
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atomic resolution map of the soluble amyloid beta assembly toxic surfaces†
Rashik
Ahmed
 a,
Michael
Akcan
a,
Adree
Khondker
b,
Maikel C.
Rheinstädter
b,
José C.
Bozelli
Jr
a,
Richard M.
Epand
a,
Vincent
Huynh
c,
Ryan G.
Wylie
c,
Stephen
Boulton
a,
Jinfeng
Huang
c,
Chris P.
Verschoor
d and
Giuseppe
Melacini
*ac
a,
Michael
Akcan
a,
Adree
Khondker
b,
Maikel C.
Rheinstädter
b,
José C.
Bozelli
Jr
a,
Richard M.
Epand
a,
Vincent
Huynh
c,
Ryan G.
Wylie
c,
Stephen
Boulton
a,
Jinfeng
Huang
c,
Chris P.
Verschoor
d and
Giuseppe
Melacini
*ac
aDepartment of Biochemistry and Biomedical Sciences, McMaster University, Hamilton, ON L8S 4M1, Canada. E-mail: melacin@mcmaster.ca
bDepartment of Physics and Astronomy, McMaster University, Hamilton, ON L8S 4M1, Canada
cDepartment of Chemistry and Chemical Biology, McMaster University, Hamilton, ON L8S 4M1, Canada
dDepartment of Health Research Methods, Evidence, and Impact (HEI), McMaster University, Hamilton, ON L8S 4M1, Canada
First published on 21st May 2019
Abstract
Soluble amyloid beta assemblies (Aβn) are neurotoxic and play a central role in the early phases of the pathogenesis cascade leading to Alzheimer's disease. However, the current knowledge about the molecular determinants of Aβn toxicity is at best scant. Here, we comparatively analyze Aβn prepared in the absence or presence of a catechin library that modulates cellular toxicity. By combining solution NMR with dynamic light scattering, fluorescence spectroscopy, electron microscopy, wide-angle X-ray diffraction and cell viability assays, we identify a cluster of unique molecular signatures that distinguish toxic vs. nontoxic Aβ assemblies. These include the exposure of a hydrophobic surface spanning residues 17–28 and the concurrent shielding of the highly charged N-terminus. We show that the combination of these two dichotomous structural transitions promotes the colocalization and insertion of β-sheet rich Aβn into the membrane, compromising membrane integrity. These previously elusive toxic surfaces mapped here provide an unprecedented foundation to establish structure-toxicity relationships of Aβ assemblies.
Introduction
While the etiology of Alzheimer's disease (AD) is complex and not fully understood, genetic mutations provide compelling evidence that the amyloid beta (Aβ) peptide plays a critical role in AD pathogenesis.1,2 Indeed, mutations in the genes encoding either the Aβ progenitor (i.e. the amyloid precursor protein or APP) or the APP processing enzyme (presenilin 1 and 2 genes) are sufficient to cause AD.1 Moreover, none of the familial AD mutations involve genes encoding for the tau protein. Instead, tau mutations enhance the deposition of neurofibrillary tangles i.e. the other neuropathological hallmark of AD, but not amyloid plaques, and lead to different neuropathological disorders.3 These genetic signatures coupled with the observation that Aβ deposition precedes other biochemical and histopathological changes, including neurofibrillary tangle formation,4 provide evidence that tau aggregation occurs downstream to Aβ aggregation. In addition, Aβ clearance is controlled by one of the most significant risk factors for late onset AD, i.e. APOE4.5 Given the genetic link between Aβ and AD, one of the main hypotheses proposed to explain AD pathogenesis is the amyloid cascade. The amyloid hypothesis posits that neuronal death in AD patients is associated with the increased production, self-association and accumulation of Aβ in the brain.2Since it was originally postulated, the generality of the amyloid cascade hypothesis has been challenged because Aβ plaque burden correlates poorly with cognitive dysfunction.6 However, this inconsistency has been reconciled by considering that soluble oligomers and protofibrils formed during the self-association cascade towards mature fibrils are neurotoxic7 and better correlate with cognitive impairment in the early stages of AD.8 Moreover, the neurotoxicity of Aβ oligomers has been linked to tau hyperphosphorylation,9 providing further evidence in support of the upstream role of soluble Aβ assemblies in the AD pathogenesis cascade.1
The central role of soluble Aβ oligomers and protofibrils in AD has prompted substantial efforts to identify the molecular determinants of neurotoxicity in soluble Aβ assemblies (Aβn, where n represents the number of Aβ molecules comprising the assembly).10–22 Unfortunately, given the transient and heterogeneous nature of Aβ intermediates, characterization of their structure and properties has been challenging. Despite these hurdles, it has been possible to delay the growth of aggregation intermediates to an extent sufficient to enable structural elucidation. For example, Ahmed et al. have shown that toxic Aβ42 oligomers stabilized through low temperature and salt conditions are largely disordered, but exhibit a turn conformation reminiscent of protofibrils and fibrils.20 In contrast, for the other major isoform of Aβ, i.e. Aβ40, toxic oligomers adopt parallel, in-register β-sheets.21 While these studies have provided an initial framework to define structural features of toxic Aβn, the location of the “toxic Aβn surfaces” remains unclear. Mapping such surface sites is critical as the exposure of toxic surfaces shared by multiple soluble Aβn species has been hypothesized to be one of the main causes of Aβn toxicity.1,23
Exposure of these toxic surfaces is thought to facilitate interactions with multiple cellular components, including membranes, which underlie key pathogenic steps in the progression of AD.1,22,24–26 In fact, extracellular Aβ oligomers are known to perturb biological and biomimetic membranes at multiple levels. The oligomers can (i) bind to membranes causing local perturbations,19,27 (ii) form annular structures that insert into the membrane and affect ion homeostasis16,18,19 and (iii) bind to membrane receptors altering signal transduction pathways.28 Similar hypotheses have been proposed to explain the neurotoxicity of Aβ protofibrils,17 although the latter have been shown to act also through detergent-like permeabilization and eventual fragmentation of the membrane.19 While these results highlight critical aspects of Aβ–membrane interactions, the “toxic surfaces” that enable key interactions with the membrane, as well as the underlying mechanism, remain elusive.
As a further step towards dissecting the molecular determinants of soluble Aβn toxicity and mapping the toxic Aβn surfaces, here we systematically investigate a library of Aβ40 assemblies sampling different degrees of cellular toxicity. To this end, we first stabilized canonical, toxic Aβ40 assemblies through desalting and low temperature29 and then treated them with a diverse set of catechins, ranging from (−)-epigallocatechin-3-gallate (EGCG), which remodels Aβ into non-toxic structures,30 to (−)-epicatechin (EC), which is expected to detoxify Aβ only partially. We then profiled our soluble Aβ library through multiple complementary techniques with different degrees of spatial resolution, including extrinsic fluorescence, electron microscopy, dynamic light scattering, wide-angle X-ray diffraction and NMR spectroscopy. Unlike previous attempts to dissect the toxicity determinants of Aβ assemblies,20,21 here we characterize representative soluble Aβ assemblies from our library both in the absence and presence of model membranes.
The comparative analysis of our soluble Aβ40 library reveals a cluster of key toxicity determinants and the associated mechanism of action. We discovered that toxicity scales proportionally to the enhanced hydrophobic exposure of Aβ40 assemblies and their ability to interact with Aβ monomers and cell membranes. The hydrophobic region spanning residues 17–28 is more accessible to monomer recognition in toxic Aβn relative to Aβn with reduced cellular toxicity. Moreover, whereas increased exposure of hydrophobic residues is required for toxicity, we find that shielding of the highly charged N-terminus, i.e. residues < 12, from Aβ monomer recognition enhances the toxicity of Aβn. These toxic Aβn surfaces are critical for the binding of Aβn to lipid membranes and for forming membrane-embedded β-sheet structures, which compromise the integrity of the cell membrane. The resulting model provides a foundation to start defining structure-toxicity relationships of Aβ assemblies.
Results and discussion
An Aβ40 assembly library that samples a cytotoxicity gradient
As a first step towards dissecting the determinants of Aβ40 toxicity, we prepared a library of soluble Aβn spanning a cytotoxicity gradient. For this purpose, we incubated canonical (non-treated) Aβn with a collection of seven distinct catechins expected to remodel to varying extents the pre-existing soluble toxic Aβn into less toxic species30–32 (ESI Fig. S1,† Methods). Out of this Aβn library, we selected a sub-set of representative Aβ assemblies (i.e. those formed in the presence of the EC, (−)-epigallocatechin (EGC) and EGCG catechins) for toxicity profiling in a human retinal pigment epithelial (RPE1) cell line. The state of the RPE1 cells was first monitored by performing PrestoBlue assays, which rely on the reductive potential of the cell as a proxy of cellular viability.33 Relative to mock (i.e. PBS delivery vehicle), canonical Aβn significantly decrease cellular viability (Fig. 1a, black vs. grey). In contrast, Aβn formed in the presence of catechins are less effective in reducing cellular viability, in the order EC (Fig. 1a, green), EGC (Fig. 1a, yellow) and EGCG (Fig. 1a, maroon), for which no significant difference is detected compared to mock (Table S1†). Only negligible changes in cellular viability were observed for cells treated with catechins alone (Fig. 1a, dark green, orange and brown).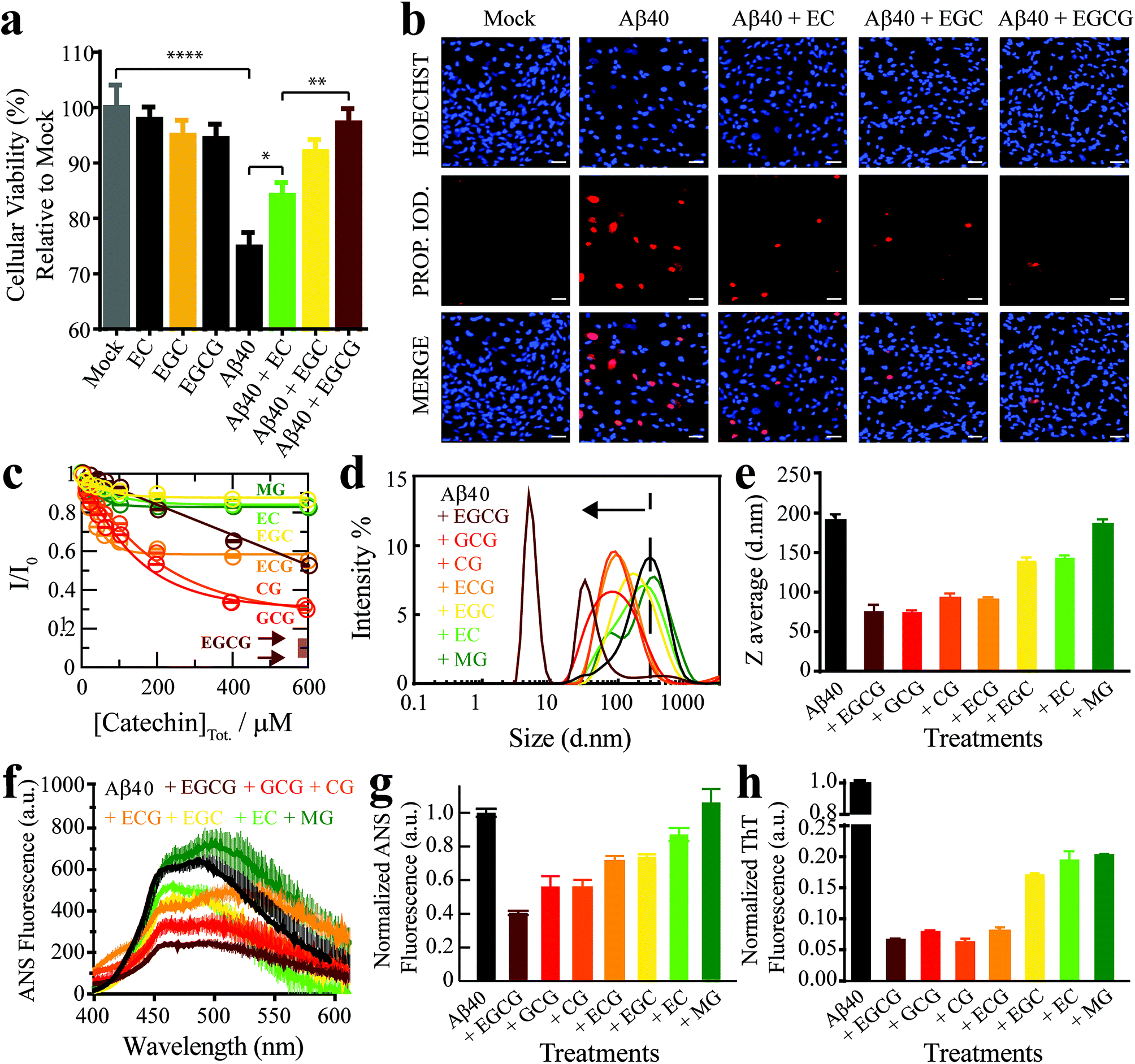 | ||
| Fig. 1 The Aβn library samples a wide-distribution of toxicity, hydrophobic exposure, cross β-sheet content and sizes. (a) Mitochondrial activity of retinal pigment epithelial (RPE1) cells after treatment with representative Aβ40 assemblies and associated controls, as monitored by the reduction of resazurin using the PrestoBlue assay.33 The data reported show the mean and standard deviation of technical replicates. One-way ANOVA and subsequent Tukey's post-hoc test was used to determine statistical significance between treatments and mock (1X PBS delivery solution), with *, ** and **** representing p-values of 0.05, 0.01 and <0.0001, respectively. (b) Representative fluorescence microscopy images of RPE1 cells (scale bar, 50 μm), showing intracellular Hoechst 33342 and propidium iodide fluorescence after incubation with selected Aβ40 assemblies. (c) Normalized Aβ40 methyl intensity losses upon catechin addition relative to the state in the absence of catechins. (d) Size distribution of Aβ40 assemblies in the absence (black) and presence of catechins (coloured as per legend) as determined by intensity measurements in dynamic light scattering experiments. (e) Z-average of the Aβ40 assemblies in (d). (f) ANS fluorescence spectra of Aβ40 assemblies in the absence (black) and presence of catechins (colour coded as per the legend). (g) ANS fluorescence intensities at 454 nm for the samples in (k), normalized to the intensity for Aβ40 alone. (h) Thioflavin T fluorescence intensities at 485 nm of Aβ40 assemblies in the absence (black) and presence of catechins (coloured as per legend) normalized to the intensity of canonical assemblies. | ||
We also stained RPE1 cells with the necrotic cell marker propidium iodide (PI), which binds to DNA in cells with severely compromised membranes.34 The RPE1 cells were also counterstained with the nuclear marker Hoechst 33342![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 35 to show that non-specific PI-staining is negligible under our conditions, as indicated by the purple vs. red fluorescence for PI in merged vs. separate panels, respectively (Fig. 1b). Fluorescence microscopy images of RPE1 cells treated with canonical Aβn indicate prominent staining with PI (Fig. 1b). In contrast, Aβn formed in the presence of catechins exhibit remarkably less PI staining (Fig. 1b), following the same EC < EGC < EGCG ranking as the cellular viability assay (Fig. 1a). Overall, these results suggest that the Aβ assemblies in our library elicit different levels of cellular dysfunction and cell death. Hence, the comparative analysis of such Aβ aggregates is anticipated to reveal key molecular determinants of soluble Aβ toxicity.
35 to show that non-specific PI-staining is negligible under our conditions, as indicated by the purple vs. red fluorescence for PI in merged vs. separate panels, respectively (Fig. 1b). Fluorescence microscopy images of RPE1 cells treated with canonical Aβn indicate prominent staining with PI (Fig. 1b). In contrast, Aβn formed in the presence of catechins exhibit remarkably less PI staining (Fig. 1b), following the same EC < EGC < EGCG ranking as the cellular viability assay (Fig. 1a). Overall, these results suggest that the Aβ assemblies in our library elicit different levels of cellular dysfunction and cell death. Hence, the comparative analysis of such Aβ aggregates is anticipated to reveal key molecular determinants of soluble Aβ toxicity.
The Aβ assembly library spans a wide distribution of sizes, hydrophobic solvent exposures and cross β-sheet contents
We first evaluated how our catechin library remodels the distribution of Aβ assemblies. For this purpose, the relative populations of the NMR visible low MW Aβ species (e.g. monomers) were gauged through residual 1H NMR intensities (Fig. 1c), while the NMR invisible Aβn were probed by dynamic light scattering (DLS) (Fig. 1d and e). While it is important to complement these data with size estimations through other means, such as TEM (vide infra), interestingly, we observed that all catechins in our library reduce the populations of both the Aβ monomers (Fig. 1c) and the Aβ assemblies at the opposite end of the molecular weight (MW) distribution (Fig. 1d and e). These results suggest that the Aβ species at the extremes of the probability distribution are converted by the catechins into Aβ species with intermediate MW. However, the extent of this remodeling is markedly catechin-dependent with (−)-catechin-3-gallate (CG) leading to large reductions in both the monomer and high MW populations (Fig. 1c–e) and methyl-3,4,5-trihydroxybenzoate (MG) causing only marginal changes (Fig. 1c–e).We also investigated the surface hydrophobicity of the Aβ assemblies formed under our conditions, as exposed hydrophobic surfaces have been associated with toxicity for another amyloidogenic system.36 The surface hydrophobicity of Aβn was probed through 8-anilino-1-naphthalenesulfonic acid (ANS) fluorescence, which exhibits a characteristic blueshift and enhancement in fluorescence intensity upon binding exposed hydrophobic sites. A substantial enhancement in ANS fluorescence was observed for canonical Aβn (Fig. 1f and g, black), whereas the extent of such enhancement is significantly reduced for most catechin-treated Aβn (Fig. 1f and g, coloured). Notably, the measurements of the catechin-treated Aβn surface hydrophobicity (Fig. 1f and g) rank in the same order as the cell toxicities (Fig. 1a), suggesting that exposed hydrophobic surfaces are a key determinant of Aβn toxicity.
Another unique signature of amyloids is the formation of extensive cross β-sheets, as reported by the fluorescent dye Thioflavin T (ThT). Canonical, toxic Aβn exhibit significant ThT fluorescence in comparison to catechin-remodeled Aβn (Fig. 1h). While the decreased ThT fluorescence in the presence of EGCG is in agreement with previous observations,30,37,38 our data on the extended catechin library reveal that other catechins also preserve the ability to destabilize intermolecular β-sheets and/or outcompete ThT. Hence, ThT-responsive β-amyloids do not appear to correlate with cytotoxicity as well as the observables reported above i.e. size and hydrophobic exposure. Indeed, solvent accessible hydrophobic moieties are one of the main drivers for Aβ–membrane interactions, which in turn have been proposed as a key determinant of the cytotoxicity associated with Aβ.39 This hypothesis is supported by our propidium iodide results, which indicate that toxic Aβn severely compromise the integrity of cell membranes (Fig. 1b). To further corroborate this hypothesis, we evaluated the interactions between a representative subset of our Aβn library and biomimetic membranes (small unilamellar vesicles, SUVs).
Toxic Aβ assemblies co-localize, bind and insert into biomimetics membranes
We profiled the membrane interactions of selected Aβ assemblies from our library that report on representative regions of our toxicity scale, i.e. the canonical as well as the EC- and EGCG-remodeled Aβn (Fig. 1a). For this purpose, SUVs composed of a mixture of DOPE:DOPS:DOPC lipids were prepared with an effective size distribution ranging from ∼10–100 nm and an average diameter of ∼34 nm (Fig. 2a and b). Prior to the addition of the Aβn library to the SUVs, we characterized the morphology of the Aβn by TEM to ensure that significant catechin-induced remodeling occurs. Indeed, compared to canonical Aβn, which primarily adopt “worm-like” protofibrils (Fig. 2c, top left panel), we observed both spherical assemblies and amorphous aggregates in the presence of EGCG (Fig. 2c, top right panel). The latter of the two species has been reported to be an intermediate in the formation of the former.23 In contrast, the EC-remodeled Aβn displays features of both canonical and EGCG-remodeled Aβn, albeit more closely resembling the canonical Aβn (Fig. 2c, top center panel). Having confirmed that catechin-induced remodeling of Aβn occurs, we then evaluated to what extent the Aβn library interacts with SUVs.
 | ||
| Fig. 2 Localization and insertion of Aβ40 assemblies into model membranes. (a) Negative-stain TEM image of 800 μM DOPE:DOPS:DOPC SUVs. (b) Size distribution of SUVs shown in (a) as determined through dynamic light scattering intensity measurements. (c) Negative-stain TEM images of Aβ40 assemblies in the absence and presence of EC and EGCG and the same assemblies treated with the SUVs in (a) and (b). All scale bars correspond to 100 nm. (d) Schematic summary of the information extracted from wide-angle X-ray diffraction experiments. (e) Complete two-dimensional intensity maps of the X-ray diffraction data with both in-plane and out-of-plane features. (f–i) In-plane (q‖) diffraction patterns (black line) and fitted Lorentzian peaks (coloured peaks) for DOPE:DOPS:DOPC lipids (green peaks) in the absence and presence of Aβ40 assemblies (blue peaks) with and without catechins (red peaks). Red lines indicate total fits derived from the summation of component peaks. (j) Normalized population of membrane-embedded β-sheet assemblies relative to canonical Aβ40 assemblies, derived through the integration of blue Aβ peaks in (f–i). (k) In-plane (q‖) diffraction patterns highlighting the cross-β inter-sheet signal intensity, which correspond to the 9.5 Å spacing between β-sheets shown in (d). (l) Out-of-plane (qz) diffraction patterns depicting the membrane lamellar spacing (panel d, dashed black and red lines corresponding to 38.7 and 52.7 Å, respectively) in the absence (black) and presence (coloured as per legend) of Aβ40 assemblies. | ||
TEM images reveal that canonical Aβn significantly colocalize with SUVs. For example, it is possible to observe select Aβn co-positioned with the lipids (Fig. 2c, bottom left panel). Similar to the canonical Aβn, EC-remodeled Aβn are also somewhat colocalized with the SUVs (Fig. 2c, bottom center panel). However, in stark contrast to both the canonical and EC-remodeled Aβn, the EGCG-remodeled Aβn are on average spatially distinct from the SUVs (Fig. 2c, bottom right panel).
To complement the TEM data on canonical vs. catechin-remodeled Aβn–membrane interactions, we performed 15N-transverse relaxation (R2), 1H-based saturation transfer difference (STD) as well as 15N-Dark State Exchange Saturation Transfer (DEST) NMR experiments, which collectively probe the interactions of Aβ with high MW (HMW) species, including SUVs, Aβn and their complexes, through the lens of the NMR visible Aβ monomers (Fig. 3a–g).29,40–48 Upon addition of SUVs to the canonical Aβn, we observed marked enhancements in R2 and STD (Fig. 3a and b), consistent with the Aβn–membrane interactions revealed by TEM (Fig. 2c). The SUV-induced changes in R2 and 1H-based saturation transfer are more pronounced for the residues in the β1 (residues 12–24) and β2 regions (residues 30–40) than for the N-terminal moiety (residues < 12), indicating that the β1 and β2 segments serve as key hot-spots of the SUV-Aβ interactions under our experimental conditions. This conclusion is independently confirmed by the comparative analysis of the 15N-DEST data (Fig. 3g–m).
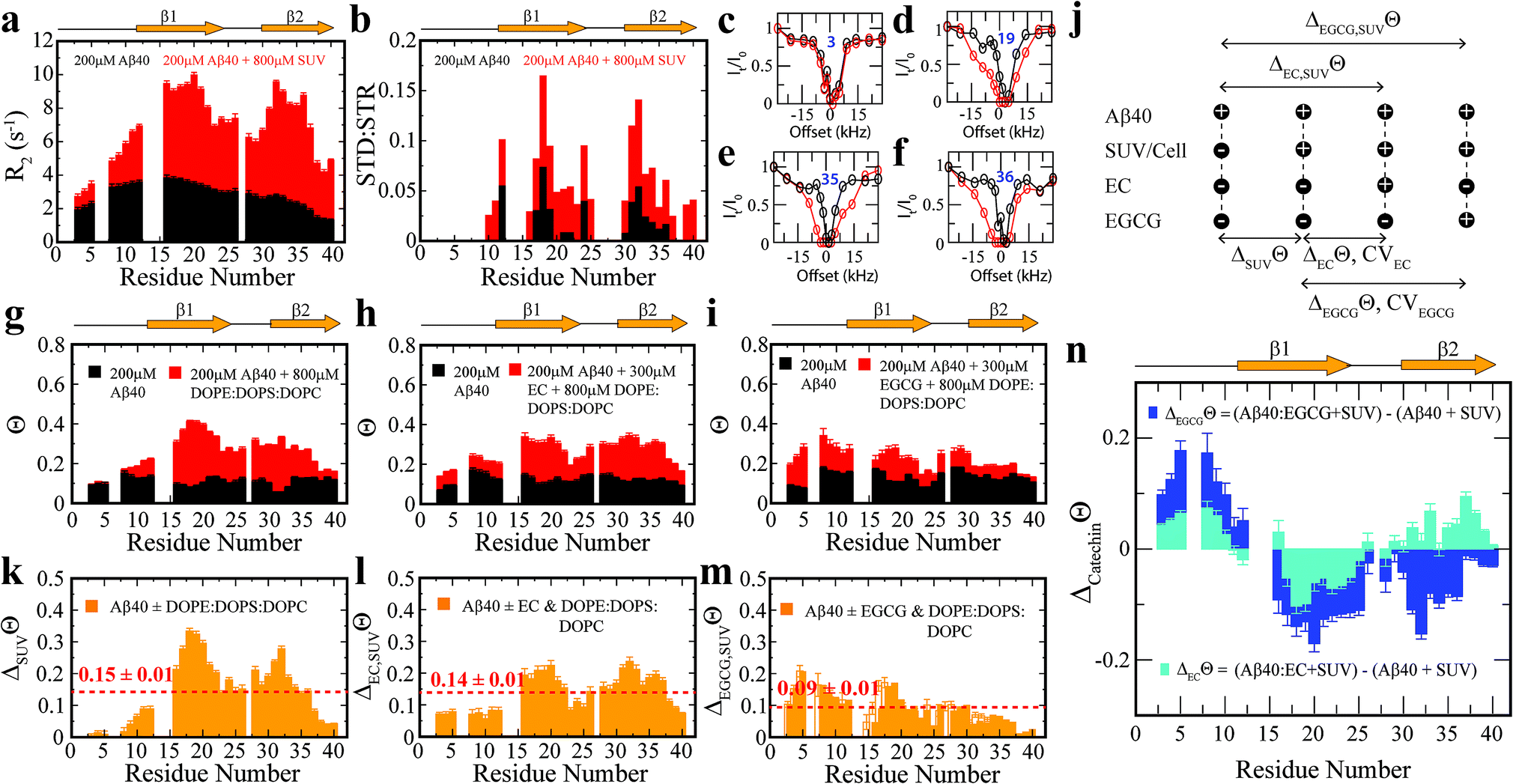 | ||
| Fig. 3 Exchange dynamics of Aβ40 monomers on the surface of soluble Aβ40 assemblies and model membranes. (a) 15N-R2 and (b) MeSTDHSQC for the canonical Aβ40 assemblies in the absence (black) and presence (red) of DOPE:DOPS:DOPC SUVs. (c–f) Representative 15N-DEST profiles for the samples shown in (a). (g) 15N-Θ profiles for the samples shown in (a), colour coding is as per legend. (h) 15N-Θ profiles for canonical Aβ40 assemblies in the absence (black) and presence (red) of EC followed by DOPE:DOPS:DOPC SUV addition. (i) 15N-Θ profiles for canonical Aβ40 assemblies in the absence (black) and presence (red) of EGCG followed by DOPE:DOPS:DOPC SUV addition. (j) Definition of key differentials in the 15N-DEST measurements and the corresponding normalized cellular viabilities. (k) Difference between the Θ profiles shown in (g). The dashed red line indicates the average Θ value. (l) Difference in the Θ profiles shown in (h). (m) Difference between the Θ profiles shown in (i). (n) 15N-Θ difference profiles for (h, red) vs. (g, red) (cyan) and (i, red) vs. (g, red) (blue). | ||
Residues in direct contact with the Aβn/SUV surface typically display an attenuation of the residual monomer DEST signal, leading to broadening of the residue-specific 15N-DEST vs. offset profile relative to amino acids for which the monomer is disengaged from the Aβn/SUV surface.31 Such broadening of the 15N-DEST profile is quantitatively measured through the Θ parameter at intermediate 15N-continuous wave (CW) offsets,40,49,50 as explained in the Methods. Consistent with the R2 and STD data (Fig. 3a and b), upon SUV addition to canonical Aβn, major DEST vs. offset profile broadening and corresponding Θ enhancements are observed for the β1 and β2 regions (Fig. 3c–g and k; ESI Fig. S3†). A similar observation applies to the addition of SUVs to EC-remodeled Aβ, which on average display a pattern comparable to canonical Aβn (Fig. 3h and lvs.Fig. 3g and k; ESI Fig. S4†). Conversely, the EGCG-remodeled Aβ do not exhibit significant β1 and β2 enhancements as compared to canonical and EC-remodeled Aβ (Fig. 3i and m; ESI Fig. S5†), in excellent agreement with the TEM observations. While the combination of our TEM and 15N-based NMR experiments reveal key differences in Aβ–membrane interactions between the less toxic EGCG-remodeled Aβ and the more toxic canonical and EC-remodeled Aβ, they do not provide direct insight about whether Aβn inserts into the membrane and about the structural features of membrane-embedded Aβn. To this end, we conducted wide-angle X-ray diffraction (WAXD) experiments in the presence of model membranes for Aβ assemblies at representative regions of our toxicity scale (Fig. 2d–l).
The WAXD two-dimensional intensity maps (Fig. 2e) were modeled with a series of Lorentzian fits (Methods) to derive structural features both in-plane (q‖, Fig. 2f–k) and out-of-plane (qz, Fig. 2l) of the membrane. For the lipid sample in the absence of Aβn, in-plane and out-of-plane Bragg peaks were observed at 1.41 Å−1 (Fig. 2f) and 0.17 Å−1 (Fig. 2l, black), respectively, corresponding to the formation of bilayer stacks with an effective bilayer width of 38.7 Å and a 5.1 Å spacing between individual lipids (Fig. 2d). Addition of canonical Aβn to these lipid bilayers results in additional in-plane features at 1.32 Å−1 (Fig. 2g, blue) and 0.76 Å−1 (Fig. 2k, red), indicating the presence of membrane-embedded Aβn adopting laminated β-sheets with 5.5 Å spacing between adjacent β-strands and 9.5 Å between β-sheet layers (Fig. 2d). Interestingly, we observe an additional peak at 1.51 Å−1 (Fig. 2g, cyan) corresponding to highly ordered lipids likely in the regions interfacing with the embedded Aβn. Moreover, an out-of-plane diffraction pattern is observed at ∼0.12 Å−1 (Fig. 2l, red) consistent with the presence of Aβ not embedded into the bilayer (Fig. 2d).
Compared to canonical Aβn, the EC- and EGCG-remodeled Aβn still preserve extended β-sheets in the membrane (Fig. 2h and i, blue), although the relative amounts are decreased in the presence of EC and EGCG, in that order (Fig. 2j). In contrast, neither of the catechin-remodelled Aβn exhibit packing of β-sheet layers (Fig. 2k, green and blue), in agreement with our ThT data (Fig. 2h). Overall, these findings suggest that the toxic Aβn formed under our conditions colocalize, interact and insert into lipid membranes wherein they adopt β-sheet structures. To identify the toxic Aβn surfaces that facilitate these multivalent interactions with the membrane, we comparatively examined the 15N-DEST differences between canonical, EC- and EGCG-remodeled Aβn in the presence of model membranes (Fig. 3n, ESI Fig. S2†).
Toxic vs. non-toxic Aβ assemblies in the membrane environment exhibit marked differences in Aβ-recognition profiles
To focus on the effects of the catechins, the canonical Aβn DEST profile (ESI Fig. S2b†) was subtracted from the catechin-remodeled Aβn DEST profiles (ESI Fig. S2c and d†). Since all profiles in ESI Fig. S2b–d† were recorded in the presence of SUVs, the resulting DEST differences (Fig. 3n) report primarily on the catechin-induced remodeling of Aβ monomer–Aβn contacts. Specifically, the EGCG-remodeled vs. canonical Aβn15N-Θ profile differences (ΔEGCGΘ) show significant decreases in Θ in the two β-strand regions typically observed in Aβ protofibrils (Fig. 3n, darkblue). These losses are consistent with the Aβ monomers being less engaged with the Aβn surface at the two β-strand sites in the presence of EGCG. However, the EGCG-induced disengagement detected for the β1 and β2 regions does not extend to the N-terminal segment, for which a significant enhancement in direct contacts is observed (Fig. 3n, darkblue). A similar N-terminal Θ DEST enhancement is observed also upon EC addition (Fig. 3n, lightblue), albeit with reduced magnitude (Fig. 3n, light vs. darkblue). Likewise, in the β1 region the EC-remodeled Aβn show Θ losses with a reduced extent compared to the EGCG-remodeled Aβn (Fig. 3n, light vs. darkblue). However, the DEST pattern observed for the N-terminal and β1 regions does not extend to the β2 segment, for which EC and EGCG result in opposite Θ changes (Fig. 3n, light vs. darkblue). These findings imply that exposure of the hydrophobic β1-turn region and concomitant shielding of the N-terminus are two key structural transitions intimately linked to toxicity, as these toxic surfaces modulate interactions with the membrane.Selection of molecular determinants of Aβn toxicity
In order to systematically isolate the Aβn features relevant for toxicity, we identified groups of coupled Aβn observables by relying on the data correlation matrix (Fig. 4a), whose elements represent the absolute Pearson's correlation coefficients (|r|) between each pair of Aβn observables (ESI†). Through agglomerative clustering of the correlation matrix, we then built a dendrogram that partitions the Aβn observables into five distinct clusters (Fig. 4b). The largest cluster, denoted as cluster 1, includes the ΔCatΘi values for residues in the 3–28 region as well as three low resolution observables, i.e. the membrane-embedded β-sheet, the size and the surface hydrophobicity. Since these measurables rank similarly to the relative toxicities (Fig. 1), we hypothesized that cluster 1 defines key molecular determinants of Aβn toxicity. This hypothesis is confirmed by two independent lines of evidence.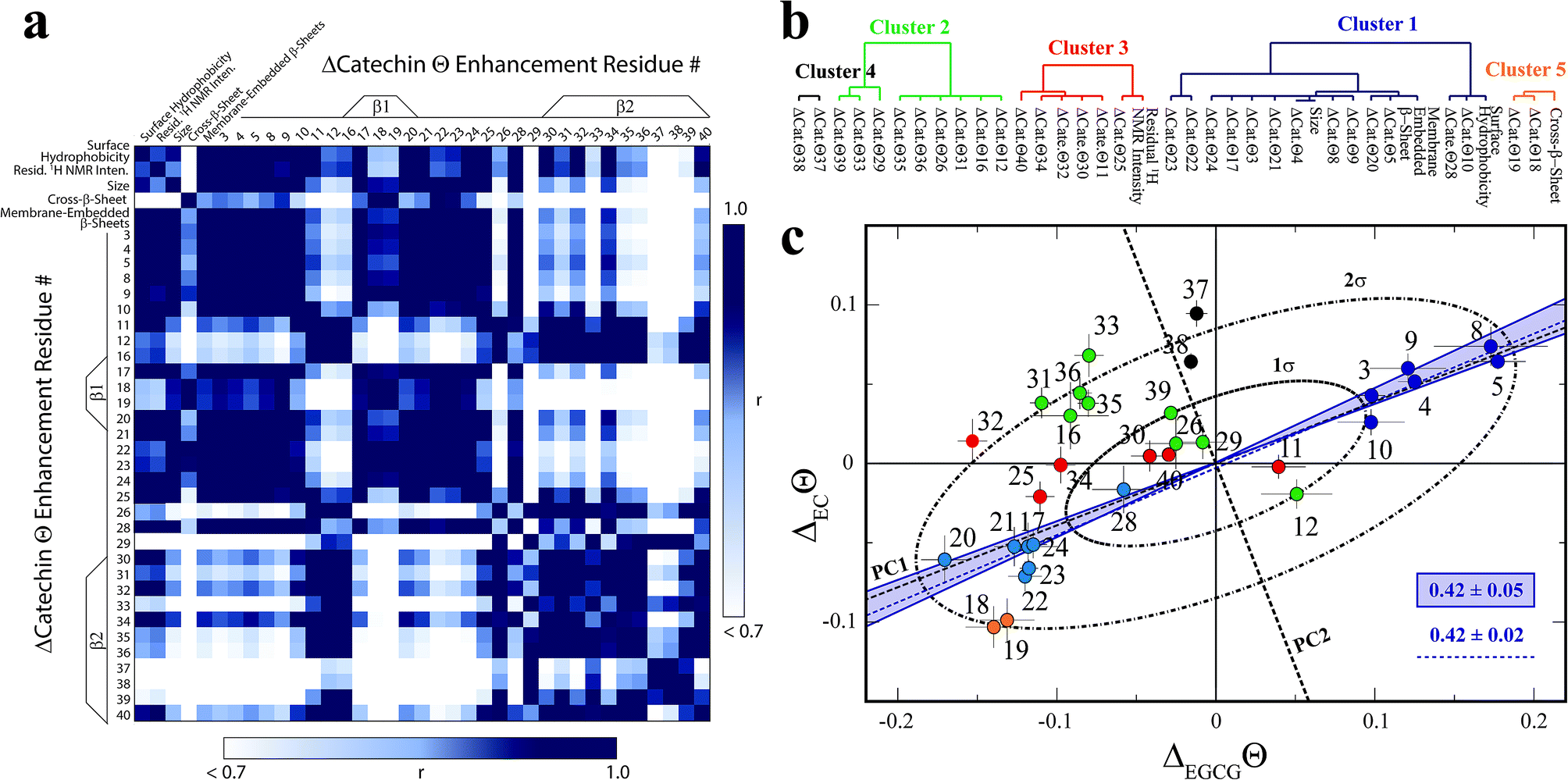 | ||
| Fig. 4 Identification of the determinants of Aβ assembly toxicity through agglomerative clustering and Singular Value Decomposition (SVD). (a) Correlation matrix for the Aβn observables from Fig. 1–3. Correlations with an absolute Pearson's correlation coefficient > 0.95 are indicated in dark blue. (b) Dendrogram displaying the clusters with an absolute Pearson's correlation coefficient > 0.9 obtained through complete linkage agglomerative clustering. (c) Singular Value Decomposition (SVD) of the 15N-DEST data. The dashed black lines indicate the first and second principal components (PC1 and PC2) obtained through the SVD of the column-mean centered (ΔECΘi, ΔEGCGΘi) matrix, where i is the residue number. The ellipsoids at one and two standard deviations for the residue scores along PC1 and PC2 are shown as black dot-dashed curves. Data for residues assigned to clusters 1, 2 and 3, 4 and 5 though agglomerative clustering are displayed as solid dark/light blue, green, red, black and orange circles, respectively, and the corresponding residue number is reported beside each circle. The solid blue lines define the region of the (ΔECΘi, ΔEGCGΘi) plane that is expected to scale with the relative cellular viability (CV) defined as (CVAβ40+EC − CVAβ40)/(CVAβ40+EGCG − CVAβ40) = 0.42 ± 0.05, based on the data of Fig. 1. The dashed blue line (slope of 0.42 ± 0.02 and correlation coefficient of 0.98) was obtained from the linear regression of the DEST data in cluster 1 (blue) and confirms that cluster 1 correlates with cellular viability. PC1 (slope of 0.39) aligns with the residues for cluster 1. | ||
First, if we re-compute the correlation matrix and agglomerative clustering after including the relative toxicities (Fig. 1a), we find that the toxicity partitions within cluster 1 (ESI Fig. S6†), confirming that the observables in this cluster scale with Aβn toxicity. Second, in the ΔEGCGΘivs. ΔECΘi plot (Fig. 4c), the cluster 1 residues fall at or near the region expected to scale with the relative EC vs. EGCG cell viability (CV) data, defined as (CVAβ40+EC − CVAβ40)/(CVAβ40+EGCG − CVAβ40) = 0.42 ± 0.05 (shaded blue area, Fig. 4c). The linear regression of ΔEGCGΘivs. ΔECΘi for cluster 1 is in fact in excellent agreement with the value expected based on the relative cellular viability (dashed blue line with slope of 0.42 ± 0.02 and correlation coefficient of 0.98; Fig. 4c). Hence, we conclude that cluster 1 (blue dendrogram in Fig. 4b and blue circles in Fig. 4c) is relevant for the toxicity of Aβn.
To gain further insight on the significance of the ΔEGCGΘivs. ΔECΘi plot and independently corroborate the residue clusters obtained through the agglomerative clustering analysis, we also performed Singular Value Decomposition (SVD) of the data in Fig. 4c. The SVD analysis reveals that the first principal component (dashed black line, Fig. 4c), which accounts for 88% of the total variance, not only resides within the range expected to scale with the relative cellular viability (i.e. within the shaded blue area in Fig. 4c), but also aligns with the residues for cluster 1. Interestingly, the SVD reveals that cluster 1 (blue circles, Fig. 4c) is composed of two distinct sub-sets that are mostly confined at opposite extremes of PC1, between the 1σ and 2σ ellipsoids (Fig. 4c). The sub-set with positive PC1 components (dark blue circles) represents the N-terminal residues that become engaged in monomer recognition, as probed by DEST, when cellular viability is enhanced. On the contrary, the cluster 1 sub-set with negative PC1 scores (light blue circles) arises from the β1-turn region residues that become engaged when cellular viability decreases.
In stark contrast to cluster 1, the other clusters obtained from the agglomerative clustering analysis (Fig. 4b, black, green, red and orange circles) fall outside the range expected to scale with cellular viability (blue shaded area, Fig. 4c) and exhibit components along PC2 that are overall higher than those observed for cluster 1 (Fig. 4c). In conclusion, the combined analyses of the correlation matrix, agglomerative clustering and SVD consistently identify the constituents of cluster 1, i.e. surface hydrophobicity, size, membrane-embedded β-sheets, N-terminal residue disengagement and β1-turn region engagement, as key molecular determinants of Aβn toxicity.
In order to verify the predictive power of the correlation between Aβn toxicity and cluster 1, we measured the relative toxicities for the Aβ assemblies not included in Fig. 1a and we compared them to those predicted by our model (Fig. 4; ESI Fig. S7†). These Aβn toxicities were not used to train our model and hence provide a critical test of its prognostic capacity. As seen in ESI Fig. S7d,† a strong linear correlation is observed between the predicted and observed toxicities (r ≥ 0.94), with a slope within error to one, thus validating the predictive power of our model.
In summary, our investigation of the Aβn library through the comparative analysis of 15N-R2 and DEST NMR combined with WAXD, TEM, DLS and extrinsic fluorescence reveals key structural differences that distinguish toxic vs. non-toxic Aβ assemblies. The integrated analyses of our data through agglomerative clustering and SVD consistently identify a cluster of molecular attributes unique to toxic Aβn (Fig. 4b, cluster 1), including surface hydrophobicity, size, membrane-embedded β-sheets, shielding of the N-terminus and simultaneous exposure of the β1-turn region to Aβ monomers, as probed through DEST NMR.
Our data shows that toxic Aβn exhibit solvent exposed hydrophobic sites accessible to ANS binding. While the relationship between surface hydrophobicity and toxicity has been observed previously for several protein systems such as the Type A/B HypF-N assemblies,51,52 the A+/A− Aβ42 oligomer pair,53 the sup35p oligomer pair,54 and others,55 here we not only systematically confirm this association for the Aβ system using a library of Aβ assemblies, but we also propose an unprecedented mechanism of Aβn toxicity probed at multiple degrees of resolution. Such mechanism reveals how hydrophobic exposure relates to Aβ–membrane interactions and Aβ monomer recognition. The combination of our TEM, DLS and 15N-DEST and R2 data collectively shows that Aβn with greater surface hydrophobicity e.g. canonical and EC-remodeled Aβn colocalize and interact with the membrane surface more effectively than the less toxic Aβn with less exposed hydrophobic sites e.g. the EGCG-remodeled Aβn (Fig. 5a).
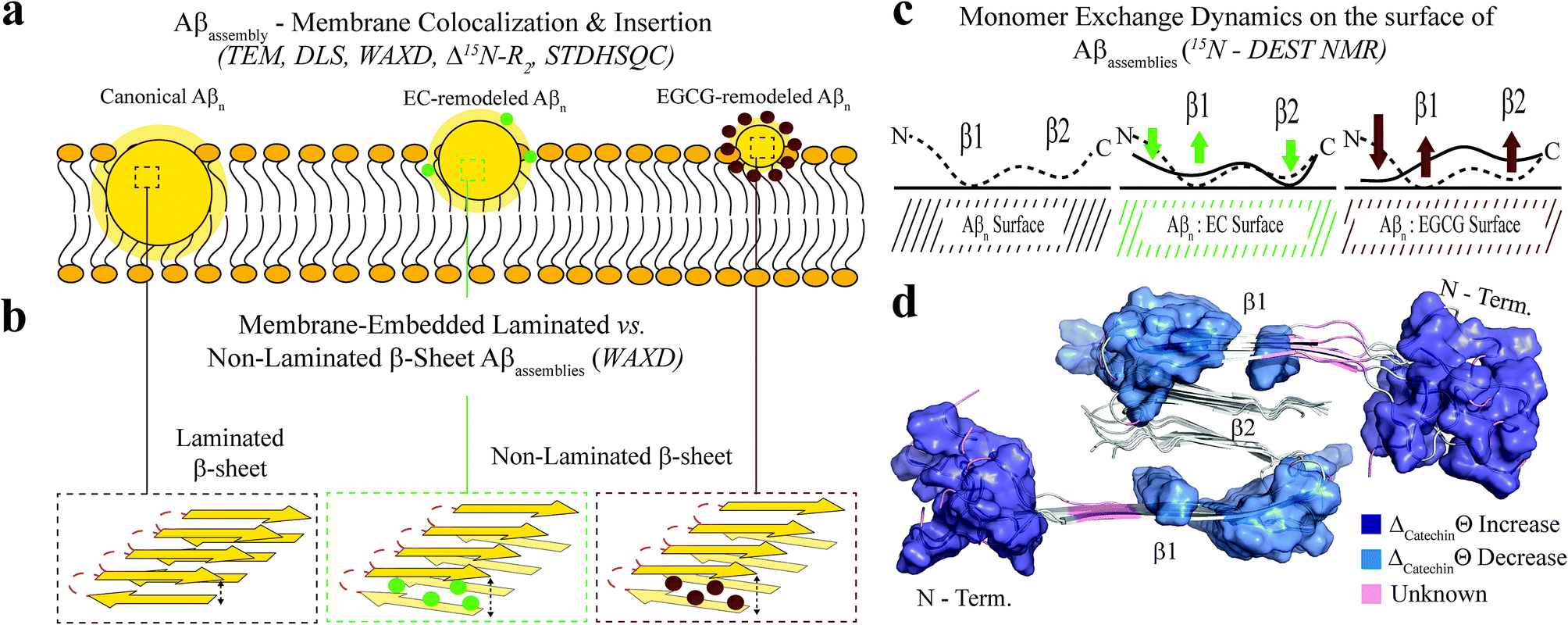 | ||
| Fig. 5 Proposed model for the molecular determinants of Aβ assembly toxicity. (a) Toxic Aβn (canonical Aβn) exhibit significant solvent exposure of hydrophobic surfaces (yellow glow surrounding Aβn). Exposed hydrophobic surfaces facilitate the colocalization, interaction and subsequent insertion of Aβn into the membrane. (b) Membrane-embedded Aβn adopt both laminated and non-laminated β-sheets, indicating that under our experimental conditions the non-laminated β-sheet signature is the minimum structural feature required for membrane insertion and induction of toxicity. (c) Toxic vs. non-toxic Aβn exhibit unique regiospecific differences in the recognition of Aβ monomers within a membrane environment. Relative to canonical Aβn (black), EC- (green) and EGCG-remodeled Aβn (maroon) exhibit progressive engagement of contacts with Aβ monomers at the N-terminus and disengagement at the β1-turn region, following the same ranking as their measured toxicities. In contrast, for the β2 region no correlation is observed between toxicity and Aβn monomer recognition. Relevant experimental techniques are indicated in parenthesis. (d) Mapping on the structure of Aβ40 fibrils57 (PDB code: 2LMN) the Aβ residues in cluster 1 (Fig. 4b and c). The N-terminal and β1-turn residues that correlate with toxicity (blue) are found in the external regions of the Aβ fibril structure. In contrast, β2 is involved in the lamination of multiple β-sheet layers and is largely inaccessible (Table S2†), explaining its ancillary role in toxicity. | ||
The surface hydrophobicity-mediated interactions with the membrane are not limited to the membrane surface, as our WAXD data show that canonical and EC-remodeled Aβn exhibit significant populations of β-sheets embedded in the membrane compared to EGCG-remodeled Aβn. The functional effect of the membrane-embedded β-sheets is recapitulated by our propidium iodide-based assay, which indicates that canonical Aβn significantly enhance the permeability of the cell membrane compared to the less toxic Aβn formed in the presence of EGCG.
Notably, we also found that cross-β-sheet structures are dispensable for membrane insertion, as only canonical Aβn exhibit cross lamination of β-sheet layers, whereas EC-remodeled Aβn, with comparable levels of membrane-embedded β-sheets, exhibit considerably reduced cross lamination, similar to EGCG remodeled Aβn (Fig. 2k and 5b). The lack of correlation between toxicity and β-sheet cross-lamination is also consistent with the variability in sheet-to-sheet pairing angles reported for oligomers of model amyloidogenic sequences stabilized by macrocyclic peptides.56
The correlation and SVD analyses also identify a cluster of residues confined to the N-terminus and β1-loop region that are key to the regulation of Aβn toxicity (Fig. 4b and c, cluster 1). The probability distribution of contacts between Aβ monomers and the Aβn/SUVs surface is markedly enhanced in the β1-loop region (residues 17–28) and concomitantly reduced at the N-terminal segment (residues 3–10) as the Aβn toxicity increases (Fig. 3n and 5c, green vs. maroon arrows). Interestingly, an unexpected decorrelation with toxicity is observed at the β2 region (residues 30–40) (clusters 2 and 4), for which the EC-remodeled Aβn, with intermediate toxicity, exhibits a further enhancement in contacts relative to the canonical Aβn (Fig. 3n and 5c, green), in stark contrast to the reduction observed for EGCG-remodeled Aβn (Fig. 3n and 5c, green vs. maroon arrows).
Notably, the N-terminus and β1-loop Aβ regions identified by the correlation and SVD analyses to be toxicity determinants (Fig. 4c, cluster 1) are located at the external surface of the Aβ40 fibril structure (Fig. 5d, blue surfaces). Furthermore, most familial AD mutations (English, Tottori, Iowa, Arctic, Dutch and Italian) that alter the biophysical properties of Aβ are observed in the N-terminal and β1 regions.1,58 Conversely, the β2 region not identified by SVD as linked to toxicity, is inaccessible to the environment (Table S2†) and is found embedded into the structural core of the fibril, where it is involved in the cross lamination of multiple β-sheet layers (Fig. 5d, grey cartoon). These observations agree with our WAXD and ThT data, consistently pointing to β-sheet lamination as accessory to toxicity induction.
Conclusions
Overall, our data indicate that Aβn toxicity is regulated by the solvent exposure of hydrophobic surfaces, wherein the hydrophobic β1-turn region is more accessible to monomer/SUV recognition, while the highly charged N-terminus is shielded from such recognition. In comparison, the role of β2 appears to be largely ancillary. These toxic surfaces enhance the colocalization, contacts and subsequent insertion of β-sheet rich Aβn into the membrane, leading to compromised membrane stability. Moreover, the proposed model is able to predict relative toxicities solely based on low-resolution measurements, such as size and surface hydrophobicity. Modulation of these properties through small-molecule treatment can be utilized as an effective strategy to reduce the toxicity associated with soluble Aβ assemblies. In addition, soluble oligomers of amyloidogenic peptides with different sequences have been suggested to share a common conformation,59 and Aβ is not only relevant for dominantly inherited AD, but also serves as a model system for a broad-range of amyloid disorders. Hence, the cluster of molecular attributes identified here to correlate with toxicity may be transferrable to other amyloidogenic systems.Author contributions
R. A. and G. M. designed research; R. A., M. A., A. K., J. B. and V. H. performed research; R. A., A. K., M. R., J. B., R. E., V. H., R. W., S. B., J. H., C. V. and G. M. analyzed data; R. A. and G. M. wrote the paper. All authors have given approval to the final version of the manuscript. This project was funded by the Natural Sciences and Engineering Research Council of Canada (NSERC RGPIN-2019-05990; RGPIN-2018-05585; RGPIN-2016-06450; RGPIN-2014-04514).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank M. Akimoto, B. VanSchouwen, N. Jafari, Y. Lu, R. Truant (McMaster U.), M. H. Lim (KAIST) and S. J. Cox (U. Michigan) for helpful discussions.Notes and references
- I. Benilova, E. Karran and B. de Strooper, Nat. Neurosci., 2012, 15, 349–357 CrossRef CAS PubMed.
- J. A. Hardy and G. A. Higgins, Science, 1992, 256, 184–185 CrossRef CAS.
- E. S. Musiek and D. M. Holtzman, Nat. Neurosci., 2015, 18, 800–806 CrossRef CAS PubMed.
- C. R. Jack, D. S. Knopman, W. J. Jagust, R. C. Petersen, M. W. Weiner, P. S. Aisen, L. M. Shaw, P. Vemuri, H. J. Wiste, S. D. Weigand, T. G. Lesnick, V. S. Pankratz, M. C. Donohue and J. Q. Trojanowski, Lancet Neurol., 2013, 12, 207–216 CrossRef CAS.
- J. M. Castellano, J. Kim, F. R. Stewart, H. Jiang, R. B. DeMattos, B. W. Patterson, A. M. Fagan, J. C. Morris, K. G. Mawuenyega, C. Cruchaga, A. M. Goate, K. R. Bales, S. M. Paul, R. J. Bateman and D. M. Holtzman, Sci. Transl. Med., 2011, 3, 89ra57 CAS.
- R. D. Terry, E. Masliah, D. P. Salmon, N. Butters, R. DeTeresa, R. Hill, L. A. Hansen and R. Katzman, Ann. Neurol., 1991, 30, 572–580 CrossRef CAS PubMed.
- W. L. Klein, G. A. Krafft and C. E. Finch, Trends Neurosci., 2001, 24, 219–224 CrossRef CAS.
- J. L. Tomic, A. Pensalfini, E. Head and C. G. Glabe, Neurobiol. Dis., 2009, 35, 352–358 CrossRef CAS PubMed.
- M. Jin, N. Shepardson, T. Yang, G. Chen, D. Walsh and D. J. Selkoe, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5819–5824 CrossRef CAS PubMed.
- Y. Miller, B. Ma and R. Nussinov, Biophys. J., 2009, 97, 1168–1177 CrossRef CAS PubMed.
- K. Ono, M. M. Condron and D. B. Teplow, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14745–14750 CrossRef CAS PubMed.
- P.-N. Cheng, C. Liu, M. Zhao, D. Eisenberg and J. S. Nowick, Nat. Chem., 2012, 4, 927–933 CrossRef CAS PubMed.
- R. Riek and D. S. Eisenberg, Nature, 2016, 539, 227–235 CrossRef.
- M. Kinoshita, E. Kakimoto, M. S. Terakawa, Y. Lin, T. Ikenoue, M. So, T. Sugiki, A. Ramamoorthy, Y. Goto and Y.-H. Lee, Phys. Chem. Chem. Phys., 2017, 19, 16257–16266 RSC.
- J. D. Gehman, R. Verardi, A. K. Mehta, G. Veglia and F. Separovic, Biophys. J., 2010, 98, 176a CrossRef.
- Y. Miller, B. Ma, C.-J. Tsai and R. Nussinov, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14128–14133 CrossRef CAS.
- H. A. Lashuel, D. Hartley, B. M. Petre, T. Walz and P. T. Lansbury, Nature, 2002, 418, 291 CrossRef CAS PubMed.
- A. Quist, I. Doudevski, H. Lin, R. Azimova, D. Ng, B. Frangione, B. Kagan, J. Ghiso and R. Lal, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10427–10432 CrossRef CAS PubMed.
- M. F. M. Sciacca, S. A. Kotler, J. R. Brender, J. Chen, D.-k. Lee and A. Ramamoorthy, Biophys. J., 2012, 103, 702–710 CrossRef CAS PubMed.
- M. Ahmed, J. Davis, D. Aucoin, T. Sato, S. Ahuja, S. Aimoto, J. I. Elliott, W. E. van Nostrand and S. O. Smith, Nat. Struct. Mol. Biol., 2010, 17, 561–567 CrossRef CAS PubMed.
- S. Chimon, M. A. Shaibat, C. R. Jones, D. C. Calero, B. Aizezi and Y. Ishii, Nat. Struct. Mol. Biol., 2007, 14, 1157–1164 CrossRef CAS PubMed.
- S. J. C. Lee, E. Nam, H. J. Lee, M. G. Savelieff and M. H. Lim, Chem. Soc. Rev., 2017, 46, 310–323 RSC.
- C. A. Ross and M. A. Poirier, Nat. Rev. Mol. Cell Biol., 2005, 6, 891–898 CrossRef CAS.
- S. A. Kotler, P. Walsh, J. R. Brender and A. Ramamoorthy, Chem. Soc. Rev., 2014, 43, 6692–6700 RSC.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2017, 86, 27–68 CrossRef CAS.
- T. Kawarabayashi, M. Shoji, L. H. Younkin, L. Wen-Lang, D. W. Dickson, T. Murakami, E. Matsubara, K. Abe, K. H. Ashe and S. G. Younkin, J. Neurosci., 2004, 24, 3801–3809 CrossRef CAS.
- E. E. Ambroggio, D. H. Kim, F. Separovic, C. J. Barrow, K. J. Barnham, L. A. Bagatolli and G. D. Fidelio, Biophys. J., 2005, 88, 2706–2713 CrossRef CAS.
- J. W. Um, H. B. Nygaard, J. K. Heiss, M. A. Kostylev, M. Stagi, A. Vortmeyer, T. Wisniewski, E. C. Gunther and S. M. Strittmatter, Nat. Neurosci., 2012, 15, 1227–1235 CrossRef CAS PubMed.
- N. L. Fawzi, J. Ying, R. Ghirlando, D. A. Torchia and G. M. Clore, Nature, 2011, 480, 268–272 CrossRef CAS PubMed.
- J. Bieschke, J. Russ, R. P. Friedrich, D. E. Ehrnhoefer, H. Wobst, K. Neugebauer and E. E. Wanker, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7710–7715 CrossRef CAS.
- E. Sironi, L. Colombo, A. Lompo, M. Messa, M. Bonanomi, M. E. Regonesi, M. Salmona and C. Airoldi, Chemistry, 2014, 20, 13793–13800 CrossRef CAS PubMed.
- C. Visentin, F. Pellistri, A. Natalello, J. Vertemara, M. Bonanomi, E. Gatta, A. Penco, A. Relini, L. de Gioia, C. Airoldi, M. E. Regonesi and P. Tortora, Hum. Mol. Genet., 2017, 26, 3271–3284 CrossRef CAS PubMed.
- M. Xu, D. J. McCanna and J. G. Sivak, J. Pharmacol. Toxicol. Methods, 2015, 71, 1–7 CrossRef CAS PubMed.
- H. Lecoeur, Exp. Cell Res., 2002, 277, 1–14 CrossRef CAS PubMed.
- S. A. Latt and G. Stetten, J. Histochem. Cytochem., 1976, 24, 24–33 CrossRef CAS PubMed.
- B. Mannini, E. Mulvihill, C. Sgromo, R. Cascella, R. Khodarahmi, M. Ramazzotti, C. M. Dobson, C. Cecchi and F. Chiti, ACS Chem. Biol., 2014, 9, 2309–2317 CrossRef CAS.
- F. L. Palhano, J. Lee, N. P. Grimster and J. W. Kelly, J. Am. Chem. Soc., 2013, 135, 7503–7510 CrossRef CAS.
- J. M. Lopez del Amo, U. Fink, M. Dasari, G. Grelle, E. E. Wanker, J. Bieschke and B. Reif, J. Mol. Biol., 2012, 421, 517–524 CrossRef CAS.
- T. L. Williams and L. C. Serpell, FEBS J., 2011, 278, 3905–3917 CrossRef CAS.
- R. Ahmed and G. Melacini, Chem. Commun., 2018, 54, 4644–4652 RSC.
- J. Milojevic, V. Esposito, R. Das and G. Melacini, J. Am. Chem. Soc., 2007, 129, 4282–4290 CrossRef CAS.
- G. Fusco, S. W. Chen, P. T. F. Williamson, R. Cascella, M. Perni, J. A. Jarvis, C. Cecchi, M. Vendruscolo, F. Chiti, N. Cremades, L. Ying, C. M. Dobson and A. de Simone, Science, 2017, 358, 1440–1443 CrossRef CAS.
- G. Fusco, T. Pape, A. D. Stephens, P. Mahou, A. R. Costa, C. F. Kaminski, G. S. Kaminski Schierle, M. Vendruscolo, G. Veglia, C. M. Dobson and A. de Simone, Nat. Commun., 2016, 7, 12563 CrossRef.
- G. Fusco, A. de Simone, T. Gopinath, V. Vostrikov, M. Vendruscolo, C. M. Dobson and G. Veglia, Nat. Commun., 2014, 5, 3827 CrossRef CAS.
- N. L. Fawzi, J. Ying, D. A. Torchia and G. M. Clore, J. Am. Chem. Soc., 2010, 132, 9948–9951 CrossRef CAS.
- C. R. Bodner, C. M. Dobson and A. Bax, J. Mol. Biol., 2009, 390, 775–790 CrossRef CAS.
- C. R. Bodner, A. S. Maltsev, C. M. Dobson and A. Bax, Biochemistry, 2010, 49, 862–871 CrossRef CAS PubMed.
- A. Ceccon, V. Tugarinov, A. Bax and G. M. Clore, J. Am. Chem. Soc., 2016, 138, 5789–5792 CrossRef CAS PubMed.
- M. Algamal, R. Ahmed, N. Jafari, B. Ahsan, J. Ortega and G. Melacini, J. Biol. Chem., 2017, 292, 17158–17168 CrossRef CAS PubMed.
- R. Ahmed, B. VanSchouwen, N. Jafari, X. Ni, J. Ortega and G. Melacini, J. Am. Chem. Soc., 2017, 139, 13720–13734 CrossRef CAS PubMed.
- S. Campioni, B. Mannini, M. Zampagni, A. Pensalfini, C. Parrini, E. Evangelisti, A. Relini, M. Stefani, C. M. Dobson, C. Cecchi and F. Chiti, Nat. Chem. Biol., 2010, 6, 140–147 CrossRef CAS.
- C. Capitini, J. R. Patel, A. Natalello, C. D'Andrea, A. Relini, J. A. Jarvis, L. Birolo, A. Peduzzo, M. Vendruscolo, P. Matteini, C. M. Dobson, A. de Simone and F. Chiti, Chem. Commun., 2018, 54, 8637–8640 RSC.
- A. R. A. Ladiwala, J. Litt, R. S. Kane, D. S. Aucoin, S. O. Smith, S. Ranjan, J. Davis, W. E. van Nostrand and P. M. Tessier, J. Biol. Chem., 2012, 287, 24765–24773 CrossRef CAS.
- R. Krishnan, J. L. Goodman, S. Mukhopadhyay, C. D. Pacheco, E. A. Lemke, A. A. Deniz and S. Lindquist, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11172–11177 CrossRef CAS.
- B. Bolognesi, J. R. Kumita, T. P. Barros, E. K. Esbjorner, L. M. Luheshi, D. C. Crowther, M. R. Wilson, C. M. Dobson, G. Favrin and J. J. Yerbury, ACS Chem. Biol., 2010, 5, 735–740 CrossRef CAS.
- J. Zheng, C. Liu, M. R. Sawaya, B. Vadla, S. Khan, R. J. Woods, D. Eisenberg, W. J. Goux and J. S. Nowick, J. Am. Chem. Soc., 2011, 133, 3144–3157 CrossRef CAS PubMed.
- A. T. Petkova, Y. Ishii, J. J. Balbach, O. N. Antzutkin, R. D. Leapman, F. Delaglio and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16742–16747 CrossRef CAS.
- C. de Jonghe, Hum. Mol. Genet., 2001, 10, 1665–1671 CrossRef CAS PubMed.
- R. Kayed, E. Head, J. L. Thompson, T. M. McIntire, S. C. Milton, C. W. Cotman and C. G. Glabe, Science, 2003, 300, 486–489 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Methods, 15N-DEST profiles, and additional statistical analyses. See DOI: 10.1039/c9sc01331h |
| This journal is © The Royal Society of Chemistry 2019 |