
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceV2O5-Based nanomaterials: synthesis and their applications
Xuyan Liu a,
Jiahuan Zenga,
Huinan Yangb,
Kai Zhoua and
Deng Pan*a
a,
Jiahuan Zenga,
Huinan Yangb,
Kai Zhoua and
Deng Pan*a
aSchool of Mechanical Engineering, University of Shanghai for Science and Technology, Shanghai 200093, China. E-mail: lxuyan@163.com; dpan@usst.edu.cn
bSchool of Energy and Power Engineering, University of Shanghai for Science and Technology, Shanghai 200093, China
First published on 23rd January 2018
Abstract
As an important part of lithium-ion batteries, the cathode material can directly affect the performance of lithium-ion batteries. However, with the increasing demand for high-energy and high-power devices, the energy and power density of electrode materials need to be further improved. Among all metal oxides, vanadium pentoxide (V2O5) is regarded as a promising candidate to serve as a cathode material for LIBs due to its high theoretical capacity. Herein, a thorough survey of the synthesis of V2O5-based nanomaterials with various structures and chemical compositions and their application as positive electrodes for LIBs is provided. This review covers V2O5 with different morphologies ranging from 1D nanorods/nanowires/nanotubes/nanofibers/nanobelts, to 2D nanosheets, and to 3D hollow and porous nanostructures. Nanocomposites consisting of V2O5 and different carbonaceous supports, e.g., amorphous carbon, carbon nanotubes, and graphene, are also investigated. The cation-doped V2O5 samples as the cathode material for LIBs are briefly discussed as well. The aim of this review is to provide an in-depth and rational understanding of how the electrochemical properties of V2O5-based cathodes can be effectively enhanced by achieving proper nanostructures with optimized chemical composition.
1. Introduction
Recently, with the shortage of traditional energy sources and fear of greenhouse gas-induced global warming, scientists and engineers are looking for renewable, highly efficient methods of creating and storing energy without disruption to the environment.1,2 With the increasing demand for green energies in the 21st century, efforts have been devoted to replace the non-renewable fossil fuels by other sustainable energies, like solar, wind, nuclear and hydroelectric power.3 Therefore, long-life, environmentally friendly, and low-cost reliable batteries are greatly needed to meet the crucial energy storage demands of modern society. Since lithium-ion batteries were first commercialized in 1991 by Sony, they have been widely used in many fields such as consumer electronics because of their high energy density and high safety.4,5 Nowadays, lithium-ion batteries (LIBs) and supercapacitors (SCs) represent two major practically applied energy storage systems; nevertheless, both have some shortcomings originating from their charge storage mechanisms.6 In this regard, rechargeable LIBs play a significant role due to their high gravimetric and volumetric energy, high power density, long cycle life and low self-discharge property.7–9 Furthermore, they have proved to be the most efficient energy storage strategy for a wide range of portable devices like cellular phones, laptops and digital electronics.10–12 Current LIB technology is well developed for portable electronic devices and has been widely used for the past twenty years. However, the employment of Li-ion batteries in hybrid electric vehicles (HEV), plug-in hybrid electric vehicles (PHEV) and pure electric vehicles (PEV) needs two to five times more energy density than the existing lithium batteries can offer (150 W h kg−1).13 Therefore, further LIB material and system developments are necessary.Fig. 1 shows a typical lithium-ion battery.25 Obviously, there are four key components in an LIB system: cathode, anode, separator and electrolyte.14 The electrolyte allows lithium ions to travel between the electrodes, and a separator keeps the anode and the cathode from making direct contact.18–21 First-generation LIBs employ the graphite as the anode, layered LiCoO2 as the cathode, and the organic liquid LiPF6/ethylene carbonate (EC)/dimethylene carbonate (DMC) as the electrolyte.15–17 The LIB performance (e.g., cell potential, capacity and energy density) is largely dependent on the intrinsic chemistry of the negative and positive electrode materials. The cathode usually consists of a metal oxide, and the anode tends to be a carbon material. At present, cathode materials for commercial LIBs are mainly transition metals oxides or active phosphates, such as LiCoO2,26,27 LiNiO2,28,29 LiMn2O4,30,31 and LiFePO4.32,33 Comparison of several kinds of cathode materials for lithium ion batteries is listed in Table 1, while graphite is commonly used as the anode active material. Lithium-ion batteries charge and discharge through a process of lithiation (lithium insertion) and de-lithiation (lithium extraction) by means of electrochemical reactions. In this process, lithium ions diffuse back and forth through the electrolyte between the anode and the cathode. During lithiation (discharging), the lithium bonded with the anode material breaks apart, producing lithium ions and electrons, and these lithium ions travel and bond with metal oxides on the cathode side and the electrons produce the electrical energy that powers devices. During de-lithiation (charging), the lithium metal oxides from the cathode break apart, producing lithium ions that pass through the electrolyte and bond with the material on the anode side with the addition of electrons.22–24
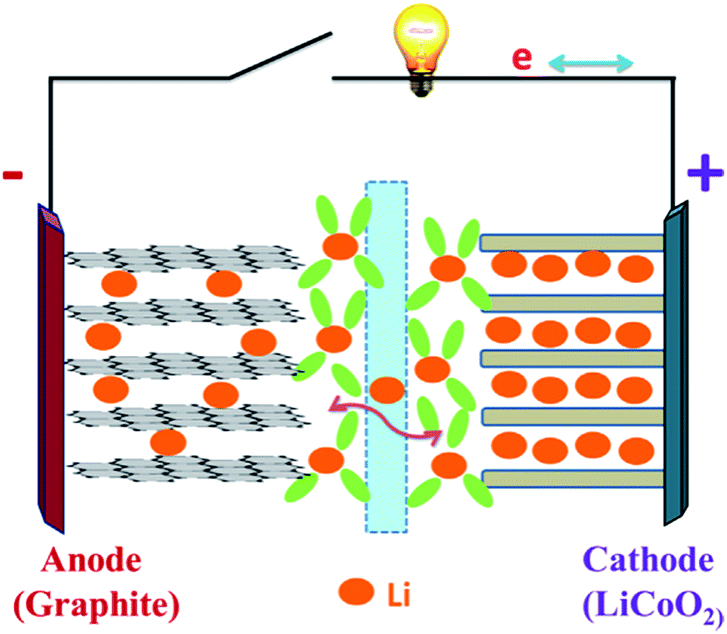 | ||
| Fig. 1 Schematic of a lithium-ion battery during the charge/discharge process.25 | ||
| Material category | LiCoO2 | LiNiO2 | LiMn2O4 | LiFePO4 |
|---|---|---|---|---|
| Operating voltage/V | 2.5–4.2 | 2.5–4.2 | 3–4 | About 3.4 |
| Theoretical capacity/mA h g−1 | 274 | 275 | 148 | 170 |
| Actual capacity/mA h g−1 | 130–150 | 150–220 | 110–130 | 135–153 |
| Electrical conductivity/S cm−1 | 10−3 | 10−1 | 10−5 | 10−9 |
| Safety | Poor | Poor | Good | Good |
| Cycle life | >500 | Poor | >500 | >500 |
| High rate discharge performance | Good | — | Good | Good |
In the past decade, because of the significant advances in nanotechnology and materials science, numerous nanostructured materials have been applied as electrode materials for LIBs. Among the potential cathode materials, layered vanadium pentoxide (V2O5) is one of the most attractive cathode materials and has been extensively studied because of its low cost, abundance, ease of synthesis, good safety and relatively high theoretical capacity of ∼437 mA h g−1, which is much higher than that of the currently used LiCoO2-based anodes (∼274 mA h g−1).34,35 In a Li/V2O5 half-cell, the Li+ intercalation and de-intercalation process can be expressed by the following equation:36
| V2O5 + xLi+ + xe− → LixV2O5 |
As Li+ are inserted into the layers of V2O5, phase transformation occurs. Theoretically, the process of Li+ intercalation into V2O5 can be divided into several stages and different phases of LixV2O5 can be formed in each stage. Fig. 2 shows the electrochemical lithium intercalation into V2O5, showing the evolution of phases with the degree of lithium intercalation.40 The structural behaviour of vanadium oxide with increased Li insertion is quite complex and it can split into several different phases. According to the amount of lithium, x, there are five phases, α-V2O5 (x < 0.01), ε-LixV2O5 (0.35 < x < 0.7), δ-LixV2O5 (x = 1), γ-LixV2O5 (x < 2) and ω-LixV2O5 (x > 2).37,38 The phases of the lithiated vanadium oxide LixV2O5 and the corresponding electrochemical reactions are listed in Table 2. Among these phases of LixV2O5, the first phase α-V2O5 has little effect on the V2O5 structure. This phase is followed by the ε-phase in which the vanadium oxide layers become more puckered. The δ-phase occurs when x = 1, and as can be seen from Fig. 2, at this phase, there is a sudden decrease in cell potential. Due to the intercalation of Li+, slight structural modifications occur, such as puckering of the layers and increasing of interlayer spacing; however, the basic layered structure is maintained. If more than one Li+ are intercalated, the δ-phase is transformed into the γ-LixV2O5 phase via an irreversible reconstruction mechanism. The γ-LixV2O5 phase can itself be reversibly cycled in the range of 0 < x < 2 while maintaining the γ-type structure. When even more Li+ are intercalated, a rock salt structure of ω-LixV2O5 is irreversibly formed. It has been reported that discharging V2O5 to the ω-LixV2O5 phase would result in a rapid capacity loss with increased cycling, which is not desirable for the cathode material. Although the theoretical capacity of V2O5 is pretty high, the practical capacity of unmodified V2O5 is far from satisfactory because of low electrical conduction, slow Li+ diffusion and irreversible phase transitions upon deep discharge.39 This would induce a huge influence in the electrode material, which leads to disintegration and loss of electric contact and eventually results in quick capacity fading upon prolonged cycling.
 | ||
| Fig. 2 Various phases associated with the electrochemical intercalation of lithium into V2O5.40 | ||
| Value of x in LixV2O5 | Phase | Electrochemical reactions |
|---|---|---|
| x < 0.01 | α-V2O5 | V2O5 + xLi+ + xe− → α-LixV2O5 |
| 0.35 < x < 0.7 | ε-LixV2O5 | V2O5 + xLi+ + xe− → ε-LixV2O5 |
| x = 1 | δ-LixV2O5 | V2O5 + xLi+ + xe− → δ-LixV2O5 |
| x < 2 | γ-LixV2O5 | V2O5 + xLi+ + xe− → γ-LixV2O5 |
| x > 2 | ω-LixV2O5 | V2O5 + xLi+ + xe− → ω-LixV2O5 |
In this review, we propose a systematic description of V2O5-based nanomaterials ranging from synthesis, to modification, structure and electrochemical application. In the first section, we will provide an overview of the formation of V2O5 materials through dimensional structure design. It includes the survey of phase-pure V2O5 with unique 1D, 2D, and 3D nanostructures. Then, we will illustrate the development of carbonaceous materials into V2O5 electrodes for LIBs. In the last section, cation-doped V2O5 samples as the cathode material for LIBs will be briefly discussed. We thus believe that this review article could serve as a good reference for V2O5-based nanomaterials.
2. Phase-pure V2O5
As a cathode material for LIBs, vanadium-based oxides have attracted worldwide attention because of their high capacity, low cost and abundance.41,42 Moreover, V2O5 has also been widely used in photocatalysts,43 sensors44,45 and electrochromic devices.46 To synthesize nanostructured V2O5, a variety of methods, including sol–gel processing, template-based methods, thermal evaporation, hydrothermal/solvothermal synthesis, reverse micelle techniques, and electrochemical deposition, have been developed. Among these methods, hydrothermal synthesis is considered the easiest and most effective way. With the assistance of surfactants, different morphologies of V2O5 are prepared by the hydrothermal method.47–50 Table 3 summarizes the electrochemical properties of vanadium pentoxide as a cathode material for various 1D, 2D and 3D nanostructures.| Precursor material | Nanostructures | Initial capacity/mA h g−1 | Current density/mA g−1 | Cycles (times)/capacity (mA h g−1) | References |
|---|---|---|---|---|---|
| V2O5 powder | Nanorod | 335 | 10 | 50/260 | 53 |
| NH4VO3 | Nanorod | 418.8 | 50 | 50/180.5 | 54 |
| NH4VO3 | Nanorod | 265 | 29.4 | ∼ | 55 |
| V2O5 powder | Nanorod | 260 | 10 | 30/240 | 56 |
| LiV3O8 powder | Nanorod | 388 | 58.8 | 70/325 | 58 |
| V2O5 powder | Nanorod | 292 | 100 | 20/286 | 59 |
| V2O5 powder | Nanorod | 338 | 101 | 50/326 | 60 |
| Ammonium metavanadate | Nanowire | 390 | 30 | 50/201 | 65 |
| Cassava starch | Nanowire | 209 | 60 | 30/198 | 69 |
| Vanadyl acetylacetonate | Nanowire | 225 | 58.8 | 60/125 | 144 |
| VOSO4·nH2O | Nanotube | 300 | ∼ | 10/160 | 72 |
| V2O5 powder | Nanotube | 457 | 30 | 10/270 | 73 |
| V2O5 powder | Nanotube | 284.7 | 60 | 10/218.5 | 74 |
| V2O5 powder | Nanotube | 204 | 20 | 4/87.7 | 75 |
| V2O5 powder | Nanotube | 275.2 | 58.8 | 50/204 | 76 |
| V2O5·nH2O | Nanofiber | 370 | 800 | 40/347 | 77 |
| Vanadium acetylacetonate | Nanofiber | 310 | 29.4 | 50/229 | 78 |
| V2O5 powder | Nanofiber | 139 | 800 | 100/133.9 | 79 |
| Vanadium hydroxylamido complex | Nanofiber | 440 | 64 | 30/200 | 80 |
| V2O5 powder | Nanobelt | 142 | 50 | 100/141 | 81 |
| NH4VO3 | Nanobelt | 288 | 50 | 50/246 | 82 |
| V2O5 powder | Nanobelt | 127.4 | 60 | 200/114.7 | 83 |
| V2O5 powder | Nanobelt | 281 | 58.8 | 50/242 | 84 |
| Ammonium metavanadate | Nanorod | 287 | 100 | 50/207 | 140 |
| NH4VO3 | Nanosheet | 185.6 | 294 | 50/179.5 | 85 |
| V2O5 crystal | Nanosheet | 290 | 59 | 50/274 | 86 |
| V2O5 powder | Nanosheet | 108 | 2940 | 200/104 | 87 |
| V2O5 powder | Nanosheet | 264(3) | 50 | 50/237 | 88 |
| NH4VO3 | Nanosheet | 147 | 100 | 100/144 | 89 |
| V2O5 powder | Nanosheet | 135 | 300 | 200/126.6 | 91 |
| V2O5 powder | Nanosheet | 251 | 500 | 100/206 | 92 |
| Vanadium oxytriisopropoxide | Nanosheet | 277 | 300 | 100/211 | 93 |
| NH4VO3 | Nanosheet | 310 | 29.4 | 50/234 | 94 |
| NH4VO3 | Hollow porous microspheres | 283 | 100 | 60/217 | 95 |
| NH4VO3 | Hollow spheres | 273 | 58.8 | 50/189 | 96 |
| NH4VO3 | Hollow microspheres | 241 | 300 | 60/190 | 97 |
| [V(acac)3] | Hollow microspheres | 286.4 | 58.8 | ∼ | 98 |
| V2O5 powder | Hollow sphere arrays | About 292 | 147 | 300/285 | 99 |
| V2O5 powder | Porous microspheres | 390 | 40 | 50/200 | 100 |
| NH4VO3 | Hierarchical and porous microspheres | 141 | 147 | 100/102 | 101 |
| NH4VO3 | Porous hierarchical octahedrons | 135 | 100 | 60/141 | 102 |
| Vanadium oxytriisopropoxide | Porous microspheres | 146.3 | 75 | 100/130 | 103 |
| V2O5 powder | Hierarchical microspheres | 275 | 294 | 200/243 | 104 |
| NH4VO3 | Hierarchical 3D microspheres | 275 | 58.8 | 50/243 | 105 |
| V2O5 powder | Hierarchical 3D microstructures | 274 | 300 | 50/219 | 106 |
| Vanadium oxytriisopropoxide | Hollow microflowers | 277 | 300 | 100/211 | 107 |
| VO(acac)2 | Hierarchical microflowers | 285 | 29.4 | 50/249 | 108 |
| NH4VO3 | Hierarchical microflowers | 145 | 200 | 150/128 | 109 |
| Vanadium(IV) acetylacetone | Yolk–shell microspheres | 280 | 58.8 | 30/220 | 110 |
| Vanadium(IV) acetylacetone | Hierarchical microspheres | 270.5(2) | 300 | 80/166.8 | 111 |
| Vanadium oxytriisopropoxide | Hollow microspheres | 291 | 100 | 6/254 | 112 |
| Vanadium oxytriisopropoxide | Box | 119 | 1500 | 400/111 | 113 |
| VOC2O4 | Hollow microspheres | 256 | 300 | 50/227 | 114 |
| NH4VO3 | Multi-shelled | 447.9 | 1000 | 100/402.4 | 115 |
| V2O5 | Hollow microspheres | 137(2) | 300 | 50/128 | 116 |
2.1 1D nanostructures
Among the many vanadium oxide nanomaterials, one-dimensional nanostructured vanadium oxide is the most studied because its preparation method is relatively simple, it has a large specific surface area compared with traditional cathode materials, and it shows good cycling stability and high specific capacity. Among the various types of nanostructures, 1D V2O5 nanostructures with nanoscale radial dimensions, including nanorods,53–60 nanowires,65–69 nanotubes,72–76 nanofibers77–80 and nanobelts,81–84 have exhibited distinct advantages in LIBs.51,52 | ||
| Fig. 3 Electrospinning reaction process for V2O5 micro/nanorods.54 | ||
Subsequently, Glushenkov et al.56 prepared nanorods of V2O5 via a two-stage procedure of ball milling and annealing in air. Commercially purchased V2O5 powder was milled in a ball mill as the first step of the synthesis. The as-milled precursor was subsequently annealed in air to produce nanorods via solid-state recrystallization. Takahashi et al.57 synthesized single-crystal V2O5 nanorod arrays using template-based electrodeposition and the electrochemical results demonstrated that the nanorod array electrodes had significantly higher current density and energy storage density than sol–gel-derived V2O5 films. Uniformly sized vanadium oxide nanorods with a length of about 10 μm and with diameters ranging from 100 to 200 nm were grown over a large area with near unidirectional alignment. This electrode exhibited high rate discharge capacity and good cycling stability.
Velazquez et al.66 prepared V2O5 nanowire arrays on silicon substrates by the thermal evaporation method. The nanowires obtained were single crystalline and highly oriented with their lengths and substrate coverage controlled by the duration of the reaction, reaction temperature, and flow velocity. The growth of these nanowire arrays has great value in the fabrication of novel battery architectures based on individual nanowires. Ramasami et al.69 synthesised a V2O5 NWC by the combustion method using cassava starch. The noteworthy features of the synthesis are that it is a simple and time-saving process, gives high yield, and uses naturally occurring fuel that doubles up as a template for the growth of nanowires/nanorods. The schematic representation of the nanowire cluster formation is given in Fig. 4. The V2O5 NWC exhibited an initial discharge and charge capacity of 209 and 206 mA h g−1, respectively with a corresponding coulombic efficiency of 96% and the obtained discharge capacity at the 30th cycle was 198 mA h g−1. The discharge capacity fading was found be negligible (with a small fading rate of 0.1% per cycle) from the beginning to the 50th cycle. The capacity retention in the 30th cycle was 95% with respect to the first cycle capacity. Compared to previous literature,70,71 the as-prepared structure behaved as a superior material with respect to capacity and cycling stability.
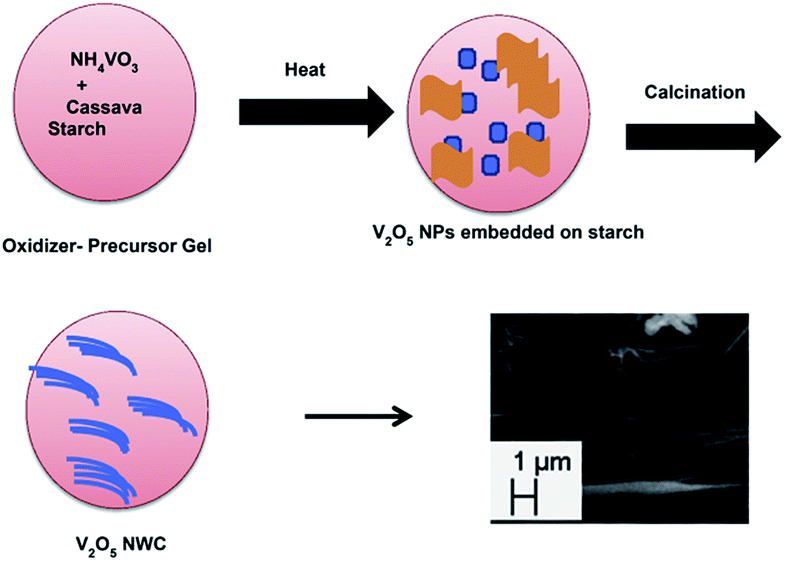 | ||
| Fig. 4 Illustration of V2O5 NWC formation.69 | ||
Nadimicherla et al.75 synthesized V2O5 nanotubes and PEG surfactant V2O5 nanotubes by a simple hydrothermal method. Batteries using V2O5 nanotube electrodes showed an initial specific capacity 192 mA h g−1, whereas the PEG surfactant V2O5 nanotubes exhibited a value of 204 mA h g−1. It was found that the PEG surfactant V2O5 nanotubes showed higher specific capacity at the initial stages and better stability as cycling progressed than the V2O5 nanotubes. This was due to the fact that polyethylene glycol occupied space between the vanadium oxide layers and contributed to the increase in Li+ intercalation. In addition, polyethylene glycol had a relatively strong interaction with the vanadium oxide layer and had a complex interaction with Li+, thus effectively alleviating the electrostatic effect of the vanadium oxide layer and Li+. Li et al.76 chose commercial V2O5 powder and oxalic acid as the raw materials to obtain a low-cost inorganic vanadium oxalate solution as the electrospinning vanadium precursor, and adopted a facile electrospinning approach followed by annealing to fabricate porous V2O5 nanotubes. The schematic of the synthesis of the porous V2O5 nanotubes is shown in Fig. 5. The sample had highly porous and hollow nanostructures of the as-prepared V2O5, and the as-prepared V2O5 had diameters in the range of 300–500 nm with many pores on the surface. In the voltage window of 2.0–4.0 V, the cathode obtained an initial discharge capacity of 275.2 mA h g−1 and 204 mA h g−1 during the first and 50th cycles at a current density of 58.8 mA g−1, respectively. Moreover, the coulombic efficiency maintained a value of >96% throughout the cycling test.
 | ||
| Fig. 5 Electrospinning preparation of porous V2O5 nanotubes: (a) electrospinning process; (b) annealing process.76 | ||
Recently, Yan's group79 has proposed a modified, facile and effective electrospinning method to synthesize V2O5 nanofibers using commercial vanadium pentoxide as the precursor material, which is illustrated in Fig. 6. Compared with commercial V2O5 at various current densities, it is evident that V2O5 nanofibers exhibited much better rate capability and cyclic stability. For instance, at a current density of 100 mA g−1, the commercial V2O5 electrode exhibited continuous capacity fading in the initial four cycles. Interestingly, the specific capacity for V2O5 nanofibers was recovered after the cathode electrode worked at various current densities for 45 cycles; this performance was significantly better than that of the commercial V2O5 electrode. Dewangan et al.80 proposed a simple mild one-step hydrothermal technique to prepare uniform V2O5 nanofiber bundles (NBs). The method used a vanadium(V) hydroxylamido complex as the vanadium source in the presence of HNO3. A bundle is made of an indefinite number of homogeneous V2O5 nanofibers with lengths up to several micrometres and widths ranging from 20 to 50 nm. The as-prepared V2O5 NBs displayed a high electrochemical performance in a non-aqueous electrolyte as a cathode material for lithium-ion batteries.
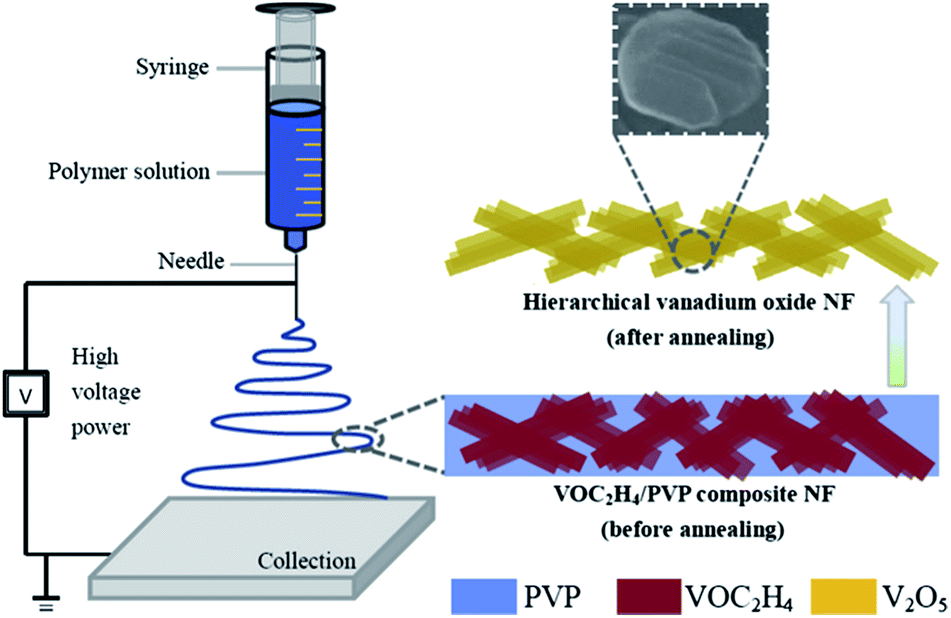 | ||
| Fig. 6 Schematic showing the strategy for fabricating hierarchical V2O5 nanofibers.79 | ||
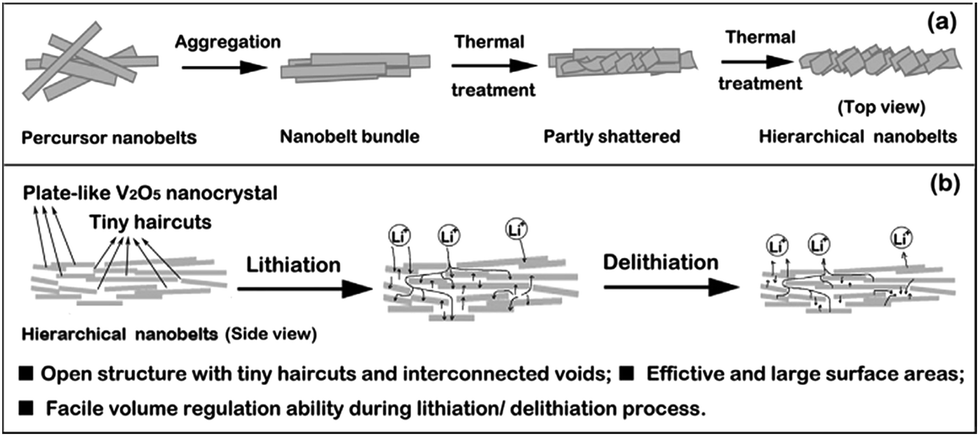 | ||
| Fig. 7 (a) Schematic of possible formation process of V2O5-HNbs, and (b) advantages of hierarchical nanobelt structures when applied in lithium-ion batteries.82 | ||
Wang and co-workers83 fabricated a self-standing V2O5 nanobelt electrode free of conductive agents, binders and current collectors by a simple one-step hydrothermal reaction. The length of the V2O5 nanobelts was up to several hundreds of micrometers and the thickness was around 40 nm. The as-prepared sample demonstrated better cycling performance than commercial conventional V2O5 electrodes with a high reversible capacity of 114.4 mA h g−1 after 200 charge–discharge cycles. The superior cycling performance of the nanobelt network electrode can be attributed to the excellent structural stability provided by the crosslinked nanobelt network. Rui et al.84 demonstrated a cost-effective and green process for the large-scale synthesis of single-crystalline V2O5 nanobelts (in kilogram scale) under ambient conditions by simply vigorous stirring the commercially available V2O5 powder in an aqueous NaCl solution. The as-prepared sample exhibited a higher reversible specific capacity of 242 mA h g−1 up to the 50th cycle with a capacity retention at 86%.
For one-dimensional nanostructured materials, the preparation methods are mostly the sol–gel method and hydrothermal method, because these processes are simple and feasible and the requirements of the experimental conditions are not too high. Common nanofibers, nanowires, nanotubes, etc. to some extent have improved the performance of vanadium oxide as a cathode material, such as shortening the diffusion distance of lithium ions and electrons, improving the charge and discharge properties of the electrode material, or increasing the effective contact area to improve the storage of lithium ions and to improve the specific capacity of the material. The superior electrochemical performance of one-dimensional nanostructures can be attributed to the following aspects.
First, one-dimensional nanostructures of V2O5 provide an effective electron transport pathway along the one-dimensional direction. Second, the one-dimensional nanostructures of V2O5 nanotubes with large electrode/electrolyte contact area and hierarchical porous channels provide short Li+ diffusion distances. Finally, the existence of pyrolyzed carbon improves the conductivity of the one-dimensional nanostructures. These nanomaterials have a high specific surface area when they are prepared alone. However, when the electrode is prepared, the one-dimensional nanostructures are greatly reduced due to the large surface energy and low mechanical stability, likely resulting in agglomeration and a significant decrease in the specific surface area. Their initial discharge specific capacity will be higher in the charge and discharge tests, but they will produce significant capacity attenuation; also, the number of cycles will not be high in the long cycle performance without further improvement.
2.2 2D nanostructures
Two-dimensional nanostructures increase the effective contact area between the electrolyte and the active material, increase the storage capacity of the lithium ions, and shorten the diffusion distance of the lithium ions to meet the demand for high charge and discharge and thus form an ideal structure for the rapid storage of lithium ions. However, the mechanical stress produced by the nanosheets in the process of circulation is easy to make the nanosheets overlap and thicken, weakening the effective contact area of the nanosheets. Therefore, the dispersibility of two-dimensional nanosheets is restricted as an excellent key factor for electrode material performance. It is helpful to effectively improve the electrochemical performance of two-dimensional vanadium pentoxide nanosheets electrode materials by introducing a conductive agent between the nanosheet and increasing the spacing of the nanosheets. Huang et al.85 developed an additive-free ultrasonic method with subsequent thermal decomposition to synthesize V2O5 self-assembled nanosheets. The loose V2O5 nanosheets were stacked as 4–6 layers and each layer (∼50 nm in thickness) was assembled using abundant nanoparticles. The as-prepared sample delivered a reversible capacity of 185.6 mA h g−1 and 179.5 mA h g−1 at the 1st and 50th cycles, respectively, corresponding to a capacity retention of 96.7%. However, NPs V2O5 only had a reversible capacity of 121.4 mA h g−1. After 50 cycles, 87.4% of the capacity (∼106.1 mA h g−1) was retained. All the results demonstrated that the unique structure of SANs V2O5 resulted in improved rate performance and high cycling stability at higher current densities, which was expected to satisfy the requirement of a long cycle life for LIBs.Rui's group86 proposed a liquid phase separation method to prepare ultrathin V2O5 nanosheets. In the range of 2.05–4 V, the first reversible specific discharge capacity of was 290 mA h g−1 at 0.2C. When the rate increased to 50C, the capacity was 117 mA h g−1. This is mainly due to the deposition of lithium ions on the surface of these ultra-thin nanosheets; ultra-thin nanosheets shorten the embedded path of lithium ions to meet the requirements of rapid charge and discharge. An et al.87 successfully prepared ultrathin V2O5 nanosheets through a supercritical solvothermal reaction followed by annealing, as illustrated in Fig. 8. As a cathode material for lithium batteries, the ultrathin V2O5 nanosheets exhibited a capacity of 108 mA h g−1 at a high rate of up to 10C at 2.4–4 V and excellent cyclability with little capacity loss after 200 cycles. The enhanced rate performance is attributed to the shortened diffusion distance and the increased electrode–electrolyte contact area of the ultrathin nanosheet structure. Subsequently, Liang's group88 fabricated nanosheet-structured vanadium pentoxide by a sol–gel method. As a cathode material, the V2O5 nanosheets exhibited enhanced cycling stability and rate capability. The as-prepared NSs showed better lithium storage properties than commercial V2O5, with a reversible capacity of 237 mA h g−1 after 50 cycles.
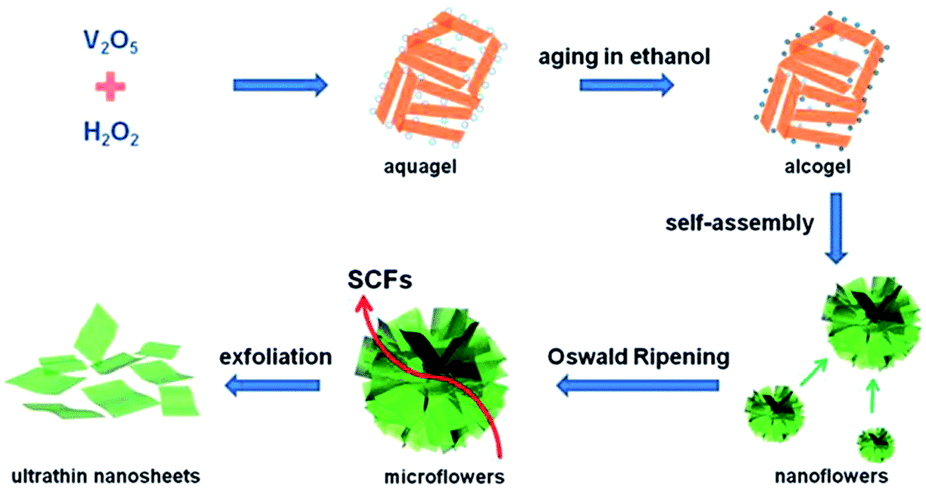 | ||
| Fig. 8 Schematic illustration of the procedure for fabricating the ultrathin vanadium oxide nanosheets.87 | ||
Song et al.89 prepared mesoporous V2O5 nanosheets by a hydrothermal method followed by instantaneous heating and calcination in air, as illustrated in Fig. 9. The as-prepared V2O5 nanosheets were composed of several well-defined porous nanosheets that assembled themselves together and formed a highly mesoporous nanosheet structure. In the voltage window of 2.5–4.0 V, the cathode obtained a reversible capacity of 147 mA h g−1 in the 2nd cycle and 144 mA h g−1 in the first and 100th cycles at a current density of 100 mA h g−1. The good performance of mesoporous V2O5 nanosheets can be attributed to the advantages of this novel structure. Xu's group90 developed a one-step polymer-assisted chemical solution method to synthesize two-dimensional V2O5 sheet networks. This unique network structure provided an interconnected transportation pathway for lithium ions. The as-prepared sample exhibited a high capacity of about 300 mA h g−1 at a current density of 100 mA g−1. Recently, Liang et al.91 proposed a bottom-up solvothermal method to synthesize ultra-large V2O5 nanosheets. The thickness of the large nanosheet was around 4 nm and the thickness of the parallel stacked nanosheets was about 3–5 nm. Moreover, the cathode obtained a capacity of is 135 mA h g−1 in the 2nd cycle and a retention of 93.8% after 200 cycles at a current density of 100 mA h g−1. The coulombic efficiency was close to 100% during the whole discharge/charge process. The excellent rate performance and remarkable cyclic stability are due to the structural advantages of this uniform 2D ultrathin nanosheet with an ultra-large lateral size: (1) the void space between the nanosheets can facilitate the penetration of electrolyte; (2) the ultra-thin feature can reduce Li+ ion diffusion distance and electron transportation distance; (3) the flexible nanosheets can accommodate the volume changes caused by repeated lithium ion insertion and removal; and (4) the ultra-large nanosheets and layer-by-layer stacking structures can better maintain the integrity of the nanosheet electrodes. Li and co-workers92 prepared 2D leaf-like V2O5 nanosheets by a novel and facile green method. The unique nanoscale characteristics, including 2D morphology, hierarchical porous structure, and large specific surface, of these 2D V2O5 nanosheets led to superior electrochemical performance in terms of their specific capacity, rate capability, and cyclability.
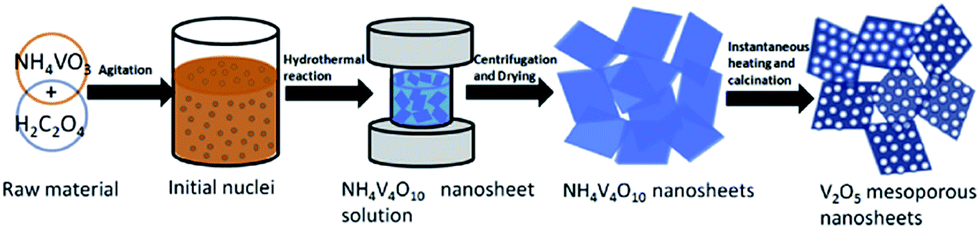 | ||
| Fig. 9 Schematic of the synthesis route of V2O5 mesoporous nanosheets in this work.89 | ||
As the cathode material of the two-dimensional nanosheet, lamellar nanostructures have a large specific surface area and good structural flexibility, which provide a larger space for the diffusion of ions and can adapt to the volume changes in the cathode material during the lithiation and de-lithiation of lithium, thus improving the cycling stability of the cathode material. If a conductive active material such as carbon nanotubes and carbon black is compounded in the structural material of the nanosheets, it is possible to prevent not only the agglomeration of the nanosheets but also the high conductivity of the active material. At the same time, this reduces the impedance of ions on nanosheets and makes the lithium ions diffuse faster in the electrode material, thus obtaining excellent charge and discharge performance, which is helpful to improve the electrochemical performance of the two-dimensional nanosheet cathode materials. Compared with one-dimensional nanostructured materials, the initial discharge specific capacity of the nanosheets is significantly lower. The reason may be that the vanadium oxide nanosheets are agglomerated during the charge and discharge process, resulting in a significant reduction in the effective contact area, which cannot effectively maintain the high specific surface area of the nanosheets. As with the one-dimensional vanadium pentoxide nanostructures, the long-term cycling performance of nanosheet cathode materials reported in the relevant literature has not improved.
2.3 3D nanostructures
In recent years, three-dimensional structures have attracted considerable attention as an important role of electrode materials. Compared with low-dimensional nanomaterials, three-dimensional nanostructured materials with desired morphologies have been prepared for their superior electrochemical properties as they not only inherit the advantages of low-dimensional nanomaterials, but also maintain the structural stability of the electrode during the Li+ intercalation and de-intercalation process. In addition, the three-dimensional nanostructures are self-supporting, and the nanocomposites are not prone to agglomeration during circulation. Also, the pore structure of the prepared electrode material is maintained for a long time. Thus, under the same electrochemical cycling test, vanadium pentoxide electrode materials have a three-dimensional or graded structure exhibiting better electrochemical performance than the low-dimensional electrode materials and better cycling stability. Compared with one-dimensional nanowires, nanotubes and two-dimensional nanosheets, the three-dimensional structure of vanadium oxide shows a variety of peculiar morphologies, such as loose nanoscale structures, and antennaed nanospheres, similar to the structure of sea urchins; these strange nanostructures can generally be prepared by adding a templating agent, such as dodecylamine and dodecyl mercaptan. For the three-dimensional nanomaterials of vanadium pentoxide, the most commonly used preparation method is the solvothermal method. | ||
| Fig. 10 (a) SEM images of V2O5 precursor.96 (b) Multilayer V2O5 hollow sphere arrays (cross-sectional SEM images in inset).99 (c) SEM images of the V2O5 product synthesized by annealing the corresponding VEG precursors in air.101 (d) FESEM images of ammonium vanadium oxide octahedrons.102 | ||
Chen and co-workers99 developed a facile polystyrene sphere template-assisted electrodeposition method for the fabrication of porous multilayer V2O5 hollow sphere arrays on graphite paper substrates. We could clearly see that a 3D porous hollow sphere structure was well formed and the individual hollow sphere exhibited a size of ∼500 nm (Fig. 10b). We also can reach the conclusion that it is reasonable for the obtained porous multilayer V2O5 hollow sphere arrays to possess impressive porous systems, as these systems will be beneficial for fast ion/electron transfer, leading to fast reaction kinetics. This is especially important for high-rate LIB applications. Wu et al.112 reported the controllable synthesis of novel rattle-type V2O5 hollow microspheres with the assistance of carbon colloidal spheres as hard templates. Carbon spheres@vanadium-precursor (CS@V) core–shell composite microspheres were first prepared through a one-step solvothermal method. V2O5 hollow microspheres with various shell architectures could be obtained after removing the carbon microspheres by calcination in air. When evaluated as a cathode material for LIBs, the rattle-type V2O5 hollow microspheres exhibited superior cycling stability and rate capability. Pan and co-workers114 proposed a one-pot template-free solvothermal method for the controllable synthesis of uniform VO2 microspheres with different complex interiors, including yolk–shell and multi-shell structures. The VO2 hollow spheres could be readily transformed into V2O5 hollow spheres without any structural deformation by calcination in air. The resulting V2O5 hollow microspheres exhibited a high initial reversible capacity of 256 mA h g−1 at a current density of 300 mA g−1 and good cycling performance over 50 cycles. Pan et al.116 also reported the synthesis of hierarchical nanosheet-constructed hollow V-glycolate microspheres by a template-free method using a cost-effective VOC2O4 solution as the precursor. After calcination in air, V2O5 hollow hierarchical microspheres with well-preserved structures could be obtained and they were found to exhibit superior rate capability and cycling stability as cathode materials for LIBs.
 | ||
| Fig. 11 Schematic of the formation of 3D porous V2O5 hierarchical microspheres.103 | ||
Recently, Dong et al.104 developed a facile two-step strategy to synthesise V2O5 microspheres with a hierarchical structure. First, they synthesized a vanadium glycolate precursor by a facile template-free and polyol-mediated solvothermal method and then, they obtained V2O5 microspheres by thermal annealing. Bai and co-workers105 facilely fabricated hierarchical 3D microspheres consisting of 2D V2O5 (vanadium pentoxide) nanosheets by a low-temperature hydrothermal method. The novel hierarchical micro-/nano-V2O5 electrode exhibited excellent electrochemical performance in terms of high-energy and high-power applications owing to its unique structural properties. These hierarchical 3D micro-/nano-V2O5 possess many unique features advantageous for LIBs: (1) 2D V2O5 nanosheets facilitate Li+ diffusion and electron transport; (2) hierarchical 3D micro-/nano-cathode structures built up by V2O5 nanosheet spheres lead to close and sufficient contact between the electrolyte and activate materials and at the same time create a buffer to accommodate volume changes during discharge/charge process; and (3) micro-scale V2O5 spheres easily induce high cell packing density beneficial for high-power batteries. Pan et al.106 developed a facile solvothermal method to synthesize hierarchical vanadium oxide with various nano/microstructures by simply varying the concentration of the precursor (VOC2O4) solution. The V2O5 prepared by solvothermal method for 2 h showed good lithium storage properties with a high reversible capacity of 219 mA h g−1 after 50 cycles. In a follow-up study based on the above experimental results, Pan107 synthesized hierarchically hollow microflowers composed of nanosheets via a one-pot solvothermal method. After annealing, the sample delivered a high initial discharge capacity of 277 mA h g−1, and the value slightly increased to 284 mA h g−1 in the second cycle probably due to improved electrolyte penetration. With a voltage window of 2.0–4.0 V, the V2O5 hollow microflowers retained a remarkable reversible capacity of 211 mA h g−1 at the end of the 100th cycle.
Chen et al.109 successfully synthesized hierarchical V2O5 microflowers by a solvothermal reaction followed by a calcination process at 350 °C. The product consisted of uniform flower-like micro-spheres with an average size of around 2 μm. After annealing, the as-prepared hierarchical V2O5 microflowers showed good lithium storage properties with a high reversible capacity of 128 mA h g−1 after 150 cycles at a current density of 200 mA g−1. It was obvious that these capacities were higher than those of bulk V2O5. Ma et al.111 developed a facile template-less approach for the synthesis of various V2O5 hierarchical structures by calcining the solvothermally prepared VO2 with different morphologies and structures, which could be simply tailored by adjusting the solvothermal reaction duration. As the cathode materials for lithium-ion batteries, the electrode delivered reversible capacities of 119.2 and 87.3 mA h g−1 at high current densities of 2400 and 3600 mA g−1, respectively, as well as a capacity retention of 78.31% after 80 cycles at 1200 mA g−1. The excellent electrochemical performance could be attributed to the purity of the phase and the synergistic effect between the yolk–shell structure and hierarchical structure of the sub-microspheres. Thus, the three-dimensional nanostructures are more stable than the low-dimensional structures. Nanocomponents are less prone to agglomeration, and the active specific surface area can be maintained for a long time. As can be seen from Table 4, the three-dimensional structures show better cycling performance over prolonged cycling than the low-dimensional structures.
| Precursor material | Nanocomposites | Initial capacity/mA h g−1 | Current density/mA g−1 | Cycles (times)/capacity (mA h g−1) | References |
|---|---|---|---|---|---|
| NH4VO3 | Mesoporous carbon foam | 268 | 29.4 | 30/220 | 117 |
| NH4VO3 | Carbon | 292 | 29.4 | 30/261 | 118 |
| Vanadium pentoxide nanofibers | Amorphous carbon | 300 | 29.4 | 50/201 | 119 |
| V2O5 powder | Mesoporous carbon | 247 | 500 | 100/163 | 120 |
| V2O5 powder | Porous carbon | 297 | 294 | 50/288 | 122 |
| V2O5 powder | Multiwalled carbon nanotube | About 152 | 25 | 50/143 | 126 |
| Vanadium oxytriisopropoxide | Multiwalled carbon nanotubes | 285 | 294 | 100/190 | 127 |
| Sodium metavanadate (NaVO3) | Single-walled carbon nanotubes | 250 | 14.7 | 30/225 | 128 |
| V2O5 powder | Multiwalled carbon nanotube | 110 | 200 | 500/about 110 | 129 |
| V2O5 powder | Multiwalled carbon nanotubes | 238 | 50 | 50/151 | 130 |
| V2O5 nanoparticles | Multi-graphitic nanotubes | 224 | 150 | 200/211 | 131 |
| VO(acac)2 | CNTs | 250 | 200 | 40/190 | 132 |
| Vanadium isopropoxide | Multiwalled carbon nanotubes | 292 | 29.4 | 50/275 | 133 |
| V2O5 powder | Multiwalled carbon nanotubes | 243 | 50 | 50/209 | 135 |
| NH4VO3 | Carbon tube-in-tube | 280 | 58.8 | 20/265 | 136 |
| V2O5 powder | Multiwalled carbon nanotubes | 402 | 100 | 50/222 | 137 |
| Vanadium oxytriisopropoxide | Single-walled carbon nanotubes | About 598 | 300 | 40/548 | 138 |
| Ammonium metavanadate | Reduced graphene oxide (RGO) | 287 | 100 | 50/207 | 140 |
| Vanadium oxytriisopropoxide | Reduced graphene oxide (RGO) | 196 | 600 | 160/102 | 141 |
| VCl4 | Graphene nanoribbons | 278 | 29.4 | 100/217 | 142 |
| NH4VO3 | Graphene oxide | 160 | 70 | 200/133 | 143 |
| Vanadyl acetylacetonate | Reduced graphene oxide (RGO) | 225 | 58.8 | 60/125 | 144 |
| Vanadyl(IV) sulfate (VOSO4) | Graphene oxide | 240 | 100 | 20/220 | 145 |
| V2O5 powder | Reduced graphene oxide (RGO) | 235 | 20 | 100/171 | 146 |
| V2O5 powder | Graphene oxide | 190.9 | 100 | 150/182.4 | 147 |
| Vanadium(IV) acetylacetone | Graphene oxide | 255 | 100 | 100/153 | 148 |
3. V2O5-Based nanocomposites
In addition to phase-pure V2O5, V2O5-based nanocomposites have also been intensively studied as positive electrodes for LIBs. In this section, composites containing V2O5 and other materials will be discussed. In this part, V2O5 with different carbonaceous supports, including amorphous carbon, carbon nanotubes (CNTs), and graphene (Gr) will be surveyed (Table 4).3.1 V2O5 with amorphous carbon
Amorphous carbon is an inexpensive, frequently used carbonaceous material that is easy to produce in industrial quantities. In LIB applications, amorphous carbon is used to produce a conductive compact outer layer on the surface of V2O5 NPs, which not only serves as a buffer layer to accommodate the volume expansion during cycling but also contributes to the formation of a stable solid electrolyte interface (SEI) layer. In past research, the use of a carbon layer as a matrix buffer has exhibited superb properties. The carbon layer can also promote the electronic conductivity of the electrodes. As early as 2013, carbon-coated V2O5 nanoparticles,117 produced via a facile and scalable method via the route illustrated in Fig. 12, demonstrated a first charge capacity of 268 mA h g−1 and a capacity of 220 mA h g−1 in the 30th cycle at a current density of 29.4 mA g−1. This carbon-coated V2O5 showed improved cycling stability and rate performance compared to nanosized V2O5. This approach can be extended to enhance the electrochemical performances of other alternative cathodes. Shin and co-workers118 reported a facile and scalable method for the synthesis of carbon-coated V2O5 nanoparticles through a carboxylic acid-assisted sol–gel method and controlled calcination. NH4VO3 was used as the vanadium source. Oxalic acid (C2O4H2), tartaric acid (C4H6O6), and citric acid (C6H8O7) were used as both the carbon sources and chelating/reducing agents for preparing three different carbon-coated V2O5 composites. In the voltage window of 2.1–4.0 V, VCA11 had a discharge capacity of 292 mA h g−1 for the first cycle and a specific capacity of 261 mA h g−1 in the 30th cycle, with a fading rate of 0.36% per cycle. Cheah et al.119 developed a plasma-enhanced chemical vapour deposition (PECVD) method to synthesize carbon-coated V2O5 nanofibers. The carbon-coated VNFs exhibited better capacity retention characteristics than bare VNFs irrespective of the initial capacity values during prolonged cycling. Such an enhancement is mainly attributed to the homogeneous carbon coating by PECVD, which improves the electronic conducting profiles of VNF and prevents undesired side-reactions with electrolyte counterparts.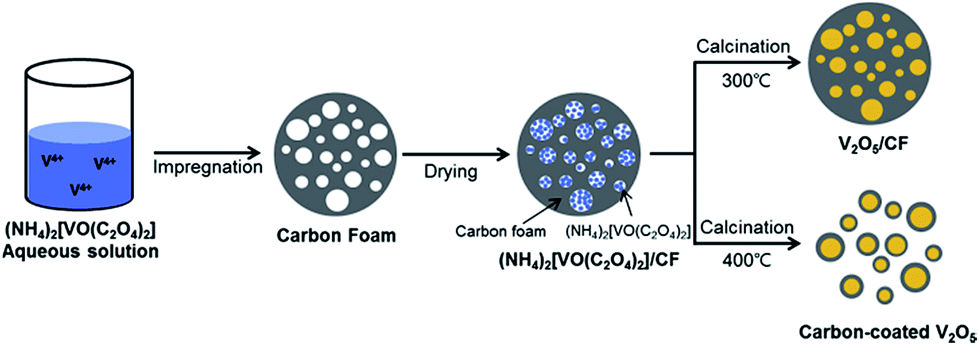 | ||
| Fig. 12 Schematic of the preparation of carbon-fabricated V2O5 samples (CF300 and CF400).117 | ||
Ihsan et al.120 have recently developed an ultrasound assisted method followed by sintering to fabricate a V2O5/mesoporous carbon composite. Mesoporous carbon, which has pore size between 2 and 50 nm, facilitates electrolyte diffusion into the bulk of the electrode material and hence provides fast transport channels for the conductive ions. Obviously, amorphous structures of carbon and lattice fringes of V2O5 are observed. After sintering, the as-prepared V2O5/mesoporous carbon showed good lithium storage properties with a high reversible capacity of 163 mA h g−1 after 100 cycles. The capacity retention of V2O5/mc was considerably higher than that of V2O5 np. Yu et al.121 rationally designed and synthesized V2O5/ordered mesoporous carbon (CMK-3) composites via an ultrasonic method. The results showed that the ultrasonic synthesis method was flexible and efficient to highly disperse V2O5 nanoparticles in CMK-3. Zhang's group122 reported that carbon-coated V2O5 nanocrystals could be obtained via a unique capillary-induced filling strategy. The obtained nanocrystals exhibited markedly enhanced rate capability and excellent cyclability when used as the cathode material for Li-ion batteries. Guo et al.123 demonstrated that the electrochemical performance and stability of V2O5 nanowires could be significantly improved by coating a thin carbon layer as the shell. The V2O5@C nanowires exhibited substantially enhanced capacitive performance compared to that of V2O5 nanowires.
3.2 V2O5 with carbon nanotubes
Carbon nanotubes (CNTs) are allotropes of carbon with a cylindrical nanostructure. They find applications in LIB electrode materials owing to their extraordinary thermal, mechanical, and electrical properties. CNTs can be categorized as single-walled nanotubes (SWCNTs) and multi-walled nanotubes (MWCNTs). SWCNTs have high conductivity (≈104 S cm−1) and provide electronic conduction without blocking electrolyte access to the active material. MWCNTs show high mechanical and chemical stability, and the mesoporous character favors the diffusion of the reacting species.124,125 CNTs are generally employed as conducting agents to replace carbon black when used in the electrode of LIBs. Nanocomposites containing V2O5 and CNTs are reported to have better lithium storage properties than bare V2O5 materials. This can be attributed to the flexible nature of the CNTs, which alleviate the internal stress caused during the charge–discharge process. With increased conductivity and surface area, such nanocomposites show enhanced lithium storage capability.By early 2011, Seng et al.126 prepared free-standing V2O5/MWCNT films by a hydrothermal technique and filtration of ultra-long nanowires. The MWCNTs not only acted as conductivity modifiers, but also contributed to forming an integrated web-like structure. Yu's group127 proposed a facile hydrothermal approach to fabricate CNTs covered by interconnected V2O5 nanosheets, as illustrated in Fig. 13. After calcination in air, flakelike nanosheets of V2O5 could be observed, which were highly interconnected and covered the CNT skeleton. The interconnected V2O5-NSs were in intimate contact with the CNTs, which was favorable for the enhancement of conductivity. The as-derived CNTs@V2O5 hybrid structure showed a larger reversible capacity over 100 cycles than the V2O5-mf sample. In the voltage window of 2.0–4.0 V, the CNTs@V2O5 electrode delivered a capacity of 285 and 190 mA h g−1 during the 1st and 100th cycle at the current rate of 1C, respectively. Zhou and co-workers130 used low-cost V2O5 powders and H2O2 as the raw materials to synthesize vanadium oxide nanosheet–MWCNT composite by a simple sol–gel method and hydrothermal process. It was found that V4+ was the predominant V element of the vanadium oxide nanosheet. The addition of carbon nanotubes improved the conductivity of the vanadium oxide nanosheets, exhibiting higher specific capacity and cycling stability than materials without carbon nanotubes. When the batteries were cycled between 1.5 V and 4 V at a constant current density of 50 mA g−1, the maximum discharge capacity of the vanadium oxide nanosheet–MWCNT composite was 238 mA h g−1 and 151 mA h g−1, respectively, after the 50th cycle. The good performance of the vanadium oxide nanosheet–MWCNT composite can be attributed to the sheet-like nanostructure having a large specific surface area and good structural flexibility, which can provide more Li+ ion intercalation sites and accommodate large volume variations.
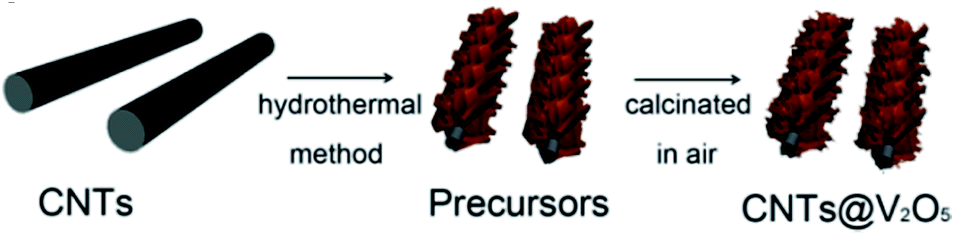 | ||
| Fig. 13 Schematic of the synthesis process for CNTs@V2O5.127 | ||
In contrast to the conventional strategy of coating the exterior and/or filling the interior of CNTs with V2O5, Chen et al.134 proposed a novel strategy to fabricate MWCNT/V2O5 core/shell nanostructures. They first used chemical vapor deposition (CVD) to grow an MWCNT sponge in a quartz tube at 860 °C, which had very low density and high porosity. Then the sponge was cut into the desired size and placed in a commercial ALD reactor, where 1000 cycles of H2O-based ALD V2O5 was deposited on it. The as-derived MWCNT/V2O5 core/shell nanostructures showed excellent electrochemical performance. Zhou's group137 have developed a simple hydrothermal route and subsequent post-sintering to fabricate a novel MWCNTs–V2O5 composite with nanosized architecture. During the hydrothermal reaction, protonated hexadecylamine acted as an intermediator. The unique porous nanoarchitecture of MWCNTs–V2O5 provided a large specific surface area and a good conductive network, which facilitated fast lithium ion diffusion and electron transfer. Additionally, the uniformly dispersed MWCNTs conducting network also behaved as an effective buffer that could relax the strain generated during charge–discharge cycles. The as-prepared sample exhibited an initial specific capacity of 402 mA h g−1, and it could remain 222 mA h g−1 after 50 cycles.
3.3 V2O5 with graphene
The use of graphene can also enhance the cycling rate and electrochemical property of V2O5. As an important 2D carbonaceous material, graphene has quickly become an important focus of material science. It has good flexibility, high surface area, and excellent thermal and chemical stability and electrical conductivity. Compared with other carbon materials like amorphous carbon, graphite, and other carbon nanomaterials, graphene nanosheets (GNSs) can provide a higher buffer property for the deformation of active anode materials due to their flexibility reaction path and high volume expansion area; graphene also exhibits a better dispersion in nanotubes and nanoparticles.139Recently, Chen et al.140 successfully prepared reduced graphene oxide (rGO)-encapsulated V2O5 nanocomposites by co-assembly between negatively charged GO and positively charged oxide nanorods (Fig. 14). The process was driven by the mutual electrostatic interactions of the two species and was followed by thermal reduction. The as-synthesized nanocomposites possessed flexible and ultrathin rGO shells that effectively enwrapped the oxide nanorods. After several electrochemical tests, the nanocomposites presented excellent results. In the voltage window of 2.0–4.0 V, the cathode achieved an initial charge and discharge capacity of 287 mA h g−1 and 207 mA h g−1 during the first and 50th cycles at a current density of 100 mA g−1, respectively. Cheng's group141 reported a simple solvothermal method to directly self-assemble V2O5 nanosheets on reduced graphene oxide (rGO). The V2O5 nanosheets/rGO hierarchical nanocomposites exhibited high reversible capacity and good rate capability compared to the bulk material by taking advantage of the synergetic effect of the two components.
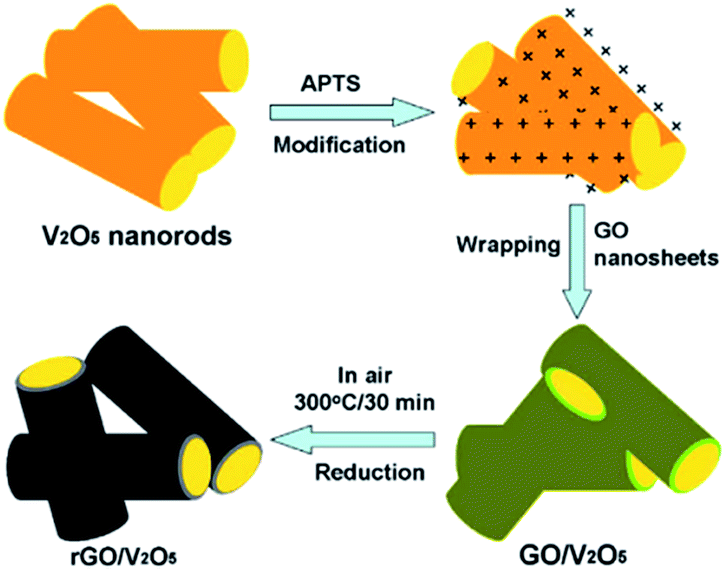 | ||
| Fig. 14 Schematic of the fabrication of rGO enwrapped V2O5 nanorods.140 | ||
Sun et al.143 developed a hydrothermal method to fabricate hydrated vanadium pentoxide nanoribbons modified with reduced graphene oxide (rGO). The intertwining network structure provided efficient electron conduction pathways and short Li+ diffusion distances. Su and co-workers147 proposed a hydrothermal method to synthesize flexible V2O5 nanowires and then V2O5/graphene composites were prepared via the dry-freezing step. It was found that the V2O5 nanowires were uniformly decorated on the surface of the graphene sheets. The unique nanocomposite expectedly exhibited an enhanced performance compared to pure V2O5 nanowires and V2O5/graphene composites. Chen et al.148 developed a facile solvothermal approach to synthesize the composite of V2O5 nanoparticles anchored on graphene. The as-prepared sample showed higher capacity and better cyclic capacity retention compared to bare V2O5, delivering a high reversible capacity of 153 mA h g−1 after 100 cycles.
4. V2O5-Metal composites
In order to overcome the shortcomings encountered in the application of V2O5 and improving its electrochemical performance, in addition to the above-mentioned nanocrystalline composites with different carbonaceous supports, cation doping has been an effective approach to improve lithium ion intercalation in V2O5, mainly because the introduction of alien ions into the V2O5 lattice may tune the ion occupation and the electronic structure of the host material.149 Recently, utilization of metal nanoparticles to improve the electrical conductivity has attracted extensive attention. Crystal structure stability is important for the V2O5 cathode, which can be stabilized by doping of metallic elements such as Al,150,151 Cu,152,158,163 Fe,153 Cr,154 Mn,155,156,159 Sn,157,164 Ag,161 Mo,160 and Ni.162 Table 5 summarizes the electrochemical properties of the cation doped V2O5 samples.| Precursor material | Doped metal | Initial capacity/mA h g−1 | Current density/mA g−1 | Cycles (N)/capacity (mA h g−1) | References |
|---|---|---|---|---|---|
| Vanadyl acetylacetonate | Al | 250 | 35 | 50/157.5 | 150 |
| V2O5 powders | Al | 216 | 150 | 50/162 | 151 |
| V2O5 powders | Cu | 229 | 100 | 60/160 | 152 |
| NH4VO3 | Fe | 255 | 58.8 | 48/195 | 153 |
| V2O5 powders | Cr | About 272 | 29 | 50/200 | 154 |
| V2O5 powders | Mn | 138 | 680 | 50/135 | 155 |
| Sodium metavanadate | Mn | 120 | 50 | 30/about 120 | 156 |
| NH4VO3 | Sn | 251.1 | 200 | 50/212 | 157 |
| NH4VO3 | Cu | 196 | 300 | 70/186 | 158 |
| VOSO4·3H2O | Mn | 251 | 300 | 50/201 | 159 |
| V2O5 powders | Mo | 203 | 44.1 | 50/about 92 | 160 |
| V2O5 powders | Ag | 266.1 | 250 | 50/276.4 | 161 |
| NH4VO3 | Ni | 262 | 300 | 50/238 | 162 |
| V2O5 powders | Cu | 266 | 58.8 | 50/226 | 163 |
| V2O5 powders | Sn | 350 | 500 | 40/355 | 164 |
Wei et al.152 proposed a precipitation method followed by heat treatment to synthesize crystalline Cu-doped V2O5. Cu0.04V2O5 showed better electrochemical performance than V2O5 because of its high electronic conductivity and good structural stability. Li and co-workers153 employed electrostatic spray deposition technique to synthesize 3D porous Fe0.1V2O5.15 thin films. The thin films were composed of rather porous spheres with diameters around 8 and 10 μm for the Fe-doped samples. In the voltage window of 2.0–4.0 V, the cathode obtained an initial charge and discharge capacity of 255 mA h g−1 and 195 mA h g−1 during the first and 48th cycles at a current density of 58.8 mA g−1, respectively. We could ascribe the better cycling performance of the Fe0.1V2O5.15 electrode to the improved stability of its layered structure. Li's group157 developed a microwave-assisted solvothermal synthesis to obtain a series of Sn-doped V2O5 microspheres. The doping of Sn4+ also induced the formation of oxygen vacancies that would allow more active sites for the intercalation/extraction reactions of Li+. Therefore, the as-prepared sample showed excellent electrochemical properties. It could be attributed primarily to the lattice expansion upon doping that led to higher lithium diffusion coefficient than that of V2O5 as well as the presence of oxygen-rich vacancies.
Zou and co-workers158 developed a simple chemical reaction combined with ultrasonic mixing to synthesize nanostructured composites of V2O5 spheres decorated by electric Cu NPs. The decorated Cu NPs resulted in good contact with active materials and facilitated transportation of the electron into the inner region of the electrode. As expected, the V2O5/Cu cathodes could afford an obviously better electrochemical performance compared to pure V2O5, with a high reversible capacity of 186 mA h g−1 after 70 cycles under a current density of 300 mA g−1 and good rate performance. Zheng's group162 developed a facile template-free nickel-mediated polyol process to prepare a high-performance V2O5 hollow microsphere for LIBs, in which nickel acted not only as the mediator to tailor the interior hollow structures of V2O5 and build a favorable 3D hierarchical nano-micron combined architecture, but also as doping units to tune vanadium valence states and improve lithium storage properties. Similarly, V2O5 doped with Cu2+ demonstrated excellent cycling stability and rate capability owing to its modified electronic conductivity and improved structural stability.163 Li et al.164 developed a sol–gel method to synthesize the homogeneous Sn-doped V2O5 sol. Preliminary three-electrode tests of the Sn-doped V2O5 revealed good cycling performance. It was believed that Sn4+ would occupy the interstitial positions between VO5 slabs and form SnO6 octahedra with oxygen, leading to a slightly expanded lattice that facilitated Li+ intercalation/extraction.
5. Conclusion and outlook
We attempt to provide a comprehensive review about V2O5-based nanomaterials as cathode materials for lithium-ion batteries. Starting with phase-pure V2O5, we have covered a wide range of nanostructures, from 1D nanorods/nanowires/nanotubes/nanofibers/nanobelts to 2D nanosheets and 3D hollow and porous structures. For each category, we have discussed about the synthesis and lithium storage properties. For the 1D nanostructured materials, the most common synthesis methods are the hydrothermal method and sol–gel method. Hence, their preparation process is simple and feasible, and the requirements of the experimental conditions are not too high. For 2D V2O5 nanosheets, the most common synthesis method is the hydrothermal method. Template-free and template-based methods are the main approaches for the synthesis of V2O5 hollow and porous structures. However, all three categories of V2O5 nanomaterials demonstrate significantly better electrochemical properties than bare V2O5 nanoparticles. These can be attributed to the following aspects. First, the nanostructures promote sufficient contact between the electrolyte and active materials. Hence, the nanostructures facilitate rapid electron transfer and mostly avoid the generation of high contact resistance, like a nanoparticle aggregated structure, thus ensuring satisfactory capacity retention even at high current densities. Finally, the existence of pyrolysed carbon improves the conductivity of the one-dimensional nanostructures.Nanocomposites consisting of V2O5 and different carbonaceous supports have also been surveyed. Amorphous carbon, graphene, and carbon nanotubes are widely used as supports for electroactive V2O5. In such systems, V2O5 is usually decorated on graphene and CNTs, while amorphous carbon is hydrothermally coated on pre-synthesized V2O5 nanostructures. As potential cathode materials for LIBs, the V2O5 and different carbonaceous composites exhibit highly reversible capacities and better cycling performance. This excellent electrochemical performance is ascribed to their unique structures, which improve electrolyte infiltration and facilitate Li ion diffusion in the electrode. In addition, cation doping of lithium-ion battery (LIBs) cathode materials is beneficial to both ion diffusion and charge transfer in the electrochemical intercalation processes and hence may improve battery rate capability. As the cathode material for lithium-ion batteries, the cation-doped V2O5 samples exhibit better electrochemical performance compared than undoped ones.
By synthesizing V2O5 nanomaterials of various structures, low electrical conduction, slow Li+ diffusion and irreversible phase transitions upon deep discharge can be alleviated to a certain extent. The cycling performance can be further improved effectively via creating unique nanocomposites. The significant progress achieved in the past decade re-assures the promising use of V2O5 as the cathode material in high-performance lithium-ion batteries. It is reasonable to confirm that V2O5-based nanocomposites with good electrochemical properties will find wide use in high-performance electrochemical energy storage devices in the near future. Nonetheless, it will still take a long time before metal oxide-based positive electrodes can be incorporated into commercial lithium-ion batteries. While the fundamental electrochemical processes are well understood for V2O5-based materials, the breakthrough will likely come from the delicate design and synthesis of nanocomposite electrode materials. Future research needs to deeply understand the structural evolution during cycling, possibly through in situ observation techniques and elaborate analysis of their atomic structure.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by a grant from the National Natural Science Foundation of China (No. 61504080 and No. 51676130).References
- N. S. Choi, Z. H. Chen, S. A. Freunberger, X. L. Ji, Y.-K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994–10024 CrossRef CAS PubMed.
- X. Y. Lai, J. E. Halperta and D. Wang, Energy Environ. Sci., 2012, 5, 5604–5618 CAS.
- V. Mathew, S. Kim, J. Kang, J. Gim, J. Song, J. Baboo, W. Park, D. Ahn, J. Han, L. Gu, Y. Wang, Y. Hu, Y. Sun and J. Kim, NPG Asia Mater., 2014, 6, 138–147 CrossRef.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- R. Marom, S. F. Amalraj, N. Leifer, D. Jacob and D. Aurbach, J. Mater. Chem., 2011, 21, 9938–9954 RSC.
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193–2203 CrossRef CAS.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS.
- F. Cheng, J. Liang, Z. Tao and J. Chen, Adv. Mater., 2011, 23, 1695–1715 CrossRef CAS PubMed.
- B. Xu, D. Qian, Z. Wang and Y. S. Meng, Mater. Sci. Eng., R, 2012, 73, 51–65 CrossRef CAS.
- J. Li, J. Klee Barillas, C. Guenther and M. A. Danzer, J. Power Sources, 2013, 230, 244–250 CrossRef CAS.
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Energy Environ. Sci., 2012, 5, 7854–7863 CAS.
- A. P. Tang, X. Y. Wang, G. R. Xu, Z. H. Zhou and H. D. Nie, Mater. Lett., 2009, 63, 1439–1441 CrossRef CAS.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacin, Adv. Mater., 2010, 22, 170–192 CrossRef PubMed.
- A. S. Aricò, P. Bruce, B. Scrosati, J. M. Tarascon and S. W. Van, Nat. Mater., 2005, 4, 366–377 CrossRef PubMed.
- H. Li, Z. X. Wang, L. Q. Chen and X. J. Huang, Adv. Mater., 2009, 21, 4593–4607 CrossRef CAS.
- X. F. Zhang, H. H. Zheng, V. Battaglia and R. L. Axelbaum, J. Power Sources, 2011, 196, 3640–3645 CrossRef CAS.
- Y. Wang, Y. Wang, E. Hosono, K. Wang and H. Zhou, Angew. Chem., Int. Ed., 2008, 47, 7461–7465 CrossRef CAS PubMed.
- L. Mai, Y. Dong, L. Xu and C. Han, Nano Lett., 2010, 10, 4273–4278 CrossRef CAS PubMed.
- B. Scrosati, J. Hassoun and Y. K. Sun, Energy Environ. Sci., 2011, 4, 3287–3295 CAS.
- N. Alias and A. A. Mohamad, J. Power Sources, 2015, 274, 237–251 CrossRef CAS.
- Y. X. Wang, B. Liu, Q. Y. Li, S. Cartmell, S. Ferrara, Z. D. Deng and J. Xiao, J. Power Sources, 2015, 286, 330–345 CrossRef CAS.
- W. Waag, C. Fleischer and D. U. Sauer, J. Power Sources, 2014, 258, 321–339 CrossRef CAS.
- S. Goriparti, E. Miele, F. D. Angelis, E. D. Fabrizio, R. P. Zaccaria and C. Capiglia, J. Power Sources, 2014, 257, 421–443 CrossRef CAS.
- M. Okubo, E. J. Hosono, J. Kim, M. Enomoto, N. Kojima, T. Kudo, H. S. Zhou and I. Honma, J. Am. Chem. Soc., 2007, 129, 7444–7452 CrossRef CAS PubMed.
- K. S. Tan, M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, J. Power Sources, 2005, 147, 241–248 CrossRef CAS.
- X. Xiong, D. Ding, Y. Bu, Z. Wang, B. Huang, H. Guo and X. Li, J. Mater. Chem. A, 2014, 2, 11691–11696 CAS.
- X. Xiong, Z. Wang, G. Yan, H. Guo and X. Li, J. Power Sources, 2014, 245, 183–193 CrossRef CAS.
- R. Saroha, A. Gupta and A. Panwar, J. Alloys Compd., 2017, 696, 580–589 CrossRef CAS.
- Q. Liu, D. Mao, C. Chang and F. Huang, J. Power Sources, 2007, 173, 538–544 CrossRef CAS.
- C. W. Sun, S. Rajasekhara, J. B. Goodenough and F. Zhou, J. Am. Chem. Soc., 2011, 133, 2132–2135 CrossRef CAS PubMed.
- V. H. Nguyen, E. M. Jin and H. B. Gu, J. Power Sources, 2013, 244, 586–591 CrossRef CAS.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- B. Kang and G. Ceder, Nature, 2009, 458, 190–193 CrossRef CAS PubMed.
- R. Baddour-Hadjean, C. Navone and J. P. Pereira-Ramos, Electrochim. Acta, 2009, 54, 6674–6679 CrossRef CAS.
- N. A. Chernova, M. Roppolo, A. C. Dillon and M. S. Whittingham, J. Mater. Chem., 2009, 19, 2526–2552 RSC.
- D. McNulty, D. N. Buckley and C. O'Dwyer, J. Power Sources, 2014, 267, 831–873 CrossRef CAS.
- J. Mendialdua, R. Casanova and Y. Barbaux, J. Electron Spectrosc. Relat. Phenom., 1995, 71, 249–261 CrossRef CAS.
- C. Delmas, H. Cognac-Auradou, J. Cocciantelli, M. Menetrier and J. Doumerc, Solid State Ionics, 1994, 69, 257–264 CrossRef CAS.
- L. Q. Ma, J. Mater. Res., 2011, 26, 2175–2185 CrossRef.
- C. Z. Wu and Y. Xie, Energy Environ. Sci., 2010, 3, 1191–1206 CAS.
- H. L. Fei, H. J. Zhou, J. G. Wang, P. C. Sun, D. T. Ding and T. H. Chen, Solid State Sci., 2008, 10, 1276–1284 CrossRef CAS.
- A. D. Raj, T. Pazhanivel, P. S. Kumar, D. Mangalaraj, D. Nataraj and N. Ponpandian, Curr. Appl. Phys., 2010, 10, 531–537 CrossRef.
- W. Zeng, W. G. Chen, Z. Y. Li, H. Zhang and T. M. Li, Mater. Res. Bull., 2015, 65, 157–162 CrossRef CAS.
- M. Kovendhan, D. P. Joseph, P. Manimuthu, A. Sendilkumar, S. N. Karthick, S. Sambasivam, K. Vijayarangamuthu, H. J. Kim, B. C. Choi, K. Asokan, C. Venkateswara and R. Mohan, Curr. Appl. Phys., 2015, 15, 622–631 CrossRef.
- A. J. Waldir, C. Ribeiro, E. R. Leite and V. R. Mastelaro, Cryst. Growth Des., 2009, 9, 3626–3631 Search PubMed.
- T. Y. Zhai, H. M. Liu, H. Q. Li and D. Golberg, Adv. Mater., 2010, 22, 2547–2552 CrossRef CAS PubMed.
- L. J. Mao, C. Y. Liu and J. Li, J. Mater. Chem., 2008, 18, 1640–1643 RSC.
- J. Liu, Y. C. Zhou, J. B. Wang, Y. Pan and D. Xue, Chem. Commun., 2011, 47, 10380–10382 RSC.
- G. M. Zhu, Z. B. Qu, G. L. Zhuang, Q. Xie, Q. Q. Meng and J. G. Wang, J. Phys. Chem. C, 2011, 115, 14806–14811 CAS.
- Y. Wang, H. J. Zhang, W. X. Lim, J. Y. Lin and C. C. Wong, J. Mater. Chem., 2011, 133, 2362 RSC.
- A. M. Glushenkov, V. I. Stukachev, M. F. Hassan, G. G. Kuvshinov, H. K. Liu and Y. Chen, Cryst. Growth Des., 2008, 8, 3661–3665 CAS.
- C. Zhu, J. Shu, X. Wu, P. Li and X. Li, J. Electroanal. Chem., 2015, 759, 184–189 CrossRef CAS.
- C. Peng, F. Xiao, J. Yang, Z. H. Li, G. T. Lei, Q. Z. Xiao, Y. H. Ding and Z. L. Hu, Electrochim. Acta, 2016, 192, 216–226 CrossRef CAS.
- A. M. Glushenkov, M. F. Hassan, V. I. Stukachev, Z. P. Guo, H. K. Liu, G. Kuvshinov and Y. I. Chen, J. Solid State Electrochem., 2010, 14, 1841–1846 CrossRef CAS.
- K. Takahashi, S. J. Limmer, Y. Wang and G. Z. Cao, J. Phys. Chem. B, 2004, 108, 9795–9800 CrossRef CAS.
- Q. Shi, J. W. Liu, R. Z. Hu, M. Q. Zeng, M. J. Dai and M. Zhu, RSC Adv., 2012, 2, 7273–7278 RSC.
- V. S. R. Channu, R. Holze and B. Rambabu, Curr. Appl. Phys., 2013, 13, 237–240 CrossRef.
- S. SarKar, H. Banda and S. Mitra, Electrochim. Acta, 2013, 99, 242–252 CrossRef CAS.
- P. Yang and C. M. Lieber, Science, 1996, 273, 1836–1840 CrossRef CAS.
- D. K. Kim, P. Muralidharan, H. W. Lee, R. Ruffo, Y. Yang, C. K. Chan, H. L. Peng, R. A. Huggins and Y. Cui, Nano Lett., 2008, 8, 3948–3952 CrossRef CAS PubMed.
- H. W. Lee, P. Muralidharan, R. Ruffo, C. M. Mari, Y. Cui and D. K. Kim, Nano Lett., 2010, 10, 3852–3856 CrossRef CAS PubMed.
- E. Hosono, H. Matsuda, T. Saito, T. Kudo, M. Ichihara, I. Honma and H. S. Zhou, J. Power Sources, 2010, 195, 7098–7101 CrossRef CAS.
- L. Q. Mai, L. Xu, C. H. Han, X. Xu, Y. Z. Luo, S. Y. Zhao and Y. L. Zhao, Nano Lett., 2010, 10, 4750–4755 CrossRef CAS PubMed.
- J. M. Velazquez and S. Banerjee, Small, 2009, 5, 1025–1029 CrossRef CAS PubMed.
- M. C. Wu and C. S. Lee, J. Solid State Electrochem., 2009, 182, 2285–2289 CrossRef CAS.
- T. Y. Zhai, H. M. Liu, H. Q. Li, X. S. Fang, M. Y. Liao, L. Li, H. S. Zhou, Y. Koide, Y. Bando and D. Golberg, Adv. Mater., 2010, 22, 2547–2552 CrossRef CAS PubMed.
- A. K. Ramasami, M. V. Reddy, P. Nithyadharseni, B. V. R. Chowdari and G. R. Balakrishna, J. Alloys Compd., 2017, 695, 850–858 CrossRef CAS.
- Y. Tang, X. Rui, Y. Zhang, T. M. Lim, Z. Dong, H. H. Hng, X. Chen, Q. Yan and Z. Chen, J. Mater. Chem. A, 2013, 1, 82–88 CAS.
- Y. Chen, G. Yang, Z. Zhang, X. Yang, W. Hou and J. J. Zhu, Nanoscale, 2010, 2, 2131–2138 RSC.
- Y. Wang, K. Takahashi, H. M. Shang and G. Z. Cao, J. Phys. Chem. B, 2005, 109, 3085–3088 CrossRef CAS PubMed.
- C. J. Cui, G. M. Wu, J. Shen, B. Zhou, Z. H. Zhang, H. Y. Yang and S. F. She, Electrochim. Acta, 2010, 55, 2536–2541 CrossRef CAS.
- J. L. Sun, Mater. Lett., 2013, 108, 193–195 CrossRef CAS.
- R. Nadimicherla, Y. L. Liu, K. Q. Chen and W. Chen, Microelectron. Eng., 2014, 127, 81–85 CrossRef CAS.
- Z. T. Li, G. X. Liu, M. Guo, L. X. Ding, S. Q. Wang and H. H. Wang, Electrochim. Acta, 2015, 173, 131–138 CrossRef CAS.
- D. M. Yu, C. G. Chen, S. H. Xie, Y. Y. Liu, K. Park, X. Y. Zhou, Q. F. Zhang, J. Y. Li and G. Z. Cao, Energy Environ. Sci., 2011, 4, 858–861 CAS.
- Y. L. Cheah, N. Gupta, S. S. Pramana, V. Aravindan, G. Wee and M. Srinivasan, J. Power Sources, 2011, 196, 6465–6472 CrossRef CAS.
- B. Yan, X. F. Li, Z. M. Bai, M. S. Li, L. Dong, D. B. Xiong and D. J. Li, J. Alloys Compd., 2015, 634, 50–57 CrossRef CAS.
- K. Dewangan, N. N. Sinha, P. G. Chavan, P. K. Sharma, A. C. Pandey, M. A. More, D. S. Joag, N. Munichandraiah and N. S. Gajbhiye, Nanoscale, 2012, 4, 645–651 RSC.
- M. L. Qin, Q. Liang, A. Q. Pan, S. Q. Liang, Q. Zhang, Y. Tang and X. P. Tan, J. Power Sources, 2014, 268, 700–705 CrossRef CAS.
- C. L. Niu, J. B. Li, H. B. Jin, H. L. Shi, Y. Q. Zhi, W. Z. Wang and M. S. Cao, Electrochim. Acta, 2015, 182, 621–628 CrossRef CAS.
- P. P. Wang, Y. X. Yao, C. Y. Xu, L. Wang, W. He and L. Zhen, Ceram. Int., 2016, 42, 14595–14600 CrossRef CAS.
- X. H. Rui, Y. X. Tang, O. I. Malyi, A. Gusak, Y. Y. Zhang, Z. Q. Liu, H. T. Tan, C. Persson, X. D. Chen, Z. Chen and Q. Y. Yan, Nano Energy, 2016, 22, 583–593 CrossRef CAS.
- J. F. Huang, X. N. Qiao, Z. W. Xu, L. Y. Cao, H. Ouyang, J. Y. Li and R. Y. Wang, Electrochim. Acta, 2016, 191, 158–164 CrossRef CAS.
- X. Rui, Z. Lu, H. Yu, D. Yang, H. H. Hng, T. M. Lim and Q. Yan, Nanoscale, 2013, 5, 556–560 RSC.
- Q. Y. An, Q. L. Wei, L. Q. Mai, J. Y. Fei, X. Xu, Y. L. Zhao, M. Y. Yan, P. F. Zhang and S. Z. Huang, Phys. Chem. Chem. Phys., 2013, 15, 16828–16833 RSC.
- S. Q. Liang, M. L. Qin, Y. Tang, Q. Zhang, X. L. Li, X. P. Tan and A. Q. Pan, Met. Mater. Int., 2014, 20, 983–988 CrossRef CAS.
- H. Q. Song, C. P. Zhang, Y. G. Liu, C. F. Liu, X. H. Nan and G. Z. Cao, J. Power Sources, 2015, 294, 1–7 CrossRef CAS.
- Y. Xu, M. Dunwell, L. Fei, E. G. Fu, Q. L. Lin, B. Patterson, B. Yuan, S. G. Deng, P. Andersen, H. M. Luo and G. F. Zhou, ACS Appl. Mater. Interfaces, 2014, 6, 20408–20413 CAS.
- S. Q. Liang, Y. Hu, Z. W. Nie, H. Huang, T. Chen, A. Q. Pan and G. Z. Cao, Nano Energy, 2015, 13, 58–66 CrossRef CAS.
- Y. W. Li, J. H. Yao, E. Uchaker, J. W. Yang, Y. X. Huang, M. Zhang and G. Z. Cao, Adv. Energy Mater., 2013, 3, 1171–1175 CrossRef CAS.
- A. Q. Pan, H. B. Wu, L. Zhang and X. W. Lou, Energy Environ. Sci., 2013, 6, 1476–1479 CAS.
- H. Y. Wu, M. L. Qin, X. L. Li, Z. Q. Cao, B. R. Jia, Z. L. Zhang, D. Y. Zhang, X. H. Qu and A. A. Volinsky, Electrochim. Acta, 2016, 206, 301–306 CrossRef CAS.
- L. Q. Mai, Q. Y. An, Q. L. Wei, J. Y. Fei, P. F. Zhang, X. Xu, Y. L. Zhao, M. Y. Yan, W. Wen and L. Xu, Small, 2014, 10, 3032–3037 CrossRef CAS PubMed.
- X. Y. Zhang, J. G. Wang, H. Y. Liu, H. Z. Liu and B. Q. Wei, Materials, 2017, 10, 77–85 CrossRef PubMed.
- E. Uchaker, N. Zhou, Y. W. Li and G. Z. Cao, J. Phys. Chem. C, 2013, 117, 1621–1626 CAS.
- A. M. Cao, J. S. Hu, H. P. Liang and L. J. Wan, Angew. Chem., Int. Ed., 2005, 44, 4391–4395 CrossRef CAS PubMed.
- M. H. Chen, X. H. Xia, J. F. Yuan, J. H. Yin and Q. G. Chen, J. Power Sources, 2015, 288, 145–149 CrossRef CAS.
- C. Q. Feng, S. Y. Wang, R. Zeng, Z. P. Guo, K. Konstantinov and H. K. Liu, J. Power Sources, 2008, 184, 485–488 CrossRef CAS.
- H. E. Wang, D. S. Chen, Y. Cai, R. L. Zhang, J. M. Xu, Z. Deng, X. F. Zheng, Y. Li, I. Bello and B. L. Su, J. Colloid Interface Sci., 2014, 418, 74–80 CrossRef CAS PubMed.
- Q. Y. An, P. F. Zhang, F. Y. Xiong, Q. L. Wei, J. Z. Sheng, Q. Q. Wang and L. Q. Mai, Nano Res., 2015, 8, 481–490 CrossRef CAS.
- C. F. Zhang, Z. X. Chen, Z. P. Guo and X. W. Lou, Energy Environ. Sci., 2013, 6, 974–978 CAS.
- Y. J. Dong, H. Y. Wei, W. Liu, Q. J. Liu, W. J. Zhang and Y. Z. Yang, J. Power Sources, 2015, 285, 538–542 CrossRef CAS.
- H. W. Bai, Z. Y. Liu, D. D. Sun and S. H. Chan, Energy, 2014, 76, 607–613 CrossRef CAS.
- A. Q. Pan, H. B. Wu, L. Yu, T. Zhu and X. W. Lou, ACS Appl. Mater. Interfaces, 2012, 4, 3874–3879 CAS.
- A. Q. Pan, H. B. Wu, L. Zhang and X. W. Lou, Energy Environ. Sci., 2013, 6, 1476–1479 CAS.
- X. F. Zhang, M. Z. Wu, S. Gao, Y. M. Xu, X. L. Cheng, H. Zhao and L. H. Huo, Mater. Res. Bull., 2014, 60, 659–664 CrossRef CAS.
- L. Chen, X. Gu, X. L. Jiang, N. N. Wang, J. Yue, H. Y. Xu, J. Yang and Y. T. Qian, J. Power Sources, 2014, 272, 991–996 CrossRef CAS.
- J. Liu, Y. C. Zhou, J. B. Wang, Y. Pan and D. F. Xue, Chem. Commun., 2011, 47, 10380–10382 RSC.
- Y. Ma, A. Huang, H. Zhou, S. Ji, S. Zhang, R. Li, H. Yao, X. Cao and P. Jin, J. Mater. Chem. A, 2017, 5, 6522–6531 CAS.
- H. B. Wu, A. Q. Pan, H. H. Hng and X. W. Lou, Adv. Funct. Mater., 2013, 23, 5669–5674 CrossRef CAS.
- X. Liu, Y. J. Hu, G. Q. Jia, H. X. Zhang, H. Jiang and C. Z. Li, J. Mater. Chem. A, 2016, 4, 12030–12035 CAS.
- A. Q. Pan, H. B. Wu, L. Yu and X. W. Lou, Angew. Chem., Int. Ed., 2013, 52, 2226–2230 CrossRef CAS PubMed.
- J. Y. Wang, H. J. Tang, L. J. Zhang, H. Ren, R. B. Yu, Q. Jin, J. Qi, D. Mao, M. Yang, Y. Wang, P. Liu, Y. Zhang, Y. R. Wen, L. Gu, G. H. Ma, Z. G. Su, Z. Y. Tang, H. J. Zhao and D. Wang, Nat. Energy, 2016, 1, 16050–16057 CrossRef CAS.
- A. Q. Pan, T. Zhu, H. B. Wu and X. W. Lou, Chem.–Eur. J., 2013, 19, 494–500 CrossRef CAS PubMed.
- J. Shin, H. Jung, Y. Kim and J. Kim, J. Alloys Compd., 2014, 589, 322–329 CrossRef CAS.
- J. Shin, T. Kim, J. Je, T. You and J. Kim, Electrochim. Acta, 2014, 139, 408–414 CrossRef CAS.
- Y. L. Cheah, R. V. Hagen, V. Aravindan, R. Fiz, S. Mathur and S. Madhavi, Nano Energy, 2013, 2, 57–64 CrossRef CAS.
- M. Ihsan, Q. Meng, L. Li, D. Li, H. Q. Wang, K. H. Seng, Z. X. Chen, S. J. Kennedy, Z. P. Guo and H. K. Liu, Electrochim. Acta, 2015, 173, 172–177 CrossRef CAS.
- L. Yu, C. X. Zhao, X. Long and W. Chen, Microporous Mesoporous Mater., 2009, 126, 58–64 CrossRef CAS.
- X. F. Zhang, K. X. Wang, X. Wei and J. S. Chen, Chem. Mater., 2011, 23, 5290–5292 CrossRef CAS.
- Y. Guo, J. Li, M. D. Chen and G. Z. Gao, J. Power Sources, 2015, 273, 804–809 CrossRef CAS.
- P. Zhou, X. Yang, L. He, Z. M. Hao, W. Luo, B. Xiong, X. Xu, C. J. Liu, M. Y. Yan and L. Q. Mai, Appl. Phys. Lett., 2015, 106, 113–116 Search PubMed.
- Z. L. An, L. He, M. Toda, G. Yamamoto, T. Hashida and T. Ono, Nanotechnology, 2015, 26, 195601–195608 CrossRef PubMed.
- K. H. Seng, J. Liu, Z. P. Guo, Z. X. Chen, D. Z. Jia and H. K. Liu, Electrochem. Commun., 2011, 13, 383–386 CrossRef CAS.
- R. X. Yu, C. F. Zhang, Q. Meng, Z. X. Chen, H. K. Liu and Z. P. Guo, ACS Appl. Mater. Interfaces, 2013, 5, 12394–12399 CAS.
- F. Carn, M. Morcrette, B. Desport and R. Backov, Solid State Sci., 2013, 17, 134–139 CrossRef CAS.
- W. Tang, X. G. Gao, Y. S. Zhu, Y. B. Yue, Y. Shi, Y. P. Wu and K. Zhu, J. Mater. Chem., 2012, 22, 20143–20145 RSC.
- X. W. Zhou, G. M. Wu, G. H. Gao, C. J. Cui, H. Y. Yang, J. Shen and B. Zhou, Electrochim. Acta, 2012, 74, 32–38 CrossRef CAS.
- D. B. Kong, X. L. Li, Y. B. Zhang, X. Hai, B. Wang, X. Y. Qiu, Q. Song, Q. H. Yang and L. J. Zhi, Energy Environ. Sci., 2016, 9, 906–911 CAS.
- Q. Guo, Z. H. Sun, M. Gao, Z. Tan, B. S. Zhang and D. S. Su, J. Energy Chem., 2013, 22, 347–355 CrossRef CAS.
- B. Sun, K. Huang, X. Qi, X. L. Wei and J. X. Zhong, Adv. Funct. Mater., 2015, 25, 5633–5639 CrossRef CAS.
- X. Y. Chen, H. L. Zhu, Y. C. Chen, Y. Y. Shang, A. Y. Cao, L. B. Hu and G. Rubloff, ACS Nano, 2012, 6, 7948–7955 CrossRef CAS PubMed.
- M. L. Qin, J. Liu, S. Q. Liang, Q. Zhang, X. L. Li, Y. Liu and M. Y. Lin, J. Solid State Electrochem., 2014, 18, 2841–2846 CrossRef CAS.
- Y. S. Hu, X. Liu, J. O. Muller, R. Schlogl, J. Maier and D. S. Su, Angew. Chem., Int. Ed., 2009, 48, 210–214 CrossRef CAS PubMed.
- X. W. Zhou, G. M. Wu, J. D. Wu, H. Y. Yang, J. C. Wang, G. H. Gao, R. Cai and Q. Y. Yan, J. Mater. Chem. A, 2013, 1, 15459–15468 CAS.
- Z. Y. Cao and B. Q. Wei, Nano Energy, 2013, 2, 481–490 CrossRef CAS.
- J. Feng, C. Wang and Y. Qian, Mater. Lett., 2014, 122, 327–330 CrossRef CAS.
- D. Z. Chen, H. Y. Quan, S. L. Luo, X. B. Luo, F. Deng and H. L. Jiang, Phys. E, 2014, 56, 231–237 CrossRef CAS.
- J. L. Cheng, B. Wang, H. L. Xin, G. C. Yang, H. Q. Cai, N. Fude and H. Huang, J. Mater. Chem. A, 2013, 1, 10814–10820 CAS.
- Y. Yang, L. Li, H. L. Fei, Z. W. Peng, G. D. Ruan and J. M. Tour, ACS Appl. Mater. Interfaces, 2014, 6, 9590–9594 CAS.
- Y. Sun, S. B. Yang, L. P. Lv, I. Lieberwirth, L. C. Zhang, C. X. Ding and C. H. Chen, J. Power Sources, 2013, 241, 168–172 CrossRef.
- D. Pham-Cong, K. Ahn, S. W. Hong, S. Y. Jeong, J. H. Choi, C. H. Doh, J. S. Jin, E. D. Jeong and C. R. Cho, Curr. Appl. Phys., 2014, 14, 215–221 CrossRef.
- V. S. R. Channu, D. Ravichandran, B. Rambabu and R. Holze, Appl. Surf. Sci., 2014, 305, 596–602 CrossRef.
- H. B. Zhao, L. Y. Pan, S. Y. Xing, J. Luo and J. Q. Xu, J. Power Sources, 2013, 222, 21–31 CrossRef CAS.
- D. Z. Su, Y. J. Zhao, D. Yan, C. H. Ding, M. Q. Ning, J. S. Zhang, J. B. Li and H. B. Jin, J. Alloys Compd., 2017, 695, 2974–2980 CrossRef CAS.
- D. Chen, R. Yi, S. Chen, T. Xu, M. L. Gordin, D. P. Lv and D. H. Wang, J. Mater. Sci. Eng. B, 2014, 185, 7–12 CrossRef CAS.
- D. Vernardou, I. Marathianou, N. Katsarakis, E. Koudoumas, I. I. Kazadojev, S. O'Brien, M. E. Pemble and I. M. Povey, Electrochim. Acta, 2016, 196, 294–299 CrossRef CAS.
- Y. L. Cheah, V. Aravindan and S. Madhavi, ACS Appl. Mater. Interfaces, 2012, 4, 3270–3277 CAS.
- K. Zhu, H. L. Qiu, Y. Q. Zhang, D. Zhang, G. Chen and Y. J. Wei, ChemSusChem, 2015, 8, 1017–1025 CrossRef CAS PubMed.
- Y. J. Wei, C. W. Ryu and K. B. Kim, J. Power Sources, 2007, 165, 386–392 CrossRef CAS.
- S. R. Li, S. Y. Ge, Y. Qiao, Y. M. Chen, X. Y. Feng, J. F. Zhu and C. H. Chen, Electrochim. Acta, 2012, 64, 81–86 CrossRef CAS.
- S. Y. Zhan, C. Z. Wang, K. Nikolowski, H. Ehrenberg, G. Chen and Y. J. Wei, Solid State Ionics, 2009, 180, 1198–1203 CrossRef CAS.
- D. M. Yu, S. T. Zhang, D. W. Liu, X. Y. Zhou, S. H. Xie, Q. F. Zhang, Y. Y. Liu and G. Z. Cao, J. Mater. Chem., 2010, 20, 10841–10846 RSC.
- H. K. Park, Solid State Ionics, 2005, 176, 307–312 CrossRef CAS.
- Z. Y. Li, C. K. Zhang, C. F. Liu, H. Y. Fu, X. H. Nan, K. Wang, X. Y. Li, W. D. Ma, X. M. Lu and G. Z. Cao, Electrochim. Acta, 2016, 222, 1831–1838 CrossRef CAS.
- M. Z. Zou, W. W. Wen, J. X. Li, H. Lai and Z. G. Huang, J. Alloys Compd., 2016, 681, 268–274 CrossRef CAS.
- H. M. Zeng, D. Y. Liu, Y. C. Zhang, K. A. See, Y. S. Jun, G. Wu, J. A. Gerbec, X. L. Ji and G. D. Stucky, Chem. Mater., 2015, 27, 7331–7336 CrossRef CAS.
- L. Q. Mai, W. Chen, Q. Xu, J. F. Peng and Q. Y. Zhu, Chem. Phys. Lett., 2003, 382, 307–312 CrossRef CAS.
- H. R. Peng, W. Z. Wu, C. Q. Zhang, G. C. Li and K. Z. Chen, Mater. Lett., 2011, 65, 3436–3439 CrossRef CAS.
- Y. Z. Zheng, H. Ding, E. Uchaker, X. Tao, J. F. Chen, Q. Zhang and G. Cao, J. Mater. Chem., 2015, 3, 1979–1985 RSC.
- H. Yu, X. Rui, H. Tan, J. Chen, X. Huang, C. Xu, W. Liu, Y. W. Denis, H. H. Hng, H. E. Hoster and Q. Yan, Nanoscale, 2013, 5, 4937–4943 RSC.
- Y. Li, J. Yao, E. Uchaker, M. Zhang, J. Tian, X. Liu and G. Cao, J. Phys. Chem. C, 2013, 117, 23507–23514 CAS.
| This journal is © The Royal Society of Chemistry 2018 |