Similarly to Aβ, CLR01 inhibited the nucleation as well as elongation phase of α-synuclein fibril formation. TEM images revealed that CLR01 prevented completely the formation of the fibrillar structure and only non-fibrillar morphology was apparent.
Another parallel to Aβ is the ability of CLR01 to disaggregate pre-existing α-synuclein fibrils: in a 60 day aggregation reaction, a 10-fold excess of CLR01 was added on day 8 (growth phase) and on day 24 (plateau). In both scenarios, ThT fluorescence gradually decreased indicating the arrest of fibril growth and dissociation of existing fibrils, which gradually disappeared in the concomitant TEM analysis.
Native gel electrophoresis revealed that CLR01 promoted the formation of high molecular weight oligomers (700–900 kDa). Higher molecular weight bands also were observed in SDS-PAGE in mixtures of α-synuclein with CLR01, but not with the negative control CLR03. We note that SDS-PAGE does not report reliably the size of such oligomers,102 but can be used to compare among experimental conditions as done in that study. These mixtures were later examined in cell culture, and shown to be non-toxic in the presence of CLR01, but not CLR03, another parallel to the tweezers' effect on Aβ aggregation.
Native ESI-MS was then employed to identify non-covalent ligand binding sites. In tandem ECD-MS/MS experiments, ligand-bound fragments could be mapped on the full-length protein sequence. In this case, prominent fragment ions strongly indicated the preferred binding of CLR01 close to the N-terminus, most likely at Lys-10 and Lys-12.
Photo-induced cross-linking of unmodified proteins (PICUP),104,105 followed by SDS-PAGE and densitometric analysis of cross-linked α-synuclein oligomers showed that CLR01 predominantly binds to monomeric α-synuclein. However, the Trp-94 fluorescence also provides a unique probe for studying the early stages of α-synuclein oligomerization. In the presence of CLR01, the Trp-94 fluorescence intensity gradually increased and thus provided experimental evidence for binding and stabilization of protein oligomers by the tweezers, consistent with earlier findings from native- and SDS-PAGE.
We tentatively propose that CLR01 binding to the two lysines at the N-terminus of α-synuclein further increases the absolute net charge of the protein (−8.8 at pH 7.0) and weakens intramolecular long-range associations between N-terminal lysines and C-terminal aspartates and glutamates, which lead to an overall compact conformation of the unstructured protein. With the doubly negatively charged CLR01 bound to N-terminal lysines, other closely located positively charged residues may rather be associated instead, and the overall conformation of α-synuclein becomes more extended and hence – diffusive. Such an increased ligand-induced reconfiguration rate may be a general principle for the design of potent α-synuclein aggregation inhibitors.
Dose-response experiments showed that CLR01 inhibited the toxicity of 20 μM α-synuclein in the PC-12 cell assay with half-maximal inhibition (IC50) = 3 ± 1 μM in MTT assay37 and 4 ± 3 μM in trypan blue assay (G. Bitan, unpublished results) demonstrating inhibition at substoichiometric concentrations. As mentioned above, the amino-acid sequence of α-synuclein contains 15 Lys residues. Thus, half-maximal inhibition occurred at a CLR01:α-synuclein concentration ratio of 1:5–1:7 and a CLR01:Lys ratio of 1:75–1:100 (not taking into account all Lys residues in other proteins and free Lys in the environment). In HEK293 cells, the addition of 1 or 10 μM CLR01 led to complete inhibition of α-synuclein-induced toxicity, suggesting that the compound could inhibit both the extracellular and intracellular toxic effects of the offending protein.37 As expected, CLR03 was inactive in all cases.
To determine whether CLR01 could prevent α-synuclein-induced toxicity, three groups of twenty-eggs each were injected with α-synuclein-containing cDNA. The developing embryos were treated with the vehicle (0 μM), 1 μM, or 10 μM of CLR01 at 8 hpf. The compound was applied simply by dissolution in the water in which the embryos developed. Treatment of the injected embryos with CLR01 resulted in a dramatic improvement in both the survival rate and the phenotype of the surviving ZF (Fig. 7A).37 Control eggs were injected with HuC-GFP cDNA and approximately 50% of the embryos in these controls survived to 24 hpf. Thereafter, all of the surviving embryos appeared to be normal. Thus, the initial mortality under the control conditions is a result of the somewhat invasive DNA microinjection and has no relevance to α-synuclein toxicity.
 | ||
| Fig. 7 CLR01 ameliorates α-synuclein (α-syn) neurotoxicity in zebrafish (ZF) and protects against ziram toxicity. (a) ZF embryos expressing human, wild-type α-synuclein were treated with CLR01 at 8 hpf (hours post fertilization) and were monitored for abnormal appearance and survival. Bright-field and fluorescence overlay images were taken at 72 hpf (A, top). Green bars represent normal-appearing embryos and red bars represent abnormal embryos (A, bottom). Reprinted with Permission from Neurotherapeutics, 2012, 9, 464–476. Copyright 2012 Springer. (b) ZF embryos were treated with 1 μM Ziram in the absence or presence of 10 μM CLR01 and survival was monitored up to 10 days (S. Prabhudesai and J. M. Bronstein, personal communication). | ||
Immunocytochemistry analysis of treated and untreated embryos revealed that in the absence of CLR01, abundant aggregated α-synuclein could be observed before cells died. In contrast, in embryos developing in the presence of CLR01, α-synuclein appeared to be soluble and dispersed uniformly in the cytoplasm. These data demonstrated that CLR01 inhibited α-synuclein toxicity not only in cell culture, but also in whole living organisms.
Surprisingly, Western blot analysis showed that in treated ZF, the total concentration level of α-synuclein was ∼20% of that of untreated fish. The reduction was in the protein levels but not in mRNA levels, suggesting that by preventing α-synuclein aggregation, CLR01 facilitated its clearance. The main mechanism of α-synuclein clearance in cell culture has been reported to be the 26S ubiquitin–proteasome system (UPS),108 suggesting that CLR01 enabled UPS degradation of α-synuclein by preventing its aggregation. Indeed, when ZF expressing α-synuclein were treated with CLR01 in the presence of the UPS inhibitor, lactacystin, though α-synuclein remained soluble, its concentration level was the same as in untreated ZF.37 Importantly, to be cleared by the UPS, α-synuclein is marked for degradation by ubiquitination on Lys residues. The experiments described above strongly suggested that the gentle, labile binding of CLR01 to the Lys residues prevented α-synuclein aggregation but not its ubiquitination, supporting the mechanism of action of CLR01 and its safety.
α-Synuclein mutations causing autosomal dominant familial PD and polymorphisms in the SNCA gene, which encodes α-synuclein, are a strong genetic risk factor for developing PD. In addition to genetic risk factors, environmental toxins, such as pesticides and fungicides, have been shown to increase PD risk significantly, and one of the most potent toxins is the fungicide ziram (zinc N,N-dimethylcarbamodithioate).109 Previously, ziram treatment has been shown to increase α-synuclein concentration levels in primary rat dopaminergic neurons, presumably due to the inhibition of clearance mechanisms.110 In a new study, ziram was found to be highly toxic in wild-type ZF embryos and the toxicity seemed to be specific to aminergic neurons (including dopaminergic neurons). The addition of CLR01 had a strong protective effect against ziram toxicity (Fig. 7B),111 presumably through a similar mechanism to that observed in the ZF expressing human α-synuclein.
To address this question, CLR01 was applied to the site of spinal-cord transection using gelfoam and synuclein accumulation and neuronal survival were examined histochemically 11-weeks post injury.39 CLR01 treatment reduced synuclein accumulation by 34% in the “poor survivor” neurons. The reduction in synuclein accumulation could be correlated with an increase from 8% to 37% survival of these neurons. In fact, a robust increase in survival was observed for each one of the individual “poor survivor” neuron type. Interestingly, the protective effect of CLR01 was stronger than that of a synuclein morpholino that suppressed synuclein expression in the same model,39 suggesting that the inhibition of formation of toxic oligomers and aggregates may be advantageous in simple suppression of protein expression.
Taken together, the molecular tweezers seem to preferentially bind to two lysines close to the α-synuclein's N-terminus. This binding event increases the protein diffusion rate and slows down aggregation. As a consequence, cell viability is restored and the simple presence of CLR01 in the water where zebra fish embryos expressing human α-synuclein develop, largely rescues the phenotype and survival of the animals. Similarly, CLR01 increases neuronal survival after spinal-cord injury in lampreys by preventing synuclein aggregation in susceptible neurons in vivo.
3.4 Inhibition of transthyretin amyloidosis
Transthyretin (TTR) is an abundant carrier protein in the blood that normally exists as a homotetramer. Over 100 mutations in the cognate gene cause amino-acid substitutions that destabilize the structure of the protein leading to tetramer dissociation, misfolding, and self-assembly of misfolded TTR into toxic oligomers and amyloid fibrils.113 Misfolding and aggregation of TTR is associated with at least three diseases. Familial amyloidotic cardiomyopathy (FAC) is a progressively debilitating, and often fatal disease affecting ∼40000 people worldwide with a mean survival time of ∼2 years from diagnosis. The main TTR mutant causing FAC is V122I, which is found in 3–4% of African–Americans (∼1.3 million people).114,115 Amyloid formation by wild-type TTR causes a similar cardiomyopathy improperly named senile systemic amyloidosis (SSA), which affects mostly Caucasian men over the age of 60116 and is a major cause of death among centenarians.117 A third disease is familial amyloidotic polyneuropathy (FAP), which affects ∼10000 people, predominantly in Portugal, Sweden, and Japan. In FAP, mutant TTR forms amyloid deposits in the kidneys, gastrointestinal (GI) tract, and peripheral nervous system (PNS), leading to progressive dysfunction and death within ∼5 years.118
CLR01 inhibits wild-type TTR aggregation, prevents TTR-induced toxicity in cell culture and reduces TTR deposition in a mouse model. CLR01 was examined for inhibition of wild-type TTR aggregation, which was induced by lowering the pH to 3, using turbidity and electron microscopy.36 Complete inhibition of aggregation was observed using turbidity at a 1:1 TTR:CLR01 concentration ratio and partial inhibition at a 100:1 concentration ratio, respectively. In agreement with the turbidity results, in the absence of CLR01, TTR formed abundant fibrils, whereas in the presence of CLR01 only amorphous structures were observed. CLR03 had no effect on the TTR aggregation rate or morphology.
Interestingly, inhibition of TTR-induced toxicity required a large excess of CLR01. Experiments using the MTT-reduction assay in differentiated PC-12 cells showed that CLR01 prevented the toxicity induced by 1 μM TTR oligomers with half-maximal inhibition (IC50) = 54 ± 19.36 PC-12 cells were used for convenience in these experiments, though they are not an accurate representation of the tissues affected by TTR in vivo. In view of the discrepancy between the substoichiometric inhibition of TTR aggregation and the excess CLR01 needed for inhibition of TTR toxicity, the question of whether or not CLR01 could have a therapeutic effect for TTR-related amyloidosis in vivo was particularly intriguing.
To answer this question, the effect of CLR01 was tested in the hTTR V30M/HSF mouse model.119 These mice express the amyloidogenic human TTR V30M variant on a hemizygote HSF1 ± background. This is an improvement on a previous model that only expressed human TTR V30M on a mouse TTR-null background. That model showed TTR deposition only in the GI tract and the deposition started at a relatively late age—11 months.120 In contrast, in the hTTR V30M/HSF mice, knock-down of HSF1 leads to early and extensive deposition of non-fibrillar TTR in different organs, including the GI tract and the PNS. TTR aggregates start to deposit at 3 months of age and evolve to fibrillar, congophilic material typically by 12–14 months of age. Neither these models, nor any other transgenic mouse strain expressing wild-type or mutant human TTR develop polyneuropathy or cardiomyopathy.121,122 Nonetheless, due to the relatively early and widespread deposition of TTR, the hTTR V30M/HSF mouse model is highly relevant for testing therapeutic strategies.
Continuous administration of 1.2 mg kg−1 per day CLR01 subcutaneously for 5 weeks using osmotic minipumps led to the robust clearance of TTR deposition and a concomitant reduction in associated markers of disease, including ER stress, apoptosis, and protein oxidation in the tissues examined, including the colon, stomach, and dorsal root ganglia (Fig. 8).123
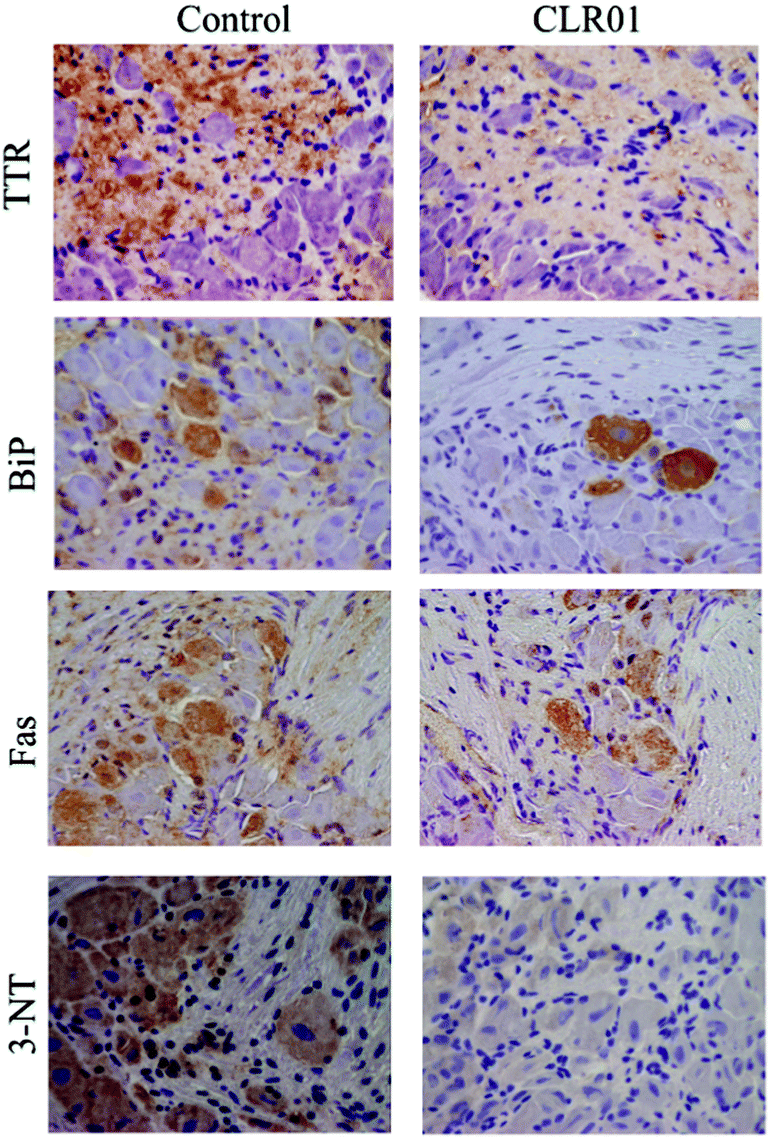 | ||
| Fig. 8 CLR01 decreases TTR burden and associated toxicity in the dorsal root ganglia (DRG) of hTTR V30M/HSF mice. Representative immunohistochemistry analysis of TTR, binding immunoglobulin protein (BiP), Fas, and 3-nitrotyrosine in DRG of mice treated with CLR01 (right panels; n = 14) and age-matched controls (left panels; n = 12); 20× magnification. Reprinted with Permission from Neurotherapeutics, 2014, 11, 450–461. Copyright 2014 Springer. | ||
These in vivo findings may seem to be initially difficult to reconcile with the concentration ratio needed for inhibition of toxicity in vitro. We propose a simple explanation. The culprit is not the native protein whose concentration is measured, but the toxic oligomers that exist at substantially lower concentrations.124 Remodeling of these oligomers by CLR01 enables their rapid clearance, as found in the α-synuclein zebrafish model,37 thereby reducing subsequent deposition of the misfolded protein. The putative mechanisms responsible for the clearance of misfolded and oligomerized/aggregated TTR from the blood and the affected tissues are largely absent in cell culture experiments, where the task of preventing toxicity is done predominantly by CLR01 itself. Therefore, substantially higher concentrations of the compound would be needed in such experiments. The data suggest that in vivo, gentle modulation of oligomerization and nucleation has a robust therapeutic impact on the pathologic process.
Again, direct interaction between CLR01 and transthyretin completely inhibited wild-type TTR aggregation, prevented TTR-induced toxicity in cell culture and finally led to a considerable reduction in TTR deposition and associated disease markers in a relevant mouse model.
3.5 CLR01 has a high safety margin
The unusual “process-specific” mode of action of molecular tweezers, i.e., binding to an amino acid rather than to a specific protein may raise concern that by binding to off targets they would cause high levels of toxicity or side effects. As discussed above, theoretically, these would be mitigated by the highly labile nature of the tweezers' binding and by the rigidity of their structures, which prevents binding to Lys residues that are not freely exposed to the solvent. Multiple lines of evidence support these assumptions.First, if molecular tweezers are indeed process-specific, the concentration ratio of molecular tweezers to protein required for inhibition of normal, regulated biological processes would be expected to be substantially higher than those needed for inhibition of abnormal protein aggregation. Indeed, the enzyme:CLR01 ratios needed for inhibiting the enzymatic activity of alcohol dehydrogenase (ADH) and poly ADP ribose polymerase 1 were found to be 1:86532 and 1:1435 (T. Schrader et al.: manuscript submitted), respectively, demonstrating that ∼3-orders of magnitude excess were needed for enzymatic inhibition in these cases. This effect is easily explained: although only a few lysine residues are found in direct proximity to ADH's active site, numerous other basic amino acids must be complexed simultaneously to reach saturation. On the PARP-1 enzyme, multiple lysines are located on the three zinc finger units; hence, all must be bound by CLR01 in order to effectively displace lesioned DNA and hence inhibit PARP-1 activity.
To test the process-specific mechanism hypothesis further, the protein:CLR01 concentration ratio needed for inhibiting aggregation of amyloid proteins was compared to the concentration required for interfering with a normal assembly process—tubulin polymerization. The protein:CLR01 concentration ratios needed for complete inhibition of different amyloid proteins were mostly in the range of 1:0.1–1:3.36 These concentration ratios had no effect on tubulin polymerization. Only at a tubulin:CLR01 concentration ratio of 1:55, some disruption, not complete inhibition, of the process was observed.125 These data support the mechanism of action of CLR01 and demonstrate that despite its ability to bind exposed Lys residues essentially on any protein, its gentle, labile binding affects amyloid proteins' self-assembly at concentrations orders of magnitude lower than those needed for disruption of normal biological processes.
The next line of evidence is cell culture experiments. Because in most of the assays used to measure the toxicity of amyloidogenic proteins the proteins were added to cells exogenously,36,37,40,41,82 the concentrations of CLR01 needed for inhibition depended on the concentrations of the protein used, and generally were in the high nM to low μM range. As mentioned above, in one case, inhibition of the toxicity of endogenously expressed α-synuclein was tested in HEK 293 cells and it was found to be inhibited completely by 1 μM CLR01.37 Remarkably, the toxicity of CLR01 itself in cell culture was only observed at 1–3 orders of magnitude higher concentrations and appeared to depend on the cell type used. In differentiated PC-12 cells, no toxicity was observed up to 200 μM and ∼15% reduced viability (MTT assay) occurred at 400 μM.36 No reduction in viability (XTT assay) was found up to 100 μM in human H1299 non-small cell lung carcinoma cells82 and up to 500 μM in the HIV reporter cell line TZM-bl.40
CLR01 also was found to be safe in vivo and did not show any signs of toxicity, such as weight loss, behavioral changes, morbidity or mortality in any of the animal experiments described above. Moreover, in the experiments using the 3 × Tg mouse model of AD, despite the proximity of Lys residues to the α- and β-secretase cleavage sites in APP, CLR01 did not affect APP processing and there was no difference in the levels of the soluble N-terminal domain (sAPP) or the C-terminal fragments, CTF-α and CTF-β.38
To further test the safety of CLR01 in mice, two studies were conducted in which CLR01 was administered intraperitoneally to 2-months-old wild-type mice either as an acute bolus at 10 or 100 mg kg−1 with subsequent analysis at 24 h, or chronically for 30 days.125 In the acute setting, administration of 100 mg kg−1 CLR01 (2500-fold above the dose used in the 3 × Tg mice, 83-fold above the dose in the TTR mice) initially caused obvious signs of distress to the mice (mainly freezing and hunching), which began to resolve after 30 min and resolved completely within 2 h. No mouse died until the time point of euthanasia at 24 h. Histological and serological analyses showed liver injury (Fig. 9), which would be expected for such a high dose, but no damage to other tissues, including the heart, lungs, kidneys, or brain.125 In the 10 mg kg−1 group, there were no signs of distress and no significant serological findings. In one out of the 8 mice in this group there were mild signs of liver degeneration. Therefore, 10 mg kg−1 per day was used as the high dose in the subsequent chronic administration experiment, whereas the low dose was 3 mg kg−1 per day. There were no behavioral or histological findings in either of the chronic-treatment groups. The only significant serological change was ∼40% decrease in blood cholesterol in the 10 mg kg−1 per day group.125 These findings suggest that CLR01 has a high safety margin and support its further development towards therapy for proteinopathies in humans.
 | ||
| Fig. 9 Liver histopathological analysis of mice 24 h following a single intraperitoneal injection of CLR01. Hepatocytes from (A) vehicle-treated, and (B) mice treated with 10 mg kg−1 CLR01 show moderate amounts of glycogen vacuolation. (C) Zone-1 hepatocytes from mice treated with 100 mg kg−1 CLR01 show glycogen vacuolation. Zone-2 hepatocytes are normal sized. Zone-3 hepatocytes are pale with a granular eosinophilic cytoplasm and some nuclei show pyknosis. Reprinted with permission from BMC Pharmacol. Toxicol., 2014, 15, 23. Copyright 2014 BioMed Central. | ||
3.6 Pharmacokinetics of CLR01
In view of the promising efficacy and safety data of CLR01, pharmacokinetic characterization of the compound has begun with the purview of generating a clinical candidate based on CLR01 itself or on a close derivative thereof.The solubility of CLR01 in different aqueous buffers is 10–15 mM and it fulfills all but one of Lipinski's recommendations for drug-like molecules.126 The molecular weight of CLR01 is 722, clearly above the recommended 500 Da cut-off. Evidently, this violation did not prevent the therapeutic effects observed in the animal studies conducted to date. The size is essential for the unique mechanism of action of CLR01 and cannot be reduced meaningfully without loss of activity. Notably, despite the negative charge of the two phosphate groups, CLR01 complies with the other Lipinski rules. It contains two H-bond donors, eight H-bond acceptors, its clogP = 2.64, and its PSA = 139.18, supporting the compound's “druggability.”
Characterization of CLR01 in standard in vitro experiments yielded the following results:125
• CLR01 was stable in human (99 ± 9%) and mouse (97 ± 2%) liver microsome preparations for 60 min, whereas testosterone, which was used as a positive control, was degraded down to 29% and 1%, respectively.
• CLR01 was stable (100 ± 5%) in both human and mouse plasma for 60 min at 37 °C.
• As would be expected based on its mechanism of action, CLR01 was 96 ± 3% and 99.0 ± 0.2% bound to human and mouse plasma proteins, respectively. These results were comparable to testosterone, (98.6 ± 0.2% and 94 ± 2%, respectively).
The IC50 values of CLR01 for inhibition of 5 major cytochrome P (CYP) 450 isoforms were all above 1 μM, suggesting no significant drug–drug interactions.127 In addition, CYP450 34A induction by CLR01 was moderate relative to rifampicin, which was used as a positive control.38
The plasma half-life of CLR01 following a 1 mg kg−1 intravenous or subcutaneous injection was ∼2.5 h,125 suggesting that mechanisms other than those tested in plasma in vitro were responsible for CLR01 clearance. Continuous administration of CLR01 at the same dose via osmotic minipumps for 21 days showed a plasma steady-state concentration of ∼140 nM.41 The subcutaneous bioavailability was 100% compared to intravenous injection.
In contrast to the high subcutaneous bioavailability, oral bioavailability by gavage following administration at 10 mg kg−1 was only ∼1%.125 In addition to the size and negative charge of CLR01, which likely restrict its oral absorption, CLR01 may be metabolized in the gastrointestinal tract. To begin to explore this possibility, we hypothesized that the most likely metabolism of CLR01 would be dephosphorylation. Therefore, the compound was incubated in vitro with alkaline phosphatase or with whole brain extracts for 60 min and the release of free phosphate was measured. p-Nitrophenylphosphate (PNP) was used as a positive control. The measurements revealed quantitative dephosphorylation of PNP, but no release of free phosphate from CLR01 in either experiment,125 probably because of its rigid structure, which confers metabolic stability, in agreement with the plasma and liver-microsome incubation experiments.
To explore the blood–brain barrier (BBB) penetration of CLR01, we spiked the compound with 3H-CLR01, which was labeled using a method that produced stable 3H labeling of aromatic carbons,128 and scintillation counting of brain extracts. Using this method, the BBB penetration was measured in young and old, wild-type and 3 × Tg mice. The BBB penetration was calculated as cpm per gram of brain relative to cpm per mL of blood. The values found were between 1 and 3% with little difference between wild-type and 3 × Tg mice or between young and old mice.125 Interestingly, although CLR01 was cleared relatively rapidly from the plasma, the radioactivity levels measured in the brain did not change significantly up to 72 h post-injection. The BBB penetration of CLR01 was not via a saturated transport system because two consecutive injections at different intervals resulted in doubling of the measured dose in the brain, and injection of 5-times the dose yielded 5-times higher penetration, without changing the brain/blood ratio.125
In the experiments using osmotic minipumps, 0.7% of the CLR01 administered was found in the blood at steady-state.41 Thus, 2% BBB penetration would lead to ∼200 fmol of CLR01 entering the brain per day in the experiment using the 3 × Tg mouse model in which the mice received 0.04 mg kg−1 per day CLR01.38 A literature search for the brain concentration levels of Aβ40 and Aβ42 in the 3 × Tg mice yielded a range of values, of which the maximum was 280 fmol total Aβ per mg brain.93 Taking into account that the mass of the 3 × Tg mouse brains we used was ∼0.5 mg, a total of ∼140 fmol Aβ would be found at a given point in the brain of these mice. Thus, the brain concentration of CLR01 likely was equal or higher than that of Aβ. Given that sub-stoichiometric CLR01 concentrations are expected to be needed in vivo, as was the case in the TTR mouse model,123 the brain levels of CLR01 appear to be sufficient and support the robust clearance of plaques and tangles observed.
4. Conclusion and outlook
To the best of our knowledge, molecular tweezers represent the first class of artificial receptor molecules that have made the way from a supramolecular host to a drug candidate with promising results in animal tests. Due to their unique structure, only lysine and arginine are well complexed with exquisite selectivity by a threading mechanism, which unites electrostatic, hydrophobic and dispersive attraction. Moderate affinities of molecular tweezers towards sterically well accessible basic amino acids with fast on and off rates protect normal proteins from potential interference with their biological function. However, peptide misfolding and subsequent pathological aggregation can be efficiently prevented even by very low tweezer concentrations in a “process-specific” manner. Thus, the early stages of abnormal Aβ, α-synuclein, and TTR assembly are redirected towards the generation of amorphous non-toxic materials that can be degraded by the intracellular and extracellular clearance mechanisms. Existing fibrils are dissociated and likewise turned into unordered aggregates amenable to clearance. Thus, specific host–guest chemistry between aggregation-prone proteins and lysine/arginine binders rescues cell viability and restores animal health in models of AD, PD, and TTR amyloidosis. In particular, the diphosphate prototype, CLR01 seems to have a high safety margin and very low toxicity, but unrivaled efficiency in counteracting neuronal lesions by toxic oligomers and fibrils. CLR01 passes the blood–brain barrier and was found in the cortex at concentrations comparable to those of Aβ. We are currently designing prodrugs with increased bioavailability and activity in order to develop disease-modifying therapy for neurological disorders.Acknowledgements
Thomas Schrader gratefully acknowledges funding from the Collaborative Research Centre 1093 “Supramolecular Chemistry on Proteins” by the DFG (Deutsche Forschungsgemeinschaft). Gal Bitan gratefully acknowledges grant 20143057 from the RGK Foundation, grant 10220 from the Michael J. Fox Foundation, grant 20152631 from the Cure Alzheimer's Fund, grant 600-6-15 from the CurePSP Foundation, grant AG050721 from NIH/NIA, and generous support from Team Parkinson/Parkinson Alliance and from the UCLA Mary S. Easton Endowment. Finally, we thank all our collaboration partners, who are cited in the references, for their important and exciting contributions to these projects.References
- C. W. Chen and H. W. Whitlock, J. Am. Chem. Soc., 1978, 100, 4921–4922 CrossRef CAS
.
- S. C. Zimmerman and C. M. Vanzyl, J. Am. Chem. Soc., 1987, 109, 7894–7896 CrossRef CAS
- J. Fleischhauer, M. Harmata, M. Kahraman, A. Koslowski and C. J. Welch, Tetrahedron Lett., 1997, 38, 8655–8658 CrossRef CAS
- F. Hof, S. L. Craig, C. Nuckolls and J. Rebek Jr., Angew. Chem., Int. Ed., 2002, 41, 1488–1508 CrossRef CAS
- A. E. Rowan, J. A. A. W. Elemans and R. J. M. Nolte, Acc. Chem. Res., 1999, 32, 995–1006 CrossRef CAS
- H. Kurebayashi, T. Haino, S. Usui and Y. Fukazawa, Tetrahedron, 2001, 57, 8667–8674 CrossRef CAS
- F. G. Klärner, J. Benkhoff, R. Boese, U. Burkert, M. Kamieth and U. Naatz, Angew. Chem., Int. Ed., 1996, 35, 1130–1133 CrossRef
- M. Fokkens, T. Schrader and F. G. Klärner, J. Am. Chem. Soc., 2005, 127, 14415–14421 CrossRef CAS PubMed
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed
- L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed
- H. J. Schneider, Acc. Chem. Res., 2013, 46, 1010–1019 CrossRef CAS PubMed
- J. M. Lehn, P. Vierling and R. C. Hayward, J. Chem. Soc., Chem. Commun., 1979, 296–298, 10.1039/c39790000296
- M. A. Hossain and H. J. Schneider, J. Am. Chem. Soc., 1998, 120, 11208–11209 CrossRef CAS
- C. P. Mandl and B. König, J. Org. Chem., 2005, 70, 670–674 CrossRef CAS PubMed
- N. Douteau-Guevel, A. W. Coleman, J. P. Morel and N. Morel-Desrosiers, J. Phys. Org. Chem., 1998, 11, 693–696 CrossRef CAS
- N. Douteau-Guevel, A. W. Coleman, J. P. Morel and N. Morel-Desrosiers, J. Chem. Soc., Perkin Trans. 2, 1999, 629–633, 10.1039/a806855k
- N. Douteau-Guevel, F. Perret, A. W. Coleman, J. P. Morel and N. Morel-Desrosiers, J. Chem. Soc., Perkin Trans. 2, 2002, 524–532, 10.1039/b109553f
- J. Dziemidowicz, D. Witt and J. Rachon, J. Inclusion Phenom. Macrocyclic Chem., 2008, 61, 381–391 CrossRef CAS
- L. Mutihac, J. H. Lee, J. S. Kim and J. Vicens, Chem. Soc. Rev., 2011, 40, 2777–2796 RSC
- S. M. Ngola, P. C. Kearney, S. Mecozzi, K. Russell and D. A. Dougherty, J. Am. Chem. Soc., 1999, 121, 1192–1201 CrossRef CAS
- T. W. Bell, A. B. Khasanov, M. G. Drew, A. Filikov and T. L. James, Angew. Chem., Int. Ed., 1999, 38, 2543–2547 CrossRef CAS
- C. T. Oberg, A. L. Noresson, H. Leffler and U. J. Nilsson, Chem. – Eur. J., 2011, 17, 8139–8144 CrossRef PubMed
- T. Schrader, Chem. – Eur. J., 1997, 3, 1537–1541 CrossRef CAS
- T. Braxmeier, M. Demarcus, T. Fessmann, S. McAteer and J. D. Kilburn, Chem. – Eur. J., 2001, 7, 1889–1898 CrossRef CAS
- V. Villari, P. Mineo, E. Scamporrino and N. Micali, Chem. Phys., 2012, 409, 23–31 CrossRef CAS
- F. G. Klärner, U. Burkert, M. Kamieth, R. Boese and J. Benet-Buchholz, Chem. – Eur. J., 1999, 5, 1700–1707 CrossRef
- M. Kamieth and F. G. Klärner, J. Prakt. Chem./Chem.-Ztg., 1999, 341, 245–251 CrossRef CAS
- F. G. Klärner, J. Panitzky, D. Blaser and R. Boese, Tetrahedron, 2001, 57, 3673–3687 CrossRef
- F. G. Klärner and B. Kahlert, Acc. Chem. Res., 2003, 36, 919–932 CrossRef PubMed
- F. G. Klärner, T. Schrader, J. Polkowska, F. Bastkowski, P. Talbiersky, M. C. Kuchenbrandt, T. Schaller, H. de Groot and M. Kirsch, Pure Appl. Chem., 2010, 82, 991–999 CrossRef
- F. G. Klärner and T. Schrader, Acc. Chem. Res., 2013, 46, 967–978 CrossRef PubMed
- P. Talbiersky, F. Bastkowski, F. G. Klärner and T. Schrader, J. Am. Chem. Soc., 2008, 130, 9824–9828 CrossRef CAS PubMed
- S. Dutt, C. Wilch, T. Gersthagen, P. Talbiersky, K. Bravo-Rodriguez, M. Hanni, E. Sanchez-Garcia, C. Ochsenfeld, F. G. Klärner and T. Schrader, J. Org. Chem., 2013, 78, 6721–6734 CrossRef CAS PubMed
- S. Dutt, C. Wilch, T. Gersthagen, C. Wölper, A. A. Sowislok, F.-G. Klärner and T. Schrader, Eur. J. Org. Chem., 2013, 7705–7714 CrossRef CAS
- T. Gersthagen, J. Hofmann, F.-G. Klärner, C. Schmuck and T. Schrader, Eur. J. Org. Chem., 2013, 1080–1092 CrossRef CAS
- S. Sinha, D. H. Lopes, Z. Du, E. S. Pang, A. Shanmugam, A. Lomakin, P. Talbiersky, A. Tennstaedt, K. McDaniel, R. Bakshi, P. Y. Kuo, M. Ehrmann, G. B. Benedek, J. A. Loo, F. G. Klärner, T. Schrader, C. Wang and G. Bitan, J. Am. Chem. Soc., 2011, 133, 16958–16969 CrossRef CAS PubMed
- S. Prabhudesai, S. Sinha, A. Attar, A. Kotagiri, A. G. Fitzmaurice, R. Lakshmanan, M. I. Ivanova, J. A. Loo, F. G. Klärner, T. Schrader, M. Stahl, G. Bitan and J. M. Bronstein, Neurotherapeutics, 2012, 9, 464–476 CrossRef CAS PubMed
- A. Attar, C. Ripoli, E. Riccardi, P. Maiti, D. D. Li Puma, T. Liu, J. Hayes, M. R. Jones, K. Lichti-Kaiser, F. Yang, G. D. Gale, C. H. Tseng, M. Tan, C. W. Xie, J. L. Straudinger, F. G. Klärner, T. Schrader, S. A. Frautschy, C. Grassi and G. Bitan, Brain, 2012, 135, 3735–3748 CrossRef PubMed
- S. M. Fogerson, A. J. van Brummen, D. J. Busch, S. R. Allen, R. Roychaudhuri, S. M. Banks, F. G. Klarner, T. Schrader, G. Bitan and J. R. Morgan, Exp. Neurol., 2016, 278, 105–115 CrossRef CAS PubMed
- E. Lump, L. M. Castellano, C. Meier, J. Seeliger, N. Erwin, B. Sperlich, C. M. Stürzel, S. Usmani, R. M. Hammond, J. von Einem, G. Gerold, F. Kreppel, K. Bravo-Rodriguez, T. Pietschmann, V. M. Holmes, D. Palesch, O. Zirafi, D. Weissman, A. Sowislok, B. Wettig, C. Heid, F. Kirchhoff, T. Weil, F. G. Klärner, T. Schrader, G. Bitan, E. Sanchez-Garcia, R. Winter, J. Shorter and J. Münch, eLife, 2015, 4, e05397 Search PubMed
- D. H. Lopes, A. Attar, G. Nair, E. Y. Hayden, Z. Du, K. McDaniel, S. Dutt, K. Bravo-Rodriguez, S. Mittal, F. G. Klärner, C. Wang, E. Sanchez-Garcia, T. Schrader and G. Bitan, ACS Chem. Biol., 2015, 10, 1555–1569 CrossRef CAS PubMed
- D. H. Williams and B. Bardsley, Angew. Chem., Int. Ed., 1999, 38, 1173–1193 CAS
- L. O. Tjernberg, C. Lilliehook, D. J. E. Callaway, J. Näslund, S. Hahne, J. Thyberg, L. Terenius and C. Nordstedt, J. Biol. Chem., 1997, 272, 12601–12605 CrossRef CAS PubMed
- W. C. Tsai, C. C. Hsu, C. Y. Chung, M. S. Lin, S. L. Li and J. H. Pang, J. Orthop. Res., 2007, 25, 1629–1634 CrossRef CAS PubMed
- R. O. Hynes, Cell, 1992, 69, 11–25 CrossRef CAS PubMed
- D. Bier, R. Rose, K. Bravo-Rodriguez, M. Bartel, J. M. Ramirez-Anguita, S. Dutt, C. Wilch, F. G. Klärner, E. Sanchez-Garcia, T. Schrader and C. Ottmann, Nat. Chem., 2013, 5, 234–239 CrossRef CAS PubMed
- T. P. Knowles, M. Vendruscolo and C. M. Dobson, Nat. Rev. Mol. Cell Biol., 2014, 15, 384–396 CrossRef CAS PubMed
- A. Alzheimer, Centralblatt für Nervenheilkunde und Psychiatrie, 1907, 30, 177–179 Search PubMed
- J. Parkinson, J. Neuropsychiatry Clin. Neurosci., 2002, 14, 223–236 CrossRef PubMed
- D. R. Rosen, T. Siddique, D. Patterson, D. A. Figlewicz, P. Sapp, A. Hentati, D. Donaldson, J. Goto, J. P. O'Regan and H. X. Deng,
et al.
, Nature, 1993, 362, 59–62 CrossRef CAS PubMed
- C. Andrade, Brain, 1952, 75, 408–427 CrossRef CAS PubMed
- M. E. MacDonald, C. M. Ambrose, M. P. Duyao, R. H. Myers, C. Lin, L. Srinidhi, G. Barnes, S. A. Taylor, M. James, N. Groat, H. MacFarlane, B. Jenkins, M. A. Anderson, N. S. Wexler and J. F. Gusella.,
et al.
, Cell, 1993, 72, 971–983 CrossRef
- V. N. Uversky, Curr. Alzheimer Res., 2008, 5, 260–287 CrossRef CAS PubMed
- G. G. Glenner and C. W. Wong, Biochem. Biophys. Res. Commun., 1984, 120, 885–890 CrossRef CAS PubMed
- N. Nukina and Y. Ihara, J. Biochem., 1986, 99, 1541–1544 CAS
- B. L. Wolozin, A. Pruchnicki, D. W. Dickson and P. Davies, Science, 1986, 232, 648–650 CAS
- M. G. Spillantini, M. L. Schmidt, V. M. Lee, J. Q. Trojanowski, R. Jakes and M. Goedert, Nature, 1997, 388, 839–840 CrossRef CAS PubMed
- P. Westermark, C. Wernstedt, E. Wilander and K. Sletten, Biochem. Biophys. Res. Commun., 1986, 140, 827–831 CrossRef CAS PubMed
- F. Chiti, P. Webster, N. Taddei, A. Clark, M. Stefani, G. Ramponi and C. M. Dobson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3590–3594 CrossRef CAS
- Y. Fezoui, D. M. Hartley, D. M. Walsh, D. J. Selkoe, J. J. Osterhout and D. B. Teplow, Nat. Struct. Biol., 2000, 7, 1095–1099 CrossRef CAS PubMed
- J. I. Guijarro, M. Sunde, J. A. Jones, I. D. Campbell and C. M. Dobson, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4224–4228 CrossRef CAS
- M. Gross, D. K. Wilkins, M. C. Pitkeathly, E. W. Chung, C. Higham, A. Clark and C. M. Dobson, Protein Sci., 1999, 8, 1350–1357 CrossRef CAS PubMed
- C. M. Dobson, Philos. Trans. R. Soc. London, Ser. B, 2001, 356, 133–145 CrossRef CAS PubMed
- C. M. Dobson, Biochem. Soc. Symp., 2001, 68, 1–26 CrossRef CAS PubMed
- F. Rahimi, A. Shanmugam and G. Bitan, Curr. Alzheimer Res., 2008, 5, 319–341 CrossRef CAS PubMed
- D. Morshedi, A. Ebrahim-Habibi, A. A. Moosavi-Movahedi and M. Nemat-Gorgani, Biochim. Biophys. Acta, 2010, 1804, 714–722 CrossRef CAS PubMed
- S. Davern, C. L. Murphy, H. O'Neill, J. S. Wall, D. T. Weiss and A. Solomon, Biochim. Biophys. Acta, 2011, 1812, 32–40 CrossRef CAS PubMed
- B. R. Groveman, A. Kraus, L. D. Raymond, M. A. Dolan, K. J. Anson, D. W. Dorward and B. Caughey, J. Biol. Chem., 2015, 290, 1119–1128 CrossRef CAS PubMed
- S. Sinha, D. H. Lopes and G. Bitan, ACS Chem. Neurosci., 2012, 3, 473–481 CrossRef CAS PubMed
- W. Li, J. B. Sperry, A. Crowe, J. Q. Trojanowski, A. B. Smith 3rd and V. M. Lee, J. Neurochem., 2009, 110, 1339–1351 CrossRef CAS PubMed
- K. Usui, J. D. Hulleman, J. F. Paulsson, S. J. Siegel, E. T. Powers and J. W. Kelly, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18563–18568 CrossRef CAS PubMed
- B. Winner, R. Jappelli, S. K. Maji, P. A. Desplats, L. Boyer, S. Aigner, C. Hetzer, T. Loher, M. Vilar, S. Campioni, C. Tzitzilonis, A. Soragni, S. Jessberger, H. Mira, A. Consiglio, E. Pham, E. Masliah, F. H. Gage and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4194–4199 CrossRef CAS
- T. J. Cohen, J. L. Guo, D. E. Hurtado, L. K. Kwong, I. P. Mills, J. Q. Trojanowski and V. M. Lee, Nat. Commun., 2011, 2, 252 CrossRef PubMed
- A. Attar and G. Bitan, Curr. Pharm. Des., 2014, 20, 2469–2483 CrossRef CAS PubMed
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353–356 CrossRef CAS PubMed
- J. A. Hardy and G. A. Higgins, Science, 1992, 256, 184–185 CAS
- Y. Xie, J. Zhang, S. Yin and J. A. Loo, J. Am. Chem. Soc., 2006, 128, 14432–14433 CrossRef CAS PubMed
- L. Hou, H. Shao, Y. Zhang, H. Li, N. K. Menon, E. B. Neuhaus, J. M. Brewer, I. J. Byeon, D. G. Ray, M. P. Vitek, T. Iwashita, R. A. Makula, A. B. Przybyla and M. G. Zagorski, J. Am. Chem. Soc., 2004, 126, 1992–2005 CrossRef CAS PubMed
- A. Attar, F. Rahimi and G. Bitan, Transl. Neurosci., 2013, 4, 385–409 Search PubMed
- T. Liu and G. Bitan, ChemMedChem, 2012, 7, 359–374 CrossRef CAS PubMed
- H. LeVine 3rd, Methods Enzymol., 1999, 309, 274–284 CAS
- G. Herzog, M. D. Shmueli, L. Levy, L. Engel, E. Gazit, F. G. Klärner, T. Schrader, G. Bitan and D. Segal, Biochemistry, 2015, 54, 3729–3738 CrossRef CAS PubMed
- R. Kayed, E. Head, J. L. Thompson, T. M. McIntire, S. C. Milton, C. W. Cotman and C. G. Glabe, Science, 2003, 300, 486–489 CrossRef CAS PubMed
- G. Bitan, M. D. Kirkitadze, A. Lomakin, S. S. Vollers, G. B. Benedek and D. B. Teplow, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 330–335 CrossRef CAS PubMed
- D. J. Selkoe, Science, 2002, 298, 789–791 CrossRef CAS PubMed
- J. J. Palop and L. Mucke, Nat. Neurosci., 2010, 13, 812–818 CrossRef CAS PubMed
- F. Rahimi, P. Maiti and G. Bitan, J. Visualized Exp., 2009, 23 DOI:10.3791/1071
- G. M. Shankar, S. Li, T. H. Mehta, A. Garcia-Munoz, N. E. Shepardson, I. Smith, F. M. Brett, M. A. Farrell, M. J. Rowan, C. A. Lemere, C. M. Regan, D. M. Walsh, B. L. Sabatini and D. J. Selkoe, Nat. Med., 2008, 14, 837–842 CrossRef CAS PubMed
- G. B. Stokin, C. Lillo, T. L. Falzone, R. G. Brusch, E. Rockenstein, S. L. Mount, R. Raman, P. Davies, E. Masliah, D. S. Williams and L. S. Goldstein, Science, 2005, 307, 1282–1288 CrossRef CAS PubMed
- T. V. Bliss and G. L. Collingridge, Nature, 1993, 361, 31–39 CrossRef CAS PubMed
- M. P. Lambert, A. K. Barlow, B. A. Chromy, C. Edwards, R. Freed, M. Liosatos, T. E. Morgan, I. Rozovsky, B. Trommer, K. L. Viola, P. Wals, C. Zhang, C. E. Finch, G. A. Krafft and W. L. Klein, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6448–6453 CrossRef CAS
- M. Townsend, G. M. Shankar, T. Mehta, D. M. Walsh and D. J. Selkoe, J. Physiol., 2006, 572, 477–492 CrossRef CAS PubMed
- S. Oddo, A. Caccamo, J. D. Shepherd, M. P. Murphy, T. E. Golde, R. Kayed, R. Metherate, M. P. Mattson, Y. Akbari and F. M. LaFerla, Neuron, 2003, 39, 409–421 CrossRef CAS PubMed
- S. Oddo, L. Billings, J. P. Kesslak, D. H. Cribbs and F. M. LaFerla, Neuron, 2004, 43, 321–332 CrossRef CAS PubMed
- A. M. Hall and E. D. Roberson, Brain Res. Bull., 2012, 88, 3–12 CrossRef CAS PubMed
- M. Kitazawa, R. Medeiros and F. M. Laferla, Curr. Pharm. Des., 2012, 18, 1131–1147 CrossRef CAS PubMed
- D. G. Flood, Y. G. Lin, D. M. Lang, S. P. Trusko, J. D. Hirsch, M. J. Savage, R. W. Scott and D. S. Howland, Neurobiol. Aging, 2009, 30, 1078–1090 CrossRef CAS PubMed
- E. Teng, V. Kepe, S. A. Frautschy, J. Liu, N. Satyamurthy, F. Yang, P. P. Chen, G. B. Cole, M. R. Jones, S. C. Huang, D. G. Flood, S. P. Trusko, G. W. Small, G. M. Cole and J. R. Barrio, Neurobiol. Dis., 2011, 43, 565–575 CrossRef CAS PubMed
- K. L. Youmans, L. M. Tai, T. Kanekiyo, W. B. Stine Jr., S. C. Michon, E. Nwabuisi-Heath, A. M. Manelli, Y. Fu, S. Riordan, W. A. Eimer, L. Binder, G. Bu, C. Yu, D. M. Hartley and M. J. LaDu, Mol. Neurodegener., 2012, 7, 8 CrossRef CAS PubMed
- R. Malik, J. Di, G. Nair, A. Attar, K. Taylor, E. Teng and G. Bitan, Methods Mol. Biol., 2016 Search PubMed
- Y. Matsuoka, M. Saito, J. LaFrancois, K. Gaynor, V. Olm, L. Wang, E. Casey, Y. Lu, C. Shiratori, C. Lemere and K. Duff, J. Neurosci., 2003, 23, 29–33 CAS
- G. Bitan, E. A. Fradinger, S. M. Spring and D. B. Teplow, Amyloid, 2005, 12, 88–95 CrossRef PubMed
- S. Acharya, B. M. Safaie, P. Wongkongkathep, M. I. Ivanova, A. Attar, F. G. Klärner, T. Schrader, J. A. Loo, G. Bitan and L. J. Lapidus, J. Biol. Chem., 2014, 289, 10727–10737 CrossRef CAS PubMed
- G. Bitan and D. B. Teplow, Acc. Chem. Res., 2004, 37, 357–364 CrossRef CAS PubMed
- D. H. Lopes, S. Sinha, C. Rosensweig and G. Bitan, Methods Mol. Biol., 2012, 849, 11–21 CAS
- R. K. Das and R. V. Pappu, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 13392–13397 CrossRef CAS PubMed
- H. Zhu and L. I. Zon, Methods Cell Biol., 2004, 76, 3–12 CrossRef CAS PubMed
- E. Emmanouilidou, L. Stefanis and K. Vekrellis, Neurobiol. Aging, 2010, 31, 953–968 CrossRef CAS PubMed
- A. G. Fitzmaurice, S. L. Rhodes, A. Lulla, N. P. Murphy, H. A. Lam, K. C. O'Donnell, L. Barnhill, J. E. Casida, M. Cockburn, A. Sagasti, M. C. Stahl, N. T. Maidment, B. Ritz and J. M. Bronstein, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 636–641 CrossRef CAS PubMed
- A. P. Chou, N. Maidment, R. Klintenberg, J. E. Casida, S. Li, A. G. Fitzmaurice, P. O. Fernagut, F. Mortazavi, M. F. Chesselet and J. M. Bronstein, J. Biol. Chem., 2008, 283, 34696–34703 CrossRef CAS PubMed
- A. Lulla, L. Barnhill, G. Bitan, M. I. Ivanova, B. Nguyen, K. O'Donnell, M. Stahl, C. Yamashiro, F.-G. Klärner, T. Schrader, A. Sagasti and J. M. Bronstein, Environ. Health Perspect., 2016 DOI:10.1289/EHP141
- D. J. Busch and J. R. Morgan, J. Comp. Neurol., 2012, 520, 1751–1771 CrossRef CAS PubMed
- M. J. Saraiva, FEBS Lett., 2001, 498, 201–203 CrossRef CAS PubMed
- J. Buxbaum, A. Alexander, J. Koziol, C. Tagoe, E. Fox and D. Kitzman, Am. Heart J., 2010, 159, 864–870 CrossRef CAS PubMed
- C. C. Quarta, J. N. Buxbaum, A. M. Shah, R. H. Falk, B. Claggett, D. W. Kitzman, T. H. Mosley, K. R. Butler, E. Boerwinkle and S. D. Solomon, N. Engl. J. Med., 2015, 372, 21–29 CrossRef PubMed
- L. H. Connors, G. Doros, F. Sam, A. Badiee, D. C. Seldin and M. Skinner, Amyloid, 2011, 18, suppl 1157–159 CrossRef PubMed
- L. S. Coles and R. D. Young, Prev. Med., 2012, 54(suppl), S9–S11 CrossRef CAS PubMed
- M. J. Saraiva, M. M. Sousa, I. Cardoso and R. Fernandes, J. Mol. Neurosci., 2004, 23, 35–40 CrossRef CAS
- S. D. Santos, R. Fernandes and M. J. Saraiva, Neurobiol. Aging, 2010, 31, 280–289 CrossRef CAS PubMed
- K. Kohno, J. A. Palha, K. Miyakawa, M. J. Saraiva, S. Ito, T. Mabuchi, W. S. Blaner, H. Iijima, S. Tsukahara, V. Episkopou, M. E. Gottesman, K. Shimada, K. Takahashi, K. Yamamura and S. Maeda, Am. J. Pathol., 1997, 150, 1497–1508 CAS
- M. H. Teng, J. Y. Yin, R. Vidal, J. Ghiso, A. Kumar, R. Rabenou, A. Shah, D. R. Jacobson, C. Tagoe, G. Gallo and J. Buxbaum, Lab. Invest., 2001, 81, 385–396 CrossRef CAS PubMed
- J. Buxbaum, C. Tagoe, G. Gallo, N. Reixach and D. French, Amyloid, 2003, 10, suppl 12–6 CrossRef
- N. Ferreira, A. Pereira-Henriques, A. Attar, F. G. Klärner, T. Schrader, G. Bitan, L. Gales, M. J. Saraiva and M. R. Almeida, Neurotherapeutics, 2014, 11, 450–461 CrossRef PubMed
- N. Reixach, S. Deechongkit, X. Jiang, J. W. Kelly and J. N. Buxbaum, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 2817–2822 CrossRef CAS PubMed
- A. Attar, W. T. Chan, F. G. Klärner, T. Schrader and G. Bitan, BMC Pharmacol. Toxicol., 2014, 15, 23 CrossRef PubMed
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS PubMed
- R. S. Obach, R. L. Walsky, K. Venkatakrishnan, E. A. Gaman, J. B. Houston and L. M. Tremaine, J. Pharmacol. Exp. Ther., 2006, 316, 336–348 CrossRef CAS PubMed
- T. Maegawa, K. Hirota, K. Tatematsu, Y. Mori and H. Sajiki, J. Org. Chem., 2005, 70, 10581–10583 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2016 |