
Direct N-formylation of nitroarenes with CO2†
Ni
Shen
 a,
Shi-Jing
Zhai
a,
Chi Wai
Cheung
*ab and
Jun-An
Ma
*ab
a,
Shi-Jing
Zhai
a,
Chi Wai
Cheung
*ab and
Jun-An
Ma
*ab
aDepartment of Chemistry, Tianjin Key Laboratory of Molecular Optoelectronic Sciences, Frontiers Science Center for Synthetic Biology (Ministry of Education), and Tianjin Collaborative Innovation Centre of Chemical Science & Engineering, Tianjin University, Tianjin 300072, P. R. of China. E-mail: zhiwei.zhang@tju.edu.cn; majun_an68@tju.edu.cn
bJoint School of National University of Singapore and Tianjin University, International Campus of Tianjin University, Binhai New City, Fuzhou 350207, P. R. China
First published on 14th July 2020
Abstract
Herein we describe a straightforward N-formylation of nitroarenes with CO2 to access N-aryl formamides exclusively in the presence of iron and hydrosilane as additives. This protocol showcases a good tolerance of a wide range of nitroarenes and nitroheteroarenes.
Carbon dioxide (CO2) constitutes an ideal C1 source in organic synthesis owing to its abundance and stability.1 In particular, CO2 serves as the best-suited substitute of more reactive C1 building blocks, such as carbon monoxide (CO), methyl iodide, and phosgene.2 Despite its high activation barrier and thermodynamic stability,2 CO2 has been widely exploited for decades to access a diverse array of functionalized compounds via carboxylation,3 carbonylation,4 or C–H bond construction.5 In this context, the incorporation of amines and CO2 towards synthesis of value-added nitrogen-containing compounds,6 especially N-formylation of amines with CO2,7–13 is among the most important chemical transformations due to the versatility of nitrogen-based compounds in academia and industry. Taking the advantage of commercially available hydrosilanes as activating and hydrogenating agents along with various catalysts, such as organic superbases,7 carbenes,8 ionic liquids and organic salts,9 inorganic salts,10 polar aprotic solvents,11 diazophospholene,12 and B(C6F5)3,13 a variety of formamides can be accessed based on amines and CO2 (Scheme 1a). Additionally, the Ding14 and Bernskoetter15 groups described the seminal Ru- and Fe-catalyzed N-formylation of alkylamines based on the CO2/H2 system, respectively (Scheme 1b). On the other hand, nitroarenes prove to be reliable aminating agents for direct synthesis of amines16 and amides17 without preforming the more reactive anilines via hydrogenation. Recently, Beller,17a Driver,17b Hu,17c Mankad,17f Wu,17d,e as well as our group17g disclosed the aminocarbonylation reactions to access aryl amides, a class of important compounds in chemical, pharmaceutical, and agrochemical industries, based on nitroarenes and CO or its surrogates. To our knowledge, the merger of CO2 and nitroarenes for straightforward amide synthesis remains unexplored. We envisioned that the use of suitable and compatible reductants and activating agents could induce the integration of both CO2 and nitroarenes for amide formation. Herein, we unveil the direct N-formylation of nitroarenes with CO2 in the presence of iron powder and hydrosilane additives (Scheme 1c). This alternative and complementary method would open a conceptually novel avenue for more step-economic and expedient access to formamides without the need of conventional anilines.
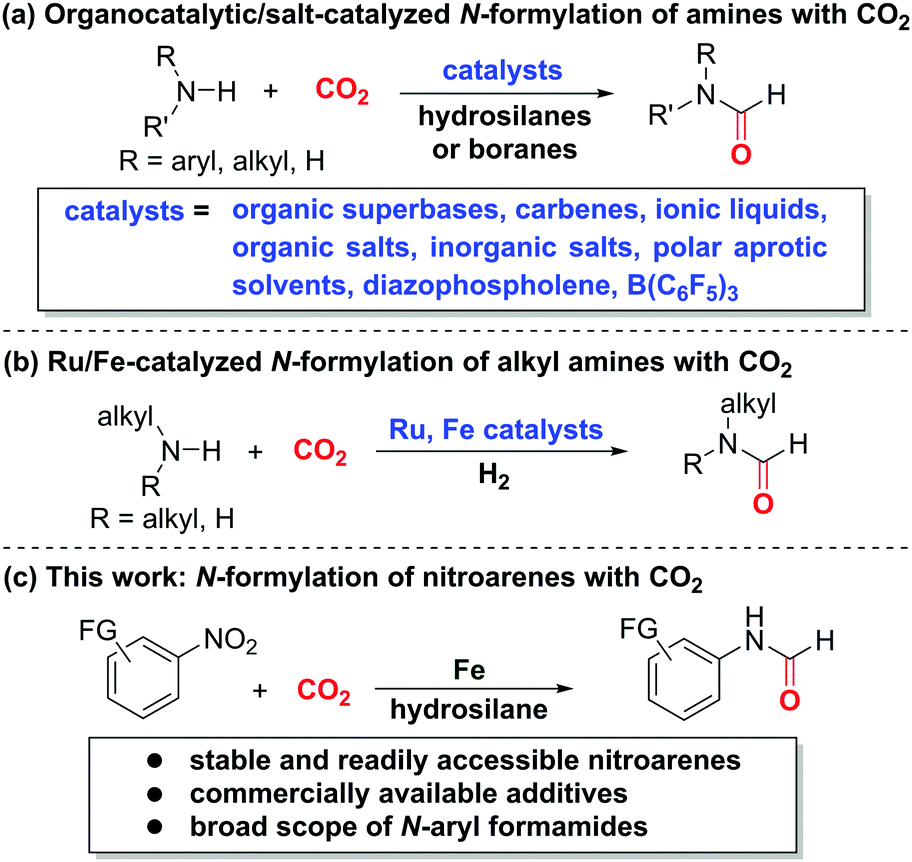 | ||
| Scheme 1 Developments of N-formylation based on CO2. | ||
We commenced the study on N-formylation of CO2 using 4-nitroanisole (1a) as the model substrate (Table 1). In the presence of 3 equivalents of Zn powder as reductant and 3 equivalents of phenylsilane as additive, 1a reacted with CO2 under balloon pressure in dimethylformamide (DMF) solvent at 110 °C to give N-4-methoxyphenyl formamide 2a exclusively in 49% yield (entry 1). In the absence of Zn or hydrosilane, no formylation took place, indicating that both metal reductant and hydrosilane are essential for reaction (entries 2 and 3). When triethoxysilane was used in place of phenylsilane, formylation underwent at 135 °C to offer 2a in 56% yield (entry 4). Further, when Fe powder was employed instead of Zn, 2a was also obtained in 51% yield (entry 5). Noteworthily, the yield could be enhanced to 63% when 10 mol% of KI was added as additive in the Fe-mediated reaction (entry 6). Subsequently, 1,1,3,3-tetramethyldisiloxane (TMDSO) was found to be the superior additive, giving 2a in 75% yield (entries 7–9). Intriguingly, the use of Fe nano powder allowed the reduction of loadings of both Fe and TMDSO to 2 equivalents in association with the yield enhancement to 83% yield (entries 10–12). Such promoting effect would be attributed to the more efficient reduction process provided by the more soluble and finely powdered Fe. In the presence of other polar aprotic solvents such as dimethylacetamide (DMA) and N-methylpyrrolidine (NMP), 2a was formed in similar yields without other side products (entries 13 and 14). Furthermore, no formamide was formed when CO2 was omitted (entry 15). These control experiments suggested that the carbonyl group of 2a originates from CO2 rather than solvent.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Metal (equiv.) | Hydrosilane (equiv.) | Additive (mol%) | Solvent | Yieldb (%) |
| a General procedure: 1a (0.5 mmol), CO2 (balloon), metal powder (1–1.5 mmol), hydrosilane (1–1.5 mmol), additive (10 mol%), solvent (1.5 mL), 135 °C, 28 h. b Isolated yield. c Reaction conducted at 110 °C. d Fe nano powder used. e Argon atmosphere without CO2. | |||||
| 1c | Zn (3) | PhSiH3 (3) | None | DMF | 49 |
| 2c | None | PhSiH3 (3) | None | DMF | 0 |
| 3c | Zn (3) | None | None | DMF | 0 |
| 4 | Zn (3) | (EtO)3SiH (3) | None | DMF | 56 |
| 5 | Fe (3) | (EtO)3SiH (3) | None | DMF | 51 |
| 6 | Fe (3) | (EtO)3SiH (3) | KI (10) | DMF | 63 |
| 7 | Fe (3) | Me(EtO)2SiH (3) | KI (10) | DMF | 25 |
| 8 | Fe (3) | PMHS (3) | KI (10) | DMF | 32 |
| 9 | Fe (3) | TMDSO (3) | KI (10) | DMF | 75 |
| 10d | Fe (3) | TMDSO (2) | KI (10) | DMF | 83 |
| 11d | Fe (2) | TMDSO (2) | KI (10) | DMF | 83 |
| 12d | Fe (1) | TMDSO (2) | KI (10) | DMF | 35 |
| 13d | Fe (2) | TMDSO (2) | KI (10) | DMA | 83 |
| 14d | Fe (2) | TMDSO (2) | KI (10) | NMP | 75 |
| 15d,e | Fe (2) | TMDSO (2) | KI (10) | DMF | 0 |
The optimized protocol (Table 1, entry 11) proved to be general in N-formylation of nitroarenes (Scheme 2). Electron-rich (1a–1f), electron-neutral (1g), and electron-deficient (1h–1m) nitroarenes all reacted with CO2 to form the corresponding N-aryl formamides in moderate to high yields. Additionally, both mono- (1a-2i, 1l) and di-substituted (1j, 1k, 1m–1o) nitroarenes could be incorporated into the formamide products 2a–2o. The sterically encumbered 1-nitro-2,4-dimethylbenzene (1o) was also suitable reaction partner. Notably, a wide range of nitro-substituted heterocycles could be utilized for formylation, including benzodioxole (1p), pyrrole (1q), pyridine (1r), N-alkylated indole (1s, 1t), benzofuran (1u), benzothiophene (1v), and benzoxazole (1w), delivering the corresponding N-heteroaryl formamides 2p–2w in 43–85% yields. Noteworthily, the employment of 3 equivalents of Fe metal (Table 1, entry 10) could improve remarkably the formylation of electron-rich nitro(hetero)arenes 1c, 1o and 1s, affording the corresponding formamides 2c, 2o and 2s in 80%, 60% and 63% yields, respectively. On the contrary, the reactions of electron-deficient nitroarenes with the additional equivalent of Fe led to substantial diminishment of yields. Gratifyingly, by adding FeCl2 (2 equiv.) and slightly increasing the loadings of TMDSO and KI, electron-deficient nitroarenes such as 1j–1m could react more efficiently to afford the formamides in higher yields (48–68%). This protocol was also amenable to multigram synthesis of 2a in synthetically useful yield.
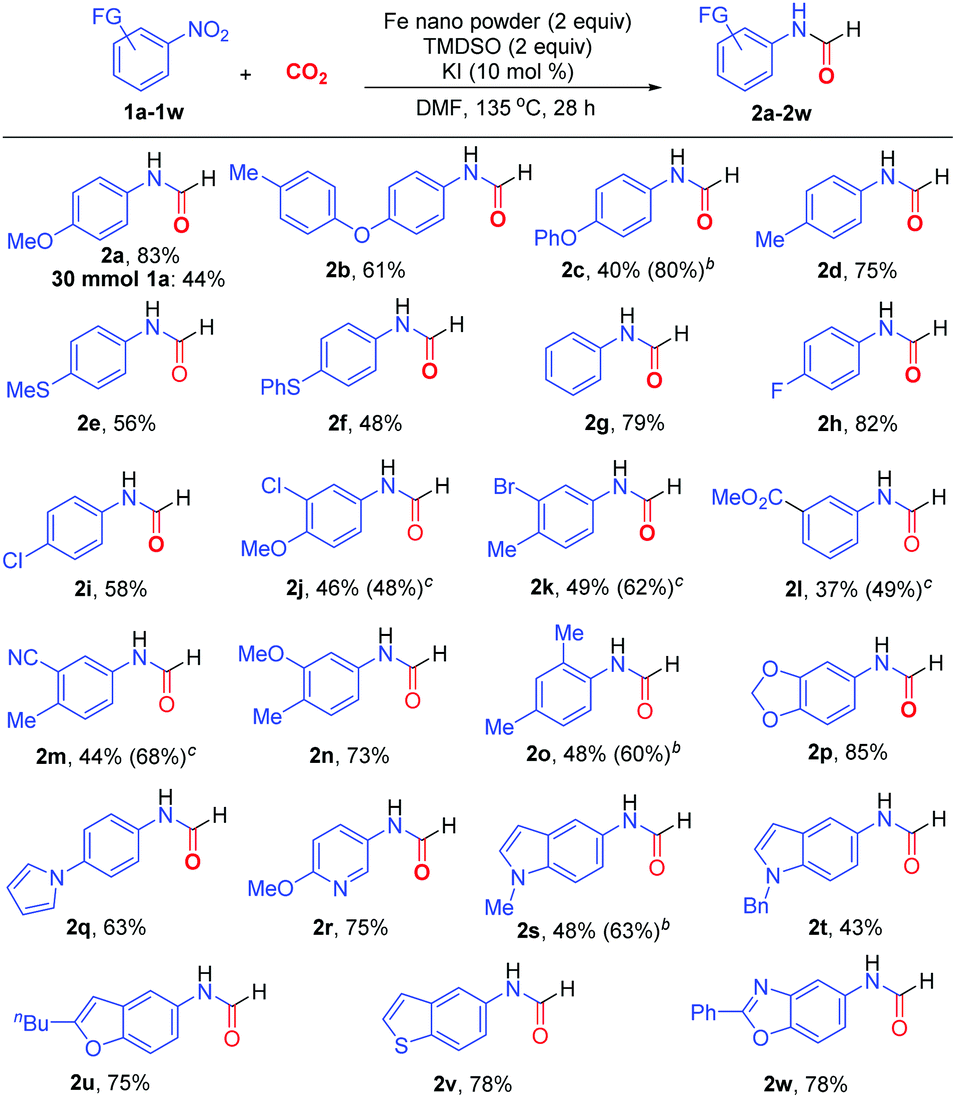 | ||
Scheme 2 Substrate scope of nitroarenes in N-formylation.a a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction conditions: nitroarene (0.5 mmol), CO2 (balloon), nano Fe (1 mmol), TMDSO (1 mmol), KI (0.05 mmol), DMF (1.5 ml), 135 °C, 28 h. Isolated yields were shown. b3 equiv. of Fe. cModified conditions: nitroarene (1 equiv.), CO2 (balloon), nano Fe (2 equiv.), FeCl2 (2 equiv.), TMDSO (3 mmol), KI (30 mol%), DMF (3 ml), 135 °C, 28 h. Reaction conditions: nitroarene (0.5 mmol), CO2 (balloon), nano Fe (1 mmol), TMDSO (1 mmol), KI (0.05 mmol), DMF (1.5 ml), 135 °C, 28 h. Isolated yields were shown. b3 equiv. of Fe. cModified conditions: nitroarene (1 equiv.), CO2 (balloon), nano Fe (2 equiv.), FeCl2 (2 equiv.), TMDSO (3 mmol), KI (30 mol%), DMF (3 ml), 135 °C, 28 h. | ||
In the course of reaction, nitroarene can be transformed to nitrosoarene, N-aryl hydroxylamine, azoxyarene, azoarene, 1,2-diaryl hydrazine, and aniline under reductive conditions.18 Thus, the reactions of these nitrogen-based species under otherwise identical conditions were examined to probe their roles in formylation (Scheme 3a). Nitrosobenzene (3a), N-phenyl hydroxylamine (3b), azoxybenzene (3c) and azobenzene (3d) all reacted to give formamide 2g in 49–64% yields. Particularly, diphenyl hydrazine (3e) reacted most efficiently to afford 2g in 70% yield. Presumably, nitrobenzene is sequentially reduced to 3a, 3b, 3c, 3d, and finally 3e,18 which acts as the ultimate intermediate to participate in the formylation reaction. Aniline 3f also reacted to give 2g in 44% yield, suggesting that it is likely the minor intermediate to induce the formylation. Furthermore, when Fe(0) was omitted, the addition of Fe(II) salt, which is a viable oxidized Fe species, could enable the formylation of nitroarene 1a in the presence of excess TMDSO to give formamide 2a in 45% yield. We surmised that hydrosilane can also act as a reductant and a hydrogen source for the reduction of nitroarene via the action of Fe-hydride species,16a,b apart from its conventional application as CO2-activating agent.6h
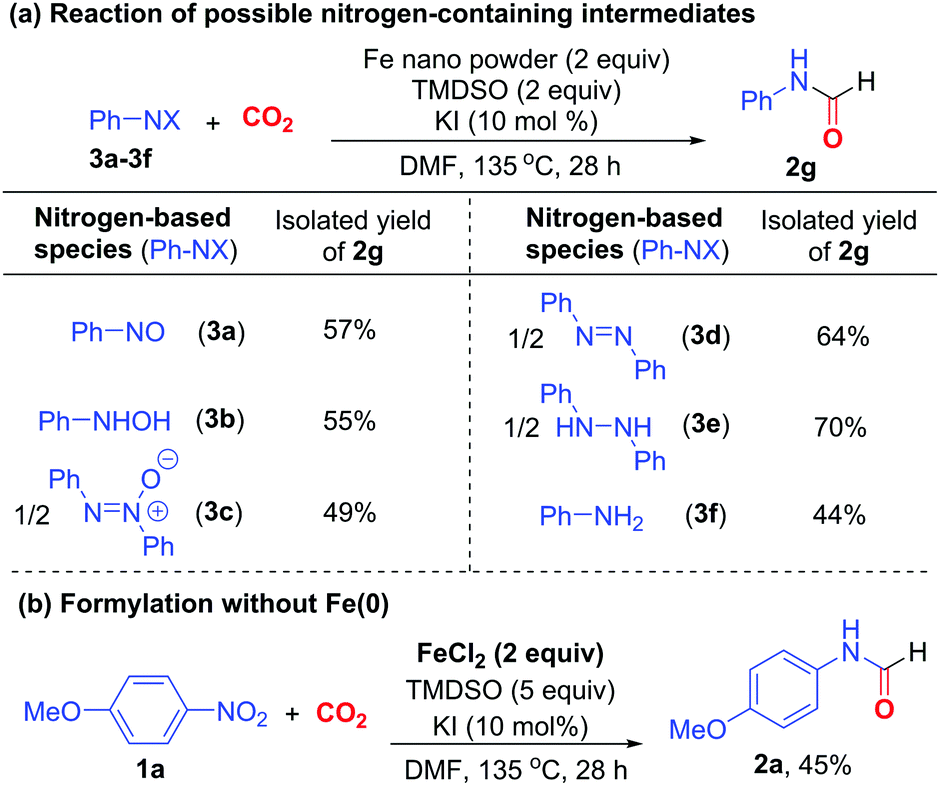 | ||
| Scheme 3 Control experiments for N-formylation. | ||
Based on the experimental results, we proposed a plausible mechanism of the formylation protocol (Scheme 4). Nitroarene 1 is sequentially reduced to 1,2-diaryl hydrazine via the intermediacy of nitrosoarene, N-aryl hydroxylamine, azoxyarene, and/or azoarene18 (pathway i). 1,2-Diaryl hydrazine then undergoes facile homolysis under heating to give aminyl radicals,17g which are then reduced to amide ion ArNH− (pathways ii and iii). Meanwhile, nitroarene can be reduced to amide dianion ArN2− followed by ArNH−, whereas Fe(0) is oxidized to Fe3O419 (pathway iv and v). A small amount of aniline can also be generated (pathway vi). In the reduction processes, Fe(0) is likely the major reducing agent, while TMDSO likely acts as both mild reducing agent and hydrogenating agent. Owing to the strong nucleophilicity of ArNH− and ArN2−, they likely activate both CO2 and TMDSO via the transition state TS-1, in which the carbamate ion formed further activates TMDSO to trigger the hydride attack to another CO2 (pathway vii). Such transition state is regarded as the lowest energy pathway for CO2 activation.6hViaTS-1, formoxysilane is formed with the concomitant regeneration of amide species (pathway viii). We proposed that iodide ion behaves as a strong nucleophile in polar aprotic solvent DMF, thereby facilitating the deformylation of formoxysilane to form a more reactive formyl iodide in conjunction with silanolate ion (pathway ix). Finally, ArNH− reacts with both formyl iodide and formoxysilane to deliver the N-aryl formamide 2 (pathways x and xi). In the same vein, ArNH2− undergoes nucleophilic substitutions with formyl iodide and formoxysilane to give amidate ion, which furnishes formamide 2 upon acidic workup (pathways xii and xiii). On the other hand, the N-formylation based on aniline may proceed but tends to be a minor pathway due to the attenuated nucleophilicity of aniline. The detailed reaction mechanism is subjected to a dedicated study in the future.
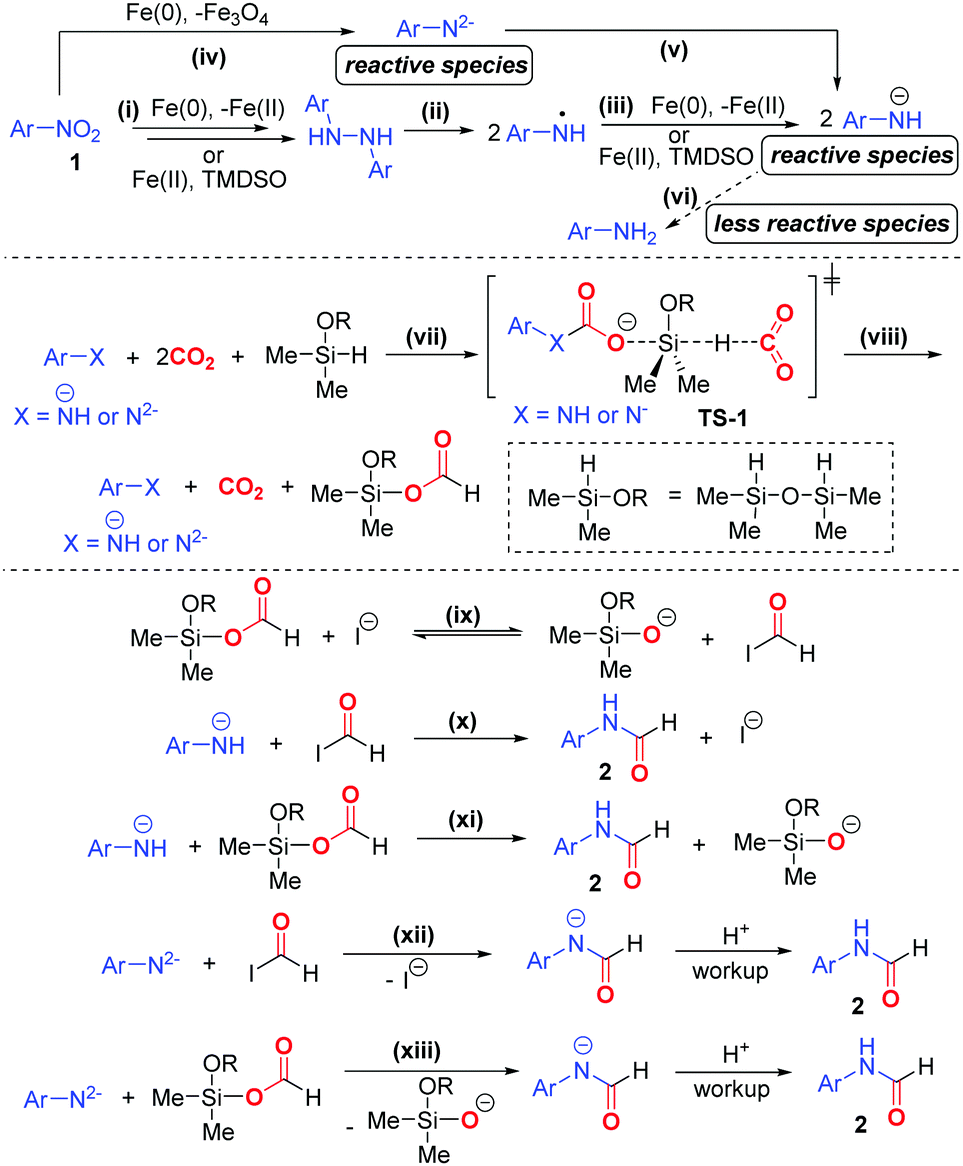 | ||
| Scheme 4 Proposed mechanism of N-formylation. | ||
Under the standard conditions, electron-rich nitro(hetero)arenes tended to react more efficiently to form higher product yields (∼50–85%) than electron-deficient and sensitive group-bearing nitroarenes (∼40–60%, Scheme 2). We rationalized that the nitrogen-based intermediates derived from the latter are more unstable, especially in the reductive reaction conditions and in the presence of basic by-products (e.g. iron oxides, silanolate ions), and likely undergo over-reduction and decomposition at varying degrees in the course of reactions. Additionally, an additional equivalent of Fe(0) was found advantageous in the reactions of electron-rich nitro(hetero)arenes but detrimental for electron-deficient ones. We speculated that the contribution of Fe(0)- and TMDSO-based reduction steps would vary with respect to the electronic effect of nitroarenes. The reduction of electron-rich nitro(hetero)arenes would mainly rely more on highly reducing Fe(0) to deliver more amide ions. Conversely, the excess Fe(0) would over-reduce the electron-deficient nitroarenes to less reactive anilines or other side products, but additional milder reduction given by Fe(II)/TMDSO system would in turn be more productive for formation of amide ions.
In conclusion, we have developed an alternative strategy to access formamides via direct N-formylation of nitroarenes with CO2. By using commercially available Fe powder and 1,1,3,3-tetramethyldisiloxane as additives, a variety of N-aryl and N-heteroaryl formamides are synthesized. Mechanistic study suggests that both Fe metal and hydrosilane can reduce nitroarenes to anionic nitrogen-based intermediates, which then activate CO2 and hydrosilane for subsequent formylation.
We acknowledged the National Key Research and Development Program of China (No. 2019YFA0905100), National Natural Science Foundation of China (No. 21772142, 21971186, and 21961142015) and Tianjin University (start-up grants) for financial support. We thank Ms Yi-lin Yang (TJU), Mr Lei Li (TJU) and Mr Shao-Peng Wang (TJU) for assistance in scope study. We especially thank the anonymous Reviewer for valuable suggestions to improve the reaction outcomes.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) E. A. Quadrelli, G. Centi, J.-L. Duplan and S. Perathoner, ChemSusChem, 2011, 4, 1194 CrossRef CAS PubMed; (b) W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703 RSC; (c) J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296 CrossRef CAS PubMed; (d) M. D. Burkart, N. Hazari, C. L. Tway and E. L. Zeitler, ACS Catal., 2019, 9, 7937 CrossRef CAS.
- (a) M. Aresta, Carbon Dioxide as Chemical Feedstock, Wiley-VCH, Weinheim, 2010 CrossRef; (b) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- (a) A. Tortajada, F. Julia-Hernandez, M. Borjesson, T. Moragas and R. Martin, Angew. Chem., Int. Ed., 2018, 57, 15948 CrossRef CAS PubMed; (b) S.-S. Yan, Q. Fu, L.-L. Liao, G.-Q. Sun, J.-H. Ye, L. Gong, Y.-Z. Bo-Xue and D.-G. Yu, Coord. Chem. Rev., 2018, 374, 439 CrossRef CAS; (c) Y.-G. Chen, X.-T. Xu, K. Zhang, Y.-Q. Li, L.-P. Zhang, P. Fang and T.-S. Mei, Synthesis, 2018, 35 Search PubMed; (d) J. Hong, M. Li, J. Zhang, B. Sun and F. Mo, ChemSusChem, 2019, 12, 6 CrossRef CAS PubMed.
- (a) K. Dong and X.-F. Wu, Angew. Chem., Int. Ed., 2017, 56, 5399 CrossRef CAS PubMed; (b) L. Wang, W. Sun and C. Liu, Chin. J. Chem., 2018, 36, 353 CrossRef CAS; (c) L. Song, Y.-X. Jiang, Z. Zhang, Y.-Y. Gui, X.-Y. Zhou and D.-G. Yu, Chem. Commun., 2020 10.1039/D0CC00547A.
- (a) A. Tlili, X. Frogneux, E. Blondiaux and T. Cantat, Angew. Chem., Int. Ed., 2014, 53, 2543 CrossRef CAS PubMed; (b) Y. Li, X. Cui, K. Dong, K. Junge and M. Beller, ACS Catal., 2017, 7, 1077 CrossRef CAS; (c) F. N. Al-Rowaili, A. Jamal, M. S. Ba Shammakh and A. Rana, ACS Sustainable Chem. Eng., 2018, 6, 15895 CrossRef CAS; (d) X.-F. Liu, X.-Y. Li and L.-N. He, Eur. J. Org. Chem., 2019, 2437 CrossRef CAS; (e) I. I. Alkhatiba, C. Garlisia, M. Pagliarob, K. Al-Alia and G. Palmisano, Catal. Today, 2020, 340, 209 CrossRef.
- (a) A. Tlili, E. Blondiaux, X. Frogneux and T. Cantat, Green Chem., 2015, 17, 157 RSC; (b) B. Yu and L.-N. He, ChemSusChem, 2015, 8, 52 CrossRef CAS PubMed; (c) J. Rintjema and A. Kleij, Synthesis, 2016, 3863 CAS; (d) N. A. Tappe, R. M. Reich, V. D’Elia and F. E. Kghn,, Dalton Trans., 2018, 47, 13281 RSC; (e) X.-F. Liu, X.-Y. Li, C. Qiao and L.-N. He, Synlett, 2018, 548 CAS; (f) J. R. Cabrero-Antonino, R. Adam and M. Beller, Angew. Chem., Int. Ed., 2019, 58, 12820 CrossRef CAS PubMed; (g) J.-Y. Li, Q.-W. Song, K. Zhang, P. Liu, J.-Y. Li, Q.-W. Song, K. Zhang and P. Liu, Molecules, 2019, 24, 182 CrossRef PubMed; (h) M. Hulla and P. J. Dyson, Angew. Chem., 2020, 132, 1014 CrossRef.
- (a) C. D. N. Gomes, O. Jacquet, C. Villiers, P. Thuery, M. Ephritikhine and T. Cantat, Angew. Chem., Int. Ed., 2012, 51, 187 CrossRef PubMed; (b) R. L. Nicholls, J. A. McManus, C. M. Rayner, J. A. Morales-Serna, A. J. P. White and B. N. Nguyen, ACS Catal., 2018, 8, 3678 CrossRef CAS; (c) G. Li, J. Chen, D.-Y. Zhu, Y. Chen and J.-B. Xia, Adv. Synth. Catal., 2018, 360, 2364 CrossRef CAS.
- (a) O. Jacquet, C. D. N. Gomes, M. Ephritikhine and T. Cantat, J. Am. Chem. Soc., 2012, 134, 2934 CrossRef CAS PubMed; (b) S. Das, F. D. Bobbink, S. Bulut, M. Soudania and P. J. Dyson, Chem. Commun., 2016, 52, 2497 RSC.
- (a) H. Zhou, G.-X. Wang, W.-Z. Zhang and X.-B. Lu, ACS Catal., 2015, 5, 6773 CrossRef CAS; (b) L. Hao, Y. Zhao, B. Yu, Z. Yang, H. Zhang, B. Han, X. Gao and Z. Liu, ACS Catal., 2015, 5, 4989 CrossRef CAS; (c) M. Hulla, F. D. Bobbink, S. Das and P. J. Dyson, ChemCatChem, 2016, 8, 3338 CrossRef CAS; (d) X.-F. Liu, X.-Y. Li, C. Qiao, H.-C. Fu and L.-N. He, Angew. Chem., Int. Ed., 2017, 56, 7425 CrossRef CAS PubMed; (e) C. Xie, J. Song, H. Wu, B. Zhou, C. Wu and B. Han, ACS Sustainable Chem. Eng., 2017, 5, 7086 CrossRef CAS; (f) Y. Hu, J. Song, C. Xie, H. Wu, Z. Wang, T. Jiang, L. Wu, Y. Wang and B. Han, ACS Sustainable Chem. Eng., 2018, 6, 11228 CrossRef CAS; (g) M. Hulla, D. Ortiz, S. Katsyuba, D. Vasilyev and P. J. Dyson, Chem. – Eur. J., 2019, 25, 11074 CrossRef CAS PubMed; (h) W. Zhao, X. Chi, H. Li, J. He, J. Long, Y. Xu and S. Yang, Green Chem., 2019, 21, 567 RSC.
- (a) K. Motokura, M. Naijo, S. Yamaguchi, A. Miyaji and T. Baba, Chem. Lett., 2015, 44, 1217 CrossRef CAS; (b) D. B. Nale and B. M. Bhanage, Synlett, 2016, 1413 CAS; (c) C. Fang, C. Lu, M. Liu, Y. Zhu, Y. Fu and B.-L. Lin, ACS Catal., 2016, 6, 7876 CrossRef CAS; (d) M.-Y. Wang, N. Wang, X.-F. Liu, C. Qiao and L.-N. He, Green Chem., 2018, 20, 1564 RSC.
- (a) J. Song, B. Zhou, H. Liu, C. Xie, Q. Meng, Z. Zhang and B. Han, Green Chem., 2016, 18, 3956 RSC; (b) H. Lv, Q. Xing, C. Yue, Z. Lei and F. Li, Chem. Commun., 2016, 52, 6545 RSC; (c) T.-X. Zhao, G.-W. Zhai, J. Liang, P. Li, X.-B. Hu and Y.-T. Wu, Chem. Commun., 2017, 53, 8046 RSC.
- C. C. Chong and R. Kinjo, Angew. Chem., Int. Ed., 2015, 54, 12116 CrossRef CAS PubMed.
- V. B. Saptal, G. Juneja and B. M. Bhanage, New J. Chem., 2018, 42, 15847 RSC.
- L. Zhang, Z. Han, X. Zhao, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2015, 54, 6186 CrossRef CAS PubMed.
- U. Jayarathne, N. Hazari and W. H. Bernskoetter, ACS Catal., 2018, 8, 1338 CrossRef CAS.
- For examples, see: (a) J. Gui, C.-M. Pan, Y. Jin, T. Qin, J. C. Lo, B. J. Lee, S. H. Spergel, M. E. Mertzman, W. J. Pitts, T. E. La Cruz, M. A. Schmidt, N. Darvatkar, S. R. Natarajan and P. S. Baran, Science, 2015, 348, 886 CrossRef CAS PubMed; (b) K. Zhu, M. P. Shaver and S. P. Thomas, Chem. Sci., 2016, 7, 3031 RSC; (c) C. W. Cheung and X. Hu, Nat. Commun., 2016, 7, 12494 CrossRef PubMed; (d) J. Xiao, Y. He, F. Ye and S. Zhu, Chem, 2018, 4, 1645 CrossRef CAS; (e) T. V. Nykaza, J. C. Cooper, G. Li, N. Mahieu, A. Ramirez, M. R. Luzung and A. T. Radosevich, J. Am. Chem. Soc., 2018, 140, 15200 CrossRef CAS PubMed; (f) S. Suárez-Pantiga, R. Hernández-Ruiz, C. Virumbrales, M. R. Pedrosa and R. Sanz, Angew. Chem., Int. Ed., 2019, 58, 2129 CrossRef PubMed; (g) L. Wang, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2019, 58, 5417 CrossRef CAS PubMed; (h) M. Rauser, R. Eckert, M. Gerbershagen and M. Niggemann, Angew. Chem., Int. Ed., 2019, 58, 6713 CrossRef CAS PubMed.
- For examples, see: (a) X. Fang, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 14089 CrossRef CAS PubMed; (b) F. Zhou, D.-S. Wang, X. Guan and T. G. Driver, Angew. Chem., Int. Ed., 2017, 56, 4530 CrossRef CAS PubMed; (c) C. W. Cheung, M. L. Ploeger and X. Hu, Chem. Sci., 2018, 9, 655 RSC; (d) J.-B. Peng, H.-Q. Geng, D. Li, X. Qi, J. Ying and X.-F. Wu, Org. Lett., 2018, 20, 4988 CrossRef CAS PubMed; (e) J.-B. Peng, D. Li, H.-Q. Geng and X.-F. Wu, Org. Lett., 2019, 21, 4878 CrossRef CAS PubMed; (f) S. Zhao and N. P. Mankad, Org. Lett., 2019, 21, 10106 CrossRef CAS PubMed; (g) N. Shen, C. W. Cheung and J.-A. Ma, Chem. Commun., 2019, 55, 13709 RSC.
- (a) H.-U. Blaser, Science, 2006, 313, 312 CrossRef CAS PubMed; (b) H.-U. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210 CrossRef CAS.
- R. Dey, N. Mukherjee, S. Ahammed and B. C. Ranu, Chem. Commun., 2012, 48, 7982 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and spectral data. See DOI: 10.1039/d0cc03098h |
| This journal is © The Royal Society of Chemistry 2020 |