
Design of small molecules targeting RNA structure from sequence†
Andrei
Ursu
a,
Jessica L.
Childs-Disney
a,
Ryan J.
Andrews
 b,
Collin A.
O’Leary
b,
Samantha M.
Meyer
a,
Alicia J.
Angelbello
a,
Walter N.
Moss
*b and
Matthew D.
Disney
*a
b,
Collin A.
O’Leary
b,
Samantha M.
Meyer
a,
Alicia J.
Angelbello
a,
Walter N.
Moss
*b and
Matthew D.
Disney
*a
aDepartment of Chemistry, The Scripps Research Institute, 130 Scripps Way, Jupiter, FL 33458, USA. E-mail: disney@scripps.edu
bRoy J. Carver Department of Biochemistry, Biophysics & Molecular Biology, Iowa State University, Ames, Iowa, USA. E-mail: wmoss@iastate.edu
First published on 16th September 2020
Abstract
The design and discovery of small molecule medicines has largely been focused on a small number of druggable protein families. A new paradigm is emerging, however, in which small molecules exert a biological effect by interacting with RNA, both to study human disease biology and provide lead therapeutic modalities. Due to this potential for expanding target pipelines and treating a larger number of human diseases, robust platforms for the rational design and optimization of small molecules interacting with RNAs (SMIRNAs) are in high demand. This review highlights three major pillars in this area. First, the transcriptome-wide identification and validation of structured RNA elements, or motifs, within disease-causing RNAs directly from sequence is presented. Second, we provide an overview of high-throughput screening approaches to identify SMIRNAs as well as discuss the lead identification strategy, Inforna, which decodes the three-dimensional (3D) conformation of RNA motifs with small molecule binding partners, directly from sequence. An emphasis is placed on target validation methods to study the causality between modulating the RNA motif in vitro and the phenotypic outcome in cells. Third, emergent modalities that convert occupancy-driven mode of action SMIRNAs into event-driven small molecule chemical probes, such as RNA cleavers and degraders, are presented. Finally, the future of the small molecule RNA therapeutics field is discussed, as well as hurdles to overcome to develop potent and selective RNA-centric chemical probes.
 Andrei Ursu | Andrei Ursu earned his PhD (2015) in Chemical Biology under the supervision of Prof. Herbert Waldmann at the Max Planck Institute of Molecular Physiology, Dortmund, Germany, focusing on chemical reprogramming of stem cells with small molecules. Since 2016, Dr. Ursu has been working as a Postdoctoral Fellow in the lab of Prof. Matthew D. Disney at The Scripps Research Institute in Jupiter, Florida. His focus is on assessing the ligandability of GGGGCC repeat expansions, that cause genetically defined amyotrophic lateral sclerosis (ALS), with small molecule chemical probes. |
 Ryan J. Andrews | Ryan J. Andrews is a PhD student in the lab of Prof. Walter Moss at Iowa State University. His research has been dedicated to the computational prediction of RNA structures in large RNA sequences. His initial research project involved scanning the whole human genome for unusually stable RNA secondary structures (compiled in the RNAStructuromeDB). More recently his research led to the development of the ScanFold method, which has since been used to analyze human messenger RNAs (mRNAs) and viral genomes in order to discover functional RNA structures as well as structured RNA drug targets. |
 Collin A. O’Leary | Collin A. O’Leary is currently pursuing his PhD at Iowa State University in the lab of Prof. Walter Moss. His research is focused on understanding the structure and function relationships of RNAs in human and pathogenic transcriptomes. Some of Collin's initial work included computational prediction of functional regions in the human mRNA MYC, followed by experimental validation of a predicted functional region. His current work is focused on combining a dimethyl sulphate (DMS)-based RNA structure sequencing protocol with the ScanFold method to yield biochemically informed, transcriptome-wide structure models. |
 Samantha M. Meyer | Samantha M. Meyer received her BS in Biochemistry and Molecular Biology from the University of Wisconsin – Eau Claire in 2019. The following fall she began doctoral studies under the guidance of Prof. Matthew D. Disney at The Scripps Research Institute in Jupiter, Florida. Her current research focuses on targeting disease-causing RNAs with small molecules, with an emphasis on expanding the versatility of Ribonuclease Targeting Chimeras (RIBOTACs). |
 Alicia J. Angelbello | Alicia J. Angelbello received her BS in Chemistry from Villanova University. She joined the Disney laboratory in 2015 as a graduate student where she works on developing small molecules to target RNA repeat expansions. |
 Walter N. Moss | Walter N. Moss is an Assistant Professor at Iowa State University whose research focuses on identifying and characterizing functional RNA structures using computational and experimental approaches. His long-term goal is to establish methodological pipelines that facilitate the discovery of structural motifs with significance to human health and disease using innovative in silico tools, biochemical and cell/molecular biological approaches. |
Key learning points• Aberrant RNA structure contributes to the pathology of numerous human diseases.• Structured, evolutionarily conserved RNA motifs can be predicted directly from sequence with the state-of-the-art computational tool, ScanFold. • Inforna decodes these evolutionarily conserved RNA 3D folds with small molecules to provide high-quality chemical probes. • Robust target engagement techniques are necessary to validate RNA-centric modes of action. • Emergent therapeutic modalities include RNA-targeted degraders and cleavers that destroy disease-causing RNAs. |
1. Introduction
Most drug discovery campaigns, both past and present, are focused on protein targets. Decades of technological advancements and scientific discoveries have been dedicated to exploring the proteome and modulating protein activity for therapeutic benefit. These efforts yielded chemical probes to test mechanistic hypotheses, uncover new biology, and manipulate biological processes. Ultimately, this knowledge has been translated into novel and safe medicines for a plethora of human diseases. However, druggable proteins are confined to a small set of families. To expand druggability and increase our understanding of disease biology, many have turned to RNA targets. RNA is best known for its role in translation, where messenger RNAs (mRNAs) are translated into proteins via the ribosome, a complex macromolecular machine composed of ribosomal RNAs (rRNAs) and proteins, in conjunction with transfer RNAs (tRNAs). The functions of RNA, however, go well beyond this critical role in biology. For example, RNA molecules encode unique secondary and tertiary structures that have biological functions on their own (acting in cis) or can recruit other factors (RNAs, proteins) to assist in their function (acting in trans).As many disease phenotypes can be traced back to dysregulation of RNA function, various approaches have been employed to target disease-causing RNAs for therapeutic benefit. The two most studied modalities are antisense oligonucleotides (ASOs) and small molecules, i.e., small molecules interacting with RNAs (SMIRNAs), which fundamentally differ in their modes of action.1 ASOs, in general, consist of modified nucleotides, either via the backbone or sugar moiety, and are designed by sequence complementarity. That is, ASOs recognize RNA primary sequence (Fig. 1A) and hybridize to cognate disease-causing RNAs to: (i) sterically block the assembly of RNA–protein or RNA–RNA interactions; or (ii) promote degradation of the disease-causing RNAs via Ribonuclease H (RNase H), an endoribonuclease that hydrolyzes the phosphodiester bonds of the RNA strand in RNA–DNA heteroduplexes. Although the design and generation of complementary ASOs for any given disease-causing RNA is rapid and straightforward, their binding sites must be accessible, i.e., unstructured. Both RNA's intramolecular (secondary and tertiary) structures and intermolecular structures with other biomolecules can affect ASO binding in cellular context.
 | ||
| Fig. 1 Overview of RNA structure and its prediction directly from sequence using ScanFold. (A) The primary structure of RNA, i.e., sequence, consists of four bases; two purines, adenine (A) and guanine (G), and two pyrimidines, cytosine (C) and uracil (U). (B) The secondary structure of an RNA consists of the non-covalent bonds that form between A and U, G and C, or G and U, bases. These pairings consist of hydrogen bonds and base stacking interactions which form stems (light green) and are often punctuated with internal loops (blue), bulges (pink), and hairpin loops (dark green). (C) The tertiary structure of RNA is largely dictated by the base pairs that form the secondary structure. Stems (light green) will form structured A-form helices and internal loops (blue), bulges (pink), and hairpin loops (dark green) will be less structured, more accessible regions that distort the more rigid helix and offer sites for trans-acting factors to bind in a sequence specific manner. Here, the dotted black line represents the single strand between the two more structured hairpins. (D) Identification of structured RNA motifs within the mRNA sequence of MYC via ScanFold. Portions of the MYC mRNA coding region and 3′ untranslated region (UTR) are depicted with overlapping ScanFold analysis windows below. In each scanning analysis window, ScanFold calculates numerous folding metrics including the minimum free energy (MFE), ensemble diversity, and z-scores which are depicted as bar graphs. It is important to note that a window will be represented by a single bar, but the downstream nucleotides (nt) (corresponding to the window size) are used to predict the metrics. ScanFold then determines the most stable and significant base pairs and uses them to generate a consensus structure (displayed as an arc diagram). Regions with highly negative z-scores and low ensemble diversity indicate regions of presumed function, with one (or few) dominating structures and that may merit further, in-depth analyses: e.g., comparative analysis, additional bioinformatics analyses, functional assays, and structure probing assays. These techniques can further characterize and validate the biological function of the structured RNA motif. | ||
In contrast to ASOs, SMIRNAs recognize unique three-dimensional (3D) RNA conformations, or structure. RNA secondary structure is dictated by its sequence, which restricts and directs the formation of intramolecular base pairing, generating helical regions interspersed with loops, bulges, and hairpins (Fig. 1B) (see ref. 2 and citations therein for a detailed description of structured RNA motifs). That is, the overall secondary structure of an RNA can be viewed as modules of structured elements, or motifs, strung together. Though built only on four nucleotide building blocks, RNA sequence encodes dynamic and sufficiently unique ensembles of 3D folds that can be targeted and/or stabilized selectively by small molecules (Fig. 1C). Importantly, RNA secondary structure can be predicted or determined accurately from RNA sequence. Secondary structure then constrains available tertiary interactions and thus tertiary structure (Fig. 1C). As tertiary structures are generally weak, they can be disrupted by small molecule binding, affecting the RNA's function.
Small molecules offer several advantages that support their use as a viable modality to target 3D folds of structured motifs within RNA. For example, structurally related analogs can be used to define structure–activity relationships (SAR), informing lead optimization for biological activity and selectivity. Moreover, SMIRNAs targeting adjacent structured RNA motifs can be covalently linked together, yielding dimeric molecules with increased binding affinity and selectivity compared to the individual compounds from which they were derived.1 Finally, SMIRNAs can be functionalized with various modules to affect direct cleavage, to induce degradation via recruitment of endogenous nucleases,3 or to image disease-causing RNAs through on-site synthesis of a Förster resonance energy transfer (FRET) pair. These features expand the mode of action of SMIRNAs to explore RNA biology and to provide therapeutic opportunities for many human diseases mediated by RNA structures.
This review highlights three key components required to design high-quality SMIRNAs with defined RNA-centric modes of action: (1) state-of-the-art approaches to identify ligandable 3D structured motifs within RNA that are evolutionarily conserved and hence likely to be functional; (2) methods to target structured motifs within RNA; and (3) RNA target validation methods. We also highlight novel modalities developed by converting occupancy-driven SMIRNAs into event-driven chemical probes (RNA cleavers and degraders) that ablate disease-causing RNAs. Finally, we offer an overview of the future challenges that need to be overcome to facilitate the design and optimization of potent and selective small molecule RNA therapeutics in a robust and rational fashion. A comprehensive review of targeting disease-causing RNAs extending beyond this tutorial can be found in ref. 4.
2. The role of RNA structure in biology & disease
RNA structure is intimately linked to both normal biology and disease pathology.5 RNA structures range from simple loops and bulges to more complex structures such as coaxial stacking, pseudoknots, and other tertiary structures. Indeed, these structures influence and dictate human biology, ranging from the regulation of translation, to splice site selection, and catalysis. RNA structure also controls viral replication and infection as well as bacterial gene expression (riboswitches). As these topics have been extensively reviewed, we direct the reader to the excellent references below for additional details.Not surprisingly, RNA mutation and aberrant expression can trigger disease by causing deregulation of normal cellular processes. For example, transcriptomic studies have revealed that microRNAs (miRNAs), small regulatory RNAs that modulate gene expression by binding to complementary mRNAs, are commonly dysregulated in tumor tissue, suggesting a mechanism by which cancer cells downregulate tumor suppressor genes or enhance expression of oncogenes. Aberrant expression of miRNAs, whether up- or down-regulated, has been linked to many other diseases, including cardiovascular disease, inflammatory and neurodevelopmental disorders and liver disease.
RNA structure has also been implicated in many neurological disorders. RNA repeat expansions cause over 30 human diseases, including Huntington's disease (HD) [r(CAG)exp], amyotrophic lateral sclerosis (ALS) [r(G4C2)exp] and myotonic dystrophy type 1 (DM1) [r(CUG)exp]. In these disorders, the repeating RNA, often found in intronic or untranslated regions (UTRs), forms hairpin structures containing repeating structured RNA motifs that interfere with normal RNA processing and function. These structures can sequester RNA-binding proteins, lead to the formation of nuclear foci, and undergo repeat-associated non-ATG (RAN) translation. This disruption in normal biology has substantial consequences, leading to disease pathologies that are both common and unique to different microsatellite disorders.
Collectively, regulation and maintenance of RNA structure is critically important to sustain normal biology, and identification of novel functional RNA structures (discussed below) featuring motifs that can be targeted with SMIRNAs will be critically important to study RNA's role in disease for therapeutic benefit.
3. Methods to identify functional RNA structures via evolutionary conservation
3.1 Overview of RNA Structure Prediction
An RNA structure is defined by the intramolecular base pairs which form as the RNA molecule folds back on itself, i.e., by the helices formed between complementary stretches of RNA. The composite strength of base pairs in a secondary structure are relatively strong compared to the weaker interactions that form an RNA's tertiary structure. Generally, the formation of RNA tertiary structure does not alter the underlying secondary structure and is instead guided by it in a hierarchical manner. Therefore, the accurate prediction of RNA secondary structure is highly valuable when defining a RNA's structured landscape in order to: (i) generate biological hypotheses about RNA structure–function relationships; and (ii) identify structured 3D folds within RNA for modulation with SMIRNAs.When predicting a single secondary structure model for a given RNA sequence, the most frequently used method is free energy minimization. This method calculates the most stable secondary structure (i.e., the structure with the most negative 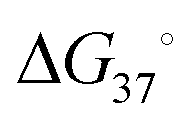 ) as evaluated from an underlying set of experimentally-derived thermodynamic parameters. The key assumption is the base pairing pattern that yields the most stable minimum free energy (MFE) secondary structure is also the best representation of the native fold. The reality of RNA folding is of course much more complicated in the cellular milieu, where a multitude of 3D conformations can not only exist, but also interconvert, depending on environmental factors and external stimuli. Therefore, the predictions made via free energy minimization methods serve only as a valuable guide for building hypotheses as to the structured RNA motifs responsible for the phenotype(s) of interest.
) as evaluated from an underlying set of experimentally-derived thermodynamic parameters. The key assumption is the base pairing pattern that yields the most stable minimum free energy (MFE) secondary structure is also the best representation of the native fold. The reality of RNA folding is of course much more complicated in the cellular milieu, where a multitude of 3D conformations can not only exist, but also interconvert, depending on environmental factors and external stimuli. Therefore, the predictions made via free energy minimization methods serve only as a valuable guide for building hypotheses as to the structured RNA motifs responsible for the phenotype(s) of interest.
The accuracy of secondary structure prediction by free energy minimization, however, decreases with sequences >700 nucleotides (such as mRNAs or viral genomes).6 For example, RNA folding algorithms performed best when the analyzed sequence length was restricted to between 100 and 150 nucleotides, thus limiting the analysis to locally stable RNA regions rather than calculating the most globally stable structure. Further, free energy minimization alone cannot clearly define whether a structured RNA motif is functional.
Recently, tools have been developed to predict structured RNA motifs throughout the transcriptome.7 These tools consider two hallmarks of functional RNAs: (i) unusual structural stability; and (ii) evolutionarily conserved base pairs. These approaches focus on finding not only well-defined, i.e., stable RNA structures, but also structured elements that are more stable than expected for their nucleotide composition (as characterized by the thermodynamic z-score eqn (1)). Further, if a specific RNA structure is likely to be functional, conservation across homologous sequences, as indicated by mutations which retain the secondary structure, should be observed.
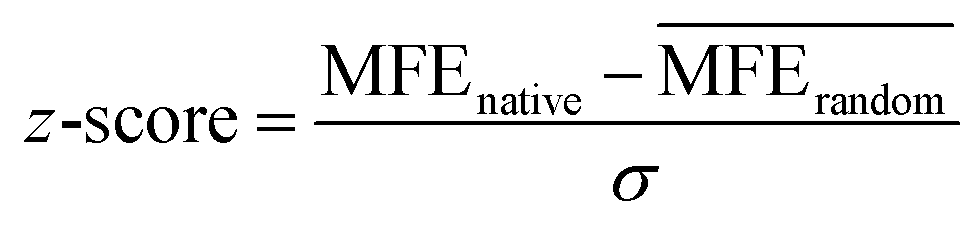 | (1) |
As shown in eqn (1), the z-score compares the MFE of a sequence within an RNA of interest (MFEnative) to the average MFE of a set of randomized RNA sequences (MFErandom), normalized by the standard deviation (SD; σ) of the MFE. That is, a native RNA sequence that is more thermodynamically stable (lower MFE) than a set of randomized sequences will yield a negative z-score and be considered to form a stable structure. The z-score reports the number of SDs the native MFE is away from the average MFE from random sequences with similar nucleotide composition.
Indeed, the most reliable tools to date for computational prediction of functional RNA secondary structures from sequence7 incorporate these strategies. The Moss Lab has recently developed a computational method which prioritizes RNA structural characterization and analysis followed by conservation analysis. This method, named ScanFold,8 characterizes the structured landscape of any large RNA sequence (Fig. 1D and Table S1, ESI†). In brief, ScanFold analyzes RNA sequences using a scanning window approach and reports the results of MFE and ensemble-based predictions across the entire sequence.
Whenever available, the predicted secondary structures are further validated with RNA structural data obtained from chemical probing experiments in cells, for example using dimethyl sulfate (DMS) or selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE).9,10 These chemical probing reagents react with non-canonically paired or single stranded nucleotides, modifying the bases in the case of DMS or the sugar moieties of dynamic nucleotides in the case of SHAPE. After modification, the RNAs are then analyzed by RNA sequencing (RNA-seq), which requires reverse transcription (RT) and polymerase chain reaction (PCR) amplification. Reaction of a nucleotide with a mapping reagent creates a unique signature during reverse transcription, either by preventing readthrough resulting in a “stop” or creating a mutation. The reactivity of each nucleotide with the chemical modifying reagent, or the extent of mutation or termination of the RT-PCR step, is calculated as normalized to untreated RNA. Increased reactivity indicates that the nucleotide is not canonically base paired. These data can then be used as checks on existing structure models or used directly during MFE calculations as a constraint on secondary structure predictions.
Base reactivities from structure probing are calculated and can be incorporated as constraints during MFE calculations in programs such as RNAfold11 (which ScanFold utilizes) or RNAstructure (Table S1),6 and are then cross-referenced with ScanFold results. Incorporating such data helps to yield biologically relevant models of RNA secondary structure(s). Notably, results from chemical probing experiments must be carefully controlled and the statistical confidence of the resulting data must be calculated, as various artifacts arising from transcriptional noise, limitations of high-throughput experimentation, and computational analysis errors can generate erroneous RNA structures.
To date, ScanFold has been applied to several genomes, including human,12 human immunodeficiency virus type 1 (HIV-1) and Zika virus (ZIKV),8 as well as mRNA sequences encoding microtubule-associated protein tau (tau)13 and α-synuclein (SNCA),14 the results of which are summarized below.
3.2 Validation of ScanFold: structured RNA Motifs within HIV-1 and ZIKV
The genomes of ZIKV and HIV-1 are composed of positive (+) sense (protein-coding) single-stranded RNA molecules: 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 807 and 9175 nucleotides (nt) in length, respectively. Their small genomes are translation-competent and, much like mRNAs, are composed of coding and non-coding regions, the former flanked by 5′ and 3′ UTRs. Because of their relevance to human health, these viruses have been studied extensively and were each found to utilize several structured RNA motifs to carry out aspects of their viral life cycles including replication, packaging, and translation. Due to the thorough structural and functional characterization of these viral RNA structures, the HIV-1 and ZIKV genomes served as ideal RNA sequences to test ScanFold's ability to detect structured RNA motifs.
807 and 9175 nucleotides (nt) in length, respectively. Their small genomes are translation-competent and, much like mRNAs, are composed of coding and non-coding regions, the former flanked by 5′ and 3′ UTRs. Because of their relevance to human health, these viruses have been studied extensively and were each found to utilize several structured RNA motifs to carry out aspects of their viral life cycles including replication, packaging, and translation. Due to the thorough structural and functional characterization of these viral RNA structures, the HIV-1 and ZIKV genomes served as ideal RNA sequences to test ScanFold's ability to detect structured RNA motifs.
The ScanFold platform, introduced in Andrews et al.,8 accurately identified all known functional structures from the HIV-1 and ZIKV genomes and revealed additional potentially structured RNA motifs throughout each. The ideal settings for detecting known structures in HIV-1 and ZIKV were optimized in this report (where a window size of 120 nt was found to best recapitulate known functional models). In a follow-up report, a detailed description of these settings were described to advise researchers using ScanFold on how to adjust settings for any RNA sequence.15 An emphasis was placed on practical usage, for quick and accurate characterization of an RNA's overall landscape of structured motifs. In this follow-up study, it was also revealed that ScanFold's characterizations of HIV-1 and ZIKV agreed with available SHAPE probing data, accurately characterizing RNA regions as either housing a uniquely structured RNA motif (where low z-score structures correlated with unambiguous experimental results and high prediction accuracy) or a more dynamic/loose structure (where more positive z-score motifs correlated to experimental results which allow more than one structural interpretation and suggest an overall unstructured nature). These results showed that while ScanFold excels at highlighting potential (and known) structured RNA motifs, it can also accurately characterize an RNA's structural landscape. Importantly, such results can be obtained quickly, easily, and using only a single sequence to point investigators towards potentially structured RNA motifs, which are likely to be biologically relevant.
4. Methods to target functional, evolutionarily conserved structures
The next challenge is to exploit the discovery of evolutionarily conserved structures to design small molecules that selectively recognize them and modulate RNA function. There are at least three critical factors for the development of SMIRNAs: (i) exploration of diverse chemical space to identify privileged chemotypes that selectively bind structured 3D folds within RNA(s); (ii) complementary exploration of structured RNA 3D folds within disease-causing RNAs that form well-defined pockets for small molecule ligands; and (iii) development of a bioinformatic platform that links (i) and (ii) and ultimately yields bioactive SMIRNAs against disease-causing RNAs.In order to gather data on the first two key factors, we developed the selection-based strategy termed two-dimensional combinatorial screening (2DCS) (Fig. 2).1,16–18 2DCS is a massively parallel screening method that probes the interaction of small molecule libraries against libraries of structured RNA motifs found within cellular RNAs. The library-vs-library screen is performed by covalently immobilizing or absorbing (dubbed AbsorbArray16) small molecules onto agarose-coated microarrays, followed by incubation with a labeled library of RNA motifs (Fig. 2). These RNA libraries contain thousands of structured RNA motifs in discrete patterns, featuring bulges, hairpins, internal loops, etc. The screen is performed in the presence of excess competitor RNAs that mimic regions common to all RNA library members, DNA and RNA base pairs, and/or tRNAs (Fig. 2). That is, the screen is completed under conditions of high oligonucleotide stringency. This screening format can be performed with structurally related small molecules such that SAR can be derived or with diverse chemical matter to expand our understanding of chemotypes that confer avidity and selectivity for RNA. This experimental approach is highly advantageous when compared to other small molecule microarray (SMM) approaches, which typically screen a single RNA target at a time.19
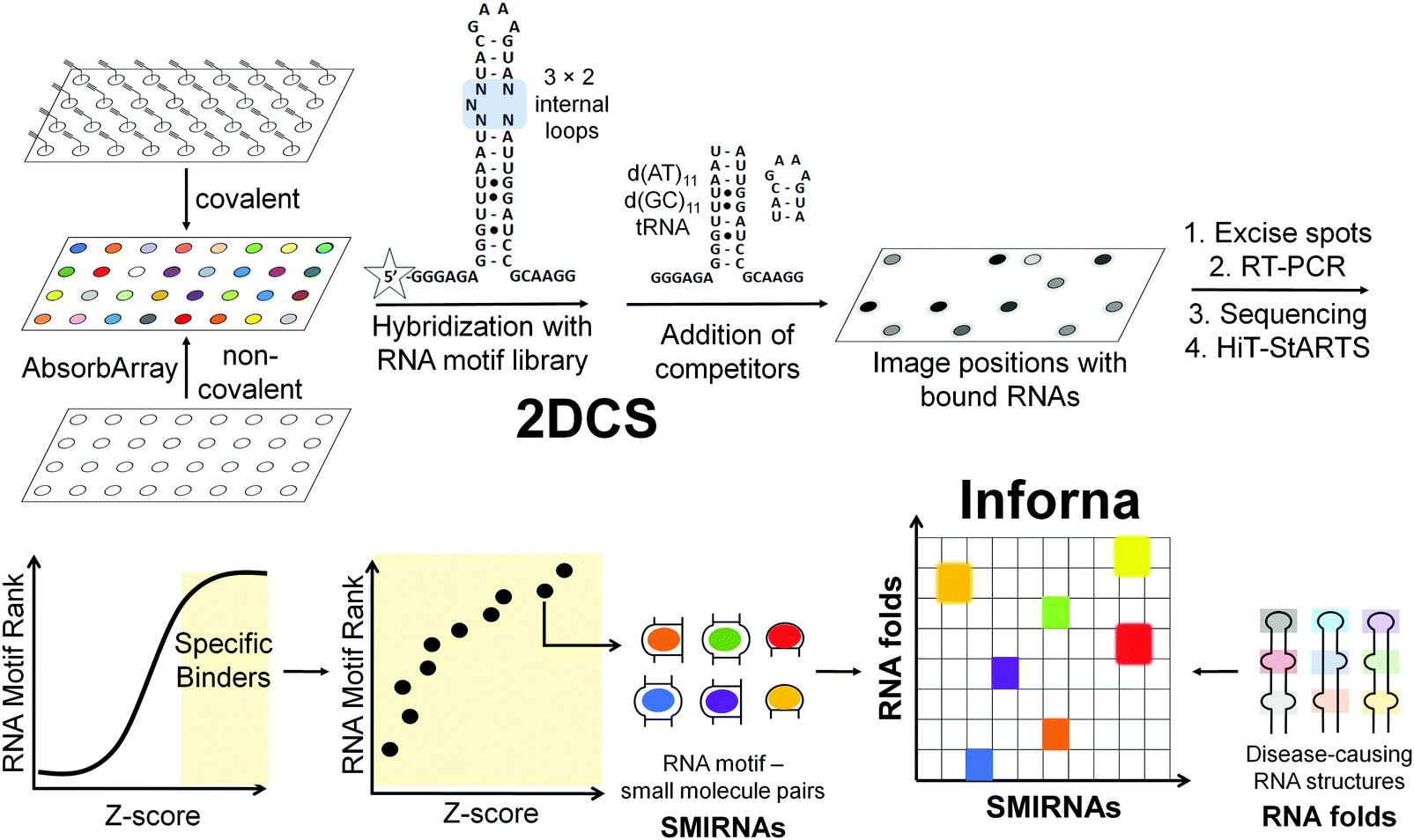 | ||
| Fig. 2 Overview of two-dimensional combinatorial screening (2DCS) and Inforna. In 2DCS, a small molecule library is spatially arrayed onto a microarray, either through covalent attachment or absorption (AbsorbArray). Compounds are then incubated with a labeled RNA motif library, e.g., 3 × 2 internal loops, containing randomized regions that form structured RNA motifs found in disease-causing RNAs. Unlabeled competitor oligonucleotides that mimic regions common to all library members, r(AU) and r(GC) base pairs, DNA oligonucleotides, and other RNAs are added to eliminate non-specific binding. Small molecules that bind RNAs are excised, amplified by RT-PCR, sequenced by RNA-seq, and analyzed by High Throughput Structure–Activity Relationships Through Sequencing (HiT-StARTS). HiT-StARTS calculates the statistical significance of the enrichment of an RNA in the selection, reported as a Z-score (Zobs). Selective small molecule-RNA motif interactions generally exhibit Zobs > 8. These small molecule-RNA motif interactions and their corresponding Z-scores comprise Inforna. Using Inforna, privileged SMIRNAs can be identified for functionally relevant RNA 3D folds within disease-causing RNAs, such as miRNAs. In addition to mining for SMIRNAs with favorable affinity landscapes for the RNA target of interest, Inforna can also predict potential off-target RNAs. | ||
RNAs that bind each small molecule are isolated from the surface of the 2DCS microarray, amplified, and subjected to RNA-seq analysis (Fig. 2).16 Simultaneously, an aliquot of the RNA library that was not incubated with the array is also amplified and analyzed by RNA-seq. The RNA-seq data undergo a rigorous statistical analysis, named High Throughput Structure–Activity Relationships Through Sequencing (HiT-StARTS), where the frequency of each structured RNA 3D fold bound to the small molecule is compared to the frequency of each structured motif in the starting library.17 A pooled population comparison calculates the statistical significance of the enrichment, reported as a Z-score (Zobs) (Fig. 2). We have shown that a Zobs > 8 represents an avid RNA motif-small molecule interaction and that the relative affinity of the interactions for a given SMIRNA directly correlates with Zobs.17 Importantly, the output of 2DCS and HiT-StARTS are privileged RNA 3D fold-small molecule interactions, i.e., the RNA affinity landscape for each small molecule, which informs ligand design and potential off-targets (Fig. 2).
The third key factor to enable the rational design of SMIRNAs is a bioinformatic pipeline to link these privileged interactions to structured 3D folds found in evolutionarily conserved regions of cellular RNAs. Indeed, our lead identification strategy, Inforna (Table S1, ESI†),20 is this pipeline and has enabled the design of many bioactive small molecules that target disease-causing RNAs, as described in Sections 7 and 8.
5. Other methods to identify small molecules that bind RNA
Various other approaches have been developed to identify small molecules that bind structured RNA motifs, particularly structure-based design. As a starting point, structure-based design uses NMR spectroscopy or X-ray crystallography to generate an ensemble of 3D structures for an RNA motif either in its free form or in complex with a small molecule. Then, virtual small molecule libraries can be docked into this ensemble and predicted hit molecules can be ranked according to the free energy of binding to the structured RNA motif. The accuracy of these predictions, however, must subsequently be validated in vitro using various biophysical techniques. Indeed, such combined approaches have been successfully implemented for viral RNAs including the hepatitis C virus (HCV) internal ribosomal entry site (IRES) and HIV transactivation response element (TAR) RNA.21Besides structure-based design, a variety of other high-throughput screening methods have been employed to identify small molecules that bind structured RNA motifs. However, in many cases, such approaches focus on a single RNA target. That is, a library of small molecules is screened against a single structured RNA motif at a time, rather than the thousands of RNA motifs probed in a target agnostic fashion as in 2DCS. These target-centric methods include SMMs,25 which have been used to identify ligands that bind the HIV TAR RNA, among others; fluorescent dye displacement assays or the use of a small molecule's intrinsic fluorescence, which was used to identify small molecules that bind the long noncoding RNA (lncRNA) metastasis associated lung adenocarcinoma transcript 1 (MALAT1);22 or monitoring the change in fluorescence of RNAs containing fluorescent nucleosides23 or end-labeled RNA constructs, which identified small molecule binders to a self-splicing group II intron.
Other emerging high-throughput screening methods for the identification of small molecules binding structured RNA motifs include automated ligand identification system (ALIS), which identifies RNA motif-small molecule binding partners through affinity-selection mass spectrometry (AS-MS), pattern recognition,24 SMM,25 and catalytic enzyme-linked click chemistry assay (cat-ELCCA), which can be used to screen for small molecule inhibitors of miRNA processing in vitro through the use of a system that amplifies chemiluminescence if processing is inhibited.26 Rational design and a variety of other screening methods have also been utilized to identify small molecules that bind RNA repeat expansions.27–29 Extensive reviews of these methods and the small molecules they identified can be found in the following ref. 30–32.
6. Target validation methods
Two challenges in identifying high-quality SMIRNAs is confirming target engagement and quantifying selectivity for the desired target relative to other RNAs featuring identical, similar, or disparate structured RNA motifs.1 Therefore, robust target validation approaches are of key importance to: (i) confirm that phenotype modulation is a direct cause of the RNA-centric mechanism of action of the SMIRNA, i.e., confirming cellular occupancy of the RNA target; and (ii) broadly profile the selectivity of SMIRNAs in a transcriptome-wide manner. Indeed, various target engagement methods have been developed and validated to assess RNA target occupancy in vitro and in cells including cross-linking and cleavage-based approaches as well as competition experiments between a SMIRNA and an ASO.1 These methods, discussed in detail below, are imperative to implement as high-quality SMIRNAs are being developed for current, as well as emerging, RNA targets.Chemical Cross-linking and Isolation by Pull-Down (Chem-CLIP) is a target validation method in which a SMIRNA is appended with nucleic acid cross-linking (e.g., chlorambucil, diazirine) and purification (e.g., biotin) modules at positions that do not affect molecular recognition (Fig. 3).1,33 In cells, the Chem-CLIP probe undergoes a proximity-induced cross-linking reaction upon binding a structured RNA motif. Total RNA is extracted and cross-linked RNAs are isolated and purified by using the purification module, enriching them in the pulled-down fraction. The RNA targets of the Chem-CLIP probe are then identified via RNA-seq or quantitative (q)RT-PCR (Fig. 3). This method can also be used in a competitive fashion (C-Chem-CLIP) to confirm the target occupancy of an unmodified SMIRNA.1 That is, in C-Chem-CLIP, the SMIRNA competes for binding to the same RNA target as the Chem-CLIP probe, which prevents crosslinking and therefore decreases enrichment of the RNA target. Additionally, the Chem-CLIP probe can be used to map binding sites of SMIRNAs in cells via Chem-CLIP-Map-Seq (Fig. 3).1,33 Here, after cross-linking, the bound RNAs isolated from cells are reverse transcribed, PCR amplified, and sequenced. The binding sites of SMIRNAs on RNA targets can then be identified by deconvolution of RT “stops”, which are proximal to the cross-linking sites.
 | ||
| Fig. 3 Methods to validate the targets of SMIRNAs, to study cellular selectivity, and to map SMIRNA binding sites within an RNA target. Schematics of target validation techniques for SMIRNAs. In ASO-Bind-Map, unmodified SMIRNAs are used to prevent hybridization of complementary ASOs, thus preventing cleavage. In Chemical Cross-Linking and Isolation by Pull-Down (Chem-CLIP) and related methods (competitive Chem-CLIP (C-Chem-CLIP) and Chem-CLIP-Map-Seq), SMIRNAs are functionalized with cross-linking (chlorambucil or diazirine) modules and a purification module (biotin) at positions that do not affect binding to the intended RNA target. In small molecule nucleic acid profiling by cleavage applied to RNA (RiboSNAP) and its competitive variant, the SMIRNA is appended with the natural product bleomycin A5. | ||
Complementary to Chem-CLIP is the cleavage-based approach named small-molecule nucleic acid profiling by cleavage applied to RNA (RiboSNAP; Fig. 3), which has been used to confirm target engagement, map binding sites, and profile off-targets of SMIRNAs in vitro and in cells.1,33 In RiboSNAP, a SMIRNA is appended to a nucleic acid cleaving module, such as bleomycin A5,34 at a position that does not contribute to the binding of the SMIRNA to the target (Fig. 3). Attachment of bleomycin A5 via its primary amino group has been shown to eliminate off-target DNA cleavage upon amide bond formation.1 Thus, the bleomycin-SMIRNA conjugate selectively cleaves sequences proximal to the structured RNA motifs engaged by the SMIRNA. Cellular targets of SMIRNAs are then identified through RNA-seq or RT-qPCR, where the abundance of targeted RNAs are reduced as a result of the RiboSNAP probe. Similarly to C-Chem-CLIP, the competitive version of RiboSNAP, coined C-RiboSNAP, can also be employed to study the parent compound (Fig. 3). SMIRNAs that compete with the RiboSNAP probe for the same RNA binding site will reduce the amount of cleavage.1 Cellular mapping of binding sites can also be accomplished with RiboSNAP probes, or RiboSNAP-Map, using RNA target-specific RT primers to identify the cleavage site.1
Although both Chem-CLIP and RiboSNAP have been robustly applied to validate engagement of SMIRNAs with various RNA targets, both require chemical functionalization of the SMIRNA, which can involve laborious, multi-step synthetic procedures. Therefore, the development of label free target validation methods that avoid chemical derivatization of SMIRNAs are highly desirable. As an example, ASO-Bind-Map18 exploits the endogenous activity of RNase H to cleave RNA–DNA heteroduplexes instead of derivatizing the SMIRNA (Fig. 3). To validate target engagement and map the binding site of a SMIRNA using ASO-Bind-Map, ASOs are designed to span the target RNA binding site such that upon RNA–DNA heteroduplex formation, RNase H efficiently cleaves the RNA target. If binding of a SMIRNA, however, thermally stabilizes the RNA binding site or triggers a conformational change that hinders the hybridization of an ASO, cleavage will be inhibited, which can be read out using RT-qPCR or RNA-seq (Fig. 3). ASO-Bind-Map is advantageous over other reagents that are used to map RNA structure and determine binding sites, such as DMS and SHAPE, which require highly resident small molecule interactions that may not be able to inhibit an irreversible reaction with the chemical modifier. Additionally, the sites that react with mapping reagents may not overlap with small molecule binding sites. Collectively, ASO-Bind-Map can confirm the binding site(s) and selectivity of SMIRNAs, both in vitro and in cells. However, unlike Chem-CLIP and RiboSNAP, this method is not target agnostic and cannot be applied across the transcriptome.
Collectively, the target validation methods presented in this section offer unparalleled accessibility to assess RNA target occupancy, profile off-targets, and map binding sites of SMIRNAs in vitro and in cells. Application of these methods early in the development of SMIRNAs is key to developing high-quality chemical probes that modulate disease biology with a defined, RNA-centric mode of action.
7. Targeting disease-causing RNAs with SMIRNAs, enabled by Inforna
Structured RNAs have long been linked to disease,5 making them ideal targets for novel SMIRNAs. For example, dysregulation of miRNA expression has been linked to cancers of the lung, prostate, and breast, cardiovascular disease, inflammatory disorders, and liver disease.35 Additionally, neurotoxic proteins such as SNCA14 and tau13 are encoded by pre-mRNAs featuring unique 3D structured RNA motifs, further substantiating the therapeutic potential of targeting disease-causing RNAs with small molecules. Our lead identification strategy, Inforna,20 can be utilized to assess the ligandability of these disease-relevant RNA 3D folds and rapidly identify privileged SMIRNAs that target these structures and affect disease biology.7.1 The RNA structurome of human miRNA precursors
As mentioned above, miRNAs regulate a myriad of biological processes and their dysregulation triggers a wide variety of human diseases.35 Thus, they are an important class of emergent therapeutic targets. Fortuitously, miRNA precursors fold into accurately predicted structures, forming well-defined structured 3D folds that can be recognized by small molecules. Indeed, blocking miRNA processing sites could directly inhibit miRNA biogenesis, i.e., reduce mature miRNA levels, and consequently deactivate signaling pathways modulated by mature miRNAs.Liu et al.,36 cataloged all structured motifs formed by human miRNA precursor hairpins in an effort to enable lead design by Inforna (Table S1, ESI†). Over 7000 motifs were cataloged, among which small loops, such as 1-nucleotide bulges and 1 × 1 internal loops, were highly represented. These bulges and loops featured various closing base pairs, increasing the overall diversity of structured RNA motifs within the miRNome and hence the ensemble of 3D folds amenable to SMIRNA targeting.
Further, 752 unique functional RNA motifs within Dicer (n = 451) and Drosha (n = 301) processing sites were reported. Among these, only 10 were identified in other highly expressed human RNAs (potential off-targets), rendering the remaining motifs highly valuable as SMIRNA binding sites. That is, there are a plethora of well-defined structured RNA motifs present within the Drosha and Dicer processing sites of miRNAs that could be selectively targeted with SMIRNAs. Access to this database of motifs present within human miRNA hairpin precursors is accessible upon request (Table S1, ESI†).
The optimal interaction from this query, as defined by inspection of affinity landscapes, was between the Drosha site of pri-miR-96, 5′U![[U with combining low line]](https://www.rsc.org/images/entities/char_0055_0332.gif) U/3′AA (1 × 1 UU internal loop), and monomeric compound 96-SM1 (Fig. 4A). We therefore studied the effects of 96-SM1 in more detail, confirming compound mode of action (inhibition of Drosha processing), de-repression of the downstream pro-apoptotic transcription factor Forkhead box protein O1 (FOXO1), and induction of apoptosis. Importantly, knock down of FOXO1 by an siRNA reduced 96-SM1's activity, providing further evidence that the observed rescue of phenotype is through the miR-96-FOXO1 circuit. Additional in cellulis selectivity studies via miRNA profiling by RT-qPCR of detectable miRNAs showed that 96-SM1 significantly affected only miR-96 levels and was as selective as an ASO antagomiR.
U/3′AA (1 × 1 UU internal loop), and monomeric compound 96-SM1 (Fig. 4A). We therefore studied the effects of 96-SM1 in more detail, confirming compound mode of action (inhibition of Drosha processing), de-repression of the downstream pro-apoptotic transcription factor Forkhead box protein O1 (FOXO1), and induction of apoptosis. Importantly, knock down of FOXO1 by an siRNA reduced 96-SM1's activity, providing further evidence that the observed rescue of phenotype is through the miR-96-FOXO1 circuit. Additional in cellulis selectivity studies via miRNA profiling by RT-qPCR of detectable miRNAs showed that 96-SM1 significantly affected only miR-96 levels and was as selective as an ASO antagomiR.
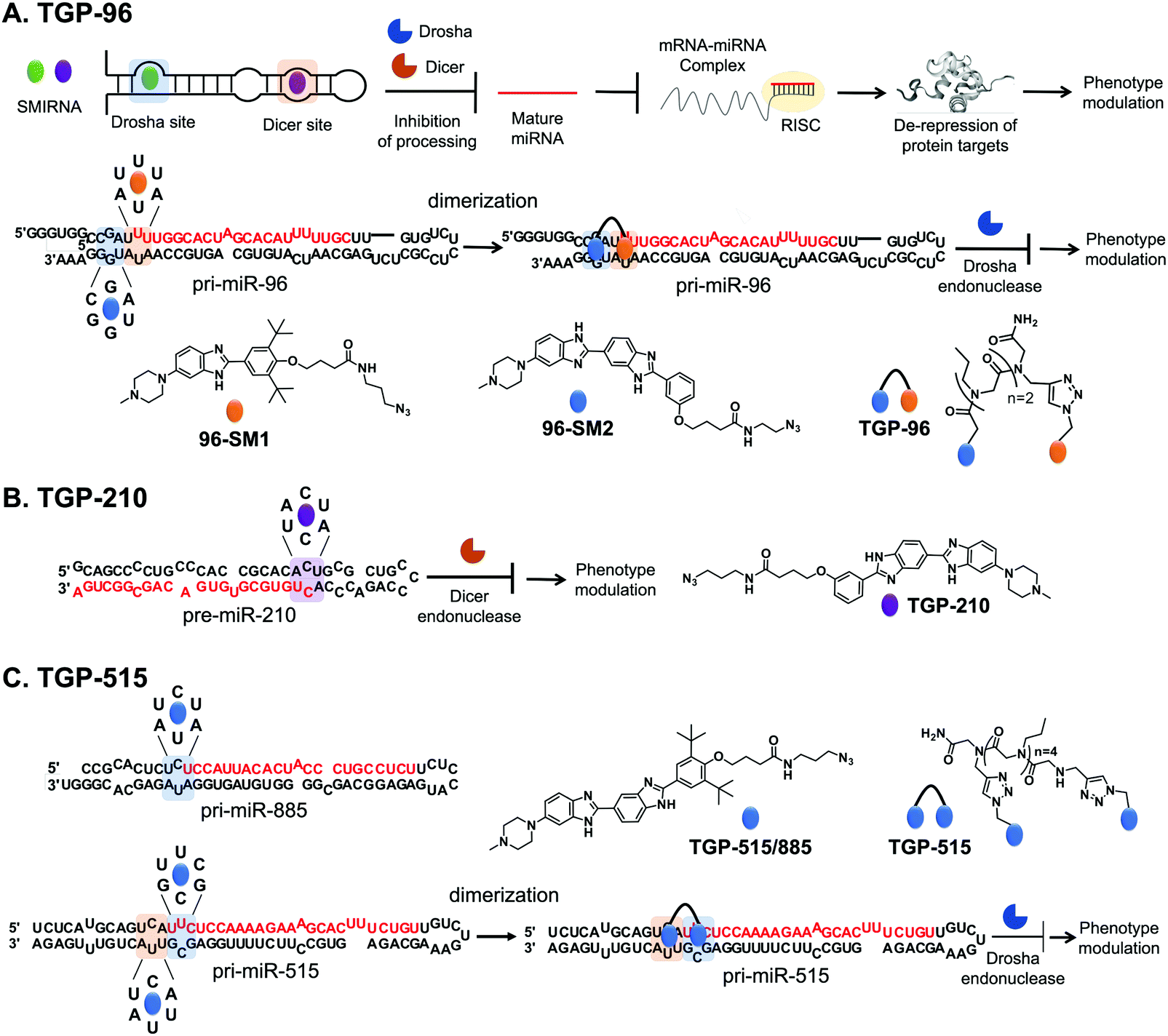 | ||
Fig. 4 Using Inforna to identify SMIRNAs targeting disease-causing miRNAs. (A) Schematic representation of miRNA biogenesis, where SMIRNA binding can inhibit processing by binding to either Drosha or Dicer sites and thereby reduce the levels of the mature miRNA. Reduction of mature miRNA levels results in decreased translational inhibition of target mRNAs by the RNA-induced silencing complex (RISC). Thus, SMIRNA inhibition of miRNA biogenesis derepresses the miRNA's protein targets, resulting in phenotype modulation. Structure of pri-miR-96 and chemical structures of monomeric compounds 96-SM1 and 96-SM2 that target 1 × 1 GG and UU internal loops (blue and orange, respectively) in the Drosha processing site. Covalent attachment of 96-SM1 to 96-SM2via a peptoid linker yields dimeric compound TGP-96, a more potent and selective SMIRNA compared to the monomeric units. Indeed, TGP-96 decreases tumor burden in a mouse xenograft model. (B) Secondary structure of pre-miR-210 and chemical structure of TGP-210, which targets a 1 × 1 CC internal loop in the Dicer processing site (highlighted in purple). (C) Secondary structure of pri-miR-885 and pri-miR-515, with the Drosha processing sites highlighted in blue and the adjacent 5′U![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) A/3′AU motif present in pri-miR-515 highlighted in orange. The chemical structures of monomeric TGP-515/885 and dimeric compound TGP-515 are also shown. TGP-515 is an example of a potent and selective SMIRNA, generated by simultaneously targeting two 1 × 1 CU internal loops near the Drosha processing site. A/3′AU motif present in pri-miR-515 highlighted in orange. The chemical structures of monomeric TGP-515/885 and dimeric compound TGP-515 are also shown. TGP-515 is an example of a potent and selective SMIRNA, generated by simultaneously targeting two 1 × 1 CU internal loops near the Drosha processing site. | ||
Although 96-SM1 inhibited miR-96 levels in cells, its cellular potency (IC50 of ∼20 μM) was not sufficient for in vivo studies. Numerous examples, including this study, have shown that covalently linking monomeric units targeting adjacent structured RNA motifs increases binding affinity and potency.1 We therefore used Inforna to identify SMIRNAs that engage motifs adjacent to pri-miR-96's Drosha site. This search yielded a small molecule binder 96-SM2 (Fig. 4A) of a nearby 5′C![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) A/3′UG (1 × 1 GG) internal loop.37 Linking 96-SM1 and 96-SM2via a peptoid linker afforded dimeric compound Targaprimir-96 (in which “Targa” indicates targets and “primiR-96” indicates pri-miR-96; TGP-96) (Fig. 4A). Notably, the optimal length of the peptoid linker was experimentally determined to mimic the precise distance between the Drosha site and the 1 × 1 GG internal loop.37 Indeed, TGP-96 bound ∼40-fold more tightly to pri-miR-96 than 96-SM1 and ∼30-fold more avidly than 96-SM2. In a triple negative breast cancer (TNBC) cell line, MDA-MB-231, TGP-96 decreased mature miR-96 levels and increased levels of pri-miR-96, as a result of inhibiting Drosha processing at a dose of 50 nM.37 As expected, TGP-96 also boosted levels of FOXO1 and triggered apoptosis, but at an 800-fold lower concentration. Importantly, in this study direct target engagement of pri-miR-96 by TGP-96 in cells was demonstrated using both Chem-CLIP and C-Chem-CLIP. The TGP-96 Chem-CLIP probe was used in a follow-up study to map the exact binding site of TGP-96 within pri-miR-96, the Drosha binding site, which was further validated by RiboSNAP-Map.
A/3′UG (1 × 1 GG) internal loop.37 Linking 96-SM1 and 96-SM2via a peptoid linker afforded dimeric compound Targaprimir-96 (in which “Targa” indicates targets and “primiR-96” indicates pri-miR-96; TGP-96) (Fig. 4A). Notably, the optimal length of the peptoid linker was experimentally determined to mimic the precise distance between the Drosha site and the 1 × 1 GG internal loop.37 Indeed, TGP-96 bound ∼40-fold more tightly to pri-miR-96 than 96-SM1 and ∼30-fold more avidly than 96-SM2. In a triple negative breast cancer (TNBC) cell line, MDA-MB-231, TGP-96 decreased mature miR-96 levels and increased levels of pri-miR-96, as a result of inhibiting Drosha processing at a dose of 50 nM.37 As expected, TGP-96 also boosted levels of FOXO1 and triggered apoptosis, but at an 800-fold lower concentration. Importantly, in this study direct target engagement of pri-miR-96 by TGP-96 in cells was demonstrated using both Chem-CLIP and C-Chem-CLIP. The TGP-96 Chem-CLIP probe was used in a follow-up study to map the exact binding site of TGP-96 within pri-miR-96, the Drosha binding site, which was further validated by RiboSNAP-Map.
Fortuitously, TGP-96 has a favorable drug metabolism and pharmacokinetic profile. In vivo studies using NOD/SCID mice injected with MDA-MB-231 cells to form breast tumors showed that TGP-96 (10 mg kg−1) reduced tumor growth by inhibiting miR-96 biogenesis and increasing FOXO1. Collectively, these studies validated Inforna as a lead identification strategy, utilizing primary RNA sequence to mine small molecules targeting structured 3D folds within disease-causing miRNA. This approach allows for the subsequent modular assembly of identified small molecules to improve the potency and selectivity of SMIRNAs. Ultimately, Inforna provides the means of directly connecting structured 3D folds with privileged small molecule interactions. Moreover, Inforna's SMIRNA predictions readily translate into biological activity in disease-relevant cell lines as a result of the RNA-centric mode of action.
Inforna identified a SMIRNA, Targapremir-210 (TGP-210; Kd ∼ 200 nM), that targets the Dicer processing site of pre-miR-210, which features a 5′AU/3′AU (1 × 1 CC) internal loop (Fig. 4B).39TGP-210 inhibited pre-miR-210 processing by Dicer in vitro and in MDA-MB-231 TNBC cells (IC50 ∼ 200 nM), as demonstrated by decreased levels of mature miR-210 and increased levels of pre-miR-210 and upon compound treatment.39 As a result of inhibiting miR-210 biogenesis, levels of GPDL1 mRNA were increased, HIF-1α mRNA levels were decreased, and apoptosis was triggered selectively in hypoxic MDA-MB-231 cells.38 That is, TGP-210 modulated the hypoxic miR-210-HIF-1α axis via GPDL1. Microarray analysis of all human miRNAs revealed that TGP-210 was selective, similar to a miR-210-targeted antagomiR. Chem-CLIP and C-Chem-CLIP studies showed direct target engagement of both the TGP-210 Chem-CLIP probe and TGP-210 itself.39 In particular, the Chem-CLIP probe selectively enriched miR-210, and this enrichment was depleted by addition of TGP-210. As a further measure of selectivity, the enrichment of other miRNAs that have motifs recognized by TGP-210 as predicted by Inforna, or RNA isoforms, was also measured. Of these 15 RNA isoforms, only miR-497 contained the same 1 × 1 CC internal loop as miR-210, while the other 14 isoforms featured motifs with weaker affinity for TGP-210. Of these 15 miRNAs, the TGP-210-Chem-CLIP-probe only enriched four, including miR-497; however, they were enriched to a lesser extent than miR-210 as they bind TGP-210 less avidly or were expressed less abundantly.39 Importantly, TGP-210 did not inhibit the biogenesis of these enriched miRNAs despite engaging them in cells because binding did not occur in a functional, i.e., Dicer or Drosha processing, site and/or these miRNAs were less abundant and contained weaker affinity motifs. Further, TGP-210 treatment decreased tumor burden in vivo using a NOD/SCID mouse model of hypoxic breast cancer.
Taken together, the study elucidated important insights into SMIRNAs targeting structured RNA motifs. For example, a SMIRNA must engage a functional RNA motif (Dicer site in the case of TGP-210; or Drosha site in the case of TGP-96) within the disease-causing miRNA, and selectivity can be obtained if the target miRNA is expressed at sufficiently higher levels than potential off-targets.
A/3′AUU (miR-515) and 5′UU/3′AA (miR-885), that bind with similar affinity to a small molecule identified by Inforna, Targaprimir-515/885 (TGP-515/885) (Fig. 4C). Further, the processing of both is inhibited to a similar extent in MCF-7 cells.20
In order to selectively target miR-515 over miR-885, Costales et al.,40 employed a modular approach to exploit the differences in the two miRNAs’ 3D folds. In particular, pri-miR-515 features an adjacent 5′UC/3′GG loop not present in pri-miR-885 (Fig. 4C). We therefore used Inforna to identify a small molecule lead for this loop. Tethering the two RNA-binding modules via a linker of precise length afforded Targaprimir-515 (TGP-515) (Fig. 4C). As compared to TGP-515/885, TGP-515 was ∼250-fold more avid and >3200-fold more selective in vitro, validating the modular assembly strategy to bolster binding affinity and selectivity.40 Interestingly, TGP-515 did not bind an RNA with only a singular binding site. This effect can be traced in part to TGP-515's self-structure, acting as a stringency clamp. The increased avidity and selectivity observed in vitro translated in cellulis, where TGP-515 inhibited biogenesis of miR-515, reducing mature levels and boosting pri-miRNA levels, while not affecting miR-885.40 This selectivity was widespread across the miRNome, as determined by RT-qPCR profiling of all miRNAs detectable in MCF-7 cells.40
A key downstream target of miR-515 is sphingosine kinase 1 (SK1) protein that synthesizes sphingosine 1-phosphate (S1P), a second messenger involved in migration. As expected, inhibition of pri-miR-515 by TGP-515 increased levels of both SK1 and S1P. Further, the compound's effect was reduced by both an siRNA directed at SK1 mRNA and a small molecule inhibitor of SK1, validating the compound's mode of action. A proteome-wide study upon TGP-515 treatment revealed that human epidermal growth factor receptor 2 (HER2) was significantly upregulated. Interestingly, MCF-7 cells are HER2-negative, and these results suggest that they may be sensitized to treatment with anti-HER2 precision medicines. Indeed, TGP-515 sensitized MCF-7 cells to Herceptin. In conclusion, this study provided a general strategy to lead optimize a dual-targeting SMIRNA into a single-target, selective compound.
7.2 Targeting the IRE within the 5′ UTR of SNCA mRNA with Synucleozid
ScanFold was used by Zhang et al.,14 to define the structured motif landscape of all human mRNAs encoding IDPs, including SNCA. In this case, ScanFold results were used to determine if these mRNAs were particularly enriched for unusually stable structures (leading to lower average z-scores across the entire mRNA sequence). While IDP-encoding mRNAs overall did not appear to be any more enriched with unusually stable structures than the average mRNA, for each IDP mRNA that was scanned, there was at least one region which contained well-defined, structured RNA motifs. The important finding of ScanFold's results was that structure-less IDPs are produced from intrinsically structured mRNAs, opening up new therapeutic modalities for diseases caused by IDPs. In the SNCA mRNA, for example, 36% of its 3,167 nt contribute to structures that generate significantly low z-scores. These nts are organized into many new structured motifs, beyond the known IRE structure that was recently targeted by Zhang et al.14
G/3′C![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) U A bulge in the IRE, along with other SMIRNAs.14 Only Synucleozid reduced SNCA protein levels in cells without affecting SNCA mRNA expression, and this reduction conferred cytoprotection against cell death caused by aggregation of pre-formed α-synuclein fibrils. Furthermore, selective inhibition of translation was observed as the compound did not affect the translation of other mRNA sequences featuring IREs with different structures in their 5′ UTRs, such as amyloid precursor protein (APP) and prion protein (PrP).
U A bulge in the IRE, along with other SMIRNAs.14 Only Synucleozid reduced SNCA protein levels in cells without affecting SNCA mRNA expression, and this reduction conferred cytoprotection against cell death caused by aggregation of pre-formed α-synuclein fibrils. Furthermore, selective inhibition of translation was observed as the compound did not affect the translation of other mRNA sequences featuring IREs with different structures in their 5′ UTRs, such as amyloid precursor protein (APP) and prion protein (PrP).
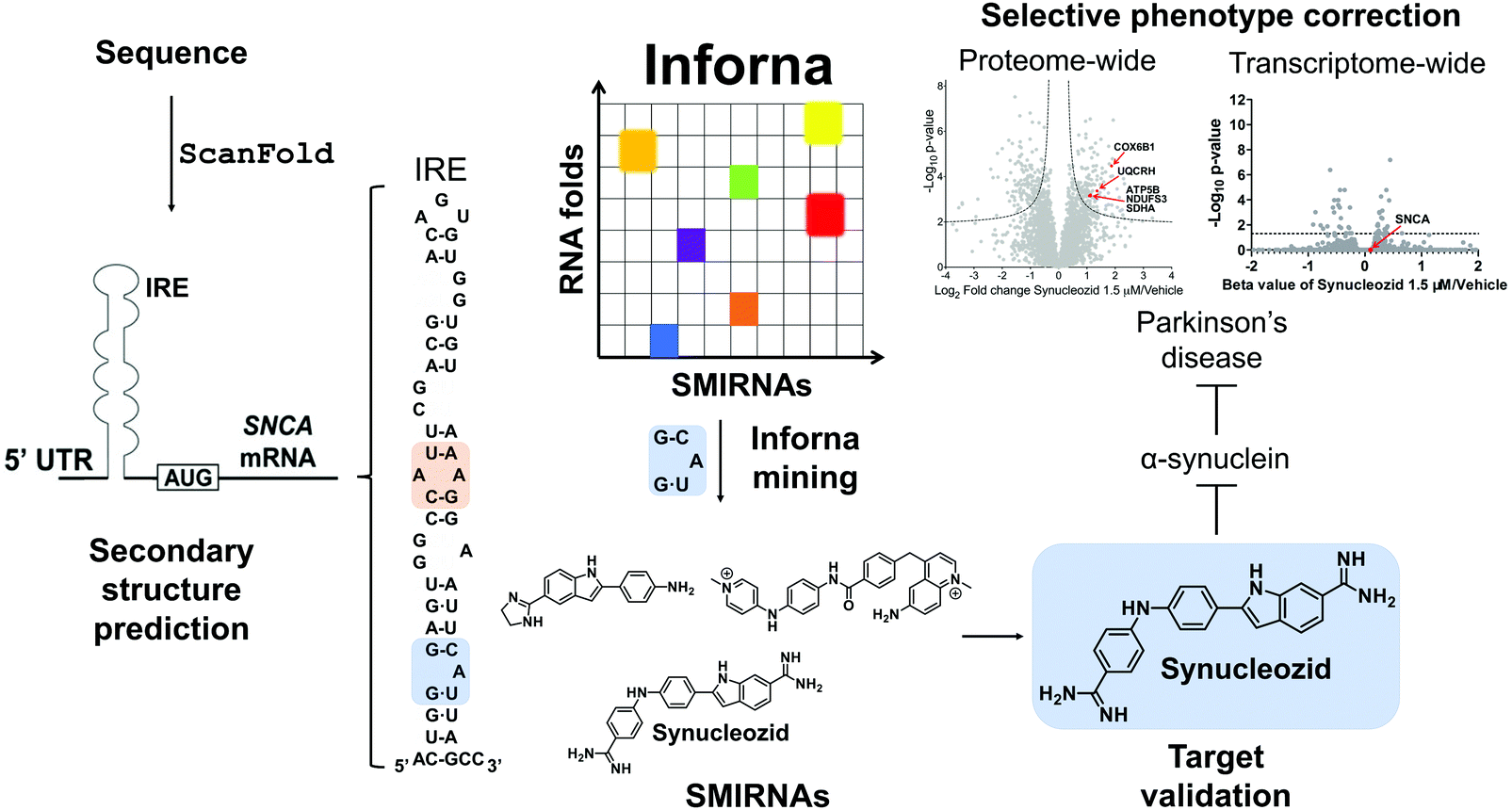 | ||
| Fig. 5 Mining Inforna to identify Synucleozid, which targets the iron responsive element (IRE) within α-synuclein's (SNCA) mRNA and inhibits translation in cellulis. The 5′ UTR of SNCA mRNA sequence contains ligandable structured RNA motifs within the IRE (highlighted in orange and blue). Mining Inforna for small molecules targeting these RNA motifs yielded potential candidates, the most potent of which named Synucleozid binds to the 5′G_G/3′CU A-bulge of the IRE. Among the 3300 proteins detectable in the proteome-wide analysis, only ∼8% were significantly down- or upregulated (p-value < 0.01) upon treatment with Synucleozid (1.5 μM). Various proteins involved in the oxidative phosphorylation pathway, such as the mitochondrial ATP synthase subunit beta (ATP5B), NADH dehydrogenase [ubiquinone] iron–sulfur protein 3 (NDUFS3), cytochrome c oxidase subunit 6B1 (COX6B1), succinate dehydrogenase [ubiquinone] flavoprotein subunit (SDHA), and cytochrome b-c1 complex subunit 6 (UQCRH), were downregulated upon Synucleozid treatment. RNA-sequencing (RNA-seq) analysis revealed limited off-target effects transcriptome-wide (99.7% of the differentially expressed genes were unchanged) following treatment with Synucleozid. Note: Synucleozid has no effect on SNCA RNA levels as its mode of action is binding the RNA and inhibiting its translation. | ||
Target engagement was demonstrated and the exact binding site of Synucleozid was defined both in vitro and in cells using ASO-Bind-Map.14 Careful design of ASOs spanning SNCA's IRE confirmed that Synucleozid targets the 5′G/3′CU structural motif both in vitro and in cells. Optical melting experiments showed that Synucleozid thermally stabilizes the IRE. Cellular mechanistic studies demonstrated that Synucleozid selectively inhibited SNCA's translation via this stabilization, which alters ribosomal loading. Furthermore, proteome- and transcriptome-wide studies showed that Synucleozid exhibited favorable selectivity at both the protein and RNA levels (Fig. 5).
Importantly, transcriptome-wide analysis of mRNAs that encode IDPs revealed that each has structured RNA motifs that could be targeted with small molecules.14 Collectively, these studies demonstrate the potential for targeting proteins with poorly defined tertiary structure at the level of their structured coding mRNAs.
7.3 Targeting MAPT pre-mRNA with SMIRNAs
Chen et al.,13 applied ScanFold to tau's pre-mRNA sequence to explore the existence of structured RNA motifs that may be functionally relevant, and potentially targetable with SMIRNAs (Fig. 6A). Novel structured RNA motifs were discovered, especially at exon–intron junctions and within the 5′ and 3′ UTRs. Twenty structured RNA regions were predicted at the exon–intron junctions. The 5′ UTR contained a single predicted region that overlaps a known IRES, while the 3′ UTR contained eight structured regions. Additional analyses of these structured RNA motifs via luciferase reporters showed their ability to affect stability and splicing of the tau pre-mRNA. In conclusion, ScanFold successfully identified previously validated structured RNA motifs within tau's pre-mRNA and predicted additional motifs that could be targeted with SMIRNAs.
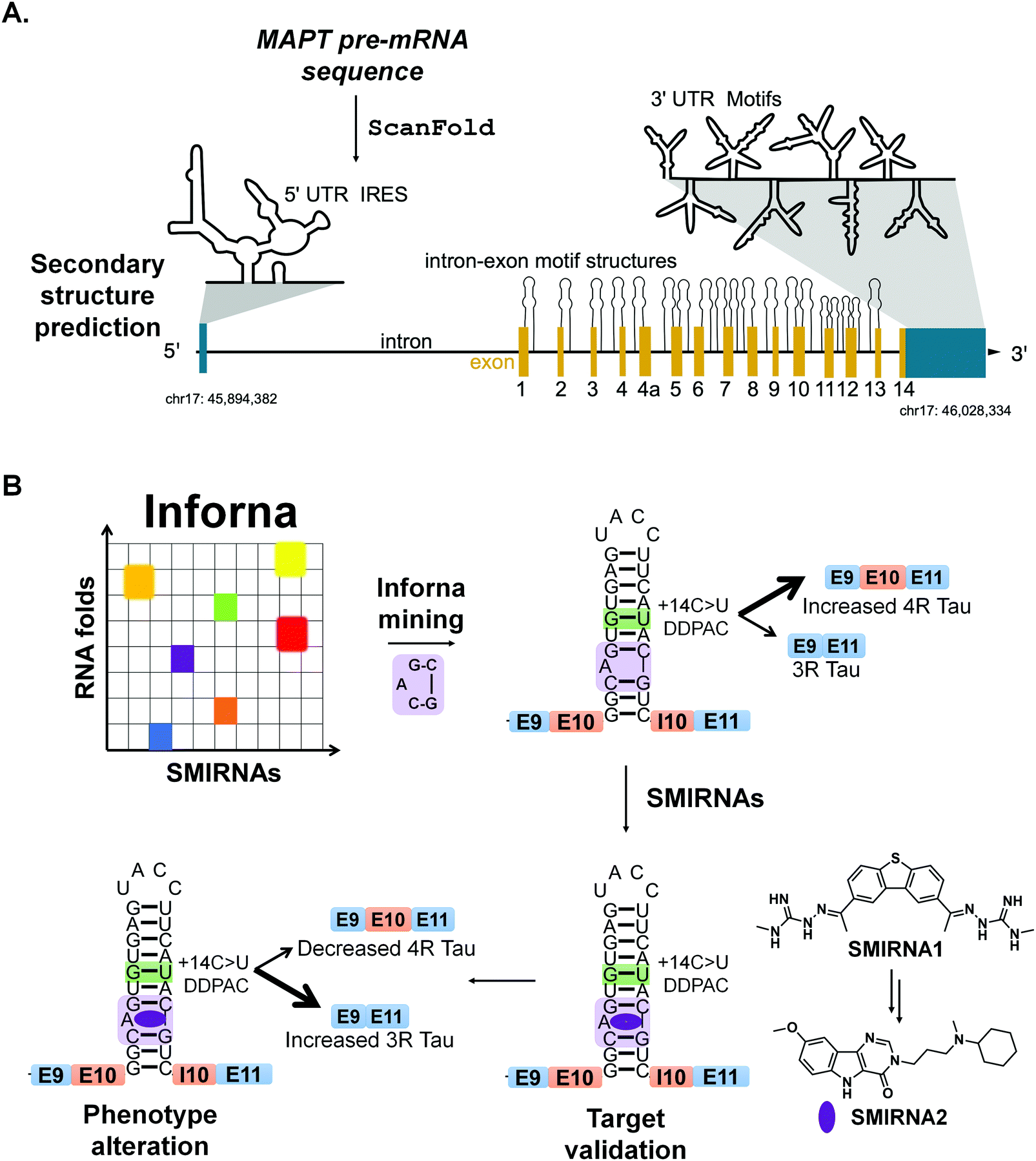 | ||
| Fig. 6 RNA structure prediction and design of SMIRNAs that target structured RNA motifs within tau's pre-mRNA. (A) Secondary structure prediction via Scanfold of microtubule associated protein tau's (MAPT) pre-mRNA sequence. The MAPT pre-mRNA is depicted with 5′ and 3′ UTRs (blue regions), introns (solid, black lines), and exons (yellow regions), along with its chromosomal location (chr17: 45,894,382-46,028,334). The 5′ UTR contains a single, large, structured region that encompasses a known internal ribosome entry site (IRES). ScanFold predicted structured RNA motifs, depicted as hairpins, at exon–intron junctions throughout the MAPT pre-mRNA. These structures are expected to affect which regions are effectively spliced out of the final mRNA product. In the 3′ UTR, eight structured regions were predicted and presumed to confer regulatory effects on mRNA processing. (B) A mutation in MAPT exon 10 (+14C > U, green box around GU pair) destabilizes a splicing regulatory element (SRE) at the exon 10-intron junction, resulting in increased inclusion of exon 10 and increased production of 4R tau. This form of tau is prone to aggregation, triggering neurotoxicity. Chemical similarity searching identified SMIRNA1 that binds the A bulge of the exon 10-intron hairpin (highlighted in purple). Further optimization of SMIRNA1 yielded compound SMIRNA2 with improved properties. Both compounds stabilize the SRE's RNA structure at the exon 10-intron junction, consequently increasing production of 3R tau and reducing production of the aggregation-prone 4R form. | ||
Recently, drug-like small molecules were identified that bind an A bulge, 5′CG/3′G, present in the exon 10-intron junction, that rescued endogenous tau splicing in the human neuroblastoma cell line Lan5 and in primary neurons from an hTau transgenic mouse model (Fig. 6B).43 These small molecules were designed from a previously Inforna-derived compound and by analysis of chemotypes that confer RNA-binding capacity as determined from the Inforna database.43
Particularly, these studies were initiated by searching for chemically similar small molecules related to the substituted 2-phenyl-1H-indole-derived compound discovered via Inforna. We were able to determine the structure of a potent compound, SMIRNA1, that bound to the exon 10-intron junction and reduced exon 10 inclusion in a cell-based reporter of exon 10 splicing (Fig. 5B). The free and bound RNA structures revealed that the A bulge was dynamic, and its conformation changed upon SMIRNA1 binding. These observations enabled a facile, high-throughput binding assay in which the A bulge was replaced with the nucleobase 2-aminopurine (2-AP), the fluorescence emission of which changes with its microenvironment, i.e., stacked or unstacked in a helix. We used this assay as well as a cell-based reporter and docking to identify three new scaffolds from chemical libraries.
As SMIRNA1 was unlikely to be blood–brain barrier (BBB) penetrant, two different hit expansion strategies were employed to identify potent SMIRNAs with favorable physiochemical properties for BBB penetrance, as determined from Central Nervous System Multiparameter Optimization (CNS-MPO) scores.44 CNS-MPO scores quantify favorable physicochemical properties for BBB penetrance, each on a scale from 0–1. These properties include: lipophilicity (clog P), distribution coefficient at pH 7.4 (clog D), molecular weight (MW), topological polar surface area (TPSA), number of hydrogen bond donors (HBD), and pKa values. The scores for each parameter are then summed; a CNS-MPO score ≥4.0 is considered promising for BBB penetrance.44 Applying this CNS-MPO score criterion early in the lead identification and optimization process increases chances of success for developing CNS clinical candidates.
In one method, a pharmacophore model was generated from SMIRNA1 and chemically similar compounds that rescued splicing in a cellular model. In the second hit expansion method, >500 analogs of the three new scaffolds were studied, selected based on their structural similarity and CNS-MPO scores. Of these, SMIRNA2 (Fig. 6B) was the most optimal with enhanced cellular potency and improved physiochemical properties. Indeed, SMIRNA2 rescued aberrant endogenous exon 10 splicing in Lan5 cells and in primary neurons from an hTau transgenic mouse model. Importantly, target engagement studies of SMIRNA2via Chem-CLIP demonstrated that it directly and selectively engaged tau pre-mRNA, as RNAs containing other bulge motifs, such as mRNAs with IREs that regulate translation and miRNAs with the same A bulge, were not enriched. Thus, Inforna can be integrated with traditional medicinal chemistry strategies for the facile lead optimization of drug-like SMIRNAs with improved physiochemical properties.
8. Emerging modalities for targeted degradation of disease-causing RNAs
The studies described above demonstrate the power of Inforna, in concert with computational methods to predict evolutionarily conserved and structured RNA motifs, to design small molecules that modulate RNA function, provided the SMIRNA binds to a functional site. There is ongoing interest, however, to develop new strategies to target any RNA, particularly if a functional site has not yet been identified or validated. Here, we describe two such strategies that employ degradation and cleavage, rather than simple binding, of the RNA target: (i) nuclease recruitment; and (ii) direct cleavage by conjugation of bleomycin A5 to SMIRNAs.1 These cleavage modes of action remove the requirement of the SMIRNA to occupy a functional site as they rid the cell of the RNA altogether. In many cases, discussed below, these small molecule degraders and cleavers are more potent and selective than the occupancy-driven compounds from which they are derived.38.1 Targeted degradation via recruitment of RNase L
Ribonuclease targeting chimeras (RIBOTACs) hijack the cell's endogenous machinery of quality control and degradation pathways to degrade RNA targets selectively.3,45 RIBOTACs are bifunctional, i.e., chimeric compounds, in which one component binds a structured RNA 3D fold and the other locally recruits endogenous RNase L to the RNA target.3,45 In inaugural studies, the RNase L recruiting module was based on RNase L endogenous activator, 2′-5′poly(A),45 but more recently has been replaced with a small molecule heterocycle.3A RIBOTAC was recently developed to target oncogenic miR-21 in cells and in vivo (Fig. 7A). MiR-21 is overexpressed in various types of cancers, and its expression negatively correlates with survival rate in triple negative breast cancer. The RIBOTAC is built on Targapremir-21 (TGP-21), a dimer that binds pre-miR-21's Dicer site and an adjacent U bulge simultaneously (Fig. 7A).3TGP-21 bound pre-miR-21 with ∼20-fold greater affinity than the monomer from which it was derived 21-SM (Kd = 20 μM for 21-SM and 1 μM for TGP-21). Treatment of MDA-MB-231 TNBC cells with TGP-21 reduced mature miR-21 levels and did so selectively across the miRNome, as assessed by miRNA profiling.3 Moreover, the expression levels of phosphatase and tensin homolog (PTEN) and programmed cell death protein 4 (PDCD4), downstream targets of miR-21, increased by ∼50% upon TGP-21 treatment, ultimately leading to reduced invasion of MDA-MB-231 cells.3
 | ||
| Fig. 7 Developing SMIRNAs into chimeric probes that degrade and cleave disease-causing miRNAs. (A) Inforna identified 21-SM that targets the Dicer processing site within pre-miR-21 (highlighted in blue). From monomeric 21-SM, the dimeric compound TGP-21 was generated to target the Dicer processing site and an adjacent bulge (highlighted in orange). A RIBOTAC probe (TGP-21 RIBOTAC) was then synthesized by appending dimeric compound TGP-21 with a small molecule that recruits endogenous RNase L. TGP-21 RIBOTAC more potently and selectively inhibits mature miR-21 levels as a result of the selective RNase L-mediated degradation of pre-miR-21. (B) Inforna identified SMIRNA3 that binds structured RNA motifs (highlighted in green, blue and orange), within the Dicer sites of pre-miR-17, -18a, and -20a in the miR-17-92 cluster. Dimeric compound SMIRNA4 was generated from monomeric SMIRNA3 units connected via a peptoid linker. SMIRNA4 simultaneously targets adjacent bulges present in pre-miR-17, -18a, and -20a, respectively. SMIRNA4 was appended with bleomycin A5 as a cleaving module, yielding SMIRNA4-bleo, and with an RNase L recruiting module, generating SMIRNA4 RIBOTAC. SMIRNA4-bleo selectively ablated the pri-miR-17-92 cluster resulting in a reduction of all mature miRNAs from this cluster. In contrast, SMIRNA4 RIBOTAC only degraded pre-miR-17, pre-miR-18a, and pre-miR-20, as RNase L is cytoplasmic and its interaction is restricted to RNAs present in the cytoplasm. | ||
To increase potency, TGP-021 RIBOTAC was synthesized by conjugating TGP-21 to a heterocyclic small molecule that recruits RNase L (Fig. 7A).3 This RIBOTAC was more potent than TGP-21in cellulis, as assessed by three different metrics: the IC50 for reducing levels of mature miR-21 (IC50 ∼ 0.05 μM for TGP-21 RIBOTACvs. 1 μM for TGP-21),3 boosting PTEN and PDCD4 levels, and rescuing phenotype (invasion). This improved potency can be traced at least partially to TGP-21 RIBOTAC's substoichiometric cleavage, degrading 26 molecules of pre-miR-21 per RIBOTAC molecule. Notably, cleavage was RNase L-dependent as indicated by both gain- and loss-of-function studies. Both miRNome- and proteome-wide studies showed that TGP-21 RIBOTAC is indeed selective.
Comparing the biological activity of TGP-21 and TGP-21 RIBOTAC allowed for direct evaluation between the two modes of action, event-driven RNA degradation of RIBOTACs vs. occupancy-driven binding of SMIRNAs. Treatment with TGP-21 RIBOTAC exhibited a faster, more active and prolonged reduction of miR-21 levels as compared to TGP-21. The selectivity of TGP-21 (dimer binder), 21-SM (monomeric ligand), and TGP-21 RIBOTAC were compared by calculating Gini coefficients from miRNome-wide profiling studies. Gini coefficients range in value from 0 to 1, indicating a non-selective and an exquisitely selective compound, respectively.46 A Gini coefficient considers the percent inhibition of each target analyzed by a small molecule, ranking the targets by the corresponding percent inhibition; that is selectivity is not scored relative to a particular target, rather over the entire target population. We point the reader to ref. 46 for details about how Gini coefficients are calculated. Generally, a compound is considered selective if the Gini Coefficient >0.75. Our studies showed that selectivity can be improved by multivalency as the Gini Coefficients for 21-SM and TGP-21 are 0.52 and 0.68, respectively. Selectivity can be further improved by converting a simple binding compound into a nuclease-recruiting probe, as the Gini Coefficient for TGP-21 RIBOTAC is 0.84.
Importantly, in a mouse model of breast cancer metastasis, TGP-21 RIBOTAC inhibited metastasis to lung, quantified by reduction of lung nodules. This reduction was due to diminished levels of pre- and mature miR-21 and increased expression of PDCD4, validating the RNA-centric mode of action of TGP-21 RIBOTACin vivo.
This study highlighted the comparison of two modes of action that affect cellular levels of mature miR-21. On one hand, occupancy-driven pharmacology exhibited by 21-SM (monomer) and TGP-21 (dimer) reduced mature miR-21 levels by interfering with the Dicer processing of pre-miR-21. On the other hand, a more potent and selective biological activity was achieved via event-driven pharmacology exhibited by RNA degrader TGP-21 RIBOTAC, as a result of degradation of pre-miR-21. Therefore, converting SMIRNAs to RIBOTACs increases potency and selectivity in cells, resulting in a more rapid, effective, and prolonged pharmacological effect in cells and in vivo. Interestingly, the catalytic nature of RIBOTACs and its prolonged effect suggest that ideal, or even perhaps very good pharmacokinetic (PK) properties might not be required to observe a therapeutic effect.
8.2 Direct cleavage of RNA targets by SMIRNAs conjugated to bleomycin A5
Another method to ablate RNA is direct cleavage through conjugation of bleomycin A5, a natural product known to cleave nucleic acids, to a SMIRNA. Through attachment of a SMIRNA at the C-terminal primary amine of bleomycin A5, DNA cleavage is reduced such that off-target DNA cleavage does not occur at concentrations required to cleave the desired RNA target.47 This bleomycin-SMIRNA conjugation strategy has been used to cleave RNA repeats in cells33 and in vivo47 and various miRNAs in cells.In one recent example, a bleomycin-conjugated SMIRNA was used to affect the biology of an entire oncogenic miRNA cluster through cleavage.48 The pri-miR-17-92 cluster is upregulated in various cancers and polycystic kidney disease with the mature miRNAs acting synergistically in some diseases.49 Thus, a method to simultaneously affect all six miRNAs within the 17-92 cluster could be advantageous. Interestingly, three of the miRNAs share a common Dicer site, 5′U/3′CA: pre-miR-17, pre-miR-18a, and pre-miR-20a (Fig. 7B). Pre-miR-17 and pre-miR-20a also share an adjacent G bulge, while pre-miR-18a contains an A bulge (Fig. 7B). Inforna identified a small molecule, SMIRNA3, that binds all three bulges with 30 μM affinity (Fig. 6B). A homodimer, SMIRNA4, was created to target the two bulges simultaneously (Fig. 7B).48 As a simple binding compound, SMIRNA4, inhibited the biogenesis of the three miRNAs in TNBC, prostate cancer, and polycystic kidney disease cells. Interestingly, cellular target engagement studies, revealed that SMIRNA4 bound both pri-miR-17-92 and pre-miR-17, pre-miR-18a, and pre-miR-20a, in agreement with its cellular localization. The dimer de-repressed the corresponding downstream protein in each disease model and rescued phenotype in the two systems in which it was studied (breast and prostate cancer).
Since the occupancy-driven compound demonstrated on-target activity and rescued disease-associated molecular defects in an RNA-centric manner, it was an excellent candidate to employ the direct cleavage approach by conjugation to bleomycin A5, which would allow for the ablation of the entire cluster (Fig. 7B). Indeed, not only did the SMIRNA-bleomycin A5 conjugate, SMIRNA4-bleo, reduce levels of all six mature miRNA in the pri-miR-17-92 cluster, but it also did so more potently than SMIRNA4 while rescuing downstream circuits in three disease models. As many miRNAs are embedded in clusters, a strategy to cleave a cluster in its entirety could have far reaching implications.
Interestingly, this study also converted SMIRNA4 into a nuclease recruiting SMIRNA4 RIBOTAC (Fig. 6B). In contrast, to the SMIRNA-bleomycin A5 conjugate, SMIRNA4 RIBOTAC was only able to cleave pre-miR-17, pre-miR-18a, and pre-miR-20. This is because RNase L is localized to the cytoplasm, meaning SMIRNA4 RIBOTAC can only cleave pre-miRs of the pri-miR-17-92 cluster that are present outside the nucleus. Thus, these studies showed that cellular localization can be used to tune compound activity.
9. Conclusions
Although the pharmaceutical industry remains focused on drugging protein targets, many companies have begun investigating the tractability of drugging RNA targets. Indeed, this movement towards RNA has been bolstered by the success of the splicing modifier risdiplam, and derivatives thereof, that treat spinal muscular atrophy.50 As a result, we are currently experiencing a boom in the identification and validation of druggable human disease-causing RNAs, made possible by advances in sequencing, computation, bioinformatics, chemical probing of RNA structure in vitro and in vivo, biophysical techniques, structural determination by X-ray crystallography and NMR spectroscopy, etc. These interdisciplinary approaches also validate the intimate connection between RNA's 3D structure and its importance in the regulation of biological processes. The ligandability of structured RNA motifs can be achieved by increasingly accurate computational prediction tools, which are easier to implement than various RNA secondary structure chemical probing methods that can be experimentally costly and laborious. For example, ScanFold8 can rapidly identify biologically relevant structured RNA motifs with high probability to form, especially when coupled with chemical probing of RNA structure in cells and in functional biological experiments. The improvement of such tools will offer the scientific community a more accessible visual perspective of RNA structure and its associated 3D folds, which will ultimately translate into establishing more rational approaches to develop SMIRNAs directly from sequence. However, continued research into the fundamental nature of RNA's 3D structure and the ensemble of conformations featured by structured RNA motifs is sure to inform even more advanced target discovery methods.In addition to fully understanding RNA structure and dynamics, an equally important aspect is the identification of chemical matter that potently and selectively interacts with structured RNA motifs, i.e., efficient charting of the chemical space for SMIRNAs. Currently available compound libraries are enriched with small molecules designed and optimized for protein targets and the fraction targeting RNA, in a selective manner, is currently unknown. Therefore, screening technologies such as 2DCS along with other methods mentioned above, will aid in identifying chemical matter that potently and selectively bind structured 3D RNA motifs within disease-causing RNAs.
Performing such campaigns by iteratively integrating chemoinformatic/machine learning/statistical approaches will help populate existing databases, such as Inforna, to: (i) improve understanding of the physicochemical properties, parameters and chemical features of small molecules that mediate RNA binding; and (ii) better design tailored-chemical libraries that are more prone to interact with structured RNA motifs.
As previously observed with small molecule chemical probes of protein targets, high potency and selectivity in vitro does not always translate into on-target activity in cells or in vivo, highlighting the fact that not all chemical matter will be biologically or therapeutically relevant. Therefore, applying target engagement techniques to probe RNA target occupancy by SMIRNAs in cells will help better prioritize chemical scaffolds to be pursued at various stages of chemical probe development. Collectively, these studies will yield the identification of potent and selective SMIRNAs. An array of techniques to assess target engagement to probe RNA-centric modes of action of SMIRNAs have been developed, including Chem-CLIP and Chem-CLIP-Map-Seq,1 RiboSNAP and RiboSNAP-Map,1 RIBOTACs,3 ASO-Bind-Map,1 and SHAPE.9,10
Notably, Chem-CLIP and its competitive version, C-Chem-CLIP, allow for direct assessment of target occupancy via covalent crosslinking reactions that either enrich or deplete, respectively, crosslinked SMIRNA-RNA motifs in pull-down fractions. This technique can be used to simultaneously conduct cellular profiling and binding studies and is advantageous over: (i) non-covalent pull down, which lacks precision in which targets are bound in the purification process; and (ii) competitive profiling with SHAPE or DMS, which leaves many sites unreactive and can generate false negatives as the labeling reaction does not occur under equilibrium.
Taken together, the use of target engagement techniques during early stages of the discovery and development process could mitigate off-target effects of SMIRNAs sooner. Although optimization of potency and selectivity in vitro is important, more relevant for the development of high-quality SMIRNAs is rescue of phenotype via an RNA-centric mode of action, i.e., potent and selective engagement of a biologically relevant structured RNA motif with minimum off-targets proteome- and transcriptome-wide.
An ongoing discussion in the field of small molecule RNA therapeutics is the drug-likeness of SMIRNAs. These semi-empirical rules were historically generated from a pool of approved drugs over a certain interval of time. However, new molecular entities (NME) that were approved since 2002 are deviating from the traditionally considered drug space. Moreover, a recent survey of the approved oral drug space indicated that parameters such as MW and hydrogen-bond acceptors (HBA) have significantly increased over the last 20 year period. Contrarily, over interpretation of ligand and/or drug-likeness metrics might filter out promising chemical candidates. “Drugging” RNA with small molecules is still in its infancy, and using parameters derived from protein-targeted drug campaigns to filter out SMIRNAs featuring “undruglike” properties might hinder the exploratory research that is necessary to advance the field of small molecule RNA therapeutics.
As previously noted, drug targets are unique; thus, the compounds that successfully target them are also unique. RNA-targeted lead and drug discovery campaigns need to be careful not to lose potential candidates due to selection guidelines that are too narrow, particularly for a field that is rapidly evolving. For example, protein–protein interactions (PPIs), featuring relatively large and flat polar surface areas, are traditionally addressed with macrocyclic compounds, that typically reside outside the “Rule of Five” (Ro5), i.e., they are “Beyond Rule of Five” (bRo5). The same principle might very well apply to RNAs, where most potent and selective SMIRNAs with in vivo activity to date are chimeric compounds, e.g., homo- and/or heterodimers. Interestingly, a survey for active ingredients in recently approved bRo5 drugs revealed several examples of chimeric compounds, including HCV NS5A homodimeric inhibitors such as Pibentrasvir, Ledispasvir, Ombitasvir, Daclatasvir, Elbasvir and Velpatasvir. Although these derivatives exhibit poor drug metabolism and pharmacokinetic (DMPK) properties, including low permeability and solubility and high plasma protein binding capacity that limit oral absorption, these liabilities are overcome by delivery to target organs by human serum proteins and their high affinity binding to the target HCV NS5A protein.
Conversely, other bRo5 approved drugs act locally, thus avoiding systemic exposure. The most recent example is Tenapanor, a sodium-proton exchange sodium/hydrogen exchanger 3 (NHE3) inhibitor, approved in 2019 for irritable bowel syndrome with constipation. Tenapanor is minimally absorbed following oral administration in human plasma (below the limit of quantification). To avoid potential systemic toxicity caused by higher doses, Tenapanor was designed to be restricted to the lumen of the gastrointestinal tract, where its target, NH3 protein, is highly expressed. Moreover, there is a growing body of evidence for the potential therapeutic application of chimeric chemical probes, such as proteolysis targeting chimeras (PROTACs), a bleomycin-SMIRNA conjugate (Cugamycin),47 and RIBOTACs.3 Consequently, charting the bRo5 chemical space is likely to reveal novel therapeutically beneficial modalities.
As we continue to identify novel, functional, conserved and structured RNA motifs, these emerging modalities will greatly expand on the types of RNAs that can be targeted with SMIRNAs. In conclusion, exciting times are ahead with the continued exploration of the potential of small molecule chemical probes targeting both functional and non-functional structured RNA motifs to explore RNA biology and affect a broad spectrum of human disorders.
Conflicts of interest
M. D. D. is a founder of Expansion Therapeutics.References
- M. D. Disney, J. Am. Chem. Soc., 2019, 141, 6776–6790 CrossRef CAS.
- D. K. Hendrix, S. E. Brenner and S. R. Holbrook, Q. Rev. Biophys., 2005, 38, 221–243 CrossRef CAS.
- M. G. Costales, H. Aikawa, Y. Li, J. L. Childs-Disney, D. Abegg, D. G. Hoch, S. P. Velagapudi, Y. Nakai, T. Khan, K. W. Wang, I. Yildirim, A. Adibekian, E. T. Wang and M. D. Disney, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 2406–2411 CrossRef CAS.
- S. M. Meyer, C. C. Williams, Y. Akahori, T. Tanaka, H. Aikawa, Y. Tong, J. L. Childs-Disney and M. D. Disney, Chem. Soc. Rev., 2020 10.1039/d0cs00560f.
- T. A. Cooper, L. Wan and G. Dreyfuss, Cell, 2009, 136, 777–793 CrossRef CAS.
- D. H. Mathews, M. D. Disney, J. L. Childs, S. J. Schroeder, M. Zuker and D. H. Turner, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7287–7292 CrossRef CAS.
- R. J. Andrews and W. N. Moss, Biochim. Biophys. Acta, Gene Regul. Mech., 2019, 1862, 194380 CrossRef CAS.
- R. J. Andrews, J. Roche and W. N. Moss, PeerJ, 2018, 6, e6136 CrossRef.
- D. Mitchell, 3rd, S. M. Assmann and P. C. Bevilacqua, Curr. Opin. Struct. Biol., 2019, 59, 151–158 CrossRef.
- C. Feng, D. Chan and R. C. Spitale, Methods Mol. Biol., 2017, 1648, 247–256 CrossRef CAS.
- A. R. Gruber, R. Lorenz, S. H. Bernhart, R. Neubock and I. L. Hofacker, Nucleic Acids Res., 2008, 36, W70–W74 CrossRef CAS.
- R. J. Andrews, L. Baber and W. N. Moss, Sci. Rep., 2017, 7, 17269 CrossRef.
- J. L. Chen, W. N. Moss, A. Spencer, P. Zhang, J. L. Childs-Disney and M. D. Disney, PLoS One, 2019, 14, e0219210 CrossRef CAS.
- P. Zhang, H.-J. Park, J. Zhang, E. Junn, R. J. Andrews, S. P. Velagapudi, D. Abegg, K. Vishnu, M. G. Costales, J. L. Childs-Disney, A. Adibekian, W. N. Moss, M. M. Mouradian and M. D. Disney, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 1457–1467 CrossRef CAS.
- R. J. Andrews, L. Baber and W. N. Moss, Methods, 2019, S1046-2023(19), 30172-0–0 Search PubMed.
- S. P. Velagapudi, M. G. Costales, B. R. Vummidi, Y. Nakai, A. J. Angelbello, T. Tran, H. S. Haniff, Y. Matsumoto, Z. F. Wang, A. K. Chatterjee, J. L. Childs-Disney and M. D. Disney, Cell Chem. Biol., 2018, 25, 1086–1094 CrossRef CAS.
- S. P. Velagapudi, Y. Luo, T. Tran, H. S. Haniff, Y. Nakai, M. Fallahi, G. J. Martinez, J. L. Childs-Disney and M. D. Disney, ACS Cent. Sci., 2017, 3, 205–216 CrossRef CAS.
- J. L. Childs-Disney, T. Tran, B. R. Vummidi, S. P. Velagapudi, H. S. Haniff, Y. Matsumoto, G. Crynen, M. R. Southern, A. Biswas, Z.-F. Wang, T. L. Tellinghuisen and M. D. Disney, Chem, 2018, 4, 2384–2404 CAS.
- C. M. Connelly, F. A. Abulwerdi and J. S. Schneekloth, Jr., Methods Mol. Biol., 2017, 1518, 157–175 CrossRef CAS.
- S. P. Velagapudi, S. M. Gallo and M. D. Disney, Nat. Chem. Biol., 2014, 10, 291–297 CrossRef CAS.
- S. S. Chavali, R. Bonn-Breach and J. E. Wedekind, J. Biol. Chem., 2019, 294, 9326–9341 CrossRef CAS.
- A. Donlic, B. S. Morgan, J. L. Xu, A. Liu, C. Roble, Jr. and A. E. Hargrove, Angew. Chem., Int. Ed., 2018, 57, 13242–13247 CrossRef CAS.
- R. W. Sinkeldam, N. J. Greco and Y. Tor, Chem. Rev., 2010, 110, 2579–2619 CrossRef CAS.
- C. S. Eubanks, J. E. Forte, G. J. Kapral and A. E. Hargrove, J. Am. Chem. Soc., 2017, 139, 409–416 CrossRef CAS.
- F. A. Abulwerdi, W. Xu, A. A. Ageeli, M. J. Yonkunas, G. Arun, H. Nam, J. S. Schneekloth, Jr., T. K. Dayie, D. Spector, N. Baird and S. F. J. Le Grice, ACS Chem. Biol., 2019, 14, 223–235 CrossRef CAS.
- A. L. Garner, D. A. Lorenz and E. E. Gallagher, in Methods Enzymol, ed. A. E. Hargrove, Academic Press, 2019, vol. 623, pp. 85–99 Search PubMed.
- A. H. Jahromi, Y. Fu, K. A. Miller, L. Nguyen, L. M. Luu, A. M. Baranger and S. C. Zimmerman, J. Med. Chem., 2013, 56, 9471–9481 CrossRef CAS.
- R. Simone, R. Balendra, T. G. Moens, E. Preza, K. M. Wilson, A. Heslegrave, N. S. Woodling, T. Niccoli, J. Gilbert-Jaramillo, S. Abdelkarim, E. L. Clayton, M. Clarke, M. T. Konrad, A. J. Nicoll, J. S. Mitchell, A. Calvo, A. Chio, H. Houlden, J. M. Polke, M. A. Ismail, C. E. Stephens, T. Vo, A. A. Farahat, W. D. Wilson, D. W. Boykin, H. Zetterberg, L. Partridge, S. Wray, G. Parkinson, S. Neidle, R. Patani, P. Fratta and A. M. Isaacs, EMBO Mol. Med., 2018, 10, 22–31 CrossRef CAS.
- J. Li, M. Nakamori, J. Matsumoto, A. Murata, C. Dohno, A. Kiliszek, K. Taylor, K. Sobczak and K. Nakatani, Chemistry, 2018, 24, 18115–18122 CrossRef CAS.
- J. Sztuba-Solinska, G. Chavez-Calvillo and S. E. Cline, Bioorg. Med. Chem., 2019, 27, 2149–2165 CrossRef CAS.
- C. M. Connelly, N. H. Moon and J. S. Schneekloth, Cell Chem. Biol., 2016, 23, 1077–1090 CrossRef CAS.
- A. Di Giorgio and M. Duca, MedChemComm, 2019, 10, 1242–1255 RSC.
- S. G. Rzuczek, L. A. Colgan, Y. Nakai, M. D. Cameron, D. Furling, R. Yasuda and M. D. Disney, Nat. Chem. Biol., 2017, 13, 188–193 CrossRef CAS.
- D. L. Boger and H. Cai, Angew. Chem., Int. Ed., 1999, 38, 448–476 CrossRef CAS.
- A. M. Ardekani and M. M. Naeini, Avicenna J. Med. Biotechnol., 2010, 2, 161–179 CAS.
- B. Liu, J. L. Childs-Disney, B. M. Znosko, D. Wang, M. Fallahi, S. M. Gallo and M. D. Disney, BMC Bioinf., 2016, 17, 112 CrossRef.
- S. P. Velagapudi, M. D. Cameron, C. L. Haga, L. H. Rosenberg, M. Lafitte, D. R. Duckett, D. G. Phinney and M. D. Disney, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5898–5903 CrossRef CAS.
- T. J. Kelly, A. L. Souza, C. B. Clish and P. Puigserver, Mol. Cell. Biol., 2011, 31, 2696–2706 CrossRef CAS.
- M. G. Costales, C. L. Haga, S. P. Velagapudi, J. L. Childs-Disney, D. G. Phinney and M. D. Disney, J. Am. Chem. Soc., 2017, 139, 3446–3455 CrossRef CAS.
- M. G. Costales, D. G. Hoch, D. Abegg, J. L. Childs-Disney, S. P. Velagapudi, A. Adibekian and M. D. Disney, J. Am. Chem. Soc., 2019, 141, 2960–2974 CrossRef CAS.
- A. Grover, H. Houlden, M. Baker, J. Adamson, J. Lewis, G. Prihar, S. Pickering-Brown, K. Duff and M. Hutton, J. Biol. Chem., 1999, 274, 15134–15143 CrossRef CAS.
- Y. Liu, E. Peacey, J. Dickson, C. P. Donahue, S. Zheng, G. Varani and M. S. Wolfe, J. Med. Chem., 2009, 52, 6523–6526 CrossRef CAS.
- J. L. Chen, P. Zhang, M. Abe, H. Aikawa, L. Zhang, A. J. Frank, T. Zembryski, C. Hubbs, H. Park, J. Withka, C. Steppan, L. Rogers, S. Cabral, M. Pettersson, T. T. Wager, M. A. Fountain, G. Rumbaugh, J. L. Childs-Disney and M. D. Disney, J. Am. Chem. Soc., 2020, 142, 8706–8727 CrossRef.
- T. T. Wager, X. Hou, P. R. Verhoest and A. Villalobos, ACS Chem. Neurosci., 2010, 1, 435–449 CrossRef CAS.
- M. G. Costales, B. Suresh, K. Vishnu and M. D. Disney, Cell Chem. Biol., 2019, 26, 1180–1186 CrossRef CAS.
- P. P. Graczyk, J. Med. Chem., 2007, 50, 5773–5779 CrossRef CAS.
- A. J. Angelbello, S. G. Rzuczek, K. K. McKee, J. L. Chen, H. Olafson, M. D. Cameron, W. N. Moss, E. T. Wang and M. D. Disney, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 7799–7804 CrossRef CAS.
- X. Liu, H. S. Haniff, J. L. Childs-Disney, A. Shuster, H. Aikawa, A. Adibekian and M. D. Disney, J. Am. Chem. Soc., 2020, 142(15), 6970–6982 CrossRef CAS.
- P. Mu, Y. C. Han, D. Betel, E. Yao, M. Squatrito, P. Ogrodowski, E. de Stanchina, A. D’Andrea, C. Sander and A. Ventura, Genes Dev., 2009, 23, 2806–2811 CrossRef CAS.
- H. Ratni, M. Ebeling, J. Baird, S. Bendels, J. Bylund, K. S. Chen, N. Denk, Z. Feng, L. Green, M. Guerard, P. Jablonski, B. Jacobsen, O. Khwaja, H. Kletzl, C.-P. Ko, S. Kustermann, A. Marquet, F. Metzger, B. Mueller, N. A. Naryshkin, S. V. Paushkin, E. Pinard, A. Poirier, M. Reutlinger, M. Weetall, A. Zeller, X. Zhao and L. Mueller, J. Med. Chem., 2018, 61, 6501–6517 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cs00455c |
| This journal is © The Royal Society of Chemistry 2020 |