
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn immunological electrospun scaffold for tumor cell killing and healthy tissue regeneration†
Xingzhi
Liu‡
a,
Hongbo
Zhang‡
bcdef,
Ruoyu
Cheng‡
a,
Yanzheng
Gu
a,
Yin
Yin
d,
Zhiyong
Sun
a,
Guoqing
Pan
a,
Zhongbin
Deng
i,
Huilin
Yang
a,
Lianfu
Deng
bc,
Wenguo
Cui
 *bc,
Hélder A.
Santos
*gh and
Qin
Shi
*aj
*bc,
Hélder A.
Santos
*gh and
Qin
Shi
*aj
aDepartment of Orthopedics, The First Affiliated Hospital of Soochow University, Orthopedic Institute, Soochow University, 708 Renmin Road, Suzhou, Jiangsu 215006, P. R. China. E-mail: shiqin@suda.edu.cn
bShanghai Key Laboratory for Prevention and Treatment of Bone and Joint Diseases, Shanghai Institute of Traumatology and Orthopaedics, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, 197 Ruijin 2nd Road, Shanghai 200025, P. R. China. E-mail: wgcui80@hotmail.com
cState Key Laboratory of Molecular Engineering of Polymers, Fudan University, No. 220 Handan Road, Shanghai 200433, P. R. China
dAnimal Experimental Center, Soochow University, 99 Renai Road, Suzhou, Jiangsu 215023, P. R. China
eDepartment of Pharmaceutical Sciences Laboratory, Åbo Akademi University, FI-00520, Finland
fTurku Center for Biotechnology, University of Turku and Åbo Akademi University, FI-00520, Finland
gDrug Research Program, Division of Pharmaceutical Chemistry and Technology, Faculty of Pharmacy, University of Helsinki, Helsinki FI-00014, Finland
hHelsinki Institute of Life Science (HiLIFE), University of Helsinki, Helsinki FI-00014, Finland. E-mail: helder.santos@helsinki.fi
iDepartment of Medicine, James Graham Brown Cancer Center, University of Louisville, 505 South Hancock Street, Louisville, KY 40202, USA
jKey Laboratory of Stem Cells and Biomedical Materials of Jiangsu Province and Chinese Ministry of Science and Technology, 199 Renai Rd, Suzhou, 215123, China
First published on 16th August 2018
Abstract
Antibody-based cancer immune therapy has attracted lots of research interest in recent years; however, it is greatly limited by the easy distribution and burst release of antibodies. In addition, after the clearance of the tissue, healthy tissue regeneration is another challenge for cancer treatment. Herein, we have developed a specific immunological tissue engineering scaffold using the agonistic mouse anti-human CD40 antibody (CD40mAb) incorporated into poly(L-lactide) (PLLA) electrospun fibers through the dopamine (PDA) motif (PLLA-PDA-CD40mAb). CD40mAb is successfully incorporated onto the surface of the electrospun fibrous scaffold, which is proved by immunofluorescence staining, and the PLLA-PDA-CD40mAb scaffold has an anti-tumor effect by locally releasing CD40mAb. Therefore, this immunological electrospun scaffold has very good potential to be developed as a powerful tool for localized tumor treatment, and this is the first to be reported in this area.
Conceptual insightsThis paper reports an immunological electrospun scaffold for tumor cell killing and healthy tissue regeneration. Herein, we have developed a specific immunological tissue engineering scaffold using the agonistic mouse anti-human CD40 antibody (CD40mAb) incorporated into a poly(L-lactide) (PLLA) membrane through the dopamine (PDA) motif (PLLA-PDA-CD40mAb). CD40mAb was successfully incorporated onto the surface of the fibrous scaffold, which was proved by immunofluorescence staining, and the PLLA-PDA-CD40mAb scaffold had an anti-tumor effect by locally releasing CD40mAb. The developed CD40mAb-functionalized electrospun PLLA-PDA fibrous scaffolds are multifunctional, which are able to specifically eliminate CD40-expressed tumor cells by recognizing CD40 receptors and regulating the expression levels of bcl-2 and bax. The scaffolds promote human dendritic cell (DC) maturation to secrete IL-12 and IFN-γ through CD40mAb cross-linking, thus activating specific immune response, and annihilate tumor cells indirectly. The CD40mAb-functionalized electrospun PLLA-PDA fibrous scaffolds are easy to fabricate, biocompatible, and multifunctional in selectively inducing tumor cell apoptosis, activating immune response and promoting healthy tissue regeneration. Therefore, these immunological electrospun fibers have very good potential to be developed as a powerful tool for localized tumor treatment and can provide sustained drug release to eliminate cancerous tissues, while restricting the drug release to avoid damage to normal tissues. |
The recurrence of malignancies at the primary site is the major cause of tumor-related deaths worldwide.1 In order to avoid the recurrence of post-surgical tumors, chemotherapy, radiotherapy or biotherapy is followed in the clinic to clear the remaining asymptomatic tumor tissues completely. Among all kinds of biotherapies, antibody-mediated immunotherapy displays enormous potential in malignancy therapy because of its high specificity and efficiency. It can drive immune cells and cytokines to eradicate tumor cells2 or directly induce tumor cell death by recognizing and interacting with tumor specific receptors.3 Compared with traditional chemotherapy or radiotherapy, antibody-based biotherapy has high specificity, low side effects, predictability, high patient compliance, and so on.4–7 However, when the antibody is delivered in vivo, it is difficult to maintain the therapeutic concentration at the local site on account of the short antibody half-life time and rapid antibody dissolution.8,9 Thus, enhancing the accumulation of antibodies at the primary tumor site or the tumor resection cavity is of great clinical importance.
In the recent decade, as a drug-eluting system, electrospun nanofibrous scaffolds are widely used in tissue engineering and regeneration medicine.10 Natural protein, synthetic polymer or hybrid material based electrospun scaffolds, such as collagen, gelatin, poly(L-lactide) (PLLA), poly(lactic-co-glycolic acid) (PLGA) and poly(ε-caprolactone) (PCL), are characterized by high surface areas and three-dimensional (3D) micro–nano structures.11 Furthermore, their unique features of good biocompatibility and biodegradability make them suitable for potential application in the clinic. However, the bioinert properties of these biomaterials significantly limit their utilization. Thus, it is of great necessity to develop bio-inspired electrospun scaffolds to replace, remodel or support damaged tissues in regenerative medicine.12 Direct electrospinning or coaxial electrospinning are common approaches to load drugs, growth factors and proteins into the scaffolds.13 However, the processing of protein integration and scaffold sterilization easily inactivated the antibodies attached to electrospun scaffolds.14,15 Recently, integrating proteins into scaffold surfaces using specific motifs has become a new strategy for bio-functioning electrospun scaffolds. The anti-CD31 antibody was immobilized onto electrospun poly(ε-caprolactone) scaffolds with hydrophobins to promote the attachment and retention of endothelial cells.16 The anti-CD34 antibody was integrated into a polyurethane (PU) surface via amine-terminated polyethylene glycol (PEG) to inhibit intimal hyperplasia in cardiovascular applications.17 Herein, to maintain the activities of antibodies on the fabricated fibers, it is of great necessity to conjugate the antibodies using specific motifs.
The polydopamine (PDA) motif arouses interest in researchers owing to its special characteristics.18 PDA is inspired from Mytilus edulis foot protein 5 (Mefp-5) in the mussel, which is rich in 3,4-dihydroxy-l-phenylalanine and lysine amino acids. It can adhere to various substrates including metallic, inorganic, and organic materials via spontaneous polymerization in mild aqueous solution.19 PDA is reactive to nucleophilic functional groups, such as amino and thiols via the catechol groups. Thus, PDA is a good candidate for intermediating antibodies to immobilize onto the surfaces of scaffolds.
CD40 is a type I membrane glycoprotein normally expressed in B cells and dendritic cells (DCs).20 As immuomodulators, CD40 and the CD40 ligand (CD40L) provide the costimulatory signal in a broad spectrum of systemic immune and inflammatory responses physiologically, including DC maturation, macrophage cytokine secretion, T-cell-dependent cellular immunity and humoral response.21–23 In biotherapy studies of antitumor response, the anti-CD40 antibody (CD40mAb) combined with the Toll-like receptor 3 (TLR3) ligand can restore DC-mediated immunity to break up the tumor suppression induced by morphine.24 CD40 signaling also primes DCs to enhance T cell response against Lewis lung carcinoma in murines.25 In addition, high expression of CD40 has been found in B cell neoplasms and many kinds of solid malignancies, suggesting that CD40 signaling is involved in the development of malignancies.26 Contact-dependent CD40 cross-linking by agonistic CD40mAb can inhibit proliferation of the myeloma cell line XG-227 and promotes the apoptosis of the gastric cancer cell line AGS.28 Remarkably, CD40 signaling can enhance tumor cell sensitivity to chemotherapy and radiotherapy.29
In the present study, we grafted agonist rat anti-human CD40mAb onto the surfaces of PLLA electrospun fibrous scaffolds through PDA motif-mediated crosslinking reaction under mild aqueous conditions. The successful incorporation of CD40mAb onto the PLLA-PDA scaffolds was demonstrated by immunofluorescence staining. We also explored whether the CD40mAb-modified scaffolds induced antitumor therapy by directly triggering CD40-expressed tumor cell apoptosis and indirectly through immunomodulation by stimulating DC activation in vitro. In addition, we also investigated the effect of the scaffolds’ structure on supporting the fibroblast proliferation (Scheme 1). At the end, we examined the anti-tumor efficacy of CD40mAb released from the scaffolds in a murine model of breast cancer.
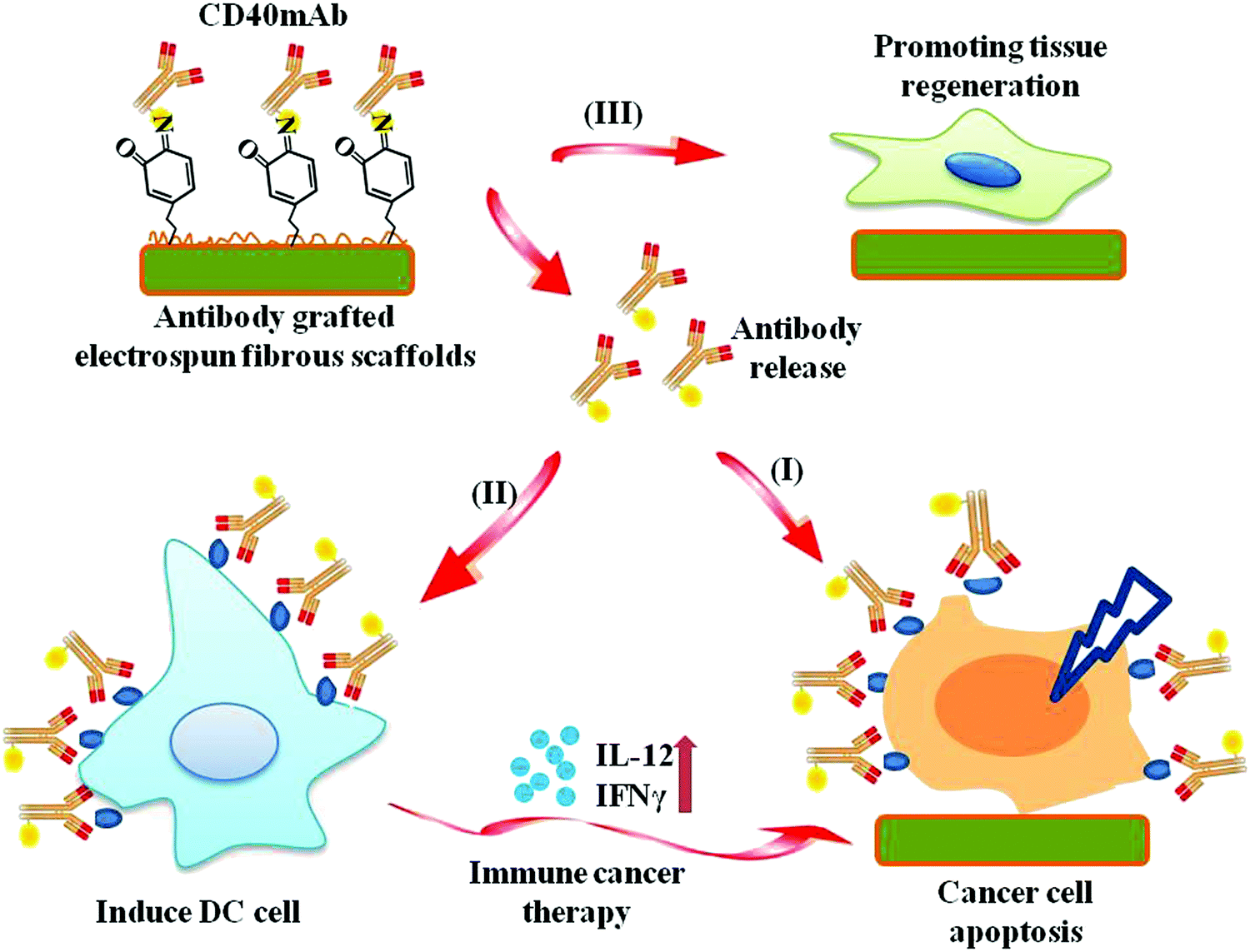 | ||
| Scheme 1 The pipeline of CD40mAb-grafted electrospun fibrous scaffolds for anti-cancer therapy and tissue regeneration. (I) Released CD40mAb directly induces tumor cell apoptosis. (II) Released CD40mAb activates dendritic cells (DCs) to initiate adaptive immune response to kill tumor cells indirectly. (III) PLLA-PDA scaffolds provide physical support for healthy cell regeneration. | ||
PLLA membranes were prepared via electrospinning as previously described.30 To prepare PLLA-PDA-mAb membranes, the scaffolds were immersed into a PDA solution for 24 h for PDA coating, and subsequently PLLA-PDA scaffolds were incorporated with agonist rat anti-human CD40mAb (PLLA-PDA-CD40mAb) or rat immunoglobulin G (IgG) (PLLA-PDA-IgG) (Fig. 1a). As shown in Fig. 1b, the fibrous morphologies of PLLA and PLLA conjugated with PDA (PLLA-PDA), PDA-IgG (PLLA-PDA-IgG), and PDA-CD40mAb (PLLA-PDA-CD40mAb) had smooth surfaces and showed no significant difference in monodispersed fibrous diameters (0.94 ± 0.21, 0.91 ± 0.36, 0.91 ± 0.17, and 0.93 ± 0.23 μm, respectively). Thus, PDA and antibody conjugation did not alter the diameters of the scaffolds.
 | ||
| Fig. 1 Characterization of the PLLA-PDA-CD40mAb scaffold. (a) The cartoon of PLLA electrospun fibrous scaffolds with the antibody grafted through PDA. (b) The morphologies of the different scaffolds under a SEM. (The insets present contact angles, which represent the polar properties of the scaffolds.) (c) XPS. (d) Stress–strain curves. (e) WCA (compared with the PLLA group *p < 0.05). | ||
Next, we examined if PDA and CD40mAb were successfully conjugated on the scaffold via X-ray photoelectron spectroscopy (XPS) (Fig. 1c). Carbon (C1s, 284.5 eV) and oxygen (O1s, 531.0 eV) peaks were observed in the PLLA electrospun fibrous scaffolds. However, a clear nitrogen peak (N1s, 399.8 eV) appeared in the electrospun fibrous scaffolds of PLLA-PDA, PLLA-PDA-IgG and PLLA-PDA-CD40mAb, suggesting the amide bonds of PDA, IgG and CD40mAb. Moreover, the areas of the N1s peaks increased after grafting with IgG or CD40mAb, indicating that the antibodies were successfully conjugated into the PLLA-PDA scaffolds.
One important parameter of the scaffolds for biomedical applications is their mechanical properties. Surface modification may change the physico-chemical properties of the fibrous scaffolds and disabled their mechanical intensity in biomedical applications. Thus, we tested the effect of antibody conjugation on the mechanical properties of the scaffolds. Fig. 1d shows the stress–strain curves of the PLLA scaffolds with or without PDA and CD40mAb conjugation, respectively. The tensile strengths were 4.3 ± 0.32 MPa, 4.2 ± 0.12 MPa, 4.41 ± 0.56 MPa and 4.51 ± 0.33 MPa, with elongation at break rates of 47.17 ± 0.23%, 46.17 ± 0.67%, 46.05 ± 0.45% and 45.71 ± 0.43% in PLLA, PLLA-PDA, PLLA-PDA-IgG and PLLA-PDA-CD40mAb, respectively. Therefore, the conjugation of PDA and the antibodies did not affect the tensile strength. Taken together, the results demonstrated that the coating approach via the PDA motif to absorb the antibodies well retained the original properties of the fibrous scaffolds.
The stability of the PLLA-PDA-CD40mA scaffolds was in vitro investigated by immersing the PLLA-PDA-CD40mA scaffolds into cell culture medium for 3 and 7 d at 37 °C and checking the microstructures of the scaffolds by SEM. As shown in Fig. S1 (ESI†), the microstructures of the scaffolds were stable. Moreover, modification of conjugated PDA and the antibody may also change the surface properties of the PLLA.14,15,17 To characterize the surface properties of the antibody-modified fibers, the wettabilities of the electrospun scaffolds were measured using a water contact angle (WCA) test. As shown in Fig. 1e, the WCA of the PLLA scaffold was 122.07 ± 2.66°, while the WCAs of the PLLA-PDA, PLLA-PDA-IgG and PLLA-PDA-CD40mAb scaffolds were close to 0°. It was indicative that the PLLA electrospun fibrous scaffolds were hydrophobic, but through PDA motif conjugation the surfaces of the fibrous scaffolds fully changed into hydrophilic. After modification, the color of the fibers turned from white to grey (Fig. S2, ESI†). Thus, the successful immobilization of PDA and CD40mAb can overcome the shortcomings of the extremely hydrophobic and non-bioactive surfaces of the PLLA electrospun fibrous scaffolds and improve cell attachment and recruitment.
Next, we proved the successful incorporation of the antibodies onto the surfaces of the PLLA scaffolds by immunofluorescence staining with the antibodies conjugated with fluorescein isothiocyanate (CD40mAb-FITC or IgG-FITC). As expected, no green fluorescent signal was detected in the PLLA and PLLA-PDA scaffolds, and neither in the PLLA scaffolds mixed with purified CD40mAb without the FITC label (Fig. S3, ESI†). In contrast, a fluorescent signal was observed in the PLLA-PDA electrospun fibrous scaffolds conjugated with FITC-labeled antibodies, demonstrating the successful immobilization of IgG or CD40mAb onto the surfaces of the scaffolds.
In comparison to a flat membrane, an electrospun scaffold has an advantage of an expanded surface because of its fibrous structure. Accordingly, the density of CD40mAb attached to the PLLA-PDA electrospun fibrous membrane is 1.49 ± 0.11 μg (cm2)−1, as calculated using eqn (1).
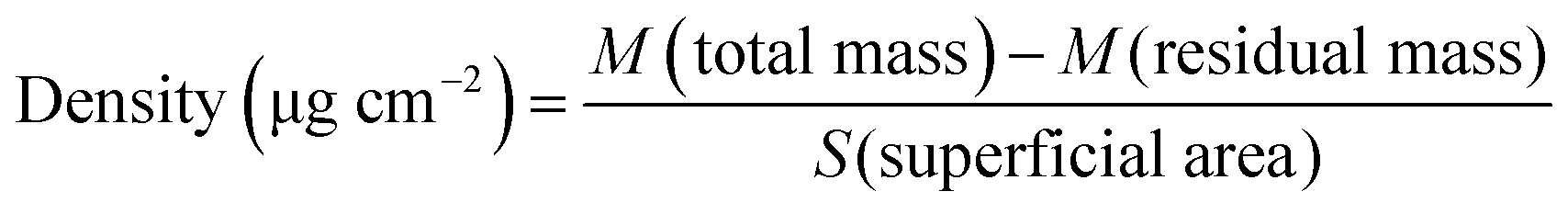 | (1) |
CD40mAb-based therapy develops anti-tumor activity via multiple mechanisms against a broad range of lymphoid and solid malignancies.31 The human breast cancer cell line MDA-MB-231 highly expresses CD40, which can be induced to apoptosis by CD40 ligation with CD40L.32 With CD40 signalling activation, MDA-MB-231 cells up-regulate the expression of the pro-apoptotic Bax protein and down-regulate the expression of the anti-apoptotic Bcl-2 protein; thus the increased Bax/Bcl-2 ratio determines the cell fate to apoptosis. The agonistic anti-CD40 antibody has been verified to induce tumor cell apoptosis and DC maturation in vitro.33 To further explore its biological function, firstly, we evaluated the proliferative effect of the PLLA-PDA-CD40mAb electrospun scaffolds on MDA-MB-231 cells. Different fibrous scaffolds were incubated in the cell culture medium for 24 h and the mediums were then harvested to culture MDA-MB-231 cells for 3 d. Cell proliferation was determined using a CCK-8 kit. Cells cultured in the release buffer from PLLA and PLLA-PDA-IgG show no statistically significant difference in optical density (OD) compared with the cells cultured in control medium (OD: 0.47 ± 0.03 and 0.47 ± 0.01 vs. 0.47 ± 0.03) (Fig. 2a). PLLA displayed good biological compatibility and PDA combined with isotype IgG had no effect on the proliferation of tumor cells. In contrast, the OD value of the MDA-MB-231 cells cultured with the release buffer from PLLA-PDA-CD40mAb decreased significantly, suggesting that the released CD40mAb in the buffer suppressed the tumor cell proliferation. To investigate whether CD40mAb could be released from the scaffolds constantly and the CD40mAb activity could last for a long period of time, MDA-MB-231 cells were cultured in the release buffer of PLLA-PDA-CD40mAb collected at 12 h, 24 h, and 48 h for 3 d. As a result, no clear time-dependent pattern was observed, which further proved that the CD40mAb released from PLLA-PDA-CD40mAb maintained its activity in the testing period (Fig. S6, ESI†).
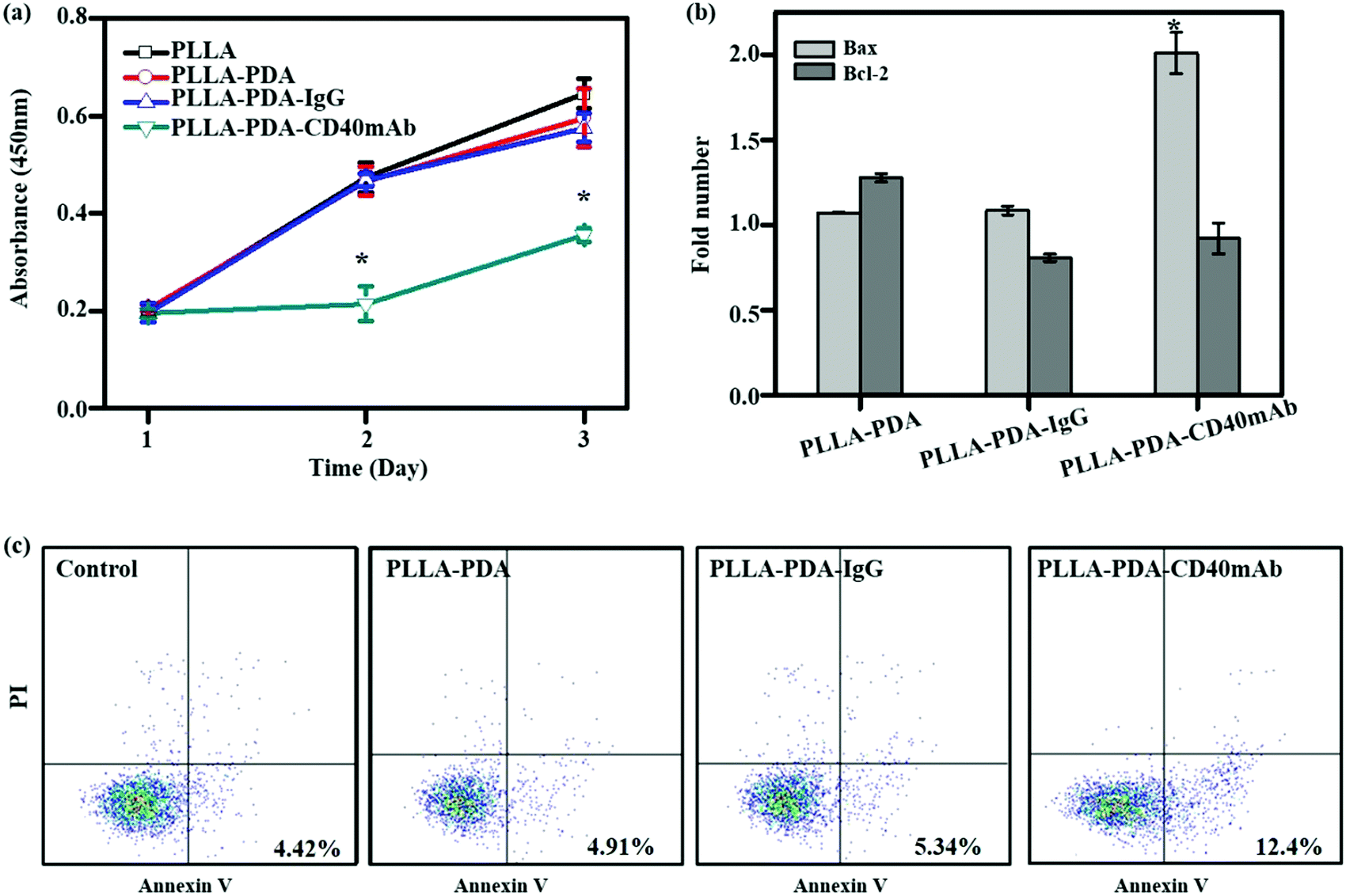 | ||
| Fig. 2 The anti-proliferation effect of CD40mAb released from PLLA-PDA-CD40mAb scaffolds for 24 h towards MDA-MB-231 cells. (a) Cell viability of MDA-MB-231. (b) The relative gene expressions of Bax and Bcl-2 of MDA-MB-231 cells (data are represented as fold changes normalized by cells cultured in cell medium). (c) Flow cytometry assay (FCA) for MDA-MB-231 cell apoptosis. Control: cells seeding onto the plate (compared with the PLLA group; *p < 0.05). | ||
Then, we seeded MDA-MB-231 cells onto the different scaffolds directly for 24 h and performed live/dead staining assay to evaluate the function of CD40mAb grafted onto the PLLA scaffolds. With a fluorescence microscope, the images displayed more dead cells (in red) on the PLLA-PDA-CD40mAb scaffolds than the ones on the PLLA-PDA and PLLA-PDA-IgG scaffolds (Fig. S7, ESI†). The SEM showed that, after 24 h, the cells grown on the PLLA-PDA-CD40mAb scaffolds became round, while the cells on the PLLA-PDA and PLLA-PDA-IgG scaffolds had branches (Fig. S8, ESI†). The data confirm that the PLLA-PDA-CD40mAb scaffold can efficiently induce MDA-MB-231 cell apoptosis and the mussel-inspired PDA coating attributes to support the mild conjugation of CD40mAb with the activity maintained for few days.
To identify the early apoptosis of MDA-MB-231 cells cultured in the release buffer (24 h released, released concentration, 1 μg ml−1), flow cytometry assay (FCA) was performed with an apoptosis staining kit. The results showed that the apoptosis of cells in the release buffers of PLLA-PDA and PLLA-PDA-IgG were similar to that of the cells with control medium after 24 h culture. These data further confirm that the PLLA and PLLA-PDA-IgG scaffolds have good compatibility. The number of apoptotic cells was clearly higher in the release buffer of the PLLA-PDA-CD40mAb scaffolds (Fig. 2c), which is consistent with the results of the CCK-8 assay, indicating that the PLLA-PDA-CD40mAb scaffolds can directly induce cancer cell apoptosis.
Bcl-2 (B-cell lymphoma 2) and Bax are critical to development, tissue homeostasis, and protection against pathogens. Bcl-2 promotes cell survival by inhibiting adaptors needed for the activation of the proteases (caspases) that dismantle the cells.34 Bax, in contrast, accelerates apoptosis by activating caspases. In our system, CD40mAb released from the fibrous scaffolds interacted with CD40 antigens on MDA-MB-231 cells, which is an initial step of CD40-mediated signal cascades and activation of caspase-dependent cell death.31 qRT-PCR results showed that the expression of Bax was statistically higher in the cells cultured with released CD40mAb than the ones with released IgG. Meanwhile, the expressions of Bcl-2 were similar in all groups (Fig. 2b). Thus it further proves that through released CD40mAb, the PLLA-PDA-CD40mAb scaffolds have a capability to “kill” tumor cells.
It is well accepted that the CD40/CD40L signal constitutes a critical co-stimulatory pathway to facilitate antigen presenting cell (APC) activation and cytotoxic T lymphocyte-mediated immunity against cancer.35 Next we further explored its co-stimulatory effect on the maturation of DCs. As an important source of antigen-presenting cells (APCs), immature DCs have a high capability of sampling the surrounding environment through endocytosis. This process promotes the maturation of DCs, which greatly enhances their ability to initiate adaptive immune response and activate antigen responsive T cells to kill target cells by perforins and granzymes.36,37 Moreover, matured DCs themselves express and stimulate other cells to express cytokines, such as IL-12 and IFN-γ, to activate nonspecific immune response to eliminate tumor cells.38,39 Thus, local application of CD40mAb via PLLA elctrospun scaffolds could play an important role in cancer therapy in the region of interest. In this study, we collected the released buffer to stimulate DC maturation in vitro. Adherent peripheral blood mononuclear cells (PBMCs) from healthy donors were incubated with free CD40mAb (1 μg ml−1) or the released buffers from the different PLLA electrospun membranes. As positive control, PBMCs were induced for mature DCs with a GM-CSF and IL-4 cocktail. After 10 d induction of DCs, all the suspended cells were photographed and harvested to perform FCA. As previous research reported,33 PBMCs with cytokines (GM-CSF and IL-4 treated) were expended and formed a dendritic shape (Fig. S9, ESI†), and had a tendency to cluster together (Fig. 3a). The expression of mature DC markers, CD11c and HLA-DR, of these cells increased distinctly (Fig. 3b). However, the cells in the released buffers from PLLA-PDA and PLLA-PDA-IgG retained the original shape, small and scattered, as the untreated control group. Remarkably, the cells cultured with both PLLA-PDA-CD40mAb released buffer and free CD40mAb displayed similar morphologies to that of mature DCs induced by the GM-CSF and IL-4 cocktail (Fig. 3a and b)40 The expression of HLA-DR in the cells with the released buffer from the PLLA-PDA-CD40mAb scaffold was similar to that of purified CD40mAb; however, it was much higher than those with the released buffers of PLLA-PDA and PLLA-PDA-IgG (Table S2, ESI†). Therefore, our results showed that the PLLA-PDA-CD40mAb scaffolds had the capability to promote the maturation of DCs by providing CD40 signaling. We speculate that matured DCs can induce the expression of cellular signals and kill tumor cells by T cell-mediated immune response. Since the activation of DCs is an important process for antitumor immunity, we also expect that the PLLA-PDA-CD40mAb platform promotes DC maturation to secrete IL-12 and IFN-γ through CD40 cross-linking, which has been a well-demonstrated pathway to annihilate cancer cells in vivo.40
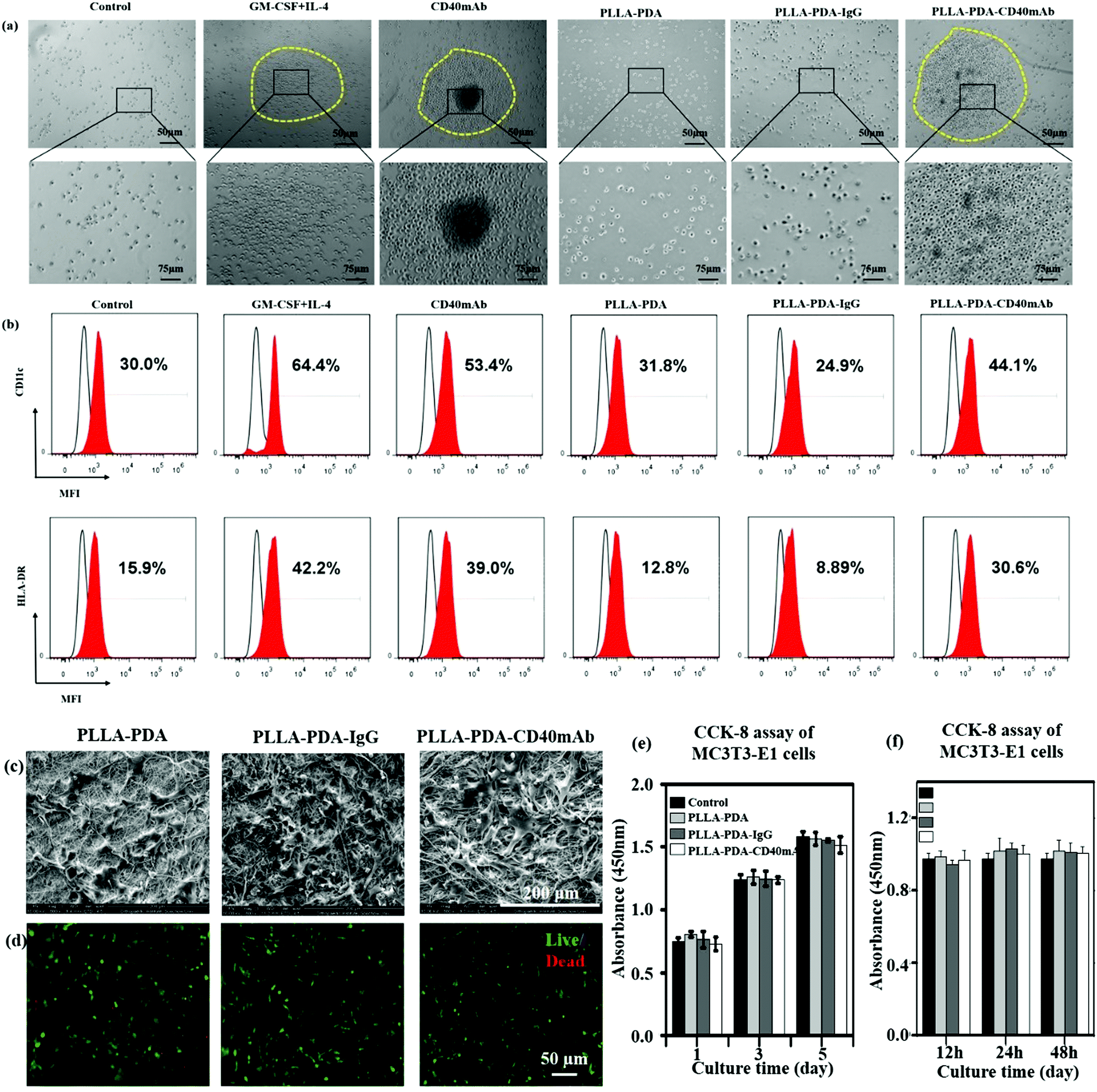 | ||
| Fig. 3 CD40mAb released from the PLLA-PDA-CD40mAb scaffold activates DCs from PBMCs. (a) The morphology of DCs and (b) the expressions of CD11c and HLA-DR. Control: cells seeded into culture medium without cytokines and antibodies. MFI: median fluorescence intensity. The proliferation effect of PLLA-PDA-CD40mAb on MC3T3-E1 cells. (c) SEM images of MC3T3-E1 cells seeded onto the electrospun PLLA-PDA, PLLA-PDA-IgG and PLLA-PDA-CD40mAb scaffolds for 3 d. (d) Live/dead staining of MC3T3-E1 cells seeded onto the electrospun PLLA-PDA, PLLA-PDA-IgG and PLLA-PDA-CD40mAb scaffolds for 3 d. (e) CCK-8 assay of MC3T3-E1 cells cultured in the released buffer (24 h) for 1, 3, and 5 d. (f) CCK-8 assay of MC3T3-E1 cells cultured in the released buffer (12, 24, 48 h) for 3 d – A: 12 h, B: 24 h, C: 48 h. Control: MC3T3-E1 cells seeded onto the plate with cell culture medium. | ||
PLLA scaffolds have been widely applied to regenerate or remodel tissue in injured regions.41 They provide an ECM-like three-dimensional (3D) microenvironment to support cell adhesion physically and proliferation.42–44 We hypothesise that in addition to cancer therapy the electrospun scaffolds can also provide physical support for the regeneration of inspired tissue. In this work, we seeded mouse preosteoblast MC3T3-E1 cells onto the fibrous scaffolds to assess the tissue regeneration capability of the PLLA scaffolds. The cellular morphology and cell–scaffold interaction were imaged with SEM. The pictures in Fig. 3c show that the cells grown on the different scaffolds had the same flat structure after 3 d and the cells connected with each other. Live/dead staining assay showed that the cells seeded onto the PLLA-PDA, PLLA-PDA-IgG and PLLA-PDA-CD40mAb scaffolds grew well with few dead cells (shown in red) detected (Fig. 3d). The live cells in green on the scaffolds displayed good activity with polygon and fusiformis shapes, connected with each other and well-spread. Thus, the CD40mAb conjugation and release process didn’t affect the properties of the scaffolds on inducing tissue regeneration.
Since the commercial CD40mAb also recognizes mouse-original CD40 molecules, we harvested the released buffers (24 h) of the different scaffolds and studied the growth effect of the released CD40mAb on MC3T3-E1 cells for 1, 3, and 5 d incubations. As a result, there was no significant difference between the different groups at the same time points (Fig. 3e). As parallel experiments, we also collected released buffers of the different scaffolds at 12 h, 24 h, and 48 h and cultured MC3T3-E1 cells for another 3 d without scaffolds. The OD values of the cells between the different groups showed no statistical difference (Fig. 3f). The results indicated that the CD40mAb conjugation and release process influenced neither the properties of the scaffolds nor the growth of MC3T3-E1 cells. MC3T3-E1 cells didn’t respond to CD40mAb under such situations because of the lack of CD40 expression on the cell surface. Thus, we proposed that the PLLA-PDA-CD40mAb scaffold was multifunctional, which not only specifically “killed” tumor cells that express CD40, but also provided physical support to promote cell proliferation in vitro.
Inspired by the anti-tumor efficiency and as a solid scaffold for cell growth of PLLA-PDA-CD40mAb scaffolds in vitro, we further examined the cancer therapeutic effect of the PLLA-PDA-CD40mAb scaffolds on the induction of tumor cell apoptosis in vivo. Severe combined immunodeficiency (SCID) mice are characterized by lack of T cells, B cells and detectable antibodies. Though they can’t induce specific immune response in vivo, they are a well-established model for implanting humanized tumor cells. In our study, human MDA-MB-231 breast cancer cells bearing tumours were developed in the axillary regions of severe SCID mice. After tumor masses were formed, parts of tumor mass were removed by surgery and the residual tumor masses (3 × 3 × 3 mm3) were left in situ. The PLLA, PLLA-PDA-IgG and PLLA-PDA-CD40mAb scaffolds were used to cover the cross-section. We also set one group as the positive control by direct injection of CD40mAb (2 μg) around the tumor mass. All the mice were euthanized and the tumours were collected 14 d after the treatment. As shown in Fig. 4a, the tumor mass without removal and treatment grew in an uncontrollable manner. As we expected, the tumor volumes in the mice treated with free CD40mAb buffer or the PLLA-PDA-CD40mAb scaffold were obviously smaller than the ones in the other two groups covered with the PLLA and PLLA-PDA-IgG scaffolds (Fig. 4b). The mean size of the tumor masses in the PLLA-PDA-CD40mAb group was the smallest among all the groups, further confirming that the PLLA-PDA-CD40mAb membranes could effectively inhibit tumor growth.
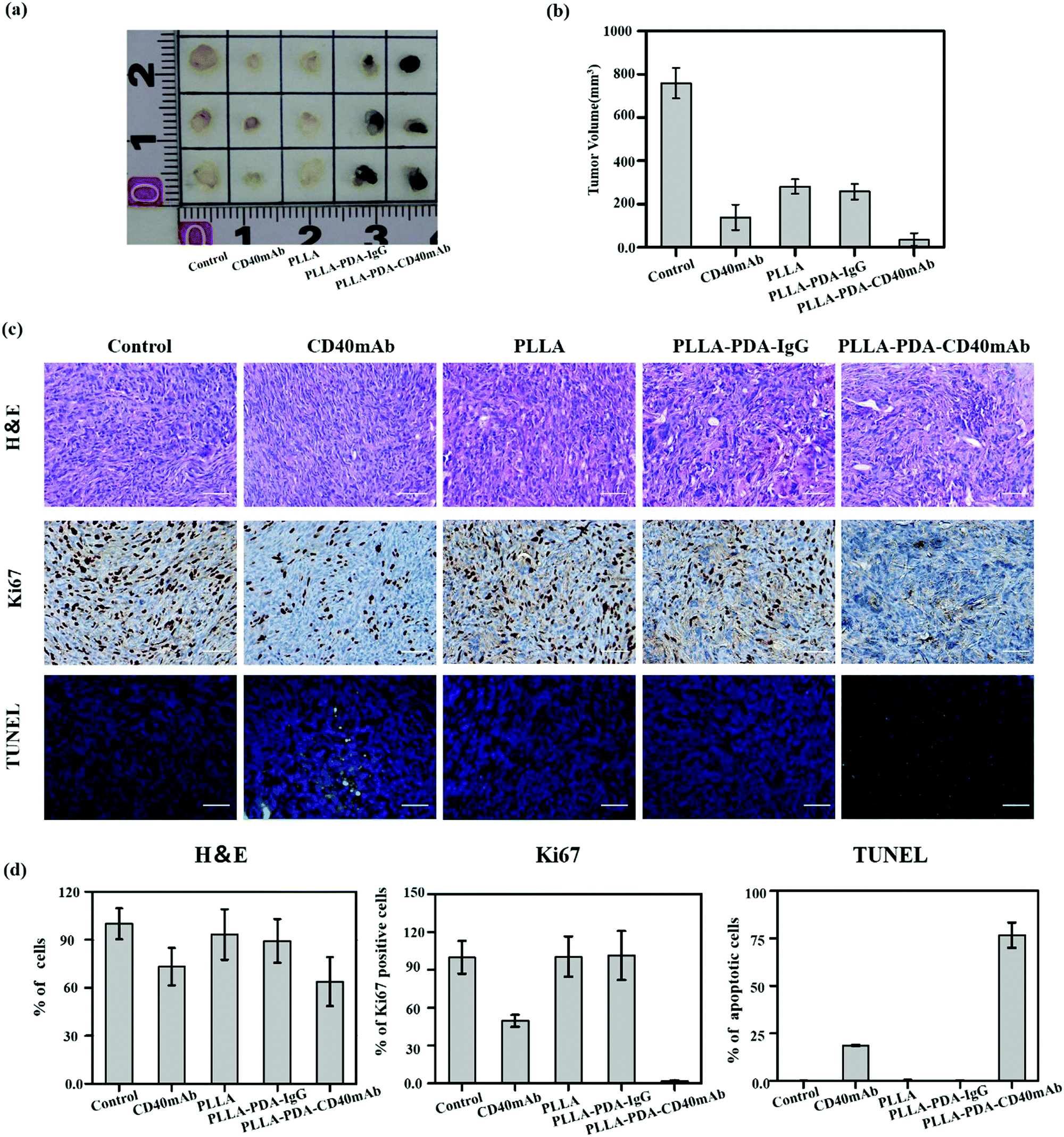 | ||
| Fig. 4 In vivo anticancer efficiency of PLLA-PDA-CD40mAb membranes. (a) The representative photographs of the tumors after various treatments and quantification of tumor masses in the different groups indicated. (b) The volumes of tumor masses. (c) H&E stained images, immunohistochemical analysis and TUNEL apoptosis assay (green: apoptotic cells; blue: nuclei) of tumor tissues after membrane-treated therapy. (d) Quantification of the expression of Ki67, TUNEL, H&E assay (***p < 0.001). | ||
We further examined the therapeutic efficacy of PLLA-PDA-CD40mAb scaffolds by the typical morphological assay (Fig. 4c). Hexatoxylin and eosin (H&E) staining images revealed that MDA-MB-231 cells were found in all groups. In the groups with the different PLLA scaffolds, non-degraded fibers were also observed in the tumor mass. Most MDA-MB-231 cells were dead in either the free CD40mAb or PLLA loaded CD40mAb-treated groups. Since Ki67 proliferation is a prognostic and predictive index in breast cancer,45 we performed Ki67 immunohistochemical staining. The Ki67 expression of MDA-MB-231 cells in the PLLA-PDA-CD40mAb group was significantly less than the ones of the other four groups. In contrast, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining assay indicated that the MDA-MB-231 cells from the PLLA-PDA-CD40mAb group were largely apoptotic; however, they were alive in the other groups. Quantitative analysis (Fig. 4d) further demonstrated that there were significant differences between the PLLA-PDA-CD40mAb group and the other groups. All of the results, from another perspective, are state-of-the-art proof that the PLLA-PDA-CD40mAb scaffolds can significantly inhibit the tumor growth by suppressing the cell proliferation and inducing the apoptosis of tumor cells in vivo. We speculate that CD40mAb attached to the scaffold activates the CD40 signaling of the tumor cell line MDA-MB-231 and “kill” the tumor cells successfully in vivo. Meanwhile, since the PLLA scaffolds absorb CD40mAb through the PDA motif, the system could avoid the side effects caused by CD40mAb burst and rapid diffusion when the free CD40mAb is applied in vivo.
In conclusion, CD40mAb is successfully conjugated to PLLA mediated by PDA and the activity of the antibody is well retained. In addition, the PDA and CD40mAb conjugation does not influence the mechanical properties, but alters the PLLA surface from hydrophobic to hydrophilic to facilitate the cell attachment. The CD40mAb-functionalized electrospun PLLA-PDA fibrous scaffolds are multifunctional, which are able to specifically eliminate CD40-expressed tumor cells by recognizing CD40 molecules and inducing cell apoptosis via a Bcl-2/Bax-dependent pathway. In addition, the scaffolds promote DC maturation via CD40mAb cross-linking in vitro, though its antitumor function needs to be further explored. More interestingly, the scaffolds provide a 3D platform to support fibroblast proliferation, suggesting that the system is favourable for inducing healthy tissue regeneration in vivo. The CD40mAb-functionalized electrospun PLLA-PDA fibrous scaffolds are easy to fabricate, biocompatible, and functional in selectively inducing tumor cell apoptosis via multiple mechanisms, and promoting tissue regeneration. As a result, they could be promising antibody-delivery systems for cancer therapy, especially for local administration to promote tumor cell death and avoid the recurrence of tumors post-surgically.
Experimental
Details on the experimental procedures are provided in the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
X. L., H. Z., and R. C. contributed equally to this work. This work was supported in part by the National Natural Science Foundation of China (81772313, 81572099, 81572131), the Shanghai Municipal Education Commission—Gaofeng Clinical Medicine Grant Support (20171906 and 17XJ11004), the Science and Technology Commission of Shanghai Municipality (18ZR1434200), the Shanghai Municipal Health and Family Planning Commission (201840027), the National Natural Science Foundation of Jiangsu Province (BK20151210), the Jiangsu Provincial Clinical Orthopedic Center, and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). Prof. Zhang acknowledges financial support from the Jane and Aatos Erkko Foundation (4704010), the Academy of Finland (297580) and the Sigrid Juselius Foundation (28001830K1). Prof. H. A. Santos acknowledges financial support from the University of Helsinki Research Funds, the HiLIFE Research Funds, the Sigrid Juselius Foundation (4704580), and the European Research Council under the European Union's Seventh Framework Programme (FP/2007–2013, 310892).Notes and references
- M. Zhou, L. Zhong, W. Xu, Y. Sun, Z. Zhang, H. Zhao, L. Yang and J. Sun, Sci. Rep., 2016, 6, 31038 CrossRef PubMed.
- L. Masarova, H. Kantarjian, G. Garcia-Mannero, F. Ravandi, P. Sharma and N. Daver, Adv. Exp. Med. Biol., 2017, 995, 73–95 CrossRef PubMed.
- M. Elmesery, J. Trebing, V. Schäfer, D. Weisenberger, D. Siegmund and H. Wajant, Cell Death Dis., 2013, 4, e916 CrossRef PubMed.
- R. Hassan, J. Zhang and I. Pastan, Lung Cancer, 2006, 54, S13–S13 CrossRef.
- P. M. Garfin and E. J. Feldman, Curr. Hematol. Malig. Rep., 2016, 11, 545–552 CrossRef PubMed.
- N. Becker and I. Benhar, Antibodies, 2012, 1, 39–69 CrossRef.
- S. Moradi-Kalbolandi, A. Hosseinzade, M. Salehi, P. Merikhian and L. Farahmand, J. Pharm. Pharmacol., 2018, 70, 841–854 CrossRef PubMed.
- S. M. Kinyanjui, D. J. Conway, D. E. Lanar and K. Marsh, Malar. J., 2007, 6, 1–8 CrossRef PubMed.
- L. C. Sandin, T. H. Tötterman and S. M. Mangsbo, OncoImmunology, 2014, 3, e27400 CrossRef PubMed.
- Q. Zhang, Y. Li, Z. Y. W. Lin, K. K. Y. Wong, M. Lin, L. Yildirimer and X. Zhao, Drug Discovery Today, 2017, 22, 1351–1366 CrossRef PubMed.
- T. Xu, Q. Yao, J. M. Miszuk, H. J. Sanyour, Z. Hong, H. Sun and H. Fong, Colloids Surf., B, 2018, 171, 31–39 CrossRef PubMed.
- M. Dodel, N. Hemmati Nejad, S. H. Bahrami, M. Soleimani and H. Hanaee-Ahvaz, Cell. Mol. Biol., 2016, 62, 16–25 Search PubMed.
- S. Hinderer, S. L. Layland and K. Schenke-Layland, Adv. Drug Delivery Rev., 2016, 97, 260–269 CrossRef PubMed.
- M. E. Soderquist and A. G. Walton, J. Colloid Interface Sci., 1980, 75, 386–397 CrossRef.
- E. Lutanie, J. C. Voegel, P. Schaaf, M. Freund, J. P. Cazenave and A. Schmitt, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 9890–9894 CrossRef.
- M. Zhang, Z. Wang, Z. Wang, S. Feng, H. Xu, Q. Zhao, S. Wang, J. Fang, M. Qiao and D. Kong, Colloids Surf., B, 2011, 85, 32–39 CrossRef PubMed.
- Y. K. Joung, I. K. Hwang, K. D. Park and W. L. Chan, Macromol. Res., 2010, 18, 904–912 CrossRef.
- J. Yu, W. Wei, M. S. Menyo, A. Masic, J. H. Waite and J. N. Israelachvili, Biomacromolecules, 2013, 14, 1072–1077 CrossRef PubMed.
- M. E. Lynge, R. van der Westen, A. Postma and B. Stadler, Nanoscale, 2011, 3, 4916–4928 RSC.
- A. Chatzigeorgiou, M. Lyberi, G. Chatzilymperis, A. Nezos and E. Kamper, BioFactors, 2009, 35, 474–483 CrossRef PubMed.
- R. J. Noelle, M. Roy, D. M. Shepherd, I. Stamenkovic, J. A. Ledbetter and A. Aruffo, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 6550–6554 CrossRef.
- C. V. Kooten and J. Banchereau, J. Leukocyte Biol., 2000, 67, 2–17 CrossRef PubMed.
- H. Dadgostar and G. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 1497–1502 CrossRef PubMed.
- M. C. Chang, Y. L. Chen, Y. C. Chiang, Y. J. Cheng, Y. W. Jen, C. A. Chen, W. F. Cheng and W. Z. Sun, Am. J. Cancer Res., 2016, 6, 157–172 Search PubMed.
- Y. Zhang, X. Hu, Y. Hu, K. Teng, K. Zhang, Y. Zheng, X. Hong, K. Yu, Y. Wang and L. Liu, J. Exp. Clin. Cancer Res., 2015, 34, 1–10 CrossRef PubMed.
- R. H. Vonderheide, K. T. Flaherty, M. Khalil, M. S. Stumacher, D. L. Bajor, N. A. Hutnick, P. Sullivan, J. J. Mahany, M. Gallagher and A. Kramer, J. Clin. Oncol., 2007, 25, 876–883 CrossRef PubMed.
- J. M. Park, X. Yang, J. J. Park, O. W. Press and M. F. Press, Hybridoma, 2000, 18, 487–495 Search PubMed.
- C. J. Qi, K. Q. Qian, Y. L. Ning, H. B. Ma, S. Z. Wang and X. G. Zhang, Cell. Immunol., 2009, 259, 135–140 CrossRef PubMed.
- Z. H. Zhou, Q. Shi, J. F. Wang, Y. J. Chen, Y. M. Zhuang, J. Z. Pan, C. S. Xu, C. J. Qi and X. G. Zhang, Apoptosis, 2005, 10, 123–134 CrossRef PubMed.
- N. Li, Y. Liu, Y. Miao, L. Zhao, H. Zhou and L. Jia, IUBMB Life, 2016, 68, 764–775 CrossRef PubMed.
- M. Remer, A. White, M. Glennie, A. Al-Shamkhani and P. Johnson, Curr. Top. Microbiol. Immunol., 2017, 405, 165–207 Search PubMed.
- G. Q. Jiang, W. U. Bin and Y. Liu, China Oncol., 2005, 15, 435–437 Search PubMed.
- Z. H. Zhou, J. F. Wang, Y. D. Wang, Y. H. Qiu, J. Z. Pan, W. Xie, L. Y. Jiang, B. Klein and X. G. Zhang, Hybridoma, 1999, 18, 471–478 Search PubMed.
- J. M. Adams and S. Cory, Science, 1998, 281, 1322–1326 CrossRef PubMed.
- C. Federmengus, E. Schultzthater, D. Oertli, W. R. Marti, M. Heberer, G. C. Spagnoli and P. Zajac, Hum. Gene Ther., 2005, 16, 348–360 CrossRef PubMed.
- P. Blanco, V. Pitard, J. F. Viallard, J. L. Taupin, J. L. Pellegrin and J. F. Moreau, Arthritis Rheumatol., 2005, 52, 201–211 CrossRef PubMed.
- C. Martin-Granados, A. R. Prescott, S. L. Sommer, I. P. Klaska, Y. Tian, E. Muckersie, C. V. Giuraniuc, L. Grant, M. Delibegovic and J. V. Forrester, J. Mol. Cell Biol., 2015, 7, 517–528 CrossRef PubMed.
- P. Brossart, F. Grünebach, G. Stuhler, V. L. Reichardt, R. Möhle, L. Kanz and W. Brugger, Blood, 1998, 92, 4238–4247 Search PubMed.
- T. Kikuchi, N. Miyazawa, M. A. Moore and R. G. Crystal, Cancer Res., 2000, 60, 6391–6395 Search PubMed.
- Z. F. Wang, G. H. Yu, Z. L. Zhu, Y. P. Zhu, F. M. Wang, J. Z. Pan, Z. J. Gu and X. G. Zhang, Chin. J. Hematol., 2003, 24, 572–575 Search PubMed.
- P. A. van der Eerden, P. J. Lohuis, A. A. Hart, W. C. Mulder and H. Vuyk, Plast. Reconstr. Surg., 2008, 122, 1747–1755 CrossRef PubMed.
- Z. Ma, C. Gao, Y. Gong and J. Shen, Biomaterials, 2005, 26, 1253–1259 CrossRef PubMed.
- A. K. Jaiswal, S. S. Kadam, V. P. Soni and J. R. Bellare, Appl. Surf. Sci., 2013, 268, 477–488 CrossRef.
- F. Yang, R. Murugan, S. Ramakrishna, X. Wang, Y. X. Ma and S. Wang, Biomaterials, 2004, 25, 1891–1900 CrossRef PubMed.
- T. Koopman, H. J. Buikema, H. Hollema, G. H. de Bock and B. van der Vegt, Breast Cancer Res. Treat., 2018, 169, 33–42 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8mh00704g |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2018 |