
Peptidomimetic toolbox for drug discovery
Elena
Lenci
 and
Andrea
Trabocchi
*
and
Andrea
Trabocchi
*
Department of Chemistry “Ugo Schiff”, University of Florence, Via della Lastruccia 13, 50019 Sesto Fiorentino, Florence, Italy. E-mail: andrea.trabocchi@unifi.it
First published on 7th April 2020
Abstract
The art of transforming peptides into drug leads is still a dynamic and fertile field in medicinal chemistry and drug discovery. Peptidomimetics can respond to peptide limitations by displaying higher metabolic stability, good bioavailability and enhanced receptor affinity and selectivity. Various synthetic strategies have been developed over the years in order to modulate the conformational flexibility and the peptide character of peptidomimetic compounds. This tutorial review aims to outline useful tools towards peptidomimetic design, spanning from local modifications, global restrictions and the use of secondary structure mimetics. Selected successful examples of each approach are presented to document the relevance of peptidomimetics in drug discovery.
 Elena Lenci | Elena Lenci received the Doctor Europaeus PhD in Chemical Sciences in 2017 from the University of Florence, after spending a research period at the University of Oxford. She is currently a postdoctoral researcher at the Department of Chemistry, University of Florence, working with Prof. Trabocchi to develop novel amino acid- and sugar-derived scaffolds for medicinal chemistry. In 2015 she was awarded the Reaxys – Italian Chemical Society Young Researcher Award, and in 2017 she received the International Award for Young Chemists “NATCHEMDRUGS”. She recently edited a book on Small Molecule Drug Discovery (Elsevier, 2019). |
 Andrea Trabocchi | Andrea Trabocchi is Associate Professor of Organic Chemistry at the University of Florence. He received training on peptide chemistry at Imperial College London, and obtained the PhD in Chemical Sciences from the University of Florence under the supervision of Prof. Guarna. After post-doctoral research, he was appointed assistant professor (2007), and associate professor (2017) in the same University. He authored a book on Peptidomimetics (Wiley, 2014) and edited two books on Diversity-Oriented Synthesis (Wiley, 2013) and Small Molecule Drug Discovery (Elsevier, 2019). Current research interests include peptidomimetic chemistry, diversity-oriented synthesis, chemoinformatics and medicinal chemistry of cancer and Alzheimer's disease. |
Key learning points1. Advantages of peptidomimetics over peptides.2. Definition and classification of peptidomimetics. 3. Synthetic strategies and molecules for local modifications, secondary structure mimetics, global restrictions. 4. Applications of peptidomimetics in drug discovery. |
1. Introduction
Peptides influence many important physiological mechanisms and control nearly all vital functions in humans, including immune defence, digestion, metabolism, reproduction, respiration and sensitivity to pain.1 Peptides show good efficacy and tolerability, as well as favourable profile in the development stages, including the knowledge of a predictable metabolism, short time to market and low attrition rates. Thanks to advances in structural optimization, formulation, and production, an increasing number of peptides are entering clinical trials and being approved as drugs.1,2 However, these molecules still suffer of several drawbacks, including (Fig. 1, left):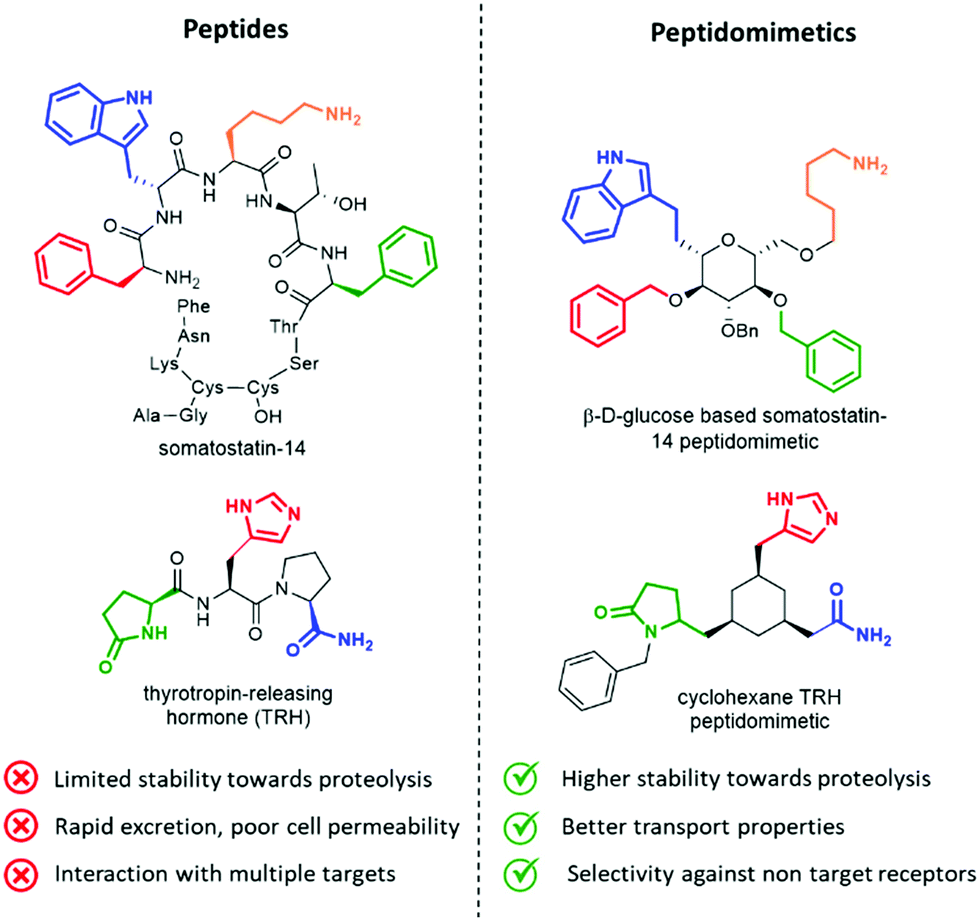 | ||
| Fig. 1 Advantages of peptidomimetics over peptides. | ||
• limited stability towards proteolysis, resulting in a half time in the order of minutes in the gastrointestinal tract and in serum;
• poor absorption and transport properties, due to relatively high molecular mass and lack of specific transport systems, often resulting in a rapid excretion through the liver and kidneys;
• interaction with multiple targets, resulting in poor selectivity and undesired side effects, due to the intrinsic flexibility addressed by N–Cα and Cα–CO rotational bonds of each amino acid;
• interaction with binding sites of antibodies, thus inducing antigenicity and an immune response in a competent host in an unpredictable way.3
Peptidomimetics have been conceived to address these limitations, as they are developed to display metabolic stability, good bioavailability, high receptor affinity and selectivity. The structure of the lead peptide is optimized by introducing functional modifications able to address the intrinsic disadvantages of peptides, while maintaining the structural features responsible for the biological activity (Fig. 1, right).4
An elegant example of a peptidomimetic compound is given by the somatostatin analogue developed by Hirschmann and Nicolaou, in which the β-D-glucose scaffold presents the four side chains responsible for the interaction in the same orientation as found in the β-turn structure of the parent somatostatin peptide (Fig. 1, top).5 Similarly, the thyrotropin-releasing hormone (TRH) mimetic developed by Olson contains a cyclohexane scaffold that replaces the peptide backbone, while presenting the three pharmacophoric groups in the correct orientation (Fig. 1, down).6 Although the peptidomimetic concept was introduced in the Eighties,7 the art of transforming peptides in new drugs is still a powerful and well-established tool in medicinal chemistry and drug discovery. In fact, the replacement of the amide bond with isosteres can reduce the rate of degradation by peptidases, and the overall pharmacological properties of the new compound can be optimized, resulting in better transport properties and increased stability against secretion.8 Besides, conformationally restricted structures that are able to mimic as much as possible the receptor-bound peptide arrangement can minimize the binding to non-target receptors, thus increasing the selectivity of the therapeutic agent.
Several peptidomimetic compounds have been already approved as drugs or are in late stage clinical trials, with application in the treatment of different pathologies, spanning from infectious diseases to cancer and rare diseases (Fig. 2).
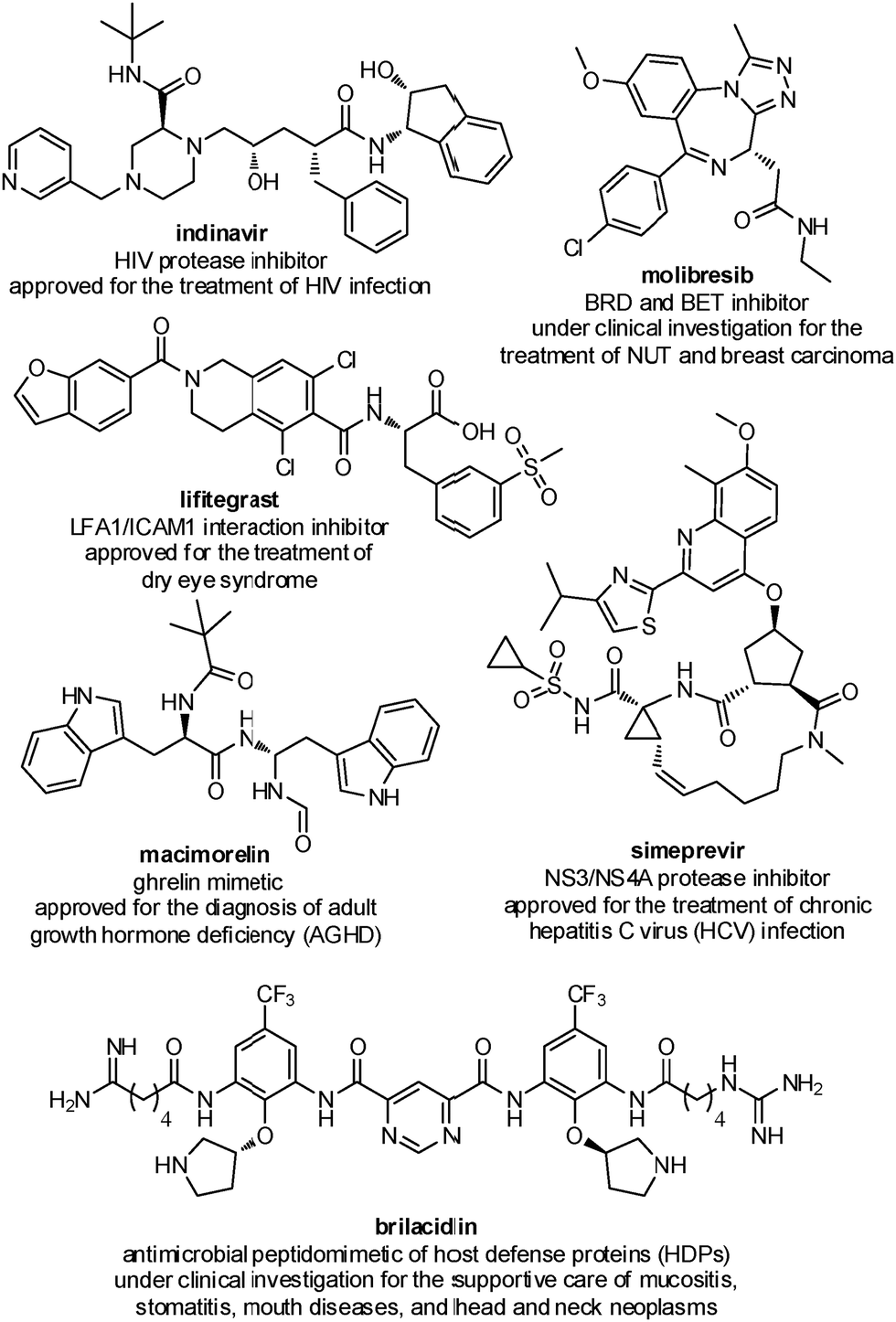 | ||
| Fig. 2 Representative examples of peptidomimetics compounds in late stage clinical trials or already approved as drugs. | ||
The field of peptidomimetics has changed largely over the last three decades. Starting from local modifications of bioactive peptides, peptidomimetics are now being developed using ad hoc rational design, with the purpose of positioning and projecting pharmacophoric elements and interacting groups in the right position. The synthetic strategies behind their development have changed significantly, too, spanning from the simple replacement of the peptide backbone, to extensive modifications of the entire structure. This review summarizes the main chemical approaches for peptidomimetic design, organized by the extent of modifications on the parent peptide, particularly delivering useful synthetic tools, with some reference to successful examples for each approach herein considered. It starts with describing local modifications, focusing on peptide bond surrogates and side chain isosteres, tethered α-amino acids and dipeptide isosteres. Then, principal secondary structure mimetics of helix, β-sheet, β-turn and β-hairpin are covered. Finally, global restriction approaches are discussed, such as the development of stapled, cross-linked and macrocyclic peptides.
2. Definition and classification
Peptidomimetics have been defined as ‘compounds whose essential elements (pharmacophore) mimic a natural peptide or protein in 3D space and which retain the ability to interact with the biological target and produce the same biological effect’.8Together with the progress made over the years, the classification of peptidomimetic compounds has evolved, from a historical arrangement into three types based on structural and functional characteristics, to a broader classification that includes new approaches based on high molecular weight foldamers and peptoids. The old classification categorizes peptidomimetics on the basis of their similarity with the native substrate:9
• Type I mimetics, or structural mimetics, show a strict analogy with the native substrate, as they carry all the functionalities in the same spatial orientation.
• Type II mimetics, or functional mimetics, do not show apparent structural analogies with the native substrate, but are able to mimic its function by interacting similarly with the target receptor or enzyme.
• Type III mimetics, or functional-structural mimetics, possess a scaffold significantly different from the native substrate, while displaying the interacting elements in the same spatial orientation.
The new classification, recently proposed by Grossmann and coworkers,10 classifies peptidomimetics on the basis of their degree of peptide character:
• Class A mimetics are the most similar to the parent peptide, as only a limited number of local modifications are introduced in the structure to stabilize the conformation and to limit the proteolysis degradation rate. This class includes former type I structural mimetics, as the backbone and side chains of these mimetics show a close alignment with the topography of the native peptide. This is the case of pentagastrin (Peptavlon), an approved drug used as a diagnostic tool for the evaluation of gastric acid secretory function, obtained by the local modification of the bioactive region of endogenous gastrin.
• Class B mimetics still possess large peptide-like character, but contain more important modifications in their structures, both in the backbone and side chains. They may include various non-natural amino acids, isolated small-molecule building blocks and backbone mimetics. This class parallels to classical type II peptidomimetics, as the analogy with the parent peptide is found on the position of the interacting elements, but includes also foldamers and peptoids, where the backbone is entirely modified, while retaining the side chains in the same topological order as found in the parent peptide.
• Class C mimetics are characterized by an increasing small-molecule character, as they have a non-peptide unnatural framework that completely replaces the backbone. The central scaffold projects the interacting elements in the same topological manner as found in the bioactive conformation of the parent peptide, but the peptide backbone is globally altered. They are closely related to traditional type III peptidomimetics, as in the case of somatostatin and TRH analogues (Fig. 1).
• Class D mimetics are the least similar to the parent peptide. These molecules mimic the mode of action of a bioactive peptide without a direct link to its side chain functionalities. They are generated through a hit-to-lead process starting from class C peptidomimetics, or they result from compound library screenings. This class parallels either traditional type II or type III peptidomimetics, depending on the degree of abstraction from the parent peptide.
3. Synthetic toolbox towards peptidomimetic design
There are several conceptually different approaches for the generation of peptidomimetics. The selection of the design strategy depends on what is known about the target protein in terms of structure, sequence, function, and the protein-binding site characteristics. When the sequence of a bioactive peptide is known, a hierarchical approach can be applied to develop the corresponding peptidomimetic (Fig. 3a).11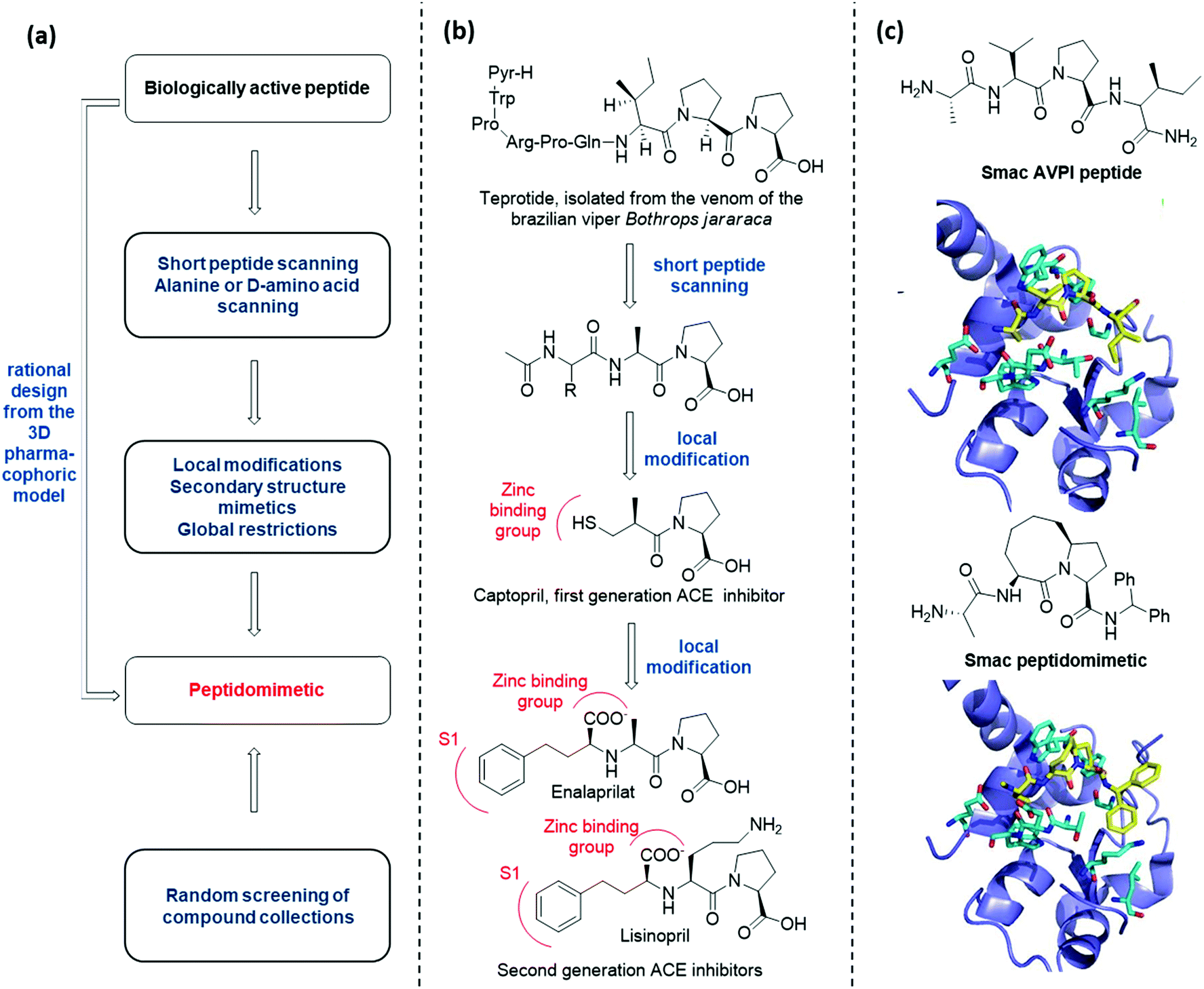 | ||
| Fig. 3 (a) Hierarchical approach to peptidomimetic design; (b) history of the development of peptidomimetic ACE inhibitors; (c) Smac peptidomimetic inhibitor of the X-linked inhibitor of apoptosis protein (XIAP) developed by rational design. | ||
At first, peptide scanning, consisting of synthesizing and testing an array of short overlapping peptides, is useful to reveal the minimal peptide sequence required for the biological interaction. Similarly, alanine or D-amino acid scanning consists of the systematic synthesis and biological evaluation of an array of peptides having only one amino acid of the parent peptide being replaced by alanine or a D-amino acid, to identify key pharmacophoric amino acids responsible for bioactivity.
Then, different synthetic approaches (classified in this review as local modifications, use of secondary structure mimetics and global restrictions) allow for the optimization of hit peptidomimetics, thus giving access to orally available drug candidates. One historical example is given by the development of Angiotensin Converting Enzyme (ACE) inhibitors, starting from the structure of teprotide, a natural bioactive peptide isolated from a Brazilian viper venom (Fig. 3b). By using peptide scanning technique, the pharmacophoric groups responsible for the activity were found to be the two proline residues at the C-terminus and the carbonyl group next to the second proline, which was found able to coordinate the catalytic Zn2+ cation. Thus, the drug Captopril was developed in the early Eighties as one of the most active ACE inhibitors. Advances in the development of ACE inhibitors to reduce side effects of Captopril were obtained by replacing the thiol with a carboxylate group, and balancing the affinity drop by introducing hydrophobic groups addressing the S1 subsite of the catalytic cleft, resulting in the development of Enalaprilat and Lisinopril.
The knowledge of the three-dimensional pharmacophoric model may allow to skip these steps, by rationally designing peptidomimetic compounds on the basis of the interacting elements responsible for the molecular recognition. An example of this approach is given by the development of Smac peptidomimetic inhibitors of the X-linked Inhibitor of Apoptosis Protein (XIAP) (Fig. 3c).12 Such compounds were developed on the basis of the crystal structure of Smac in complex with XIAP BIR3 domain, resulting in a series of conformationally constrained bicyclic Smac mimetics.
On the other hand, when the structure of the bioactive peptide or the pharmacophoric model is not known, the only possible approach is the random screening of large peptidomimetic libraries (Fig. 3a).
3.1 Local modifications
Modifying a peptide through minimal variations at specific positions is the most conservative approach and successful strategy for the design of first-generation peptidomimetics and protease inhibitors. This procedure consists of replacing specific chemical moieties by isosteres, that comprise atoms, ions or molecular fragments showing approximately the same electronic distribution and similar physical properties. Generally, local modifications are restricted to single amino acids, and are grouped into backbone, side chain and dipeptide isostere replacements. A wide range of peptide bond isosteres have been reported over the years, including the replacement of the amide bond with thioamide, alkene, triazole ring, or chemical groups able to mimic the tetrahedral transition state resulting from the cleavage of the scissile peptide bond by proteolytic enzymes, such as the hydroxyethylene moiety. Also, several strategies for the local modification of amino acid side chains have been developed. Apart from the simple C-alkylation to achieve quaternary amino acids, and the homologation with β- and γ-amino acids, the array of locally modified amino acids for developing peptidomimetics includes cyclic compounds with added conformational restriction. Finally, the simultaneous modification of two contiguous amino acids with the use of dipeptide isosteres, including δ-amino acids, is another well-established approach for the development of peptidomimetics with improved peptide stability and enhanced biological activity. | ||
| Fig. 4 Panel of (a) peptide bond isosteres, and (b) transition state analogues used in the design of peptidomimetic compounds. | ||
A special case among amide bond isosteres is offered by the replacement of the amide bond with fragments containing a hydroxyl group. These moieties have been conceived as transition-state surrogates, since they are stable and non hydrolysable functional groups able to mimic the tetrahedral intermediate that occurs when a peptide bond is cleaved (Fig. 4b). The hydroxyethylamine moiety is a homologous function maintaining the hydrogen-bonding features of the transition-state intermediate, and it is found in several FDA-approved HIV protease inhibitors.16 Other transition state analogues are the hydroxyethylene moiety, where the nitrogen is replaced with a CH2 group, as found in the synthetic renin inhibitor aliskiren for the treatment of hypertension, and β-hydroxy-γ-amino acids, also called statines, that are found in the natural aspartic protease inhibitor pepstatin. Other examples of transition state analogues that have been successfully used to design renin and HIV protease inhibitors include phosphinates, α-hydroxy-β-amino acids, trifluoroethylamines, methyleneamino moieties and ketomethylenes.
An important modification in the context of side chain isosteres is the possibility of using non-natural amino acids with appendages that are created to enhance the binding affinity of the interacting elements with hot spots on the biological target surface. In this category, much efforts have been directed for the substitution of aromatic side chains with heterocycles or larger aromatic groups to increase the size and hydrophobicity for van der Waals and π-stacking interactions (Fig. 5a).17 Also, several surrogates of basic and acidic side chains have been developed to reduce or enhance their basicity/acidity and to modulate their strong ionic interactions. For example, the guanidino group of arginine is a highly basic group present in several synthetic and natural inhibitors of trypsine-like serine proteases and integrin receptors. Despite its importance in biological interactions, this group is often responsible of low selectivity and poor oral availability of the resulting compound. For these reasons, several guanidine isosteres have been conceived to modulate the basicity of this group, including amino-imidazoline or amino-pyridine heterocycles, and the replacement of the adjacent CH2 with a carbonyl moiety, such as in the keto-arginine (Fig. 5b). Similarly, several isosteres of the carboxylic group of aspartic and glutamic acids have been proposed to modulate the acidity and the lipophilicity, as for integrin and angiotensin II receptor agonists.18 The carboxylic acid group can be replaced with phosphonate (more acidic), acylsulphonamides (less acidic), hydroxamic acid (useful to chelate metals) or the tetrazole ring, as this heterocycle shows similar planar structure and molecular electrostatic potential and hydrogen-bonding profile (Fig. 5c).
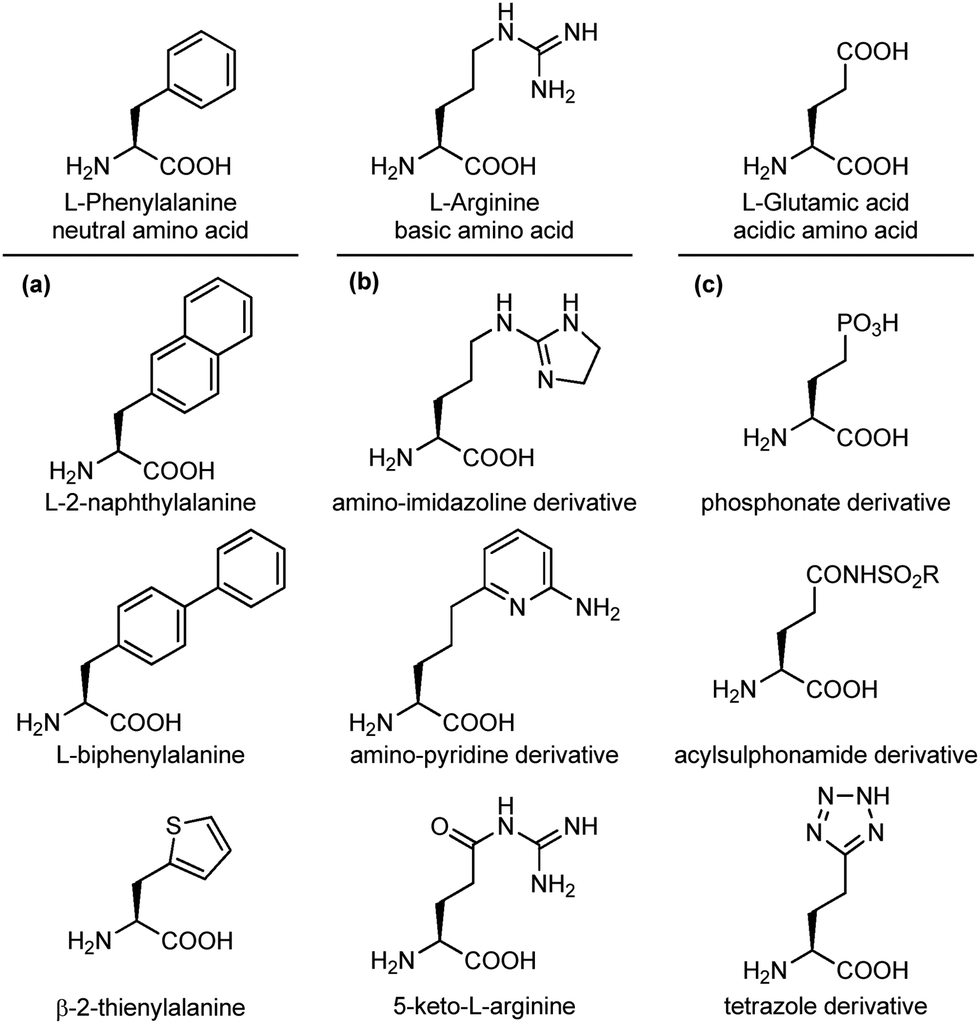 | ||
| Fig. 5 Side chain isosteres of (a) neutral, (b) basic or (c) acidic amino acids. | ||
Another important peptidomimetic tool for introducing local modifications around side chains is to adjust the flexibility on all the rotatable bonds present in the amino acid unit to modulate the conformational profile of the overall peptide. These are called ‘tethering’ strategies, and over the years several approaches addressing the dihedral torsion angles of the amino acids have been proposed (ϕ, ψ, χ, Fig. 6). The simplest method consists of alkylating the Cα-atoms to achieve quaternary carbon atoms (Fig. 6a). α-Substituted amino acids have a reduced rotational freedom around N–Cα and Cα–CO bonds, so that their insertion in a peptide contributes to reducing the rotational freedom around backbone bonds of about 90%. α-Me-alanine is the most widely studied α-alkylated amino acid, which is able to restrict the ϕ, ψ dihedral angles present in α or 310 helices. A similar approach is offered by N-alkylation (Fig. 6b), which results in constraining the ϕ, ψ angles without influencing the χ angle. N-Methylation is a traditional approach typically used for understanding the role of each amino acid within a peptide sequence, as it affects both the hydrogen-bonding capability and the conformational profile of the amino acid. On the other hand, the alkylation at the β-position of the side chain (if present) is an approach that results in constraining χ1 and χ2 dihedral angles, without affecting significantly the backbone angles, and it is particularly useful for selecting preferentially one rotamer in the case of aromatic amino acids (Fig. 6c). Although the reduction of the conformational flexibility of peptides has been largely focused on constraining backbone dihedral angles, reducing the side chain rotational freedom is equally important for the development of biologically active peptidomimetics, especially in the case of those side chains directly involved in the biological interaction. The control of χ1 (around the Cα–Cβ bond) and χ2 (around the Cβ–Cγ bond) dihedral angles was described by Hruby as the ‘design in χ-space’19 and is generally perceived as the most difficult to achieve in peptidomimetic design. Such restriction can be obtained by introducing a double bond or a cyclopropane ring, resulting in the use of α,β-unsaturated α-amino acids, also called dehydro-amino acids, or cyclopropyl-amino acids, by selecting the appropriate E or Z isomer to block the desired position of the side chain (Fig. 6d). The tethered approach has been applied also to produce aromatic amino acid mimetics, by developing conformationally constrained bicyclic α-amino acids. For example, 2-aminotetralin-2-carboxylic acid is a constrained phenylalanine analogue that has been used largely for the synthesis of opioid peptides since the Nineties, in the search of non-addictive analgesics with a prolonged duration of action and reduced side effects (Fig. 6d). Similarly, the formation of Nα–Cα-cyclized amino acids results in constraining both ϕ and χ dihedral angles, and reducing the conformational flexibility around ψ angle.
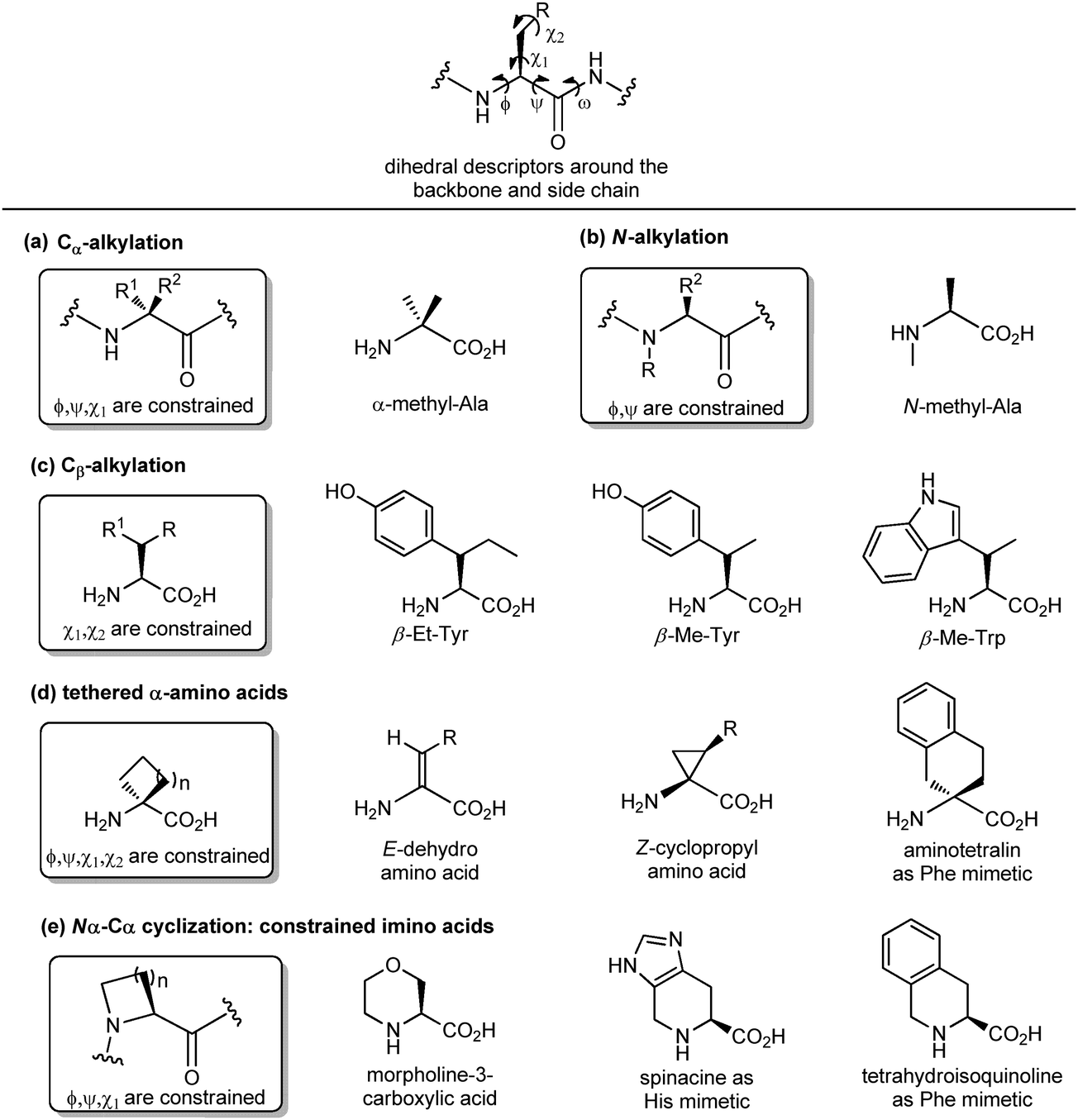 | ||
| Fig. 6 Panel of different synthetic approaches for constraining the backbone and side chain dihedral angles: (a) Cα-alkylation, (b) N-alkylation, (c) Cβ-alkylation, (d) formation of tethered α-amino acids and (e) Nα–Cα cyclization. | ||
This is due to the steric hindrance between the cycle and the adjacent residues, as well as the non-bonded interactions between the carbonyl group and the ring. These constrained imino acids,20 have been developed firstly to mimic the key proline residue, by changing the ring size or introducing heteroatoms and substituents at positions 3, 4 and 5, and then to mimic aromatic amino acids. For example, spinacine is a constrained imino acid histidine mimetic that has been used successfully for the synthesis of δ-opioid receptor ligands. Also, tetrahydroisoquinoline mimicking phenylalanine has been used for the synthesis of several protease inhibitors, including farnesyl transferase, acetylcholine esterase and renin (Fig. 6e).
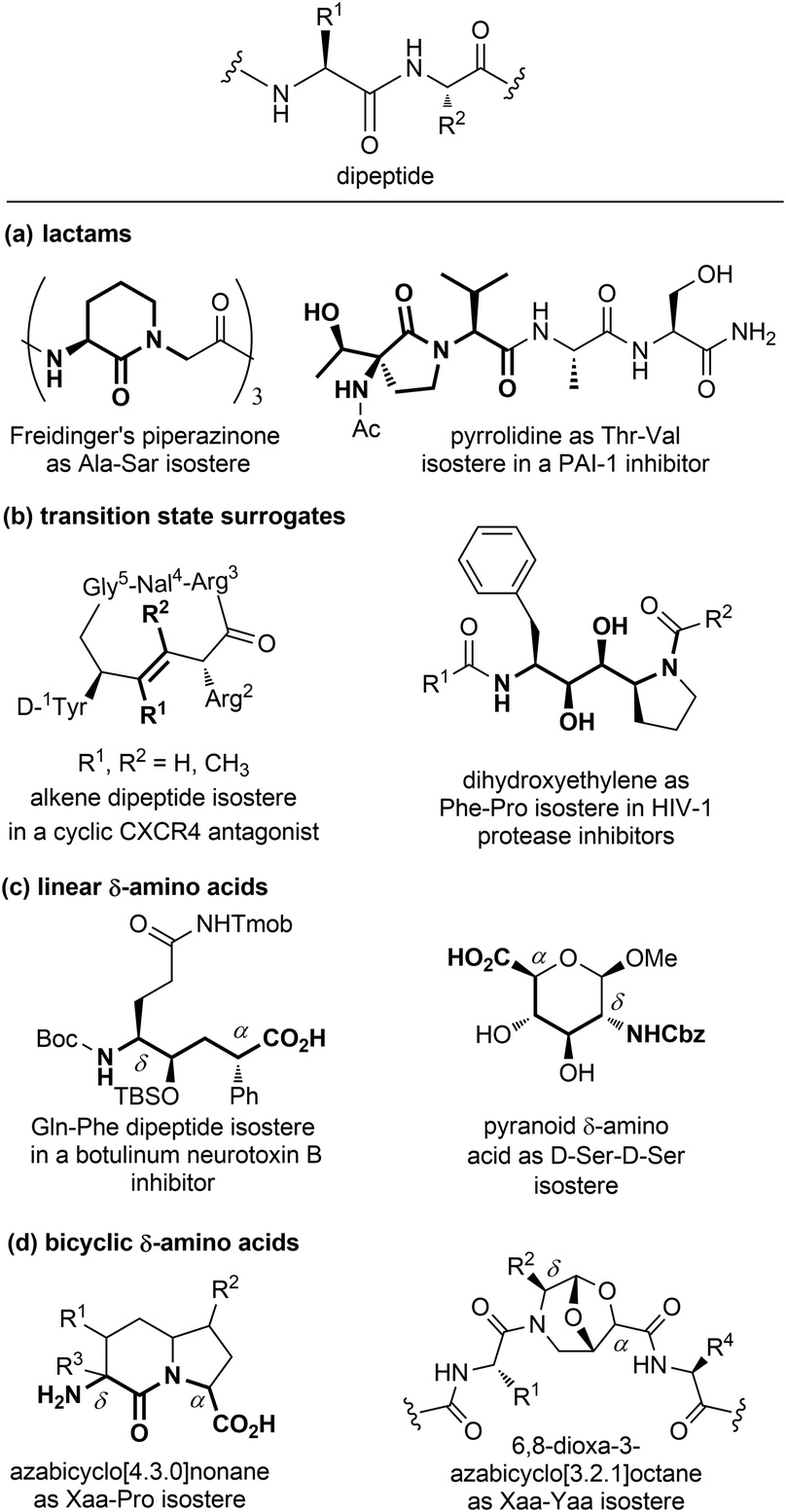 | ||
| Fig. 7 Panel of isosteres of the dipeptide unit, classified as: (a) lactams, (b) transition state surrogates, (c) linear and (d) bicyclic δ-amino acids. | ||
Among monocyclic δ-amino acids, furanoid and pyranoid δ-sugar amino acids are the most relevant examples, especially in the field of β-turn mimetics. These compounds not only constrain the peptide chain thanks to the sugar ring, but possess other useful features of carbohydrates, such as the stereochemical richness and the presence of multiple hydroxyl groups useful for modulating the hydrophilic nature of the template. In particular, pyranoid δ-amino acids can be considered as constrained D-Ser-D-Ser mimetics, and have been used for the synthesis of Leu-enkephalin and somatostatin analogues (Fig. 7c).24 An important sub-group of δ-amino acids are covered by bicyclic compounds, especially for replacing the i + 1 and i + 2 residues in β-turn systems, as described in Section 3.2.2. As for fused pyrrolidine bicycles, the most important examples are the azabicyclo[x.3.0]alkanes as mimetics of the Xaa-Pro dipeptide unit. Among the group of bridged bicycles, an example is provided by 6,8-dioxa-3-azabicyclo[3.2.1]octane-7-carboxylic acids, that show an atom-by-atom correlation with a dipeptide moiety (Fig. 7d).25
3.2 Secondary structure mimetics
Protein–protein interactions (PPIs) are involved in numerous biological processes and play a key role in several pathological conditions. Thus, the development of molecules able to target PPIs possess a significant therapeutic potential. However, small molecules hardly target PPIs, as the binding surfaces between proteins are usually large and shallow, involving many polar and hydrophobic interactions. This stimulated the search for alternative strategies involving the identification of peptide binding epitopes derived from protein interaction sites that serve as a starting point for the design of PPI inhibitors. Such epitopes are defined usually by secondary structures that aligns amino acid side chains in a well-defined manner, specifically α-helices, β-sheets and β-strands, characterized by specific, repetitive torsion angle ranges, and turns and loops that are characterized by non-repetitive irregular wide range of torsion angles. Bullock and coworkers analysed the full set of helical protein interfaces in the Protein Data Bank and found that about 62% of the helical interfaces contribute to protein–protein interactions.26 However, peptides likely lose their secondary structure when free in solution and this flexible nature can result in a limited stability towards proteolysis and low target selectivity. For these reasons, the development of compounds able to mimic peptides in their exact bioactive conformation is an established goal in this field. Several strategies have been developed to yield peptide secondary structures that enable a projection of side chain functionalities, in analogy to peptide secondary structures, as presented in the next sections.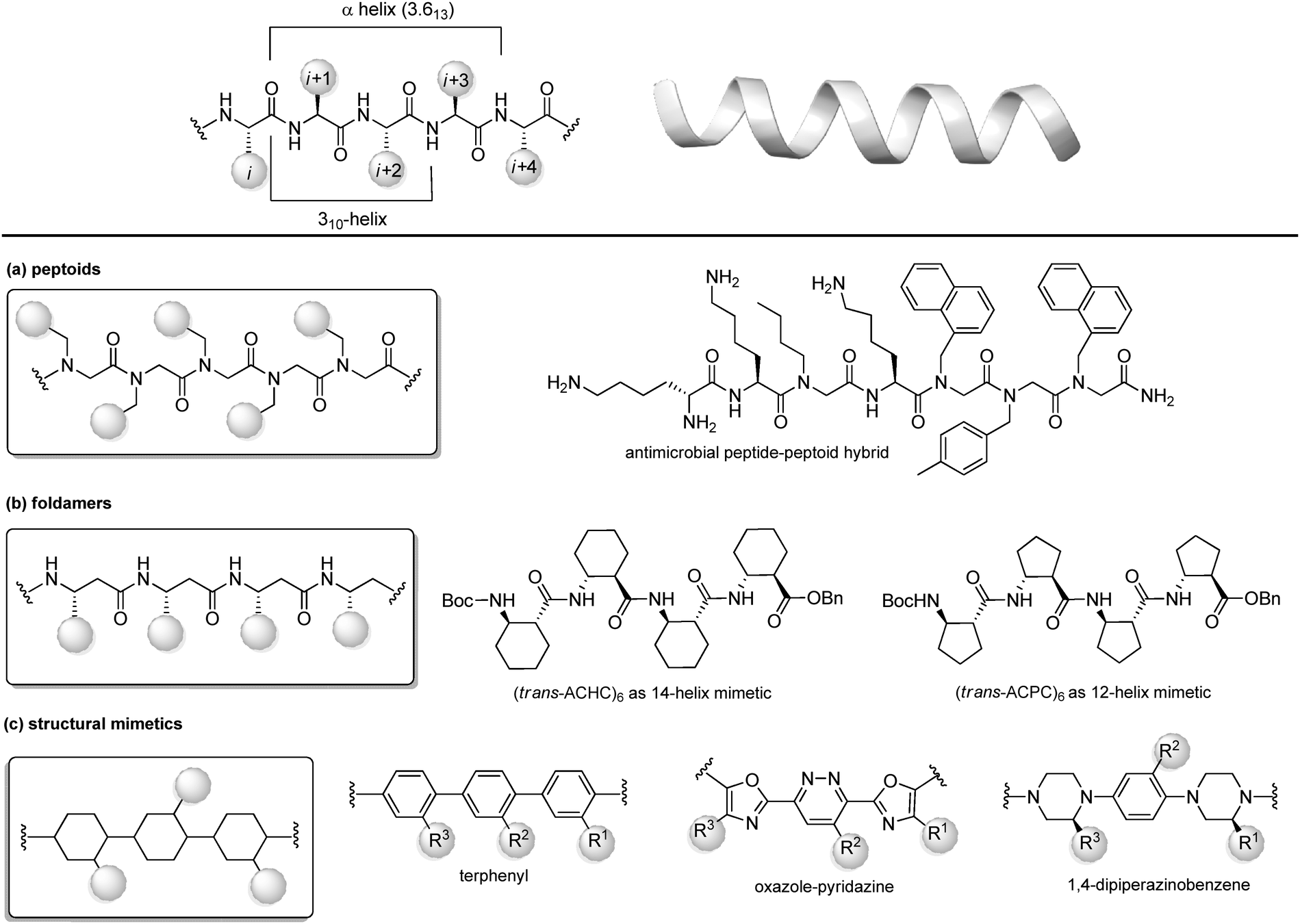 | ||
| Fig. 8 Chemical approaches for stabilizing peptide mimetics of α-helices. (a) peptoids; (b) foldamers; (c) use of structural mimetics. | ||
(a) Peptoids. Peptoids can be described as α-peptide mimetics, in which the side chains are attached to the amide nitrogen atom rather than to the α-carbon atom, thus resulting in a formal shift of the side chain position, with a significant improvement of the resistance to enzymatic hydrolysis and cell permeability (Fig. 7a). In the first peptoids, reported in 1992,27 the monomers were N-substituted glycines, including N-(1-phenylethyl)Gly as β-Me-Phe mimetic or N-(methylimidazole)Gly as Homo-His mimetic. In the same year, the submonomer approach was introduced by Zuckermann, consisting of alternating acylation and nucleophilic displacement reactions using α-haloacetic acid and primary amines, respectively.28 Peptoids fold into chiral helices similarly to type I polyproline helix, and the amide bonds adopt a cis geometry, with the macrodipole being oriented opposite to the α-helical patterns. Thus, the resulting helical conformation is stable even in different solvents and/or at different pH values. However, as peptoid backbones lack of chiral centers and hydrogen-bond donor atoms, the folding of these molecules driven by intramolecular interactions can be challenging. In fact, they can accommodate distorted cis and trans amide conformers, as well as tight turns. To solve this problem, hybrid structures containing N-alkyl-β-alanine monomers (as β-amino acid mimetics) have been developed to construct β-peptoids, β-peptides (also called foldamers, see next) or hybrid α/β-peptoids. Another strategy for stabilizing the peptoid structure consists of constraining the linear peptoid into a macrocyclic structure, with head-to-tail cyclization, or side chain-to-side chain cross-links (see Section 3.3). Peptoids have important applications in the field of antimicrobial peptides (AMPs). In fact, these small amphiphilic peptides possess a cationic region important for the bacterial cytoplasm membrane permeabilization, while being selective for the neutral mammalian cell membrane. Much effort has been focused on developing peptoids able to mimic these structures, to increase the poor pharmacokinetic profile of AMPs and to access the drug market.29 Recently, a novel peptide–peptoid hybrid able to fight bacterial pathogens involved in canine skin infections has been developed (Fig. 8a), including Staphylococcus pseudintermedius and Pseudomonas aeruginosa, that are resistant to other antimicrobial compounds.30
(b) Foldamers. Firstly introduced by Gellman as ‘any polymeric structure capable of adopting a specific folded conformation’,31 foldamers are now defined as non-natural compact oligomeric structures characterized by a well-defined spatial organization.32 They can be composed by nucleotides (in case of nucleotidomimetics), aromatic compounds (abiotics) or synthetic monomers mimicking amino acids (as in the case of peptidomimetics). Although peptidomimetic foldamers can potentially adopt every secondary structure, depending on the monomeric units used and the ratio between them, they play a relevant role for the development of helix mimetics. In particular, they are classified depending on the different peptidomimetic units that are used to mimic α- to δ-peptide backbones. Foldamers of α-peptides include the previously described peptoids, as well as N-permethylated peptides, azapeptides, and oligomers composed of 1,2-diaminoethanes, oxazolidin-2-ones, and pyrrolinones. γ-Peptides can be mimicked by using vinylogous peptides, urea and carbamates, while foldamers of δ-peptides are carbopeptoids and amide-linked sialooligomers. The most exhaustively studied foldamers are β-peptides, as they show well-characterized folding properties and a high structural similarity to α-peptides. They are synthetized by the consecutive coupling of β2-, β3-(bearing a side chain at position C2 or C3, respectively) or β2,3-amino acids (with two side chains at both C2 and C3 positions), as well as coupling cyclic derivatives, such as trans-aminocyclopentane carboxylic acid (ACPC) and trans-aminocyclohexane carboxylic acid (ACHC) (Fig. 8b). These foldamers, initially thought to possess high flexibility, show a more pronounced tendency to fold into a helix as compared to their α-peptide analogues, and it is proved that the driving force to the folding process is due to the flexibility of the Cα–Cβ bond rather than to the hydrogen-bonding network. The preference for each helical conformation can be obtained by modulating the content of monomers. For example, ACHC favours the 314-helix formation, while the ACPC favours the 2.512-helix (Fig. 8b). An important class of foldamers is presented by heterogenous or hybrid foldamers, such as α,β-peptides, constituted by the combination of α and β amino acids. These structures play a key role in developing peptidomimetics, as the presence of α-amino acids guarantee the surface recognition and interaction as in the parent peptide, while the introduction of β amino acids, especially when rigid, cyclic or disubstituted, can be used to improve the resistance to proteolysis and the tendency to give a helical conformation. α,β-Peptides with patterns such as ‘ααβααβ’ or ‘αααβ’ proved to be relevant for designing various PPI inhibitors. For example, Gellman and coworkers33 developed a heterogenous α/β-2 foldameric peptide possessing a ‘ααβαααβ’ backbone, mimicking the α-helix domain of BH3 protein, responsible for disrupting the PPI between BH3 PUMA and Mcl-1, two important proteins of the BCL-2 family that play a key role in mediating apoptosis.
(c) Structural mimetics. While foldamers and peptoids still possess a peptide character, another strategy for designing helix mimetics is the use of molecular scaffolds capable of replacing the essential conformational components of helices.34 The first example in this field, reported by Hamilton in the early 2000s, is a terphenyl scaffold that projects the substituents at positions i, i + 4 and i + 7 of the α-helix, thanks to the steric interactions between the ortho substituents (Fig. 8c). However, terphenyl compounds are highly hydrophobic and require long and tedious synthetic routes, thus other scaffolds with rod-like structure have been proposed since then, in order to improve the water solubility and to reduce the synthetic complexity. Five or six-membered heteroaromatic rings were conceived, as in the case of the oxazole-pyridazine derivative, where the presence of the heteroatoms at one side of the helix resulted in increasing the polarity of the structure, forming amphiphilic water-soluble scaffolds.
Also, oligoamides, such as the terpyridyl scaffold or the 3-O-alkylated oligobenzamide, possessing rigid frameworks and an intricated network of hydrogen-bonds, showed a pronounced curvature and an eclipsed disposition of the substituents. Generally, oligoamides are more synthetically accessible and easier to functionalize, as well as being more water-soluble, although showing reduced activity as PPI inhibitors. Other hydrogen-bond-guided scaffolds are terephthalamides, benzoylureas, thioester-substituted arylamides and biphenyl-4′,4-dicarboxamides. Finally, 1,4-dipiperazinobenzene oligomers, possessing chiral backbones, proved to be valuable rod-like structures to mimic the α-helix when adopting the trans conformation. These structures, characterized by a highly structured arrangement, show high binding specificity and inhibitory potential, and a good cell permeability, revealing the importance of the three-dimensional complexity in regulating the interaction with the biological target.
Two other approaches involving global restrictions used to stabilize helices are peptide stapling and the introduction of hydrogen-bond surrogates. These approaches are discussed in detail in Section 3.3.1.
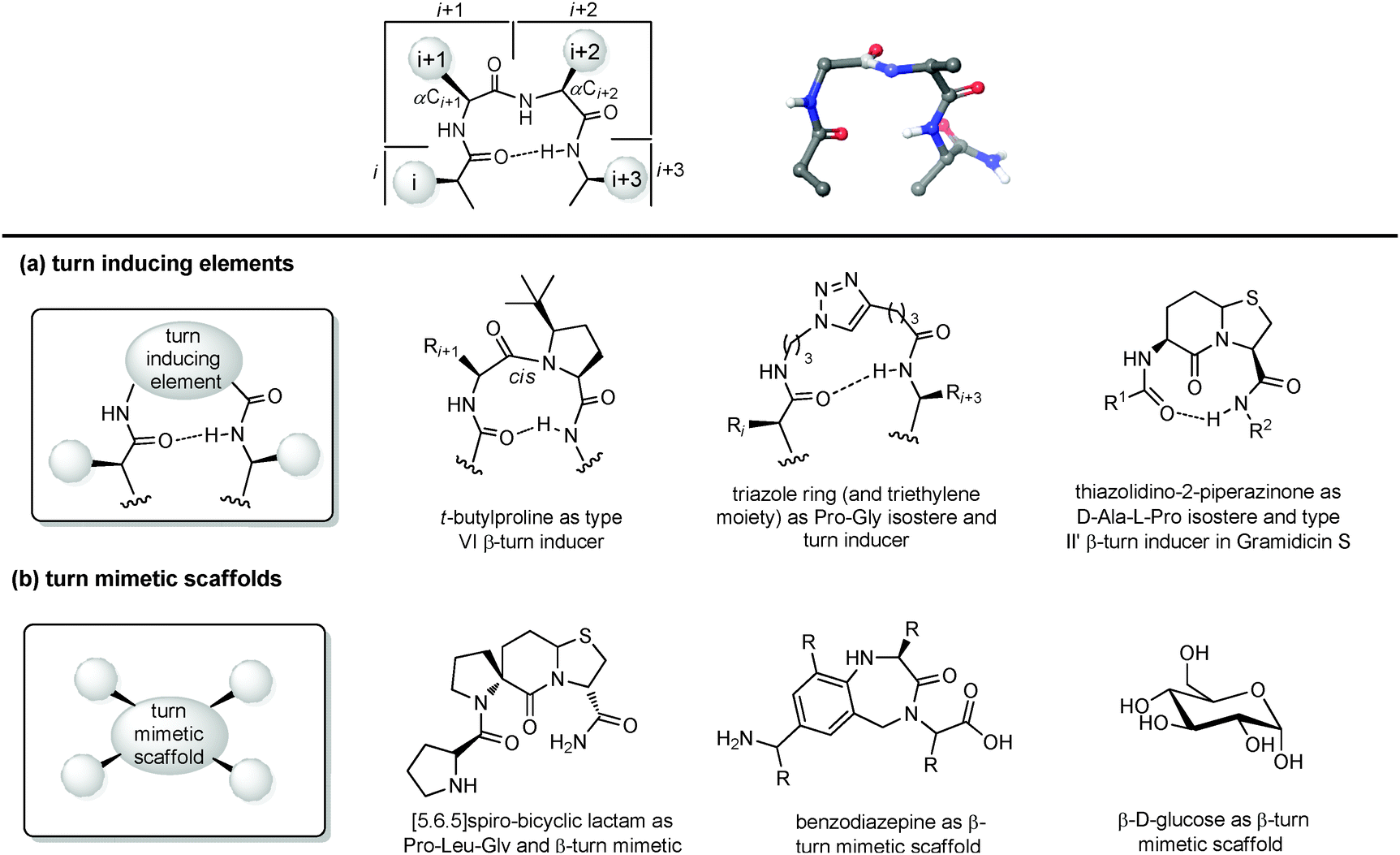 | ||
| Fig. 9 Chemical approaches for mimicking β turn: (a) use of turn inducing elements, (b) use of small molecular scaffold as structural mimetics. | ||
(a) Turn inducing elements. This approach consists of replacing the amino acid at i + 1 and/or i + 2 with a suitable element that can induce the formation of the turn and reduce the conformational freedom of that sequence (Fig. 9a). In fact, even in nature, β-turns are often populated by proline residues, as this secondary amino acid has unique properties in directing the folding of the polypeptide chain by controlling the cis–trans equilibrium. In particular, two different cases should be distinguished: in type I β-turns, proline is found as a turn inducer at position i + 1, generating a trans amide bond at the X-Pro sequence, whereas in type VI β-turns proline is present at position i + 2 establishing a cis amide bond at the X-Pro sequence. In both cases, the use of proline analogues can stabilize the β-turn. For example, the inclusion of D-proline at position i + 1 induces the formation of type I and type II′ β-turns, whereas 5-tert-butylproline forces the amide bond to assume a cis conformation, thus stabilizing a type VI β-turn. Other frequently applied turn inducing elements are α-aminoisobutyric acid (Aib), pipecolic- and morpholine-based heterocyclic compounds and the triazole ring. Also, constrained dipeptide isosteres, such as bicyclic proline-based δ-amino acids, as described in Section 3.1.3, can be used to replace the amino acids at positions i + 1 and i + 2 and to fold the peptide sequence into a β-turn. Hanessian and Lubell reported an extensive overview of tethered prolines to generate bicyclic compounds of different size and possessing different heteroatoms able to mimic the side chain positions of the central dipeptide of a β-turn,35 such as the thiazolidino-2-piperidinone scaffold, that has been used to mimic the D-Ala-L-Pro type II′ β-turn conformation in Gramicidin S.
(b) Small molecular scaffolds as structural mimetics. Another approach consists of using a rigid scaffold to replace the entire peptide backbone and aligning the side chains in a spatial arrangement according to peptide turn residues (Fig. 9b). These compounds resemble the central dipeptide sequence present at positions i + 1 and i + 2, but do not show any amide bonds, so they are more stable and constrained. Usually, they have the amino and the carboxyl groups at opposite positions of the scaffold, so they are ready to be inserted into a peptide and show the same hydrogen-bond as found in the β-turn.
Several scaffolds have been proposed over the years, including spirocyclic lactams, such as the spirobicyclic[5.6.5]lactam exploited as a Pro-Leu-Gly mimetic for the modulation of dopamine receptor activity, and the benzodiazepine scaffold.36
Also, glucose has been used as a β-turn scaffold mimetic, as proved by the development of a somatostatin agonist, as shown in Fig. 1.5 β-turn can be also stabilized by adding chemical tethers that constrain the structure. The various macrocyclization approaches are reported in Section 3.3.1.
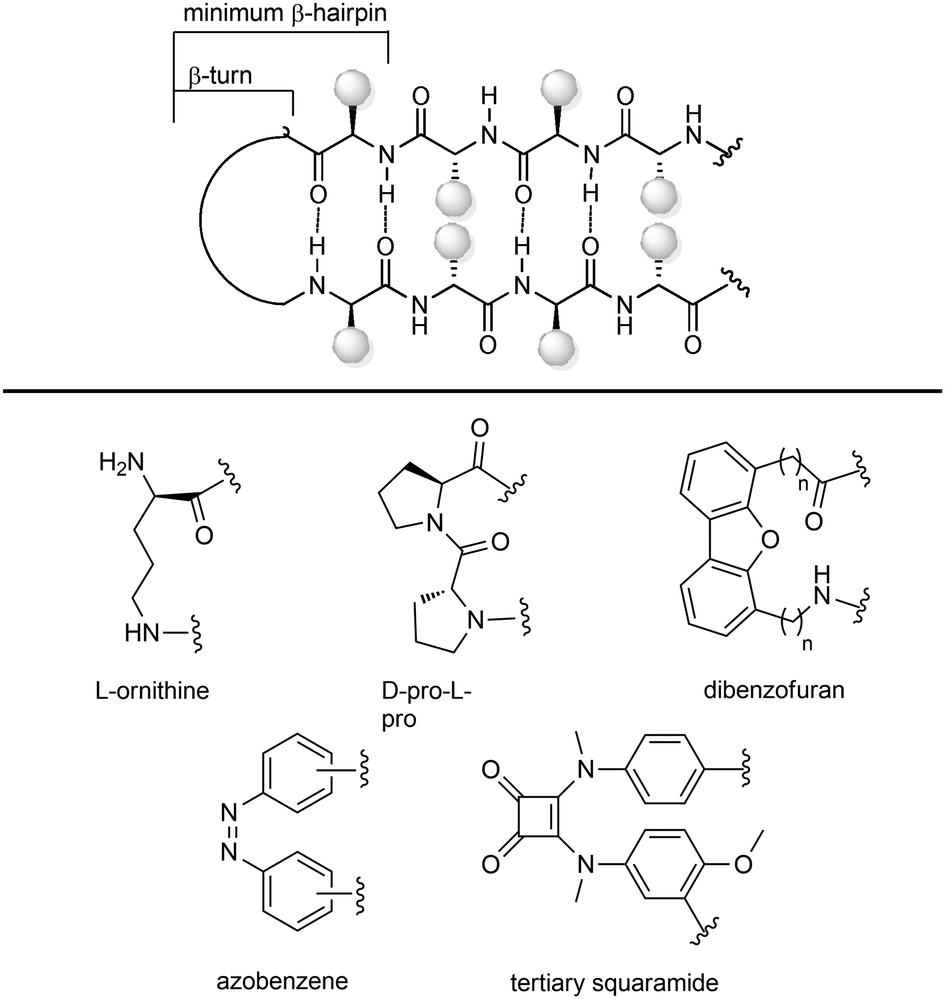 | ||
| Fig. 10 Stabilization of β-hairpin structures using turn inducing motifs. | ||
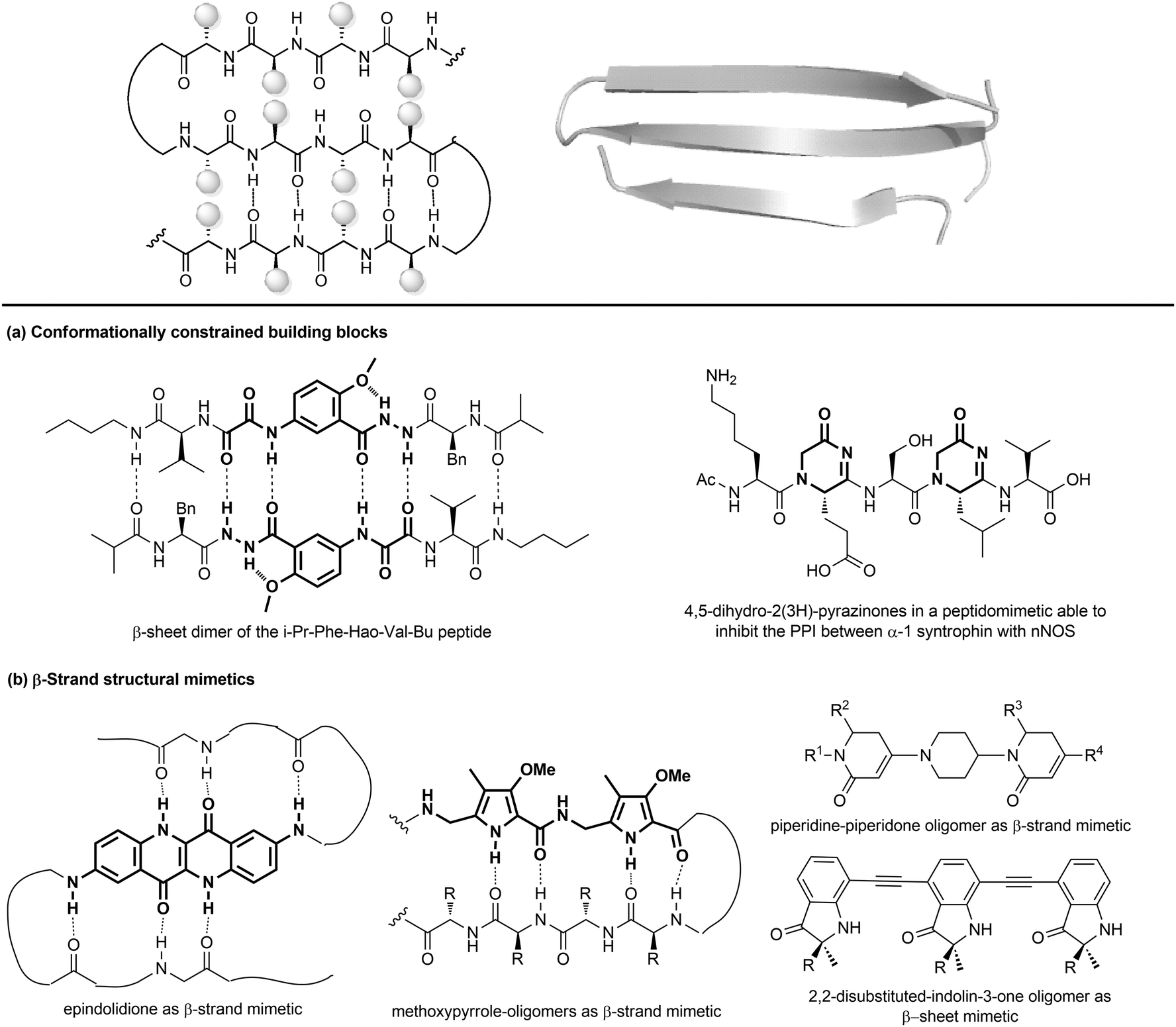 | ||
| Fig. 11 Stabilization of β-sheets structures using (a) conformationally constrained building blocks and (b) β-strand structural mimetics. | ||
Given their key role in the formation of the three-dimensional structure in proteins, β-sheets, β-strands and β-hairpins represent interesting structural elements for the development of peptidomimetics, especially in the field of CNS diseases, as the supramolecular association of β-sheets has been implicated in formation of protein aggregates, such as the amyloid fibrils in the Alzheimer's disease. Also, considering that the structure and stability of these motifs are still not as well understood as α-helices, developing β-sheet-like peptidomimetics is interesting to study the folding propensities of these structures in peptides and proteins. The following entries describe the main approaches to develop β-sheet mimetics.39
(a) Conformationally constrained building blocks to stabilize β-strands. Constrained amino acid mimetics can be used to reproduce the typical hydrogen-bonding pattern and the side chain functionality of a β-strand, stabilizing the overall β-sheet structure (Fig. 11a). A typical example of this strategy is presented by the use of the unnatural amino acid Hao (5-hydrazino-2-methoxybenzoic acid), which is able to mimic the hydrogen-bonding edge of a tripeptide in a β-strand conformation, promoting the overall folding into a β-sheet structure, especially when used in the sequence Phe-Hao-Val.39 Other frequently used β-strand-enforcing amino acids are the 1,6-dihydro-3(2H)-pyridinone (Ach), the D-Pro-DADME (1,2-diamino-1,1-dimethylethane) and the 4,5-dihydro-2(3H)-pyrazinone moiety. Two units of pyrazinone have been used in a pentapeptide that was found able to inhibit the interaction of the PDZ domain of α-1 syntrophin with nNOS more tightly than the parent peptide.40
(b) Flat small molecule scaffolds as structural mimetics. Many different molecular scaffolds have been developed, although the suitable functionalization and diversification of β-sheet structural mimetics is still a challenge, as compared to other secondary structure mimetics (Fig. 11b).39
So far, only the β-strand can be mimicked using structures that are characterized by hydrogen-bond donors and acceptors in exact positions, in order to replicate the hydrogen-bonding network found between β-strands. For example, aromatic scaffolds such as epindolidione and methoxypyrrole-based amino acid sequence can be used to induce β-sheet-like structures when used to replace one segment of the peptide backbone. Also, oligomers composed of 2,2-disubstituted-indolin-3-ones and the piperidine-piperidinone, possessing limited rotational freedom, can be used to mimic a β-strand.
As for β-hairpins, β-sheets secondary structures can be stabilized by using covalent and non covalent interactions between the strands or between the N-terminus of one strand to the C-terminus of the other one.
3.3 Global restrictions
Transforming a linear peptide into a macrocycle is an attractive strategy for generating a conformationally-reduced peptidomimetic molecule. These approaches are different from the introduction of cyclic scaffolds, as they do not modify the parent peptide locally but involve the modification of the overall conformational profile of the target peptide. Macrocyclic peptides have gained wide attention as therapeutic agents, especially for controlling PPI interactions, which are not easily addressed by small molecules. Also, they show higher metabolic resistance as compared to linear peptides, and can fold into a well-defined conformation, thus resulting in improved receptor selectivity and binding affinity. In fact, they contain a high proportion of cis amide bonds, do not contain free C- and N-termini, and possess limited conformational freedom. They are widespread in nature, and examples include somatostatin, the macrocyclic peptide hormone regulating the release of glucagon and insulin, its natural mimetic octreotide, calcitonin, cyclosporine A, nisin and colistin. Cyclization strategies are classified into four different groups (Fig. 12):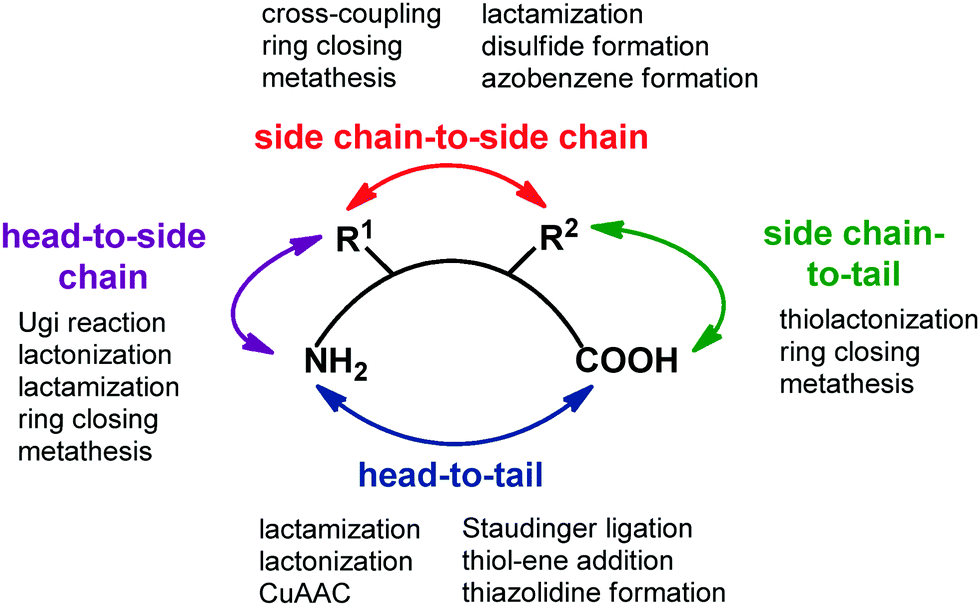 | ||
| Fig. 12 General synthetic approaches for peptide macrocyclization and most commonly used reactions for each approach. | ||
1. Head-to-tail (between N- and C-terminal ends).
2. Side chain-to-side chain (by exploiting reactive functional groups on the side chain of natural or non natural amino acids).
3. Head-to-side chain (between N-terminal end and a side chain functional group of an amino acid).
4. Side chain-to-tail (between C-terminal end and a functional group of an amino acid).
Over the years, many synthetic approaches have been developed to cyclize a linear peptide into a macrocycle.41 Among them, lactamization, lactonization, formation of disulfide bridges between cysteines, triazole formation by CuAAC click chemistry reaction and ring closing metathesis are the most used approaches. Macrocyclization are best performed under high dilution to minimize the unwanted intermolecular processes of oligo- and polymerization. This is often achieved by anchoring the linear peptide to an insoluble polymer. Ring size is the most important factor that governs the success of peptide macrocyclization. Usually, peptides containing more than seven amino acids can be cyclized easily, whereas smaller peptides are rather troublesome. In this case, the incorporation of turn-inducing structural elements, as internal conformational helper, can improve the success of the macrocyclization, by favouring the accommodation of the backbone in the transition state conformation with the least amount of strain. If internal conformational elements cannot be used, external reagents that assist peptide macrocyclization can be exploited. Examples include carbosilane dendrimeric carbodiimides, metal ions such as lithium, caesium and silver(I) salts, and non ribosomal enzymes, such as thioesterase or butelase-1, that are found to be unique in cyclizing a range of peptide substrates in nearly quantitative yields and short reaction times.
Peptide stapling consists of linking two amino acids lying on the same face of a helix at certain positions (specifically i, i + 4, i + 7). Stapled peptides show enhanced α-helicity, improved protease stability and, in many cases, increased cellular uptake, although sometimes cell permeability may result decreased.42 There is no single optimal stapling strategy for all cases, as the choice of the stapling technique depends on both the desired α-helical structure and the nature of the PPI that needs to be studied. In general, both the location and the linker length have to be chosen carefully to avoid interference with target binding and to facilitate efficient helix stabilization. In fact, it has been demonstrated that the stapling element can no longer be considered as an inert structural motif, as it is known to play a role in the overall physicochemical properties of stapled peptides, and sometimes can even interact with the biological target. The most widely used approaches are the ring closing metathesis (also called hydrocarbon stapling) and lactamization. The hydrocarbon stapling technique involves the formation of an olefin-based cross-link which is established by ring closing metathesis between two α,α-disubstituted amino acids bearing olefinic side chains.43 An example of a stapled peptide obtained with this approach is ATSP-7041 (Fig. 13a),44 a potent dual inhibitor of both MDM2 and MDMX, whose derivative ALRN-6924, developed by Aileron Therapeutics, is currently being evaluated in multiple clinical trials as an antitumor compound for p53-positive cancers. Multicomponent reactions proved to be powerful tools for peptide macrocyclization and stapling, as they can increase the molecular diversity and the length of the linkers, providing a fine-tuning of specific conformations and physicochemical properties of the resulting peptide. Most widely used multicomponent reactions for peptide stapling approaches are those based on imines (Strecker, A3 and Petasis reactions) and on isocyanides (Passerini, Yudin, Ugi and Ugi-Smiles reactions).45
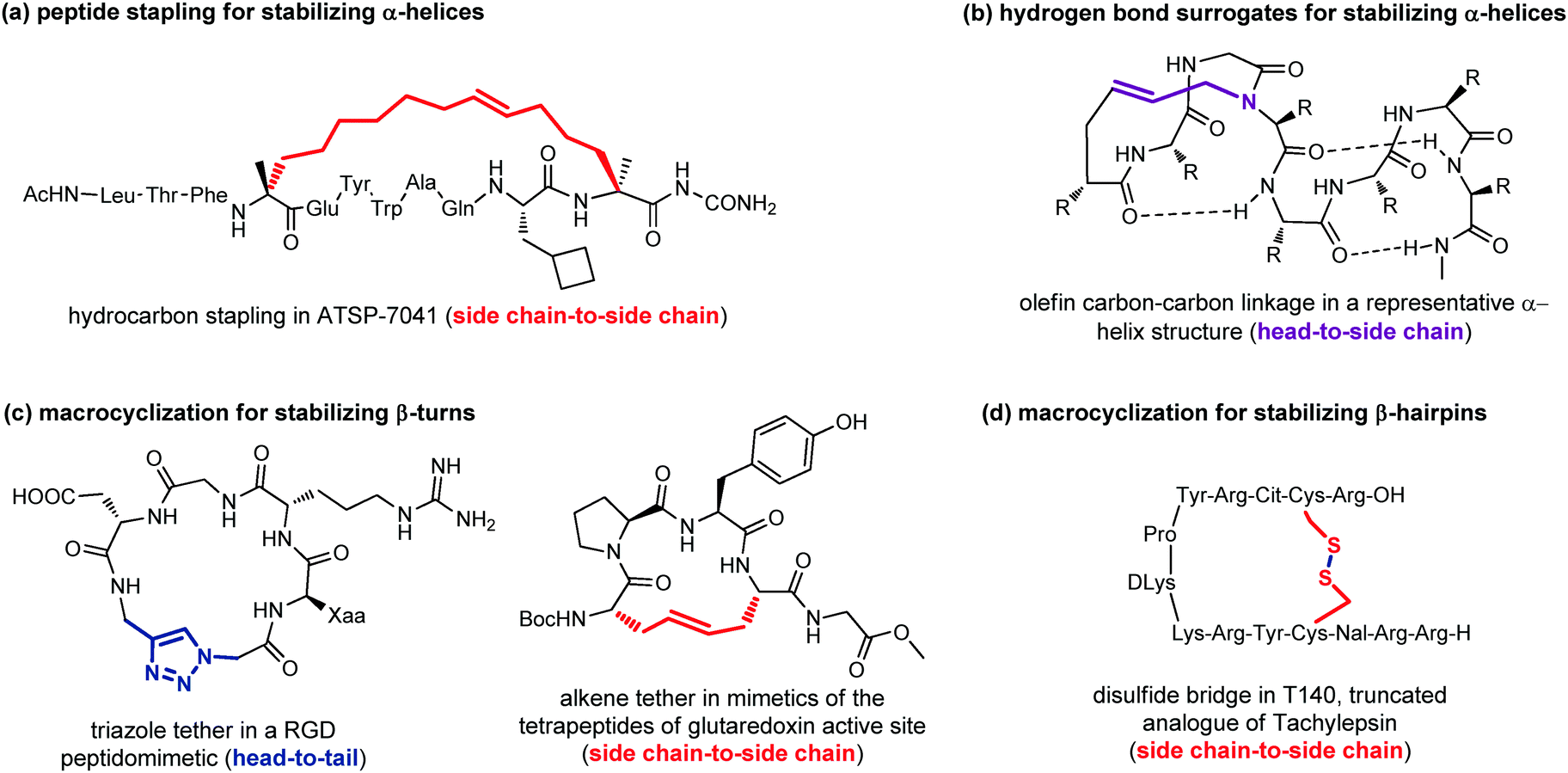 | ||
| Fig. 13 Representative examples of macrocyclization used to stabilize: (a) α-helices with peptide stapling; (b) α-helices with hydrogen-bond surrogates; (c) β-turns; (d) β-hairpins. | ||
Hydrogen-bond surrogates are used to stabilize N-terminal caps and help the nucleation of α-helices. Generally, at the N-terminal position of a α-helix an amino acid such as aspartic acid or asparagine is found, being able to form a strong hydrogen-bonding interaction with backbone amides at position i + 2 or i + 3. The main idea of this head-to-side chain approach is to replace this hydrogen-bond with a covalent linker between the N-terminal amino acid and a side chain of the amino acid at position i + 3, in order to stabilize the helix, as helix nucleation is the energetically most demanding step. Various linker types have been developed over the years,46 but a commonly used strategy is the ring closing metathesis reaction to form a double bond cross-link between 4-pentenoic acid (used as a terminal amino acid surrogate) and a N-allyl dipeptide monomer inserted at the end of the interactive sequence (Fig. 13b). This approach, previously developed to stabilize α-helices, has been recently applied to stabilize β-hairpins, too, even though only few examples have been reported, so far.47
Another important approach for the stabilization of β-turns and β-hairpins is presented by the generation of cyclic peptidomimetics through the formation of a chemical tether. For example, the CuAAC reaction was used to obtain a RGD peptidomimetic, containing the triazole ring as a turn stabilizing element in place of an amino acid, thus obtaining a cyclo[Arg-Gly-Asp-(triazole)-Gly-Xaa] sequence that showed cytotoxic activity in the submicromolar range and a conserved β-turn profile (Fig. 13c).48 Similarly, the ring-closing metathesis was used by Grubbs and coworkers to replace the disulfide bridge in some tetrapeptides present in the glutaredoxin active site that are naturally folded as β-turns (Fig. 13c).49 An example of the use of the disulphide bridge between two cysteines is found in the amphiphilic peptides of Tachylepsin family and their truncated analogue, such as T140 peptide, which is known to inhibit the T-cells infection of HIV-1, by blocking the interaction with the CXCR4 chemokine receptor (Fig. 13d).50
4. Conclusions and future directions
The development of peptidomimetics is a powerful tool in medicinal chemistry, due to the importance of transforming the non orally available and proteolitically unstable peptides into compounds with improved bioactivity and pharmacokinetics.During last decades, the basic concepts and approaches to peptidomimetic compounds have evolved, and many different strategies have been developed to reduce the conformational flexibility and the peptide character of the overall structure. Even the classification has evolved, from an historical arrangement based on structural and functional characteristics of the compound, to a broader classification that includes new approaches based on high molecular weight foldamers and peptoids. In this tutorial review, main chemical approaches have been organized by the entity of modifications on the parent peptide. Local modification are those synthetic tools that can be used to replace a peptide bond, a side chain or a dipeptide unit with a conformationally constrained and proteolitically stable isostere group. These tools are used especially for the development of enzyme inhibitors and receptor ligands. On the other hand, the use of secondary structure mimetics, able to reproduce the folding of helices, turns and β-sheets, play a key role for the development of PPI modulators, as the binding surfaces between proteins are usually large and shallow, involving numerous polar and hydrophobic interactions. Finally, global restrictions represent useful approaches for generating either conformationally-reduced peptidomimetic small molecule and for stabilizing peptides in their secondary structures folding. In particular, peptide stapling and hydrogen bond surrogates can be used to support α-helices, whereas the introduction of chemical tethers proved useful to enhance the stability of β-turns and β-hairpins. Although the panorama of synthetic tools may seem already rich and vast, it is expected that in the future new approaches for the development of peptidomimetics will be developed. This field is moving towards the development of high molecular weight peptidomimetic with a significant peptide-character, although the development of small molecule peptidomimetics will likely remain timely, in line with the increasing interest to peptide research and development in both pharma and biotech taking advantage of multidisciplinary science and technology expanding peptide chemical and target spaces.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge MIUR-Italy (“Progetto Dipartimenti di Eccellenza 2018–2022” allocated to Department of Chemistry “Ugo Schiff”) and Fondazione Cassa di Risparmio di Pistoia e Pescia (Bando Giovani@Ricerca scientifica 2018) for financial support.References
- K. Fosgerau and T. Hoffmann, Drug Discovery Today, 2015, 20, 122–128 CrossRef CAS PubMed
.
- N. Qvit, S. J. S. Rubin, T. J. Urban, D. Mochly-Rosen and E. R. Gross, Drug Discovery Today, 2017, 22, 454–462 CrossRef CAS PubMed
- M. H. Van Regenmortel, Biologicals, 2001, 29, 209–213 CrossRef CAS PubMed
-
A. Trabocchi and A. Guarna, Peptidomimetics in Organic and Medicinal Chemistry: The Art of Transforming Peptides in Drugs, John Wiley & Sons Ltd, Chichester, UK, 2014 Search PubMed
- R. Hirschmann and K. C. Nicolaou, Acc. Chem. Res., 2009, 42, 1511–1520 CrossRef CAS PubMed
- G. L. Olson, D. R. Bolin, M. P. Bonner, M. Bös, C. M. Cook, D. C. Fry, B. J. Graves, M. Hatada, D. E. Hill, M. Kahn, V. S. Madison, V. K. Rusiecki, R. Sarabu, J. Sepinwall, G. P. Vincent and M. E. Voss, J. Med. Chem., 1993, 36, 3039–3049 CrossRef CAS PubMed
- P. S. Farmer and E. J. Ariëns, Trends Pharmacol. Sci., 1982, 3, 362–365 CrossRef CAS
- J. Vagner, H. Qu and V. J. Hruby, Curr. Opin. Chem. Biol., 2008, 12, 292–296 CrossRef CAS PubMed
- A. S. Ripka and D. H. Rich, Curr. Opin. Chem. Biol., 1998, 2, 441–452 CrossRef CAS PubMed
- M. Pelay-Gimeno, A. Glas, O. Koch and T. N. Grossmann, Angew. Chem., Int. Ed., 2015, 54, 8896–8927 CrossRef CAS PubMed
- G. R. Marshall, Tetrahedron, 1993, 49, 3547–3558 CrossRef CAS
- H. Sun, J. A. Stuckey, Z. Nikolovska-Coleska, D. Qin, J. L. Meagher, S. Qiu, J. Lu, C.-Y. Yang, N. G. Sait and S. Wang, J. Med. Chem., 2008, 51, 7169–7180 CrossRef CAS PubMed
- For a comprehensive review on amino acid and peptide bond isosteres, see: I. Avan, C. Dennis Hall and A. R. Katritzky, Chem. Soc. Rev., 2014, 43, 3575–3594 RSC
- S. Roesner, G. J. Saunders, I. Wilkening, E. Jayawant, J. V. Geden, P. Kerby, A. M. Dixon, R. Notman and M. Shipman, Chem. Sci., 2019, 10, 2465–2472 RSC
- For a comprehensive review on triazole-containing peptidomimetics, see: Y. L. Angell and K. Burgess, Chem. Soc. Rev., 2007, 36, 1674–1689 RSC
- A. K. Ghosh, H. L. Osswald and G. Prato, J. Med. Chem., 2016, 59, 5172–5208 CrossRef CAS PubMed
- L. Moroder and H.-J. Musiol, J. Pept. Sci., 2020, 26, e3232 CrossRef CAS PubMed
- P. Stefanic and M. S. Dolenc, Curr. Med. Chem., 2004, 11, 945–968 CrossRef CAS PubMed
- V. J. Hruby, G. Li, C. Haskell-Luevano and M. Shenderovich, Biopolymers, 1997, 43, 219–266 CrossRef CAS PubMed
- For a comprehensive review on constrained imino acids, see: O. Van der Poorten, A. Knuhtsen, D. Sejer Pedersen, S. Ballet and D. Tourwe, J. Med. Chem., 2016, 59, 10865–10890 CrossRef CAS PubMed
- R. M. Freidinger, J. Med. Chem., 2003, 46, 5553–5566 CrossRef CAS PubMed
- K. Kobayashi, S. Oishi, R. Hayashi, K. Tomita, T. Kubo, N. Tanahara, H. Ohno, Y. Yoshikawa, T. Furuya, M. Hoshino and N. Fujii, J. Med. Chem., 2012, 55, 2746–2757 CrossRef CAS PubMed
- B. E. Haug and D. H. Rich, Org. Lett., 2004, 6, 4783–4786 CrossRef CAS PubMed
- For a comprehensive review on sugar amino acids based scaffolds, see: T. K. Chakraborty, S. Jayaprakash and S. Ghosh, Comb. Chem. High Throughput Screening, 2002, 5, 373–387 CrossRef CAS PubMed
- A. Trabocchi, G. Menchi, F. Guarna, F. Machetti, D. Scarpi and A. Guarna, Synlett, 2006, 331–353 CAS
- B. N. Bullock, A. L. Jochim and P. S. Arora, J. Am. Chem. Soc., 2011, 133, 14220–14223 CrossRef CAS PubMed
- R. J. Simon, R. S. Kania, R. N. Zuckermann, V. D. Huebner, D. A. Jewell, S. Banville, S. Ng, L. Wang, S. Rosenberg and C. K. Marlowe, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 9367–9371 CrossRef CAS PubMed
- R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS
- N. Molchanova, P. R. Hansen and H. Franzy, Molecules, 2017, 22, 1430 CrossRef PubMed
- I. Greco, A. P. Emborg, B. Jana, N. Molchanova, A. Oddo, P. Damborg, L. Guardabassi and P. R. Hansen, Sci. Rep., 2019, 9, 3679 CrossRef PubMed
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS
- For a comprehensive review on foldamers, see: D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4012 CrossRef CAS PubMed
- B. J. Smith, E. F. Lee, J. W. Checco, M. Evangelista, S. H. Gellman and W. D. Fairlie, ChemBioChem, 2013, 14, 1564–1572 CrossRef CAS PubMed
- For a comprehensive review on α-helices structural mimetics, see: J. M. Davis, L. K. Tsou and A. D. Hamilton, Chem. Soc. Rev., 2007, 36, 326–334 RSC
- S. Hanessian, G. McNaughton-Smith, H.-G. Lombart and W. D. Lubell, Tetrahedron, 1997, 53, 12789–12854 CrossRef CAS
- E. M. Khalil, W. H. Ojala, A. Pradhan, V. D. Nair, W. B. Gleason, R. K. Mishra and R. L. Johnson, J. Med. Chem., 1999, 42, 628–637 CrossRef CAS PubMed
- For a comprehensive review on β-hairpin peptidomimetics, see: J. A. Robinson, Acc. Chem. Res., 2008, 41, 1278–1288 CrossRef CAS PubMed
- J. Atcher, A. Nagai, P. Mayer, V. Maurizot, A. Tanatani and I. Huc, Chem. Commun., 2019, 55, 10392–10395 RSC
- J. S. Nowick, Acc. Chem. Res., 2008, 41, 1319–1330 CrossRef CAS PubMed
- M. C. Hammond, B. Z. Harris, W. A. Lim and P. A. Bartlett, Chem. Biol., 2006, 13, 1247–1251 CrossRef CAS PubMed
- For a comprehensive review on peptide macrocyclization strategies, see: C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS PubMed
- For a comprehensive review on peptide stapling techniques, see: Y. H. Lau, P. de Andrade, Y. Wu and D. R. Spring, Chem. Soc. Rev., 2015, 44, 91–102 RSC
- Y.-W. Kim, T. N. Grossmann and G. L. Verdine, Nat. Protoc., 2011, 6, 761–771 CrossRef CAS PubMed
- Y. S. Chang, B. Graves, V. Guerlavais, C. Tovar, K. Packman, K. H. To, K. A. Olson, K. Kesavan, P. Gangurde, A. Mukherjee, T. Baker, K. Darlak, C. Elkin, Z. Filipovic, F. Z. Qureshi, H. Cai, P. Berry, E. Feyfant, X. E. Shi, J. Horstick, D. A. Annis, A. M. Manning, N. Fotouhi, H. Nash, L. T. Vassilev and T. K. Sawyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3445–3454 CrossRef PubMed
- L. Reguera and D. G. Rivera, Chem. Rev., 2019, 119, 9836–9860 CrossRef CAS PubMed
- A. B. Mahon and P. S. Arora, Drug Discovery Today: Technol., 2012, 9, 57–62 CrossRef PubMed
- N. Sawyer and P. S. Arora, ACS Chem. Biol., 2018, 13, 2027–2032 CrossRef CAS PubMed
- Y. Liu, L. Zhang, J. Wan, Y. Li, Y. Xu and Y. Pan, Tetrahedron, 2008, 64, 10728–10734 CrossRef CAS
- S. J. Miller, H. E. Blackwell and R. H. Grubbs, J. Am. Chem. Soc., 1996, 118, 9606–9614 CrossRef CAS
- H. Tamamura, M. Sugioka, Y. Odagaki, A. Omagari, Y. Kan, S. Oishi, H. Nakashima, N. Yamamoto, S. C. Peiper, N. Hamanaka, A. Otaka and N. Fujii, Bioorg. Med. Chem. Lett., 2001, 11, 359–362 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2020 |