
Recent advances in alkoxy radical-promoted C–C and C–H bond functionalization starting from free alcohols
Xinxin
Wu
a and
Chen
Zhu
 *ab
*ab
aKey Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, 199 Ren-Ai Road, Suzhou, Jiangsu 215123, China. E-mail: chzhu@suda.edu.cn
bKey Laboratory of Synthetic Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China
First published on 19th July 2019
Abstract
Direct functionalization of inert C(sp3)–C(sp3) and C(sp3)–H bonds represents one of the most valuable synthetic tactics, yet large obstacles remain in terms of reactivity and selectivity. Alkoxy radicals enable C–C bond scission via β-C elimination and C(sp3)–H bond cleavage through a hydrogen atom transfer (HAT) process, thus providing an efficient method to address these problems. In view of atom- and step-economy, the direct use of abundant alcohols as alkoxy radical precursors in radical transformations is of high synthetic value. This feature article summarizes our recent achievements in (a) C(sp3)–C(sp3) bond cleavage via ring-opening of cycloalkanols and (b) site-specific C(sp3)–H functionalization of unprotected aliphatic alcohols, along with the reconstruction of various new chemical bonds, e.g. C–halogen, C–C, C–N, C–S, and C–Se bonds.
 Xinxin Wu | Xinxin Wu obtained a BS degree from Nanjing Tech University in 2011, and a PhD degree from East China University of Science and Technology in 2016 under the supervision of Prof. Jinxing Ye. Then he moved to Chen Zhu's group at Soochow University as a postdoctoral research fellow. His current research focuses on photochemistry, radical chemistry and asymmetric synthesis and their applications in synthesis of biologically important compounds. |
 Chen Zhu | Chen Zhu received a BS degree from Xiamen University in 2003, and a PhD degree from Shanghai Institute of Organic Chemistry in 2008 under the supervision of Prof. Guo-Qiang Lin. After postdoctoral research in Gakushuin University, Japan with Prof. Takahiko Akiyama, he moved to the University of Texas Southwestern Medical Center, working with Prof. John R. Falck and Prof. Chuo Chen as postdoctoral fellow. He was appointed as professor at Soochow University, China in Dec. 2013. His current research interests mainly focus on radical-mediated transformations, in particular C–C and C–H bond functionalization. |
1. Introduction
Owing to the ubiquity of inert chemical bonds such as C–C and C–H bonds in nature, direct transformation of inert chemical bonds into other valuable functional groups represents an ideal synthetic tactic with the upmost atom- and step-economy.1,2 The past few decades have witnessed rapid ascent in this area, regardless of the large obstacles remaining in terms of reactivity and selectivity. Complementary to transition-metal catalysis that has made remarkable progress in C–C and C–H bond activation, controllable radical reactions have also proven to be a powerful toolkit showcasing the distinct potentials to regulate the reactivity as well as chemo/regio-selectivities.The radical translocations enabled by reactive heteroatom-centered radicals such as N- and O-radicals have long been exploited in organic chemistry, such as the Hofmann–Löffler–Freytag reaction3 and Barton nitrite ester photolysis.4 Apart from the applications in C(sp3)–H activation via hydrogen atom abstraction, O-radicals also display a unique feature which induces β-C fragmentation to cleave C(sp3)–C(sp3) bonds. Alkoxy radicals, devoid of stabilization by inductive or delocalization effects, are a class of highly active intermediates normally hard to rein. In addition, direct formation of alkoxy radicals from free alcohols is formidably challenging due to the strong bond dissociation energy (BDE, ∼105 kcal mol−1)5 and high oxidation potential of alcoholic O–H bonds. These factors impede the wide exploitation of alkoxy radicals in synthetic chemistry. Traditionally, alkoxy radicals could be circuitously obtained from the elaborated alcohol derivatives, e.g. nitrite esters,4 lead(IV) alkoxides,6 peroxides,7 sulfonates,8 hypohalites,9N-alkoxylpyridine-2-thiones,10 and N-alkoxyphthalimides (Scheme 1).11 From the perspective of atom/step-economy and synthetic efficiency, in comparison, the alkoxy radical-mediated reactions starting from free alcohols are of higher value, and have received much attention recently.
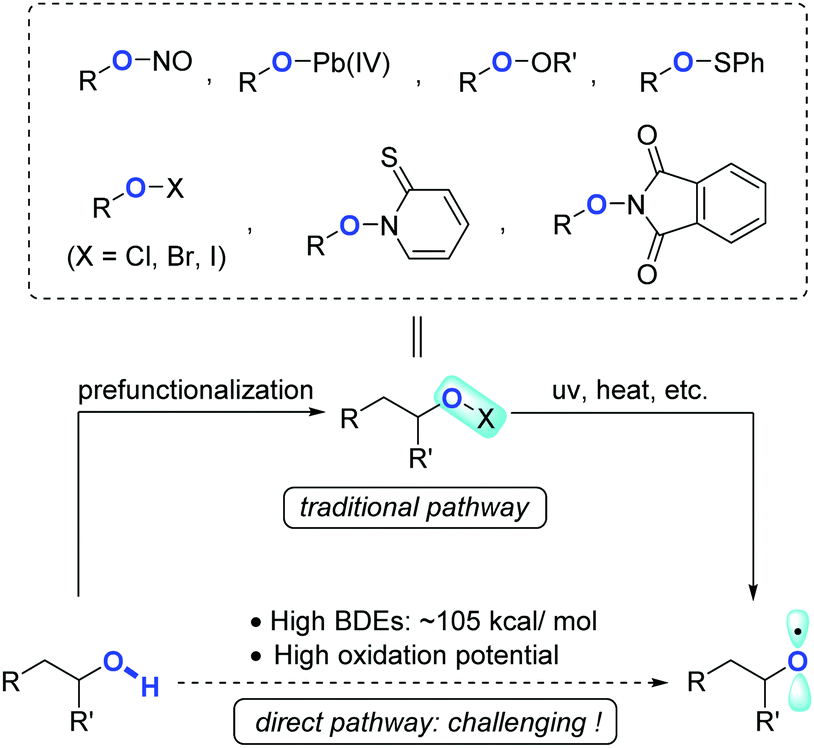 | ||
| Scheme 1 Generation of alkoxy radicals. | ||
Over the past few years, our group has been devoted to the formation of alkoxy radicals directly from free alcohols under mild conditions and the subsequent transformations. This feature article summarizes our recent achievements in (a) C(sp3)–C(sp3) bond cleavage via ring-opening of cycloalkanols and (b) site-specific C(sp3)–H functionalization of unprotected aliphatic alcohols, along with the reconstruction of various new chemical bonds, e.g. C–halogen, C–C, C–N, C–S, and C–Se bonds.
2. C(sp3)–C(sp3) bond cleavage via ring-opening functionalization of cycloalkanols
a. Formation of C–halogen bonds
Despite a number of elegant synthetic methods toward C(sp3)–C(sp3) bond cleavage, transition metal-mediated C(sp3)–C(sp3) activation still encounters tremendous obstacles, attributed to the fact that (a) the congested environment around the C–C σ-bond and the highly directed nature of the C–C σ-bond cause a poor interaction between the metal and the symmetric σ-orbital of C(sp3)–C(sp3) bonds, and (b) the resultant alkylmetal species by metal insertion is apt to undergo a β-hydride elimination rather than formation of new chemical bonds.Relying on strain energy, cyclopropanols and cyclobutanols bearing small-sized rings are frequently employed as privileged precursors in radical-mediated ring opening to synthesize the distally substituted ketones. The in situ generated alkoxy radicals trigger β-C fragmentation, offering a complementary approach for C(sp3)–C(sp3) bond cleavage to transition-metal catalysis.
Given that incorporating a fluorine atom into organic compounds remarkably improves the chemical, physical, and biological properties, the development of novel fluorination methods is crucial for multiple fields of pharmaceuticals, materials, and agrochemicals.12 In 2015, we disclosed the silver-catalyzed ring-opening fluorination of cyclobutanols and cyclopropanols for the first time, affording a variety of γ- and β-fluorinated ketones respectively.13 Selectfluor was used as the fluorinating agent and AgI salt was used as a catalyst. The investigation of substrate scope demonstrated that, regardless of electronic properties, either aryl or alkyl substituted cycloalkanols proceeded smoothly to give the corresponding products at room temperature (Scheme 2). Notably, the transformation of ring-fused benzocyclobutenols led to medium-sized cyclic fluoroketones, otherwise hard to make, through ring expansion.
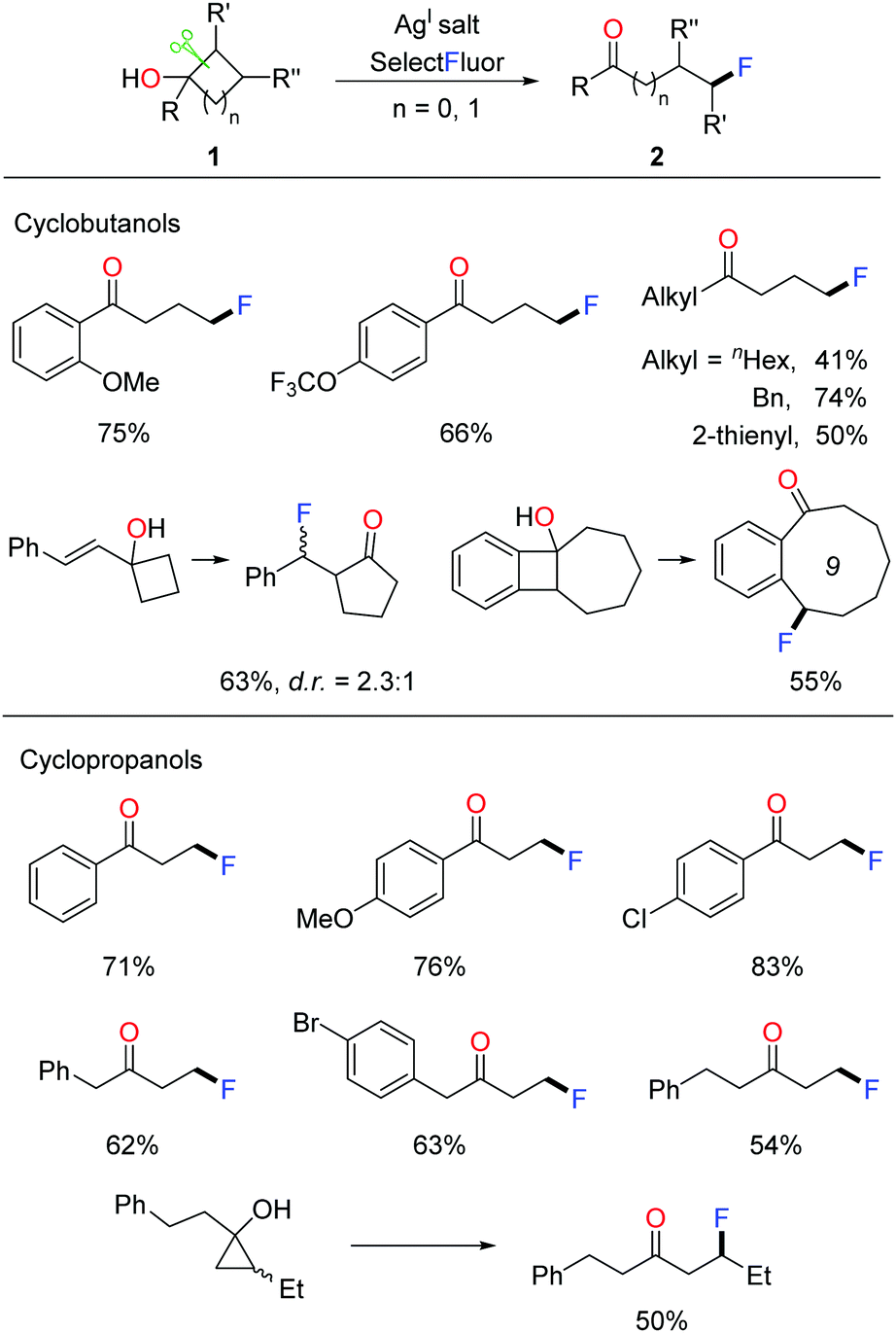 | ||
| Scheme 2 Ring-opening fluorination of cycloalkanols. | ||
A set of experiments were carried out to elucidate the mechanism. The results ruled out the pathways of electrophilic fluorination and silver-mediated reductive elimination, and supported the radical pathway. Accordingly, a plausible mechanism is depicted in Scheme 3.14 Initially, coordination of cycloalkanol 1 with AgI salt affords the complex a, which is then converted to the F-AgIII species b through the oxidation by Selectfluor. Homolysis of the intermediate b generates the cylcoalkoxy radical c along with the F-AgII complex. The ring-opening of cycloalkoxy radical c gives rise to the ring-opened alkyl radical d, which interacts with the F-AgII species to furnish the fluorinated product 2 and to regenerate the AgI species.
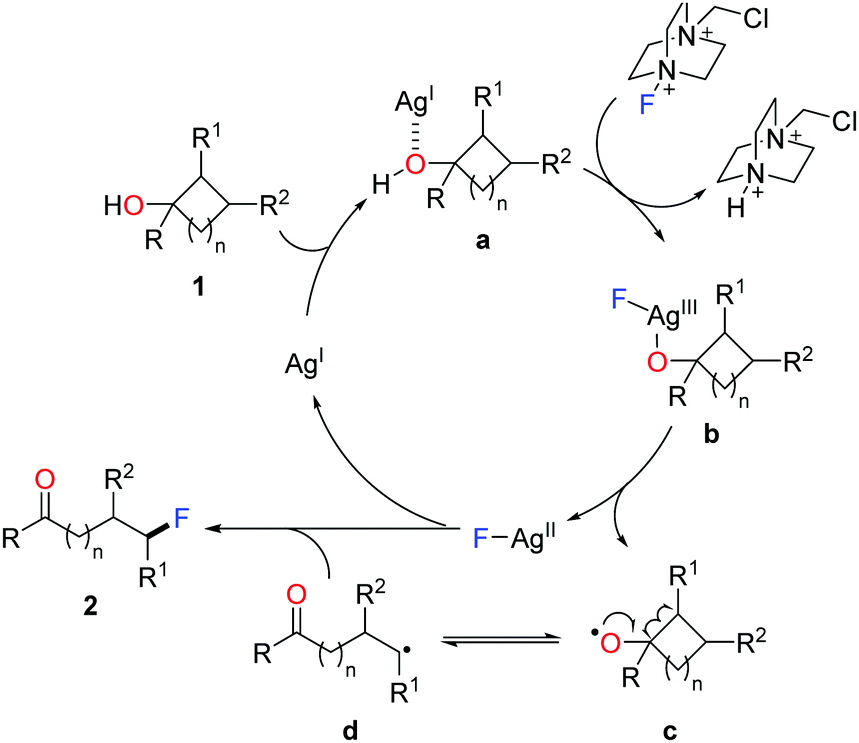 | ||
| Scheme 3 Proposed mechanism for ring-opening fluorination of cycloalkanols. | ||
Afterwards, radical mediated ring-opening fluorination of cycloalkanols was also reported by Murakami, Loh, and Lectka, respectively.15
Halogenated (Cl, Br, I) aliphatic ketones extensively serve as versatile building blocks in synthetic chemistry. However, the distally halogenated ketones are not easy to access via the direct halogenation at remote positions of alkyl ketones. Encouraged by the ring-opening fluorination, the silver-catalyzed protocol was further applied to the radical ring-opening chlorination.16 The combination of AgNO3 and K2S2O8 exhibited a good catalytic efficiency. With the use of NCS (N-chlorosuccinimide) as the chlorinating reagent, a portfolio of distally chlorinated ketones were obtained from the corresponding cycloalkanols under mild conditions (Scheme 4). The substrate scope covered all the three-, four-, five-, six-, and seven-membered cycloalkanols. The reaction could be performed at a gram-scale, indicating the practicality of the method. In addition, a set of distally brominated ketones were also afforded with the use of NBS (N-bromosuccinimide) in lieu of NCS.
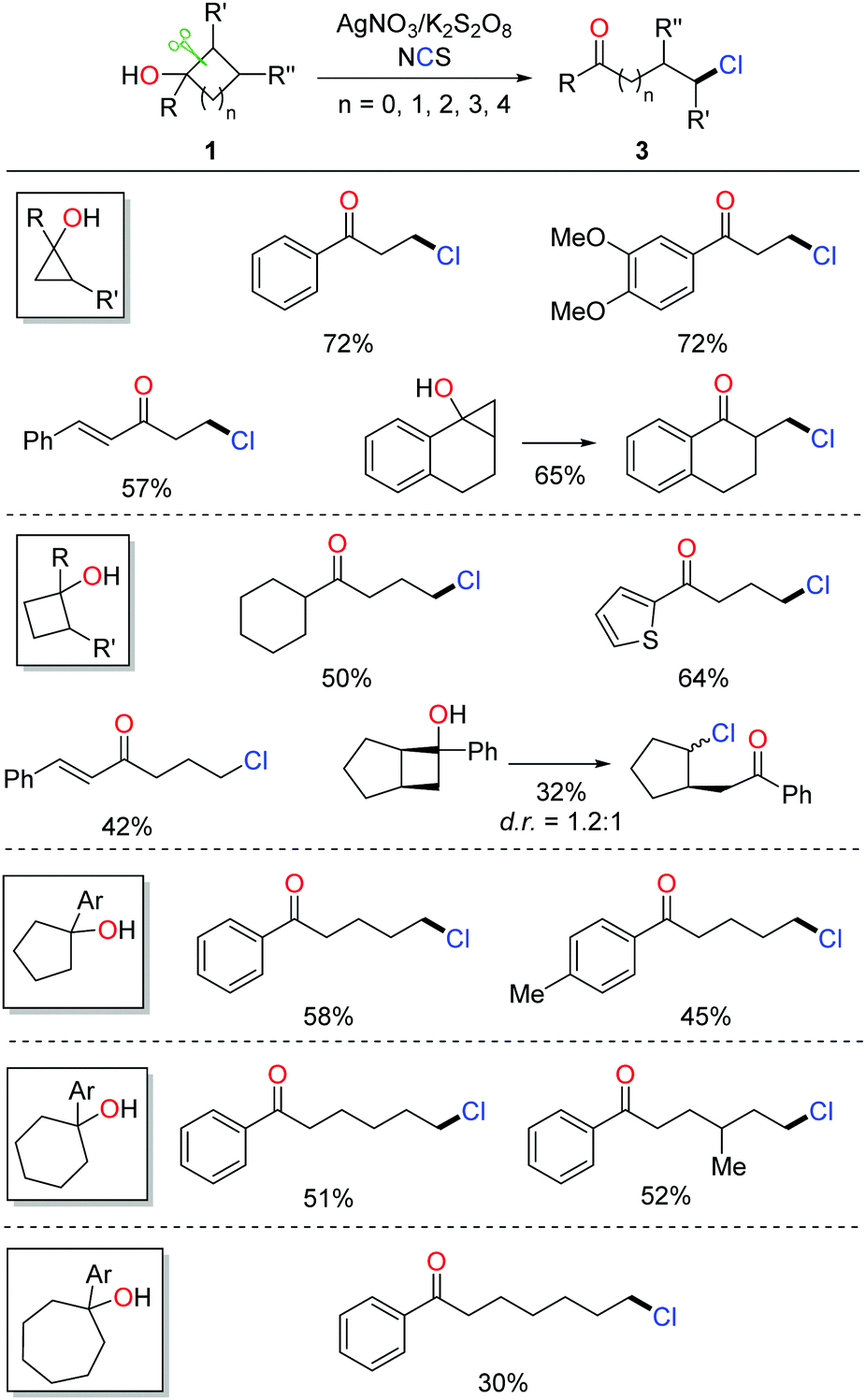 | ||
| Scheme 4 Ring-opening chlorination of cycloalkanols. | ||
The proposed mechanism is shown in Scheme 4. The generation of alkoxy radical a is probably from the single-electron oxidation of cycloalkanol by the AgI/K2S2O8 system (path a) or hydrogen atom abstraction of an O–H bond by the imidyl radical (path b). Ring opening of the cycloalkoxy radical a leads to alkyl radical b, which then interacts with NCS to afford the chlorinated ketone 3 and regenerate the imidyl radical (Scheme 5).
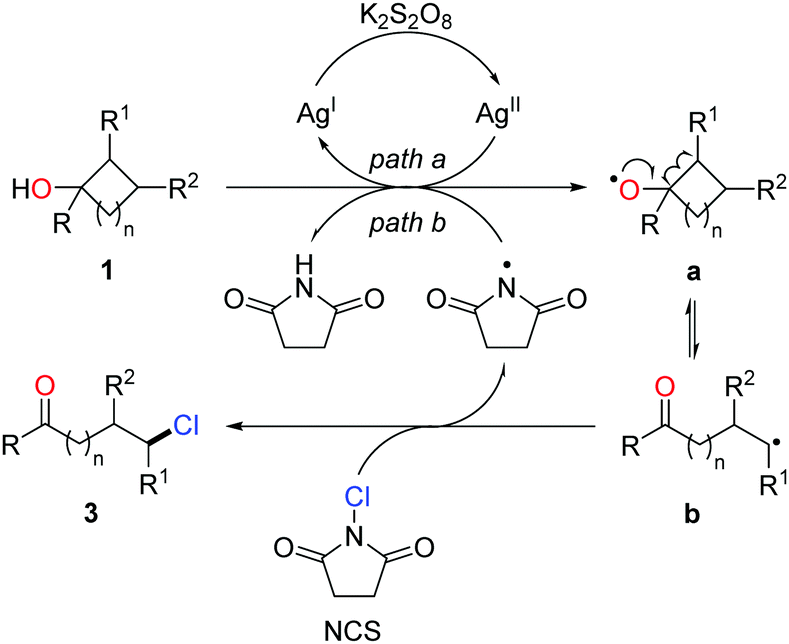 | ||
| Scheme 5 Proposed mechanism for ring-opening chlorination of cycloalkanols. | ||
A similar approach to γ-chloroketones was also achieved by the manganese-catalyzed ring-opening of cyclobutanols (Scheme 6).17 Inexpensive MnCl2 catalyst and nucleophilic chlorine source TMSCl were employed in lieu of Ag salt and NCS, affording comparable yields.
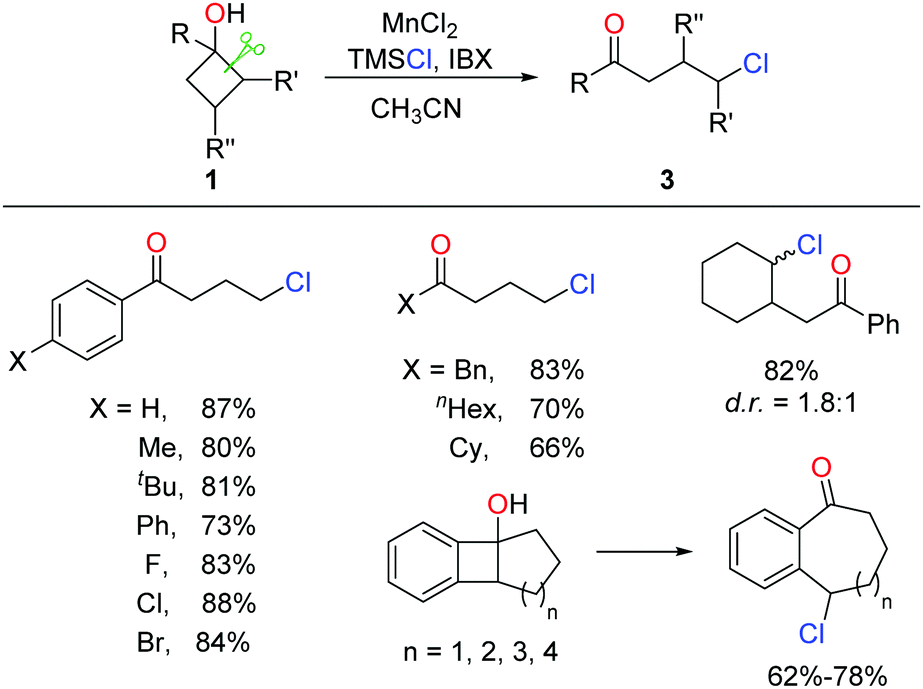 | ||
| Scheme 6 The manganese-catalyzed ring-opening chlorination of cyclobutanols. | ||
Analogous ring-opening chlorination of cyclobutanols mediated by AgOTf and tBuOCl was subsequently developed by Zhang and coworkers.18
The ring-opening functionalization of medium- and large-sized rings with low strain energy remains a formidable challenge. Recently, Knowles et al. disclosed the photocatalytic generation of alkoxy radicals via proton coupled electron transfer (PCET), and accomplished the ring opening of unstrained cycloalkanols under mild conditions.19 We also applied the photocatalytic method to the ring-opening bromination of unstrained cycloalkanols (Scheme 7). Optimization of the reaction conditions revealed that by using NBS as the bromine source and PIDA (phenyliodine diacetate) as an additive the desired δ-bromoketone was readily yielded from cyclopentanol under visible-light irradiation.20 This protocol was suitable for the ring-opening bromination of less-strained five-, six-, seven-, twelve-, and fifteen-membered cycloalkanols. The brominated ketones could be easily transformed to other useful compounds, indicating the value of the protocol. Beyond these, the unprecedented ring-opening cyanation and alkynylation of large-sized cycloalkanols were also achieved under the photo-induced conditions.
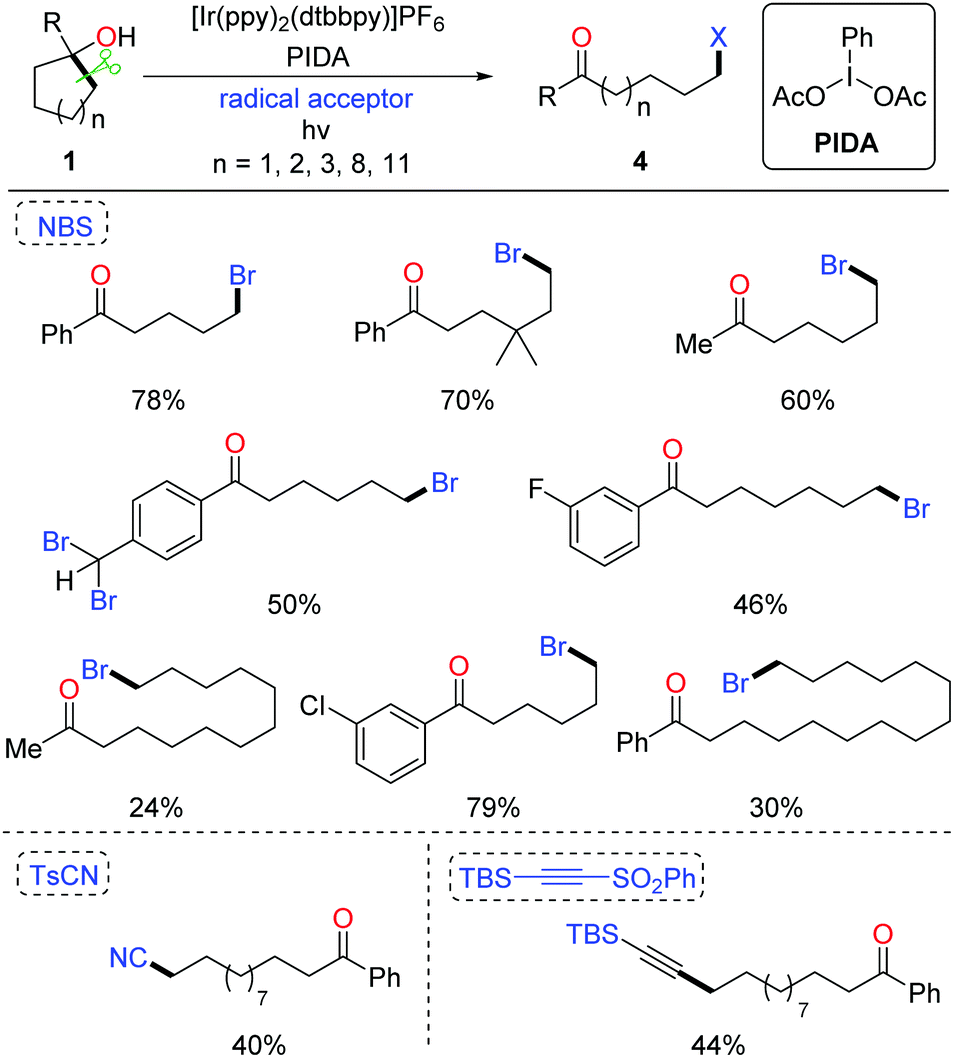 | ||
| Scheme 7 The photo-induced ring-opening functionalization of less-strained cycloalkanols. | ||
According to the mechanistic studies including the Stern–Volmer experiments and cyclic voltammograms, a plausible mechanism is outlined in Scheme 8. The photo-induced generation of alkoxy radical a might go through two possible pathways, the photocatalytic PCET process (path a) and homolysis of the O–I bond of intermediate c derived from the interaction of PIDA and cycloalkanol (path b). Subsequent β-C fragmentation of intermediate a delivers the ring-opened alkyl radical b, which is captured by NBS to form the final product 4.
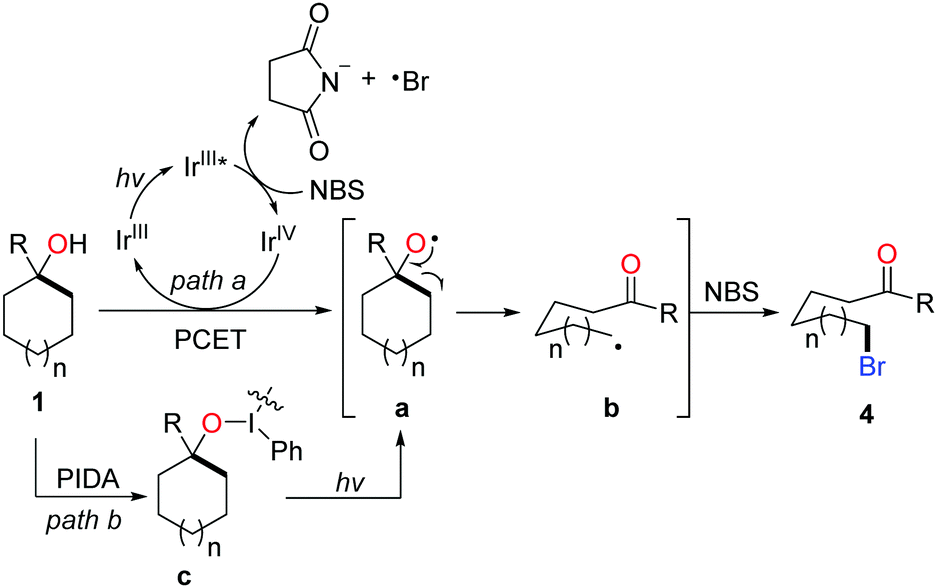 | ||
| Scheme 8 Plausible mechanism for the photo-induced ring-opening bromination of less-strained cycloalkanols. | ||
b. Formation of C–C bonds
Tetralone derivatives usually serve as versatile building blocks in synthetic chemistry. However, development of rapid and efficient access to 1-tetralones still remains underexplored. In the absence of extra radical scavengers, the cascade ring-opening of cyclobutanols and intramolecular cyclization readily afforded 1-tetralones.21 In the presence of AgNO3 and K2S2O8, a portfolio of tetralones were obtained in synthetically useful yields at room temperature (rt) (Scheme 9, top). The DFT studies also verified the feasibility of the reaction. A plausible mechanism is postulated (Scheme 9, bottom). Single-electron oxidation of cyclobutanol with the AgNO3/K2S2O8 system produces alkoxy radical a that then undergoes cyclic C–C bond cleavage to give alkyl radical b. Different from the previous works, the alkyl radical would intramolecularly add to the arene, affording the radical intermediate c which undergoes oxidation or hydrogen abstraction to furnish the tetralone product.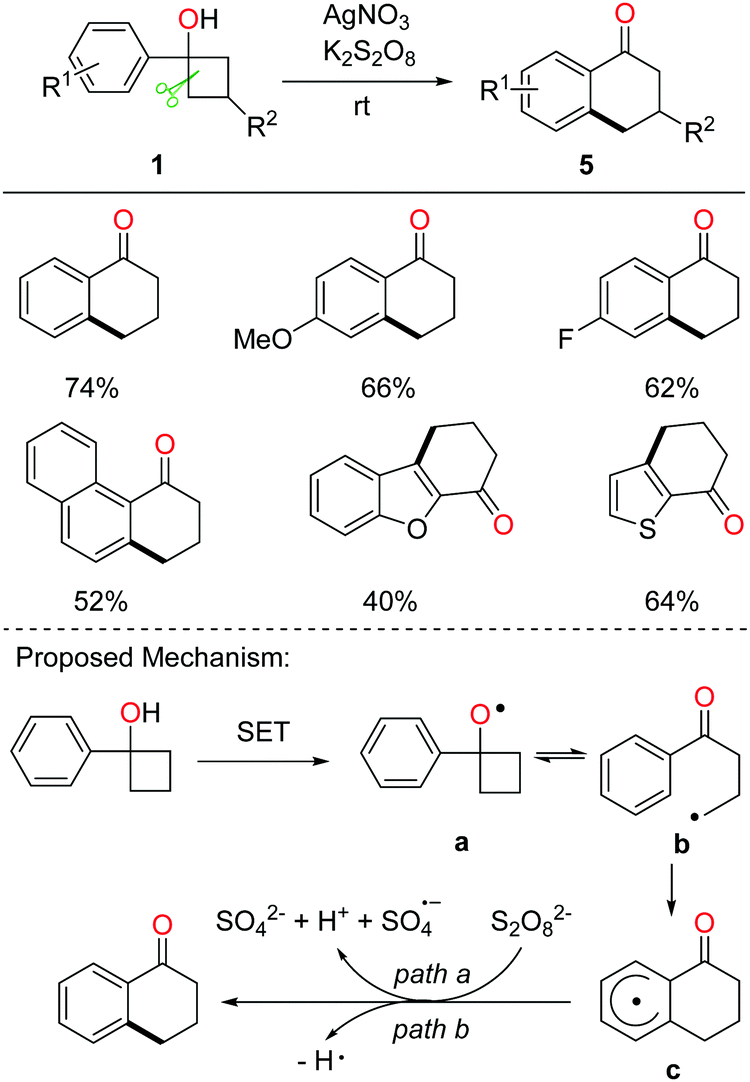 | ||
| Scheme 9 Synthesis of 1-tetralones and the proposed mechanism. | ||
The intermolecular C–C bond formation could also be accomplished by ring-opening functionalization of cyclobutanols. The corresponding γ-cyanated and alkynylated ketones were obtained via the manganese-catalyzed ring-opening of cyclobutanols, in which TsCN was used as a C1 unit and the TBS-protected phenylsulfonyl ethyne was used as a C2 unit, respectively (Scheme 10).22 Alternatively, the phenylsulfonyl alkyne substituted by phenyl or alkyl groups was also apt to afford the corresponding alkynylated products. Moreover, with the use of allyl phenyl sulfone as an allylating reagent, the allylated ketone was afforded in synthetically useful yields.
 | ||
| Scheme 10 The manganese-catalyzed ring-opening cyanation and alkynylation of cyclobutanols. | ||
The proposed mechanism for manganese-catalyzed ring-opening of cyclobutanols is depicted in Scheme 11. Initially, the oxidation of MnIII complex by a hypervalent iodine reagent (PIDA or DMP, DMP = Dess–Martin periodinane) affords the high-valent MnV complex, which then incorporates with cyclobutanol to furnish the intermediate a. The SET between MnV and O-atom results in alkoxy radical b and MnIV complex. The subsequent ring-opened alkyl radical c is intercepted by using TsCN or phenyl sulfone to generate the corresponding product.
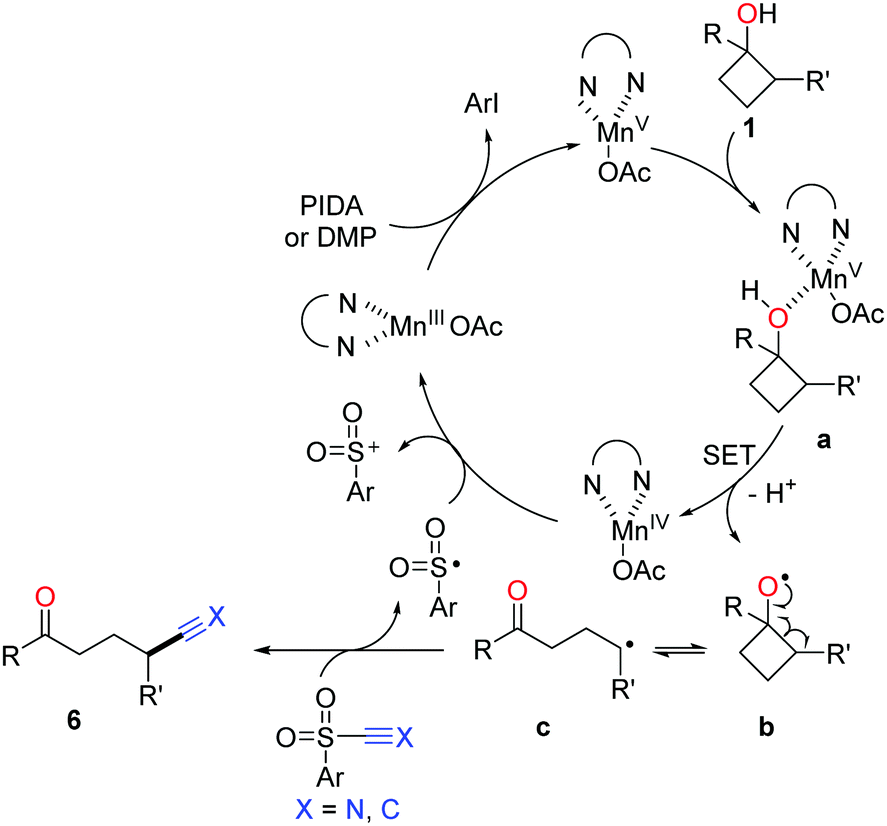 | ||
| Scheme 11 Proposed mechanism for the manganese-catalyzed ring-opening functionalization of cyclobutanols. | ||
Very recently, the successful merging of visible light-promoted ring-opening of cyclopropanols and functional group migration (FGM)23 furnished the unsymmetric 1,8-dicarbonyl compounds via the consecutive C–C bond scission and construction (Scheme 12).24 The reactions were performed under visible-light irradiation, and the use of BF3 as an additive significantly improved the yields. The transformation faced two challenges: (1) unmatched radical polarity. The addition of nucleophilic alkyl radicals to the electron-rich unactivated alkenes is kinetically unfavorable; (2) identical property of the alkyl radical intermediates a and b makes the radical addition step thermodynamically unfavored as well. The reaction was facilitated by the intramolecular FGM, which enabled the challenging radical addition step. This photocatalytic protocol featured mild conditions, good practicality, and high product diversity.
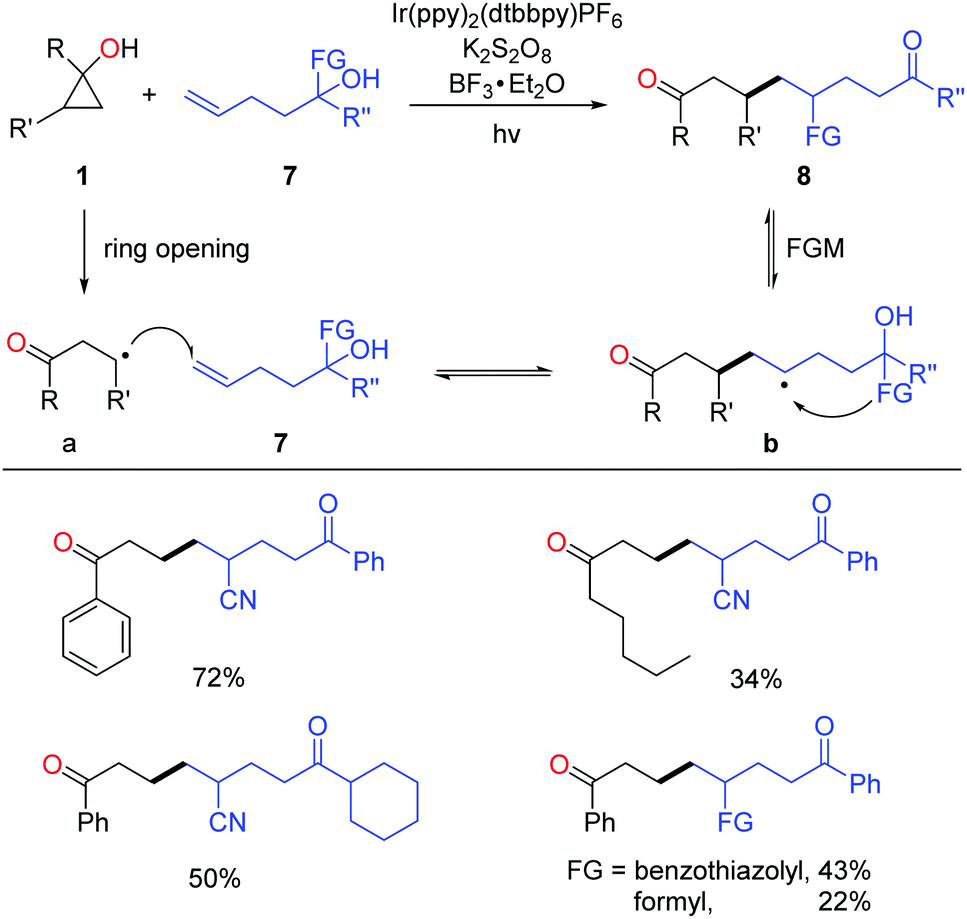 | ||
| Scheme 12 Synthesis of unsymmetric 1,8-dicarbonyl compounds. | ||
Chen et al. reported the reaction of cyclic and linear alkanols with cyclic iodine(III) reagent BI-OAc to furnish the alkynylated and olefinated products via photo-induced β-C scission.25 The benziodoxole/alcohol complex derived from the interaction between alkanol and BI-OAc was oxidized by the RuIII complex to provide alkoxy radicals that initiated the following transformation.
Zuo et al. disclosed a cerium-catalyzed cycloaddition of secondary cycloalkanols with electron-deficient alkenes through the synergistic cerium-mediated photocatalysis and photo-induced electron transfer catalysis, affording a variety of bridged-lactone products.26 The cerium complex-mediated LMCT process proposed by the authors enabled the bond homolysis to generate an alkoxy radical that triggered the β-C scission and subsequent transformations. Additionally, continuous flow for scale-up synthesis of the polycyclic core of nepalactones was conducted, indicating the synthetic utility of the protocol.
Very recently, Knowles et al. reported a photocatalytic ring expansion of cycloalkanols enabled by the PCET process.27 In the reaction, the alcoholic O–H bond was activated via the PCET process, leading to an alkoxy radical intermediate that undergoes subsequent β-C fragmentation to afford an alkyl radical. The following addition of the alkyl radical to the intramolecular alkene proceeded to afford valuable medium-sized cyclic ketones.
c. Formation of C–N bonds
Owing to the importance of N-containing compounds and their extensive occurrence, the application of the ring-opening strategy to construct C–N bonds is of synthetic use. By means of manganese catalysis, the ring-opening azidation of cyclobutanols was achieved with the use of TMSN3 as a nitrogen source and hypervalent iodine reagent BI-OH as an oxidant (Scheme 13).28 Despite electronic and steric effects, a range of structurally diverse cyclobutanols were readily converted to the corresponding γ-azido substituted ketones.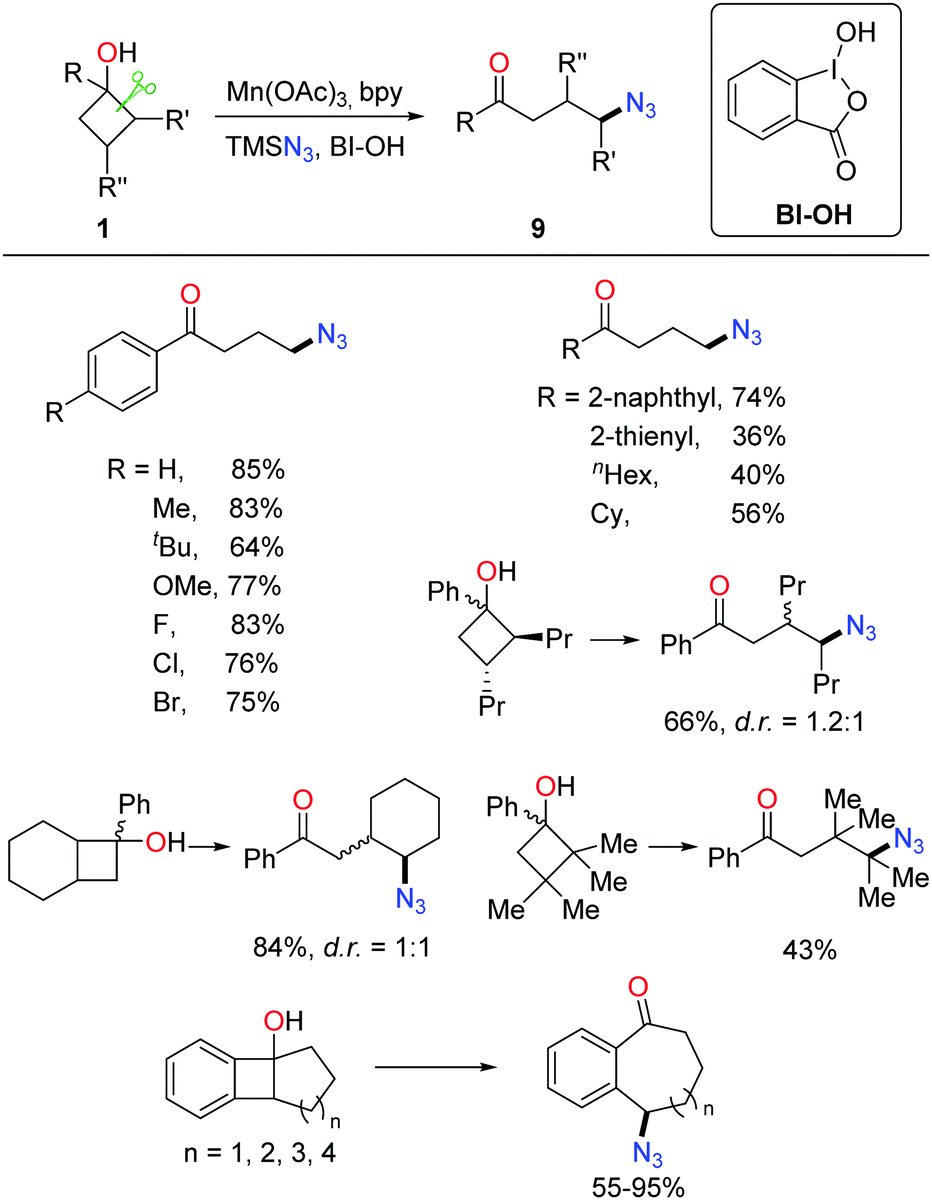 | ||
| Scheme 13 The manganese-catalyzed ring-opening azidation of cycloalkanols. | ||
As shown in Scheme 14, mechanistically, the interaction of Mn(OAc)3, TMSN3, and BI–OH gives rise to the high-valent MnV–N3 complex a. Incorporation of cyclobutanol 1 with the complex a generates complex b which then undergoes an SET process to furnish alkoxy radical c and the MnIV–N3 complex e. The transfer of azide from complex e to the ring-opened alkyl radical d affords the final product 9.
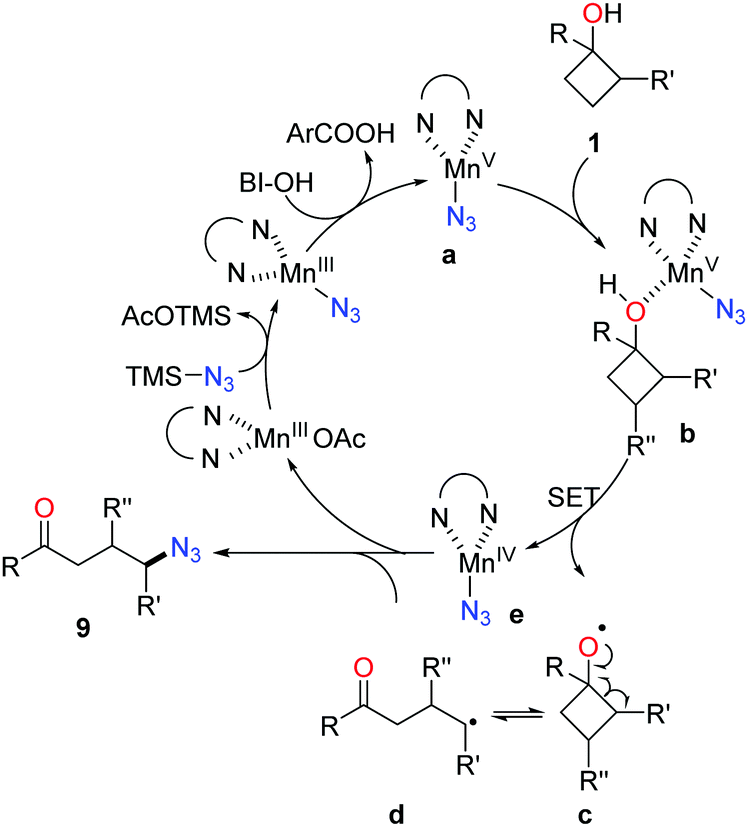 | ||
| Scheme 14 Proposed mechanism for the manganese-catalyzed ring-opening azidation of cycloalkanols. | ||
A similar strategy was further applied to the manganese-promoted ring-opening amination of cyclobutanols, in which DBAD (di-tert-butyl azodicarboxylate) was used as the aminating reagent (Scheme 15).29 A variety of alkyl hydrazines were generated in useful yields and exclusive regioselectivity at rt. Regarding the catalytic cycle, alkoxy radical a is generated initially from the single electron oxidation of cyclobutanol 1 by Mn(OAc)3 or the MnV species in situ formed from the interaction of Mn(OAc)3 with BI–OH. The radical intermediate a readily undergoes cyclic C–C bond cleavage to afford the alkyl radical b that then couples with DBAD to generate the N-centered radical intermediate c. Hydrogen abstraction of c from cyclobutanol affords the final product 10, regenerates alkoxy radical a, and perpetuates the catalytic cycle.
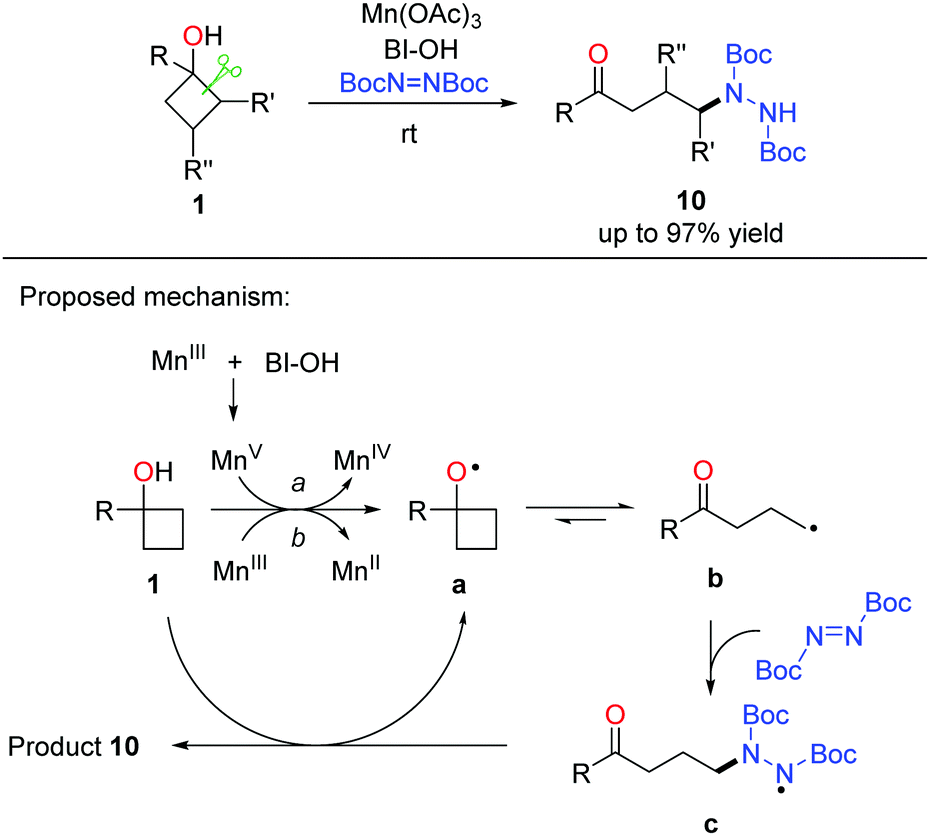 | ||
| Scheme 15 The manganese-catalyzed ring-opening hydrazination of cycloalkanols. | ||
Afterwards, Zuo et al. disclosed the cerium(III)-mediated photocatalytic hydrazination of less-strained cycloalkanols.30 Mechanistic studies indicated that the cerium(III) chloride was assisted by a chloride anion, showing good absorption in the blue-light region and leading to more efficient photoexcitation and higher catalytic efficiency. Another function of the cerium(III) chloride complex is to coordinate with an O-atom, which promotes the β-C scission to give the ring-opened alkyl radicals.
d. The formation of C–S and C–Se bonds
To further exploit the practicability of the method, the formation of C(sp3)–S and C(sp3)–Se bonds via the manganese-catalyzed ring-opening of cyclobutanols was also disclosed (Scheme 16).31,32 Commercially available reagents RS-SR (R = Ar, Me, Bn) and RSe-SeR (R = Ar, Bn) were used as the radical acceptors. The survey of the substrate scope demonstrated a good functional group tolerance; many susceptible groups such as alkynyl and alkenyl were compatible with the conditions. The protocol furnished a variety of γ-thiolated and selenylated ketones (11 and 12), paving a new avenue for the introduction of sulphides and selenides into organic compounds.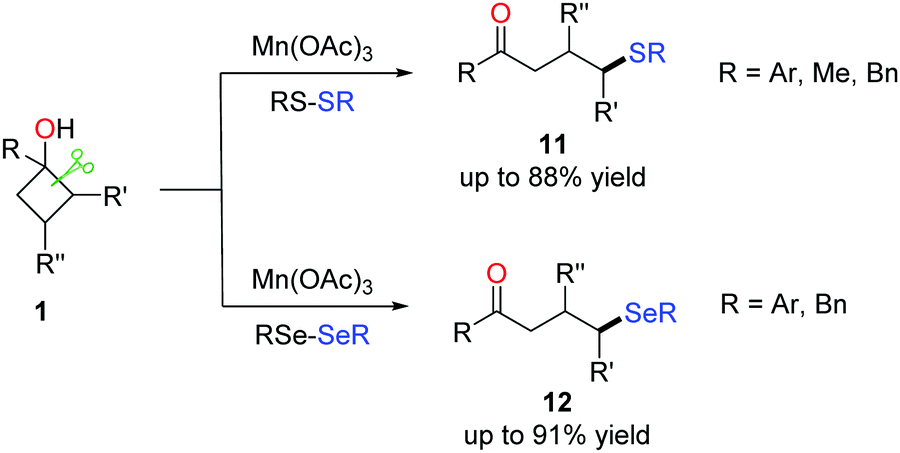 | ||
| Scheme 16 The manganese-mediated ring-opening C(sp3)–S/C(sp3)–Se bond formation. | ||
3. Remote C(sp3)–H functionalization via alkoxy radical-promoted HAT process
In the 1960s, the pioneering work on alkoxy radical-mediated HAT to achieve remote functionalization on an unreactive aliphatic site was reported by Barton and coworkers, namely the Barton nitrite ester reaction.4a Afterwards, numerous approaches were developed over the past half century for transformation of alcohols into suitable precursors to generate alkoxy radicals.4,6–11 Recently, the C(sp3)–H bond allylation and alkenylation promoted by alkoxy radicals, which were generated from the preformed N-alkoxyphthalimides, were disclosed by Chen et al.33 Soon after, Meggers et al. applied a similar strategy to the asymmetric alkylation of α,β-unsaturated N-acylpyrazoles.34 It was found that, conventionally, the formation of alkoxy radicals mostly relied on the transformation of alcohols into various alkoxy radical precursors.35 Hence, direct and efficient generation of alkoxy radicals from unmodified alcohols is in demand. Given the high BDEs and strong oxidation potential of alcoholic O–H bonds, the alkoxy radical-promoted HAT process involving free alcohols still remains challenging.Stimulated by the previous works on radical-mediated remote functional group migration (FGM),23 we disclosed tertiary-alcohol-directed remote C(sp3)–H heteroarylation via sequential HAT and distal heteroaryl migration (Scheme 17).36 Unprotected alcohols 13 were directly employed as the alkoxy radical precursors under photocatalytic conditions. Concerning the importance and ubiquity of heteroaryls in pharmaceuticals, a series of heteroaryls, e.g. benzothiazolyl, thiazolyl, and pyridyl, were surveyed, which showcased the migratory aptitude in the C(sp3)–H heteroarylation. Both secondary and tertiary C(sp3)–H bonds were readily functionalized with unique regioselectivity. The reactivity of different C(sp3)–H bonds followed the order: 3° > 2° ∼ benzylic >1° C–H. Mechanistic studies suggested that the generation of alkoxy radicals might be facilitated by a PCET process, which initiated the cascade of HAT process and intramolecular heteroaryl migration.
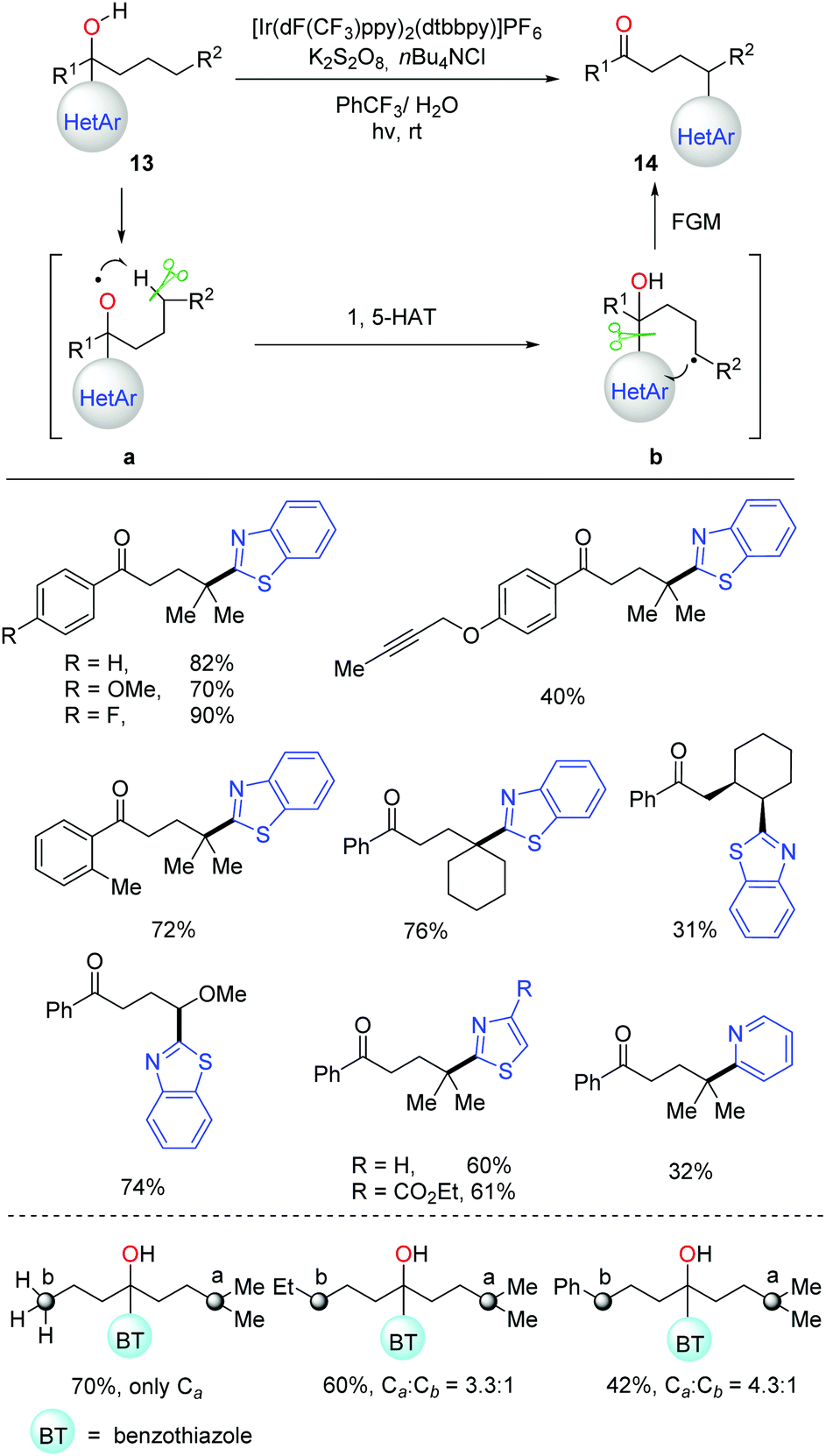 | ||
| Scheme 17 Free alcohol-directed heteroarylation of remote C(sp3)–H bonds. | ||
The photocatalytic protocol was then applied to the tertiary-alcohol-directed cyanation of remote C(sp3)–H bonds. The reaction proceeded in a cascade of HAT and remote cyano migration, similarly to the above C–H heteroarylation pathway (Scheme 18).37 A wide range of cyanohydrins 15 bearing various functional groups were tolerated in the reaction. Mechanistic investigation such as the quantum yield (φ = 4.7) indicated that the overall reaction outcome might be the integration of the photocatalytic pathway and radical-chain reaction. The products 16 were further converted to many valuable compounds, e.g. amines, carboxylic acids, and heterocycles, manifesting the utility of alkyl nitriles.
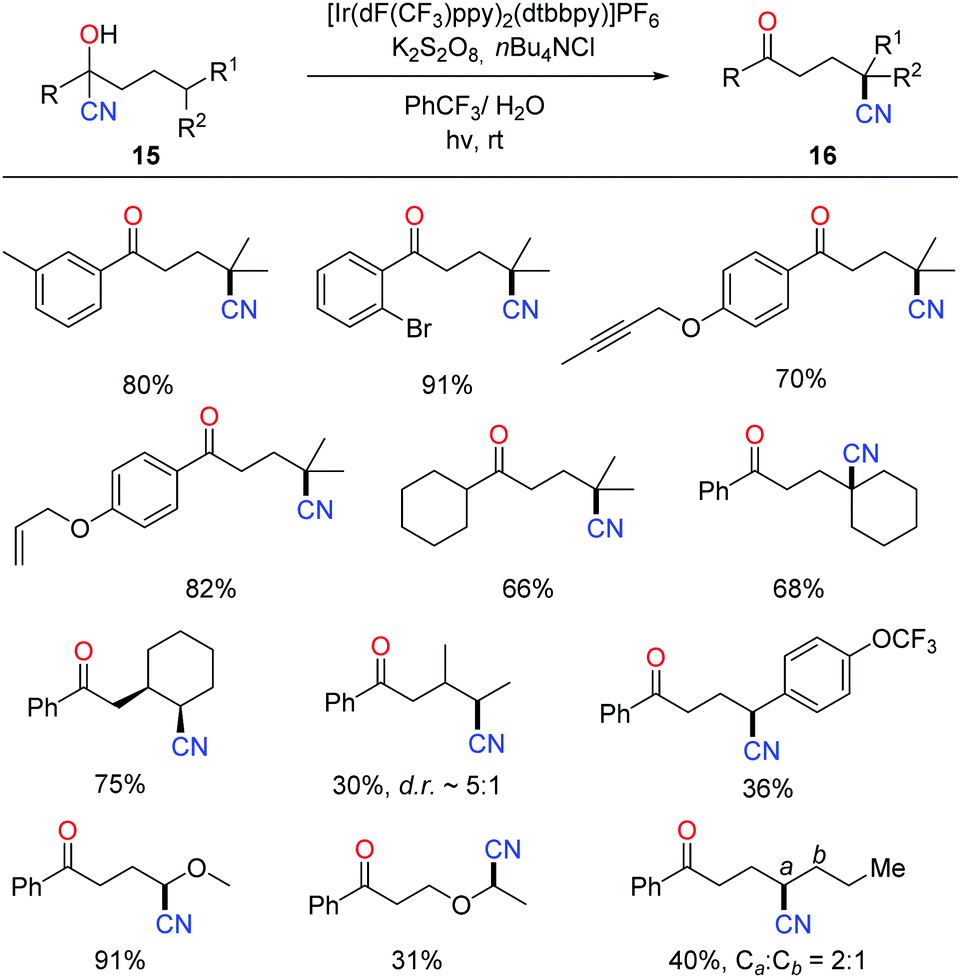 | ||
| Scheme 18 Free alcohol-directed cyanation of remote C(sp3)–H bonds. | ||
Recently, a metal-free visible light-enabled intermolecular C(sp3)–H heteroarylation of aliphatic alcohols was disclosed (Scheme 19).38 Heteroaryl was readily incorporated at the δ-position of free alcohol with a good yield and an exclusive regioselectivity. Phenyliodine bis(trifluoroacetate) (PIFA) was used as the only reagent. This Minisci-type reaction featured mild conditions and broad substrate scope. All the primary, secondary, and tertiary alcohols were suitable substrates. A portfolio of heteroaryls, e.g. quinoline, isoquinoline, pyridine, phenanthridine, acridine, pyrazine, pyrimidine, quinoxaline, and 4-hydroxyquinazoline, were eligible acceptors for the alkyl radicals generated from the 1,5-HAT process. Furthermore, this strategy could be applied to the late-stage modification of complex natural products and drugs.
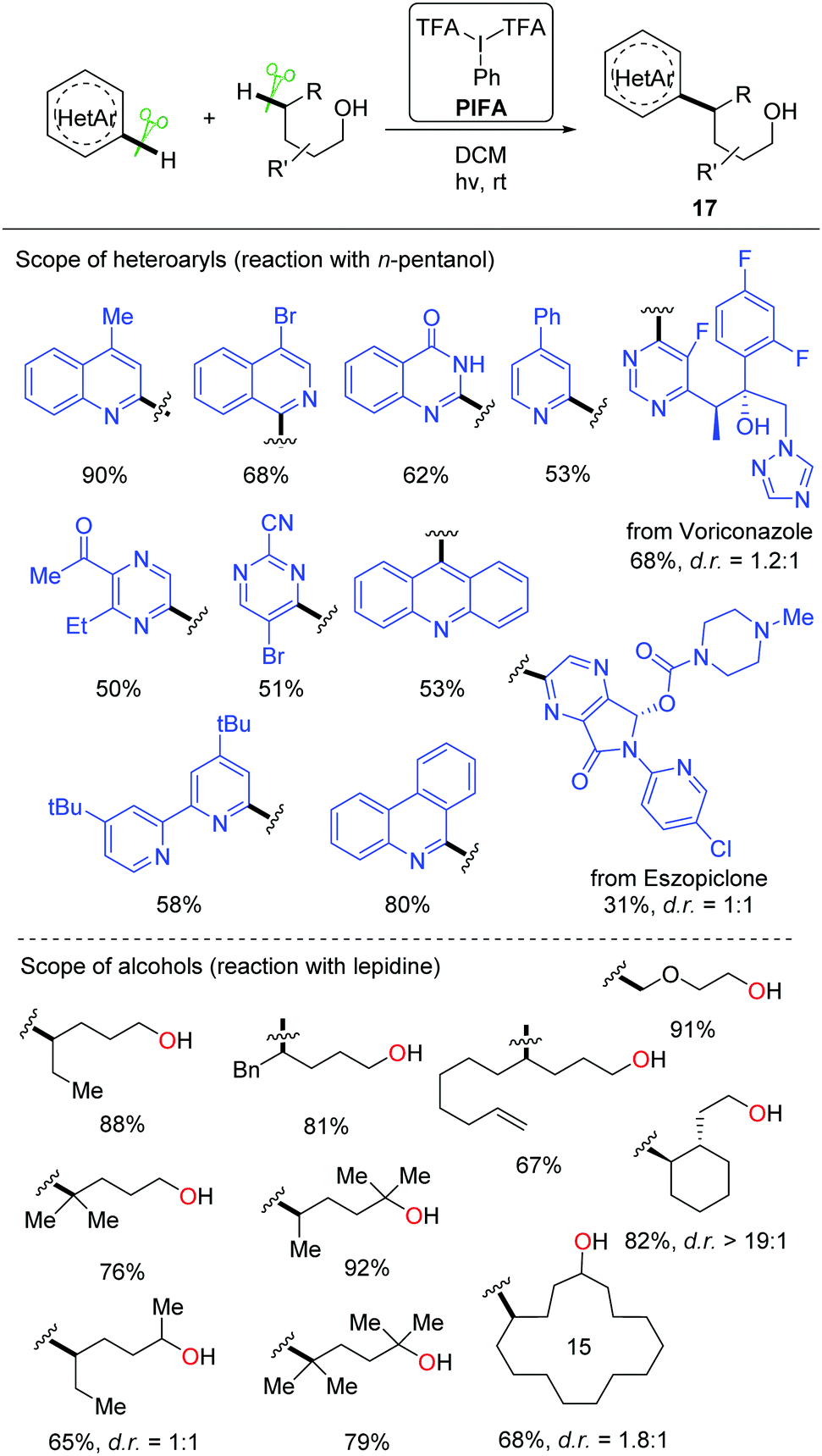 | ||
| Scheme 19 Free alcohol-directed intermolecular heteroarylation of C(sp3)–H bonds. | ||
A set of experiments were conducted to elucidate the mechanism. The NMR experiments verified that the interaction of PIFA and alcohol gave the dialkoxyiodo benzene intermediate a (Scheme 20). The radical-clock experiments supported the radical process. Homolysis of the intermediate a proceeds under visible-light irradiation, affording the alkoxy radical b that undergoes 1,5-HAT at the δ-position of aliphatic alcohol to give the alkyl radical intermediate c. The addition of alkyl radical c to the protonated N-heteroaryl d gives rise to the radical cation e, which is oxidized by PIFA or in situ formed iodanyl radical to provide the final product 17.
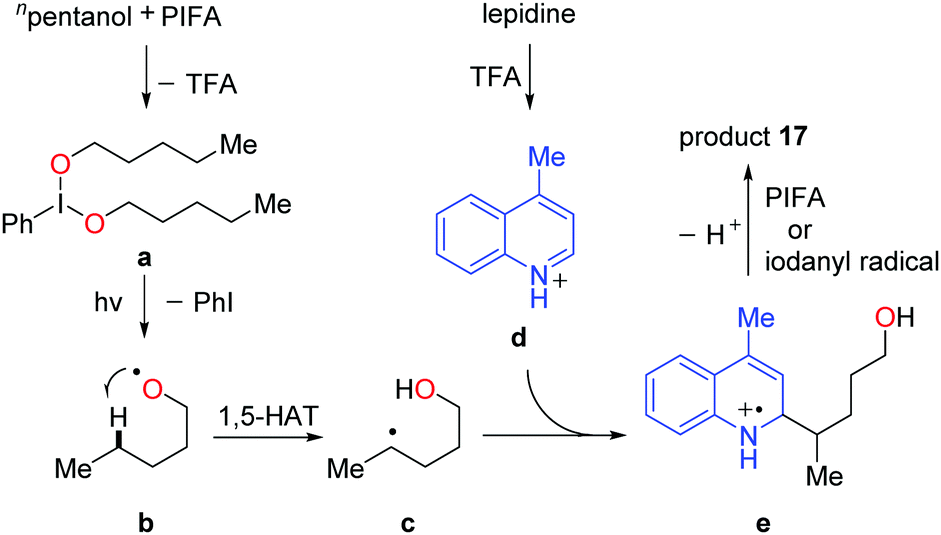 | ||
| Scheme 20 Plausible mechanism for the alcohol-directed intermolecular heteroarylation of C(sp3)–H bonds. | ||
Afterwards, Chen and He et al. reported a photoredox-catalyzed C(sp3)–H heteroarylation of primary and secondary alcohols. In the reaction, Ru(bpy)3Cl2 was employed as a photosensitizer and hypervalent iodine reagent PFBI-OH (perfluorinated hydroxybenziodoxole) was used as an oxidant.39
Zuo et al. reported a cerium salt-catalyzed δ-hydrazination of primary alcohols under visible-light irradiation. The alkoxy radicals were generated from alcohols via the ligand-to-metal charge transfer (LMCT) process.40 A variety of primary alcohols were efficiently functionalized with unique regioselectivity. Mechanistically, the coordination of Ce(IV) complex with alcohol excited by visible-light irradiation affords the alkoxy radical and Ce(III) complex through the homolysis of Ce(IV)-OR species. The alkyl radical generated from the alkoxy radical-mediated 1,5-HAT process is captured by DBAD to form a new C–N bond.
Jiao et al. disclosed a silver salt-catalyzed regioselective C(sp3)–H bond functionalization of primary and secondary alcohols.41 AgNO3 was used as a catalyst, and K2S2O8 was used as an oxidant. The well-designed oximonitrile-based sulfones functioned as the alkyl radical acceptors. A set of δ-oximonitrile substituted alcohols were afforded at 50 °C in moderate yields. In the reaction, an alkoxy radical was generated from the homolysis of nascent AgII–O species.
4. Conclusion and outlook
Recent advances in alkoxy radical-mediated C(sp3)–C(sp3) and C(sp3)–H bond functionalization are summarized. Two parts of content are discussed: (1) C(sp3)–C(sp3) bond cleavage via ring-opening functionalization of cycloalkanols, and (2) regioselective C(sp3)–H functionalization of free alcohols. The alkoxy radicals are generated direct from the free alcohols under mild conditions, demonstrating atom- and step-economy and high synthetic efficiency. These protocols have opened new vistas for the functionalization of cyclic C(sp3)–C(sp3) bonds and C(sp3)–H bonds.Despite the great achievements over the past few years, there are still many challenges in this area. For example, asymmetric construction of new chemical bonds via the alkoxy radical mediated β-C fragmentation or HAT process is urgently desired. In addition, the radical-mediated ring-opening transformation based on non-strained rings is underexplored. Non-trivial synthetic strategies are needed to deal with the inherent poor reactivity. Furthermore, the combination of HAT and FGM opens a novel avenue for remote C(sp3)–H functionalization, which will attract continuous interest from the synthetic community. Future efforts are still anticipated on these aspects.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. Z. is grateful for the financial support from the National Natural Science Foundation of China (Grant no. 21722205), the Project of Scientific and Technologic Infrastructure of Suzhou (SZS201708), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). X. W. acknowledges the China Postdoctoral Science Foundation (no. 2018M632359).Notes and references
- For selected reviews on C–C bond activation, see: (a) A. K. Sadan, R. K. Saini and W. E. Billups, Chem. Rev., 2003, 103, 1539 CrossRef PubMed; (b) T. Seiser and N. Cramer, Org. Biomol. Chem., 2009, 7, 2835 RSC; (c) T. Seiser, T. Saget, D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 7740 CrossRef CAS PubMed; (d) A. Flores-Gaspar and R. Martin, Synthesis, 2013, 563 CAS; (e) L. Souillart, E. Parker and N. Cramer, Top. Curr. Chem., 2014, 346, 163 CrossRef CAS; (f) T. Xu, A. Dermenci and G. B. Dong, Top. Curr. Chem., 2014, 346, 233 CrossRef CAS; (g) I. Marek, A. Masarwa, P. O. Delaye and M. Leibeling, Angew. Chem., Int. Ed., 2015, 54, 414 CrossRef CAS PubMed; (h) L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410 CrossRef CAS PubMed.
- For selected reviews on C–H bond activation: (a) X. Chen, K. M. Engle, D. H. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (c) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS PubMed; (d) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed.
- (a) A. W. Hofmann, Chem. Ber., 1883, 16, 558 CrossRef; (b) K. Löffler and C. Freytag, Chem. Ber., 1909, 42, 3427 CrossRef.
- (a) D. H. R. Barton, J. M. Beaton, L. E. Geller and M. M. Pechet, J. Am. Chem. Soc., 1960, 82, 2640 CrossRef CAS; (b) D. H. R. Barton and J. M. Beaton, J. Am. Chem. Soc., 1960, 82, 2641 CrossRef CAS; (c) D. H. R. Barton, J. M. Beaton, L. E. Geller and M. M. Pechet, J. Am. Chem. Soc., 1961, 83, 4076 CrossRef CAS.
- For selected reviews, see: (a) X. Xue, P. Ji, B. Zhou and J.-P. Cheng, Chem. Rev., 2017, 117, 8622 CrossRef CAS PubMed; (b) S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255 CrossRef CAS PubMed.
- (a) K. Heusler and J. Kalvoda, Angew. Chem., Int. Ed. Engl., 1964, 3, 525 CrossRef; (b) M. L. Mihailović and Ž. Čeković, Synthesis, 1970, 209 CrossRef; (c) J. Kalvoda and K. Heusler, Synthesis, 1971, 501 CrossRef CAS.
- (a) Ž. Čeković and M. M. Green, J. Am. Chem. Soc., 1974, 96, 3000 CrossRef; (b) Ž. Čeković, L. Dimitrijević, G. Djokić and T. Srnić, Tetrahedron, 1979, 35, 2021 CrossRef; (c) Ž. Čeković and M. Ĉvetković, Tetrahedron Lett., 1982, 23, 3791 CrossRef; (d) R. Kundu and Z. T. Ball, Org. Lett., 2010, 12, 2460 CrossRef CAS PubMed.
- (a) A. L. J. Beckwith, B. P. Hay and G. M. J. Williams, J. Chem. Soc., Chem. Commun., 1989, 1202 RSC; (b) Ž. Čeković and G. Petrović, Tetrahedron, 1999, 55, 1377 CrossRef; (c) G. Petrović, R. N. Saičić and Ž. Čeković, Synlett, 1999, 635 CrossRef.
- (a) C. Walling and A. Padwa, J. Am. Chem. Soc., 1961, 83, 2207 CrossRef CAS; (b) C. Walling and D. Bristol, J. Org. Chem., 1972, 37, 3514 CrossRef CAS; (c) C. R. Walling and T. Clark, J. Am. Chem. Soc., 1974, 96, 4530 CrossRef CAS; (d) A. Martín, J. A. Salazar and E. Suárez, J. Org. Chem., 1996, 61, 3999 CrossRef PubMed; (e) A. Martín, I. Pérez-Martín and E. Suárez, Org. Lett., 2005, 7, 2027 CrossRef PubMed.
- (a) A. L. J. Beckwith and B. P. Hay, J. Am. Chem. Soc., 1988, 110, 4415 CrossRef CAS; (b) J. Hartung and F. Gallou, J. Org. Chem., 1995, 60, 6706 CrossRef CAS.
- (a) S. Kim, T. A. Lee and Y. Song, Synlett, 1998, 471 CrossRef CAS; (b) H. Zhu, J. G. Wickenden, N. E. Campbell, J. C. T. Leung, K. M. Johnson and G. M. Sammis, Org. Lett., 2009, 11, 2019 CrossRef CAS PubMed; (c) M. Zlotorzynska and G. M. Sammis, Org. Lett., 2011, 13, 6264 CrossRef CAS PubMed.
- For reviews, see: (a) , Fluorine in the Life Sciences, ChemBioChem, 2004, 5, 557 CrossRef , (special issue); (b) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (d) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS; (e) M. Cametti, B. Crousse, P. Metrangolo, R. Milani and G. Resnati, Chem. Soc. Rev., 2012, 41, 31 RSC.
- H. Zhao, X. Fan, J. Yu and C. Zhu, J. Am. Chem. Soc., 2015, 137, 3490 CrossRef CAS PubMed.
- (a) F. Yin, Z. Wang, Z. Li and C. Li, J. Am. Chem. Soc., 2012, 134, 10401 CrossRef CAS PubMed; (b) Z. Li, Z. Wang, L. Zhu, X. Tan and C. Li, J. Am. Chem. Soc., 2014, 136, 16439 CrossRef CAS PubMed.
- (a) N. Ishida, S. Okumura, Y. Nakanishi and M. Murakami, Chem. Lett., 2015, 44, 821 CrossRef CAS; (b) S. Ren, C. Feng and T.-P. Loh, Org. Biomol. Chem., 2015, 13, 5105 RSC; (c) S. Bloom, D. Bume, C. R. Pitts and T. Lectka, Chem. – Eur. J., 2015, 21, 8060 CrossRef CAS PubMed.
- X. Fan, H. Zhao, J. Yu, X. Bao and C. Zhu, Org. Chem. Front., 2016, 3, 227 RSC.
- L. Huan and C. Zhu, Org. Chem. Front., 2016, 3, 1467 RSC.
- F. Q. Huang, J. Xie, J. G. Sun, Y. W. Wang, X. Dong, L. W. Qi and B. Zhang, Org. Lett., 2016, 18, 684 CrossRef CAS PubMed.
- H. G. Yayla, H. Wang, K. T. Tarantino, H. S. Orbe and R. R. Knowles, J. Am. Chem. Soc., 2016, 138, 10794 CrossRef CAS PubMed.
- D. Wang, J. Mao and C. Zhu, Chem. Sci., 2018, 9, 5805 RSC.
- J. Yu, H. Zhao, S. Liang, X. Bao and C. Zhu, Org. Biomol. Chem., 2015, 13, 7924 RSC.
- R. Ren, Z. Wu, Y. Xu and C. Zhu, Angew. Chem., Int. Ed., 2016, 55, 2866 CrossRef CAS.
- For recent reviews, see: (a) X. Wu, S. Wu and C. Zhu, Tetrahedron Lett., 2018, 59, 1328 CrossRef CAS; (b) X. Wu and C. Zhu, Chin. J. Chem., 2019, 37, 171 CAS . For selected examples from our group, see: ; (c) Z. Wu, R. Ren and C. Zhu, Angew. Chem., Int. Ed., 2016, 55, 10821 CrossRef CAS PubMed; (d) M. Ji, Z. Wu, J. Yu, X. Wan and C. Zhu, Adv. Synth. Catal., 2017, 359, 1959 CrossRef CAS; (e) R. Ren, Z. Wu, L. Huan and C. Zhu, Adv. Synth. Catal., 2017, 359, 3052 CrossRef CAS; (f) M. Ji, J. Yu and C. Zhu, Chem. Commun., 2018, 54, 6812 RSC; (g) Z. Wu, D. Wang, Y. Liu, L. Huan and C. Zhu, J. Am. Chem. Soc., 2017, 139, 1388 CrossRef CAS PubMed; (h) Y. Xu, Z. Wu, J. Jiang, Z. Ke and C. Zhu, Angew. Chem., Int. Ed., 2017, 56, 4545 CrossRef CAS PubMed; (i) D. Chen, M. Ji, Y. Yao and C. Zhu, Acta Chim. Sin., 2018, 76, 951 CrossRef; (j) S. Wu, X. Wu, D. Wang and C. Zhu, Angew. Chem., Int. Ed., 2019, 58, 1499 CrossRef CAS PubMed; (k) S. Yang, X. Wu, S. Wu and C. Zhu, Org. Lett., 2019, 21, 4837 CrossRef CAS PubMed.
- M. Ji, Z. Wu and C. Zhu, Chem. Commun., 2019, 55, 2368 RSC.
- K. Jia, F. Zhang, H. Huang and Y. Chen, J. Am. Chem. Soc., 2016, 138, 1514 CrossRef CAS PubMed.
- A. Hu, Y. Chen, J.-J. Guo, N. Yu, Q. An and Z. Zuo, J. Am. Chem. Soc., 2018, 140, 13580 CrossRef CAS PubMed.
- K. Zhao, K. Yamashita, J. E. Carpenter, T. C. Sherwood, W. R. Ewing, P. T. W. Cheng and R. R. Knowles, J. Am. Chem. Soc., 2019, 141, 8752 CrossRef CAS PubMed.
- R. Ren, H. Zhao, L. Huan and C. Zhu, Angew. Chem., Int. Ed., 2015, 54, 12692 CrossRef CAS PubMed.
- D. Wang, R. Ren and C. Zhu, J. Org. Chem., 2016, 81, 8043 CrossRef CAS PubMed.
- J.-J. Guo, A. Hu, Y. Chen, J. Sun, H. Tang and Z. Zuo, Angew. Chem., Int. Ed., 2016, 55, 15319 CrossRef CAS PubMed.
- R. Ren, Z. Wu and C. Zhu, Chem. Commun., 2016, 52, 8160 RSC.
- M. Wang, Z. Wu and C. Zhu, Org. Chem. Front., 2017, 4, 427 RSC.
- J. Zhang, Y. Li, F. Y. Zhang, C. C. Hu and Y. Chen, Angew. Chem., Int. Ed., 2016, 55, 1872 CrossRef CAS PubMed.
- C. Y. Wang, K. Harms and E. Meggers, Angew. Chem., Int. Ed., 2016, 55, 13495 CrossRef CAS PubMed.
- For recent reviews, see: (a) J.-J. Guo, A. Hu and Z. Zuo, Tetrahedron Lett., 2018, 59, 2103 CrossRef CAS; (b) K. Jia and Y. Chen, Chem. Commun., 2018, 54, 6105 RSC . For selected examples, see: ; (c) W. S. Trahanovsky, M. G. Young and P. M. Nave, Tetrahedron Lett., 1969, 2501 CrossRef CAS; (d) M. P. Doyle, L. J. Zuidema and T. R. Bade, J. Org. Chem., 1975, 40, 1454 CrossRef CAS; (e) H. Guan, S. Sun, Y. Mao, R. Lu and L. Liu, Angew. Chem., Int. Ed., 2018, 57, 11413 CrossRef CAS; (f) X. Bao, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2019, 58, 2139 CrossRef CAS; (g) I. Kim, B. Park, G. Kang, J. Kim, H. Jung, H. Lee, M.-H. Baik and S. Hong, Angew. Chem., Int. Ed., 2018, 57, 15517 CrossRef CAS PubMed.
- X. Wu, M. Wang, L. Huan, D. Wang, J. Wang and C. Zhu, Angew. Chem., Int. Ed., 2018, 57, 1640 CrossRef CAS.
- M. Wang, L. Huan and C. Zhu, Org. Lett., 2019, 21, 821 CrossRef CAS.
- X. Wu, H. Zhang, N. Tang, Z. Wu, D. Wang, M. Ji, Y. Xu, M. Wang and C. Zhu, Nat. Commun., 2018, 9, 3343 CrossRef.
- G.-X. Li, X. Hu, G. He and G. Chen, Chem. Sci., 2019, 10, 688 RSC.
- A. Hu, J.-J. Guo, H. Pan, H. Tang, Z. Gao and Z. Zuo, J. Am. Chem. Soc., 2018, 140, 1612 CrossRef CAS PubMed.
- Y. Zhu, K. Huang, J. Pan, X. Qiu, X. Luo, Q. Qin, J. Wei, X. Wen, L. Zhang and N. Jiao, Nat. Commun., 2018, 9, 2625 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2019 |