
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photochromic spiropyran- and spirooxazine-homopolymers in mesoporous thin films by surface initiated ROMP†
F.
Krohm
a,
J.
Kind
b,
R.
Savka
c,
M.
Alcaraz Janßen
b,
D.
Herold
b,
H.
Plenio
c,
C. M.
Thiele
b and
A.
Andrieu-Brunsen
*a
aErnst-Berl Institut für Technische und Makromolekulare Chemie, Technische Universität Darmstadt, Alarich-Weiss-Str. 4, D-64287 Darmstadt, Germany. E-mail: brunsen@cellulose.tu-darmstadt.de
bClemens-Schöpf-Institut für Organische Chemie und Biochemie, Technische Universität Darmstadt, Alarich-Weiss-Str. 16, D-64287 Darmstadt, Germany
cEduard-Zintl-Institut für Anorganische und Physikalische Chemie, Technische Universität Darmstadt, Alarich-Weiss-Str. 12, 64287 Darmstadt, Germany
First published on 9th February 2016
Abstract
The control of ionic permselectivity in porous films is an interesting aspect in the context of lab-on-chip devices and μ-electronics. Especially, visible light triggered ionic permselectivity control is of relevance because control by light can be maintained externally without changing internal system parameters. In addition, light is a sustainable energy source if sunlight is used. Here, we present the first mesoporous films modified with two different photochromic homopolymers by surface-initiated ring opening metathesis polymerization (SI-ROMP). Spiropyran- and spirooxazine functionalized norbornene monomers and the corresponding ROMP homopolymers are synthesized in solution and in mesopores and compared concerning their optical properties such as photochromic conversion kinetics, photostability, and the ratio of converted molecules. Optical properties are investigated using UV/VIS spectroscopy and 1H-NMR spectroscopy. Especially, spirooxazine, whose surface functionalization has not been studied in detail, shows fast switching properties and higher ratios of photochromically interconverted molecules. After grafting spiropyran- and spirooxazine norbornene homopolymers into mesopores, a slightly faster photochromic interconversion of polymers located inside the mesopores is observed compared to the solution polymers.
Introduction
For the design of technological innovation, nature is a multifaceted source of inspiration. For example, wetting on surfaces,1 release,2 or complex transport control through pores3–7 is inspired by nature. In the context of sustainability, it is desirable to re-use resources like water or reduce energy consumption in technological applications. One approach towards more sustainable energy consumption is the use of sunlight. Combining research on transport control through pores and responsivity towards sunlight, the combination of photochromic spiropyran- or spirooxazine polymers as functional components with ceramic mesopores as structural components can be one versatile approach.The photochromic reaction of spiropyran was first reported in 1952.8 Recently, the mechanism of photochromism was investigated with respect to its ultrafast dynamics9 and merocyanine isomers by using DFT calculations.10 Since then, two main areas of interest related to spiropyrans have developed: One is related to the complexation of ions accompained by an ion-specific color change and thus a sensor or release response.11,12 Secondly, spiropyran is used for surface functionalization to manipulate and switch surface properties such as wetting. The challenge of switching surface wettability is related to the relatively low changes in the contact angle which usually are below 15°.13–15 In addition to molecular functionalization, many groups are studying spiropyran containing polymer coatings on surfaces for sensor or cell adhesion studies.15–19 Thereby, changes in surface properties are often limited by the spiropyran content. Using radical polymerization spiropyran monomers can only be copolymerized with non-spiropyran containing monomers and thus the spiropyran contents reported are mostly in the range of 10–15 mol%.15,20,21 Only a few studies report on the synthesis of spiropyran homopolymers accessible by ring-opening metathesis polymerization.22–24 These studies report the modification of surfaces with ROMP homopolymers carrying a spiropyran in the side chain. Interestingly, nitrospiropyran dominates the literature related to surface functionalization and photochromic behaviour of surface properties. To the best of our knowledge, there are no studies related to spirooxazine homopolymer synthesis at surfaces or even in the confined space of pores. Spirooxazine is an interesting photochromic compound because it shows much faster switching kinetics than spiropyran. Especially, in terms of transport control, different switching kinetics are of potential interest because they could allow the adjustment of transport kinetics.
In this context, the functionalization of porous materials with photochromic units is very fascinating to mimic light-gated ion channels. So far, mesoporous silica or single pores have been functionalized using azobenzenes25,26 or spiropyran molecules observing a modulation of probe molecule transport upon irradiation.27–29 In addition, spiropyran copolymers are reported to show an effect on membrane permeability.30 Transport modulation is ascribed either to electrostatic27 or to hydrophobic/hydrophilic29 interactions. Interestingly though, spiropyran homopolymers or even spirooxazine homopolymers have not been used to improve understanding.
Here, the first steps towards photo-responsive pores based on the spiropyran- and spirooxazine homopolymer functionalization of mesoporous silica thin films are reported. A synthesis strategy for spiropyran and spirooxazine monomers for SI-ROMP polymerization is developed and these monomers are applied to graft ROMP homopolymers from silica mesoporous films. The photochromic behavior in terms of the ratio of molecules that respond to light and the interconversion kinetics is investigated by using in situ irradiation NMR31 in solution and UV/VIS-spectroscopy. By combining both methods, extinction coefficients of spiropyran and merocyanine forms are accessible. Furthermore, this is the first study that examines the photochromic behavior of homopolymers using NMR spectroscopy. The presented synthetic approach and the deep understanding of photo-responsive behavior are a strong basis for further investigations towards sustainable transport control by using visible light and sunlight.
Experimental
The synthesis of all applied compounds (Fig. 1) including their characterization is described in detail within the ESI.†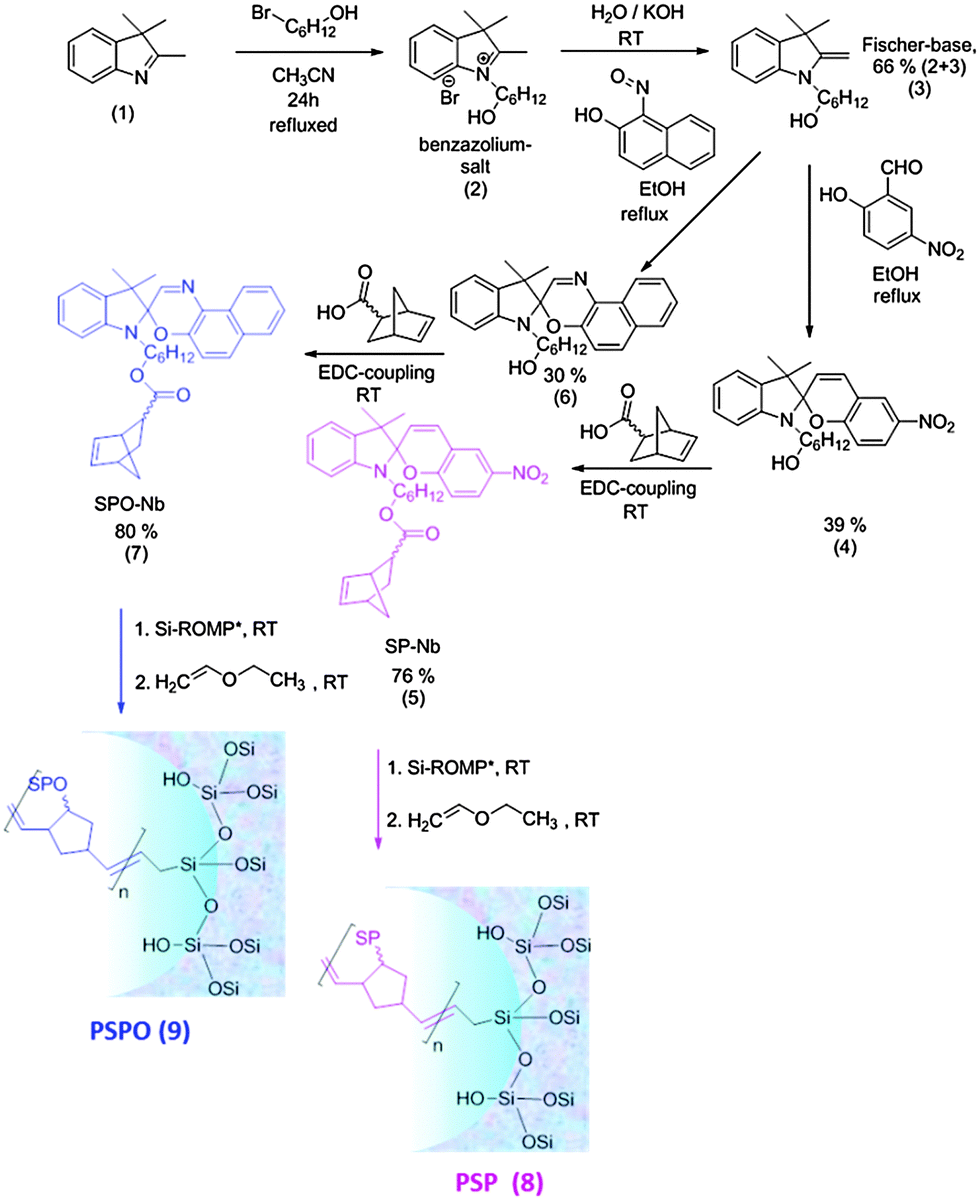 | ||
| Fig. 1 Synthetic strategy for both monomers SP-Nb (5) and SPO-Nb (7) and the synthetic strategy for surface initiated ROMP on mesoporous allylsilica thin films. | ||
Infrared (IR) spectroscopy
IR measurements are performed on a Spectrum One (PerkinElmer) instrument in attenuated total reflection (ATR) mode. Mesoporous films are scratched from the substrate to record IR spectra in the range from 4000 to 600 cm−1. The measured spectra are background corrected and normalized to the Si–O–Si band at 1080 cm−1.UV/VIS-spectroscopy
UV/VIS absorption and kinetic measurements of spiropyran and spirooxazine containing compounds are carried out using a Cary 60 UV/VIS-spectrometer (Agilent). The recording of measured data is done using the software package Agilent Cary WinUV-Software. All data are background corrected prior to each measurement. The used solvent is dried carefully and stored above molecular sieves. UV and VIS irradiation is performed by using a Model Superlite 410 with different cut-off filters (λUVA = 320 bis 400 nm (I = 11.8 mW cm−2) and λgreen = 550 nm (I = 30.6 mW cm−2)) (LUMATEC GmbH Deisenhofen, Germany). The samples are irradiated with a distance of 3.0 cm at an angle of 45° to the quartz glass (d = 1.0 cm) cuvette.NMR
All employed compounds and precursors for the attachment on mesoporous silica films are characterized by NMR-spectroscopy using a Bruker DRX 500 or a Bruker AC 300 spectrometer. A sample concentration of 10 mg substance per 1 mL deuterated solvent is used and 1H-NMR and 13C-NMR are recorded. All NMR-spectra are evaluated using MestReNova.All in situ irradiation NMR experiments are performed on a Bruker Avance III HD 700 MHz NMR-spectrometer equipped with a QCI (1H, 13C, 19F, 15N) probe. Measurements and processing of the resulting spectra are performed using Bruker Topspin 3.2.
Kinetic measurements are carried out as single scan pseudo-2D proton experiments with a 90° excitation angle, 64 K FID points, a sweep width of 20 ppm and a variable delay between consecutive scans. All resulting data planes are apodized by exponential multiplication (LB 0.3 Hz) and Fourier transformed along F2. Further pseudo-2D spectra are transformed into 1D datasets. Fractions of different species are calculated from the integrals of corresponding resonances.
The irradiation setup is based on the setup published by Feldmeier et al.31 The samples are prepared in 5 mm amberized NMR tubes (Wilmad LabGlass). After dissolving the sample in DMF-d7 or DMSO-d6 (Sigma Aldrich) (∼1 mg mL−1) a coaxial stem insert (WGS-5BL-SP – Wilmad LabGlass) containing the silica wave guide (BFH 48-1000, Thorlabs) with a roughened tip is inserted into the sample solution. In the case of spiropyran solutions, irradiation is applied using UV LEDs (Nichia SMD LED UV NCSU275 or SMD LED UV NCSU276A), whereas spirooxazine solutions are irradiated using the Lumatec Superlite 410. Irradiation power is measured using an AVASPHERE-50-IRRAD integration sphere and an avaspec ULS3648 UV/VIS detector. For the setup with the Lumatec Superlite 410 lamp an irradiation power of 4.7 mW cm−2 is measured at the tip of the waveguide. For the setup with the UV LED an irradiation power of 0.65 mW cm−2 is measured. For the green LED an irradiation power of 16.4 mW cm−2 is obtained.
Extinction coefficient
The extinction coefficients for spiropyran and merocyanine forms are determined by NMR and UV/VIS spectroscopy.32 From 1H-NMR spectra of highly concentrated samples, measured with 32 or 64 scans, a sweep width of 20 ppm, 64 K FID points and a relaxation delay of 20 s, fractions of spiropyran and merocyanine are obtained by integration of the corresponding signals leading to thermal equilibrium fractions. From these, extinction coefficients of the corresponding species can be calculated from absorption in UV/VIS spectra.32 UV-spectra are recorded on a Jasco V-630 UV/VIS spectrometer with 0.1 nm resolution and a wavelength range from 320 to 800 nm. The samples are prepared in DMF and measured in 1.0 mm quartz crystal cuvettes (Hellma Analytic).Transmission electron microscopy (TEM)
TEM measurements are performed using a FEI CM20 TEM microscope with a maximum resolution of 2.3 Å, equipped with a LAB-6 cathode and a CCD camera (Olympus), using an acceleration voltage of 200 kV. The samples are scratched from glass substrates and dispersed in some drops of ethanol. The suspension is placed on a copper TEM grid.Plasma treatment
The CO2-plasma-etching for the selective destruction of allyl groups on the external mesoporous film surface is carried out using a capacitively coupled radiofrequency-plasma device (Diener, model: Femto, Germany, 13.56 MHz, radiofrequency energymax = 200 W). The samples are evacuated in the chamber to 0.2 mbar for 5 min. Subsequently, CO2 plasma is generated for 12 s at 20% of the maximum radiofrequency energy with a pressure of 0.2 mbar.Results and discussion
Synthesis of spiropyran and spirooxazine monomers
To ensure a relatively high functional density, spiropyran- and spirooxazine homopolymers are used to prepare photo-responsive mesopores. Spiropyran-norbornene (SP-Nb) (5) and spirooxazine-norbornene (SPO-Nb) (7) monomers are synthesized in a four-step procedure summarized in Fig. 1 (details can be found in the ESI†). The synthesis concept for SP-OH (4) and the active ester mediated coupling of the norbornene polymerizable unit (5) to SP-OH (4) are based on previously reported studies.33–35 The NMR analysis shows a pure SP-Nb monomer (6) with a total yield of around 20% after four reaction steps, which is close to the literature yield of 25%.33 This synthetic approach is successfully transferred to SPO-Nb (7) with a comparable total yield.Polymerization of spiropyran- and spirooxazine norbornene
Ring opening metathesis polymerization of the monomers SP-Nb (5) and SPO-Nb (7) is performed in the presence of catalyst-modified36 mesoporous allylsilica films resulting in the formation of photochromic homopolymers PSP (8) and PSPO (9) in solution and covalently attached to the mesoporous films (see below). The formation of PSP (8) and PSPO (9) homopolymers in solution is probably due to the gentle purification procedure, used in the grafting approach (see below) resulting in non-bound catalyst being still present which then leads to homopolymer generation in solution. After precipitation in methanol, solution homopolymers with a molecular weight of 6.6 × 104 g mol−1 for PSP (8) and 1.4 × 104 g mol−1 for PSPO (9) (ESI†) and a molar mass distribution with a PDI around 1.6 and 1.2 were obtained, respectively.Photochromism of SP-Nb, SPO-Nb, PSP and PSPO
The bulk homopolymers PSP and PSPO as well as the SP-Nb (5) and SPO-Nb (7) monomers are characterized concerning their photochromic behavior.Molecular spiropyran as well as molecular spirooxazine is known to interconvert from a closed form into an open merocyanine (MC) form upon irradiation with UV-light (Fig. 2). The merocyanine form shows a characteristic absorption at 568 nm in the case of SP-Nb and PSP (Fig. 2a and c) and 608 nm in the case of SPO-Nb and PSPO (Fig. 2b and d). Merocyanine can be described by various mesomeric structures, only one of which is zwitterionically charged. We included this fact as a zwitterionic and quinoidal form into the scheme in Fig. 2. The extent to which charge separation is actually present depends on the solvent polarity.10,37 It is reported in the literature that in polar solvents the uncharged mesomeric forms dominate.10,37,38 Heating or visible light irradiation results in relaxation of merocyanine into the closed SP or SPO state. In addition to photochromic behavior, SP and SPO show acidochromic39 or even mechanochromic40,41 properties (Fig. S1, ESI†) resulting in proton induced ring opening reactions. Therefore, absorption can be affected by the solution pH.42 In this study, all experiments related to photochromism were performed in DMF (NMR:DMF-d7) because precipitation during irradiation is prevented under these conditions. The shift in the absorption wavelength of spiropyrans and the corresponding merocyanines is caused by a severe change in the electronic structure upon ring opening. This difference in the electronic structure likewise induces a severe difference in the chemical shifts of spiropyrans and merocyanines.43 This can be exploited to examine the molar ratios of spiropyrans and merocyanines during irradiation with UV light or in photostationary states (PSSs). For spiropyrans, the signals of the two geminal methyl groups can be used to determine the ratios of spiropyran and merocyanine forms, as in the case of the closed spiro form both methyl groups are anisochronous, while they are isochronous for the merocyanine form. A significant difference in chemical shift between spiropyrans and merocyanines is observed allowing for individual integration.
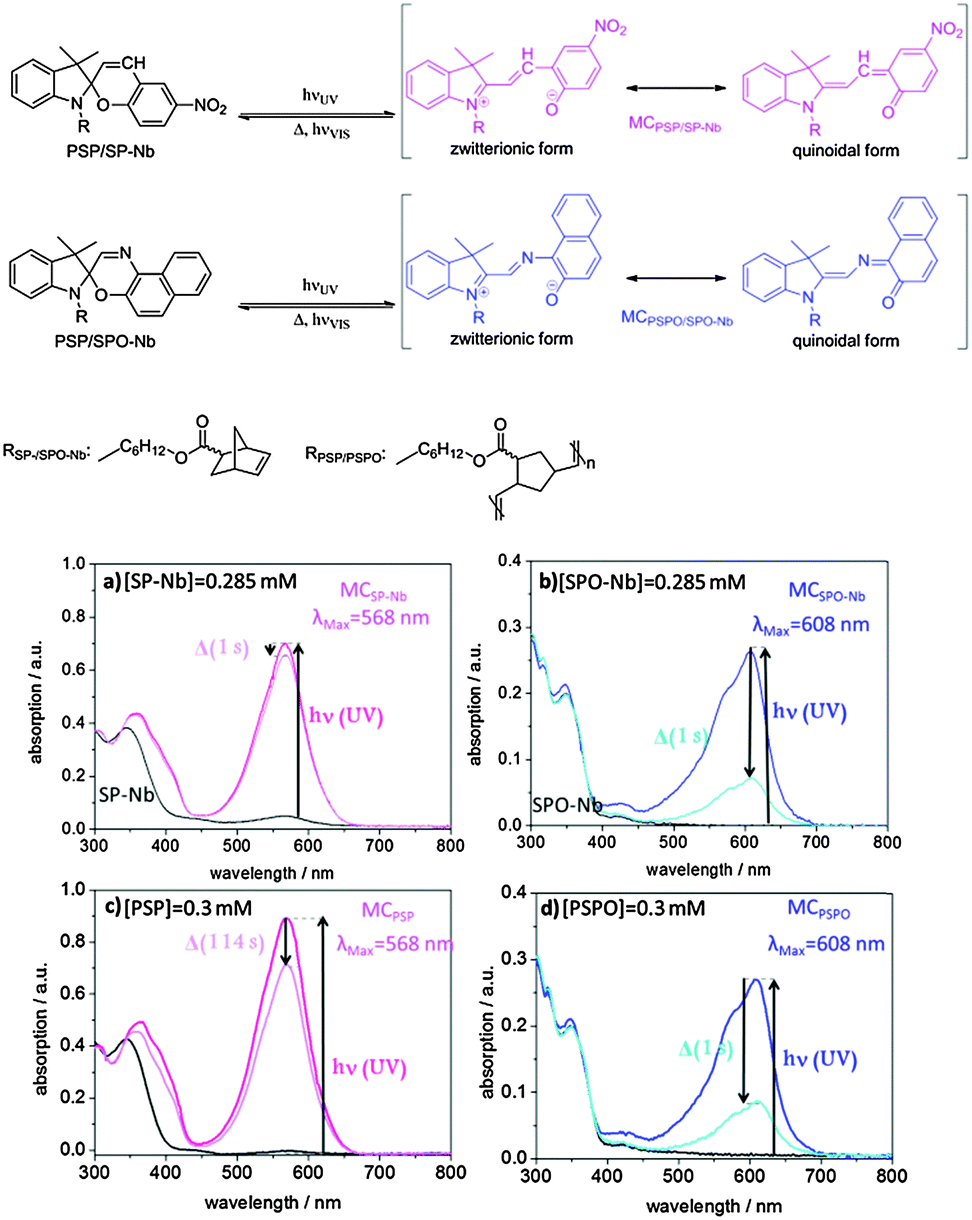 | ||
| Fig. 2 Schematic description of the photochromic mechanism in spiropyran- and spirooxazine containing species and UV/VIS-spectroscopic measurements in DMF of (a) SP-Nb, (b) SPO-Nb, (c) PSP and (d) PSPO in DMF (dry) under a nitrogen atmosphere. The absorption spectra before irradiation are shown in black. The absorption spectra directly after irradiation are shown in dark blue for SPO and in pink for SP. The absorption spectra after irradiation and in darkness at room temperature are shown in light blue and pink for SPO and SP, respectively. | ||
For a detailed investigation of the photochromic process, absorption spectra of the monomers, polymers and the mesoporous films are recorded using UV/VIS spectroscopy. Fig. 2a–d shows the UV/VIS spectra of the monomers and bulk polymers before and after UV irradiation. The absorption maxima of SP-Nb (5) and SPO-Nb (7) in DMF are at 345 and 348 nm, respectively (Fig. 2a (black), b (blue)). The absorption maxima of the corresponding merocyanine are located at 568 nm for MCSP-Nb and 608 nm for MCSPO-Nb. Comparing the UV-VIS absorption spectra of SP-Nb and PSP (Fig. 2a and c) and SPO-Nb (7) and PSPO (9) (Fig. 2b and d) before and after UV-irradiation, no significant difference can be observed between the monomer and the corresponding homopolymer. Comparing SP-Nb (5) and SPO-Nb (7) or PSP (8) and PSPO (9) the expected significant difference in time dependence is confirmed by comparing the absorption spectra directly after irradiation and one second later. This trend is not affected by generating a PSPO homopolymer.
This observation of much faster thermal relaxation kinetics for spirooxazine compounds is supported by kinetic UV/VIS measurements (Fig. 3). Under UV-irradiation with an energy density of 106.2 mJ cm−2 and a wavelength range of 320–400 nm the intensity of the merocyanine absorbance in DMF at 608 nm increases as the irradiation time increases until a photostationary state (PSS) is reached after less than 3 s in the case of the spirooxazine monomer (SPO-Nb) and polymer (PSPO). The spiropyran polymer (PSP) reaches the photostationary state after approximately 18 s, whereas it takes the monomer (SP-Nb) approximately 30 s of UV-irradiation. After excitation with UV-light, thermal relaxation and induced relaxation by irradiation with visible light were investigated. Both, excitation with UV and induced relaxation with visible light irradiation, appear to follow first order kinetics for the monomer as well as for the homopolymers in solution. This is consistent with reported observations for spiropyran-based molecules and co-polymers.15,44 Fitting the time dependent merocyanine absorption (Fig. 3a) using first order rate laws, assuming relatively low concentrations of absorbing species, yields photon-flux-dependent rate constants. Small deviations of measured data from first order rate laws are observed, the reason for which is not yet understood. More detailed kinetic investigations will be performed in the future. These kUV are observed in the following order: SPO-Nb > PSPO ≫ PSP > SP-Nb. All determined rate constants are summarized in Table 2.
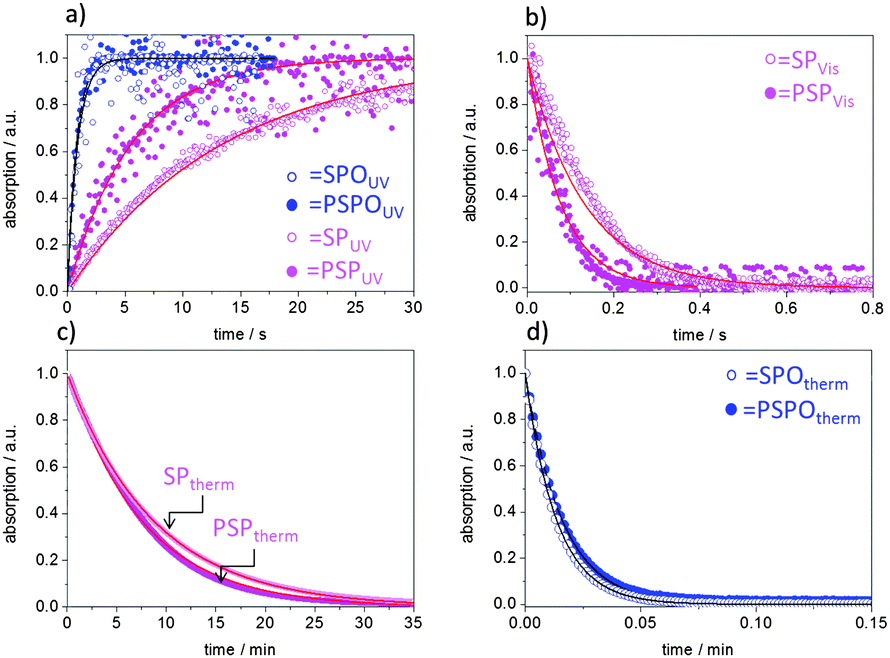 | ||
| Fig. 3 Normalized absorption by irradiation with constant flux densities of (a) UV and (b) visible light for SP-Nb (pink empty circles), SPO-Nb (blue empty circles), PSP (pink filled circles), and PSPO (blue filled circles). (c and d) The normalized absorption as a function of time at ambient temperature without irradiation for SP-Nb and PSP (c) and SPO-Nb and PSPO (d). Circles correspond to the measured values, and the solid lines represent fits according to a first order rate law. | ||
Based on the molar extinction coefficients for SP-Nb and SPO-Nb determined in deuterated DMF based on NMR experiments (ESI†), the merocyanine ratios upon UV-light irradiation, as obtained by UV-VIS spectroscopy, are calculated and summarized in Table 1. For SP-Nb 9–13% and for PSP 6–11% of all spiropyran units are converted into the open merocyanine form. Based on the UV/VIS experiments the ratio of photochromically converted functional groups is significantly higher in the case of SPO-Nb and PSPO. Around 50% of spirooxazine units are converted into the open merocyanine form. This shows a more effective photochromic response in the case of spirooxazine compared to spiropyran which most probably can be explained by the electronic states that participate in the photochromic process. In the case of spirooxazine, in contrast to spiropyran, no triplet state is involved in the photochromic reaction9,38,45 which could result in a higher efficiency of the photochromic reaction. NMR experiments reveal a comparable ratio under identical irradiation conditions in the case of SP-Nb at 300 K compared to UV-VIS experiments. NMR experiments are discussed in more detail below.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 L m mol−1, εMcSP = 2.796 × 106 L m mol−1. The calculation was done based on Lambert–Beer's-law: cMC = A/εMC × d at room temperature unless otherwise indicated. The values for PSP and PSPO are based on the monomer extinction coefficients
000 L m mol−1, εMcSP = 2.796 × 106 L m mol−1. The calculation was done based on Lambert–Beer's-law: cMC = A/εMC × d at room temperature unless otherwise indicated. The values for PSP and PSPO are based on the monomer extinction coefficients
| Sample and concentration | Absorption/a.u. | c (MC)/μM | c (MC):c(sample)/% UV/VIS | c (MC):c(sample)/% NMR |
|---|---|---|---|---|
| a Value obtained by UV/VIS spectroscopy under NMR irradiation conditions. | ||||
| SP-Nb (0.285 mM) | 0.70 | 25 | 9/12a | 13 (300 K); 17 (240 K) |
| SPO-Nb (0.285 mM) | 0.26 | 144 | 51 | 60 (240 K) |
| PSP (0.3 mM) | 0.89 | 32 | 11 | 6 (300 K) |
| PSPO (0.3 mM) | 0.27 | 150 | 50 | 17 (240 K) |
| k UV/s−1 | k therm/s−1 | k Vis/s−1 | |
|---|---|---|---|
| a Value obtained by UV/VIS spectroscopy under NMR irradiation conditions. | |||
| SP-Nb | 0.066 ± 0.009 | 0.0019 ± 0.0001 | 0.116 ± 0.009 |
| SP-Nba | 0.0035 ± 0.00005 | — | — |
| SP-Nb NMR | 0.0032 ± 0.00007 | 0.0022 ± 0.00002 | 0.0066 ± 0.00009 |
| PSP | 0.217 ± 0.011 | 0.0020 ± 0.0001 | 0.238 ± 0.022 |
| PSP NMR | 0.0049 ± 0.000122 | 0.0022 ± 0.00003 | — |
| SPO-Nb | 1.500 ± 0.043 | 1.257 ± 0.048 | — |
| PSPO | 1.341 ± 0.056 | 1.079 ± 0.041 | — |
| PSP@mesopore | 0.204 ± 0.004 | — | 0.170 ± 3 × 10−4 |
| PSP@mesopore, plasma | 0.247 ± 0.005 | 0.001 ± 0.0000005 | — |
| PSPO@mesopore | 1.080 ± 0.054 | 0.770 ± 0.003 | — |
| PSPO@mesopore, plasma | 1.195 ± 0.306 | 0.910 ± 0.0004 | — |
Upon subsequently irradiating the same solution with visible light (Fig. 3b), the merocyanine absorbance decreases monoexponentially. The decrease of merocyanine absorption in the case of SPO-Nb and PSPO at room temperature is so fast (Table 2) that an induced relaxation with VIS irradiation (λgreen = 550 nm, I = 30.6 mW cm−2) under the applied experimental conditions is only possible for SP-Nb and PSP. Zero absorption is again reached after 18 s for the SP-Nb monomer and after 12 s for the PSP polymer. The determined rate constants are summarized in Table 2. For thermal relaxation into the closed SP form under ambient conditions, a much longer time of approximately 30 min is needed to reach zero merocyanine absorption for SP-Nb and SPO-Nb (Table 2). In contrast to SP-Nb and PSP, thermal relaxation back into the closed SPO form is very fast for both SPO-Nb and PSPO. The monomers and homopolymers are converted into the colorless closed form within less than 3 s (Table 2).
NMR experiments
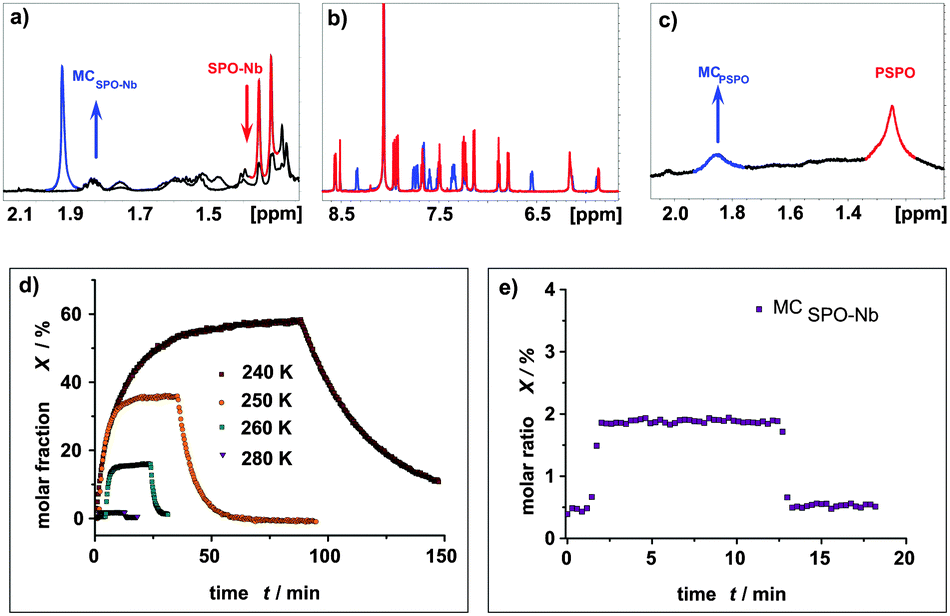
In addition, NMR measurements with in situ irradiation are performed to determine the molar ratios of the spiropyran and merocyanine forms during irradiation, photostationary states (PSSs) and relaxation for the SP-Nb, SPO-Nb systems and the corresponding polymers. Proton NMR spectra of SP-Nb as well as for the PSP polymer prior to and during irradiation with UV light (λ = 365 nm) are shown in Fig. 4a and b.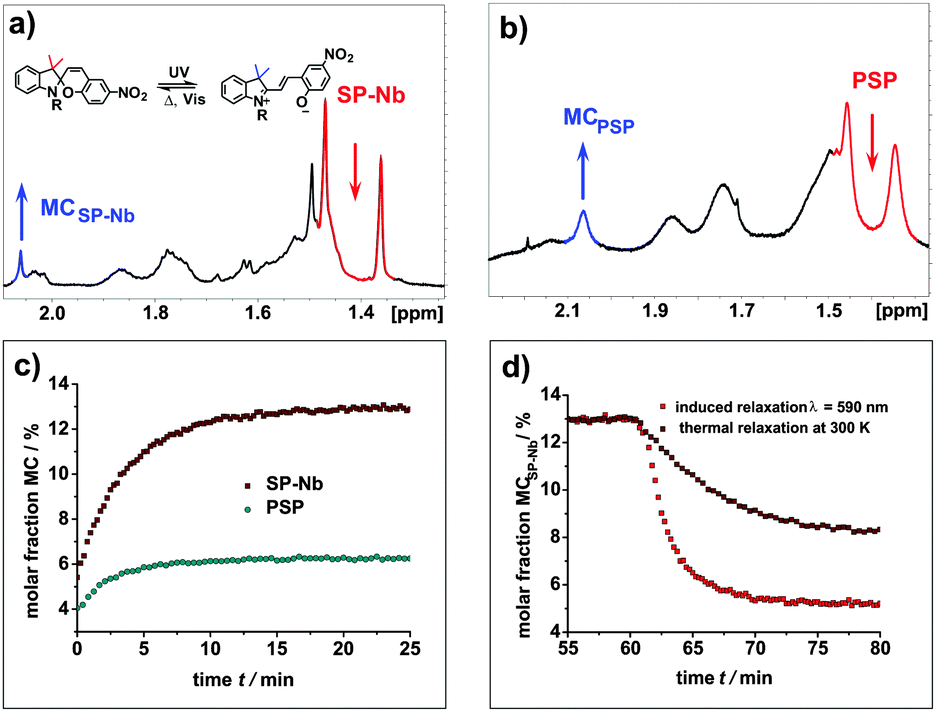 | ||
| Fig. 4 (a) 1H-NMR spectra of SP-Nb solution in DMF-d7 during irradiation with UV light. During irradiation, intensities of both methyl group resonances (red) decrease while the intensity for the methyl groups of MCSP-Nb (blue) increases. (b) 1H-NMR spectra of PSP polymer solution in DMF-d7 at 280 K during irradiation with UV light, with marked resonances of the methyl groups in SP (red) and MCSP (blue) moieties. (c) Molar fractions of MCSP-Nb and MCPSP in DMF-d7 at 300 K during irradiation with UV light and (d) thermal and induced (λ = 520 nm) relaxation of the MCSP-Nb monomer at 300 K. | ||
The molar ratios of SP-Nb and MCSP-Nb during irradiation are calculated from the integrals of the methyl groups (1.38 ppm and 2.92 ppm) obtained in a series of 1H-NMR spectra. In comparison with the previously shown UV/VIS data, a slower saturation, and thermal relaxation on the minute time scale, is observed (Fig. 4c). This can be explained by the reduced photon flux, and thus the reduced excitation probability, which is caused by the in situ irradiation setup used compared to the direct irradiation of the sample in the UV/VIS measurements.
In addition, a higher concentration of SP-Nb is necessary for the NMR experiments and thereby quenching effects could play a more important role. Nevertheless, from 320 K down to 240 K excitation of MCSP-Nb, PSSs and thermal and induced relaxation can be monitored. In PSSs, fractions of MCSP-Nb of 4.5% at 320 K, 13% at 300 K, 14% at 260 K and 17% at 240 K are observed. As in the UV/VIS measurements, monoexponential excitation, thermal relaxation and induced relaxation behaviors are observed (Table 2). For the thermal relaxation a comparable rate constant ktherm (SP-Nb and PSP) can be obtained for NMR and UV/VIS measurements. In contrast to UV/VIS measurements, in NMR experiments it becomes clear that irradiation with visible light does not give the thermal equilibrium composition; in fact the fraction of MCSP-Nb is reduced below the equilibrium value. This has already been observed for other spiropyrans46 and it shows that MCSP-Nb is forced into the closed SP form under visible light irradiation.
For the polymers much broader NMR signals are observed in 1H-NMR spectra in comparison to the SP-Nb and SPO-Nb monomers (Fig. 4b and 5c). Nevertheless, molar fractions of SP and MCSP moieties within the polymers can be estimated by integrating methyl group resonances at 1.35 ppm (SP-PSP) and 1.85 ppm (MC-PSP) for PSP and 1.25 ppm (SP-PSPO) and 1.85 ppm (MC-PSPO) for PSPO. In agreement with UV/VIS spectroscopy, monoexponential excitation and relaxation behavior is observed for the PSP polymer. In contrast to the SP-Nb monomer at 300 K, the total amount of MCPSP in PSP is only 6% under the applied experimental conditions. This is a smaller ratio than observed for the monomer under identical conditions which might indicate a hindrance of interconversion into the open merocyanine form in the case of confining SP-Nb into a polymeric structure.
 | ||
| Fig. 5 1H-NMR spectra of SPO-Nb solution in DMF-d7 measured at 240 K prior to (red) and during irradiation (blue) with UV light (λ = 365 nm). (a) Aliphatic region with a severe signal overlap of the two methyl group resonances of the SPO-Nb and resonances of the N-bound aliphatic side chain. (b) Aromatic region with well separated signals for SPO-Nb and MCSPO-Nb. (c) 1H-NMR spectrum of the PSPO polymer in DMF-d7 at 240 K during irradiation with UV light with highlighted signals of SPO (red) and MCSPO (blue). (d) Molar fractions of MCSPO-Nb at different temperatures approximated from the integrals of corresponding resonances in a series of 1H-NMR spectra. (e) Molar fraction of MCSPO-Nb at 280 K. | ||
Unfortunately, for SPO-Nb the geminal methyl groups are not suitable for integration due to a severe signal crowding caused by the signals of the aliphatic side chain (Fig. 5a). Nevertheless, extraction of molar fractions is possible based on the well separated signals in the aromatic region (Fig. 5b). At room temperature, in situ irradiation of SPO-Nb does not give an observable fraction of MCSPO-Nb with the described irradiation setup, sample concentration and NMR parameters, as the fraction of MCSPO-Nb is below the limit of detection (Fig. 5d). By stepwise reducing the temperature down to 240 K, increasing fractions of MCSPO-Nb up to 60% can be obtained (Fig. 5c). At 280 K, only a fraction of 2% MCSPO-Nb can be observed. This temperature dependence of SP/MC photochromism is more pronounced for SPO-Nb compared to SP-Nb. A possible explanation can be seen in the different electronic transitions involved in the photochromic transition which passes through a triplet state for SP but not for SPO.9,38,45,47 Furthermore, the temperature dependent first order rate constants for the thermal relaxation of SPO-Nb show a typical Arrhenius-type behavior (Fig. S5, ESI†).
Irradiating SPO-Nb with UV light leads to a PSS within one minute and a rapidly occurring thermal relaxation can be observed after switching off the UV light. These observations match those obtained from UV/VIS spectroscopy.
For the PSPO polymer (Fig. 5c) line broadening was even more pronounced than for the PSP polymer (Fig. 4b). Consequently, signals in the aromatic region are not suitable for integration and molar fraction determination. In contrast to the SP-Nb and PSP, no signal separation for the methyl groups can be observed at all. Nevertheless, a rough approximation of the molar ratios can be made based on the integration of the methyl group signals of PSPO and MCPSPO moieties (Fig. 5c). For the PSPO polymer a MCPSPO fraction of about 17% is estimated. This is a reduction of a factor of 3 compared to the SPO-Nb monomer and is in agreement with the observed MC fraction of PSP in comparison to SP-Nb.
Thus, the MC fraction is smaller for PSP and PSPO homopolymers compared to the corresponding monomers in DMF and the excitation and thermal relaxation are slower for polymers than for monomers as already observed by UV/VIS measurements at ambient temperature. While the reason for this observation remains unclear, one could speculate that the molecular rearrangement in the homopolymers compared to the monomers in solution is restricted or that quenching effects due to the spatial proximity of photochromic units in homopolymers are effective. The different ratios between UV/VIS and NMR experiments result from the different irradiation intensities between the two setups.
Mesoporous films functionalized with photochromic polymers
To graft PSP and PSPO ROMP homopolymers to a mesoporous surface allyltriethoxysilane was co-condensed into the mesoporous silica walls (Fig. 6a). Mesoporous films containing 20 mol% allyltriethoxysilane with a porosity of approximately 20 vol% according to ellipsometry and effective medium theory48 are obtained. TEM images (Fig. 6b) support the presence of a porous structure with pore sizes smaller than 6 nm corresponding to the Pluronic® F127 template. In a consecutive step the ROMP catalyst is attached to the double bond located at the mesopore wall.22 Catalyst binding is indirectly proven by stable polymer attachment and reference experiments that did not show stable polymer attachment without the presence of catalyst or surface attached allyl groups. The activated catalyst is then able to perform the ROMP polymerization of SP-Nb and SPO-Nb resulting in surface grafted PSP and PSPO. To distinguish between the polymer grafted inside the porous matrix and the one on the outer surface we used a CO2-based plasma treatment before catalyst binding. This CO2-based plasma destroys the double bonds at the external surface (ESI†). Consequently, non-plasma treated samples are functionalized within the mesopores and on the external planar surface, and the plasma treated samples are exclusively functionalized within the mesoporous matrix. Infrared spectroscopy (Fig. 4c) comparing unmodified mesoporous allylsilica (black) and PSP modified allylsilica (pink) with the solution polymer reveals the presence of the allyl groups based on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C-vibration at 1636 cm−1 and the presence of polymer visible in the carbonyl-vibration at 1728 cm−1. All spectra are normalized to the Si–O–Si band at 1080 cm−1 showing the presence of the mesoporous silica film structure after polymer functionalization. Comparing the UV-VIS absorption spectra for surface grafted PSP and PSPO (Fig. 7a–d) identical absorption bands as for the monomer and polymer in solution are observed at 568 nm for surface grafted PSP and at 608 nm for surface grafted PSPO. In addition, the much faster switching of PSPO back into the closed form compared to PSP is observed in accordance with the respective monomer and homopolymer in DMF solution.
C-vibration at 1636 cm−1 and the presence of polymer visible in the carbonyl-vibration at 1728 cm−1. All spectra are normalized to the Si–O–Si band at 1080 cm−1 showing the presence of the mesoporous silica film structure after polymer functionalization. Comparing the UV-VIS absorption spectra for surface grafted PSP and PSPO (Fig. 7a–d) identical absorption bands as for the monomer and polymer in solution are observed at 568 nm for surface grafted PSP and at 608 nm for surface grafted PSPO. In addition, the much faster switching of PSPO back into the closed form compared to PSP is observed in accordance with the respective monomer and homopolymer in DMF solution.
 | ||
| Fig. 6 (a) Schematic representation of the mesoporous allyltriethoxysilane containing silica mesoporous thin films and the binding of the ROMP catalyst. (b) Representative TEM images of mesoporous silica thin films containing 20 mol% allyltriethoxysilane. (c) Infrared spectra for mesoporous allylsilica containing 20 mol% allyltriethoxysilane (black) and PSP- (pink) or PSPO-modified mesoporous allylsilica films (blue). | ||
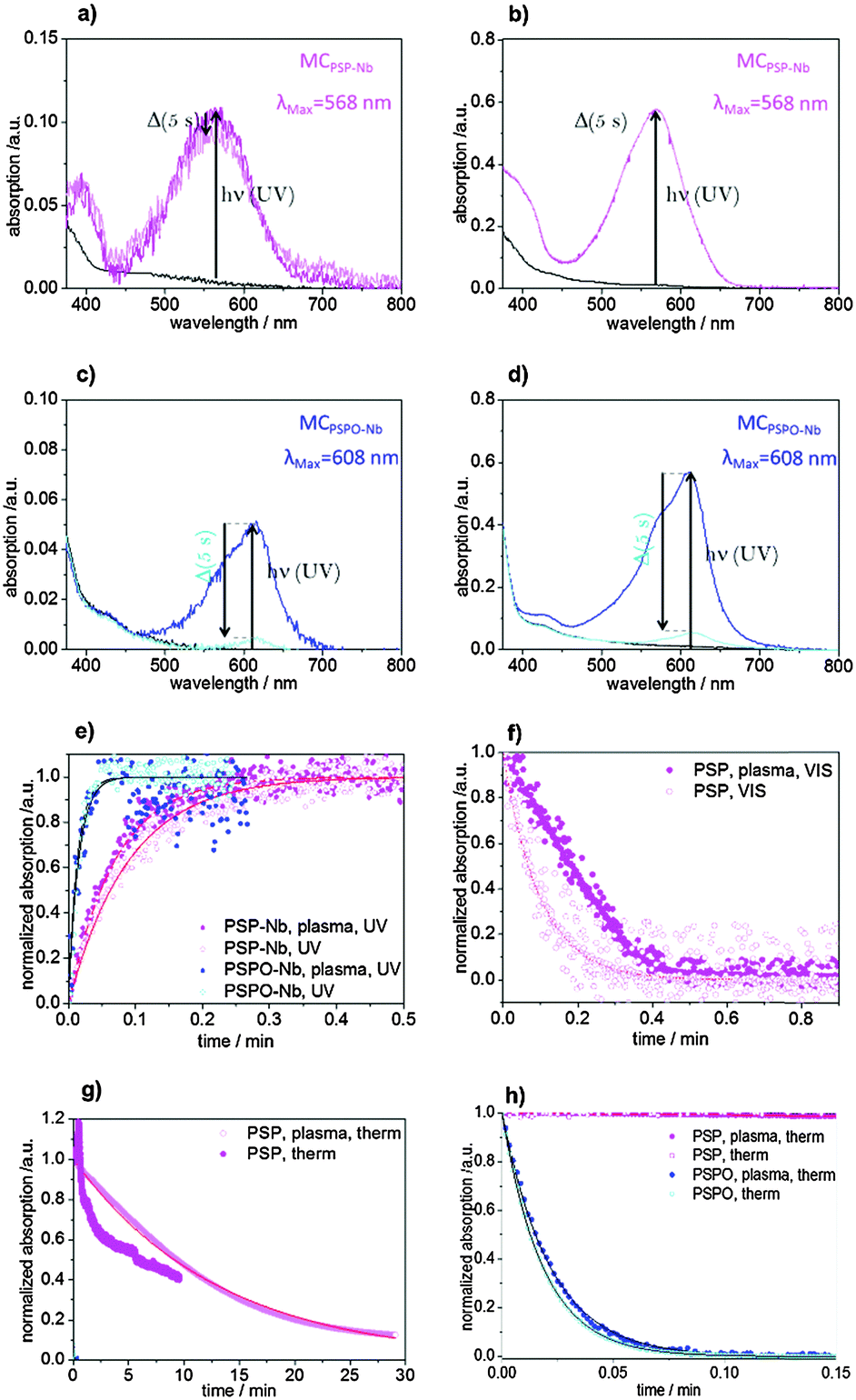 | ||
| Fig. 7 UV-VIS absorption spectra of mesoporous allylsilica films functionalized with PSP with (a) and without plasma treatment (b) and functionalized with PSPO with (c) and without (d) plasma treatment in DMF (dry) under a nitrogen atmosphere. (e–h) Normalized absorption changes with time upon irradiation with constant flux densities of UV (e) and visible light (f) and at ambient temperature without irradiation (g and f) for PSP (pink) and PSPO (blue) with (filled circles) and without (empty circles) plasma treatment. Circles correspond to the measured values, and the solid lines represent the first-order kinetic fits. The kinetic data in (e) for PSP-Nb, plasma, UV (blue filled circles) were smoothened to exclude the effect of the UV-irradiation lamp (for details see the ESI†). | ||
The time-dependent absorption increases, and decreases monoexponentially (Fig. 7e) upon UV-irradiation and thermal relaxation (Fig. 7g and h). As observed for the monomer and polymer in solution, the grafted PSPO interconverts into the closed from within a few seconds at room temperature. Thus, the visible-light induced relaxation into the closed spiro form can be measured for surface grafted PSP under the applied experimental conditions (Fig. 7f) but not for grafted PSPO. Comparing the rate constants between surface grafted PSP and PSPO with and without plasma treatment it seems that the polymer inside the mesopores (plasma treated samples) interconverts slightly faster into the open merocyanine form upon UV-irradiation compared to the polymer on the external surface (non-plasma treated samples) (Fig. 7e). This might be due to the lower molecular weight inside the pores as observed for RAFT polymerization49 or due to interaction with the silica pore walls. The determined rate constants (kUV) for grafted PSP and PSPO at plasma treated mesoporous surfaces and non-plasma treated mesoporous surfaces are summarized in Table 2. In comparison to the free polymer in solution the grafted PSPO interconversion under UV-irradiation is slower if grafted to the mesoporous surface for both plasma treated and untreated samples. For PSP on the other hand, the UV-excitation of the polymer grafted into the mesopores is slower than for the free polymer in solution, whereas the surfaces with grafted PSP at the external surface and within mesopores switch slightly faster than the free polymer in solution. Switching back from the open colored merocyanine form into the closed spiro form using visible light revealed a monoexponential time constant of kVIS 0.17 s−1 for non-plasma treated samples. This value is in between the observed time constant of the monomer (SP-Nb) and the polymer PSP in DMF solution. Time dependent absorption changes upon VIS irradiation for PSP-modified mesoporous films after plasma treatment could not be fitted using first order rate laws (Fig. 7f). They show an initial linear decrease of absorption. The thermal interconversion from the open merocyanine into the closed spiro form (Fig. 6g and h) is again much faster for grafted PSPO compared to grafted PSP. Plasma treated samples with grafted PSP show a ktherm of 0.001 s−1, whereas plasma treated surfaces with grafted PSPO show an almost 3 orders of magnitude higher ktherm (ktherm 0.91 s−1) under the applied experimental conditions. This corresponds to the observation of homopolymers in DMF solution (Fig. 3c and d).
Without plasma treatment, and thus with PSPO grafted also at the external surface, ktherm is slightly slower compared to the plasma treated films. Thus, both kUV and ktherm, seem to be faster within the mesopores compared to the external surface. Comparing the thermal relaxation from the merocyanine into the closed spiro form a slower interconversion for surface grafted polymers as for the solution polymer is observed under the applied experimental conditions. This gives a first insight into the potential effects of spatial confinement in mesopores on photochromic properties of PSP and PSPO homopolymers.
To systematically correlate switching constants to spatial confinement in pores or at planar surfaces, molecular weight, polymer density and surface chemistry would have to be comparable between solution, porous and planar surfaces. This will be part of our future studies. Nevertheless, it is clear that SP and SPO species in the form of monomers or polymers in solution or grafted to surfaces show different switching kinetics which might be beneficial for transport modulation in porous devices for example if transport velocity should be adjusted.
Photostability of PSP, PSPO, SP-Nb, and SPO-Nb
In addition to switching kinetics, switching fatigue and thus the behavior over several switching cycles are important for later potential transport control through pores. The photochromic interconversion process as well as the stability of the monomers and polymers during irradiation, the time-dependent absorption in several switching cycles for monomers and polymers in DMF as well as the surface grafted polymers in contact with DMF have been analyzed (Fig. 8).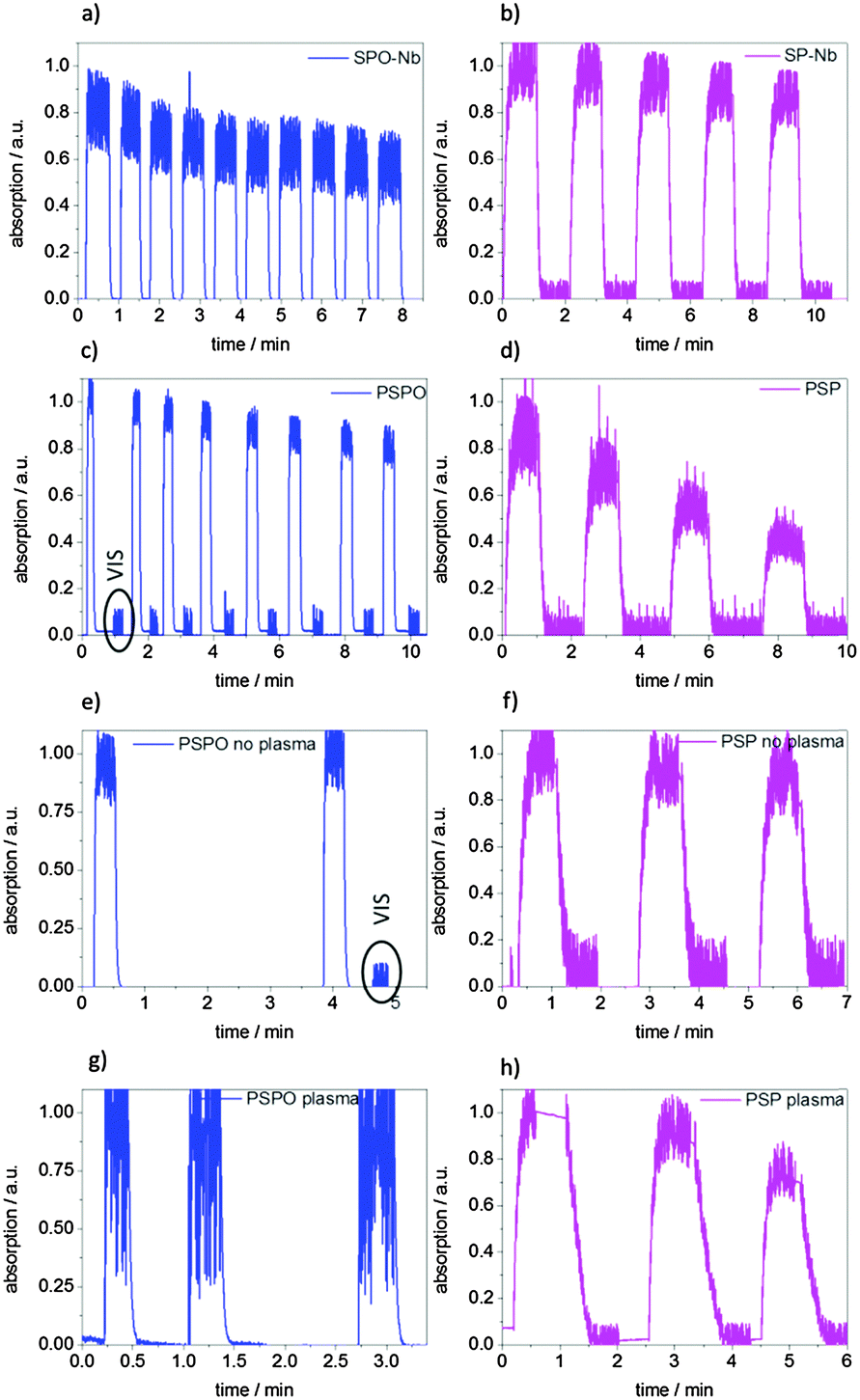 | ||
| Fig. 8 Multiple switching cycles by alternating irradiation with UV and visible light for (a) SPO-Nb, (b) SP-Nb, (c) PSPO, (d) PSP dissolved in DMF and (f) PSP grafted to mesoporous allylsilica films without and (h) with plasma treatment as well as for (e) PSPO grafted to mesoporous allyl silica films without and (g) with plasma treatment. Total irradiation times and irradiation energies are summarized in the ESI† (Table S2). | ||
The photochromic behavior is reversible for multiple cycles for SP-Nb, PSP, SPO-Nb and PSPO. However, the number of possible cycles is limited by fatigue as a result of the intense UV irradiation. In accordance with reported studies, a constant exposure to UV-light leads to decomposition and thus to a decrease in absorption (data not shown).38,50 Upon alternating UV- and visible irradiation or UV- and thermal influence, three to five UV-VIS irradiation cycles were measured for SP-Nb, PSP and surface attached PSP. Because of the fast thermal relaxation of SPO-Nb and PSPO from the merocyanine into the closed spiro form at ambient temperature, two to ten UV–thermal cycles were analyzed for SPO-Nb and PSPO. It can be observed that PSPO in contrast to SPO-Nb does not interconvert completely into the colorless closed form, visible by the merocyanine absorption that decreases to 0.02 a.u. (2%) but not to zero. Because of this a VIS irradiation step was included before subsequently irradiating with UV-light and starting the next switching cycle (Fig. 8c). All samples were irradiated until the merocyanine absorption reaches an equilibrium value (PSS). After in total 10.2 min (SPO-Nb and PSPO-bulk) UV-irradiation (corresponding to 10 irradiation cycles) SPO-Nb absorption decreases to 80% of the original value. The PSPO-Nb (Fig. 8b) shows a slightly lower loss of absorption intensity of 15% in eight switching cycles and a total UV irradiation time of 2.1 min (1.5 J cm−2). The SP-Nb absorption decreases about 15% within five switching cycles and a total irradiation time of 5 min, whereas the PSP shows a much stronger decrease of 50% within 4 cycles and a total UV-irradiation time of almost 4 min (2.8 J cm−2).
SPO-Nb and SP-Nb only show slight differences in switching stability at room temperature within 5 cycles of each 1 min UV-irradiation. Comparing the two polymers, this changes and PSPO is significantly more stable than PSP. The better photostability of PSPO in comparison to PSP is in accordance with the literature.38
It is reported that spiropyrans can undergo side reactions (e.g., oxidation reactions or reactions driven by radical formation) and that photofatigue is facilitated by dimer formation.15,38,51,52 In contrast to SP, SPO does not pass through a triplet state, which is susceptible to side reactions, but only through a singlet state upon UV-excitation.9,38 This might explain the observed better photostability of SPO-Nb and PSPO compared to SP-Nb and PSP. Besides the difference in switching kinetics this better photostability in subsequent irradiation cycles favors the use of spirooxazine derivatives instead of spiropyran derivatives in photochromic devices.
Comparing the photo-fatigue upon photochromic conversion for the surface grafted polymer (Fig. 8e and f) with the bulk polymer (Fig. 8c and d) no significant differences can be observed for up to three switching cycles corresponding to a total irradiation time of 3 min for grafted PSPO and grafted PSP. Comparing the plasma and non-plasma treated mesoporous films it seems that the grafted polymer inside the mesopores in the case of PSP (Fig. 8h) as well as for PSPO (Fig. 8g) seems to show a slightly stronger absorption decrease within 3 switching cycles than the non-plasma treated samples (Fig. 8e and f). This could be related to the proximity of photoswitchable units in the spatial confinement of mesopores which might facilitate aggregate formation and thus photofatigue.
Conclusions
Based on a synthetic strategy for spiropyran-functionalized norbornene monomers and the derived ROMP homopolymers, we have developed a synthetic strategy for spirooxazine-functionalized norbornene-based monomers and the corresponding SI-ROMP homopolymers. Spiropyran and spirooxazine substituted polynorbornenes were prepared in solution or grafted from mesoporous silica thin films using surface initiated ROMP. Based on UV/VIS- and NMR spectroscopic data, spiropyran- and spirooxazine homopolymers show very different photochromic behaviors and switching kinetics than the related molecular species. Different kinetics are expected to be useful for subsequent photochromic control of ion transport through pores. Based on NMR spectroscopy, the molar attenuated extinction coefficient (ε) was determined as well as the relative amount of species undergoing photochromic transition upon irradiation. This ratio varies between 6% and 60% under identical irradiation conditions and is significantly lower for homopolymers compared to the behavior of the respective monomer solutions. Interestingly, spiropyran- and spirooxazine homopolymers show slightly faster responses to irradiation when located inside the mesoporous matrix rather than in solution or on the external surface of a mesoporous silica film. This might be attributed to the influence of the spatial confinement on the photochromic behavior itself or on a lower molecular weight of polymers inside the pores. The latter was observed for example for iniferter-initiated polymerization by our group and can be attributed to the potential influence of spatial confinement on the polymerization. A tuning of photochromic rate constants should offer interesting possibilities in the sensing of confinement effects, control of release and ion transport characteristics. Consequently, our synthetic strategy and the understanding of switching properties in solution and inside mesopores offer a good basis towards further investigation of photochromically controlled ionic transport through mesopores. To achieve relevant transport rates the photochromic behavior of the functional polymers inside the pores and the wetting properties need to be optimized. This will constitute the next step towards light-controlled ionic transport through mesopores.Acknowledgements
We gratefully acknowledge Heike Herbert for GPC measurements and the NMR service of the Chemistry Department at the Technische Universität Darmstadt for routine NMR spectral measurements. This work was financially supported by Fonds der Chemischen Industrie, Boehringer Ingelheim Stiftung, Adolf-Messer-Stiftung, the European Research Council (ERC starting grant No. 257041) and Landes-Offensive zur Entwicklung wissenschaftlich-ökonomischer Exzellenz (LOEWE) of the State of Hessen through the research initiative “Soft Control”.References
- Z. Chu and S. Seeger, Chem. Soc. Rev., 2014, 43, 2784–2798 RSC.
- L. Chen, W. Wang, B. Su, Y. Wen, C. Li, Y. Zhou, M. Li, X. Shi, H. Du, Y. Song and L. Jiang, ACS Nano, 2014, 8, 744–751 CrossRef CAS PubMed.
- M. Tagliazucchi and I. Szleifer, Mater. Today, 2015, 18, 131–142 CrossRef CAS.
- S. Alberti, G. J. A. A. Soler-Illia and O. Azzaroni, Chem. Commun., 2015, 51, 6050–6075 RSC.
- T. Li, X. He, P. Yu and L. Mao, Electroanalysis, 2015, 27, 879–883 CrossRef CAS.
- D. J. Chung, Y. Ito and Y. Imanishi, J. Appl. Polym. Sci., 1994, 51, 2027–2033 CrossRef.
- Y. S. Park, Y. Ito and Y. Imanishi, Macromolecules, 1998, 31, 2606–2610 CrossRef CAS.
- E. Fischer and Y. Hirshberg, J. Chem. Soc., 1952, 4522–4530 CAS.
- J. Kohl-Landgraf, M. Braun, C. Öscoban, D. P. N. Goncalves, A. Heckel and J. Wachtveitl, J. Am. Chem. Soc., 2012, 134, 14070–14077 CrossRef CAS PubMed.
- G. Balasubramanian, J. Schulte, F. Müller-Plathe and M. C. Böhm, Chem. Phys. Lett., 2012, 554, 60–66 CrossRef CAS.
- X. Wang, J. Hu, G. Liu, J. Tian, H. Wang, M. Gong and S. Liu, J. Am. Chem. Soc., 2015, 137, 15262–15275 CrossRef CAS PubMed.
- J. Hu and S. Liu, Macromolecules, 2010, 43, 8315–8330 CrossRef CAS.
- J. Groten, C. Bunte and J. Rühe, Langmuir, 2012, 28, 15038–15046 CrossRef CAS PubMed.
- D. Dattilo, L. Armelao, G. Fois, G. Mistura and M. Maggini, Langmuir, 2007, 23, 12945–12950 CrossRef CAS PubMed.
- H. Schenderlein, A. Voss, R. W. Stark and M. Biesalski, Langmuir, 2013, 29, 4525–4534 CrossRef CAS PubMed.
- K. H. Fries, J. D. Driskell, G. R. Sheppard and J. Locklin, Langmuir, 2011, 27, 12253–12260 CrossRef CAS PubMed.
- J. Edahiro, K. Sumaru, Y. Tada, K. Ohi, T. Takagi, M. Kameda, T. Shinbo, T. Knamori and Y. Yoshimi, Biomacromolecules, 2005, 6, 970–974 CrossRef CAS PubMed.
- M. Natali and S. Giordani, Chem. Soc. Rev., 2012, 41, 4010–4029 RSC.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- L. Florea, D. Diamond and F. Benito-Lopez, Macromol. Mater. Eng., 2012, 297, 1148–1159 CrossRef CAS.
- T. Wu, G. Zou, J. Hu and S. Liu, Chem. Mater., 2009, 21, 3788–3798 CrossRef CAS.
- L. Florea, A. Hennart, D. Diamond and F. Benito-Lopez, Sens. Actuators, B, 2012, 175, 92–99 CrossRef CAS.
- L. Hauser, A.-C. Knall, M. Roth, G. Trimmel, M. Edler, T. Griesser and W. Kern, Monatsh. Chem., 2012, 143, 1551–1558 CrossRef CAS PubMed.
- S. Samanta and J. Locklin, Langmuir, 2008, 24, 9558–9565 CrossRef CAS PubMed.
- N. Liu, D. R. Dunphy, P. Atanassov, S. D. Bunge, Z. Chen, G. P. López, T. J. Boyle and C. J. Brinker, Nano Lett., 2004, 4, 551–554 CrossRef CAS.
- N. G. Liu, Z. Chen, D. R. Dunphy, Y. B. Jiang, R. A. Assink and C. J. Brinker, Angew. Chem., Int. Ed., 2003, 42, 1731–1734 CrossRef CAS PubMed.
- G. Wang, A. K. Bohaty, I. Zharov and H. S. White, J. Am. Chem. Soc., 2006, 128, 13553–13558 CrossRef CAS PubMed.
- M. Zhang, X. Hou, J. Wang, Y. Tian, X. Fan, J. Zhai and L. Jiang, Adv. Mater., 2012, 24, 2424–2428 CrossRef CAS PubMed.
- I. Vlassiouk, C. D. Park, S. A. Vail, D. Gust and S. Smirnov, Nano Lett., 2006, 6, 1013–1017 CrossRef CAS PubMed.
- A. Nayak, H. L. Liu and G. Belfort, Angew. Chem., Int. Ed., 2006, 45, 4094–4098 CrossRef CAS PubMed.
- C. Feldmeier, H. Bartling, E. Riedle and R. M. Gschwind, J. Magn. Reson., 2013, 232, 39–44 CrossRef CAS PubMed.
- F. M. Raymo, S. Giordani, A. J. P. White and D. J. Williams, J. Org. Chem., 2003, 68, 4158–4169 CrossRef CAS PubMed.
- S.-R. Keum, S.-M. Ahn, S. J. Roh and S.-Y. Ma, Dyes Pigm., 2010, 86, 74–80 CrossRef CAS.
- F. M. Raymo and S. Giordani, J. Am. Chem. Soc., 2001, 123, 4651–4652 CrossRef CAS PubMed.
- J. M. Galvin and G. B. Schuster, Supramol. Sci., 1998, 5, 89–100 CrossRef CAS.
- L. H. Peeck, R. D. Savka and H. Plenio, Chem. – Eur. J., 2012, 18, 12845–12853 CrossRef CAS PubMed.
- D. G. Patel, M. M. Paquette, R. A. Kopelman, W. Kaminsky, M. J. Ferguson and N. L. Frank, J. Am. Chem. Soc., 2010, 132, 12568–12586 CrossRef CAS PubMed.
- J. Biteau, F. Chaput and J.-P. Boilot, J. Phys. Chem., 1996, 100, 9024–9031 CrossRef CAS.
- S. Wan, Y. Zheng, J. Shen, W. Yang and M. Yin, ACS Appl. Mater. Interfaces, 2014, 6, 19515–19519 CAS.
- A. D. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martínez, S. R. Whte, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef PubMed.
- L. Metzler, T. Reichenbach, O. Brügner, H. Komber, F. Lombeck, S. Müllers, R. Hanselmann, H. Hillebrecht, M. Walter and M. Sommer, Polym. Chem., 2015, 6, 3694–3707 RSC.
- G. Mistlberger, M. Pawlak, E. Bakker and I. Klimant, Chem. Commun., 2015, 51, 4172–4175 RSC.
- J. B. J. Flannery, J. Am. Chem. Soc., 1968, 90, 5660–5671 CrossRef CAS.
- S. Stitzel, R. Byrne and D. Diamond, J. Mater. Sci., 2006, 41, 5841–5844 CrossRef CAS.
- A. K. Chibisov and H. Görner, J. Phys. Chem. A, 1999, 103, 5211–5216 CrossRef CAS.
- C. Wolff, J. Kind, H. Schenderlein, H. Bartling, C. Feldmeier, R. M. Gschwind, M. Biesalski and C. M. Thiele, Magn. Reson. Chem., 2016 DOI:10.1002/mrc.4403.
- N. Tamai and H. Masuhara, Chem. Phys. Lett., 1992, 191, 189–194 CrossRef CAS.
- C. Boissiere, D. Grosso, S. Lepoutre, L. Nicole, A. B. Bruneau and C. Sanchez, Langmuir, 2005, 21, 12362–12371 CrossRef CAS PubMed.
- L. Silies, H. Didzoleit, C. Hess, B. Stühn and A. Andrieu-Brunsen, Chem. Mater., 2015, 27, 1971–1981 CrossRef CAS.
- G. Baillet, G. Giusti and R. Guglielmetti, Bull. Chem. Soc. Jpn., 1995, 68, 1220–1225 CrossRef CAS.
- G. Such, R. Evans, L. Yee and T. Davis, J. Macromol. Sci., Polym. Rev., 2003, 43, 547–579 CrossRef.
- A. Radu, R. Byrne, N. Alhashimy, M. Fusaro, S. Scarmagnani and D. Diamond, J. Photochem. Photobiol., A, 2009, 206, 109–115 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tc04054j |
| This journal is © The Royal Society of Chemistry 2016 |