
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-omics and temporal dynamics profiling reveal disruption of central metabolism in Helicobacter pylori on bismuth treatment†
Bingjie
Han
a,
Zhen
Zhang
a,
Yanxuan
Xie
a,
Xuqiao
Hu
b,
Haibo
Wang
b,
Wei
Xia
 a,
Yulan
Wang
c,
Hongyan
Li
b,
Yuchuan
Wang
*a and
Hongzhe
Sun
*ab
a,
Yulan
Wang
c,
Hongyan
Li
b,
Yuchuan
Wang
*a and
Hongzhe
Sun
*ab
aSchool of Chemistry, Sun Yat-sen University, Guangzhou, 510275, P. R. China. E-mail: wangych55@mail.sysu.edu.cn
bDepartment of Chemistry, The University of Hong Kong, Hong Kong, P. R. China. E-mail: hsun@hku.hk
cCAS Key Laboratory of Magnetic Resonance in Biological Systems, State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics, Wuhan Institute of Physics and Mathematics, Chinese Academy of Sciences, Wuhan, 430071, P. R. China
First published on 25th July 2018
Abstract
Integration of multi-omics enables uncovering cellular responses to stimuli or the mechanism of action of a drug at a system level. Bismuth drugs have long been used for the treatment of Helicobacter pylori infection and their antimicrobial activity was attributed to dysfunction of multiple proteins based on previous proteome-wide studies. Herein, we investigated the response of H. pylori to a bismuth drug at transcriptome and metabolome levels. Our multi-omics data together with bioassays comprehensively reveal the impact of bismuth on a diverse array of intracellular pathways, in particular, disruption of central carbon metabolism is systematically evaluated as a primary bismuth-targeting system in H. pylori. Through temporal dynamics profiling, we demonstrate that bismuth initially perturbs the TCA cycle and then urease activity, followed by the induction of oxidative stress and inhibition of energy production, and in the meantime, induces extensive down-regulation in H. pylori metabolome. The present study thus expands our knowledge on the inhibitory actions of bismuth and provides a novel systematic perspective of H. pylori in response to a clinical drug that sheds light on enhanced therapeutic methodologies.
Introduction
Helicobacter pylori (H. pylori), a highly prevalent human pathogen, is associated with a variety of human gastrointestinal diseases such as chronic gastritis, peptic ulcer and gastric carcinoma in severe cases.1,2 It is reported that H. pylori infects over 50% of the world's population with the characteristics of extremely rare spontaneous eradication and increased antibiotic resistance.3,4 Following the identification of H. pylori in 1982,5 extensive research on this bacterium has been carried out in an effort to make clear the way of its survival under acidic conditions in the human stomach,6–8 to elucidate the relationship between H. pylori infection and the development of gastric cancer,9 and importantly, to fully understand the underlying mechanisms of current treatment regimens10,11 for better therapeutic options.The commonly used therapy for H. pylori infection based on proton pump inhibitors (PPI) and a combination of antibiotics is frequently challenged for its decreased eradication rates, primarily owing to the increasing prevalence of antibiotic resistance.3 Bismuth-containing quadruple therapy, with the addition of bismuth to the standard antibiotic regimens, has been recommended as first-line therapy and shown improved cure rates even towards resistant strain-caused infections.12 As a borderline metal ion, Bi(III) shows high affinity towards biological thiols and is able to replace essential metals such as Ni(II), Zn(II) and Fe(III) from metalloproteins.13–16 According to previous studies, bismuth-based drugs can utilize certain iron-transport pathways to enter H. pylori cells and functionally disrupt a number of key enzymes through multi-targeted mode of action.10,17,18 However, the inhibitory mechanisms of bismuth against the bacterium were mostly studied at the proteome level. Further exploration of cellular responses of H. pylori to bismuth drugs at transcriptome and metabolome levels may provide more insights into the cellular events and molecular changes.
To depict a complete cellular response of an organism under stress, integration of multiple-omics data is necessary and is an active research area with many successful applications.19,20 Transcriptomics enables identification of genes involved in an affected physiological state. At the metabolome level, due to the more direct relationship of the metabolic profiles with the phenotype of an organism,21 metabolomics represents a sensitive and systemic approach to reveal the alterations of endogenous low molecular weight compounds that reflects the interventions on cellular physiology. Furthermore, metallomics and metalloproteomics are emerging research areas aiming at the entirety of metals and metalloids within a cell or tissue to study their physiological functions.22–24 Different from traditional quantitative proteomics, metalloproteomics aims to recognize the relationship between biometals and cellular proteins as well as the functional alterations of proteins upon metal binding, which significantly facilitates the mechanistic understanding of metals in biology and medicine and opens a new horizon for unveiling the role of biometals at the proteome-wide scale.10
In this study, we extensively examined the cellular responses of H. pylori under the stress of a bismuth drug i.e., colloidal bismuth subcitrate (CBS) at transcriptome and metabolome levels by RNA-sequencing and a combination of NMR and GC-MS-based metabolic profiling, respectively. By integration of the results of transcriptomics and metabolomics with metalloproteomics, we demonstrate that bismuth influences multiple metabolic pathways and suppresses energy production in H. pylori through disruption of the central carbon metabolism of the bacterium. We further provide the temporal dynamics profile for describing the adaptive and toxic responses of H. pylori exposed to bismuth at the metabolome and phenotype levels. The inhibitory mechanisms of bismuth against H. pylori are comprehensively discussed based on the findings from multiple omics analyses and bioassays, yielding a much deeper understanding than either approach alone.
Results
CBS transcriptionally impairs central metabolism in H. pylori
Given that H. pylori generally grows much more slowly than other laboratory bacteria (Fig. S1A†), we first examined the growth curves of H. pylori in the absence and presence of CBS under the condition as described in the experimental section. As shown in Fig. S1A (ESI†), treatment of CBS led to slightly increased OD600 values of H. pylori culture, indicating that CBS is bacteriostatic. To fully reveal the inhibitory effects of CBS on H. pylori, we therefore analyzed the transcriptome profile of H. pylori upon CBS treatment for 12 h (ca. 1.5 generation time), during which good quality RNAs were extracted, further verifying that the bacteria were not in a severely damaged state. Compared to the control group, the transcription levels of 920 genes were significantly altered by CBS, among which, 588 genes including 286 up- and 302 down-regulated genes were contributed by both 20 and 80 μg mL−1 CBS treatment groups (Fig. 1A). Moreover, hierarchical clustering analysis revealed that the transcription pattern of the altered genes in the CBS-treated groups was different from that of the control group (Fig. S1B†).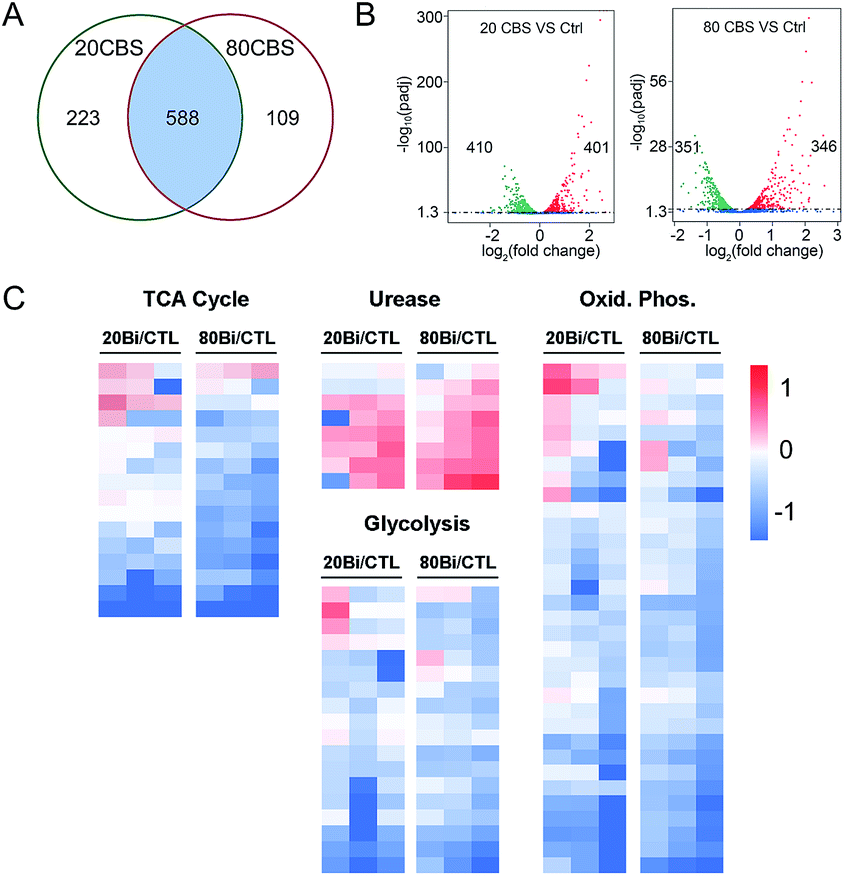 | ||
| Fig. 1 Transcriptomic profiling reveals altered gene transcription patterns in H. pylori upon CBS-treatment. (A) Venn diagram showing the number of altered genes in the 20 and 80 μg mL−1 CBS treated groups compared to the control group. (B) Volcano plots showing the altered genes in the 20 and 80 μg mL−1 CBS treated groups compared to the control group. Colored dots represent each gene that was significantly up-regulated (red) or down-regulated (green). (C) Gene transcription profiles from H. pylori cells treated with 20 and 80 μg mL−1 CBS for 12 h. Values of three biological replications reported as log2 fold changes over untreated control (CTL) in the transcription level of the TCA cycle, oxidative phosphorylation, glycolysis, and urease related genes. | ||
Significantly down-regulated genes were found to enrich in several central carbon metabolic pathways such as the TCA cycle, glycolysis/gluconeogenesis and oxidative phosphorylation (Fig. 1C and S1C†). Apart from that, the down-regulated genes in CBS treatment groups were also related to glutathione metabolism, microbial metabolism in diverse environments, epithelial cell signalling in H. pylori infection and a two-component system (Fig. S2B and D†), while the genes related to urease maturation, nucleotide excision repair, porphyrin and chlorophyll metabolism, biosynthesis of amino acids, and ribosome were up-regulated (Fig. 1C, S2A and C†), reflecting the complicated response of H. pylori towards CBS stress.
CBS induces global changes of the metabolome in H. pylori
To further explore the global responses of H. pylori to CBS treatment, we examined the CBS-induced metabolic changes in H. pylori in detail. Firstly, 1H NMR spectroscopy was employed to analyze the metabolites of CBS treated groups at a concentration of 20 or 80 μg mL−1 for 12 h and the untreated group (as a control) (Fig. S3A†). In total, 29 metabolites (Table S2†) were obtained and further confirmed by a series of 2D NMR spectra, including COSY, TOCSY, HSQC and HMBC. The identified metabolites consisted of a range of amino acids, such as alanine, arginine, glycine, ornithine and proline; organic acids, such as lactate, acetate, and fumarate; and a number of nucleosides and nucleotides, such as inosine, adenosine, uracil, and nicotinamide adenine dinucleotide (NAD+). Multivariate data analyses including PCA and OPLS-DA were subsequently performed to further analyze the variance among the three groups and screen significantly altered metabolites. The PCA scores plots (Fig. S3B†) of the 1H NMR spectral data showed clear separations between the profiles of the CBS-treated and untreated cells. OPLS-DA models were used to further evaluate metabolic changes induced by CBS through limiting correlation coefficient values (Fig. 2). The model quality was indicated by the values of R2 and Q2, which represents the quality of fit and predictability of the model, respectively. Compared to the untreated cells, organic acids including lactate, acetate, nicotinate and formate showed a marked increase in CBS treated groups, while reduction in the levels of certain amino acids including alanine, arginine, glutamate and ornithine was observed, which may lead to slowing down of the urea cycle. Meanwhile, the changes of other metabolites including pyruvate, succinate, betaine, hypoxanthine, acetamide and NAD+ indicated that CBS could affect the energy metabolism and nucleotide metabolism in H. pylori.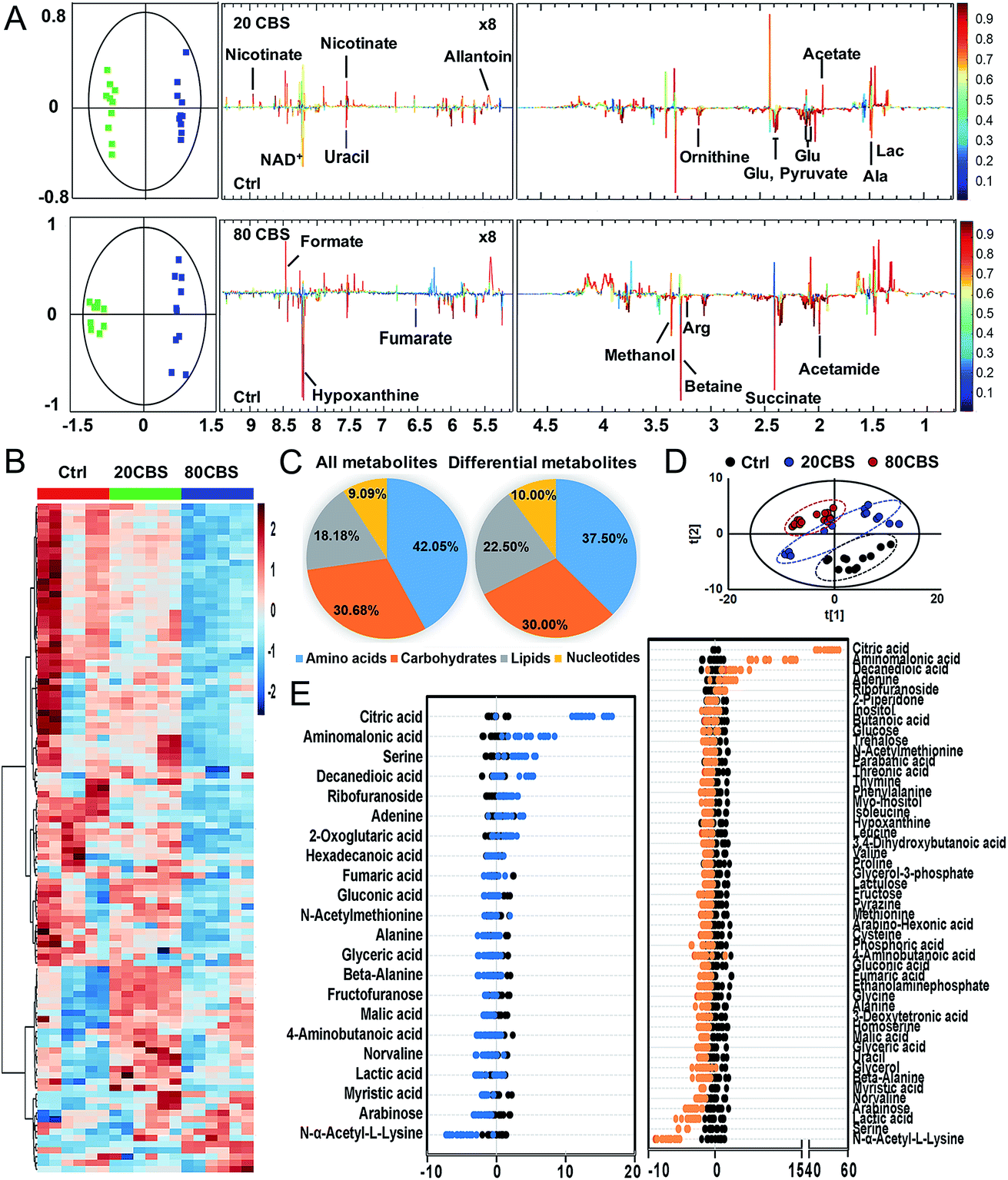 | ||
| Fig. 2 Metabolic profiling of H. pylori upon CBS treatment. (A) OPLS-DA scores plots (left) and coefficient-coded loading plots (right), discriminating the 600 MHz 1H NMR profiles between the untreated (green squares) and 20 or 80 μg mL−1 CBS-treated H. pylori cells (blue squares) (n = 10). These models are cross-validated with CV-ANOVA, p < 0.05. (B) Heat map for relative abundances of all identified metabolites of H. pylori in the control and 20 and 80 μg mL−1 CBS treated groups through GC-MS analysis. (C) Pie chart showing the distribution of all identified metabolites and differential metabolites (upon 80 μg mL−1 CBS treatment) in four major categories. (D) PCA analysis of GC-MS profiles obtained from the control and 20 and 80 μg mL−1 CBS-treated H. pylori metabolite extracts. Each dot represents metabolite profiling data from one individual sample and colors indicate samples in different treatment groups. (E) Z-score plot of differential metabolites based on the control, corresponding to data in (B). Each point represents one metabolite in one technical repeat, and the points are colored by sample types (black, control; blue, 20 μg mL−1 CBS treatment; orange, 80 μg mL−1 CBS treatment). | ||
Furthermore, we also examined the metabolome profiles of H. pylori in response to CBS by GC-MS. After removal of the internal standard (ribitol), 74 metabolites with reliable signals were detected for each sample. Representative total ion current chromatograms from the three groups are shown in Fig. S4A (ESI†). The correlation coefficient of two technical repeats was 0.996–0.999, indicative of the reliability of the data (Fig. S4B†). The metabolomic profiles of the three groups are shown as a heat map in Fig. 2B, which clearly indicates that CBS modulated the H. pylori metabolome and the abundance of the majority of metabolites decreased. In total, 22 and 49 metabolites with differential abundance were identified in CBS treated groups at a concentration of 20 and 80 μg mL−1, respectively. Z-Scores with values of −5.38 to 53.83 displayed variations of these metabolites based on the control (Fig. 2E). According to the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, 74 identified metabolites could be classified into four major categories, i.e., amino acids (42.05%), carbohydrates (30.68%), lipids (18.18%), and nucleotides (9.09%) (Fig. 2C). After treatment of H. pylori with CBS for 12 h, 49 differential metabolites showed a decrease in amino acids to 37.50% and an increase in lipids to 22.50%. The control and CBS treated groups were clearly separated in three different quadrants of the PCA scores plot (Fig. 2D). Furthermore, OPLS-DA models were used for the multivariate analysis, revealing a much clearer separation between the control and CBS treated metabolic profiles (Fig. S4C†).
We then jointly evaluated the metabolomics results from these two techniques. 21 metabolites (6 increased and 15 decreased) identified by NMR with a correlation coefficient |r| > 0.602 and 49 metabolites (6 increased and 43 decreased) identified by GC-MS with a VIP value >1 and p-value < 0.05 were considered to be significantly different in abundance after CBS treatment. Among these, 13 metabolites were identified by both techniques: 8 metabolites including alanine, fumarate, glutamate, lactate, ornithine, succinate, uracil and hypoxanthine were identified with decreased abundance by two techniques, whereas the abundance of glycine, proline, valine and beta-alanine was found to be decreased only in GC-MS analysis; while no changes in abundance of aspartate were observed as revealed by both techniques (Fig. S5A†). Six pathways were identified to be enriched by combined analysis of the differential metabolites by both NMR and GC-MS, including aminoacyl-tRNA biosynthesis, methane metabolism, citrate cycle, pyruvate metabolism, glyoxylate and dicarboxylate metabolism and glycine, serine and threonine metabolism (Fig. S5B†).
CBS treatment leads to decreased activities in the H. pylori TCA cycle
We then evaluated the effects of CBS on H. pylori by integrating the transcriptomics and metabolomics data in this study with our previous metalloproteomics data.10 By mapping the identified CBS-regulated genes, proteins (including 63 Bi-binding proteins) and metabolites to their respective biochemical pathways outlined in KEGG, we identified the TCA cycle as a common enriched pathway from all three omics analyses (Fig. 3A), which may serve as a key pathway that contributes to the toxicity of CBS in H. pylori. Five metabolites involved in the H. pylori TCA cycle were identified by GC-MS, generally showing a decreased abundance upon CBS treatment, except the obvious increase in citrate level (Fig. 3B), which may be caused by the citrate ligand in the Bi drug. We also detected the decreased abundance of acetyl-CoA, the starting compound in the TCA cycle. According to the changes of metabolites in the citrate cycle, we speculate that CBS may slow down the central carbon metabolism and reduce the activity of the TCA cycle. We thus analyzed the gene transcription levels of the TCA cycle by qRT-PCR and measured the activities of enzymes in the TCA cycle, i.e., citrate synthase, aconitase, isocitrate dehydrogenase, fumarase and malate dehydrogenase. As shown in Fig. 3B, activities of the tested enzymes were generally decreased by CBS in a dose-dependent manner. Among them, the decline in the activities of fumarase and malate dehydrogenase was most obvious, and the relevant genes including gltA, frdA, frdB, frdC, fumC and mqo showed a consistent downward trend after 12 h CBS treatment (Fig. S5C†). It was worth noting that the increased abundance of citrate due to the supplementation of CBS did not promote enzyme activities in the cycle.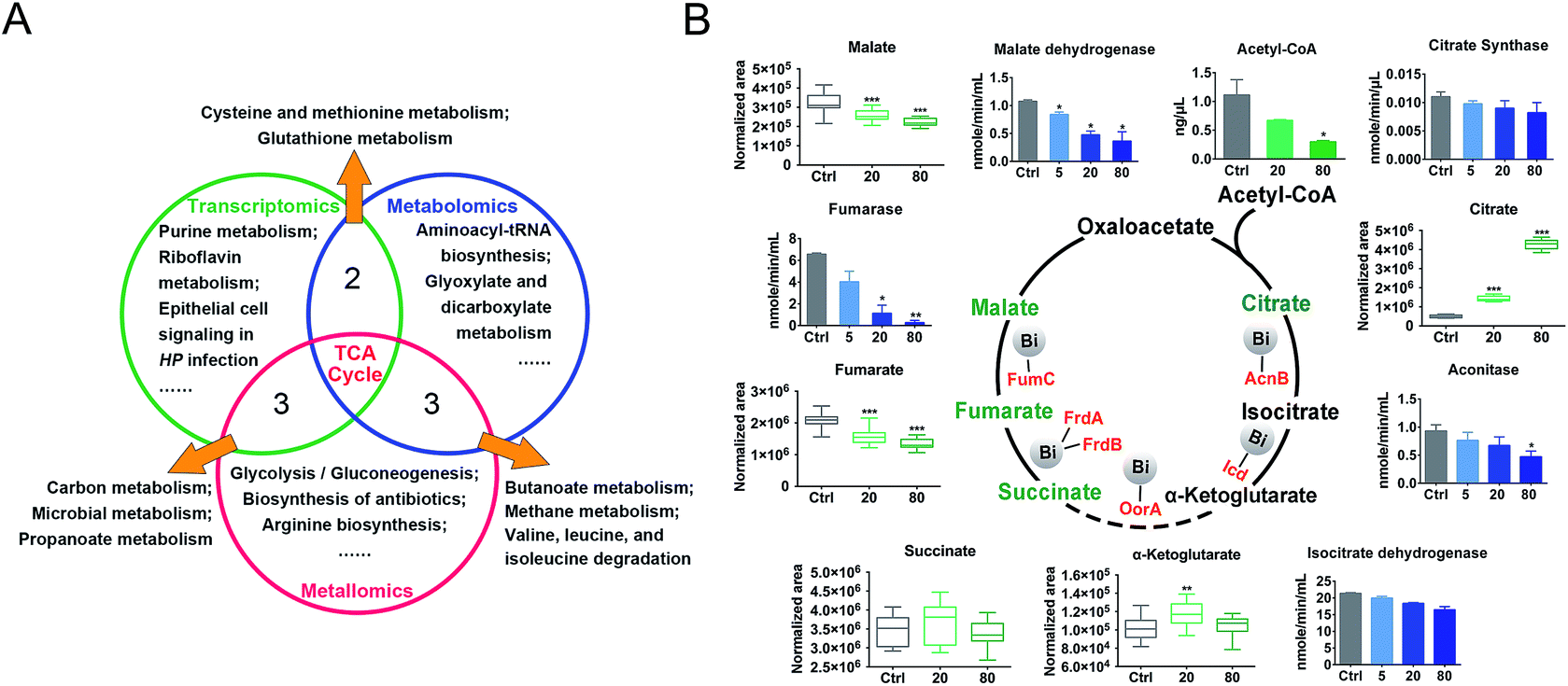 | ||
| Fig. 3 Enriched KEGG pathways and activities of key enzymes in the H. pylori TCA cycle: (A) comparison of the functional pathways enriched by Bi-altered genes, metabolites, and Bi-binding and regulated proteins, as revealed by transcriptomics, metabolomics and metalloproteomics studies. (B) Effect of CBS in regulation of the enzyme activities and metabolite abundance in the H. pylori TCA cycle. The asterisks indicate significant difference from the control group, as compared by two-tailed t-test. (*0.01 < p < 0.05; **0.001 < p < 0.01; ***p < 0.001). | ||
Temporal dynamics response of H. pylori towards CBS treatment
Due to the multi-targeted mode of action of Bi(III) (as CBS) in H. pylori,25 and as revealed by our multi-omics data, many cellular pathways in H. pylori including the citrate cycle, oxidative phosphorylation, amino acid metabolism and nucleotide metabolism could be affected by CBS. However, mapping only adverse outcomes of a toxicant may fall short of describing the adaptive response. We thus extracted H. pylori metabolites at a series of time points from 15 min to 8 h and analyzed the changes in the metabolic profiles. We found that H. pylori responded to CBS treatment quickly, and metabolic changes were observable within 15 min of exposure, followed by a clearly up-regulated metabolite abundance up to the time point of 4 h. At 30 min, the abundance of most differential metabolites increased while an extensive decrease in abundance was observed at 8 h (Fig. S6A†) and 12 h (Fig. 2B), possibly suggesting that the bacterium mounted a defense response to combat continuous drug exposures from 30 min to 4 h, while a decrease in metabolic rates occurred at 8 h. Growth of H. pylori on agar plates could be observed after 8 h CBS treatment (Fig. S6B†), indicating the survival of H. pylori at the studied time points. As shown in Fig. 4A, the control and CBS treatment groups showed an obvious separation in the PCA profile at 15 min, 30 min, 4 h and 8 h of CBS exposure. | ||
| Fig. 4 CBS-induced physiological responses of H. pylori at different time points. (A) PCA analysis of GC-MS profiles of control and CBS-treated H. pylori metabolite extracts at a series of time points. (B) Relative transcriptions of TCA cycle genes in H. pylori treated with 80 μg mL−1 CBS for 0, 15, 30 and 60 min. Gene transcriptions were normalized against 16S rRNA and the untreated control. (C) Abundance of ATP in H. pylori. (D) Relative ROS levels in H. pylori. (E) Urease activities of H. pylori, defined as nmol ammonia produced per min (per mg total protein). (F–H) Boxplots of normalized abundance of metabolites at different time points. (F) Fumarate. (G) Adenine. (H) Aminomalonic acid. Data are presented as mean ± SEM of triplicate samples from three biological replicates. The two-tailed t-test was used for all comparisons between two groups. The asterisks in (B) and (D) indicate significant changes of the measured values from that in H. pylori at the starting time point. The asterisks in (C) and (E) indicate significant difference from the untreated control group of the same time point. (*0.01 < p < 0.05; **0.001 < p < 0.01 and ***p < 0.001). | ||
We further determined the ATP and ROS levels in H. pylori upon CBS treatment at different time points. We found that ATP production was significantly decreased at 4 h, and almost completely stopped at 8 h and 12 h of CBS exposure (Fig. 4C), in accordance with the time points where extensive metabolome decreases occurred, indicative of decreased metabolic rates and reduced energy output of H. pylori after 8 h of CBS treatment. Significant increases in intracellular ROS levels were detected at 4 h and thereafter (Fig. 4D), suggesting elevated oxidative stress caused by CBS.
Alterations of ATP and ROS levels also suggested the disturbance in the bacterial TCA cycle and cellular respiration.26 We speculate that the TCA cycle in H. pylori serves as a primary target of the Bi drug. As urease has long been considered as an important target of CBS to inhibit H. pylori,27–29 we thus compared the transcription levels of major genes involved in these two systems upon CBS treatment by qRT-PCR. We observed that most of the genes coding for TCA cycle enzymes such as gltA, acnB, icd, frdA, frdB, frdC, fumC and mqo were down-regulated at 30 min exposure (Fig. 4B), while alteration of urease related genes ureA, ureB, ureE, ureF, ureG and ureH was noted mostly at the time point of 2 h (Fig. S6C†), consistent with the apparently decreased urease activity detected after 2 h (Fig. 4E). These results indicated that CBS treatment affected the H. pylori TCA cycle prior to the urease system.
Discussion
In this study, we unveiled alterations of several H. pylori metabolic pathways in response to CBS at both the transcriptional and metabolic levels. Our combined data from transcriptomics, metabolomics and metalloproteomics allowed more detailed mechanistic insights of CBS toxicity to be uncovered.Iron and nickel homeostasis
The survival and colonization of H. pylori depend strongly on the proper homeostasis of intracellular essential metals.30 Effective bismuth uptake is the premise of CBS toxicity in H. pylori. A recent study on tracking metal uptake in single H. pylori cells suggested the competition between bismuth and iron transport into H. pylori.17 Among the proposed iron trafficking proteins in H. pylori,31 more than half of the genes or proteins were regulated by bismuth, such as the up-regulated genes encoding ferrous iron uptake protein (feoB), the ferric iron uptake Fec system (fecA, fecD), the iron uptake energy transduction protein (tonB) and the iron-sensing global regulatory protein (fur). As a major iron storage protein, ferritin (Pfr) is essential for the survival of H. pylori in infected hosts and protects bacteria against oxidative stress and lethal iron starvation.32 Under 80 μg mL−1 CBS stress, about 5-fold up-regulation of gene pfr was detected in our study. Importantly, HpPfr was identified as a direct Bi-binding protein by a Bi(III)-coordinated fluorescent probe.10 It is likely that bismuth binding may disrupt the function of HpPfr in iron storage, which could be important for H. pylori, owing to the large iron requirements of metalloenzymes such as [Ni,Fe]-hydrogenase,33 catalase,34 SOD35 and cytochrome systems.36Concomitantly, we also observed nickel homeostasis being interfered by Bi(III). The transcription level of nixA, encoding the high-affinity nickel transport protein, showed a significant decrease after CBS treatment; genes encoding nickel chaperone proteins (hypA, hypB, hypF, slyD, hspA, ureE, ureG, ureF, and ureH) and a nickel storage protein (hpnl) were also regulated, implying the interference of nickel uptake and transport. H. pylori urease is a crucial metalloenzyme for its colonization at acidic pH. Its maturation is highly dependent on the proper insertion of nickel ions into its active site via a battery of accessory proteins. We have demonstrated very recently that the inhibition of urease activity by Bi(III) was achieved through binding to UreG, leading to disruption of nickel incorporation into apo-urease.27 Dynamic analysis of urease activity showed that a significant decrease of urease activity occurred at the time point of 2 h after CBS treatment, consistent with the qRT-PCR results (Fig. S6B†) of the regulated transcriptional levels of genes encoding urease and its accessory proteins (ureA, ureB, ureE, ureF, ureG and ureH) at 2 h. Furthermore, amino acids related to urea metabolism37,38 such as arginine, citrulline, ornithine, glutamate and glutamine identified in this study were down-regulated in H. pylori in response to CBS, which may correlate with the inhibited urease activity.
Central carbon metabolism
Glucose appears to be the main carbohydrate utilized by H. pylori.39,40 The entry of glucose into H. pylori is sodium-linked via a highly specific transporter protein GluP located in the bacterial membrane.41,42H. pylori is able to metabolize glucose by using glycolysis, the pentose phosphate pathway, and the Entner–Doudoroff pathway.43 Genes glk, tpiA, and HP1346 in glycolysis and zwf, pgl, and rpe in the pentose phosphate pathway were down-regulated in response to CBS, and bindings of bismuth to glycolytic enzymes fructose-bisphosphate aldolase (FbpA) and enolase (Eno) were detected,10 indicative of suppressed glucose catabolism by the metallodrug. This corroborated well with the decreased abundance of lactate and gluconate (1.8-fold and 1.4-fold, respectively), which were reported to be the major products of glucose catabolism in H. pylori,39,40 and the decreased abundance of pyruvate, which represents the end point for glycolysis and the Entner–Doudoroff pathway.44Acetyl-CoA is a key metabolite which is at the branch point of several metabolic routes. In H. pylori, acetyl-CoA is formed through both the direct conversion of acetate catalyzed by acetyl-CoA synthetase (AcsA),45 and the oxidative decarboxylation of pyruvate catalyzed by highly oxygen labile enzyme pyruvate-ferredoxin oxidoreductases (POR).46 The decrease in acetyl-CoA abundance in response to CBS treatment was possibly a result of its inhibited formation and accelerated consumption via the fatty acid biosynthesis pathway, as evidenced by the accumulation of acetate, decreased transcription of genes por and acsA, and up-regulation of genes accA and accD encoding acetyl-CoA carboxylase which catalyzes the conversion of acetyl-CoA to form malonyl-CoA, the first step of fatty acid biosynthesis.47
The H. pylori TCA cycle is proposed to be a noncyclic branched pathway directed toward α-ketoglutarate in the tricarboxylic oxidative branch and toward succinate in the dicarboxylic reductive branch.48 Although evidence suggests the presence of a cyclic TCA cycle in H. pylori,49–51 it should not be efficiently circulated as in other bacteria such as E. coli, since the reduction of fumarate to succinate was not easily reversed.52,53 As the starting compound for the TCA cycle, the decreased acetyl-CoA abundance further indicated an inhibited TCA cycle flux. In support of this, decreased activities of TCA cycle enzymes in response to Bi(III) were detected at 12 h (Fig. 3B), and alternations of TCA cycle metabolites (citrate, succinate, and α-ketoglutarate) and genes were observed as early as 30 min after bismuth treatment (Fig. 4B and Table S3†). Indeed, binding of Bi(III) to enzymes in the TCA cycle, i.e., AcnB, Icd, FrdA, FrdB and FumC was identified by metalloproteomics10,54 and inactivation of fumarase activity as a result of bismuth-binding was demonstrated in a previous study.55
The importance of a reductive dicarboxylic pathway is that fumarate, reduced by fumarate reductase, serves as a terminal electron acceptor in the electron transport chain and provides an important source for ATP production in anaerobic bacteria.52H. pylori uses a simple respiratory chain composed of cytochrome c reductase, cytochrome c-553 and cb-type cytochrome c oxidase as terminal oxidase, with NADPH being the most likely electron donor53 and menaquinones as electron carriers.56 We observed extensive down-regulations of genes encoding complex II to V in oxidative phosphorylation and enzymes related to menaquinone biosynthesis60,61 after 12 h CBS treatment (Fig. 1C, Table S1†), indicative of the inhibited respiration in H. pylori. Under CBS stress, decreased ATP content in H. pylori was initially observed at 4 h and almost complete ATP depletion occurred at the later time points (Fig. 4C), confirming the disrupted ATP generation via oxidative phosphorylation. The significant ATP reduction upon bismuth treatment was also reported previously.62 The phenotype of ATP content was also consistent with the metabolic profile in that extensive metabolite decrease in H. pylori occurred at the time points of ATP depletion (Fig. S6A†), indicating that CBS induces irreversible damage in H. pylori and the energy is not sufficient to maintain inner balance for normal bacterial metabolism. Moreover, the decreased abundance of fumarate was only detected after 8 h CBS treatment (Fig. 4F), implying the reduced ability of fumarate to serve as a biosynthetic and bioenergetic precursor in H. pylori central carbon metabolism. Although an immediate response of the H. pylori TCA cycle was noted upon CBS treatment (Fig. 4B), it was not promptly leading to disturbance in ATP generation, indicating there exist certain adaptive mechanisms in H. pylori in response to early Bi(III) stress. Overall, the changes at transcriptomic, metallomic and metabolomic levels as discussed above suggest that CBS suppresses energy production in H. pylori by targeting and disrupting the central carbon metabolism as one of its toxic mechanisms.
Amino acid, fatty acid and nucleotide metabolism
Amino acids are important sources of carbon, nitrogen and energy in H. pylori. The bacterium is able to utilize urea nitrogen for amino acid synthesis following hydrolysis of urea to ammonia,37 and deamination of amino acids such as glutamine and asparagine would lead to the formation of central intermediary metabolites and ammonium ions that participate in the organism's carbon and nitrogen metabolism.57,58 According to the temporal dynamics analysis, the severe influence of bismuth on H. pylori was observed after 8 h treatment, corresponding to the time point of saturated Bi(III) uptake by H. pylori.17 Therefore, right after CBS treatment, the bacterium may enter a state that requires a major metabolic shift to adapt to bismuth-induced stress, which could be inferred by the increased metabolite levels of H. pylori exposed to CBS at the time point of 30 min, including the regulation of a number of amino acids such as isoleucine, glycine, threonine, phenylalanine, tryptophan and tyrosine. Interestingly, the essential amino acids that are strictly required for H. pylori growth59i.e., valine, leucine, isoleucine, methionine and phenylalanine detected by GC-MS did not show decreased abundance before the time point of 8 h (Table S3†). As H. pylori is unable to synthesize these amino acids due to lack of the corresponding genes in their biosynthetic pathways,60,61 our results indicate that the bacterium tends to regulate the transport and maintain normal levels of essential amino acids to adapt to early bismuth stress. The significant reduction of amino acid abundance upon prolonged CBS treatment may lead to deficient bacterial growth.Moreover, three saturated fatty acids including sebacic acid, palmitic acid and stearic acid also increased significantly at the time point of 30 min (Table S3†). Fatty acids are important components of lipids that play a role in cell membrane organization, signal transduction, and energy storage.63 Stearic acid and palmitic acid in Bacillus species are able to improve resistance against decreased membrane potential caused by protonophores.64 Palmitic acid is a common fatty acid involved in protein palmitoylation, an important protein modification for mediating protein localization, protein–protein interactions65 and promoting the invasiveness of infectious bacteria.66 In view of the crucial role of bacterial fatty acids, their increased abundance at the time point of 30 min may thus represent an adaptive response of H. pylori to early bismuth stress. After 12 h CBS treatment, fatty acid metabolism-related pathways enriched by the bismuth-regulated genes were identified by transcriptome analysis (Fig. S2†), along with the generally decreased fatty acid abundance (Table S3†), indicating that bismuth affects the metabolism of fatty acids in H. pylori, possibly leading to the reduced utilization of fatty acids as sources of metabolic energy and effector molecules.
Purine and pyrimidine nucleotides play a central role as building blocks of nucleic acids, elements of energy metabolism, constituents of coenzymes, etc. Tracing the incorporation rates of nucleotide precursors into H. pylori reveals that the bacterium utilizes a de novo pathway and a less active salvage pathway for the biosynthesis of pyrimidine nucleotides,67 while for purine nucleotide biosynthesis, it takes salvage pathways as the ordinary routes.68,69 Under bismuth stress, extensive genes in purine and pyrimidine metabolism pathways were regulated, such as the down-regulation of genes pyrB, pyrG, ndk and apt encoding key nucleotides biosynthesis enzymes aspartate transcarbamoylase, CTP synthetase, nucleoside diphosphokinase and adenine phosphoribosyltransferase; while at the metabolome level, we observed decreased abundance of uracil, uridine, thymine, and hypoxanthine, indicating the influenced nucleotide metabolism by bismuth. Significant up-regulation of adenine abundance was noted upon 30 min bismuth treatment (Fig. 4G), which also implies an adaptive response of the bacterium, in view of the reported role of adenine in supporting H. pylori growth and proliferation.70
Antioxidant response
H. pylori is continuously exposed to attack from reactive oxygen/nitrogen species when colonizing in gastric mucosa, as its infection could induce an inflammatory response, resulting in an oxidative burst. To counter the deleterious oxidative attack, the bacterium has developed a range of defensive strategies, including antioxidant enzymes, inhibitors of oxidant generation and DNA repair systems.71 We have demonstrated in previous studies that bismuth impairs the oxidative defense systems in H. pylori by binding and functionally disrupting key enzymes such as catalase, arginase and peroxiredoxin.10,72 Herein, we observed at the transcriptome level the down-regulation of genes tpx, trxA and trxB encoding key antioxidant enzymes,73 and genes mutS and recA encoding DNA repair enzymes,74,75 further corroborated the reduced oxidative defense ability of H. pylori upon CBS treatment. Moreover, we detected elevated levels of intracellular ROS induced by CBS (Fig. 4D).10 Generation of ROS is considered a common mechanism of killing bacteria by antibiotics, which primarily results from the interaction of antibiotics with their cellular targets, leading to disruption of the TCA cycle and hyperactivation of the electron transport chain that induces the formation of superoxide and hydroxyl radicals.76,77 Considering the multi-targeted mode of action of CBS against H. pylori, it is difficult to delineate the specific series of intracellular events of bismuth-induced ROS production based on current results. As the elevated ROS level in H. pylori was detected at the same time point of decreased ATP production, i.e., after 4 h CBS treatment, the disturbed proton gradient in the respiratory chain may contribute to the elevated ROS, which was also proposed to be the reason for Ag(I)-mediated ROS generation in bacteria and algae.20,78 Meanwhile, impairment of the oxidative defense system by bismuth disabled the bacterium to counterbalance the increased oxidative stress. At the metabolome level, we observed the significant up-regulation of aminomalonic acid (Ama) after 12 h CBS treatment (Fig. 4H). As a potential marker of oxidative damage to proteins,79 the accumulation of Ama may directly correlate with the elevated ROS levels caused by CBS. Moreover, we observed the significantly decreased level of betaine as analyzed by 1H NMR (Table S2†), which could be due to its excessive consumption in counteracting free radicals and repairing the damaged membrane caused by ROS.80Conclusions
The complex, dynamic responses of living cells to external stimuli make it difficult to obtain a comprehensive understanding of the precise cellular events. Here, we decipher the complicated inhibitory mechanisms of bismuth to H. pylori by integrating multiple omics data and bioassays, which deepens our knowledge of the actions of bismuth on various intracellular pathways at systemic levels (Fig. 5). We uncover the adaptive and toxic responses of H. pylori exposed to CBS at different treatment periods. The severe damage of H. pylori by the metallodrug was observed after 8 h treatment, characterized by the depleted energy production and extensively down-regulated H. pylori metabolome, which possibly occurred due to the functional perturbation of multiple cellular proteins as a result of accumulated bismuth binding. Moreover, disruption of the central carbon metabolism by CBS in H. pylori is proposed for the first time and systematically investigated, which is a unique target of bismuth in H. pylori that cannot be inhibited by other components of the clinical bismuth-containing quadruple therapy. Importantly, bacterial central metabolism has recently been regarded as a crucial yet largely unexplored druggable system in view of its importance in pathogenesis and less likelihood to develop mutational resistance.81,82 Bismuth drugs may therefore show the advantage in targeting the central carbon metabolism in H. pylori and suppress its energy production. These insights could lead to enhanced therapeutic options in the clinical battle against bacterial infection and guide drug design in the future.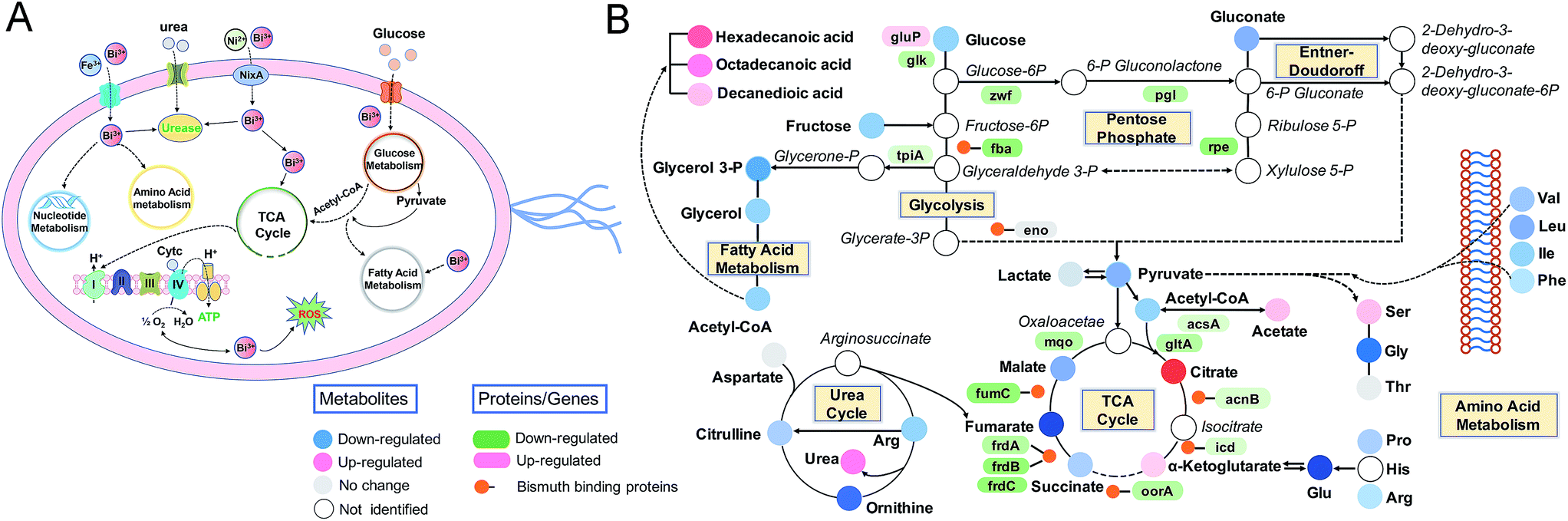 | ||
| Fig. 5 The underlying inhibitory mechanisms of bismuth in H. pylori, as derived from linking transcriptome, metalloproteome and metabolome responses to physiological effects. (A) Schematic representation of biological pathways in H. pylori affected by bismuth. (B) Detailed alternations of relative metabolites and genes or proteins in the representative pathways. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21601209 and 21671203), the Natural Science Foundation of Guangdong Province (2017A030313063), the Research Grants Council of Hong Kong (17305415P, 17333616P and 17307017P), the Fundamental Research Funds for the Central Universities, a starting fund from Sun Yat-sen University, and the University of Hong Kong (for an e-SRT on Integrative Biology). We thank Dr Zhigang Liu and Manjun Yang for helpful comments on metabolomics studies.Notes and references
- K. M. Fock, D. Y. Graham and P. Malfertheiner, Nat. Rev. Gastroenterol. Hepatol., 2013, 10, 495–500 CrossRef PubMed.
- D. Y. Graham, Gastroenterology, 2015, 148, 719–731 CrossRef PubMed.
- B. N. Dang and D. Y. Graham, Nat. Rev. Gastroenterol. Hepatol., 2017, 14, 383–384 CrossRef PubMed.
- P. Malfertheiner, F. Megraud, C. A. O'Morain, J. P. Gisbert and E. M. El-Omar, et al. , Gut, 2017, 66, 6–30 CrossRef PubMed.
- J. R. Warren and B. Marshall, Lancet, 1983, 321, 1273–1275 CrossRef.
- C. Montecucco and R. Rappuoli, Nat. Rev. Mol. Cell Biol., 2001, 2, 457–466 CrossRef PubMed.
- M. H. Yuen, Y. H. Fong, Y. S. Nim, P. H. Lau and K. B. Wong, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1–9 CrossRef PubMed.
- J. A. Bugaytsova, O. Björnham, Y. A. Chernov, P. Gideonsson and T. Borén, et al. , Cell Host Microbe, 2017, 21, 376–389 CrossRef PubMed.
- M. Sigal, C. Y. Logan, M. Kapalczynska, H. J. Mollenkopf, H. Berger, B. Wiedenmann, R. Nusse, M. R. Amieva and T. F. Meyer, Nature, 2017, 548, 451–455 CrossRef PubMed.
- Y. Wang, L. Hu, F. Xu, Q. Quan, Y. T. Lai, W. Xia, Y. Yang, Y. Y. Chang, X. Yang, Z. Chai, J. Wang, I. K. Chu, H. Li and H. Sun, Chem. Sci., 2017, 8, 4626–4633 RSC.
- E. A. Marcus, G. Sachs and D. R. Scott, Aliment. Pharmacol. Ther., 2015, 42, 922–933 CrossRef PubMed.
- M. P. Dore, H. Lu and D. Y. Graham, Gut, 2016, 65, 870–878 CrossRef PubMed.
- R. Wang, T. P. Lai, P. Gao, H. Zhang, P. L. Ho, P. C. Woo, G. Ma, R. Y. Kao, H. Li and H. Sun, Nat. Commun., 2018, 9, 439, DOI:10.1038/s41467-018-02828-6.
- N. Yang, H. Zhang, M. Wang, Q. Hao and H. Sun, Sci. Rep., 2012, 2, 999, DOI:10.1038/srep00999.
- W. Xia, H. Li and H. Sun, Chem. Commun., 2014, 50, 1611–1614 RSC.
- Y. Wang, L. Hu, X. Yang, Y. Y. Chang, X. Hu, H. Li and H. Sun, Metallomics, 2015, 7, 1399–1406 RSC.
- C. N. Tsang, K. S. Ho, H. Sun and W. T. Chan, J. Am. Chem. Soc., 2011, 133, 7355–7357 CrossRef PubMed.
- Y. Wang, C. N. Tsang, F. Xu, P. W. Kong, L. Hu, J. Wang, I. K. Chu, H. Li and H. Sun, Chem. Commun., 2015, 51, 16479–16482 RSC.
- M. Y. Hirai, M. Yano, D. B. Goodenowe, S. Kanaya, T. Kimura, M. Awazuhara, M. Arita, T. Fujiwara and K. Saito, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10205–10210 CrossRef PubMed.
- S. Pillai, R. Behra, H. Nestler, M. J.-F. Suter, L. Sigg and K. Schirmer, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 3490–3495 CrossRef PubMed.
- L. M. Raamsdonk, B. Teusink, D. Broadhurst, N. Zhang and S. G. Oliver, et al. , Nat. Biotechnol., 2001, 19, 45–50 CrossRef PubMed.
- H. Sun and Z. F. Chai, Annu. Rep. Prog. Chem., Sect. A: Inorg. Chem., 2010, 106, 20–38 RSC.
- S. Mounicou, J. Szpunar and R. Lobinski, Chem. Soc. Rev., 2009, 38, 1119–1138 RSC.
- Y. Wang, H. Wang, H. Li and H. Sun, in Essential and non-essential metals: carcinogenesis, prevention and cancer therapeutics, ed. A. Mudipalli and J. T. Zelikoff, Humana Press, 2017, pp. 199–222 Search PubMed.
- R. Ge, Z. Chen and Q. Zhou, Metallomics, 2012, 4, 239–243 RSC.
- M. A. Lobritz, P. Belenky, C. B. M. Porter, A. Gutierrez and J. J. Collins, et al. , Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8173–8180 CrossRef PubMed.
- X. Yang, M. Koohi-Moghadam, R. Wang, Y. Y. Chang, P. C. Y. Woo, J. Wang, H. Li and H. Sun, PLoS Biol., 2018, 16, e2003887, DOI:10.1371/journal.pbio.2003887.
- H. Li and H. Sun, Curr. Opin. Chem. Biol., 2012, 16, 74–83 CrossRef PubMed.
- R. Ge and H. Sun, Acc. Chem. Res., 2007, 40, 267–274 CrossRef PubMed.
- A. Danielli and V. Scarlato, FEMS Microbiol. Rev., 2010, 34, 738–752 CrossRef PubMed.
- R. Ge and X. Sun, BioMetals, 2012, 25, 247–258 CrossRef PubMed.
- B. Waidner, S. Greiner, S. Odenbreit, H. Kavermann and S. Bereswill, et al. , Infect. Immun., 2002, 70, 3923–3929 CrossRef PubMed.
- M. R. Leach and D. B. Zamble, Curr. Opin. Chem. Biol., 2007, 11, 159–165 CrossRef PubMed.
- P. C. Loewen, X. Carpena, C. Rovira, A. Ivancich, R. Perez-Luque, R. Haas, S. Odenbreit, P. Nicholls and I. Fita, Biochemistry, 2004, 43, 3089–3103 CrossRef PubMed.
- C. Spiegelhalder, B. Gerstenecker, A. Kersten, E. Schiltz and M. Kist, Infect. Immun., 1993, 61, 5315–5325 Search PubMed.
- M. A. Smith, M. Finel, V. Korolik and G. L. Mendz, Arch. Microbiol., 2000, 174, 1–10 CrossRef PubMed.
- C. L. Williams, T. Preston, M. Hossack, C. Slater and K. E. L. McColl, FEMS Immunol. Med. Microbiol., 1996, 13, 87–94 CrossRef PubMed.
- G. L. Mendz and S. L. Hazell, Microbiology, 1996, 142, 2959–2967 CrossRef PubMed.
- G. L. Mendz, S. L. Hazell and B. P. Burns, J. Gen. Microbiol., 1993, 139, 3023–3028 CrossRef PubMed.
- A. D. R. Peter, A. Chalk and W. M. Blows, Microbiology, 1994, 140, 2085–2092 CrossRef PubMed.
- B. P. B. George, L. Mendz and L. Stuart, Biochim. Biophys. Acta, 1995, 1244, 269–276 CrossRef.
- G. Psakis, M. Saidijam, K. Shibayama, J. Polaczek and P. J. F. Henderson, et al. , Mol. Microbiol., 2009, 71, 391–403 CrossRef PubMed.
- S. L. Hazell and G. L. Mendz, Helicobacter, 1997, 2, 1–12 CrossRef PubMed.
- G. L. Mendz, S. L. Hazell and B. P. Burns, Arch. Biochem. Biophys., 1994, 312, 349–356 CrossRef PubMed.
- J. F. Tomb, O. White, A. R. Kerlavage, R. A. Clayton and J. C. Venter, et al. , Nature, 1997, 388, 539–547 CrossRef PubMed.
- N. J. Hughes, P. A. Chalk, C. L. Clayton and D. J. Kelly, J. Bacteriol., 1995, 177, 3953–3959 CrossRef PubMed.
- B. P. Burns, S. L. Hazell and G. L. Mendz, Microbiology, 1995, 141, 3113–3118 CrossRef PubMed.
- S. M. Pitson, G. L. Mendz, S. Srinivasan and S. L. Hazell, Eur. J. Biochem., 1999, 260, 258–267 CrossRef PubMed.
- I. E. Corthésy-Theulaz, G. E. Bergonzelli, H. Henry, D. Bachmann, D. F. Schorderet, A. L. Blum and L. N. Ornston, J. Biol. Chem., 1997, 272, 25659–25667 CrossRef.
- N. J. Hughes, C. L. Clayton, P. A. Chalk and D. J. Kelly, J. Bacteriol., 1998, 180, 1119–1128 Search PubMed.
- B. Kather, K. Stingl, M. E. van der Rest, K. Altendorf and D. Molenaar, J. Bacteriol., 2000, 182, 3204–3209 CrossRef PubMed.
- Z. Ge, Expert Opin. Ther. Targets, 2002, 6, 135–146 CrossRef PubMed.
- M. Chen, L. P. Andersen, L. Zhai and A. Kharazmi, FEMS Immunol. Med. Microbiol., 1999, 24, 169–174 CrossRef PubMed.
- R. Ge, X. Sun, Q. Gu, R. M. Watt, J. A. Tanner, B. C. Wong, H. H. Xia, J. D. Huang, Q. Y. He and H. Sun, J. Biol. Inorg Chem., 2007, 12, 831–842 CrossRef PubMed.
- Z. Chen, Q. Zhou and R. Ge, BioMetals, 2012, 25, 95–102 CrossRef PubMed.
- S. W. Marcelli, H. T. Chang, T. Chapman, P. A. Chalk, R. J. Miles and R. K. Poole, FEMS Microbiol. Lett., 1996, 138, 59–64 CrossRef PubMed.
- R. M. Stark, M. S. Suleiman, I. J. Hassan, J. Greenman and M. R. Millar, J. Med. Microbiol., 1997, 46, 793–800 CrossRef PubMed.
- E. F. Miller and R. J. Maier, J. Bacteriol., 2014, 196, 3074–3081 CrossRef PubMed.
- D. J. Reynolds and C. W. Penn, Microbiology, 1994, 140, 2649–2656 CrossRef PubMed.
- P. Doig, B. L. de Jonge, R. A. Alm, E. D. Brown and T. J. Trust, et al. , Microbiol. Mol. Biol. Rev., 1999, 63, 675–707 Search PubMed.
- A. Marais, G. L. Mendz, S. L. Hazell and F. Mégraud, Microbiol. Mol. Biol. Rev., 1999, 63, 642–674 Search PubMed.
- M. V. Bland, S. Ismail, J. A. Heinemann and J. I. Keenan, Antimicrob. Agents Chemother., 2004, 48, 1983–1988 CrossRef PubMed.
- Y. Fujita, H. Matsuoka and K. Hirooka, Mol. Microbiol., 2007, 66, 829–839 CrossRef PubMed.
- S. E. Diomandé, C. Nguyen-The, M. H. Guinebretière, V. Broussolle and J. Brillard, Front. Microbiol., 2015, 6, 813, DOI:10.3389/fmicb.2015.00813.
- J. T. Dunphy and M. E. Linder, Biochim. Biophys. Acta, 1998, 1436, 245–261 CrossRef.
- J. Sobocińska, P. Roszczenko-Jasińska, A. Ciesielska and K. Kwiatkowska, Front. Immunol., 2018, 8, 2003, DOI:10.3389/fimmu.2017.02003.
- G. L. Mendz, B. M. Jimenez, S. L. Hazell, A. M. Gero and W. J. O'Sullivan, J. Appl. Bacteriol., 1994, 77, 1–8 CrossRef PubMed.
- G. L. Mendz, B. M. Jimenez, S. L. Hazell, A. M. Gero and W. J. O'Sullivan, J. Appl. Bacteriol., 1994, 77, 674–681 CrossRef PubMed.
- G. Liechti and J. B. Goldberg, J. Bacteriol., 2012, 194, 839–854 CrossRef PubMed.
- G. L. Mendz, A. J. Shepley, S. L. Hazell and M. A. Smith, Arch. Microbiol., 1997, 168, 448–456 CrossRef PubMed.
- A. Stent, A. L. Every and P. Sutton, Am. J. Physiol.: Gastrointest. Liver Physiol., 2012, 302, G579–G587 CrossRef PubMed.
- Y. Y. Chang, T. Cheng, X. Yang, L. Jin, H. Sun and H. Li, J. Biol. Inorg Chem., 2017, 22, 673–683 CrossRef PubMed.
- S. L. Comtois, Microbiology, 2003, 149, 121–129 CrossRef PubMed.
- G. Wang, P. Alamuri, M. Z. Humayun, D. E. Taylor and R. J. Maier, Mol. Microbiol., 2005, 58, 166–176 CrossRef PubMed.
- S. A. Thompson and M. J. Blaser, Infect. Immun., 1995, 63, 2185–2193 Search PubMed.
- M. A. Kohanski, D. J. Dwyer, B. Hayete, C. A. Lawrence and J. J. Collins, Cell, 2007, 130, 797–810 CrossRef PubMed.
- H. V. Acker and T. Coenye, Trends Microbiol., 2017, 25, 456–466 CrossRef PubMed.
- H. J. Park, J. Y. Kim, J. Kim, J. H. Lee, J. S. Hahn, M. B. Gu and J. Yoon, Water Res., 2009, 43, 1027–1032 CrossRef PubMed.
- M. Rykaer, B. Svensson, M. J. Davies and P. Hagglund, J. Proteome Res., 2017, 16, 3978–3988 CrossRef PubMed.
- T. Chen, J. Sheng, Y. Fu, M. Li, J. Wang and A. Q. Jia, J. Proteome Res., 2017, 16, 824–830 CrossRef PubMed.
- P. Murima, J. D. McKinney and K. Pethe, Chem. Biol., 2014, 21, 1423–1432 CrossRef PubMed.
- W. R. Bishai, Nat. Chem. Biol., 2017, 13, 925–926 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, supplementary figures and omics data. See DOI: 10.1039/c8sc01668b |
| This journal is © The Royal Society of Chemistry 2018 |