
Palladium-catalysed ligand-free reductive Heck cycloisomerisation of 1,6-en-α-chloro-enamides†
Yangyang
Hou‡
a,
Jing
Ma‡
a,
Hongyi
Yang
a,
Edward A.
Anderson
 b,
Andy
Whiting
c and
Na
Wu
*ac
b,
Andy
Whiting
c and
Na
Wu
*ac
aState Key Laboratory for the Chemistry and Molecular Engineering of Medicinal Resources, School of Chemistry and Pharmaceutical Science, Guangxi Normal University, Gulin, Guangxi 541004, China. E-mail: wuna07@gxnu.edu.cn
bChemistry Research Laboratory, Oxford University, 12 Mansfield Road, Oxford, OX1 3TA, UK
cDepartment of Chemistry, Durham University, South Road, Durham DH1 3LE, UK. E-mail: na.wu@durham.ac.uk
First published on 4th March 2019
Abstract
The first example of an intramolecular hydroarylation of 1,6-en-α-chloro-enamides was achieved by a palladium-catalysed ligand-free reductive Heck cycloisomerisation with no competing Heck-cyclised by-product.
Reductive processes in metal-catalysed organic synthesis are often well understood, involving common reductants such as dihydrogen, formates, formic acid and activated alcohols.1 Similarly, palladium-catalysed hydroarylation (reductive Heck reactions) between arylhalides and alkenes typically involve: (i) alkylamines, formates and activated alcohols as the hydride sources in the process;2 (ii) neutral or anionic aryl–Pd complexes, and electron-poor olefins and styrenes (preferred olefin substrates for insertion);2a,3 and (iii) key aryl-Pd species, coordinatively saturated with ligands (phosphines, N-heterocyclic carbenes, halides and acetates) to inhibit β-H-Pd elimination side-reactions.4
Herein, we report a palladium-catalysed reductive Heck cyclisation of 1,6-enynamides. In contrast to the common features of reductive Heck reactions, we report here that: (i) hydroarylation of styrene occurred through an intramolecular hydride transfer5 and an indolyl alkylpalladium(II)-species was reduced through an intermolecular hydride transfer likely from i-PrOH (or 1,4-dioxane6) as a H-donor, confirmed by D-isotope exchange studies; (ii) chloride dissociation of an electrophilic α-chloro-enamide was realised in the absence of alkylammonium salts as halide abstractors and a cationic Pd(II)-enamide Heck coupling proceeded with both electron-neutral and electron-rich styrenes;7 (iii) interestingly, the key enamide-Pd species was free from ligand saturation;8 and (iv) no β-H-Pd elimination by-product was observed.
Ynamides and enamides are versatile functional groups that find use as fascinating building blocks for the synthesis of nitrogen-containing compounds.9 Recently, Sarpong reported intermolecular Heck coupling reactions of bench-stable α-halo eneformamides in DMF or 1,4-dioxane10 and Tang reported a reduction of α-halo-enamides to enamides using Et3N as a reductant (Scheme 1).11 In order to explore a balance of reactivity and stability of α-halo-enamides, we prepared more electrophilic α-chloro tosylmides 7a and employed Sarpong's Heck conditions to test the potential intramolecular cyclisation of 7a. However, our approach was distinct from Sarpong's Heck in that a reductive Heck cyclised 8a was obtained exclusively, rather than a Heck cyclised 8a′ (Table 1, entries 1–3). Alternatively, using activated alcohols as solvents (which were employed in alkenylpalladative reduction of ynamides by Anderson12), 8a was also afforded in satisfactory yields (entries 4–13). Surprisingly, when electron-rich palladium ligands were employed, which were expected to prohibit β-H-Pd elimination via coordinative saturation of Pd(II) and Pd(0), the reductive Heck cyclisation was supressed (entries 14–18). In general, PdCl2 is more sustainable in 1,4-dioxane (entries 1 and 2) compared to in i-PrOH, as the precipitation of palladium black is immediately observed in i-PrOH.
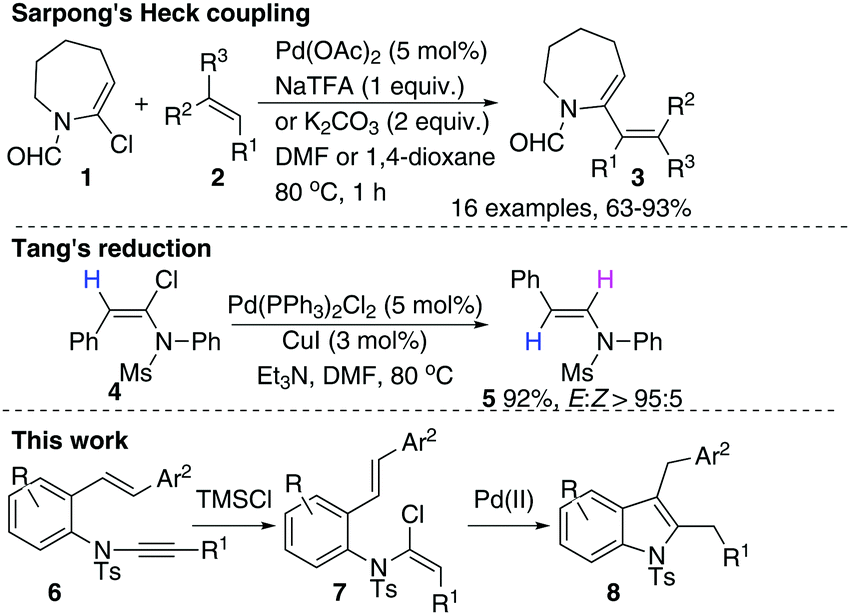 | ||
| Scheme 1 Heck coupling and reduction of chloro-enamides. | ||
|
|
|||
|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | Yieldb (%) |
| a 7a (0.15 mmol), Pd catalyst, K2CO3 (0.3 mmol), solvent (4 mL), 80 °C, 6 h, N2. b Isolated yield. c Without K2CO3. d N.R. = no reaction. e dec. = decomposition. f TMTU = tetramethyl thiourea. g Bipy = 2,2′-bipyridine. | |||
| 1 | PdCl2 (2.5) | 1,4-Dioxane | 81 |
| 2 | PdCl2 (20) | 1,4-Dioxane | 80 |
| 3 | PdCl2 (20)c | 1,4-Dioxane | N.R.d |
| 4 | PdCl2 (20) | i-PrOH | 93 |
| 5 | Pd(TFA)2 (10) | i-PrOH | 81 |
| 6 | None | i-PrOH | N.R.d |
| 7 | PdCl2 (20) | MeOH | Trace |
| 8 | PdCl2 (20) | EtOH | N.R.d |
| 9 | PdCl2 (20) | t-BuOH | dec.e |
| 10 | PdCl2 (20) | Toluene | dec.e |
| 11 | PdCl2 (10) | i-PrOH | 60 |
| 12 | PdCl2 (5) | i-PrOH | 44 |
| 13 | PdCl2 (2.5) | i-PrOH | 44 |
| 14 | PdCl2 (10), TMTUf (20) | i-PrOH | N.R.d |
| 15 | PdCl2 (10), bipyg (10) | i-PrOH | dec.e |
| 16 | Pd(PPh3)4 (5) | 1,4-Dioxane | N.R.d |
| 17 | Pd(PPh3)4 (5), (n-Bu)3P (10) | 1,4-Dioxane | N.R.d |
| 18 | Pd(PPh3)4 (5), (t-Bu)3P (10) | 1,4-Dioxane | dec.e |
| 19 | Pd2(dba)3 (2.5) | i-PrOH | Trace |
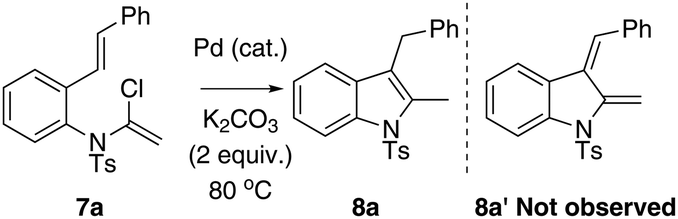
Based on the optimised results, entries 2 and 3 (Table 1) were applied to explore the substrate scope using 2-styryl-α-chloro-enamide derivatives 7 (Table 2). In order to overcome the hydrolytic lability of α-chloro-enamides, these species were prepared from in situ generated HCl and addition to enynamides 6, which were used directly without further isolation.13
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Starting material 6 | 8 | Yielda (%) | |||
| R | EWG | R1 | Ar2 | |||
| a Isolated yield. b Isopropanol was utilized as the solvent. c 1,4-Dioxane was utilized as the solvent. d Demethylated by-product was observed. e The reaction was conducted at 100 °C for 18 hours. | ||||||
| 1 | H | Ts | H | Ph | 8a | 79b; 50c |
| 2 | H | Ts | H | p-MeC6H4 | 8b | 58b; 18c |
| 3d | H | Ts | H | p-OMeC6H4 | 8c | 73b; 42c |
| 4 | H | Ts | H | p-ClC6H4 | 8d | 56b; tracec |
| 5 | H | Ts | Ph | Ph | 8e | 77b; 70c |
| 6 | H | Ts | p-MeC6H4 | Ph | 8f | 47b; 50c |
| 7 | H | Ts | p-t-BuC6H4 | Ph | 8g | 70b; 43c |
| 8 | H | Ms | H | Ph | 8h | 49b; 10c |
| 9 | H | Ns | H | Ph | 8i | 82b; 37c |
| 10d | H | Ts | H | o-OMeC6H4 | 8j | 23b; 10c |
| 11 | H | Ts | H | m-Furanyl | 8k | 24b; 10c |
| 12e |
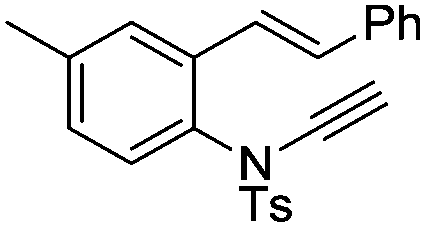
|
8l | 67b; 23c | |||
| 13d,e |
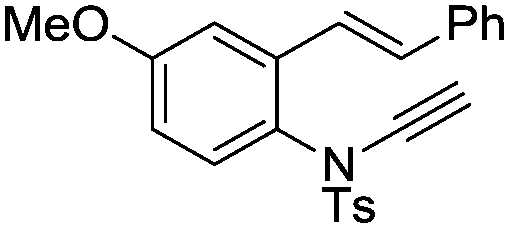
|
8m | 53b; 11c | |||
| 14 |
|
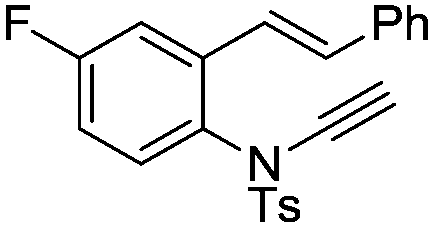
8n | 51b; N.R.c | |||
| 15 |
|
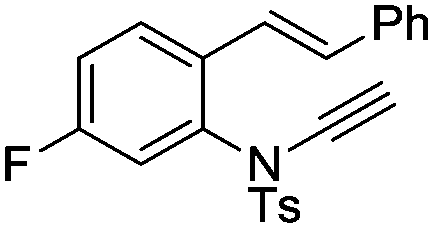
8o | 49b; N.R.c | |||
| 16e |
|
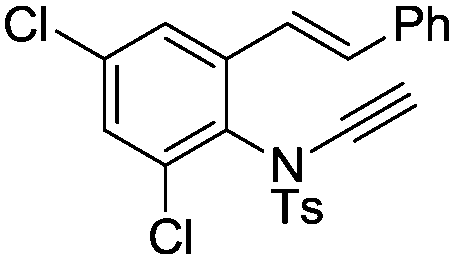
8p | 39b; N.R.c | |||
| 17 |
|
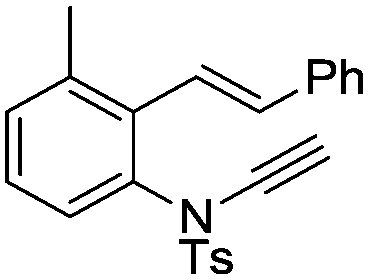
8q | 20b; N.R.c | |||
| 18 |
|
|
||||
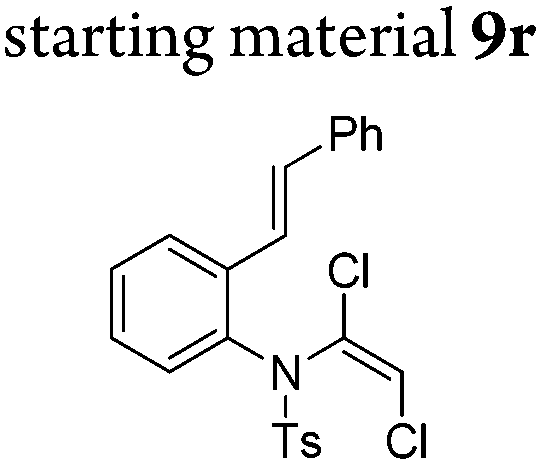
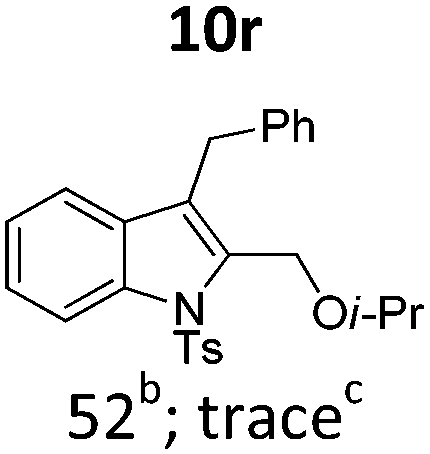
In general, the one-pot, sequential cyclisation afforded 3-benzylindoles 8 in higher yields in i-PrOH than in 1,4-dioxane (Table 2). When the styryl group contained electron-donating groups (entries 2 and 3, Table 2) and mildly electron-deficient groups (entry 4, Table 2), the reductive Heck process proceeded to deliver products 8 in moderate to good yields. As for the ynamide fragment, terminal and internal aryl ynamides were tolerated (entries 1, 5–9, Table 2). When the tosylamide was replaced with Ms- and Ns-amides, a better yield was obtained for the Ns-variant in i-PrOH (entry 9, Table 2). However, the substrates 6 were restricted to para-substituted aryl groups, and the cyclisation of in situ generated 7 containing ortho- and meta-substituted aryl groups was more complicated with slow conversion (20 h), accompanied by complex mixtures (entries 10 and 11, Table 2). Noticeably, the substrates 6 bearing electron-poor styrenes and electron-poor ynamide moieties were incompatible with the reaction conditions, where complex mixtures were formed. Replacing styryl and ynamidyl fragments of 6 with alkyl-substituted alkenes and alkyl ynamides, respectively, also led to complex mixtures.14 We next assessed the benzene rings of indoles. The reactions (entries 12–17, Table 2) proceeded well to deliver products bearing electron-donating or electron-withdrawing groups on the aniline ring, although the yield dropped to 20% when C-4 was substituted (entry 17). Noticeably, when en-α,β-dichloro-enamide 9 was employed, an unusual competing C–O coupling was found to give an isopropoxide 10 (entry 18).15
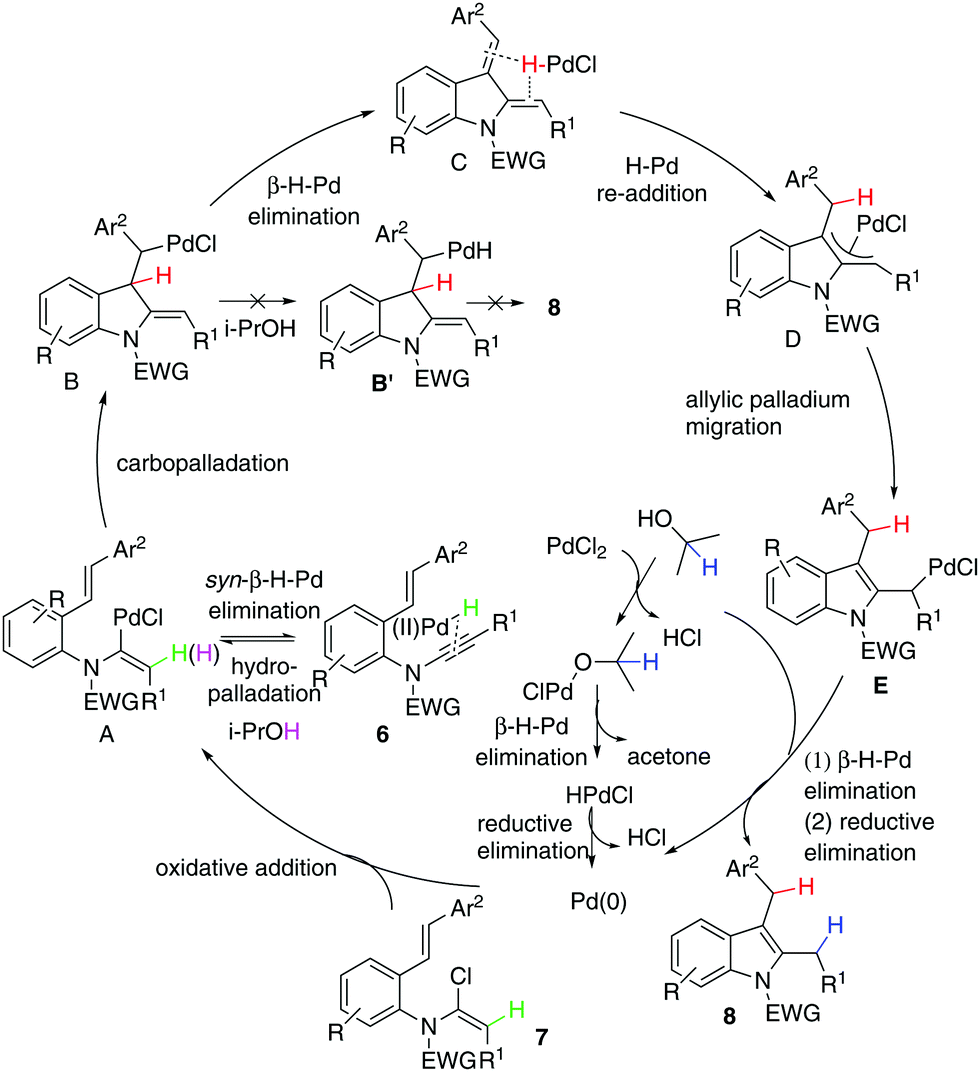
Given that the configuration of 8o was confirmed by X-ray diffraction analysis (Fig. 1),16 we are able to propose that a novel Pd(0)-catalysed reductive Heck cycloisomerisation mechanism (Scheme 2) explains these observations. This is initiated by oxidative addition of Pd(0), generated by β-hydride elimination and reductive elimination via coordination of PdCl2 with i-PrOH17 or 1,4-dioxane.6 The intramolecular coordination of the styrene may facilitate dissociation of the chloride anion to form the cationic Pd(II)-enamide species A from the highly electrophilic α-chloro-enamide 7.7
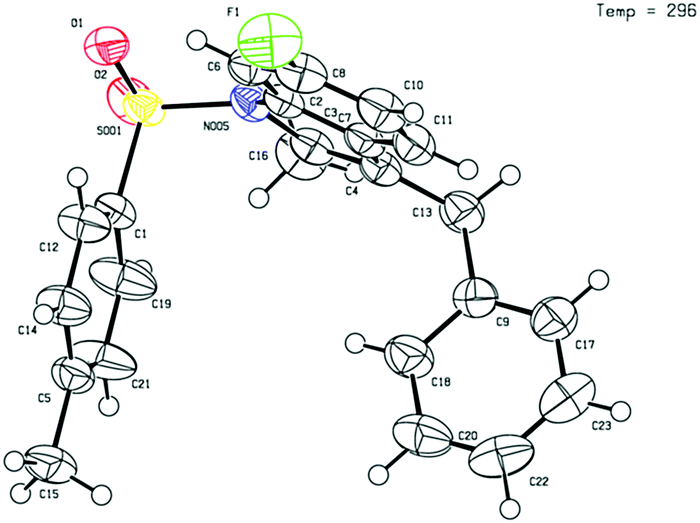 | ||
| Fig. 1 X-ray crystal structure of 8o. | ||
 | ||
| Scheme 2 Proposed overall mechanistic scheme. | ||
This is followed by an ionic Heck enamidation of the electron-rich styrene to afford diene C, which could be understood as arising from the styrene acting as a Lewis base attacking the electrophilic palladium(II).7 There is a driving force involved in aromatisation through a pseudo-intramolecular reversible re-addition of the Pd(II)–H species to dienyl indoline,18 which ligates Pd–H, to deliver the indolyl palladium species D. Upon alkene migration via the allylpalladium species, E was obtained. Then, there is a preference for Pd(II) to transfer methylene hydrogen from 1,4-dioxane or methanetriyl hydrogen from i-PrOH through its coordination with the solvent/β-hydride elimination/reductive elimination to irreversibly afford 8. If the cycle is not fast enough, reversible syn-β-H-Pd elimination of A and subsequent hydropalladation of ynamide 6 will occur,19 allowing proton exchange between substrate 7 and the solvent.
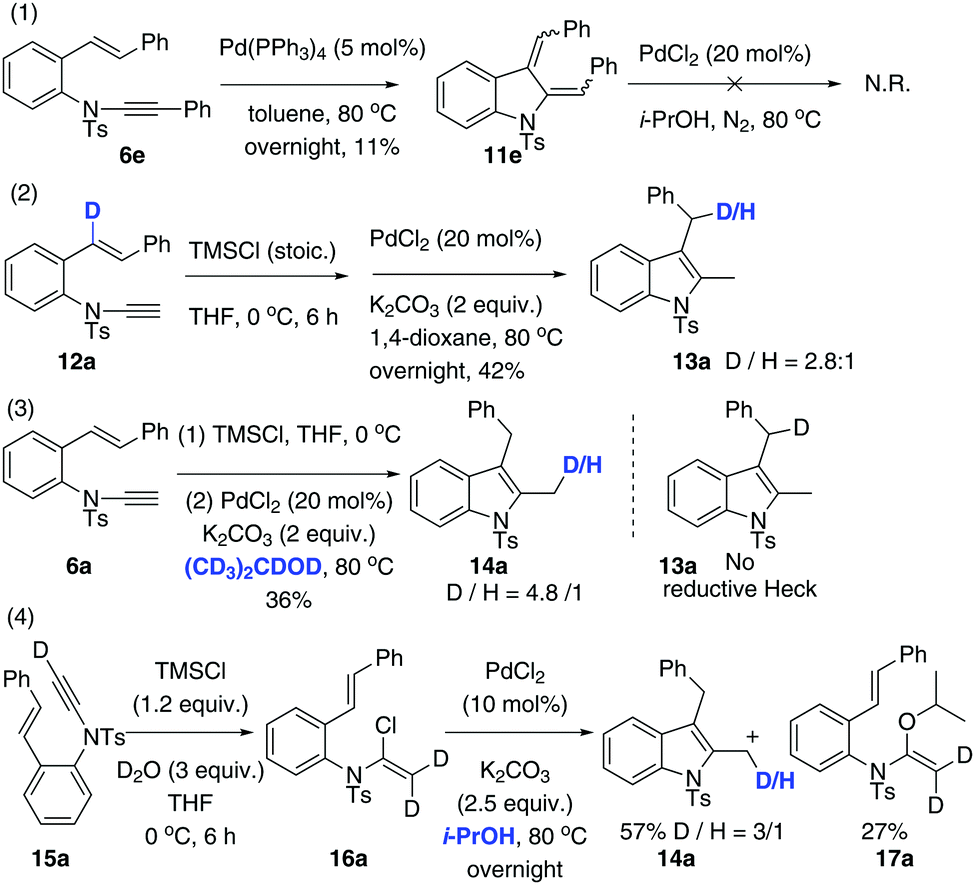
Our next focus was to seek potential reductants and determine whether they contribute to the proposed reductive Heck cycloisomerisation sequence. Firstly, the dienyl indoline 11e, acting as the presumptive intermediate C in Scheme 1, was prepared via cycloisomerisation of enynamide 6e. When it was subjected to PdCl2-catalysed, ligand-free conditions in i-PrOH, no reductive product 8e was obtained, implying that the reductive process was not initiated by an intermolecular H-Pd species generated from PdCl2 and i-PrOH. Secondly, to determine the source of the incoming hydrogen atom for the hydroenamidation of the styrene, we conducted a labelling experiment using 12a with deuterium labeled at the styryl moiety. Interestingly, 13a was obtained with deuterium migrating to the benzylic position, which elucidates that in the reduction of the styrene, the hydride source comes from the intramolecular H-Pd species, generated by β-H-Pd elimination and re-addition to the styrene.
Next, through various deuterium solvent screenings (1,4-dioxane-d8 and DMF-d7), we found that 6a was converted to the mono-deuterated product in 2-propanol-d8, without deuteration at the benzylic carbon. This indicates that before the reductive elimination of the C–Pd(II)–D bond, palladium is located at the methylene position, rather than at the benzylic position, which excludes the possibility of a pathway to form 8viaB′. Furthermore, this result confirmed that the solvent was indeed involved as a hydride donor in the reduction of the terminal alkylpalladium(II) species (Scheme 3).
 | ||
| Scheme 3 Deuterium labelling study. | ||
Finally, isotopomer 16a, with two deuterium atoms on the β-carbon of the α-chloro-enamide, was subjected to the reductive cycloisomerisation conditions. Interestingly, the indole 14a was obtained with one deuterium replaced with a hydrogen atom, accompanied by a C–O coupled isopropoxide 17a. This reveals that syn-β-D-Pd elimination of A and re-addition to ynamide 6 occurs reversibly, allowing D–H exchange of deuterated A with i-PrOH to take place.20
In conclusion, a palladium-catalysed ligand-free reductive Heck cycloisomerisation of aromatic 1,6-enynamides has been realised using in situ generated 1,6-en-α-chloro-enamides in a one-pot, stepwise protocol. Deuterium isotope labeling studies revealed that intramolecular hydride transfer and intermolecular hydride donation from the solvent were observed. Moreover, this indicates that there was a hydride exchange between chloroenamide and i-PrOH. The mild, straightforward experimental conditions will heighten valuable potential for the synthesis of complex azacyclic target compounds from acyclic units in both academic and industrial research settings.
This work was supported by the Royal Society – Newton International Fellowship (170322), the National Natural Science Foundation of China (21462004), the State Key Laboratory for the Chemistry and Molecular Engineering of Medicinal Resources (CMEMR2014-A04) and GXNU (2014ZD004).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) M. Ahlquist, G. Fabrizi, S. Cacchi and P. Norrby, J. Am. Chem. Soc., 2006, 128, 12785 CrossRef CAS PubMed; (b) S. Raoufmoghaddam, S. Mannathan, A. J. Minnaard, J. G. de Vries and J. N. H. Reek, Chem. – Eur. J., 2015, 21, 18811 CrossRef CAS PubMed; (c) K. Wu, N. Sun, B. Hu, Z. Shen, L. Jin and X. Hu, Adv. Synth. Catal., 2018, 360, 3038 CrossRef CAS.
- (a) G. Yue, K. Lei, H. Hirao and J. Zhou, Angew. Chem., Int. Ed., 2015, 54, 6531 CrossRef CAS PubMed; (b) Z.-M. Zhang, B. Xu, Y. Qian, L. Wu, Y. Wu, L. Zhou, Y. Liu and J. Zhang, Angew. Chem., Int. Ed., 2018, 57, 10373 CrossRef CAS PubMed; (c) L. Jin, J. Qian, N. Sun, B. Hu, Z. Shen and X. Hu, Chem. Commun., 2018, 54, 5752 RSC.
- (a) B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 79 CrossRef CAS PubMed; (b) X. Qin, M. W. Y. Lee and J. S. Zhou, Angew. Chem., Int. Ed., 2017, 56, 12723 CrossRef CAS PubMed.
- (a) Z. Wang, Z. Zhang and X. Lu, Organometallics, 2000, 19, 775 CrossRef CAS; (b) N. Wu, L. Deng, L. Liu, Q. Liu, C. Li and Z. Yang, Chem. – Asian J., 2013, 8, 65 CrossRef CAS PubMed.
- For Pd-catalysed hydride transfer, see (a) H. Nakamura, S. Onagi and T. Kamakura, J. Org. Chem., 2005, 70, 2357 CrossRef CAS PubMed; (b) H. Nakamura, M. Ishikura, T. Sugiishi, T. Kamakura and J.-F. Biellmann, Org. Biomol. Chem., 2008, 6, 1471 RSC; (c) N. Wu, A. Messinis, A. S. Batsanov, Z. Yang, A. Whiting and T. B. Marder, Chem. Commun., 2012, 48, 9986 RSC; (d) K. Shekarrao, P. P. Kaishap, S. Gogoi and R. C. Boruah, Adv. Synth. Catal., 2015, 357, 1187 CrossRef CAS.
- (a) S. Y. W. Lau, N. G. Andersen and B. A. Keay, Org. Lett., 2001, 3, 181 CrossRef CAS PubMed; (b) J. A. M. Torre, P. Espinet and A. C. Albéniz, Organometallics, 2013, 32, 5428 CrossRef.
- (a) J. Mo, L. Xu and J. Xiao, J. Am. Chem. Soc., 2005, 127, 751 CrossRef CAS PubMed; (b) J. Mo and J. Xiao, Angew. Chem., Int. Ed., 2006, 45, 4152 CrossRef CAS PubMed; (c) Z. Hyder, J. Ruan and J. Xiao, Chem. – Eur. J., 2008, 14, 5555 CrossRef CAS PubMed; (d) J. Ruan, J. A. Iggo, N. G. Berry and J. Xiao, J. Am. Chem. Soc., 2010, 132, 16689 CrossRef CAS PubMed.
- For palladium-catalysed ligand-free reactions, see (a) C. Zhu, B. Yang and J.-E. Bäckvall, J. Am. Chem. Soc., 2015, 137, 11868 CrossRef CAS PubMed; (b) C. Zhu, B. Yang, T. Jiang and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2015, 54, 9066 CrossRef CAS PubMed.
- (a) G. Evano, N. Blanchard, G. Compain, A. Coste, C. S. Demmer, W. Gati, C. Guissart, J. Heimburger, N. Henry, K. Jouvin, G. Karthikeyan, A. Laouiti, M. Lecomte, A. Martin-Mingot, B. Métayer, B. Michelet, A. Nitelet, C. Theunissen, S. Thibaudeau, J. Wang, M. Zarca and C. Zhang, Chem. Lett., 2016, 45, 574 CrossRef CAS; (b) X.-N. Wang, H.-S. Yeom, L.-C. Fang, S. He, Z.-X. Ma, B. L. Kedrowski and R. P. Hsung, Acc. Chem. Res., 2014, 47, 560 CrossRef CAS PubMed; (c) Y. Zhang, K. A. DeKorver, H. Y. Li, A. G. Lohse, R. Hayashi, Z. J. Lu and R. P. Hsung, Chem. Rev., 2010, 110, 5064 CrossRef PubMed; (d) G. Evano, A. Coste and K. Jouvin, Angew. Chem., Int. Ed., 2010, 49, 2840 CrossRef CAS PubMed.
- T. K. Beng, S. M. Wilkerson-Hill and R. Sarpong, Org. Lett., 2014, 16, 916 CrossRef CAS PubMed.
- W. Cao, P. Chen, L. Wang, H. Wen, Y. Liu, W. Wang and Y. Tang, Org. Lett., 2018, 20, 4507 CrossRef CAS PubMed.
- (a) R. L. Greenaway, C. D. Campbell, H. A. Chapman and E. A. Anderson, Adv. Synth. Catal., 2012, 354, 3187 CrossRef CAS; (b) C. D. Campbell, R. L. Greenaway, O. T. Holton, P. R. Walker, H. A. Chapman, C. A. Russell, G. Carr, A. L. Thomson and E. A. Anderson, Chem. – Eur. J., 2015, 21, 12627 CrossRef CAS PubMed.
- K. Ohashi, S. Mihara, A. H. Sato, M. Ide and T. Iwasawa, Tetrahedron Lett., 2014, 55, 632 CrossRef CAS.
- For unreactive substrate table, see ESI†.
- For alkoxylation of 2-(chloromethyl)-1H-indole derivative, see: J.-R. Huang and C. Bolm, Angew. Chem., Int. Ed., 2017, 56, 15921 CrossRef CAS PubMed.
- 1890685 (8o). The single crystal X-ray structure of product 8o is included in the ESI†.
- (a) R. L. Greenaway, C. D. Campbell, H. A. Chapman and E. A. Anderson, Adv. Synth. Catal., 2012, 354, 3187 CrossRef CAS; (b) B. Ding, Z. Zhang, Y. Liu, M. Sugiya, T. Imamoto and W. Zhang, Org. Lett., 2013, 15, 3690 CrossRef CAS PubMed; (c) P. R. Melvin, D. Balcells, N. Hazari and A. Nova, ACS Catal., 2015, 5, 5596 CrossRef CAS.
- (a) S. F. Vice, H. N. Carvalho, N. G. Taylor and G. I. Dmitrienko, Tetrahedron Lett., 1989, 30, 7289 CrossRef CAS; (b) X. Chen, G. Zheng, Y. Li, G. Song and X. Li, Org. Lett., 2017, 19, 6184 CrossRef CAS PubMed.
- G. Liu, W. Kong, J. Che and G. Zhu, Adv. Synth. Catal., 2014, 356, 3314 CrossRef CAS.
- For detailed mechanistic study, see ESI†.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1890685 (8o). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc00537d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |