
Transition-metal-carbene-like intermolecular insertion of a borylene into C–H bonds†
Siyuan
Liu
abc,
Marc-André
Légaré
 bc,
Alexander
Hofmann
bc,
Theresa
Dellermann
bc and
Holger
Braunschweig
*bc
bc,
Alexander
Hofmann
bc,
Theresa
Dellermann
bc and
Holger
Braunschweig
*bc
aSchool of Materials Science and Engineering, China University of Petroleum, Qingdao, Shandong, 266580, P. R. China
bInstitute for Inorganic Chemistry, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@uni-wuerzburg.de
cInstitute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 20th May 2020
Abstract
We demonstrate that, in analogy to transition-metal carbene chemistry, [(OC)5Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) BN(SiMe3)2] facilitates intermolecular transfer of the borylene [:BN(SiMe3)2], which ultimately undergoes insertion into C–H bonds under very mild conditions. The one-pot multiple functionalization of the cyclopentadienyl rings of tungstenocene dihydride is demonstrated using this approach.
BN(SiMe3)2] facilitates intermolecular transfer of the borylene [:BN(SiMe3)2], which ultimately undergoes insertion into C–H bonds under very mild conditions. The one-pot multiple functionalization of the cyclopentadienyl rings of tungstenocene dihydride is demonstrated using this approach.
As a response to the synthetic challenge of directly functionalising C–H bonds, free carbenes have emerged as an important class of compounds because of their high reactivity in several C–C bond forming mechanisms.1 Carbenes have been studied intensively in recent decades, not only in fundamental investigations, but in studies geared toward finding concrete applications in the synthetic sciences. Although most free carbenes are high-energy transient species that often exhibit uncontrollable and unselective reactivity on their own,2 the use of transition metal catalysts or scaffolds can make carbenes substantially more useful for a variety of reactions. Among these, the metal-catalysed insertion of carbenes into C–H bonds is becoming a useful method for the direct and atom-economical formation of C–C bonds. As the key step of this process, a transition-metal (TM) carbene complex (LnM
CR1R2) reacts directly with an organic substrate, ultimately leading to the insertion of the CR1R2 unit into a C–H bond. This type of reaction has been used extensively for the synthesis of a growing number of compounds through chemo- and even stereoselective processes.1b,3
 | ||
| Scheme 1 Carbene complexes (I) and TM borylene complexes (II). | ||
An intriguing analogy between terminal TM borylene4 and TM carbene complexes (Scheme 1), based on their common ME double bonds (E = B or C), suggests the exciting prospect of using the former in synthetically useful reactions that mimic the reactivity of the latter. In fact, TM complexes and catalysts5 are already powerful tools for C–B-bond-forming reactions, such as C–H borylation,6 hydroboration7 and diboration,8 which are well studied and extensively used in synthesis. However, these reactions generally rely on TM boryl complexes, and the synthetic potential of carbene-analogous borylene complexes remains very underdeveloped in comparison.
With our sights set on harnessing the attractive synthetic possibilities suggested by the MB double bond in borylene complexes and their analogy with carbenes, we and others have explored the reactivity of these complexes in metathesis,9 borylene transfer,10 and cycloaddition11 reactions with nucleophiles and electrophiles,12 while in 2008 we reported a single example of photolytic, intermolecular borylene insertion into a C–H bond.13 Despite the exciting potential of mild borylene insertion into C–H bonds, no further example of such a reaction has been reported since. Reinforcing the analogy with carbenes, transient free borylene ligands – and particularly their base adducts – have recently proven to be highly reactive towards many substrates.14 Analogously to free carbenes, for example, these high-energy species tend to undergo uncontrolled intramolecular C–H insertion, a process often considered undesirable.10d,e,14e,15 However, these studies are yet to uncover simple intermolecular mechanisms for the formation of C–B bonds that would be relevant to organic synthesis. In this Communication, we demonstrate that terminal borylene complexes are promising reagents for C–H functionalisation through a facile borylene insertion reaction that is unlike other types of TM–boron complexes, instead being reminiscent of TM carbene reactivity.
As part of our continuing studies of TM borylene complexes,4,5f we reacted the molybdenum aminoborylene complex [(OC)5MoBN(SiMe3)2] (1) with tungstenocene derivatives. Indeed, the tungstenocene framework has a well-established coordination chemistry with boron that includes stable boryl and diboryl complexes relevant to hydroboration.16 When equimolar amounts of 1 and Cp2WH2 (2) were reacted in deuterated benzene at room temperature, a slow reaction was observed, as evidenced by the disappearance of the 11B NMR signal associated with 1 (δ = 92) from the spectrum of the solution over approximatively two days, along with the emergence of a new broad signal at δ = 39. The 1H NMR spectrum of the reaction mixture allowed us to identify the formation of a major product featuring signals corresponding to the protons of a N(SiMe3) group (δ = 0.32) and cyclopentadienyl (Cp) ligands (δ = 4.74 (2H), 4.34 (5H), 3.92 (2H)). This signal pattern associated with the Cp ligands is consistent with mono-substitution of a single Cp ring of precursor 2. Indeed, the main product of the reaction was characterised as [Cp(C5H4B(H)N(SiMe3)2)WH2] (3), which arises from the direct insertion of borylene fragment [(Me3Si)2NB] into a C–H bond of one of the Cp ligands of 2 (Scheme 2). It is interesting to note that this insertion process occurs at a C–H site known to have nucleophilic character; the Cp ligands of tungstenocene are known to be capable of interacting with Lewis acids.16d–g This observation may help to guide future development of methods for the insertion of borylene fragments into organic substrates.
 | ||
| Scheme 2 Analogy between carbene complexes (I) and TM borylene complexes (II). | ||
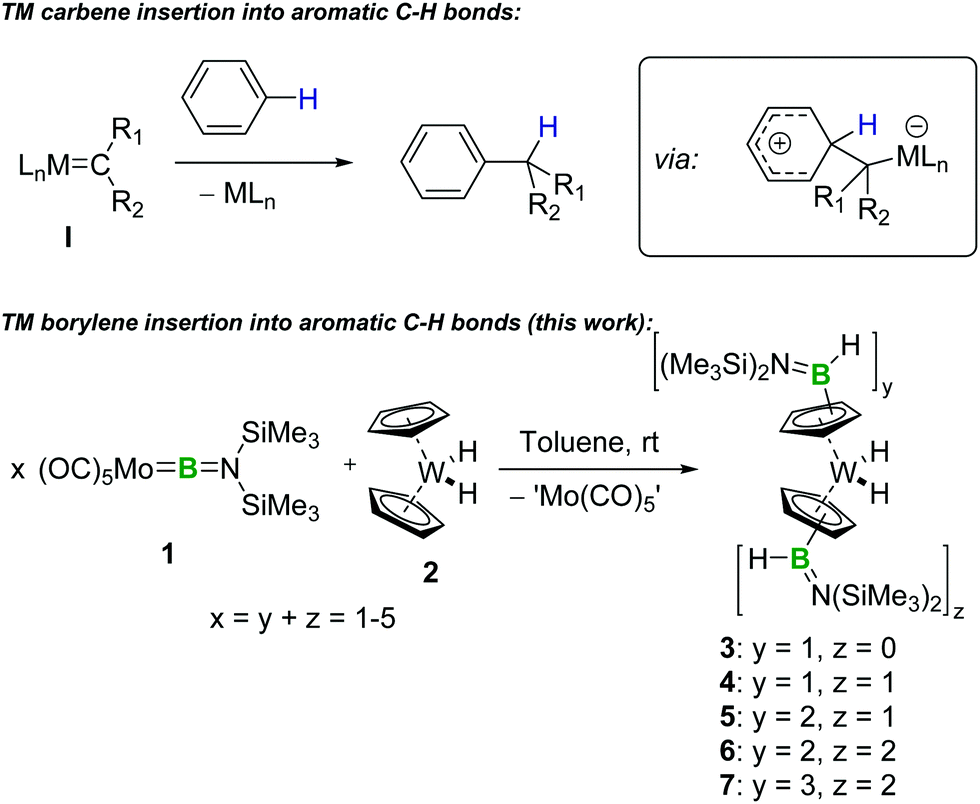
The C–H functionalization product 3 was isolated from the molybdenum-containing byproducts of the reaction (likely including [Mo(CO)6], the presence of which was confirmed by single-crystal X-ray diffraction (XRD), as well as elemental molybdenum)17 by repeated crystallisation of the latter from cold pentane, followed by recovery of 3 as yellow crystals by slow evaporation of the resulting pentane supernatant solutions (see ESI†). Unfortunately, while NMR spectroscopy indicates that the reaction of 2 to form 3 proceeds with high conversion, the difficulties associated with selectively crystallising these organometallic species decreases the isolated yields. Nevertheless, the identity of 3 was confirmed by high-resolution mass spectrometry (HRMS) and by single-crystal XRD (Fig. 1).
 | ||
| Fig. 1 Crystallographically-determined solid-state structure of 3. Atomic displacement ellipsoids depicted at 50% probability level. Carbon ellipsoids and most hydrogen atoms are omitted for clarity. | ||
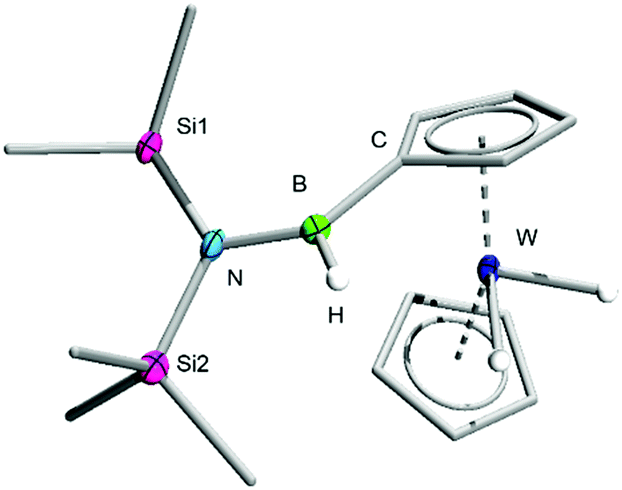
Interestingly, twofold 1,1′-functionalisation of 2 is also possible under similar conditions. Stirring a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 1 and 2 in toluene leads to the formation of a new product within 48 h that is virtually indistinguishable from 3 on the basis of 11B NMR spectroscopy. Its 1H NMR spectrum, which displays two distinct signals for the Cp rings, allows its conclusive identification as [(C5H4B(H)N(SiMe3)2)2WH2] (4), a complex that arises from the insertion of a borylene in a C–H bond of each Cp ring of 2. The elemental composition of 4 was confirmed by HRMS and its solid-state structure elucidated by single-crystal XRD (Fig. 2).
:1 mixture of 1 and 2 in toluene leads to the formation of a new product within 48 h that is virtually indistinguishable from 3 on the basis of 11B NMR spectroscopy. Its 1H NMR spectrum, which displays two distinct signals for the Cp rings, allows its conclusive identification as [(C5H4B(H)N(SiMe3)2)2WH2] (4), a complex that arises from the insertion of a borylene in a C–H bond of each Cp ring of 2. The elemental composition of 4 was confirmed by HRMS and its solid-state structure elucidated by single-crystal XRD (Fig. 2).
 | ||
| Fig. 2 Crystallographically-determined solid-state structure of 4. Atomic displacement ellipsoids depicted at 50% probability level. Carbon ellipsoids and most hydrogen atoms are omitted for clarity. | ||
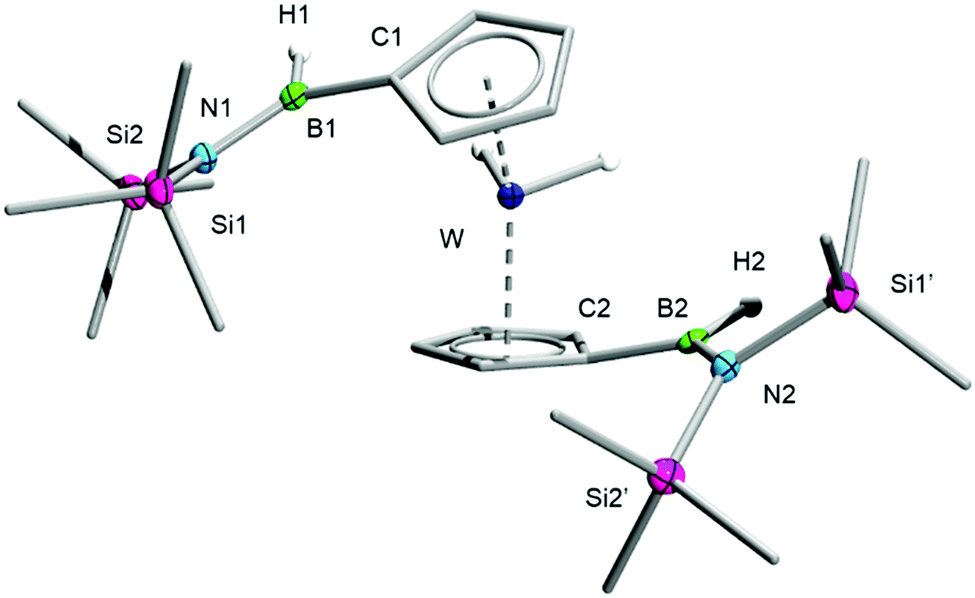
The selectivity of the twofold borylene C–H insertion in 2, which favours a single functionalization at each of the Cp ligands and does not seem to lead to double insertion of a single Cp ring, is easily rationalised on electronic grounds. Indeed, as a π-accepting substituent, the [–B(H)N(SiMe3)2] group introduced by the first insertion process plausibly acts as a deactivating agent for the other C–H bonds of the same Cp ring, as previously observed by Siebert in the electrophilic borylation of ferrocene by BBr3.18 In fact, the relative coplanarity of borane substituents of 3 and 4 with their attached Cp rings (angles between planes ca. 20° for 3 and 4) suggests that the former groups truly act as π-accepting substituents. Nevertheless, given the prodigious borylene-transfer ability of 1 proven in previous work,4 we suspected that 1 may be a sufficiently potent borylene source to effect the multiple functionalization of a single Cp ring. To this end, we reacted a 3:1 mixture of 1 and 2 in toluene. While the single and double borylene insertion into C–H bonds of 2 reported above proceeded to completion at room temperature, we found in this case that complete consumption of borylene 1 required heating the mixture to 60 °C for 96 hours. The resulting mixture after this period was found to be complex – perhaps because of the difficulties associated with using exact quantities of 1 and 2 in small scales – but we were able to confirm that C–H functionalization occurred under these conditions. NMR spectroscopy and HRMS allowed us to confirm the presence of small amounts of the bis-functionalised complex 4 in the reaction mixture, in addition to new borylene insertion products. The main species of these was the threefold-functionalised complex [(1,3-C5H3(B(H)N(SiMe3)2)2)W(C5H4B(H)N(SiMe3)2)H2] (5), which we were unfortunately unable to separate from the other substituted tungstenocene complexes. An intriguing tetraboryl tungstenocene dihydride (6) was also identified by 1H NMR spectroscopy and HRMS as a significant product (Scheme 3).
 | ||
| Scheme 3 Structures of 5 and 6 indicated by 1H NMR spectroscopy and proposed structure of the pentaboryl product 7. | ||
In order to target this unusual highly borylated compound, we reacted five equivalents of 1 with Cp2WH2 (2). After heating the mixture to 60 °C for 96 hours, we were able to purify the tetraboryl complex and to obtain it as a brown oil in low yield. Analysis of its 1H NMR spectrum allowed us to characterise the new complex as [(1,3-C5H3(B(H)N(SiMe3)2)2)WH2] (6). The isolation of 6 as an oil unfortunately precluded its structural characterisation by XRD, but its composition was confirmed by HRMS. Interestingly, we also detected the presence of trace amounts of a pentaboryl tungstenocene dihydride (7) as a minor contaminant (Scheme 3 and Fig. S13, ESI†). This suggests that under more forcing conditions, it could be possible to obtain even higher degrees of C–H insertion through the use of borylene 1, although the instability of this borylene complex at higher temperatures may hamper such a reaction.
In summary, we find that the molybdenum borylene complex 1 is a powerful source of a borylene fragment, which facilitates a TM carbene-like intermolecular insertion of a borylene moiety into the C–H bonds of tungstenocene dihydride (2). Mono-, bis-, tris- and tetrakis-functionalization of 2 is observed under mild conditions, as well as the formation of small amounts of a pentaboryl complex, despite the presumed deactivating properties of the boryl groups now bound to the Cp ligands. These results suggest that transition metal borylene complexes could be used as potent “C–H borylenation” agents for organic molecules, an exciting prospect that is being explored in our research group.
The authors thank the Deutsche Forschungsgemeinschaft (DFG) for financial support of this work. S. L. thanks the China Scholarship Council for a doctoral scholarship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) W. V. E. Doering, R. G. Buttery, R. G. Laughlin and N. Chaudhuri, J. Am. Chem. Soc., 1956, 78, 3224 CrossRef CAS; (b) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704 CrossRef CAS PubMed.
- (a) J. March, Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley-Interscience, New York, 6th edn, 2007 Search PubMed; (b) H. Zollinger, Diazo Chemistry II: Aliphatic, Inorganic and Organometallic Compounds, Wiley-VCH, New York, 1995 CrossRef; (c) Reactive Intermediate Chemistry, ed. R. A. Moss, M. S. Platz and M. Jones Jr., Wiley-Interscience, New York, 2004 Search PubMed.
- (a) M. P. Doyle, M. A. McKervey and T. Ye, Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, Wiley, New York, 1998 Search PubMed; (b) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS PubMed; (c) H. M. L. Davies and A. R. Dick, Top. Curr. Chem., 2009, 292, 303 CrossRef PubMed; (d) M. R. Fructos, M. Besora, A. A. C. Braga, M. M. Díaz-Requejo, F. Maseras and P. J. Peres, Organometallics, 2016, 36, 172 CrossRef; (e) Y. Liu, Z. Liu, Z. Luo, J. Z. Zhang, L. Liu and F. Xia, J. Phys. Chem. A, 2016, 120, 1925 CrossRef CAS PubMed.
- (a) H. Braunschweig, R. D. Dewhurst and V. H. Gessner, Chem. Soc. Rev., 2013, 42, 3197 RSC; (b) H. Braunschweig, R. D. Dewhurst and A. Schneider, Chem. Rev., 2010, 110, 3924 CrossRef CAS PubMed; (c) H. Braunschweig, C. Kollann and D. Rais, Angew. Chem., Int. Ed., 2006, 45, 5254 CrossRef CAS PubMed; (d) H. Braunschweig, C. Kollann and U. Englert, Angew. Chem., Int. Ed., 1998, 38, 3179 CrossRef; (e) H. Braunschweig, Q. Ye and K. Radacki, Chem. Commun., 2012, 48, 2701 RSC; (f) D. Vidovic, G. Pierce and S. Aldridge, Chem. Commun., 2009, 1157 RSC; (g) D. L. Coombs, S. Aldridge, A. Rossin, C. Jones and D. J. Willock, Organometallics, 2004, 23, 2911 CrossRef CAS.
- (a) H. Nöth and G. Schmid, Angew. Chem., Int. Ed. Engl., 1963, 2, 623 CrossRef; (b) R. T. Baker, J. C. Calabrese and S. A. Westcott, J. Organomet. Chem., 1995, 498, 109 CrossRef CAS; (c) K. Burgess and M. Jaspars, Organometallics, 1993, 12, 4197 CrossRef CAS; (d) R. T. Baker, J. C. Calabrese, S. A. Westcott and T. B. Marder, J. Am. Chem. Soc., 1995, 117, 8777 CrossRef CAS; (e) G. Schmid, Angew. Chem., Int. Ed. Engl., 1970, 9, 819 CrossRef CAS; (f) S. Aldridge and D. L. Coombs, Coord. Chem. Rev., 2004, 248, 535 CrossRef CAS; (g) J. T. Goettel and H. Braunschweig, Coord. Chem. Rev., 2019, 184 CrossRef CAS.
- (a) T. Ishiyama and N. Miyaura, J. Organomet. Chem., 2003, 680, 3 CrossRef CAS; (b) I. A. I. Mkhalid, D. N. Coventry, D. Albesa-Jove, A. S. Batsanov, J. A. K. Howard, R. N. Perutz and T. B. Marder, Angew. Chem., Int. Ed., 2006, 45, 489 CrossRef CAS PubMed; (c) G. A. Chotana, M. A. Rak and M. R. Smith, J. Am. Chem. Soc., 2005, 127, 10539 CrossRef CAS PubMed; (d) J. F. Hartwig, K. S. Cook, M. Hapke, C. D. Incarvito, Y. Fan, C. E. Webster and M. B. Hall, J. Am. Chem. Soc., 2005, 127, 2538 CrossRef CAS PubMed; (e) D. N. Coventry, A. S. Batsanov, A. E. Goeta, J. A. K. Howard, T. B. Marder and R. N. Perutz, Chem. Commun., 2005, 2172 RSC; (f) T. Ishiyama, Y. Nobuta, J. F. Hartwig and N. Miyaura, Chem. Commun., 2003, 2924 RSC; (g) J.-Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka and M. R. Smith, Science, 2002, 295, 305 CrossRef CAS PubMed; (h) H. Chen, Science, 2000, 287, 1995 CrossRef CAS PubMed.
- (a) C. M. Crudden and D. Edwards, Eur. J. Org. Chem., 2003, 4695 CrossRef CAS; (b) T. Ishiyama and N. Miyaura, J. Organomet. Chem., 2000, 611, 392 CrossRef CAS; (c) T. B. Marder and N. C. Norman, Top. Catal., 1998, 5, 63 CrossRef CAS; (d) I. Beletskaya and A. Pelter, Tetrahedron, 1997, 53, 4957 CrossRef CAS; (e) H. Wadepohl, Angew. Chem., Int. Ed. Engl., 1997, 36, 2441 CrossRef CAS; (f) K. Burgess and M. J. Ohlmeyer, Chem. Rev., 1991, 91, 1179 CrossRef CAS; (g) D. Männig and H. Nöth, Angew. Chem., Int. Ed. Engl., 1985, 24, 878 CrossRef.
- T. Ishiyama, N. Matsuda, M. Murata, F. Ozawa, A. Suzuki and N. Miyaura, Organometallics, 1996, 15, 713 CrossRef CAS.
- (a) H. Braunschweig, M. Burzler, K. Radacki and F. Seeler, Angew. Chem., Int. Ed., 2007, 46, 8071 CrossRef CAS PubMed; (b) J. Bauer, H. Braunschweig, A. Damme, J. O. C. Jimenez-Halla, T. Kramer, K. Radacki, R. Shang, E. Siedler and Q. Ye, J. Am. Chem. Soc., 2013, 135, 8726 CrossRef CAS PubMed.
- (a) H. Braunschweig, Q. Ye, A. Vargas, R. D. Dewhurst, K. Radacki and A. Damme, Nat. Chem., 2012, 4, 563 CrossRef CAS PubMed; (b) H. Braunschweig, M. Colling, C. Kollann, H. G. Stammler and B. Neumann, Angew. Chem., Int. Ed., 2001, 40, 2359 CrossRef; (c) H. Braunschweig, M. Forster and K. Radacki, Angew. Chem., Int. Ed., 2006, 45, 2132 CrossRef CAS PubMed; (d) H. Braunschweig, R. D. Dewhurst, F. Hupp, M. Nutz, K. Radacki, C. W. Tate, A. Vargas and Q. Ye, Nature, 2015, 522, 327 CrossRef CAS PubMed; (e) H. Braunschweig, I. Krummenacher, M.-A. Légaré, A. Matler, K. Radacki and Q. Ye, J. Am. Chem. Soc., 2017, 139, 1802 CrossRef CAS PubMed.
- (a) S. Aldridge, C. Jones, T. Gans-Wichler, A. Stasch, D. L. Kays, N. D. Coombs and D. J. Willock, Angew. Chem., Int. Ed., 2006, 45, 6118 CrossRef CAS PubMed; (b) H. Braunschweig, Q. Ye, K. Radacki and A. Damme, Angew. Chem., Int. Ed., 2012, 51, 7839 CrossRef CAS PubMed; (c) H. Braunschweig, T. Herbst, D. Rais and F. Seeler, Angew. Chem., Int. Ed., 2005, 44, 7461 CrossRef CAS PubMed.
- (a) H. Braunschweig, Q. Ye, A. Vargas, R. D. Dewhurst and K. Radacki, J. Am. Chem. Soc., 2014, 136, 9560 CrossRef CAS PubMed; (b) H. Braunschweig, Q. Ye, K. Radacki and A. Damme, Angew. Chem., Int. Ed., 2012, 51, 7839 CrossRef CAS PubMed.
- H. Braunschweig, R. D. Dewhurst, T. Herbst and K. Radacki, Angew. Chem., Int. Ed., 2008, 47, 5978 CrossRef CAS PubMed.
- (a) M.-A. Légaré, C. Pranckevicius and H. Braunschweig, Chem. Rev., 2019, 119, 8231 CrossRef PubMed; (b) M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896 CrossRef PubMed; (c) M.-A. Légaré, M. Rang, G. Bélanger-Chabot, J. Schweizer, I. Krummenacher, R. Bertermann, M. C. Holthausen and H. Braunschweig, Science, 2019, 363, 1329 CrossRef PubMed; (d) M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10282 CrossRef CAS PubMed; (e) B. Pachaly and R. West, Angew. Chem., Int. Ed. Engl., 1984, 23, 454 CrossRef; W. J. Grigsby and P. P. Power, J. Am. Chem. Soc., 1996, 118, 7981 Search PubMed; A. Meller, U. Seebold, W. Maringgele, M. Noltemeyer and G. M. Sheldrick, J. Am. Chem. Soc., 1989, 111, 8299 Search PubMed; (f) P. Bissinger, H. Braunschweig, K. Kraft and T. Kupfer, Angew. Chem., Int. Ed., 2011, 50, 4704 CrossRef CAS PubMed.
- (a) Y. Wang and G. H. Robinson, Inorg. Chem., 2011, 50, 12326 CrossRef CAS PubMed; (b) P. Bissinger, H. Braunschweig, A. Damme, R. D. Dewhurst, T. Kupfer, K. Radacki and K. Wagner, J. Am. Chem. Soc., 2011, 133, 19044 CrossRef CAS PubMed; (c) M. Arrowsmith, J. Böhnke, H. Braunschweig, H. Gao, M.-A. Légaré, V. Paprocki and J. Seufert, Chem. – Eur. J, 2017, 23, 12210 CrossRef CAS PubMed.
- (a) J. F. Hartwig and S. R. De Gala, J. Am. Chem. Soc., 1994, 116, 3661 CrossRef CAS; (b) J. F. Hartwig and X. He, Organometallics, 1996, 15, 5350 CrossRef CAS; (c) J. F. Hartwig and X. He, Angew. Chem., Int. Ed. Engl., 1996, 35, 315 CrossRef CAS; (d) H. Braunschweig and C. Kollann, Z. Naturforsch., B: J. Chem. Sci., 1999, 54, 839 CAS; (e) H. Braunschweig and T. Wagner, Chem. Ber., 1994, 127, 1613 CrossRef CAS; (f) X. Xu, Z. Sun, X. Zhang, L. Meng and X. Li, Inorg. Chim. Acta, 2019, 495, 119018 CrossRef CAS; (g) L. H. Doerrer, A. J. Graham, D. Haussinger and M. L. Green, J. Chem. Soc., Dalton Trans., 2000, 813 RSC.
- H. Braunschweig, M. Forster and K. Radacki, Angew. Chem., Int. Ed., 2006, 45, 2132 CrossRef CAS PubMed.
- (a) W. Ruf, T. Renk and W. Siebert, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1976, 31, 1028 Search PubMed; (b) W. Ruf, M. Fuller and W. Siebert, J. Organomet. Chem., 1974, 64, C45 CrossRef CAS; (c) T. Renk, W. Ruf and W. Siebert, J. Organomet. Chem., 1976, 120, 1 CrossRef CAS; (d) B. Wrackmeyer, U. Dörfler and M. Herberhold, Z. Naturforsch., B: J. Chem. Sci., 1993, 48, 121 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of new compounds and crystallographic details. CCDC 1999138 and 1999139. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc03075a |
| This journal is © The Royal Society of Chemistry 2020 |