
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
(Photo)electrocatalysis of molecular oxygen reduction by S-doped graphene decorated with a star-shaped oligothiophene†
Anastasios
Stergiou
*,
Dimitris K.
Perivoliotis
 and
Nikos
Tagmatarchis
*
and
Nikos
Tagmatarchis
*
Theoretical and Physical Chemistry Institute, National Hellenic Research Foundation, 48 Vassileos Constantinou Avenue, 11635 Athens, Greece. E-mail: astergiou@eie.gr; tagmatar@eie.gr
First published on 19th March 2019
Abstract
Heteroatom-doped graphene-based materials attract great interest as non-metal electrocatalysts for the oxygen reduction reaction (ORR). In this work, a straightforward approach was described to prepare nanoensembles of star-shaped oligothiophene 1 supramolecularly immobilized on sulfur-doped graphene sheets (SG). The 1/SG ensemble was comprehensively characterized by Raman and IR spectroscopy and morphologically imaged by HR-TEM, while the loading of 1 onto SG was estimated by TGA under an inert atmosphere. Based on detailed electrochemical and electrocatalytic assays, 1/SG was proved to be a highly efficient and stable electrocatalyst toward the ORR. The high catalytic activity of 1/SG was attributed to the (a) presence of chemical defects, induced by the insertion of electron rich sulfur within the lattice of SG, (b) existence of structural defects, due to the generation of vacancies along the carbon lattice in SG, and (c) high and homogeneous coverage of the SG surface by the sulfur-rich star-shaped oligothiophene 1. In addition, the optical properties of 1/SG were screened by UV-Vis and steady-state and time-resolved PL and the development of strong photoinduced intra-ensemble electronic interactions within the ensemble was revealed. Exploiting the latter, by photoirradiating 1/SG, a significantly improved photoelectrocatalytic activity towards the ORR was observed.
Introduction
The “oxygen reduction reaction” (ORR) is a fundamental reaction in plant cells catalyzed by specialized metalloenzymes adsorbing the oxygen molecules and dissociating the O–O bond producing sustainable energy for the growth of plants. Modern technology exploits natural mechanisms1en route to meet the emerging demands on energy production and environmental protection by designing bioinspired materials based on the advances in materials science and engineering.2 Over the last decade, the rise of chemistry and engineering of carbon allotropes, especially graphene, has offered great potential to introduce non-metal nanostructured catalysts as alternative robust and highly reactive cathodes for ORR electrocatalysis.3It has already been established that graphene oxide (GO) based nanomaterials could challenge metal-based cathodes4 in terms of efficiency and durability and especially due to the significantly lower cost compared to metal-based electrodes.3a The generation of ORR active sites within the surface of graphene, mainly through elemental doping with sulfur and/or nitrogen atoms, is a critical aspect towards functional graphene-based cathodes.5 A great effort has already been made in this field unveiling that the enhanced ORR efficiency of doped graphene is related both to chemical defects, induced by the insertion of electron rich elements (i.e. sulfur), and the presence of structural defects (i.e. generation of vacancies along the carbon lattice).6 The insertion of heteroatoms across the graphitic lattice breaks the aromaticity of the extended conjugated network of graphene and provides unpaired electrons enhancing conductivity,7 and generates structural and chemical defects resulting in an enhancement of O2 physisorption.8 Focusing on S-doped graphene (SG), it is evident that the d-orbitals of the embedded sulfur atoms are soft nucleophiles and the local strain, due to the bigger size of S atoms compared to C atoms, favors the ORR reactivity around these sites. The latter is supported by the dramatic difference in ORR efficiency even in the case of nanocarbon-based materials with less than 1% w/w S-loading.9 In sharp contrast, undoped GO and exfoliated graphene, which also adsorb molecular oxygen onto their lattice, lack effective active sites for the electrocatalytic reaction. In addition, SG has an increased spin density around the doped regions, arising from the S–C bond polarization, due to the different electron densities of the S and C atoms, resulting in higher activity compared to intact graphene.8,10
Although the mechanisms of O2 adsorption and its electroreduction over S-functionalities are yet to be fully revealed, the C–S–C bonds, in the form of thiophene rings within the heteroatomic lattice of SG, are considered the most reactive sites for the ORR.11 Thiol groups12 and sulfur oxides11 at the edges of the doped graphitic network were also found to assist the overall ORR output.
Enriching graphene with embedded thiophene rings is an objective carried out in different ways by annealing GO in the presence of excess p-toluenesulfonic acid,13 Na2S,14 CS2,15 H2S,16 SO2,17 (NH4)2SO4,18 Lawesson's reagent19 and benzyl disulfide.20 Another strategy to access S-doped graphene is the pyrolysis of S-rich resins under an ambient atmosphere,21 or polymerization/S-doping of small molecules.22 Elevated temperature is necessary to force the substitution of oxygen atoms by sulfur and subsequently undergo oxidative cyclization reactions of sulfur with the neighboring carbon atoms. Other strategies to access S-enriched nanocarbons involve flame pyrolysis of thiophene23 or preparation of graphene-grafted conjugated microporous polymers based on thiophene functionalities.24 Beyond experimental observations, theoretical calculations additionally show that the incorporated S-atoms could be found either in the form of thiophene as a 5-membered ring at structurally defected sites or in the “graphitic” form as a 6-membered ring in the case of one atom substitution (i.e. C by S). Studies on SG lattice models have shown that the formation of the former is thermodynamically favorable in contrast to the latter. Furthermore, studies on the density of states revealed that in the “graphitic” thiopyran conformation the energy states at the Fermi level are occupied with electrons resulting in an excellent conductivity, while the “thiophene” conformation generates a bandgap.7 Apart from S-doping of GO, the generation of C–S–C active sites was accomplished by amorphous or microporous S-enriched carbons starting from sulfur or thiophene rich precursors.25
Focusing on the development of SG by reacting GO with Lawesson's reagent, the insertion of sulfur within the graphene lattice accompanied by thermally-induced healing of the disrupted electronic network takes place. In such a way, the presence of sulfur as an electroactive site is combined with the extended sp2 graphene domains capable of hosting π-conjugated molecules via numerous π–π interactions. In fact for the latter, theoretical first principles studies showed the self-assembly of sulfur-rich poly(3-hexyl-thiophene) (P3HT) onto a graphitic scaffold, favoring a face-on orientation of the sulfur atoms of P3HT with the lattice.26
Considering the aforementioned discussion, exploring the nature and the role of S-functionalities in the ORR efficiency is of great interest for developing metal-free carbon-based nanostructured cathodes useful for fuel cells and batteries. In this work, a 2D-hybrid material based on a well-defined star-shaped conjugated oligothiophene immobilized onto S-doped graphene was prepared and the impact of the oligothiophene/graphene interactions towards (photo)electrocatalysis for the ORR was studied. The non-covalent decoration of SG by the synthesized oligothiophene 1 was found to be beneficial for lowering the onset potential for the ORR compared to pristine SG, while in the presence of light a further improvement was witnessed by 60% increment of the cathodic current and the significant 34 mV drift of the onset potential to more positive values.
Results and discussion
The presence of three hexyl-chains in a terthiophene-functionalized planar trithienobenzene (TTB) material 1 benefits enhanced solubility in common organic solvents upon interaction with SG, which otherwise forms unstable dispersions. In addition, these solubilizing chains are beneficial for allowing smoother processing of the 1/SG ensemble. Furthermore, recently we showed that linear oligothiophenes, possessing three or nine conjugated rings carrying similar hexyl chains, can be efficiently assembled onto exfoliated graphene via π–π stacking interactions contributing to the development of strong intrahybrid electronic communication upon photoillumination.27 On top, the star-shape structure of 1 is essential for increasing the surface contact with the graphene network due to the presence of the planar TTB core. This particular branched structure possesses a larger surface than a linear conjugated thiophene chain and consequently an increased contact area with the SG sheets, a beneficial property for the development of more efficient intra-ensemble van der Waals interactions. As a result, oligothiophene 1 molecules were effectively physisorbed around the electrocatalytic active regions of SG, where chemical and structural defects, due to S-atoms and healing of the distorted graphene lattice, respectively, coexist. Furthermore, the visible light harvesting ability of oligothiophene 1 allowed performing ORR photoelectrocatalytic studies.Star-shaped oligothiophene 1, consisting of a terthiophene-functionalized planar trithienobenzene core, was obtained by a five-step chemical synthesis route as presented in Scheme 1. The synthetic process initiated from a microwave-assisted Stille coupling reaction between 2,5-dibromo-3-hexylthiophene and 2-tributyl-stannylthiophene afforded terthiophene 2. Markedly, microwave irradiation aided the acceleration of the otherwise slow coupling reaction, while simultaneously impeding competing side reactions. Then, 2 was treated with N-bromosuccinimide (NBS) in the dark to yield mono-brominated terthiophene 3, which is actually a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of terthiophenes carrying the hexyl-chain either at C-3 or C-4 of the central thiophene core. The presence of structural isomers in 3 neither interferes with the electronic and structural properties of the compound nor affects those of the target material 1, since the position of the alkyl chains at C-3 or C-4 do not disturb the effective conjugation length. Next, two sequential Sonogashira coupling reactions were performed to obtain the alkyne-terminated terthiophene 4, which was then attached to 1,3,5-triiodo-2,4,6-trichlorobenzene28 affording the star-shaped template 5, the precursor of the target star-shaped conjugated oligothiophene 1. Finally, nucleophilic attack of sulfur ions from sodium sulfide to the carbon–carbon triple-bond of 5, followed by hetero-cyclization in a two-step-one-pot reaction29 yielded the fused heterocyclic TTB core in material 1.
:1 mixture of terthiophenes carrying the hexyl-chain either at C-3 or C-4 of the central thiophene core. The presence of structural isomers in 3 neither interferes with the electronic and structural properties of the compound nor affects those of the target material 1, since the position of the alkyl chains at C-3 or C-4 do not disturb the effective conjugation length. Next, two sequential Sonogashira coupling reactions were performed to obtain the alkyne-terminated terthiophene 4, which was then attached to 1,3,5-triiodo-2,4,6-trichlorobenzene28 affording the star-shaped template 5, the precursor of the target star-shaped conjugated oligothiophene 1. Finally, nucleophilic attack of sulfur ions from sodium sulfide to the carbon–carbon triple-bond of 5, followed by hetero-cyclization in a two-step-one-pot reaction29 yielded the fused heterocyclic TTB core in material 1.
 | ||
| Scheme 1 Synthesis of star-shaped oligothiophene 1. Reagents and conditions: (i) 2-Tributyl-stannylthiophene, CuI, Pd(PPh3)4, 120 W, 120 °C, dry DMF, O2 free. (ii) N-Bromosuccinimide, dry DCM, in the dark, 0 °C to r.t. (iii) Trimethyl-ethynylsilane, CuI, Pd(PPh3)4, dry Et3N, dry THF, under N2, 100 °C. (iv) Excess KF, THF/MeOH, r.t. (v) 1,3,5-Tribromo-2,4,6-trichlorobenzene, CuI, Pd(PPh3)4, dry Et3N, dry DCM. (vi) Excess Na2S, NMP, 180 °C. Inset: UV-Vis spectrum of 1 recorded in THF. | ||
The structures of 1 and of all thiophene-based precursors 2–5 were verified by 1H and 13C NMR spectroscopy (ESI, Fig. S1–S6†). Briefly, in the 1H NMR spectrum of 1 (ESI, Fig. S6†) well resolved doublets, due to the outer thiophene ring at 7.22, 7.16 and 7.11 ppm with a 1:1:1 ratio, were identified, while also the proton doublets of the three thiophene rings fused to the TTB core at 7.07 ppm and the characteristic signal of the first methylene group of the alkyl chain attached to the thiophene at 2.70 ppm were evident. As far as the 13C NMR spectrum is concerned, two major regions of aliphatic and conjugated carbons are present at 1–32 ppm (6 major equivalent signals) and 123–137 ppm (16 equivalent carbon signals), respectively (ESI, Fig. S6†). Additional verification for the structure of 1 arose from MALDI-TOF-MS, showing the presence of the molecular ion at 1237 amu (ESI, Fig. S7†). The electronic absorption spectrum of 1 has a broad intense absorption in the visible region centered at 440 nm (inset of Scheme 1). The latter arises from the unique star-shaped structure, since analogous alkyl-substituted terthiophenes are poor absorbers above 350 nm.28
In parallel, thermal treatment of commercially available GO in diglyme with Lawesson's reagent, as both the sulfur source and reducing agent,30 afforded SG. Markedly, SG is a promising substrate electrocatalyst for the ORR, attributed to both the incorporation of sulfur-active sites and the thermal self-healing of the graphitic lattice occurred during the heating period of the thionation reaction.31 During the process, partial recovery of the sp2 framework takes place, resulting in a conductive network doped with embedded sulfur atoms. Next, the as-prepared SG was added to a THF solution of 1 and the mixture was briefly sonicated and then stirred for 18 hours. After that period, it was centrifuged, the supernatant was decanted and the precipitate was isolated and washed with dichloromethane to remove completely any unbound 1. Eventually, this process gave rise to the isolation of 1/SG upon the assembly of oligothiophene 1 over the surface of SG nanosheets (Fig. 1a).
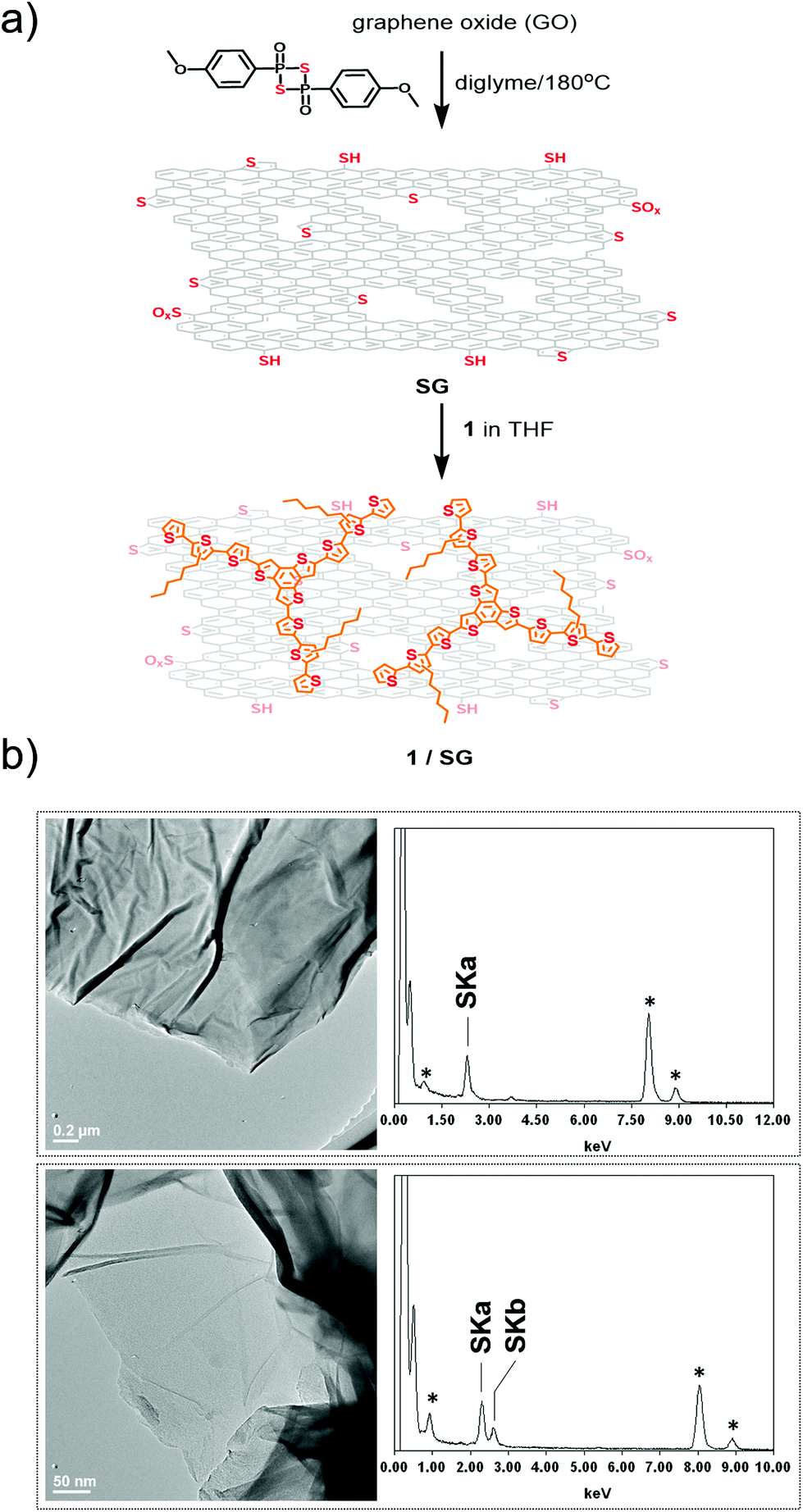 | ||
| Fig. 1 (a) Preparation of the 1/SG ensemble featuring oligothiophene 1 immobilized onto S-doped graphene via π–π interactions. (b) Representative HR-TEM images and EDX analysis of SG (upper panel) and 1/SG (lower panel). With asterisks are marked EDX signals from the copper mesh grid. | ||
High resolution transmission electron microscopy (HR-TEM) imaging of SG revealed the presence of extended graphene sheets with lateral size in the micrometer order, while energy-dispersive X-ray spectroscopy (EDX) revealed the presence of sulfur in the lattice of SG, validating further the successful doping with sulfur atoms (Fig. 1b). HR-TEM and EDX analysis of 1/SG nanoensembles revealed the preservation of the morphology of SG and the existence of sulfur, respectively (Fig. 1b).
Vibrational spectroscopy has shed light on the structural characteristics of SG and 1/SG. Concerning SG nanosheets, Raman spectroscopy revealed the D-band at around 1350 cm−1 related to structural defects in sp3-hybridized carbon and the G-band at around 1600 cm−1 attributed to the in-plane vibration of the resonant sp2 carbon–carbon lattice (Fig. 2a). In general, the latter mode is sensitive to chemical doping, and therefore, has been commonly employed to detect the type of doping in nanocarbon materials and graphene in particular. The incorporation of S-atoms within the graphitic sp2 network induces n-doping to SG,32 as indicated by the down-shift of the G-band by 20 cm−1versus the value registered for GO (Fig. 2b). Moreover, the relative intensity of the D/G ratio was employed as a means to evaluate the disorder degree. Evidently, the D/G ratio for SG was increased as compared to the one registered for GO (2.1 vs. 1.53, respectively), an observation directly related to the self-healing/reduction of graphene during the thionation reaction.13 The latter observation further points to an increased defect density in graphene sheets due to S-doping.33 The supramolecular interactions of 1 with SG were similarly expected to further shift the G-band to lower frequencies due to charge transfer phenomena from the thiophene-rich 1 to the graphene lattice. Indeed, an additional 3 cm−1 shift for 1/SG, compared to SG was observed. In the reference material 1/GO, in which 1 interacts with GO, a down-shift of 18 cm−1versus the value registered for GO was identified, certifying the occurrence of charge transfer processes from the fused oligothiophene species to graphene in GO and SG. Interestingly the shape of the 2D band in SG and 1/SG was found to be sharper and more intense due to the higher crystallinity of SG as compared to the distorted GO nanosheets.34
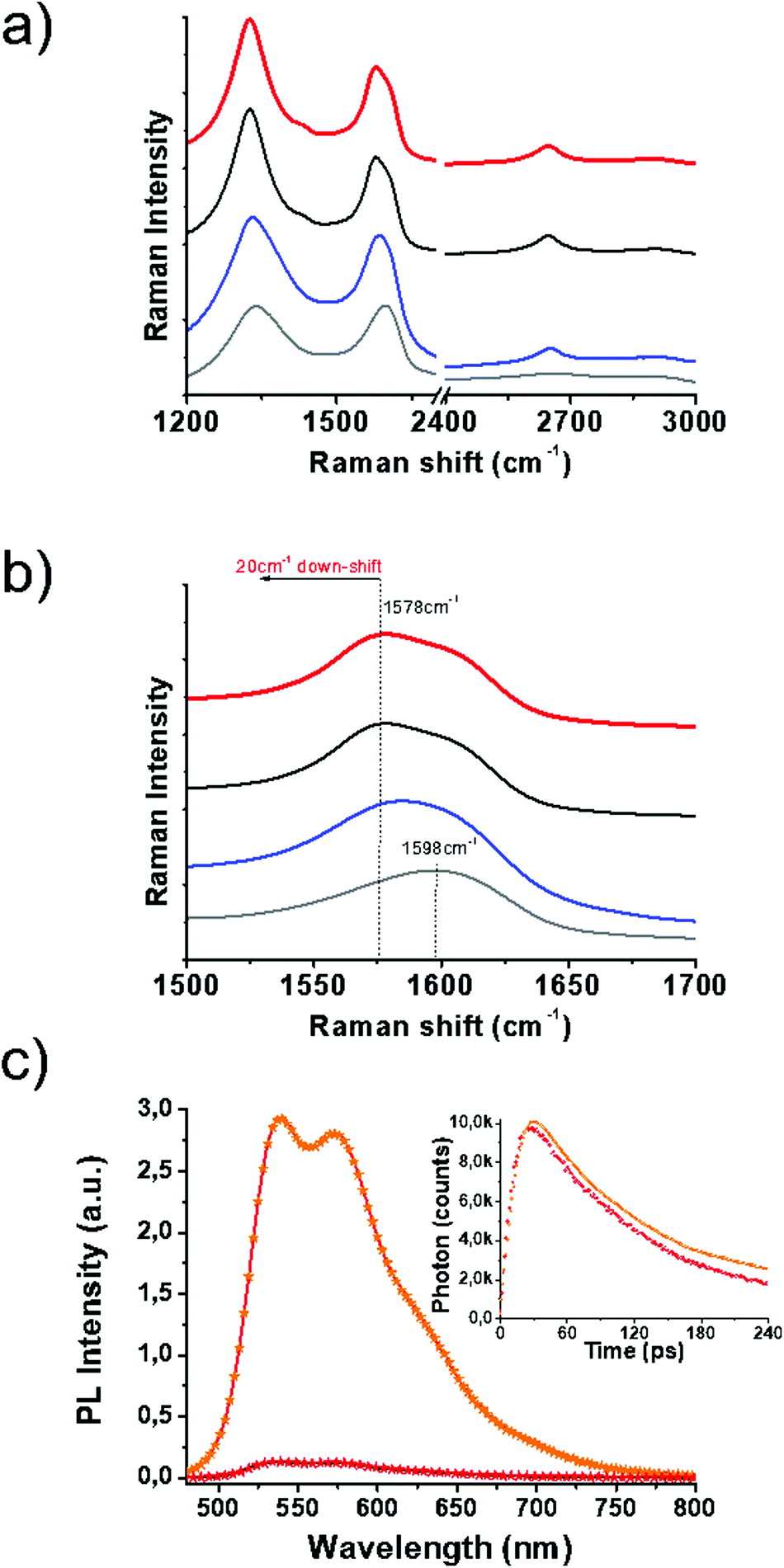 | ||
| Fig. 2 (a) Full Raman spectra (λexc 633 nm) and (b) expansion of the G-band of 1/SG (red), GO (grey), SG (black) and 1/GO (blue). (c) Photoluminescence spectra (λexc 441 nm) of 1 (orange) and 1/SG (red) obtained in benzonitrile. Inset: Photoluminescence lifetime decay graphs. | ||
The ATR-IR spectrum of SG in comparison to that of GO possesses a more intense band associated with the stretching vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds at 1655 cm−1 as a result of the partial restoration of the sp2 conjugated carbon network. Furthermore, indicative of the reduction of oxygen functionalities in SG were the diminutive intensities at 1040 cm−1 (C–O) and 1720 cm−1 (carbonyl CO). Additional distinct modes owing to C–S (624 cm−1), C–S–C (1080 cm−1) and –SH (2662 cm−1) functionalities in the IR spectrum of SG were also evident (ESI, Fig. S8a†). IR spectroscopy was also employed to identify the presence of the star-shaped oligothiophene 1 within the 1/SG nanoensemble. In this context, the characteristic vibrations of 1 at 2921, 1461, 788 and 683 cm−1 were also present in 1/SG (ESI, Fig. S8b†), indicating the successful formation of the ensemble.
C bonds at 1655 cm−1 as a result of the partial restoration of the sp2 conjugated carbon network. Furthermore, indicative of the reduction of oxygen functionalities in SG were the diminutive intensities at 1040 cm−1 (C–O) and 1720 cm−1 (carbonyl CO). Additional distinct modes owing to C–S (624 cm−1), C–S–C (1080 cm−1) and –SH (2662 cm−1) functionalities in the IR spectrum of SG were also evident (ESI, Fig. S8a†). IR spectroscopy was also employed to identify the presence of the star-shaped oligothiophene 1 within the 1/SG nanoensemble. In this context, the characteristic vibrations of 1 at 2921, 1461, 788 and 683 cm−1 were also present in 1/SG (ESI, Fig. S8b†), indicating the successful formation of the ensemble.
Thermogravimetric analysis (TGA) assays performed under an inert atmosphere are in line with the IR observations. For GO, a mass loss of 50% was observed in the temperature range 180–600 °C, related to the decomposition of the oxygen functionalities, while the mass loss occurred at higher temperatures above 600 °C is related to the structural deformation of the graphene sheet (ESI, Fig. S9a†). In contrast, SG showed higher thermal stability, losing 3% and 20% of mass at 296 °C and 806 °C (ESI, Fig. S9b†), respectively, due to the less content of oxygen functionalities as a result of the thermally-induced thiolation reaction. Furthermore, we calculated a 20% mass loading of the physisorbed oligothiophene 1 on SG from the corresponding TGA curve of 1/SG, where two major mass loss steps were evident at 336 °C and 579 °C (ESI, Fig. S9c†) attributed to the thermal decomposition of the immobilized organic molecules of 1 onto the SG nanosheets.
The immobilization of 1 over SG was further evidenced by UV-Vis absorption and photoluminescence spectroscopy assays performed in benzonitrile. The absorption maximum of oligothiophene 1 within 1/SG was found at 438 nm blue-shifted by 8 cm−1 compared to the value registered at 446 nm for the free molecules of 1 in solution (ESI, Fig. S10a†). Furthermore, the fluorescence spectrum of physisorbed 1 was found to be quantitatively quenched by SG within the 1/SG ensemble (Fig. 2c and ESI, Fig. S10b†), for samples exhibiting equal absorption at the excitation wavelength (441 nm). Analogous strong interactions at the excited state were revealed for the 1/GO reference material (ESI, Fig. S10d†), although ground state interactions were diminutive (ESI, Fig. S10c†). Further insight on the electronic interplay between the two species within the 1/SG ensemble was delivered by time-resolved fluorescence spectroscopy. In this frame, based on time correlated single photon counting spectroscopy, the fluorescence emission decay of the immobilized oligothiophene component within 1/SG decayed 20 ps faster (t1/SG = 65 ps) compared to the corresponding free molecules of 1 (t1 = 85 ps) (Fig. 2c, inset). In conjunction with the fluorescence emission quenching observed in the steady-state assays, these results are supportive of electron and/or energy transfer as the decay mechanism of the formation of the singlet excited state 11*.
Prior to conducting the electrochemical studies concerning the electrocatalytic ORR activity of 1/SG, cyclic voltammetry (CV) assays, in nitrogen-saturated benzonitrile with TBAPF6 as the electrolyte, to evaluate the redox properties of 1/SG in comparison with those of the individual components 1 and SG (ESI, Fig. S11†) were performed. In more detail, 1 revealed two reversible one-electron oxidation processes at +0.64 and +0.97 V vs. Hg/HgO, while within 1/SG the oxidation owing to the oligothiophene species was cathodically shifted by 10 mV and registered at +0.63 V, indicating easier oxidation due to intra-ensemble interactions with SG. In addition, a broad reduction located at −0.69 V was attributed to SG. The latter value was 6 mV more positive and lower in intensity as compared to the reduction process registered for GO in the corresponding reference material 1/GO. From these fundamental redox data, it is clear that the electrochemical window for the ORR (i.e. +50 to −400 mV vs. Hg/HgO) is not affected by the intra-ensemble redox processes.
Initially SG, oligothiophene 1 and the 1/SG ensemble were subjected to CV runs in an oxygen-saturated environment with an aqueous 0.1 M KOH electrolyte. As presented in Fig. 3, all materials showed the characteristic cathodic current related to the oxygen reduction and the same stands for the corresponding LSV studies (ESI, Fig. S12 and S13†). The electroreduction of dissolved oxygen, with a reduction potential peak close to GO performance at −390 mV and a slightly more negative value by ∼7 mV onset potential, was catalyzed by oligothiophene 1 (ESI, Table S1†). The Ep of 1/SG appeared at −320 mV, being 57 mV positively shifted compared to pristine SG. Taken as a reference, the Ep for the non-covalently interacting 1 with GO was registered at −345 mV, being 45 mV positively shifted compared to pristine GO. The Ep of 1/SG was very close to the value registered for the commercial 5% Pt/C at around −315 mV. The electrochemical data of 1/SG, compared with those owing to 1, GO, 1/GO and 5% Pt/C materials, are collectively presented in ESI, Table S1.† Based on these results, it is clearly demonstrated that the electrocatalytic output for the ORR efficiency of 1/SG was not an overlay of the two individual components, 1 and SG, but rather attributed to a synergistic effect in which the enhanced overall performance is due to the presence of the conjugated thiophene rings in close proximity to the S-doped graphene.
 | ||
| Fig. 3 Cyclic voltammographs of (a) SG, (b) 1 and (c) 1/SG in nitrogen (dotted) and oxygen (solid) saturated aqueous 0.1 M KOH electrolytes. | ||
The electrocatalytic properties of 1/SG toward the ORR were further screened by linear sweep voltammetry (LSV) acquired by the rotating disk electrode (RDE) at different rotation rates 400–3600 rpm (ESI, Fig. S14†) and chronoamperometry (CA) assays in an oxygen-saturated aqueous 0.1 M KOH electrolyte. Fig. 4a shows ORR polarization curves for 1/SG and SG obtained at a rotation rate of 1600 rpm, where, in both cases, the characteristic plateau of the 4-electron mechanism is absent, indicating a major 2-electron reduction pathway and minor 4-electron reduction, as previously shown.31 Although the oxygen electroreduction abides by the indirect 2-electron mechanism in both 1/SG and SG, the immobilization of 1 onto SG induces significant improvement in the ORR performance. Indeed, the ORR Eonset for 1/SG was found at −210 mV, which is 40 mV lower compared to the Eonset for SG (i.e. −250 mV), while the half-wave potential for 1/SG was −330 mV, which is 50 mV lower compared to the corresponding value registered for SG (−380 mV) with a slight increment in the diffusion-limiting current density (jd) by 10%. Next, the kinetic current density (jk) for 1/SG was determined at −270 mV vs. Hg/HgO by using the Koutecky–Levich (K–L) equation and it was found to be 0.33 mA cm−2 (Fig. 4b), being 3.3 times greater than that of SG. Further information about the ORR kinetics can be extracted by constructing the mass transfer corrected Tafel plots (Fig. 4c). The Tafel slope for 1/SG was determined to be −45 mV dec−1 in the low current density region (region I) and −155 mV dec−1 at high current densities (region II), being clearly lower than those determined for SG (−70 and −166 mV dec−1, in regions I and II, respectively). The latter result implies higher catalytic activity towards the ORR for 1/SG as the overpotential increases faster with the current density.35 Overall, these findings, summarized in Table 1, highlight the critical role of thiophene rings in 1 for the electrocatalytic performance of the 1/SG ensemble.
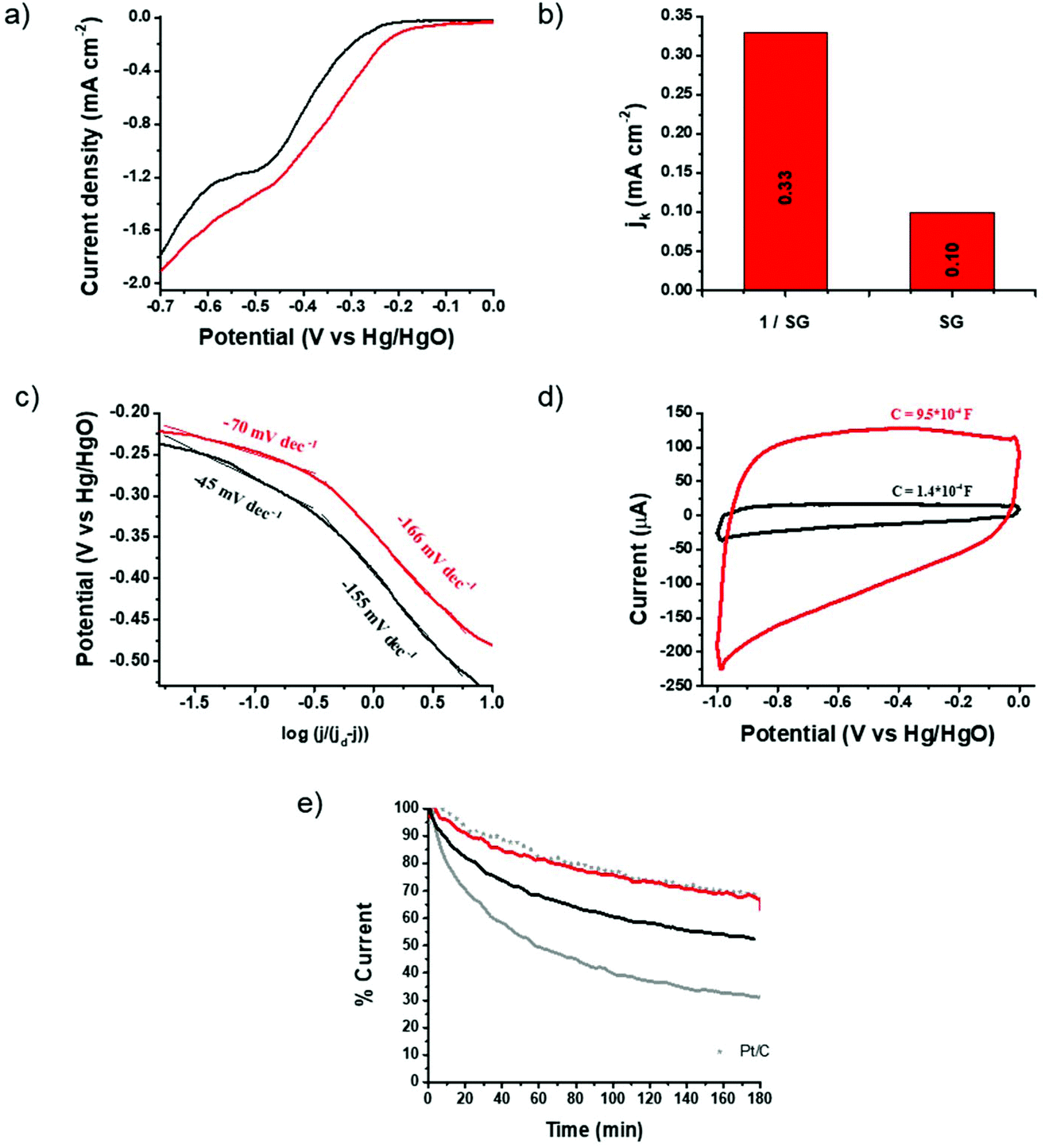 | ||
| Fig. 4 (a) ORR polarization curves at 1600 rpm for SG (black) and 1/SG (red) recorded in the O2 saturated aqueous 0.1 M KOH electrolyte. (b) The corresponding kinetic current density values obtained at −270 mV vs. Hg/HgO via the K–L equation. (c) Tafel plots for SG (black) and 1/SG (red). Data derived from Fig. 3a and b. (d) Capacitance curves for SG (black) and 1/SG (red) recorded in the N2 saturated aqueous 0.1 M KOH electrolyte at a scan rate of 0.2 V s−1. (e) Chronoamperometry graphs of 1/SG (red), SG (black), 1/GO (grey) and commercial Pt/C (grey scatter) recorded in the O2 saturated aqueous 0.1 M KOH electrolyte at −0.3 V vs. Hg/HgO. | ||
| Electrochemical parameters | Material | |
|---|---|---|
| SG | 1 /SG | |
| Onset potential (mV vs. Hg/HgO) | −250 | −210 |
| Half-wave potential (mV vs. Hg/HgO) | −380 | −330 |
| Diffusion-limited current density (mA cm−2) | 1.15 | 1.27 |
| Kinetic current density (mA cm−2) | −0.10 | −0.33 |
| Tafel slopes (mV dec−1) | −70/−166 | −45/−155 |
| Capacitance (F) | 1.4 × 10−4 | 9.5 × 10−4 |
| Activity loss (within 3 hours) | >50% | 23% |
The strong intra-ensemble electronic interactions witnessed by UV-Vis, Raman and photoluminescence spectroscopy may further polarize the C–S bonds of SG furnishing more efficient active sites for the ORR.8,10 Considering that the active site density is closely related to the capacitance,36 a possible explanation of the enhancement mechanism is given below. It is known that the insertion of sulfur functionalities across the graphene lattice results in enhanced capacitance, possibly, arising from narrow micropores within the lattice created during the doping/healing process.37 Performing CV runs for the pristine SG nanosheets and the 1/SG ensemble, a 7-fold increment in the 1/SG capacitance, being 9.5 × 10−4 F, over that of SG, being 1.4 × 10−4 F, by integrating the average graph-area derived by voltammographs at different scan rates, was observed (ESI, Fig. S15a and b†). Both values are higher than the capacitance of the starting GO, being 0.8 × 10−4 F (ESI, Fig. S15c†). The insertion of sulfur functionalities and healing of the lattice are responsible for the increased capacitance of SG, while the 7-fold increment to 1/SG is attributed to the presence of the adsorbed star-shaped oligothiophene 1 onto S-doped graphene (Fig. 4a and ESI, Fig. S14†). As a result, the conjugated thiophenes were beneficial for the electrosorption of the dissolved oxygen and electrolyte ions, consequently enhancing the ORR output as previously described. Notably, the immobilization of 1 onto SG does not generate new micropores, however, we hypothesize that the electronic interactions between the thiophene rings and the conducting S-doped graphene are responsible for the enhanced activity.
The durability of 1/SG was evaluated versus continuous current flow in an oxygen-saturated aqueous 0.1 M KOH electrolyte at −0.3 V at 1600 rpm. Chronoamperometry assays for 1/SG revealed a current loss of 23% after 3 hours, a value identical to the corresponding one due to the commercial 5% w/w Pt/C (Fig. 4e). Markedly, in reference 1/GO the current loss exceeded 70% and probably this is owing to the electroreduction of oxygen species on the nanocarbon's surface.
A synopsis of the data acquired for the recently developed S-enriched nanocarbon ORR electrocatalysts is provided in Table 2. Generally, the kinetic current density value (jk), determined in the kinetic region of the LSV curve (i.e. close to the onset potential), is among the most important parameters considered for the ORR activity assessment of different electrocatalysts. In this context, S-doped graphene produced by thermal treatment of GO/porous silica sheets with H2S16b exhibited a jk value of 0.15 mA cm−2 while S-doped graphene synthesized by thiophene pyrolysis24 exhibited a jk value of 0.40 mA cm−2. Furthermore, S-enriched conjugated polymer nanosheets unveiled high jk values ranging between 0.21 and 0.60 mA cm−2.25,26 Based on these data, the currently prepared and examined 1/SG ensemble can be classified among the top-rated S-enriched nanocarbon catalysts for oxygen electroreduction.
| Sulfur (% w/w) | Kinetic current (mA cm−2) 1600 rpm | Tafel slopes (mV dec−1) | E onset (V) | n | Ref. | |
|---|---|---|---|---|---|---|
| S-Doped graphene oxide-porous silica sheets | 1.2–1.7 | 0.15 (at −0.28 V vs. Ag/AgCl, KCl 3 M) | n.a. | n.a. | 3.5 to 3.2 (−0.3 to −0.8 V vs. Ag/AgCl, KCl 3 M) | 16b |
| Sulfur-enriched conjugated polymer nanosheet | 2.2–8.65 | 0.60 (at +0.6 V vs. RHE) | n.a | n.a. | 2.8 (+0.4 V vs. RHE) | 23 |
| S-Doped carbon by thiophene pyrolysis | 3.4 | 0.40 (at −0.28 V vs. Ag/AgCl, sat. KCl) | n.a. | −0.16 (Ag/AgCl, sat. KCl) | 2.2 (−0.4 to −0.9 V Ag/AgCl, sat. KCl) | 24 |
| Graphene-based conjugated microporous polymers | 7.7 | 0.21 (at −0.22 V vs. Ag/AgCl, KCl 3 M) | n.a. | −0.15 (Ag/AgCl, KCl 3 M) | 4 (−0.45 to −0.9 V vs. Ag/AgCl, KCl 3 M) | 25 |
| SG | 0.30 | 0.10 (at −0.27 V vs. Hg/HgO) | −70, −166 | −0.25 vs. Hg/HgO | 2 | This work |
| 1 /SG | 0.35 | 0.33 (at −0.27 V vs. Hg/HgO) | −45, −155 | −0.21 vs. Hg/HgO | 2 | This work |
Finally, taking into account the light harvesting properties of oligothiophene 1 in the visible region and the evidenced photoinduced intra-ensemble electronic interactions within 1/SG, we investigated the impact of light irradiation on ORR electrocatalysis. The 1/SG ensemble was deposited on transparent fluorine-doped tin oxide glass substrates, immersed in the O2 saturated aqueous 0.1 M KOH electrolyte and illuminated with a conventional 500 W halogen lamp (the band-gap of 1/SG was calculated to be 2.37 eV by absorption spectroscopy, see ESI, Fig. S16†). Performing LSV runs, the generated photocurrent at −0.7 V for 1/SG, being −3.10 mA, was found to be increased by 60% compared to the current recorded under dark conditions being −1.87 mA (Fig. 5). Furthermore, the Eonset of the photoexcited ensembles, registered at −213 mV, was found to be 34 mV more positive compared to the value obtained under dark conditions, suggesting easier reduction of the dissolved oxygen molecules in the presence of light. It is evident that analogous photoresponse was absent in the case of pristine oligothiophene 1 and SG employed as reference materials in the study (Table 3).
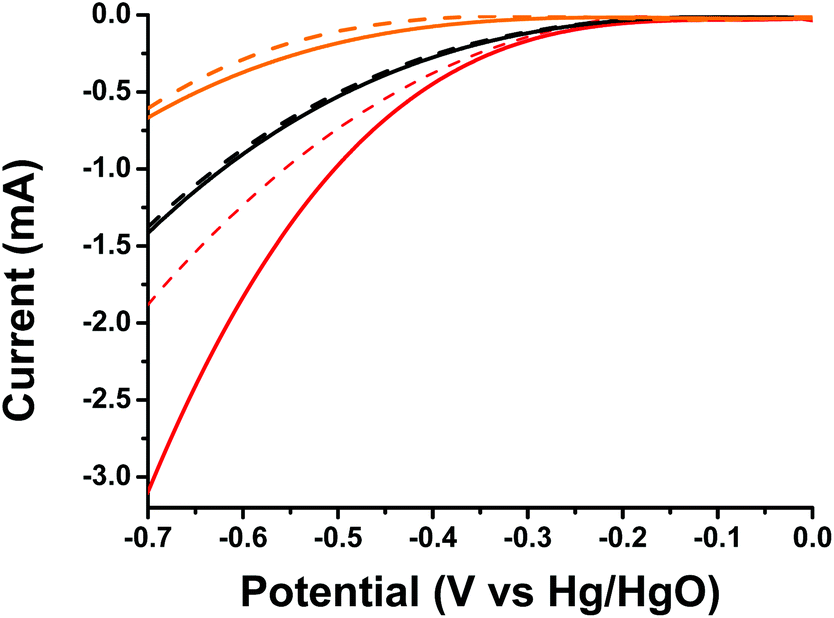 | ||
| Fig. 5 LSV curves of oligothiophene 1 (orange), SG (black) and 1/SG (red), recorded under light illumination (solid) and dark (dashed) conditions, in the O2 saturated aqueous 0.1 M KOH electrolyte. | ||
| Material | E onset (mV)/I (mA) at −0.7 V | |
|---|---|---|
| Dark | Light irradiation | |
| 1 | −411/−0.6 | −380/−0.65 |
| SG | −265/−1.36 | −263/−1.39 |
| 1 /SG | −247/−1.87 | −213/−3.10 |
As manifested by Raman spectroscopy as well as steady state and time resolved photoluminescence spectroscopy, under visible light illumination strong interactions take place between the physisorbed photoexcited oligothiophene molecules and S-doped graphene within 1/SG. This is to say that the singlet excited state 11* is formed and deactivated via charge/energy-transfer leading to charge-separation, similar to recent reports on fullerene-based materials.38,39 Then, in the presence of dissolved oxygen molecules, the intra-ensemble charge separated state decays via transfer of electrons to the electrocatalytic cycle of oxygen reduction. The latter mechanism is in line with the evidenced amplified current for 1/SG under irradiation conditions, since it contributes to higher electron flow, from the photoexcited oligothiophene 1 to oxygen viaSG. Considering the absence of an analogous observation for pristine oligothiophene 1, it is highlighted that the presence of SG nanosheets is critical for the stabilization of the photoinduced charge separation within 1/SG and the electron transfer to oxygen, thus improving the overall ORR photoelectrocatalysis.
Experimental
General
All chemicals were commercially available and used without further purification unless otherwise stated. Steady state UV-Vis electronic absorption spectra were recorded on a PerkinElmer (Lambda 19) UV-Vis-NIR spectrophotometer. Steady-state emission spectra were recorded on a Fluorolog-3 Jobin Yvon-Spex spectrofluorometer (model GL3-21). Picosecond time-resolved fluorescence spectra were measured by the time-correlated single photon counting (TCSPC) method on a Nano-Log spectrofluorometer (Horiba JobinYvon), by using a laser diode as an excitation source (NanoLED, 375 nm) and a UV-vis detector TBX-PMT series (250–850 nm) by Horiba JobinYvon. Lifetimes were evaluated with the DAS6 Fluorescence-Decay Analysis Software. Micro-Raman scattering measurements were performed at room temperature in backscattering geometry using a RENISHAW inVia Raman microscope equipped with a CCD camera and a Leica microscope. A 2400 lines per mm grating was used for all measurements, providing a spectral resolution of ±1 cm−1. As an excitation source the He/Ne laser (633 nm) was used. Measurements were carried out with 60 seconds of exposure time at varying numbers of accumulations. The laser spot was focused on the sample surface using a long working distance 50× objective. Raman spectra were collected on numerous spots on the sample and recorded with a Peltier cooled CCD camera. The data were collected and analyzed with Renishaw Wire and Origin software. Electrochemical measurements were carried out in a standard three-compartment electrochemical cell using a rotating disk electrode (RDE) setup from Metrohm Autolab connected to an EG&G Princeton Applied Research potentiostat/galvanostat (Model PARSTAT 2273A) connected to a personal computer running PowerSuite software. As a counter electrode, a platinum wire was used, and as a reference a Hg/HgO (aqueous 0.1 M KOH electrolyte) electrode was placed into a Luggin capillary. The working electrode was a RDE with a glassy carbon (GC) disk (geometric surface area, 0.071 cm2) or a static GC electrode. The working electrode was cleaned before each experiment through polishing with a cloth and 6, 3 and 1 mm diamond paste. The ORR measurements were realized at room temperature in oxygen-saturated aqueous 0.1 M KOH. Linear sweep voltammetry (LSV) measurements on the RDE of different materials were conducted at different rotation rates recorded with a scan rate of 5 mV s−1. The kinetic current densities (jk) were calculated using the Koutecky–Levich (K–L) equation:| 1/j = 1/jd + 1/jk | (1) |
| jk = j/(jd − j) | (2) |
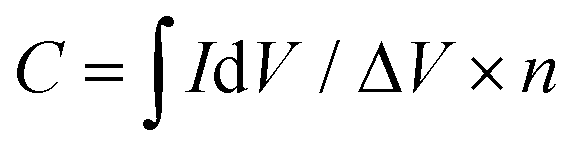 | (3) |
 (C) is the integrated area of the CV curve; ΔV (V) is the potential window and v is the scan rate (V s−1).
(C) is the integrated area of the CV curve; ΔV (V) is the potential window and v is the scan rate (V s−1).
The voltammographs shown in Fig. S10† were recorded using a GC working electrode and platinum wires were used as counter and pseudo-reference electrodes (Fc/Fc+ as an internal reference) and 0.1 M TBAPF6 in acetonitrile as the electrolyte. TBAPF6 (98%) was recrystallized three times from acetone and dried in a vacuum at 100 °C before being used as the electrolyte. Before each experiment, the cell was purged with Ar for 30 seconds. For the photoelectrocatalytic studies all materials were deposited on FTO-coated glass substrates (surface resistivity ∼7 Ω Sq−1) from benzonitrile solutions and dried under vacuum. As a light source a conventional linear (118 mm) 500 W halogen lamp was used. Mid-IR spectra in the region of 550–4000 cm−1 were obtained on an FTIR spectrometer (Equinox 55 from Bruker Optics) equipped with a single reflection diamond ATR accessory (Dura-Samp1IR II by SensIR Technologies). 1H and 13C NMR spectra were acquired with a Varian 300 MHz spectrometer. HR-TEM measurements were carried out using a JEM-2100F (JEOL) high-resolution field-emission gun TEM operated at 80 keV at room temperature and under a pressure of 10−6 Pa. HR-TEM images were recorded with a charge-coupled device with an exposure time of typically 1 second.
:20 mass ratio were mixed in diglyme (50 mL) and heated under nitrogen for 3 hours at 200 °C. Then the reaction mixture was allowed to reach r.t. and filtered through a PTFE membrane under vacuum. The solid residue was washed thoroughly with toluene, methanol and dichloromethane. The as-derived filter cake was dispersed in methanol with the aid of a sonication bath and centrifuged affording SG as a black fine powder.
Conclusions
Summarizing, the synthesis and immobilization of star-shaped oligothiophene 1 onto S-doped graphene sheets, yielding 1/SG ensembles, as efficient electrocatalysts for the ORR, was accomplished. Employing complementary spectroscopic, thermal and microscopy imaging techniques the success of preparation was proved. In addition, detailed electrochemical and electrocatalytic assays revealed improved activity and stability towards the ORR in alkaline media for 1/SG, outperforming not only the individual components of the ensemble 1 and SG but also GO and 1/GO tested as references. The high catalytic activity of 1/SG, evidenced by the increased kinetic current density and low Tafel slope values, was attributed to the (a) presence of chemical defects, induced by the insertion of the electron rich sulfur within the lattice of SG, (b) existence of structural defects, due to the generation of vacancies along the carbon lattice in SG, and (c) high and homogeneous coverage of the SG surface by the sulfur-rich star-shaped oligothiophene 1. Further analysis on the ORR kinetics showed a major 2-electron mechanism for 1/SG.Strong photoinduced intra-ensemble electronic interactions were also witnessed in 1/SG. Hence, the light harvesting properties of 1 were exploited towards ORR photoelectrocatalysis, where a better performance for 1/SG in terms of lower Eonset and amplified current generated was observed under light irradiation. In contrast, analogous studies on oligothiophene 1 and pristine SG did not show further catalytic improvement. Collectively, the critical role of SG nanosheets in the stabilization of the light-induced charge separation within 1/SG and the subsequent electron transfer to oxygen is highlighted. Overall, the current findings can serve as significant milestones for the future design of high-performance ORR non-metal doped-graphene-based electrocatalysts.40 This in turn will lead to the development of alternative non-precious cathode electrocatalysts to replace platinum in energy-related applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support of this work by the project “Advanced Materials and Devices” (MIS 5002409) which is implemented under the “Action for the Strategic Development on the Research and Technological Sector”, funded by the Operational Programme “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014–2020) and co-financed by Greece and the European Union (European Regional Development Fund). Financial support through a Ph.D. scholarship by the General Secretariat for Research and Technology (GSRT) – Hellenic Foundation for Research and Innovation (HFRI) to D. K. P. (Grant 95) is also acknowledged. We are indebted to Prof. M. Prodromidis, at the Department of Chemistry, University of Ioannina, Greece, for kindly providing the RDE system, to Prof. A. G. Coutsolelos and Dr G. Charalambidis at the Department of Chemistry, University of Crete, Greece, for kind assistance with the MALDI-TOF-MS acquisition, and to Prof. H. Shinohara and his group at the Department of Chemistry, Nagoya University, Japan, for kindly providing access to perform the HR-TEM and EDX measurements.Notes and references
- F. Möller, S. Piontek, R. G. Miller and U.-P. Apfel, Chem. – Eur. J., 2018, 24, 1471 CrossRef PubMed.
- D. Grumelli, B. Wurster, S. Stepanow and K. Kern, Nat. Nanotechnol., 2013, 4, 2904 Search PubMed.
- (a) X. Liu and L. Dai, Nat. Rev. Mater., 2016, 1, 16064 CrossRef CAS; (b) D. He, H. Tang, Z. Kou, M. Pan, X. Sun, J. Zhang and S. Mu, Adv. Mater., 2017, 29, 1601741 CrossRef.
- (a) M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594 CrossRef CAS; (b) A. A. Gewirth, J. A. Varnell and A. M. DiAscro, Chem. Rev., 2018, 118, 2313 CrossRef CAS.
- (a) K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760 CrossRef CAS; (b) X. Wang, G. Sun, P. Routh, D.-H. Kim, W. Huang and P. Chen, Chem. Soc. Rev., 2014, 43, 7067 RSC.
- (a) C. Wang, L. Ma, L. Liao, S. Bai, R. Long, M. Zuo and Y. Xiong, Sci. Rep., 2013, 3, 2580 CrossRef; (b) D. Higgins, M. A. Hoque, M. H. Seo, R. Wang, F. Hassan, J.-Y. Choi, M. Pritzker, A. Yu, J. Zhang and Z. Chen, Adv. Funct. Mater., 2014, 24, 4325 CrossRef CAS.
- A. G. Garcia, S. E. Baltazar, A. H. Romero Castro, J. F. Perez Robles and A. Rubio, J. Comput. Theor. Nanosci., 2008, 5, 2221 CrossRef CAS.
- L. Zhang, J. Niu, M. Li and Z. Xia, J. Phys. Chem. C, 2014, 118, 3545 CrossRef CAS.
- S.-A. Wohlemuth, R. J. White, M.-G. Willinger, M.-M. Titirici and M. Antonietti, Green Chem., 2012, 14, 1514 Search PubMed.
- I.-Y. Jeon, S. Zhang, L. Zhang, H.-J. Choi, J.-M. Seo, Z. Xia, L. Dai and J.-B. Baek, Adv. Mater., 2013, 25, 6138 CrossRef CAS.
- (a) W. Kiciński, M. Szala and M. Bystrzejewski, Carbon, 2014, 68, 1 CrossRef; (b) H. Shen, E. Gracia-Espino, J. Ma, K. Zang, J. Luo, L. Wang, S. Gao, X. Mamat, G. Hu, T. Wagberg and S. Guo, Angew. Chem., Int. Ed., 2017, 56, 13800 CrossRef CAS.
- C. K. Chua and M. Pumera, ACS Nano, 2015, 9, 4193 CrossRef CAS.
- Y. Li, J. Wang, X. Li, D. Geng, M. N. Banis, Y. Tang, D. Wang, R. Li, T.-K. Sham and X. Sun, J. Mater. Chem., 2012, 22, 20170 RSC.
- (a) Y. Chen, J. Li, T. Mei, X. Hu, D. Liu, J. Wang, M. Hao, J. Li, J. Wang and X. Wang, J. Mater. Chem. A, 2014, 2, 20174 Search PubMed; (b) L. Chen, X. Cui, Y. Wang, M. Wang, R. Qiu, Z. Shu, L. Zhang, Z. Hua, F. Cui, C. Wei and J. Shi, Dalton Trans., 2014, 43, 3420 RSC.
- J. Park, Y. J. Jang, Y. J. Kim, M. Song, S. Yoon, D. H. Kim and S.-J. Kim, Phys. Chem. Chem. Phys., 2014, 16, 103 RSC.
- (a) C. Liang, Y. Wang and T. Li, Carbon, 2015, 82, 506 CrossRef CAS; (b) S. Yang, L. Zhi, K. Tang, X. Feng, J. Maier and K. Müllen, Adv. Funct. Mater., 2012, 22, 3634 CrossRef CAS.
- H. L. Poh, P. Šimek, Z. Sofer and M. Pumera, ACS Nano, 2013, 7, 5262 CrossRef CAS.
- D. He, Z. Kou, Y. Xiong, K. Cheng, X. Chen, M. Pan and S. Mu, Carbon, 2014, 66, 312 CrossRef CAS.
- H. Liu, L. Zhang, Y. Guo, C. Cheng, L. Yang, L. Jiang, G. Yu, W. He, Y. Liu and D. Zhu, J. Mater. Chem. C, 2013, 1, 3104 RSC.
- Z. Yang, Z. Yao, G. Li, G. Fang, H. Nie, Z. Liu, X. Zhou, X. Chen and S. Huang, ACS Nano, 2012, 6, 205 CrossRef CAS PubMed.
- (a) Y. Zhang, M. Chu, W. Deng, Y. Tang, M. Ma and Q. Xie, Chem. Commun., 2014, 50, 6382 RSC; (b) X. Liu and M. Antonietti, Adv. Mater., 2013, 25, 6284 CrossRef CAS.
- Y. Su, Z. Yao, F. Zhang, H. Wang, Z. Mics, E. Cánovas, M. Bonn, X. Zhuang and X. Feng, Adv. Funct. Mater., 2016, 26, 5893 CrossRef CAS.
- S. Inamdar, H.-S. Choi, P. Wang, M. Y. Song and J.-S. Yu, Electrochem. Commun., 2013, 30, 9 CrossRef CAS.
- Z. Zhuang, F. Zhang, D. Wu, N. Forler, H. Liang, M. Wagner, D. Gehrig, M. R. Hansen, F. Laquai and Z. Feng, Angew. Chem., Int. Ed., 2013, 52, 9668 CrossRef.
- D. Liu, L. Dai, X. Lin, J.-F. Chen, J. Zhang, X. Feng, K. Müllen, X. Zhu and S. Dai, Adv. Mater., 2019, 31, 1804863 CrossRef.
- D. H. Kim, H. S. Lee, H.-J. Shin, Y.-S. Bae, K.-H. Lee, S.-W. Kim, D. Choi and J.-Y. Choi, Soft Mater., 2013, 9, 5355 RSC.
- A. Stergiou, H. B. Gobeze, I. D. Petsalakis, S. Zhao, H. Shinohara, F. D'Souza and N. Tagmatarchis, Nanoscale, 2015, 7, 15840 RSC.
- M. Sonoda, A. Inaba, K. Itahashi and Y. Tobe, Org. Lett., 2001, 3, 2419 CrossRef CAS.
- T. Kashiki, S. Shinamura, M. Kohara, E. Miyazaki, K. Takimiya, M. Ikeda and H. Kuwabara, Org. Lett., 2009, 11, 2473 CrossRef CAS.
- D. K. Perivoliotis, Y. Sato, K. Suenaga and N. Tagmatarchis, ACS Appl. Energy Mater., 2018, 1, 3869 CrossRef CAS.
- A. Ambosi, C. K. Chua, A. Bonanni and M. Pumera, Chem. Rev., 2014, 114, 7150 CrossRef.
- (a) Q. Su, S. Pang, V. Alijani, C. Li, X. Feng and K. Müllen, Adv. Mater., 2009, 21, 3191 CrossRef CAS; (b) A. Stergiou and N. Tagmatarchis, ACS Appl. Mater. Interfaces, 2016, 8, 21576 CrossRef CAS; (c) H. Liu, Y. Liu and D. Zhu, J. Mater. Chem., 2011, 21, 3335 RSC.
- X. Zhang, J. Zhu, C. S. Tiwary, Z. Ma, H. Huang, J. Zhang, Z. Lu, W. Huang and Y. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 10858 CrossRef CAS.
- S. Abdolhosseinzadeh, H. Asgharzadeh and H. S. Kim, Sci. Rep., 2015, 5, 10160 CrossRef CAS.
- Y. Zheng, S. Zhao, S. Liu, H. Yin, Y.-Y. Chen, J. Bao, M. Han and Z. Dai, ACS Appl. Mater. Interfaces, 2015, 7, 5347 CrossRef CAS.
- J. Benson, Q. Xu, P. Wang, Y. Shen, L. Sun, T. Wang, M. Li and P. Papakonstantinou, ACS Appl. Mater. Interfaces, 2014, 6, 19726 CrossRef CAS.
- M. Seredych and T. J. Bandosz, J. Mater. Chem. A, 2013, 1, 11717 RSC.
- S. Bellani, A. Ghadirzadeh, L. Meda, A. Savoini, A. Tacca, G. Marra, R. Meira, J. Morgado, F. Di Fonzo and M. R. Antognazza, Adv. Funct. Mater., 2015, 25, 4531 CrossRef CAS.
- R. M. Girón, J. Marco-Martínez, S. Bellani, A. Insuasty, H. C. Rojas, G. Tullii, m. r. Antognazza, S. Filipone and N. Martin, J. Mater. Chem., 2016, 4, 14284 RSC.
- I. S. Amiinu, J. Zhang, Z. Kou, X. Liu, O. K. Asare, H. Zhou, K. Cheng, H. Zhang, L. Mai, M. Pan and S. Mu, ACS Appl. Mater. Interfaces, 2016, 8, 29408 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic and electrochemical data. See DOI: 10.1039/c9nr01620a |
| This journal is © The Royal Society of Chemistry 2019 |