
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh elasticity, chemically recyclable, thermoplastics from bio-based monomers: carbon dioxide, limonene oxide and ε-decalactone†
Leticia Peña
Carrodeguas
,
Thomas T. D.
Chen
,
Georgina L.
Gregory
 ,
Gregory S.
Sulley
and
Charlotte K.
Williams
*
,
Gregory S.
Sulley
and
Charlotte K.
Williams
*
Chemistry Research Lab, Department of Chemistry, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: charlotte.williams@chem.ox.ac.uk
First published on 3rd November 2020
Abstract
One solution to problems with petroleum derived plastics is to design polymers for the circular economy. In this regard, polymer chemistries, like ester or carbonate linkages, which are closer to equilibrium are very promising but to use these materials requires improvements to their properties and methods of manufacture. Here, efficient polymerization catalyses are used to transform wastes and bio-sourced monomers into thermoplastics which combine high elasticity and strength and which can be degraded to allow for some chemical recycling. The plastics are prepared from carbon dioxide, limonene oxide (from waste citrus fruit peel) and ε-decalactone (from triglycerides). These monomers are polymerized, using catalyzed controlled polymerizations with high conversion efficiencies, to selectively form ABA block polymers (A = high Tg polycarbonate, B = low Tg polyester). The series of 5 poly(limonene carbonate)-b-poly(ε-decalactone)-b-poly(limonene carbonate) (PLC-PDL-PLC) samples allow for systematic variations in the overall molar masses (Mn = 50–100 kg mol−1) and hard-block compositions (21–63 wt% PLC). All the polymers are fully characterized using a range of spectroscopies, gel permeation chromatography, thermal and tensile mechanical measurements. The leading plastic combines tensile strength (stress at break, σb = 21.2 MPa, Young's Modulus, Ey = 321 MPa) and high elasticity (elongation at break, εb = 400%) – an enhancement of more than 20× in elongation at break and tensile toughness over poly(limonene carbonate), overcoming the well-known brittleness and processing limitations of PLC. It undergoes selective, catalyzed depolymerization to limonene oxide, carbon dioxide and the precursor polyester providing a future chemical recycling and upcycling opportunity.
Introduction
Today's plastics have linear economies: the vast majority are sourced from petroleum, are applied in a myriad of applications but at end-life, recycling is not efficiently implemented and there is a growing problem of environmental pollution from waste plastics.1–3 In terms of thermoplastic chemistry, current structures are not optimized for end-of-life recycling with mechanical reprocessing often rapidly deteriorating properties and devaluing applications.4 Chemical recycling, i.e. depolymerization to monomer, should allow for increased circularity without compromising the materials’ properties but is complicated by the additives needed to enhance polymer properties/processing and its high energy requirement, as common polymers feature thermodynamically stable C–C bonds.5 To design polymers for a future circular economy, many researchers have focussed on polymer chemistries which are closer to equilibrium such as polyesters and polycarbonates.5 These materials have the added advantage that they may be prepared from bio-based raw materials, such as agricultural wastes or waste industrial gases like carbon dioxide and their use may reduce the greenhouse gas emissions associated with polymer manufacturing.6–10 To facilitate the transition from oil-based plastics, the production of both bio-based monomers and polymers must be efficient and conform to the principles of green chemistry.2,11 Despite significant success in the syntheses, bio-derived polymers still show material property limitations.12,13 For example, many have low tensile strength and are highly brittle; they require the addition of plasticizers for subsequent material processing and use.13 Unfortunately, using plasticizers presents toxicological and environmental concerns, as well as complicating recycling.14 One solution may be to develop polymers which reduce, or even obviate, plasticizer use. Another benefit would be to design polymers which facilitate chemical recycling to monomers, especially where depolymerization occurs with lower energy than is needed for hydrocarbons.5,15–17 Naturally there is a trade-off between the requirement for sufficient high temperature stability to allow for conventional processing and use, and the need for energy-efficient chemical recycling by thermally activated depolymerization chemistry.5 Here, a series of ABA block polymers, featuring carbonate and ester linkages, are designed to meet the following criteria: (1) They are fully bio-derived and maximize use of industrial wastes; (2) They improve upon existing bio-derived plastics’ properties, specifically tackling the reduction in plasticizer use and (3) They allow for selective and catalyzed chemical recycling. The structures are fully amorphous poly(limonene carbonate)-b-poly(ε-decalactone)-b-poly(limonene carbonate) (PLC-PDL-PLC), where the PC functions as the hard, high Tg A-block and PDL is the soft, low Tg B-block.The hard block is prepared by the ring-opening copolymerization (ROCOP) of epoxides and carbon dioxide – a rare example of an efficient and economically viable carbon dioxide utilization process amenable to large-scale operation.18 The polymerization catalyst is essential for efficient and cost-effective processes with various homogeneous metal complexes showing good performances.19–23 The process may also work using captured carbon dioxide and could be coupled with carbon capture and sequestration.24 So far, epoxide/CO2 ROCOP is generally applied with petrochemical epoxides, but when bio-derived epoxides are used, it's feasible to produce fully renewable polycarbonates.25,26 One of the most successful examples is poly(limonene carbonate) (PLC) prepared from (R)-limonene oxide, naturally occurring as a mixture of cis and trans isomers, which is extracted from waste citrus fruit peel.27 In 2004, Coates and co-workers reported the first catalyst for limonene oxide/CO2 copolymerization, a zinc β-diiminate complex, and the properties of poly(limonene carbonate) (PLC).28 Later, Coates and co-workers investigated PLC stereocomplexes prepared by co-crystallization of PLC enantiomers.29,30 Others used similar zinc catalysts to increase the molar mass of PLC (Mn > 50 kg mol−1) and investigated it as an engineering plastic, antimicrobial material (after post-functionalization) or gas permeable film.31–33 Higher molar mass PLC is distinctive, compared to other bio-based plastics, because of its unusually high glass transition temperature (Tg = 130 °C) and its high tensile strength (σ = 55 MPa), but its applications are severely limited by its poor elongation at break values of ∼15% – i.e. it is very brittle.32,34 One other process limitation is that the zinc-β-diiminate catalysts only polymerize trans-limonene oxide, thereby giving rise to significant left-over cis-limonene oxide (∼40%).28 Kleij and co-workers reported an Al-(amine)tris(phenolate) catalyst applied in conjunction with an ionic co-catalyst, PPNCl, which copolymerizes both cis- and trans-LO.35,36 So far, this Al-catalyst system yields only low molar mass PLC,35,36 which can be used to make networks and resins, but cannot deliver useful properties on its own.37–41
One strategy to improve the properties of higher molar mass PLC would be to blend it with other polymers, but because most polymers are mutually incompatible, PLC containing block polymers would be required both as blend compatibilizers or to increase molar mass and optimize properties.42–52 For example, phase separated ABA block polymers, where A = hard, high Tg polymer and B = soft, low Tg polymer, are commercial thermoplastic elastomers, plastomers and blend compatibilizers.7 Greiner and co-workers reported a higher molar mass block polycarbonate, PLC-b-PCHC [PCHC = poly(cyclohexene carbonate)], which undergoes phase separation, but its properties are sub-optimal since both PCHC and PLC are high Tg polymers – i.e. the plastic remains very brittle.42 Rieger and co-workers reported PLC-b-PBL [PBL = poly(β-butyrolactone)], prepared using a switchable catalytic method, to achieve a material showing 18% elongation at break using ∼50 wt% PBL.49,53 This block polymer does not significantly overcome the property limitations either but signals that B-block polymers with considerably lower Tg and greater flexibility than atactic PBL (Tg = 5 °C) should be explored. In this context, poly(ε-decalactone) (PDL) is promising since it is both bio-sourced, from castor oil, and has a very low glass transition temperature (Tg = −60 °C).7,43,54–59 Hillmyer and co-workers have pioneered ABA block polyesters, where A = polylactide (PLA) and B = PDL, as thermoplastic elastomers.7,43,54–57,60 Our group, have studied ABA polymers, where A = poly(cyclohexene phthalate), or poly(cyclohexene carbonate) and B = PDL, as thermoplastic elastomers, rigid plastics and adhesives.43,61,62 Given the property improvements enabled by the use of PDL, ABA materials combining poly(limonene carbonate) and poly(ε-decalactone) merit investigation and, here, PLC-b-PDL-b-PLC are presented.
Results
The synthesis of PLC-b-PDL-b-PLC block polymers requires the combination of DL ring opening polymerization (ROP) and the LO/CO2 ROCOP. Whilst many catalysts are able to efficiently perform lactone ROP, there are far fewer options for LO/CO2 ROCOP. In this work, the Al catalyst, previously reported by Kleij and co-workers, was chosen due its ability to enchain both cis- and trans-LO.36 However, a limitation is its failure to perform DL ROP. To overcome this problem, PDL was synthesized by DL ROP (conversion >90%), using a dizinc macrocyclic catalyst, [LZn2Ph2] (1), initiated using trans-1,2-cyclohexane diol (CHD) at 100 °C (Scheme 1, (i)). This catalyst was selected as it is highly active and selective for DL ROP.52,60 Furthermore, the catalyst has a non-initiating co-ligand (phenyl) which is important because when combined with the diol co-initiator it selectively yields only hydroxyl telechelic PDL.62,63 The resulting hydroxyl-telechelic PDL shows molar masses up to 67 kg mol−1, with narrow dispersities (Đ ≤ 1.13). Using the hydroxyl telechelic PDL as a macroinitiator for LO/CO2 ROCOP, using just the Al-catalyst, (2), was unsuccessful and the polymerization failed to initiate. It was hypothesized that the co-catalyst, i.e. bis(triphenylphosphine)iminium chloride (PPNCl), was essential for reactivity as a control experiment utilizing 2 for LO/CO2 ROCOP, in the absence of PPNCl, also failed to convert any monomer. For block polymer synthesis, a PPNCl co-catalyst should be avoided because as the chloride ion would compete with the diol as an initiator which would result in a mixture of α-chloride-ω-hydroxyl and α,ω-dihydroxyl terminated polymer chains and hence a mixture of different block polymer structures (Fig. S1†). To overcome this issue, the PDL chain ends were reacted to afford PPN-carboxylate end-groups, [PPN]2[PDL] (Scheme 1, (ii)–(iv)).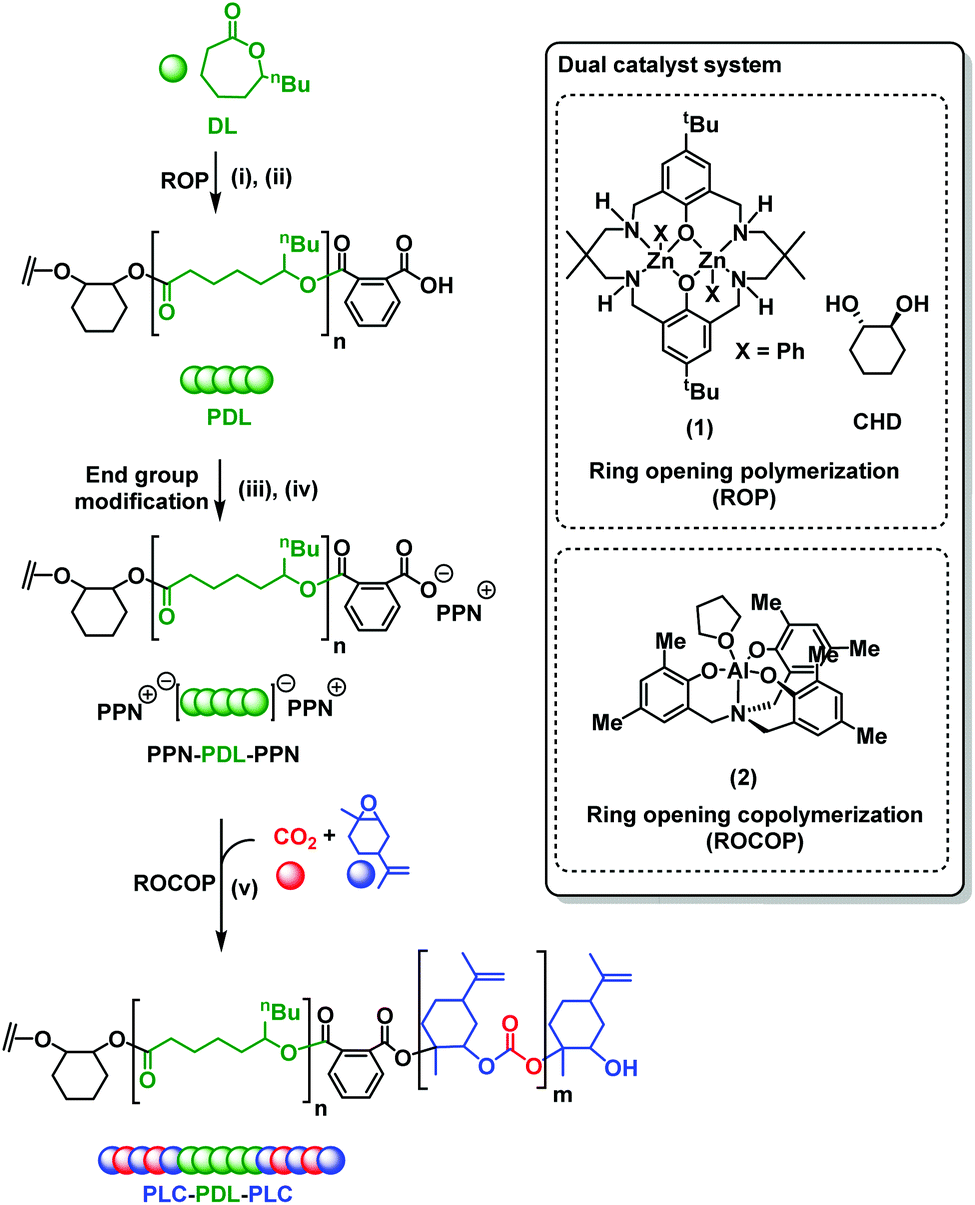 | ||
Scheme 1 Block polymer synthesis. (i) 1,2-cyclohexane diol (CHD), ε-decalactone (DL), 2 h, 100 °C, toluene. (ii) Phthalic anhydride (PA), 16 h, 100 °C, molar ratio: [1]/[CHD]/[PA]/[DL] = 1/4/100/350–1000. (iii) NaOH, 2 h, 25 °C, chloroform. (iv) PPNI, 2 h, 40 °C. Molar ratio: [diacid end group]/[NaOH]/[PPNI] = 1/1/1. (v) [2]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[PPN-PDL-PPN]:[LO] = 1:0.25:125 (LO used as monomer and solvent), 24 h, 20 bar CO2, 45 °C. :[PPN-PDL-PPN]:[LO] = 1:0.25:125 (LO used as monomer and solvent), 24 h, 20 bar CO2, 45 °C. | ||
The synthesis involved reaction of the hydroxyl terminated PDL with two equivalents of phthalic anhydride (PA) to obtain diacid terminated PDL (Scheme 1, (ii)). This was followed by deprotonation, using NaOH, and subsequent cation exchange with bis(triphenylphosphine)iminium iodide (PPNI) (Scheme 1, (iii)–(iv), see ESI† for experimental details). PPNI was employed (instead of PPNCl) as control studies suggested the iodide ions cannot initiate the ROCOP of LO/CO2. Using the [PPN]2[PDL]/2 catalyst system, the ROCOP of LO/CO2 (20 bar CO2 pressure) was performed at 45 °C to obtain PLC-b-PDL-b-PLC (Scheme 1, (v)).
Using this strategy, five different fully renewable ABA-polymers were synthesized with molar mass up to 115 kg mol−1 and with controllable compositions (expressed as wt% PLC) (Table 1). By adjusting the monomer concentrations and reaction times, the compositions was varied from 21–60 wt% PLC, whilst keeping the overall molar mass constant (∼80 kg mol−1) (Table 1, entries 1–3). To understand the influences of polymer molar mass on properties, two samples with high PLC content (63 and 42 wt%) but differing molar masses of 50 and 115 kg mol−1 were prepared (Table 1, entries 4 and 5).
| PDLa | PLC-b-PDL-b-PLCb | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| # | DL (equiv.) | M n (kg mol−1) [Đ]c | M n (kg mol−1) [Đ]c | wt% PLCd | DPe | ABA NAMEf |
T
gg (°C) |
T
d,5%i (°C) |
Yield (%) |
|
a ROP reaction conditions: [1]:[CHD] = 1:4, 100 °C, toluene.
b ROCOP reaction conditions: [2]:[PPN-PDL-PPN]:[LO] = 1:0.25:125, 20 bar CO2, 45 °C.
c
M
n and Đ measured by SEC (THF eluent, calibrated using polystyrene standards).
d Weight % of PLC block, determined from the 1 H NMR spectra, by analysis of the integrals of PLC and PDL main chain peaks at 5.1–5.2 ppm and 4.85 ppm, respectively.
e Degree of polymerization calculated from Mn and molar weight of the repeating unit.
f Triblock polymer naming system (total Mn by SEC, wt% PLC by NMR integration).
g Glass transition temperature obtain from DSC (third heating cycle, 10 °C min−1 heating rate).
h Upper Tg measured by DMTA from peak in tanδ (n.d. = not determined).
i Thermal decomposition temperature at 5% weight loss, measured by TGA (25–500 °C, 10 °C min−1 heating rate, N2 flow).
|
|||||||||
| 1 | 500 | 33.4 [1.28] | 86.9 [1.17] | 60 | 133–196–133 | ABA (87,60) | −50, 50h | 197 | 22 |
| 2 | 750 | 40.1 [1.08] | 81.0 [1.09] | 48 | 94–236–94 | ABA (81,48) | −47, n.d. | 239 | 19 |
| 3 | 1000 | 62.7 [1.13] | 84.8 [1.04] | 21 | 56–368–56 | ABA (85,21) | −40, n.d. | 178 | 24 |
| 4 | 350 | 25.1 [1.19] | 50.7 [1.06] | 63 | 63–147–63 | ABA (51,63) | 0, n.d. | 184 | 41 |
| 5 | 1000 | 61.9 [1.14] | 114.6 [1.14] | 42 | 134–364–134 | ABA (115,42) | −51, n.d. | 177 | 28 |
The triblock samples were purified by precipitation of a dichloromethane solution containing the reaction mixture into pentane three times. In terms of polymer characterization data, the 1H and 13C{1H} NMR spectra show peaks corresponding to both the blocks (Fig. S2 and 3†). No transesterification/carbonylation between the blocks was observed, as apparent from the 13C{1H} NMR spectrum which shows only PLC and PDL carbonyl peaks with no additional cross-peaks. Selectivity for carbon dioxide uptake is very high (>99%) with no ether linkages (at 3.00–3.50 ppm) being observed. Polymerizations are well controlled as indicated by a shift in molar mass values and retention of narrow dispersities as the reactions progress. For instance, the SEC analysis of ABA (81,48) showed an Mn increase from 40.1 to 81.0 kg mol−1 with retention of a narrow dispersity of ∼1.09 (Fig. S4†). Similar shifts in molar mass and retention of narrow dispersity are seen for the other samples (Fig. S5–8†). In some cases, the SEC traces show low intensity ‘shoulder’ peaks which are tentatively assigned to PLC homopolymer, possibly due to low quantities of water leading to limonene diol formation and its functioning as an initiator.23,64 This lower molar mass PLC can be separated from the desired triblock polymers by repeated pentane precipitations. The 31P{1H} NMR spectroscopy end group analysis of the PDL showed the characteristic hydroxyl end groups, at 147.1 ppm (Fig. S9†). After reaction with PA, the hydroxyl end groups disappeared and a new peak at 135.5 ppm appeared. This NMR data suggests all the PDL chains are end capped with di-acid groups. After the LO and CO2 ROCOP, the di-acid end group peak disappeared and a series of peaks, centred at 141.5 ppm, are observed. The peaks are identical to a spectrum of PLC and the slightly differing chemical shifts arises from the regioirregularity.
Thermal properties
The polymers’ thermal properties were analyzed by differential scanning calorimetry (DSC), dynamic mechanical thermal analysis (DMTA) and thermogravitmetric analysis (TGA). For ABA (87,60), DSC measurements clearly revealed a Tg at −50 °C, attributed to the ‘soft’ PDL segment (Fig. 1A) and closely comparable to PDL homopolymer (−58–51 °C). The block polymer value is far from that calculated for fully miscible PLC and PDL blocks (∼27 °C),57,65 which suggests partial block phase separation. From previous work, using DSC to realiably observe the upper Tg is challenging, perhaps due to the low χ value in this class of triblock polymers. To observe the upper glass transiton temperature the more sensitive technique of DMTA has proved more successful.61,62 DMTA of a sample of ABA (87,60) clearly showed the upper Tg at +50 °C, as the maximum in tan(δ) (ratio between the storage and loss moduli) (Fig. 1B), and is assigned to the hard PLC-rich segment. The Tg values for the series of triblock samples, of differing molar mass and composition, all show similar behavior by DSC analysis (Table 1, Fig. S10†). Due to the soft character of these polymer films, it was not always possible to reliably determine the upper Tg by DMTA. For the lowest molar mass sample, ABA (51,63) (Table 1, entry 4), a Tg value of 0 °C was observed by DSC indicative of significant block miscibility (Tg,calc for miscible blocks = 7 °C).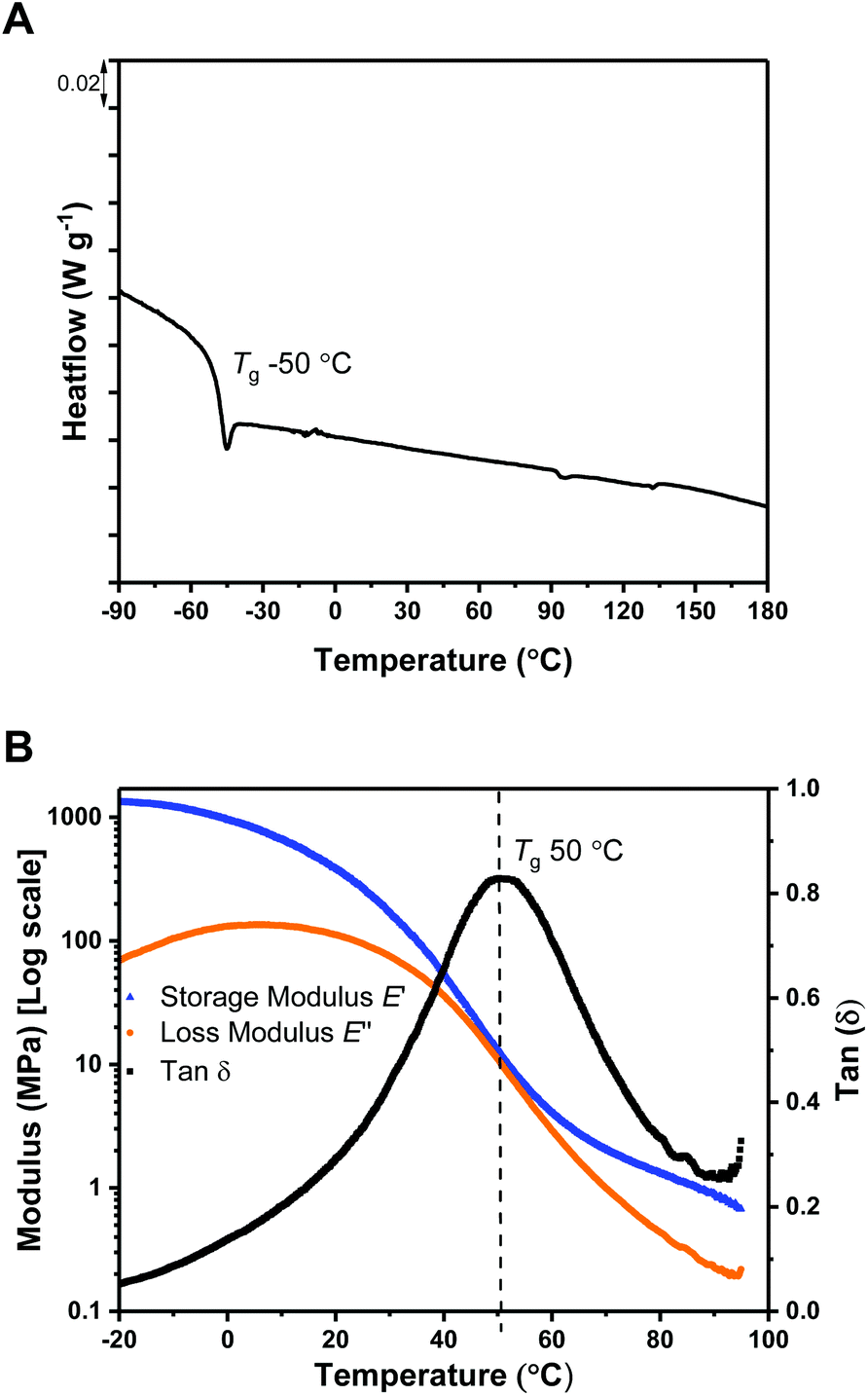 | ||
| Fig. 1 Thermal Characterisation of ABA (87,60) by DSC (A) and DMTA (B). | ||
The thermal stability of a polymer is critical for any application because it sets the processing temperature limits. To evaluate the processing viability of these materials, samples were subjected to thermal gravimetric analysis (TGA) to identify the polymer degradation temperature via measurement of the mass lost against temperature. For ABA (87,60) (Table 1, entry 1), the temperature at which 5% of the initial polymer mass is lost, Td, 5%, was determined to be 197 °C. Given its moderate upper Tg value (ca. 50 °C), this material has a broad processing temperature window. The degradation curve shows two distinct steps, with the relative mass loss at each stage corresponding to the block composition (Fig. 2). This behavior supports thermal degradation from the PLC chain-ends (Fig. S11–14†).61,62
 | ||
| Fig. 2 Thermal gravimetric analysis data for ABA (87,60). | ||
Mechanical properties
Each of the block polymers were cast into PTFE molds, from methylene chloride solutions (30 wt%). The solvent was allowed to evaporate, under ambient conditions for 48 h, and the films were further dried, in vacuo and at 50 °C, for 24 h. Transparent, colorless, freestanding films with thicknesses of around 120 μm were obtained, except in the case of ABA (85,21) which could not be processed into a freestanding film owing to its softness (Fig. S15†).Uniaxial extension measurements were performed on the four block polymer samples in order to determine the stress–strain relationships. Dumbbell-shaped specimen bars were cut from the solvent cast films, according to ISO 527-2 type 5B, and measurements were conducted, at 10 mm min−1 cross-head speed, in accordance with ISO 527. As expected, the tensile mechanical properties of the polymers varied with both the relative PLC weight fraction and the overall molar mass of the triblock.
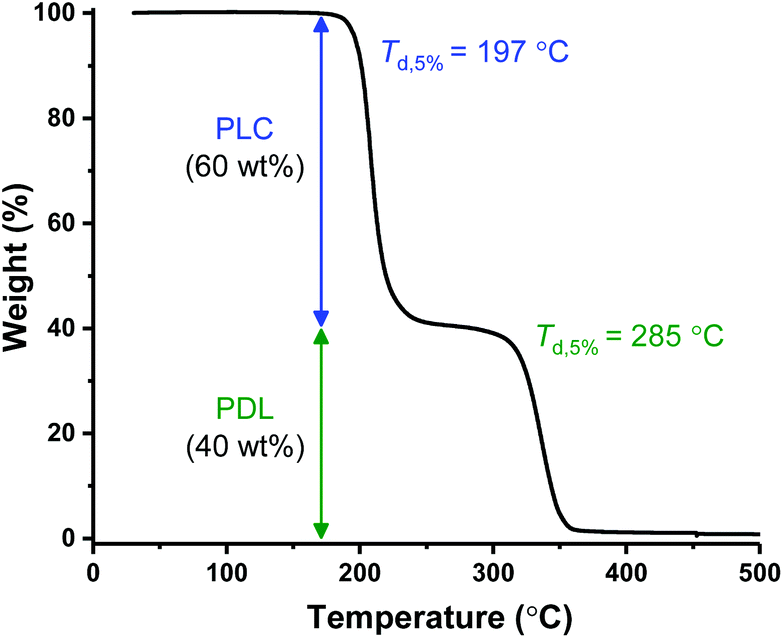
ABA (87,60) showed behavior typical of a ductile plastic, with a diffuse yield point at around 10% strain, beyond which the material showed characteristic plastic deformation (Fig. 3A and inset). The material showed moderate/high tensile strength (σ = 21.2 MPa) and high elongation at break (εb = 400%) (Table 2, entry 3). Its tensile toughness (UT) was determined as 62.3 MJ m−3 by integrating the stress–strain data. ABA (51,63) has a similar composition but significantly lower molar mass (51 kg mol−1), it showed a much lower tensile strength (σ = 5.6 MPa) (Fig. 3A; Table 2, entry 2). This was accompanied by a significant decrease in the Young's modulus (Ey = 165 MPa) and only a minor increase in the elongation at break (εb = 452%). This result highlights the importance of maximizing the polymer molar mass to enhance the mechanical properties by increasing soft block entanglement and hard domain physical crosslinking.66
 | ||
| Fig. 3 Mechanical Properties of PLC-b-PDL-b-PLC triblock polymers. (A) Stress–strain curves for uniaxial extension measurements. Failure points marked with an “X”. Inset: enlargement of the 0–100% strain region. (B) Ashby plot of tensile strength vs. elongation at break allowing comparison of the properties of ABA (87,60) with commercial and literature samples (see ESI Table S1† for further details on samples used in comparison). | ||
| Entry | Polymer | PLC wt % | M n (kg mol−1) | E y (MPa) | σ b (MPa) | ε b (%) |
|---|---|---|---|---|---|---|
| a Young's modulus. b Stress at break. c Elongation at break. Mean values from measurements conducted independently on at least 5 specimens. d SEC in CHCl3. ABA polymer tensile specimens were cut from a solvent cast film (30 wt% in DCM) conforming to dimensions for ISO 527-2 type 5B. Uniaxial tensile measurements conducted at 10 mm min−1 crosshead speed. e Sample did not yield a free-standing film as it was too soft and hence mechanical data not reported. | ||||||
| 132 | PLC | 100 | 53.4d | 950 | 55 | 15 |
| 2 | ABA (51,63) | 63 | 50.7 | 165 | 5.6 | 452 |
| 3 | ABA (87,60) | 60 | 86.9 | 321 | 21.2 | 400 |
| 4 | ABA (81,48) | 48 | 81.0 | 2.0 | 1.1 | 2563 |
| 5 | ABA (115,42) | 42 | 114.6 | 2.1 | 0.32 | 3962 |
| 6 | ABA (85,21)e | |||||
In contrast, polymers with lower hard block weight fractions, i.e. ABA (81,48) and ABA (115,42), both with 40–50 wt% PLC exhibited behaviors akin to elastomers. No yield point could be determined and the specimens showed extensions to high elongations (εb > 2500%), however, minimal stress was required to break both (σ < 1.2 MPa) consistent with their low strengths (Fig. 3, Table 2, entries 4 and 5). Hysteresis experiments (0–200% strain, 10 cycles) performed on the stronger of the two elastomers, ABA (81,48), showed good elastic recovery of around 84% (Fig. S16†). The maximum stress reached by the specimen decreased noticeably between the first and second cycles compared to only a slight reduction with each successive cycle thereafter, which attests to some degree of plastic deformation.
As a control experiment, attempts to solvent cast a 50:50 blend of the two constituent polymers, i.e. PDL and PLC, resulted in the formation of a waxy substance which could not be removed from the mold and was unsuited for any further testing (Fig. S15†). This result emphasizes the importance of the triblock polymers in providing materials with useful properties.
Chemical recycling
These fully renewable triblock polymers also present an interesting potential for PLC block chemical recycling back to the component monomers: LO and CO2. The depolymerization of the PLC block was investigated by dissolving ABA (87,60) in toluene, at 80 °C, with the same dizinc catalyst, 1, used in DL ROP (Fig. 4A). The depolymerization reaction was monitored by the regular removal of aliquots for NMR and SEC analysis. The successful PLC depolymerization and quantitative reformation of LO was observed after 6 h (using mesitylene as an internal standard). 1H NMR spectroscopy shows the complete disappearance of the PLC resonances and concomitant increase in the LO peaks (Fig. 4B & S18, S21†). Kinetic studies show a linear decrease in polycarbonate concentration vs. time, indicative of a zero order rate dependence in PLC concentration (Fig. 4C, blue squares). The depolymerization reaction does not occur in the absence of catalyst, 2 (Fig. 4C, red circles), and, under these conditions, the concentration of PLC remains constant over the timescale of the experiment. For the degradation experiments (Fig. 4C, blue squares), SEC analysis showed a continual decrease in polymer molar mass, whilst retaining narrow Đ values (1.1–1.2), from 87 kg mol−1 (Mn of the block polymer before depolymerization) to 33 kg mol−1, the molar weight of the starting PDL (Fig. 4D, S19†). This data supports the selective and controlled (step-wise) depolymerization of the PLC end-blocks. To investigate whether depolymerization or transesterification could occur in the PDL block, the reaction was deliberately heated for an extended period of time (2 days), but the PDL retained the same values for molar mass and dispersity. The residual PDL can be further degraded via dilute acid catalyzed hydrolysis (p-toluenesulfonic acid, 0.01 M) at 60 °C. Under these conditions, the PDL molar mass decreased to ∼6% of its oringal value in 40 h (Fig. S22†).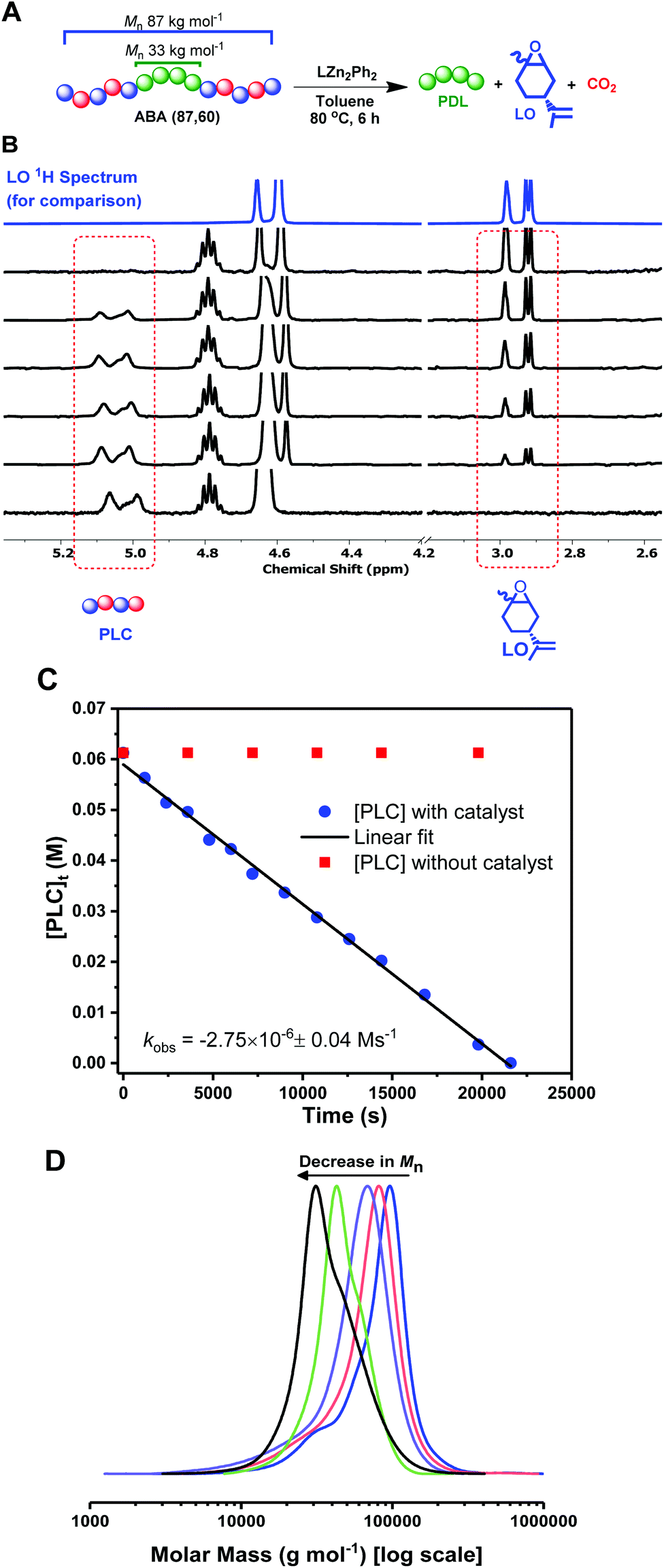 | ||
| Fig. 4 Deploymerization reactions of ABA (87,60). (A): 1.6 mM of 1; (B) Stacked 1H NMR spectra (CDCl3) taken at various timepoints throughout the depolymerization (LO 1H NMR spectrum is added for comparison); (C) Kinetic analysis with catalyst 1 (blue circles) and no catalyst (red squares); (D) SEC analysis (THF eluent, vs. PS standards) of aliquots taken at various time-points (see also Fig. S19†). | ||
Discussion
Sustainable block polymers
Conventional plastic production relies on unsustainable fossil fuel sources and the majority of these materials are neither recycled nor degradable.1,6 Polyesters and polycarbonates are interesting alternatives as their chemistry is intrinsically degradable and/or recyclable, e.g. by hydrolysis of ester/carbonate linkages and many of the monomers can also be obtained from renewable sources.5,6,16 For instance, PDL has been shown to readily degrade under acidic conditions, which may be suitable for recycling.61,62 DL is also a non-toxic monomer that can be obtained from castor oil.7,55,62 Currently, commercial polycarbonates are produced via the polycondensation of bisphenol A and phosgene, both of which are toxic.67 On the other hand, the monomers that make up PLC (LO and CO2) are non-hazardous and bio-renewable.68LO is derived from limonene,25,36 an abundant natural terpene obtained from citrus fruit peel waste or produced by algae. Limonene has an estimated global production capacity of 70000 to 100000 tonnes per year and its repurpose as feedstock for polymer production is highly promising.27,69 In addition, the CO2 usage is appealing as it is an abundant, non-hazardous and fully renewable resource and a waste product of many industrial and agricultural processes.18 The synthesis of PLC directly consumes CO2 as a feedstock and its production may contribute towards the reduction of greenhouse gas emissions by both the photosynthesis of the citrus fruit plant and the opportunity to store CO2 in value-added materials.69,70
A recent techno-economics study estimated the cost for PLC production to be between $1.36–$1.51 per kg, which is comparable to existing petrochemical plastics such as polystyrene ($1.2–$1.6 per kg).69 Thus, the synthesis of PLC-b-PDL-b-PLC (DL, LO and CO2) appears to meet many of the criteria for sustainability, including re-use of industrial wastes, use of bio-based monomers, efficient conversions and the delivery of better plastic properties. Because the monomers are already commercially available and used at scale in other processes, there is also improved potential for delivery of these products at scale. Naturally, more detailed analyses of process conditions, scale and costs, as well as thorough life cycle assessments would be needed to validate the opportunities for these products.
Mechanical property comparisons
One important aspect of the new triblock polymers is the improvements to the thermal and mechanical properties afforded by combining PLC and PDL into triblock structures. By tuning the polymer's molar mass values and/or the relative weight fractions of the constituent blocks, it is possible to obtain readily processable polymers that show characteristics of elastomers (ABA (81,48) and ABA (115,42)) at lower PLC wt % or toughened plastics (ABA (51,63) and ABA (87,60)) at higher PLC wt %.Of the samples, the most promising in terms of mechanical properties is the ductile thermoplastic, ABA (87,60). The other example in this material class, ABA (51,63), shows a significantly reduced tensile strength and minimal improvement in elongation. The remaining two materials, examples of elastomers, show very high elongations at break but have the shortcoming of low tensile strength. Nonetheless, these high strain-low stress materials may be of interest in future as natural fibre mimics, e.g. elastins.71
The enhanced performance of ABA (87,60) arises primarily from the toughening imparted by incorporating the softer PDL block. The tensile toughness of the material (UT = 62.3 MJ m−3) is greatly increased when compared to bio-based PLC (UT estimated at 3.15 MJ m−3), which fails to exceed 20% strain and exhibits brittle failure.32
Comparisons to other literature materials enable benchmarking of the properties of ABA (87,60) and highlight the need for bio-based materials with improved mechanical performances. We recently reported ABA triblock polymers, based on poly(cyclohexene carbonate) (PCHC), derived from CO2, and PDL, with the structure PCHC-b-PDL-b-PCHC.62 Compared to an equivalent composition sample, ABA (87,60) has a lower tensile toughness (PCHC-b-PDL-b-PCHC with 50 wt% PCHC, UT = 112 MJ m−3) but both polymers show similar values for tensile strength (σ = 21.2 vs. 20 MPa) (Fig. 3B above). The advantage of ABA (87,60), compared to the sample containing PCHC, is the ability to increase the renewable content and to make use of industrial wastes (limonene). Another well-established bio-based polymer is polylactide (PLA) which when applied without additives is also very brittle. Compared to a purposefully toughened poly(lactide) (PLA) sample, with σ = 25.8 MPa, ABA (87,60) shows equivalent tensile strength and greater toughness (Fig. S17†).72
Recently, a series of block polymers incorporating PLC as the hard block and poly(hydroxybutyrate) (PHB) as the soft block were reported.53 Block polymers, containing 54 wt% PHB, at two different molar mass values (‘low’ Mn = 90 and ‘high’ Mn = 211 kg mol−1) were compared and both samples showed slightly lower tensile strength and Young's moduli compared to pure PLC. However, only the high molar mass sample showed an improvement in elongation at break, with the maximum tensile strain at 18%. Whilst the tensile strengths of these PLC-b-PHB are higher than ABA (87,60), the elongation at break and toughness are much lower.
One future research direction could be to explore other soft blocks to toughen PLC or other brittle, aliphatic polycarbonates, e.g. PCHC. Poly(ε-caprolactone) (PCL) would be desirable given its low glass transition temperature and the commercial availability at low cost of ε-CL. The crystallinity of the PCL block would, however, need to be supressed and strategies to achieve this include its copolymerization with other lactones, e.g. δ-valerolactone.73 Other soft block polymers could be produced by the ROP of functionalized lactones or by the ROCOP of epoxides, like propene oxide or alkyl glycidyl ether, and anhydrides, like succinic or glycolic anhydride.54,74,75 Polycarbonate soft-block polymers could include poly(trimethylene carbonate) or materials derived from the ROCOP of alkylene oxides and carbon dioxide.76
Another area for future development is to exploit the pendent double bond, in the repeat unit of PLC, for post-functionalization. The alkene groups could be cross-linked or substituted with moieties that undergo intermolecular interactions so as to enhance microphase separation, and increase tensile strength and toughness.
Depolymerization
To better design plastics for chemical recycling requires selective, catalytic depolymerization chemistries, ideally under conditions accessible to scale-up, and the delivery of the true monomers.5 This latter point is very important as any proposed recycling process should avoid unnecessary derivatives or neutralization steps. Another consideration is that the thermal treatment of many polycarbonates, prepared from ROCOP of CO2/epoxides, often leads to formation of the thermodynamically favoured cyclic carbonate product. There are only a few examples of depolymerization to the corresponding epoxide and these processes were recently reviewed by Darensbourg.77 Base- or metal-catalyzed depolymerization of poly(cyclopentene carbonate) and poly(indene carbonate) yield both epoxide and cyclic carbonate products.78–80 Depolymerization of poly(indene carbonate) typically only gives indene oxide as a minor product, whereas a high selectivity for cyclopentene oxide formation (92%) was reported when using a catalyst comprising (Salen)CrCl/n-Bu4NN3 ([repeat unit]:[Cr]:[base] = 50:1:2). For the latter reaction, a temperature of 110 °C was necessary and the complete degradation of the poly(cyclopentene carbonate) was only observed after 30 h. The rapid and complete depolymerization of 1-benzyloxycarbonyl-3,4-epoxy pyrrolidone (BEP), to CO2 and the starting N-hetero-epoxide, was reported using a dinuclear chromium-salen complex operating at 100 °C.81 Although this novel polycarbonate is fully recyclable the synthesis of BEP requires several steps. In the context of these prior investigations, the complete depolymerization of PLC to LO is particularly noteworthy. Koning and coworkers previously reported PLC depolymerization to LO with no observable cyclic carbonate,38 using 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) to deprotonate the hydroxyl end-groups and triggering chain backbiting. Under these conditions, a polycarbonate repeat unit to catalyst loading ([M]:[TBD]) of 25:1, enabled quantitative recovery of LO after 16 h at 110 °C (no depolymerization was observed at 80 °C). A significant broadening of the polymer Đ to ∼2 was also noted and attributed to a deprotonation/protonation equilibrium. This work represents an improvement on Koning's pioneering prior work because the use of the di-zinc catalyst, 1, allows for faster depolymerization, with complete reaction to LO monomer occurring in just 6 h ([M]:[1] = 27:1) and at a slightly lower temperature (80 °C). The reaction proceeds controllably and the residual polymer retains a narrow Đ throughout – this finding potentially offers opportunities to control forward and reverse reactions in future. In addition to full recovery of LO, the selective unzipping of just the PLC end-blocks in these triblock polymers, whilst leaving the PDL block unaffected, is conceptually interesting. The remaining PDL could be used to initiate polymerization with other monomers readily allowing access to different materials and fulfilling concepts of upcycling.16
Conclusions
The successful and efficient copolymerization of three commercial bio-based monomers, carbon dioxide, limonene oxide and ε-decalactone, yielded ABA triblock polymers. The polymers comprise PLC blocks, with high Tg and rigidity (A), flanking PDL blocks, with low Tg and high flexibility (B-blocks). The materials compositions and molar masses were straightforward to control since living polymerizations were applied. The materials were fully characterized, including by thermal and tensile mechanical measurements; the most promising sample shows significant improvement in properties compared to poly(limonene carbonate). It shows a 20× increase in elongation at break, retains good tensile strength and is 20× tougher than PLC. It shows a processing temperature window of >140 °C and produces optically transparent toughened plastic films. After use, it was efficiently chemically recycled to limonene oxide, carbon dioxide and poly(ε-decalactone) pre-polymer. Overall, this work demonstrates a generally applicable method to effectively combine bio-based monomers to yield plastics showing higher value properties and with potential for straightforward chemical recycling. Future investigations into the applications for these block polymers as toughened plastics and substitutes for petrochemicals are recommended. In terms of polymer chemistry, the PLC block features alkene substituents which could be further reacted, e.g. through cross-linking processes or with other functional groups.39 The principles presented here are expected to be broadly applicable to other bio-based monomer combinations, including terpene derived anhydrides and epoxides, carbohydrate-derived lactones and epoxides and waste carbon dioxide.Conflicts of interest
CKW is a director of econic technologies.Acknowledgements
Professor Clive Siviour and research team are thanked for allowing use of the DMTA instrumentation. The EPSRC (EP/S018603/1; EP/L017393/1) Faraday Institution (Project: SOLBAT), Oxford Martin School (Future of Plastics Programme) and UK Catalysis Hub (EP/R027129/1) are acknowledged for research funding.References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef.
- T. Keijer, V. Bakker and J. C. Slootweg, Nat. Chem., 2019, 11, 190–195 CrossRef CAS.
- J. H. Clark, T. J. Farmer, L. Herrero-Davila and J. Sherwood, Green Chem., 2016, 18, 3914–3934 RSC.
- I. A. Ignatyev, W. Thielemans and B. Vander Beke, ChemSusChem, 2014, 7, 1579–1593 CrossRef CAS.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- Y. Q. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS.
- M. A. Hillmyer and W. B. Tolman, Acc. Chem. Res., 2014, 47, 2390–2396 CrossRef CAS.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS.
- R. Auvergne, S. Caillol, G. David, B. Boutevin and J. P. Pascault, Chem. Rev., 2014, 114, 1082–1115 CrossRef CAS.
- Z. K. Wang, L. Yuan and C. B. Tang, Acc. Chem. Res., 2017, 50, 1762–1773 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- M. M. Reddy, S. Vivekanandhan, M. Misra, S. K. Bhatia and A. K. Mohanty, Prog. Polym. Sci., 2013, 38, 1653–1689 CrossRef CAS.
- S. Farah, D. G. Anderson and R. Langer, Adv. Drug Delivery Rev., 2016, 107, 367–392 CrossRef CAS.
- K. J. Groh, T. Backhaus, B. Carney-Almroth, B. Geueke, P. A. Inostroza, A. Lennquist, H. A. Leslie, M. Maffini, D. Slunge, L. Trasande, A. M. Warhurst and J. Muncke, Sci. Total Environ., 2019, 651, 3253–3268 CrossRef CAS.
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS.
- C. Jehanno, M. M. Pérez-Madrigal, J. Demarteau, H. Sardon and A. P. Dove, Polym. Chem., 2019, 10, 172–186 RSC.
- A. Sangroniz, J. B. Zhu, X. Tang, A. Etxeberria, E. Y. Chen and H. Sardon, Nat. Commun., 2019, 10, 3559 CrossRef.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS.
- C. M. Kozak, K. Ambrose and T. S. Anderson, Coord. Chem. Rev., 2018, 376, 565–587 CrossRef CAS.
- Y. Y. Wang and D. J. Darensbourg, Coord. Chem. Rev., 2018, 372, 85–100 CrossRef CAS.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- D. J. Darensbourg, Green Chem., 2019, 21, 2214–2223 RSC.
- A. M. Chapman, C. Keyworth, M. R. Kember, A. J. J. Lennox and C. K. Williams, ACS Catal., 2015, 5, 1581–1588 CrossRef CAS.
- S. J. Poland and D. J. Darensbourg, Green Chem., 2017, 19, 4990–5011 RSC.
- M. Selva, A. Perosa, G. Fiorani and L. Cattelan, in Green Synthetic Processes and Procedures, The Royal Society of Chemistry, 2019, pp. 319–342, 10.1039/9781788016131-00319.
- R. Ciriminna, M. Lomeli-Rodriguez, P. D. Cara, J. A. Lopez-Sanchez and M. Pagliaro, Chem. Commun., 2014, 50, 15288–15296 RSC.
- C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 CrossRef CAS.
- F. Auriemma, C. De Rosa, M. R. Di Caprio, R. Di Girolamo, W. C. Ellis and G. W. Coates, Angew. Chem., Int. Ed., 2015, 54, 1215–1218 CrossRef CAS.
- F. Auriemma, C. De Rosa, M. R. Di Caprio, R. Di Girolamo and G. W. Coates, Macromolecules, 2015, 48, 2534–2550 CrossRef CAS.
- O. Hauenstein, M. M. Rahman, M. Elsayed, R. Krause-Rehberg, S. Agarwal, V. Abetz and A. Greiner, Adv. Mater. Technol., 2017, 2, 1700026 Search PubMed.
- O. Hauenstein, M. Reiter, S. Agarwal, B. Rieger and A. Greiner, Green Chem., 2016, 18, 760–770 CAS.
- O. Hauenstein, S. Agarwal and A. Greiner, Nat. Commun., 2016, 7, 11862 CrossRef CAS.
- S. Neumann, L.-C. Leitner, H. Schmalz, S. Agarwal and A. Greiner, ACS Sustainable Chem. Eng., 2020, 8, 6442–6448 CrossRef CAS.
- N. Kindermann, A. Cristofol and A. W. Kleij, ACS Catal., 2017, 7, 3860–3863 CrossRef CAS.
- L. P. Carrodeguas, J. Gonzalez-Fabra, F. Castro-Gomez, C. Bo and A. W. Kleij, Chem. – Eur. J., 2015, 21, 6115–6122 CrossRef.
- T. Stosser, C. L. Li, J. Unruangsri, P. K. Saini, R. J. Sablong, M. A. R. Meier, C. K. Williams and C. Koning, Polym. Chem., 2017, 8, 6099–6105 RSC.
- C. L. Li, R. J. Sablong, R. van Benthem and C. E. Koning, ACS Macro Lett., 2017, 6, 684–688 CrossRef CAS.
- C. L. Li, M. Johansson, R. J. Sablong and C. E. Koning, Eur. Polym. J., 2017, 96, 337–349 CrossRef CAS.
- C. L. Li, R. J. Sablong and C. E. Koning, Angew. Chem., Int. Ed., 2016, 55, 11572–11576 CrossRef CAS.
- C. L. Li, T. Veldhuis, B. Reuvers, R. J. Sablong and C. E. Koning, Polym. Int., 2019, 69, 24–30 CrossRef.
- J. Bailer, S. Feth, F. Bretschneider, S. Rosenfeldt, M. Drechsler, V. Abetz, H. Schmalz and A. Greiner, Green Chem., 2019, 21, 2266–2272 RSC.
- Y. Q. Zhu, M. R. Radlauer, D. K. Schneiderman, M. S. P. Shaffer, M. A. Hillmyer and C. K. Williams, Macromolecules, 2018, 51, 2466–2475 CrossRef CAS.
- Y. Y. Zhang, G. W. Yang and G. P. Wu, Macromolecules, 2018, 51, 3640–3646 CrossRef CAS.
- Y. Y. Zhang, G. W. Yang, Y. Y. Wang, X. Y. Lu, G. P. Wu, Z. S. Zhang, K. Wang, R. Y. Zhang, P. F. Nealey, D. J. Darensbourg and Z. K. Xu, Macromolecules, 2018, 51, 791–800 CrossRef CAS.
- M. Reiter, A. Kronast, S. Kissling and B. Rieger, ACS Macro Lett., 2016, 5, 419–423 CrossRef CAS.
- S. Paul, C. Romain, J. Shaw and C. K. Williams, Macromolecules, 2015, 48, 6047–6056 CrossRef CAS.
- M. R. Kember, J. Copley, A. Buchard and C. K. Williams, Polym. Chem., 2012, 3, 1196–1201 RSC.
- S. Kernbichl, M. Reiter, F. Adams, S. Vagin and B. Rieger, J. Am. Chem. Soc., 2017, 139, 6787–6790 CrossRef CAS.
- C. Romain and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 1607–1610 CrossRef CAS.
- C. Romain, Y. Zhu, P. Dingwall, S. Paul, H. S. Rzepa, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2016, 138, 4120–4131 CrossRef CAS.
- T. T. D. Chen, Y. Q. Zhu and C. K. Williams, Macromolecules, 2018, 51, 5346–5351 CrossRef CAS.
- S. Kernbichl, M. Reiter, J. Mock and B. Rieger, Macromolecules, 2019, 52, 8476–8483 CrossRef CAS.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2016, 49, 2419–2428 CrossRef CAS.
- D. K. Schneiderman, E. M. Hill, M. T. Martello and M. A. Hillmyer, Polym. Chem., 2015, 6, 3641–3651 RSC.
- S. Lee, K. Lee, Y. W. Kim and J. Shin, ACS Sustainable Chem. Eng., 2015, 3, 2309–2320 CrossRef CAS.
- M. T. Martello, D. K. Schneiderman and M. A. Hillmyer, ACS Sustainable Chem. Eng., 2014, 2, 2519–2526 CrossRef CAS.
- P. Olsen, T. Borke, K. Odelius and A. C. Albertsson, Biomacromolecules, 2013, 14, 2883–2890 CrossRef CAS.
- M. P. F. Pepels, R. A. C. Koeken, S. J. J. van der Linden, A. Heise and R. Duchateau, Macromolecules, 2015, 48, 4779–4792 CrossRef CAS.
- Y. Zhu, C. Romain and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 12179–12182 CrossRef CAS.
- G. L. Gregory, G. S. Sulley, L. P. Carrodeguas, T. T. D. Chen, A. Santmarti, N. J. Terrill, K.-Y. Lee and C. K. Williams, Chem. Sci., 2020, 11, 6567–6581 RSC.
- G. S. Sulley, G. L. Gregory, T. T. D. Chen, L. P. Carrodeguas, G. Trott, A. Santmarti, K.-Y. Lee, N. J. Terrill and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 4367–4378 CrossRef CAS.
- C. Romain, J. A. Garden, G. Trott, A. Buchard, A. J. P. White and C. K. Williams, Chem. – Eur. J., 2017, 23, 7367–7376 CrossRef CAS.
- G.-P. Wu and D. J. Darensbourg, Macromolecules, 2016, 49, 807–814 CrossRef CAS.
- J.-O. Lin, W. Chen, Z. Shen and J. Ling, Macromolecules, 2013, 46, 7769–7776 CrossRef CAS.
- J. D. Tong and R. Jerome, Macromolecules, 2000, 33, 1479–1481 CrossRef CAS.
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC.
- S. Paul, Y. Q. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- D. Zhang, E. A. del Rio-Chanona, J. L. Wagner and N. Shah, Sustain. Prod. Consum., 2018, 14, 152–160 CrossRef.
- N. von der Assen and A. Bardow, Green Chem., 2014, 16, 3272–3280 RSC.
- M. Vatankhah-Varnosfaderani, A. N. Keith, Y. Cong, H. Liang, M. Rosenthal, M. Sztucki, C. Clair, S. Magonov, D. A. Ivanov, A. V. Dobrynin and S. S. Sheiko, Science, 2018, 359, 1509 CrossRef CAS.
- P. A. Delgado and M. A. Hillmyer, RSC Adv., 2014, 4, 13266 RSC.
- D. K. Schneiderman, E. M. Hill, M. T. Martello and M. A. Hillmyer, Polym. Chem., 2015, 6, 3641–3651 RSC.
- P. Olsén, K. Odelius and A.-C. Albertsson, Biomacromolecules, 2016, 17, 699–709 CrossRef.
- T. Stosser, G. S. Sulley, G. L. Gregory and C. K. Williams, Nat. Commun., 2019, 10, 2668 CrossRef.
- S. J. Poland and D. J. Darensbourg, Green Chem., 2017, 19, 4990–5011 RSC.
- D. J. Darensbourg, Polym. Degrad. Stab., 2018, 149, 45–51 CrossRef CAS.
- D. J. Darensbourg, A. D. Yeung and S.-H. Wei, Green Chem., 2013, 15, 1578–1583 RSC.
- D. J. Darensbourg, S.-H. Wei and S. J. Wilson, Macromolecules, 2013, 46, 3228–3233 CrossRef CAS.
- D. J. Darensbourg, S.-H. Wei, A. D. Yeung and W. C. Ellis, Macromolecules, 2013, 46, 5850–5855 CrossRef CAS.
- Y. Liu, H. Zhou, J.-Z. Guo, W.-M. Ren and X.-B. Lu, Angew. Chem., Int. Ed., 2017, 56, 4862–4866 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc02295k |
| This journal is © The Royal Society of Chemistry 2020 |