
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwenty-five years of polymersomes: lost in translation?
Simon
Matoori†
 * and
Jean-Christophe
Leroux
* and
Jean-Christophe
Leroux
Institute of Pharmaceutical Sciences, Department of Chemistry and Applied Biosciences, ETH Zurich, HCI H 301, Vladimir-Prelog-Weg 3, 8093 Zurich, Switzerland. E-mail: jleroux@ethz.ch; Fax: +41 44 633 13 14; Tel: +41 44 633 73 10
First published on 24th January 2020
Abstract
Soon after the discovery of polymeric vesicles (polymersomes), reports of their high membrane stability raised hopes for the development of next generation vesicles for drug delivery and diagnostic applications. Twenty-five years later, however, liposomes remain the only clinically tested colloidal vesicular formulations. To highlight the translational challenges faced by polymersomes, we critically reviewed a selection of polymersome formulations with a focus on their pharmacokinetic and pharmacodynamic aspects.
 Simon Matoori | Simon Matoori is a pharmacist by training and performed his PhD studies under the supervision of Jean-Christophe Leroux at ETH Zurich, Switzerland, on novel vesicular formulations for therapeutic and diagnostic applications. He is currently a postdoctoral fellow in the laboratory of David J. Mooney at the John A. Paulson School of Engineering and Applied Sciences at Harvard University. His research interests are in biomaterials, drug delivery, and clinical radiology. |
 Jean-Christophe Leroux | Jean-Christophe Leroux is full professor of Drug Formulation and Delivery at the Institute of Pharmaceutical Sciences at ETH Zurich, Switzerland. He has made important fundamental and applied contributions to the fields of biomaterials and drug delivery, and has been involved in the development of innovative bio-detoxification systems for the treatment of intoxications. He is a fellow of the AAPS and the CRS, and the co-founder of the start-up pharmaceutical companies Versantis AG and Inositec AG. |
1.1 Polymersome definition
Polymersomes (polymersomes, polymeric vesicles, Fig. 1A) are defined as vesicular macromolecular assemblies whose bilayer membrane is composed of amphiphilic polymers (block, dendronized, graft, or alkylated copolymers).1–4 The hydrophilic corona of the membrane faces the aqueous core and outer aqueous phase. The hydrophobic layer of the membrane separates the inner from the outer medium. The molecular composition and length of the hydrophobic and hydrophilic blocks determine different polymersome properties such as membrane rigidity, size, and stability.5,6 Polymersomes have been the subject of several recent review articles.7–9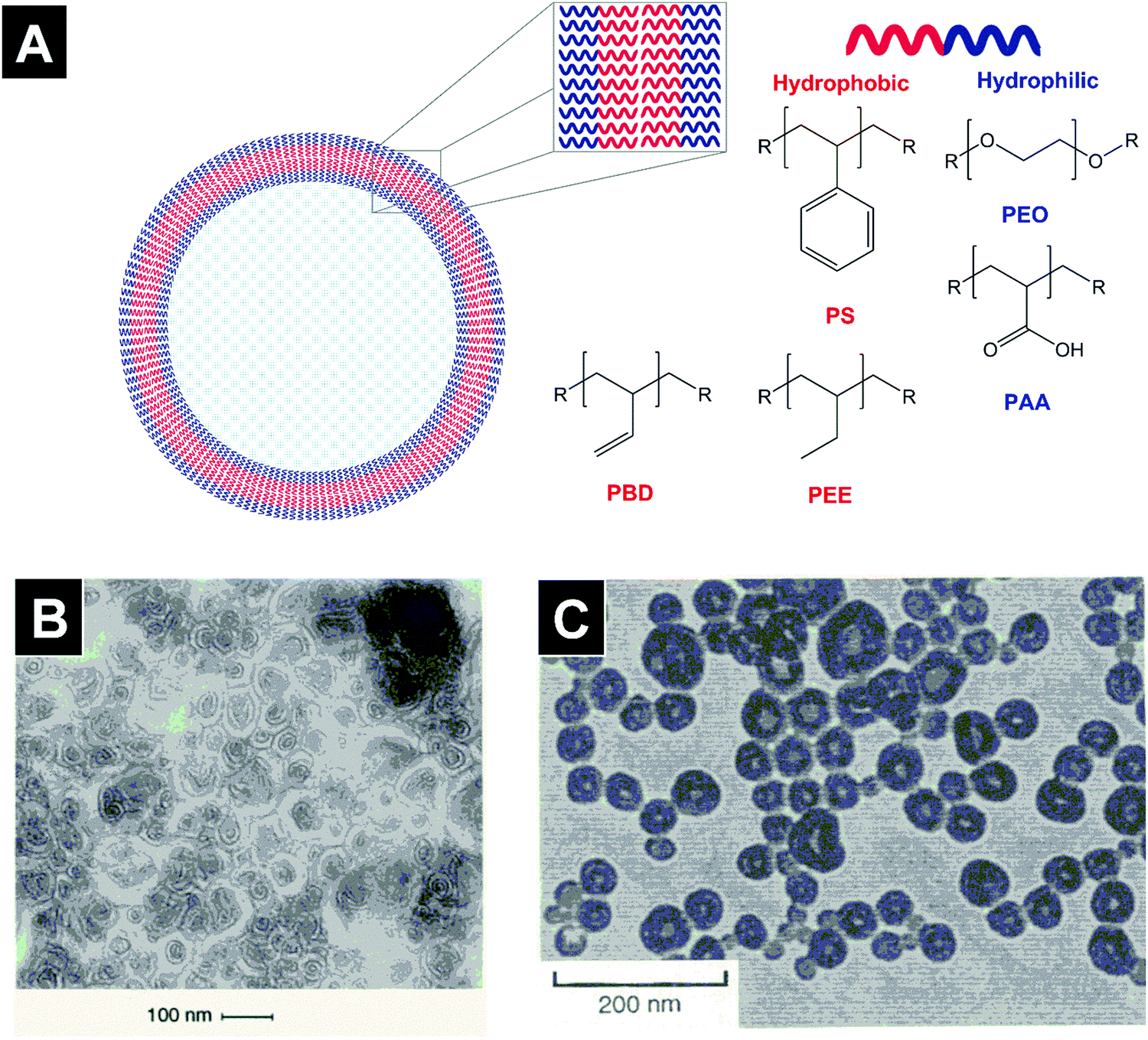 | ||
| Fig. 1 Schematic depiction and first reported TEM images of polymeric vesicles in aqueous media. Polymersomes are vesicular structures made of amphiphilic co-polymers (A). In 1995, van Hest et al. demonstrated the vesicular morphology of macromolecular assemblies made of PS-dendr-(NH2)8 on TEM images in aqueous solution (B). Soon afterwards, Zhang and Eisenberg showed that PS-b-PAA diblock copolymers formed vesicular structures in a water–DMF mixture using TEM imaging (C). Figures adapted from ref. 1 and 2 with permission. PS: poly(styrene); PBD: poly(butadiene), PEE: poly(ethylethylene), PEO: poly(ethylene oxide), PAA: poly(acrylic acid). | ||
1.2 Historical perspective
In 1995, two seminal papers in Science advanced our understanding of polymeric macromolecular assemblies.1,2 In the first published study, van Hest et al. described different macromolecular structures formed by amphiphilic polymers composed of a poly(styrene) (PS) block and a poly(propylene imine) dendrimer (dendr) in aqueous solution. While PS-dendr-(NH2)8 formed vesicles with diameters below 100 nm (Fig. 1B), PS-dendr-(NH2)16 formed micellar rods (∼12 nm in diameter) and PS-dendr-(NH2)32 spherical micelles (∼10–20 nm).2 In the subsequent Science issue, an article by Zhang and Eisenberg reported the morphological diversity of macromolecular assemblies of PS-b-poly(acrylic acid) (PS-b-PAA) diblock copolymers in a N,N-dimethylformamide–water mixture. Upon decreasing the PAA block length, transmission electron microscopy (TEM) images revealed a transition from spherical (PS200-b-PAA21, 26 nm in diameter) to rod-like micelles (PS200-b-PAA15, 23 nm), to vesicles (PS200-b-PAA8, 100 nm, Fig. 1C), and large spherical aggregates (PS200-b-PAA4, up to 1.2 μm). These studies revealed that amphiphilic copolymers were able to form vesicular structures in low molecular weight solvents, and that tuning the hydrophilic to hydrophobic ratio of block copolymers allowed the formation of different macromolecular assemblies. A subsequent study by Zhang and Eisenberg revealed that the macromolecular morphology of a diblock copolymer can be altered by modifying the ionic strength of the medium.10The notion that polymersome membranes are much tougher than liposomal ones was first put forward by Discher et al. who showed that poly(ethyl ethylene) (PEE)37-b-poly(ethylene oxide)40 (PEO, also referred to as poly(ethylene glycol), PEG) polymersomes resisted higher areal strains than liposomes made of the unsaturated phospholipid 1-stearoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine (SOPC, phase transition temperature Tm ∼ 6 °C) in micropipette aspiration experiments.11 A subsequent study by the same group demonstrated that poly(butadiene)46 (PBD)-b-PEO26, PBD55-b-PEO50, PBD125-b-PEO80, and PBD250-b-PEO150 polymersomes ruptured at an areal expansion of ∼10–40%, while SOPC liposomes ruptured at ∼4%.5 This study is the origin of the widely propagated notion that polymersomes are considerably more stable than liposomes.8,12–16 While the evidence put forward by Discher et al. clearly showed superior mechanical properties of the investigated polymersomes over the liposomal control, comparisons with additional control formulations need to be performed as clinically used liposome formulations differ greatly from SOPC vesicles both in terms of phospholipid phase transition temperature and cholesterol content.17 In liposomes, hydrogenation of the phospholipid improves membrane packing and thus leads to increased toughness and lower permeability.18 Evaluating stability differences among polymersome systems with different glass transition temperatures and biodegradability profiles may further nuance this statement. PBD250-b-PEO150 polymersomes (membrane thickness ∼21 nm) ruptured at lower areal strength than PBD125-b-PEO80 polymersomes (∼15 nm), indicating that neither hydrophobic block length nor membrane thickness are the only determining factors of membrane strength.5 Interestingly, poly(ethyl ethylene) (PEE)-b-PEO polymersomes showed similar resistance to areal strain (rupture at ∼10%) as PBD-b-PEO of a similar molecular weight.5 Furthermore, investigations of the elastic moduli of various polymersome and liposome formulations demonstrated the strong influence of block copolymer/phospholipid composition and did not show a general superiority of polymersomes.5,19–27 Finally, the question, which clinical applications necessitate tougher membranes than liposomal ones, also needs to be addressed.
1.3 Preparation methods
Polymersomes can be prepared by various methods. In general, the polymer is dissolved in an organic solvent to form an organic phase that is added to an aqueous solution (aqueous phase). The mixing allows a fine dispersion of the polymer in the water phase and the subsequent formation of polymersomes. Different techniques may be employed for the mixing step: nanoprecipitation, emulsification, or film rehydration. In order to compare these methods quantitatively, the polymersome yield or at least the polymer concentration of the dispersion would be needed. Unfortunately, this information is generally lacking. Furthermore, preclinical and clinical studies necessitate scaled up preparation procedures with high reproducibility and acceptable amounts of residual organic solvents and degradation products. For liposomes, retaining the same physicochemical properties and toxicity profile after scale up remains challenging despite the multitude of FDA-approved formulations.28–30 The ample industrial experience on tackling the challenges of large-scale liposome production will be highly valuable for polymersome systems, especially if similar procedures (e.g., film rehydration, ethanol injection, extrusion) can be applied.1.3.1 Nanoprecipitation
Nanoprecipitation is a widely used method for polymersome production.6,31,32 The polymers are dissolved in a suitable water-miscible organic solvent, to which the aqueous phase is slowly added under stirring, usually with a syringe pump. This method is associated with engineering challenges regarding loading efficiency, scalability, and reproducibility.33 To address some of these challenges, flash nanoprecipitation, a method used to prepare solid-core nanoparticles from block copolymers, was recently adapted to polymersome preparation.33 In this method, multi-stream mixers are employed to mix an amphiphilic block copolymer-containing water-miscible organic solvent with an aqueous solution under turbulent conditions, and to subsequently introduce this mixture into an aqueous reservoir.33 This method enabled the preparation of poly(propylene sulfide)-b-PEO polymersomes loaded with hydrophilic (green fluorescent protein, alkaline phosphatase) and hydrophobic cargoes (rapamycin), and promises to yield a scalable polymersome preparation platform once validated for a range of diblock copolymers and larger batch volumes. Unfortunately, flash nanoprecipitation has not yet been shown to solve another issue of nanoprecipitation-based methods, the necessity of diluted conditions (generally below 1 wt%)6,31–33 and thus additional concentration steps to prepare concentrated polymersome dispersions.The complete removal of the water-miscible organic solvents used in nanoprecipitation is of high importance in order to avoid toxicity and vesicular instability, as organic solvents have been reported to act as a plasticizer in the polymersome membrane.34 Thorough purification procedures such as dialysis or cross-flow filtration coupled with a quantification of residual solvents would be needed.
1.3.2 Emulsification
In the oil-in-water single emulsion method, the polymer-containing, at least partly water-immiscible organic solvent is mixed with the aqueous phase under sonication, homogenization, or vigorous stirring.35,36 The solvent is then removed by evaporation or filtration methods.35–37 This method was shown to allow the preparation of PS-b-PEO polymersomes with high concentrations (up to 10% w/v) and low solvent residues, making it suitable for in vivo experiments.37 In addition, emulsification-based methods can relatively easily be scaled up and may be performed in a continuous process.38,39In the double-emulsion method, polymersomes form in a water-oil-in-water double emulsion containing an aqueous inner phase, a polymer-containing, at least partly water-immiscible organic solvent in the middle phase, and an aqueous outer phase.40–42 This method can be elegantly carried out with a microfluidic system with which narrow size distributions in the low micrometer size range can be achieved.40–42 Microfluidics-based systems for mass production of vesicles were proposed but have yet to prove their capacity to provide large volumes of highly concentrated vesicular dispersions at industrial scale.43,44
1.3.3 Film rehydration
In the film rehydration method, the polymer is dissolved in an organic solvent and subsequently dried, allowing the formation of a thin polymer film.45–47 The polymers self-assemble into vesicles upon addition of the aqueous phase and thorough mixing. This method is widely used in the preparation of liposomes as it yields highly concentrated dispersions with high yields and low residual solvent amounts.48 The latter are generally not reported for polymersomes prepared with this method,45–47 and its applicability to vesicles with highly hydrophobic blocks is limited.491.3.4 Cross-linking
Cross-linking the hydrophobic copolymer fragments generally aims at increasing the stability of the polymersome membrane. Cross-linking procedures have been reported for many polymersome systems such as PBD-b-PEO or PEO-b-PAA-b-poly(N-isopropylacrylamide).50–52 However, the removal of toxic residues from the cross-linking procedure could be challenging as they may be retained in the membrane. If the drug is already loaded, performing a cross-linking procedure could potentially chemically modify the drug.1.3.5 Size control
The vesicle size affects the uptake by the mononuclear phagocytic system (which influences circulation time), extravasation, and organ distribution.53 Upon oral application, it may further impact the systemic availability due to size-dependent differences in diffusion across the mucin layer and M-cell uptake.54 Membrane extrusion, sonication, and size-exclusion chromatography can be used to control the polymersome size.34,55–60 Similarly to liposomes, the size of polymersomes composed of polymers with low glass transition temperature hydrophobic fragments (e.g., poly(propylene oxide), PBD) can readily be decreased applying freeze–thaw and/or extrusion cycles, yielding polymersomes of less than 100 nm in diameter.55–59 However, for polymers with a high glass transition such as PS-b-PEO, decreasing the size of polymersomes with filtration and sonication has only been reported in the presence of considerable volume fractions of organic solvent.34 The solvent probably acts as a plasticizer of the semi-crystalline membrane, facilitating macromolecular rearrangements during filtration or sonication. Size-exclusion chromatography was further used to separate fractions of PBD-b-PEO polymersomes of different sizes.60 For more information on controlling polymersome size, the reader is referred to a recent review on this subject.81.4 Drug loading
As their lipidic counterparts, polymersomes offer the possibility to host hydrophilic and hydrophobic cargo in the aqueous core and hydrophobic part of the membrane, respectively.1.4.1 Hydrophilic drugs and membrane proteins
Hydrophilic substrates can be passively or actively loaded into vesicles. In passive loading strategies, the cargo is added to the aqueous phase in which the vesicles form (e.g., film rehydration) or to the inner water phase (e.g., microfluidics). If the substrate is ionizable and efficiently diffuses across the membrane (i.e., low molecular weight and moderately polar weak acids or bases), an active loading method based on a transmembrane pH gradient can be employed. In this approach, the uncharged species of the drug diffuses across the membrane into the vesicular core. The substrate becomes charged and subsequently trapped if the core pH is low (basic cargo) or high (acidic cargo).61 Very high encapsulation efficiencies and a stable retention can be achieved with this method.17,62,63Leaky membranes impair the stability of the transmembrane pH-gradient and may result in low loading efficiencies and drug leakage after loading. The thicker membranes of polymersomes could therefore be an advantage over the liposomal ones for this remote loading procedure if they remain permeable enough for a sufficiently rapid diffusion of the substrate. Doxorubicin encapsulation efficiencies of ∼25% and ∼60% were reported for poly(2,4,6-trimethoxybenzylidenepentaerythritol carbonate) (PTMBPEC)-b-PEO polymersomes64 and polymersomes made of a blend of PBD-b-PEO (75 wt%) and poly(L-lactic acid)-b-PEO (25 wt%).65 In comparison, liposomes made of saturated phospholipids can reach encapsulation efficiencies of up to almost 100%.66,67 The drug loading in terms of doxorubicin weight per weight of polymer or lipid was ∼8 wt% for PTMBPEC-b-PEO polymersomes,64 ∼5 wt% for polymersomes made of PBD-b-PEO blended with poly(L-lactic acid)-b-PEO,65 ∼5 wt% for PEGylated liposomes made of the saturated phospholipid 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC),68 and ∼13 wt% for the Food and Drug Administration (FDA)-approved liposomal doxorubicin formulation Doxil© (Table 1).69,70
| Trade name | Active pharmaceutical ingredient | Liposome composition (mol%) | First FDA-approved indication (year of approval) |
|---|---|---|---|
Ambisome®![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 71–73 71–73 |
Amphotericin B | Hydrogenated soy phosphatidylcholine (HSPC), cholesterol, distearoyl phosphatidylglycerol (53:26:21) |
Fungal infections and visceral leishmaniasis (1997) |
| Doxil®69,70,72 |
Doxorubicin | HSPC, cholesterol, mPEO(2000)-distearoyl-sn-glycero-3-phosphoethanolamine (55:40:5) |
AIDS-related Kaposi's sarcoma (1995) |
| Marqibo®74 |
Vincristine | Sphingomyelin (SM) and cholesterol (55:45) |
Acute lymphoblastic leukemia (2012) |
The investigation of doxorubicin leakage during storage at 4 °C revealed strong differences between polymersomes made of a PBD-b-PEO and poly(L-lactic acid)-b-PEO blend and the liposomal Doxil® formulation. While these polymersomes showed a release of ∼5% doxorubicin after one month at 4 °C,65 the amount of free doxorubicin in Doxil® did not change over six months.17 With a leakage rate of 5% per month, only ∼36% of the initially encapsulated drug would remain in the core of the polymersomes after twenty months in the fridge (the shelf-life of Doxil®). To enhance the retention of the encapsulated cargo, polymersomes containing highly hydrophobic blocks such as PS may be a better (albeit non-biodegradable) option. PS-b-PEO polymersomes showed a leakage rate of only 1.5% per month of rhodamine B in surfactant-containing solutions75 and retained a transmembrane pH gradient over five months at 4 °C.37
Over thirty years of experience with the transmembrane pH-gradient loading method in liposomes yielded a range of investigative and FDA-approved liposome formulations with high encapsulation efficiency, drug loading, and very low drug leakage rates.66,76 Therefore, optimized liposomal formulations are highly suitable for the transmembrane pH gradient-based active loading method and able to retain drugs in the core for prolonged periods of time. Clear advantages of the polymersome formulations reported to date are not obvious for this procedure in terms of encapsulation efficiency, drug loading, and drug leakage.
Conjugating small and large molecules to the polymersome-forming block copolymer can be used to encapsulate drugs and decorate polymersomes with targeting ligands.77–79 Incorporating drugs using a biodegradable linker (analogously to drug–lipid conjugation for liposomes80,81) may provide prolonged release kinetics due to an improved retention in the vesicle.82
To increase the permeability of polymersomes for hydrophilic molecules and ions in a selective manner, transmembrane channel proteins can be incorporated into the membrane. In enzymatic cascade reactions, for instance, enzymes that are encapsulated in channel protein-containing membranes can be spatially separated from inhibiting factors while preserving their access to the substrate, as shown for cytidine-monophosphate-N-acetylneuraminic acid synthesis with PMOXA-b-PDMS-b-PMOXA polymersomes containing the channel protein OmpF G119D.83 Moreover, the incorporation of an Escherichia coli glycerol transporter protein into PMOXA-b-PDMS-b-PMOXA polymersomes led to an enhanced diffusion of the sugar alcohol ribitol and its enzymatic biosensing in the polymersome core.84 Transmembrane protein incorporation in polymersomes is further used to provide cell-mimicking systems with different membrane environments than those encountered in liposomes as recently reviewed.85 In more fundamental membrane protein studies, the chemical versatility of polymersome membranes was used to tune their mechanical properties, allowing for an improvement of membrane protein insertion.86 As most of these channel proteins have evolved for lipid membranes, however, the thickness of the membrane may theoretically limit the insertion of transmembrane proteins. Interestingly, polymersome membranes seem to adapt their conformation to allow the incorporation of biopores smaller than the hydrophobic membrane layer as shown for PMOXA-b-PDMS-b-PMOXA systems.87
1.4.2 Hydrophobic drugs
A major difference between lipid and polymeric vesicles is the membrane thickness which is limited to ∼3–5 nm for liposomes, while polymersomes can have membranes of up to ∼50 nm in thickness when long block copolymers are used.75,80,88 Therefore, polymersomes could in theory accommodate larger and higher amounts of hydrophobic molecules than liposomes. In addition, thicker membranes may result in slower release rates of hydrophobic substrates due to higher diffusional distances (Fig. 2). The hydrophobic cargo is generally added to the organic phase and incorporated into the membrane upon vesicle formation (e.g., nanoprecipitation, film rehydration, microfluidics).88–90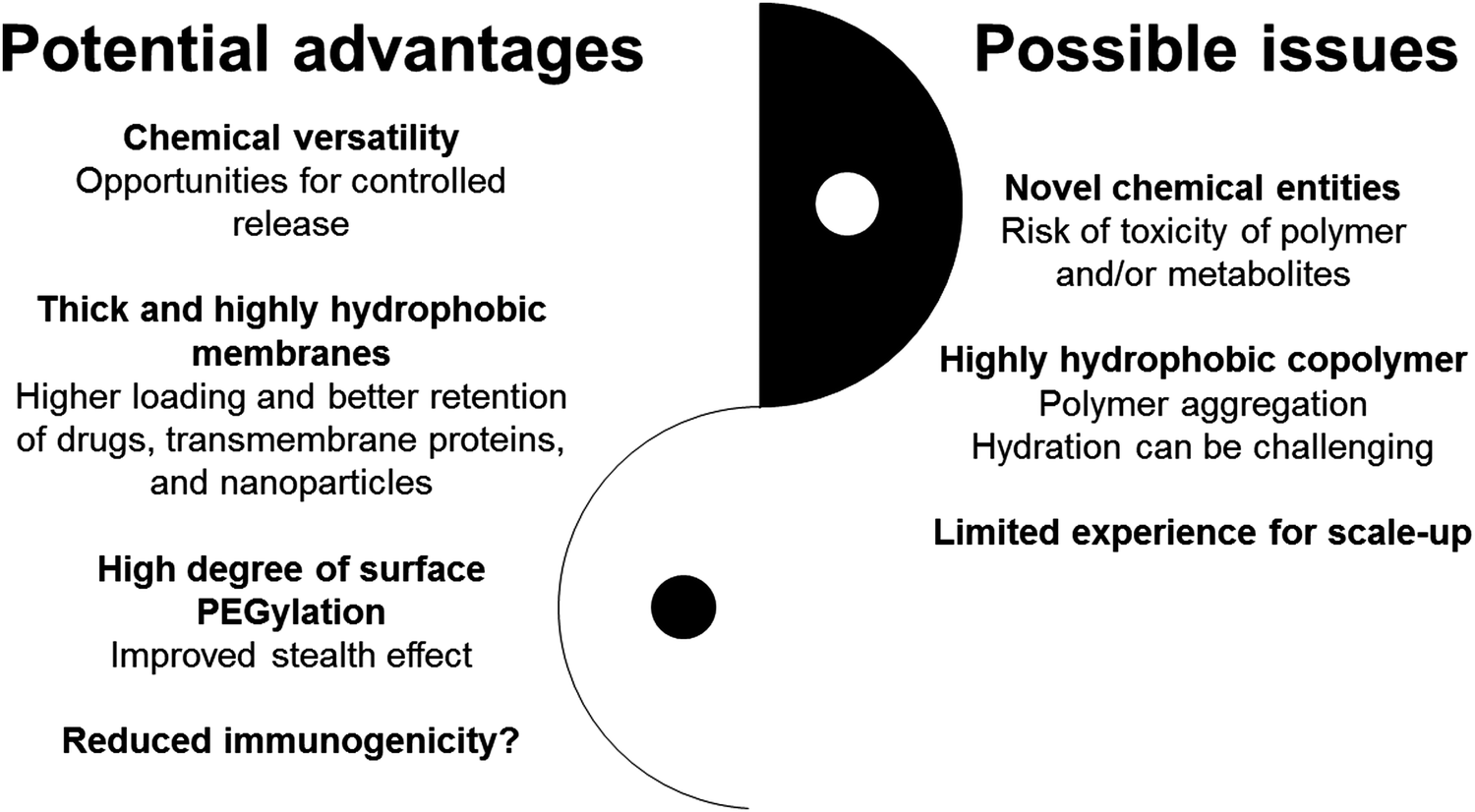 | ||
| Fig. 2 Potential advantages and possible disadvantages of polymersomes vs. liposomes. | ||
Using nanoprecipitation at pH 10.5, doxorubicin was loaded into poly(trimethylene carbonate)-b-poly(L-glutamic acid) polymersomes with an encapsulation efficiency of 78% and a drug loading of 47 wt%.90 Transmission electron microscopy indicated that the high drug loading was related to the formation of doxorubicin nanoparticles at the alkaline pH (pKa 8.3), which were incorporated in the hydrophobic layer of the membrane.90 Hydrophobically modified iron oxide nanoparticles with a radius of ∼3–5 nm were also incorporated into the membrane of poly(trimethylene carbonate)-b-poly(L-glutamic acid) and PEO-b-poly(D,L-lactic acid) polymersomes.31,91 Interestingly, ∼5 nm-thick liposomal membranes made of the saturated phospholipid 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) can also accommodate oleic acid-derivatized hydrophobic iron oxide nanoparticles of 5 nm in radius.92 Therefore, the membrane thickness does not seem to be the only determining factor in the loading of large hydrophobic substrates such that liposomal controls could be relevant even in the case of large hydrophobic substrates.
When loading substrates into the membrane, the risk of low drug retention due to the low diffusion distance in the membrane needs to be considered. Even though polymersome membranes are generally thicker than liposomal ones, the diffusion distance remains small, resulting in a risk of rapid cargo release. Indeed, paclitaxel-loaded hydrophobic polymeric nanoparticles with a diameter of ∼180 nm showed a fast release of their hydrophobic cargo due to diffusion.93 To determine the degree of retention in the membrane, thorough studies on the release kinetics of membrane-loaded substrates are needed in biorelevant media and under sink conditions or, preferably, in vivo. Moreover, the use of biodegradable polymers (e.g., for systemic administration) could further impact the release profile in vivo, as the degradation of the polymer could destabilize the membrane and result in accelerated cargo release.
1.5 In vivo studies
1.5.1 Systemic administration
As the membrane strength and surface PEGylation of liposomal systems influence their circulation half-life and leakiness,66 the initial reports on the high membrane stability of polymersomes raised hopes for longer circulation times and a greater drug retention in vivo than their lipidic counterparts.For liposomes, increasing the membrane stability with cholesterol and high phase transition temperature phospholipids (e.g., DSPC, HSPC) allowed to prolong the circulation half-life and improve the retention of luminal cargo.94,95 Therefore, in the case of polymersomes, high glass transition temperature polymers may be the preferable hydrophobic polymeric segments when long circulation times and high drug retention are sought. The phase and glass transition temperature of commonly used phospholipids and hydrophobic block are listed in Table 2.
| Lipid | T m (°C) |
|---|---|
| 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC, [18:1]2a) |
−17100 |
| 1,2-Dipalmitoyl-sn-glycero-3-PC (DPPC, [16:0]2a) |
41100 |
| 1,2-Distearoyl-sn-glycero-3-phosphocholine (DSPC, [18:0]2a) |
55100 |
| Hydrogenated soy phosphocholine (HSPC, mainly 18:0,a ∼11% 16:0a) |
∼49100 |
| Sphingomyelin (SM, [18:1/16:0]a) |
41b101 |
| 1-Stearoyl-2-oleoyl-sn-glycero-3-phosphocholine (SOPC, [18:0/18:1]a) |
6100 |
In PEGylated liposomes, the binding of opsonins to the liposomal surface was reduced, the uptake by opsonin-recognizing cells of the mononuclear phagocyte system lowered, and the circulation time prolonged compared with non-PEGylated liposomes.102 Due to their higher surface PEO density (100 mol% vs. generally 0.1–5 mol% for PEGylated liposomes48,63,103,104), PEO-containing polymersomes might exhibit a longer circulation half-life than liposomes. This hypothesis is debatable, however, because a PEO content of more than 5 mol% did not improve liposomal surface PEO coverage, blood protein adsorption, and circulation longevity with PEGylated liposomes.103,105
PEGylated liposomes elicit an immune response with anti-PEO immunoglobulin M antibodies. These antibodies led to an accelerated clearance upon repeated injection of PEGylated liposomes.69,106,107 Similar immune responses with an impact on the pharmacokinetic profile were reported for PEGylated polymeric nanoparticles after multiple applications.107 Interestingly, in a recent study investigating intravenous weekly administrations of poly(propylene sulfide)-b-PEO polymersomes in non-human primates, increases in anti-PEO immunoglobulin M or G antibodies were not observed.108 More studies on immune responses against repeatedly administered PEGylated polymersomes are warranted to confirm and mechanistically understand these findings. The immunogenicity of other hydrophilic fragments (e.g., poly(oxazoline), PAA) is less well understood.109
The number of pharmacokinetic studies on polymersomes is limited. One of the most cited ones investigated the circulation time of four non-biodegradable polymersomes made of PBD-b-PEO or PEE-b-PEO.45 To assess their circulation time in vivo, these polymersomes were stained with the hydrophobic fluorescent dye PKH26.45 This dye is conjugated to a C14 and a C22 chain and generally used for cell staining as the aliphatic chains insert into the lipid bilayer.110 An investigation of the dye retention in the membrane was unfortunately not reported in this study. The circulation half-lives of the polymersomes (∼120 nm) upon intravenous injection in rats ranged between 16 and 28 h, with longer circulation times observed for the longer diblock copolymers PBD54-b-PEO50 (∼28 h) and PBD130-b-PEO80 (∼26 h) than for PBD46-b-PEO26 (∼16 h).45 Interestingly, PEE37-b-PEO40 (∼19 h) polymersomes did not show a longer half-life than their PBD counterparts, even though hydrogenation theoretically leads to a higher membrane stability due to better membrane packing. In another pharmacokinetic study, 111Indium-labeled PBD70-b-PEO22 polymersomes of 90 nm showed a circulation half-life of ∼20 h in mice as determined with single-photon emission computed tomography.59 Unfortunately, a direct comparison to PEGylated liposomes was not performed in these studies.45,59 In the literature, PEGylated liposomes of similar size showed half-lives of 15 to 35 h in rodents.106,111–115 Therefore, clear evidence of a longer circulation time of polymersomes compared with PEGylated liposomes is currently missing. The impact of the high surface PEO density of polymersomes on half-life remains questionable. Furthermore, further studies on the impact of immunological reactions on the half-life of repeatedly administered polymersomes are warranted.
To address the non-biodegradability of PBD-b-PEO vesicles, polymersomes made of a blend of PBD125-b-PEO80 (75 wt%) and poly(L-lactic acid)56-b-PEO109 (25 wt%) were loaded with doxorubicin in the core and paclitaxel in the membrane (3 mg kg−1 doxorubicin, 7.5 mg kg−1 paclitaxel) and assessed for their antitumor efficacy. This formulation led to a prolonged tumor doxorubicin exposure and accelerated tumor regression in tumor-bearing nude mice compared with the free drugs (1.5 mg kg−1 doxorubicin, 1.0 mg kg−1 paclitaxel).116 Doxorubicin tumors levels of the polymersome formulation peaked after one day and decreased to ∼40% and ∼0% after two and four days, respectively (Fig. 3A).116 Unfortunately, paclitaxel levels were neither assessed in the tumor nor in plasma and liposomal controls were not included in this study. The clinically used liposomal doxorubicin formulation Doxil® could serve as an appropriate positive control due to its high stability (saturated phospholipids, high cholesterol content) and stealth properties (PEGylation).17,70,117 In a study on Doxil® in tumor-bearing mice, the liposomal formulation resulted in a much greater and prolonged exposure of the tumor to doxorubicin compared with the free drug (Fig. 3B).118 The maximum concentration was achieved after two days, remained stable until day five, and was decreased to ∼70% on day seven after injection.118 This lower and shorter tumor doxorubicin exposure in the case of the polymersomes could be due to a shorter plasma half-life of the polymersomes or a higher leakage of doxorubicin before reaching the tumor. Unfortunately, the plasma half-life of the PBD-b-PEO/poly(L-lactic acid)-b-PEO polymersome system was not calculated in the study.116 Comparing the half-life of similar polymersomes (PBD130-b-PEO80, ∼26 h45) with Doxil® liposomes (∼35 h,112 both in rats), strongly different plasma clearance profiles in mice would not be expected. Furthermore, a comparison of the absolute tumor accumulation of the doxorubicin-loaded vesicles cannot be made because the tumor doxorubicin levels were not quantified in terms of amount of drug per amount of tumor tissue in the polymersome study.116 This example, in which a liposomal formulation led to a longer exposure of the tumor to the chemotherapeutic agent, underlines the importance of liposomal controls in the evaluation of novel polymersome formulations.
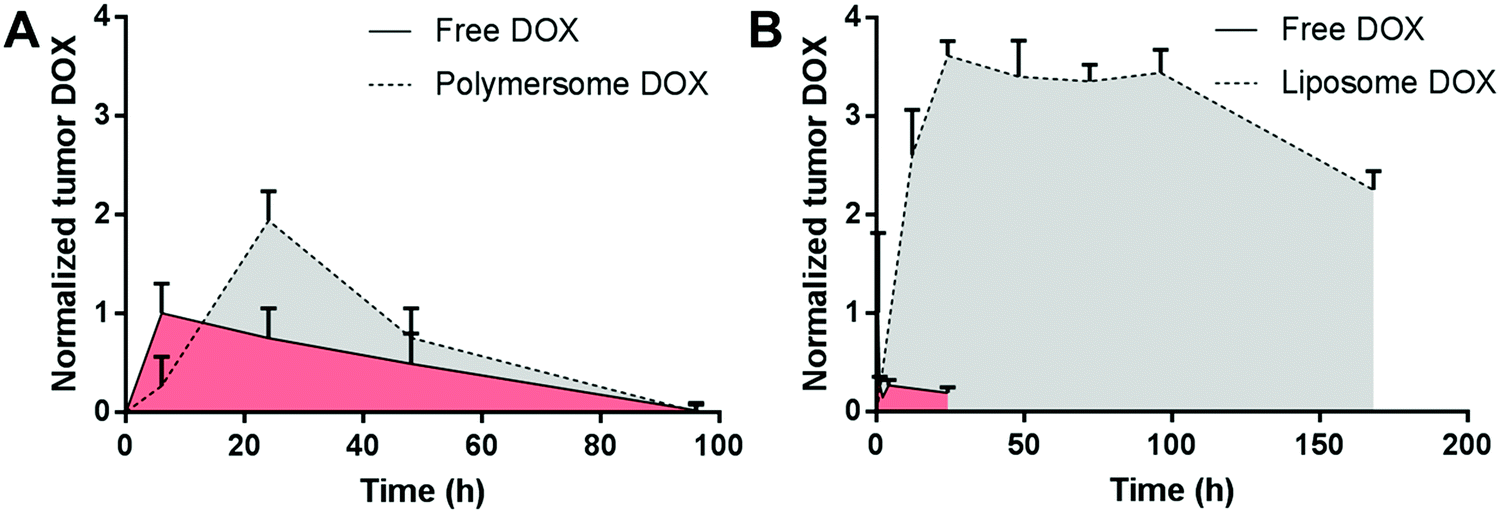 | ||
| Fig. 3 Comparison of the normalized tumor doxorubicin deposition in mice receiving polymersome or liposome formulations. Polymersome encapsulation led to higher doxorubicin levels compared with the free drug at 24 h (A), while liposomal doxorubicin (Doxil©) led to prolonged tumor exposure to doxorubicin for at least seven days (B). Plot A: free and polymersome-encapsulated doxorubicin administered in nude mice bearing MDA-MB231 human Caucasian breast adenocarcinoma tumors of 0.5 cm2, dose not mentioned, n = 2 per time point.116 Plot B: free doxorubicin and Doxil© at 9 mg kg−1 administered ten days after 4T1 mouse mammary carcinoma tumor inoculation in female BALB/c mice (tumor size not indicated), n = 3–5 per time point.118 Data normalized in both plots to the highest mean tumor doxorubicin level in the respective free doxorubicin group. DOX, doxorubicin. | ||
To demonstrate the polymersomes’ capacity to host large hydrophobic substrates in their membrane, oligo(porphyrin) near-infrared dyes were incorporated in PBD-b-PEO polymersomes.88 These highly rigid and hydrophobic dyes were added to the polymer film and encapsulated in the membrane during rehydration.88 While the polymersomes could incorporate up to pentameric oligo(porphyrin), liposomes made of SOPC could only accommodate dimers in their membrane.88 The authors speculated that the trimeric to pentameric oligo(porphyrin) were too large (∼3–5 nm) for the liposomal (∼3.5 nm) compared to the polymersome membrane (∼10 nm).88 However, other factors could have influenced the loading procedure as the membrane of DPPC liposomes was capable of incorporating hydrophobic nanoparticles of ∼5 nm.92 Upon intratumoral injection into subcutaneous glioma-bearing rats, a strong fluorescence signal of the oligo(porphyrin)-loaded polymersomes was observed in the investigated 20 min post-injection interval.88 In view of the missing positive and negative controls in the in vivo study, the added value of a polymersome-based delivery system over other platforms cannot be judged.
To target the hyaluronic acid-binding glycoprotein CD44 on cancer cells, a study used hyaluronic acid as the hydrophilic copolymer fragment. Doxorubicin-containing poly(γ-benzyl-L-glutamate)-b-hyaluronic acid polymersomes were taken up by CD44-expressing breast cancer cells in vitro.119 These polymersomes decreased the tumor size of breast cancer-bearing rats in vivo more than free doxorubicin119 but a liposomal doxorubicin control formulation (e.g., bearing surface-exposed hyaluronic acid120) was missing. The encapsulation of doxorubicin into polymersomes further decreased its cardiotoxicity119 which was already reported for liposomal doxorubicin in 1984.121
Cytosol-targeting polymersomes were developed for the delivery of cyclic dinucleotide agonists of the immunomodulatory protein stimulator of interferon genes for cancer immunotherapy.122 These chemotherapeutic drugs exhibit an unfavorable pharmacokinetic profile due to rapid systemic clearance and low cell permeability.122 The polymersomes were made of PEO-b-poly(2-(diethylamino)ethyl methacrylate-co-butyl methacrylate-co-pyridyl disulfide ethyl methacrylate) (PEO-b-poly[DEAEMA-co-BMA-co-PDSMA]) copolymers.122 The PDSMA block allowed for an improved cargo retention after crosslinking via disulfide bridge formation while providing a redox-responsive element which is destabilized in the supposedly increasingly reducing environment of the endosome.122 However, the extent of endosomal bio-reduction is disputed.123–125 Another destabilizing moiety in the hydrophobic part of the membrane are the DEAEMA units whose amines get protonated upon acidification in the endosome.122 In combination with the hydrophobic BMA block, the positive charges aimed at destabilizing the endosomal membrane to promote endosomal escape.122 Intratumoral injection of these polymersomes into B16–F10 melanoma-bearing mice decreased the tumor growth and induced remission in approx. one third of the animals over the 60 day study.122 Systemic administration led to lower antitumor effects, and remission was only achieved in combination with immune checkpoint blockade.122 This example provided an interesting attempt to use the versatility of the hydrophobic polymer fragment to increase drug retention while promoting polymersome destabilization in the endosome and endosomal escape. A liposomal system with these membrane properties is difficult to imagine due to the lower versatility of phospholipids.
Another polymersome system with a disulfide-bond crosslinked membrane was recently proposed for the treatment of ulcerative colitis.126 The polymersomes were composed of PEO-b-poly(trimethylene carbonate-co-dithiolane trimethylene carbonate)-b-poly(ethyleneimine) and PEO-b-poly(trimethylene carbonate-co-dithiolane trimethylene carbonate) modified with a macrophage-targeting peptide.126 The polymers self-assembled into asymmetric vesicles with of the cationic poly(ethyleneimine) facing the aqueous core, leading to an almost neutral surface charge.126 The polymersomes were loaded with a silencing ribonucleic acid against tumor necrosis factor-alpha and dexamethasone sodium phosphate.126 In a biodistribution study, the polymersomes with the macrophage-targeting peptide homed to the colon to a significantly higher extent than the peptide-free vesicle control after intravenous application.126 In an ulcerative colitis mouse model, the double-loaded polymersomes led to a significant downregulation of tumor necrosis factor-alpha and attenuated several markers of ulcerative colitis disease activity.
Polymersomes with reversible sugar-binding capacity were developed for diabetes therapy.127 PEO-b-poly[(7-(2-methacryloyloxyethoxy)-4-methylcoumarin)-stat-2-(diethylamino)ethyl methacrylate-stat-(α-D-glucopyranosyl)ethyl methacrylate] (PEO-b-poly[CMA-stat-DEA-stat-GEMA]) glycopolymersomes were loaded with the leptin ConA to enable glucose binding.127 ConA was retained in the membrane due to its affinity for glucopyranosyl, electrostatic interactions with DEA, and the cross-linking of the CMA membrane fragment.127 Because of the higher affinity of ConA for glucose than glucosyl group, these ConA-loaded glycopolymersomes took up and released glucose depending on the sugar concentration of the outer phase.127 Intravenous administration of ∼5 mg kg−1 glycopolymersomes with ∼30 mg kg−1 ConA in type I diabetic mice led to a decrease in blood glucose to normal levels for approximately 36 h. Unfortunately, the glucose-binding capacity of the formulation was not reported such that theoretical calculations on the administered dose and observed glucose-lowering effects cannot be carried out. In vivo studies on dose finding and dependence were also missing. Further studies are warranted to investigate the system's performance in rapid glucose bursts (e.g., postprandial glucose peaks) and potential immune-mediated effects on circulation time and sugar-binding capacity upon repeated administration.
Iodine-loaded polymersomes were investigated for computed tomography imaging and radioisotope therapy of breast cancer.128 After intravenous injection to mice, 125I-radiolabeled poly(iodinated carbonate)-b-PEO polymersomes (approx. 100 nm) mainly distributed in organs of the mononuclear phagocytic system and in 4T1 murine breast cancer tumors.128 Using a two-compartment pharmacokinetic model, a blood circulation half-life of over 10 h in the second phase was calculated.128 In contrast, free sodium iodate accumulated in the thyroid and was rapidly cleared by the kidneys.128 A linear signal increase in the tumor was observed over 20 h on computed tomography after intravenous application of 125I-radiolabeled polymersomes.128 Substituting 125I for the beta emitter 131I led to the development of polymersomes with antitumoral properties.128 This study illustrates how the chemical versatility of polymersomes can be used to design systems with built-in therapeutic and diagnostic moieties.
In conclusion, convincing evidence that polymersomes circulate longer while retaining higher amounts of cargo than liposomes is still lacking for most systems and applications. With regard to the established clinical safety profile of liposomes, undertaking the challenging process of bringing polymersomes to the clinic seems only worthwhile if two conditions are fulfilled: the identification of applications necessitating polymersome-specific features and a clear demonstration of these advantages for the selected polymersome formulation over relevant liposomal controls. Unfortunately, head-to-head comparisons of polymersomes with well-established liposome formulations (Table 1) with high membrane toughness (high cholesterol content, saturated lipids) and stealth properties (PEGylation) are not reported in most of the reviewed studies.
1.5.2 Oral administration
The gastrointestinal (GI) tract is a harsh environment for vesicles.129 High bile salts concentrations, strong osmolarity and pH changes, and high enzymatic activity generally lead to rapid destabilization of vesicular structures.129–133 Bile salts impaired the membrane integrity of liposomes by insertion into the outer leaflet of the membrane and by the partitioning of the lipids into bile salt micelles, which may lead to cargo release.133–138 Hypo- and hyperosmolar environments, which can range from 100 to 600 mOsmol kg−1 in the small intestine,139–141 induce morphologic changes (i.e., shrinking and swelling, respectively) of vesicles,142,143 and potentially impair the structural integrity of the membrane. Polyester-based biodegradable polymersomes, surface-exposed peptide-based targeting ligands, and phospholipid-based liposomes may further be hydrolyzed in the acidic environment of the stomach or due to enzymatic action (e.g., pepsin, (chymo)trypsin, phospholipase A2 for phospholipids).130,144,145 The environment of the colon is also destabilizing for vesicles as the water resorption thickens the chyme and increases the osmolarity.37 As even PEGylated liposomes with high phase transition temperature phospholipids and cholesterol content do not resist these conditions,37 oral delivery could be a promising field for highly stable polymersome formulations.PS-b-PEO polymersomes were investigated by our group as an oral ammonia scavenger to treat hyperammonemia, a serious complication of liver disease.37 The weak base ammonia can be efficiently and selectively captured by transmembrane pH-gradient vesicles with an acidic core.37,48,146,147,161 While neither cholesterol-containing PEGylated DPPC and DSPC liposomes nor PBD-b-PEO polymersomes were resistant to physiologically relevant bile salt concentrations, PS-b-PEO polymersomes preserved their ammonia capture capacity in bile salt-containing media at extreme osmolarity levels and in digestive enzyme-containing fluids.37 Despite performing well in commonly used GI-simulating fluids, these polymersomes proved to be unstable in dietary fiber-based hydrogels and failed to decrease plasma ammonia levels in hyperammonemic rats. This study underlines differences in stability between liposomes and polymersomes and among polymersomes. It further points to shortcomings of commonly used simulated GI fluids in the evaluation of vesicles for oral delivery,138,148,149 and highlights the need to account for the colonic environment in vesicle-based oral detoxification.
To demonstrate the solubility enhancement of a poorly soluble small molecular drug with a polymersome formulation, the multikinase inhibitor sorafenib was encapsulated in the hydrophobic part of the membrane of PBD-b-PEO polymersomes and orally administered to healthy mice.150 The area under the plasma concentration vs. time curve of the polymersome formulation was higher compared to the control formulation (sorafenib suspension).150 As PBD-b-PEO polymersomes are not stable in surfactant-containing media,151 differences in luminal bile salt concentrations may impact on the sorafenib release profile. Other drug delivery systems such as pH-sensitive Eudragit® nanoparticles or poly(N-vinylpyrrolidone-vinyl acetate)-containing tablets also showed high oral bioavailability for sorafenib and could serve as alternatives to polymersomes for this drug.152,153
A study reported that orally applied insulin-containing dextran-b-poly(D,L-lactide-co-glycolide) polymersomes led to appreciable systemic insulin levels and decreased glucose levels in diabetic mice.154 In view of the difficulty of reaching the systemic circulation with macromolecular drugs due to the mucus layer and the tight gut epithelium,54 an in-depth investigation of the translocation from the gut lumen into the systemic compartment is needed to fully understand these results.
1.6 Polymersomes as reaction compartments
As different hydrophobic blocks impact on membrane rigidity and hydrophobicity, the polymersome membrane can be tuned for selective permeability.56,155,156 Polymersomes have therefore been developed as selectively permeable reaction compartments for diagnostic use and synthetic applications. We illustrate this application with two polymersome systems described above whose use was extended to the diagnostic field. For a detailed discussion of the use of polymersomes as micro- or nanoreactors and their use in diagnostics and theranostics, the reader is kindly referred to recent reviews.157–160The usefulness of transmembrane pH-gradient PS-b-PEO polymersomes to quantify ammonia in solution was recently demonstrated by our group.37 The influx of the weakly basic ammonia into the acidic vesicular core led to an increase in core pH which was quantified by an encapsulated pH-sensitive fluorescent dye.37 The high hydrophobicity of the PS-b-PEO polymersome membrane allowed for a high selectivity to ammonia compared to other weakly basic metabolites and drugs.37 In contrast, the permeability of cholesterol-containing PEGylated DPPC liposomes towards weakly basic drugs was higher.147
The reversibly glucose-sequestering PEO-b-poly(CMA-stat-DEA-stat-GEMA) glycopolymersomes described above were further used as a glucose assay.127 As they swelled from 400 to 800 nm upon exposition to glucose solutions (“glucose sponge”), the authors proposed a particle size-based glucose-sensing system.127 This system was neither validated in blood or plasma nor selective to glucose in the presence of mannose.127 Moreover, the practicality and reliability of particle size measurements compared with well-established point-of-care glucose tests remains questionable.
1.7 Conclusion
Polymeric vesicles were initially described in two seminal publications by van Hest et al. and Zhang and Eisenberg in 1995. Four years later, Discher et al. showed that PEE-b-PEO polymersomes had tougher membranes than low phase-transition temperature liposomes made of SOPC. This study raised high hopes for the development of a new generation of vesicular drug delivery systems that are superior to liposomes in terms of stability, circulation time, and drug loading capacity. Twenty years later, there are still open questions about the generalizability of the higher membrane strength beyond the tested formulations. The very few studies on circulation time pointed to similar half-lives of polymersomes and PEGylated liposomes. A clear superiority in the loading capacity for commonly used hydrophilic or hydrophobic drugs has not been established for polymersomes, and the impact of membrane biodegradability on plasma half-life, drug release, and polymersome stability in general remains poorly investigated. Furthermore, finding upscalable polymersome preparation procedures leading to very high polymersome concentrations and low amounts of residual solvent remains challenging, especially if the experience on the industrial production of liposome cannot be applied (e.g., for highly hydrophobic copolymers). In our opinion, more systematic studies comparing polymersomes of various composition against strong liposome controls are needed to demonstrate the proposed superiority of polymersomes. Only when such advantages are clearly established and relevant clinical applications necessitating these properties are identified, can it be judged if the challenging clinical development of an alternative vesicular drug delivery platform to liposomes, whose clinical safety profile is well established, is warranted.Abbreviations
| DOPC | 1,2-Dioleoyl-sn-glycero-3-phosphocholine |
| DPPC | 1,2-Dipalmitoyl-sn-glycero-3-phosphocholine |
| DSPC | 1,2-Distearoyl-sn-glycero-3-phosphocholine |
| FDA | Food and Drug Administration |
| HSPC | Hydrogenated soy phosphocholine |
| PAA | Poly(acrylic acid) |
| PBD | Poly(butadiene) |
| PEE | Poly(ethylethylene) |
| PEO | Poly(ethylene oxide) |
| PLA | Poly(lactic acid) |
| PLGA | Poly(lactic-co-glycolic acid) |
| PTMBPEC | Poly(2,4,6-trimethoxybenzylidenepentaerythritol carbonate) |
| PS | Poly(styrene) |
| SM | Sphingomyelin |
| SOPC | 1-Stearoyl-2-oleoyl-sn-glycero-3-phosphocholine |
| TEM | Transmission electron microscopy |
Conflicts of interest
JCL and SM are co-inventors on patents/patent applications related to liposome and polymersome formulations for treating and diagnosing hyperammonemia. These patents have been licensed to Versantis AG. JCL is a shareholder of Versantis AG.Acknowledgements
We thank Prof. Dr. Peter Walde, Dr. Yinyin Bao, Aaron Schmidt, Dr. Anastasia Spyrogianni, and Dr. Charlotte Gourmel for insightful discussions and their critical reading of the manuscript. S. M. gratefully acknowledges a doctoral scholarship from the Swiss Chemical Industry (SSCI). We dedicate this article to Prof. Dr. Bruno Gander, whose passion for drug formulation and critical thinking inspired numerous generations of pharmacists at ETH Zurich, and wish him all the best for his future endeavors.References
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CrossRef CAS PubMed.
- J. C. van Hest, D. A. Delnoye, M. W. Baars, M. H. van Genderen and E. W. Meijer, Science, 1995, 268, 1592–1595 CrossRef CAS PubMed.
- H. J. Lee, S. R. Yang, A. E. Jung An and J.-D. Kim, Macromolecules, 2006, 39, 4938–4940 CrossRef CAS.
- F. Le Devedec, A. Won, J. Oake, L. Houdaihed, C. Bohne, C. M. Yip and C. Allen, ACS Macro Lett., 2016, 5, 128–133 CrossRef CAS.
- H. Bermudez, A. K. Brannan, D. A. Hammer, F. S. Bates and D. E. Discher, Macromolecules, 2002, 35, 8203–8208 CrossRef CAS.
- S. Hocine, D. Cui, M.-N. Rager, A. Di Cicco, J.-M. Liu, J. Wdzieczak-Bakala, A. Brûlet and M.-H. Li, Langmuir, 2013, 29, 1356–1369 CrossRef CAS PubMed.
- T. Chidanguro, E. Ghimire, C. H. Liu and Y. C. Simon, Small, 2018, 14 Search PubMed.
- J. Leong, J. Y. Teo, V. K. Aakalu, Y. Y. Yang and H. Kong, Adv. Healthcare Mater., 2018, 7, 1701276 CrossRef PubMed.
- J. Siepmann, A. Faham, S. D. Clas, B. J. Boyd, V. Jannin, A. Bernkop-Schnürch, H. Zhao, S. Lecommandoux, J. C. Evans, C. Allen, O. M. Merkel, G. Costabile, M. R. Alexander, R. D. Wildman, C. J. Roberts and J. C. Leroux, Int. J. Pharm., 2019, 558, 128–142 CrossRef CAS PubMed.
- L. Zhang, K. Yu and A. Eisenberg, Science, 1996, 272, 1777–1779 CrossRef CAS PubMed.
- B. M. Discher, Y. Y. Won, D. S. Ege, J. C. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS PubMed.
- M.-H. Li and P. Keller, Soft Matter, 2009, 5, 927 RSC.
- K. Letchford, Eur. J. Pharm. Biopharm., 2007, 65, 259–269 CrossRef CAS PubMed.
- D. E. Discher, V. Ortiz, G. Srinivas, M. L. Klein, Y. Kim, D. Christian, S. Cai, P. Photos and F. Ahmed, Prog. Polym. Sci., 2007, 32, 838–857 CrossRef CAS PubMed.
- L. Guan, L. Rizzello and G. Battaglia, Nanomedicine, 2015, 10, 2757–2780 CrossRef CAS PubMed.
- M. Mohammadi, M. Ramezani, K. Abnous and M. Alibolandi, Int. J. Pharm., 2017, 519, 287–303 CrossRef CAS.
- Y. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- J. De Gier, J. G. Mandersloot and L. L. M. Van Deenen, Biochim. Biophys. Acta, Biomembr., 1968, 150, 666–675 CrossRef CAS.
- H. Bermúdez, D. A. Hammer and D. E. Discher, Langmuir, 2004, 20, 540–543 CrossRef PubMed.
- H. V. Ly, D. E. Block and M. L. Longo, Langmuir, 2002, 18, 8988–8995 CrossRef CAS.
- N. P. Kamat, M. H. Lee, D. Lee and D. A. Hammer, Soft Matter, 2011, 7, 9863 RSC.
- E. Mabrouk, D. Cuvelier, L.-L. Pontani, B. Xu, D. Lévy, P. Keller, F. Brochard-Wyart, P. Nassoy and M.-H. Li, Soft Matter, 2009, 5, 1870 RSC.
- K. Allen Rodowicz, H. Francisco and B. Layton, Chem. Phys. Lipids, 2010, 163, 787–793 CrossRef CAS.
- K. Akashi, H. Miyata, H. Itoh and K. Kinosita, Biophys. J., 1996, 71, 3242–3250 CrossRef CAS PubMed.
- E. Farge and P. F. Devaux, J. Phys. Chem., 1993, 97, 2958–2961 CrossRef CAS.
- S. D. Shoemaker and T. K. Vanderlick, Biophys. J., 2002, 83, 2007–2014 CrossRef CAS PubMed.
- Y. Takechi-Haraya, K. Sakai-Kato, Y. Abe, T. Kawanishi, H. Okuda and Y. Goda, Langmuir, 2016, 32, 6074–6082 CrossRef CAS PubMed.
- M. Sala, K. Miladi, G. Agusti, A. Elaissari and H. Fessi, Colloids Surf., A, 2017, 524, 71–78 CrossRef CAS.
- C. Charcosset, A. Juban, J. P. Valour, S. Urbaniak and H. Fessi, Chem. Eng. Res. Des., 2015, 94, 508–515 CrossRef CAS.
- A. Wagner and K. Vorauer-Uhl, J. Drug Delivery, 2011, 1–9 CAS.
- C. Sanson, O. Diou, J. Thévenot, E. Ibarboure, A. Soum, A. Brûlet, S. Miraux, E. Thiaudière, S. Tan, A. Brisson, V. Dupuis, O. Sandre and S. Lecommandoux, ACS Nano, 2011, 5, 1122–1140 CrossRef CAS.
- R. S. M. Rikken, H. Engelkamp, R. J. M. Nolte, J. C. Maan, J. C. M. van Hest, D. A. Wilson and P. C. M. Christianen, Nat. Commun., 2016, 7, 12606 CrossRef CAS.
- S. Allen, O. Osorio, Y. G. Liu and E. Scott, J. Controlled Release, 2017, 262, 91–103 CrossRef CAS.
- Y. Men, F. Peng, Y. Tu, J. C. M. van Hest and D. A. Wilson, Polym. Chem., 2016, 7, 3977–3982 RSC.
- F. Meng, C. Hiemstra, G. H. M. Engbers and J. Feijen, Macromolecules, 2003, 36, 3004–3006 CrossRef CAS.
- Y. Lee, J.-B. Chang, H. K. Kim and T. G. Park, Macromol. Res., 2006, 14, 359–364 CrossRef CAS.
- S. Matoori, Y. Bao, A. Schmidt, E. J. Fischer, R. Ochoa-Sanchez, M. Tremblay, M. M. Oliveira, C. F. Rose and J.-C. Leroux, Small, 2019, 15, e1902347 CrossRef PubMed.
- C. Charcosset, I. Limayem and H. Fessi, J. Chem. Technol. Biotechnol., 2004, 79, 209–218 CrossRef CAS.
- R. A. Williams, S. J. Peng, D. A. Wheeler, N. C. Morley, D. Taylor, M. Whalley and D. W. Houldsworth, Chem. Eng. Res. Des., 1998, 76, 902–910 CrossRef CAS.
- J. Thiele, A. R. Abate, H. C. Shum, S. Bachtler, S. Förster and D. A. Weitz, Small, 2010, 6, 1723–1727 CrossRef CAS PubMed.
- H. C. Shum, Y. Zhao, S.-H. Kim and D. A. Weitz, Angew. Chem., 2011, 123, 1686–1689 CrossRef.
- E. Lorenceau, A. S. Utada, D. R. Link, G. Cristobal, M. Joanicot and D. A. Weitz, Langmuir, 2005, 21, 9183–9186 CrossRef CAS PubMed.
- M. K. Mulligan and J. P. Rothstein, Microfluid. Nanofluid., 2012, 13, 65–73 CrossRef CAS.
- D. Carugo, E. Bottaro, J. Owen, E. Stride and C. Nastruzzi, Sci. Rep., 2016, 6, 25876 CrossRef CAS.
- P. J. Photos, L. Bacakova, B. Discher, F. S. Bates and D. E. Discher, J. Controlled Release, 2003, 90, 323–334 CrossRef CAS.
- J. C.-M. Lee, H. Bermudez, B. M. Discher, M. A. Sheehan, Y.-Y. Won, F. S. Bates and D. E. Discher, Biotechnol. Bioeng., 2001, 73, 135–145 CrossRef CAS.
- E. A. Scott, A. Stano, M. Gillard, A. C. Maio-Liu, M. A. Swartz and J. A. Hubbell, Biomaterials, 2012, 33, 6211–6219 CrossRef CAS.
- V. Agostoni, S. H. Lee, V. Forster, M. Kabbaj, C. R. Bosoi, M. Tremblay, M. Zadory, C. F. Rose and J.-C. Leroux, Adv. Funct. Mater., 2016, 26, 8382–8389 CrossRef CAS.
- C. Fetsch, J. Gaitzsch, L. Messager, G. Battaglia and R. Luxenhofer, Sci. Rep., 2016, 6, 33491 CrossRef.
- A. Jofre, J. B. Hutchison, R. Kishore, L. E. Locascio and K. Helmerson, J. Phys. Chem. B, 2007, 111, 5162–5166 CrossRef CAS.
- H. Xu, F. Meng and Z. Zhong, J. Mater. Chem., 2009, 19, 4183–4190 RSC.
- D. Gräfe, J. Gaitzsch, D. Appelhans and B. Voit, Nanoscale, 2014, 6, 10752–10761 RSC.
- G. J. R. Charrois and T. M. Allen, Biochim. Biophys. Acta, Biomembr., 2003, 1609, 102–108 CrossRef CAS.
- E. Moroz, S. Matoori and J.-C. Leroux, Adv. Drug Delivery Rev., 2016, 101, 108–121 CrossRef CAS.
- F. Li, L. H. J. de Haan, A. T. M. Marcelis, F. A. M. Leermakers, M. A. Cohen Stuart and E. J. R. Sudhölter, Soft Matter, 2009, 5, 4042 RSC.
- R. Rodríguez-García, M. Mell, I. López-Montero, J. Netzel, T. Hellweg and F. Monroy, Soft Matter, 2011, 7, 1532 RSC.
- E. Cabane, V. Malinova, S. Menon, C. G. Palivan and W. Meier, Soft Matter, 2011, 7, 9167 RSC.
- F. Ahmed, R. I. Pakunlu, A. Brannan, F. Bates, T. Minko and D. E. Discher, J. Controlled Release, 2006, 116, 150–158 CrossRef CAS.
- R. P. Brinkhuis, K. Stojanov, P. Laverman, J. Eilander, I. S. Zuhorn, F. P. J. T. Rutjes and J. C. M. van Hest, Bioconjugate Chem., 2012, 23, 958–965 CrossRef CAS PubMed.
- J. E. Bartenstein, J. Robertson, G. Battaglia and W. H. Briscoe, Colloids Surf., A, 2016, 506, 739–746 CrossRef CAS.
- L. D. Mayer, M. B. Bally and P. R. Cullis, Biochim. Biophys. Acta, Biomembr., 1986, 857, 123–126 CrossRef CAS.
- L. D. Mayer, P. R. Cullis and M. B. Bally, J. Liposome Res., 1994, 4, 529–553 CrossRef.
- M. Roveri, A. Pfohl, P. Jaaks, N. Alijaj, J.-C. Leroux, P. Luciani and M. Bernasconi, Nanomedicine, 2017, 12, 1135–1151 CrossRef CAS.
- W. Chen, F. Meng, R. Cheng and Z. Zhong, J. Controlled Release, 2010, 142, 40–46 CrossRef CAS.
- F. Ahmed, R. I. Pakunlu, G. Srinivas, A. Brannan, F. Bates, M. L. Klein, T. Minko and D. E. Discher, Mol. Pharmaceutics, 2006, 3, 340–350 CrossRef CAS.
- D. C. Drummond, O. Meyer, K. Hong, D. B. Kirpotin and D. Papahadjopoulos, Pharmacol. Rev., 1999, 51, 691–743 CAS.
- L. D. Mayer, L. C. L. Tai, M. B. Bally, G. N. Mitilenes, R. S. Ginsberg and P. R. Cullis, Biochim. Biophys. Acta, Biomembr., 1990, 1025, 143–151 CrossRef CAS.
- N. Dos Santos, K. A. Cox, C. A. McKenzie, F. van Baarda, R. C. Gallagher, G. Karlsson, K. Edwards, L. D. Mayer, C. Allen and M. B. Bally, Biochim. Biophys. Acta, Biomembr., 2004, 1661, 47–60 CrossRef CAS.
- T. Ishida, M. Ichihara, X. Wang, K. Yamamoto, J. Kimura, E. Majima and H. Kiwada, J. Controlled Release, 2006, 112, 15–25 CrossRef CAS.
- S. A. Abraham, D. N. Waterhouse, L. D. Mayer, P. R. Cullis, T. D. Madden and M. B. Bally, Methods Enzymol., 2005, 391, 71–97 CAS.
- M. Iman, Z. Huang, F. C. Szoka and M. R. Jaafari, Int. J. Pharm., 2011, 408, 163–172 CrossRef CAS PubMed.
- H.-I. Chang and M.-K. Yeh, Int. J. Nanomed., 2012, 7, 49–60 CAS.
- D. Bobo, K. J. Robinson, J. Islam, K. J. Thurecht and S. R. Corrie, Pharm. Res., 2016, 33, 2373–2387 CrossRef CAS PubMed.
- D. A. Thomas, H. M. Kantarjian, W. Stock, L. Heffner, S. Hirabayashi, B. Lu, S. R. Deitcher and S. O’Brien, Blood, 2008, 112, 2930 Search PubMed.
- Z. Song, Y. Huang, V. Prasad, R. Baumgartner, S. Zhang, K. Harris, J. S. Katz and J. Cheng, ACS Appl. Mater. Interfaces, 2016, 8, 17033–17037 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS PubMed.
- N. Christian, M. Milone, S. Ranka, G. Li, P. Frail, K. Davis, F. Bates, M. Therien, P. Ghoroghchian, C. June and D. Hammer, Bioconjugate Chem., 2007, 18, 31–40 CrossRef CAS PubMed.
- S. Egli, M. G. Nussbaumer, V. Balasubramanian, M. Chami, N. Bruns, C. Palivan and W. Meier, J. Am. Chem. Soc., 2011, 133, 4476–4483 CrossRef CAS.
- J. Lin, P. Ghoroghchian, Y. Zhang and D. Hammer, Langmuir, 2006, 22, 3975–3979 CrossRef CAS.
- R. D. Signorell, A. Papachristodoulou, J. Xiao, B. Arpagaus, T. Casalini, J. Grandjean, J. Thamm, F. Steiniger, P. Luciani, D. Brambilla, B. Werner, E. Martin, M. Weller, P. Roth and J.-C. Leroux, Int. J. Pharm., 2018, 536, 388–396 CrossRef CAS.
- A. Pratsinis, S. Zuercher, V. Forster, E. J. Fischer, P. Luciani and J.-C. Leroux, Biomaterials, 2017, 145, 128–137 CrossRef CAS PubMed.
- R. D. Signorell, P. Luciani, D. Brambilla and J.-C. Leroux, Eur. J. Pharm. Biopharm., 2018, 128, 188–199 CrossRef CAS PubMed.
- L. Klermund, S. T. Poschenrieder and K. Castiglione, ACS Catal., 2017, 7, 3900–3904 CrossRef CAS.
- X. Zhang, M. Lomora, T. Einfalt, W. Meier, N. Klein, D. Schneider and C. G. Palivan, Biomaterials, 2016, 89, 79–88 CrossRef CAS.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018, 47, 8572–8610 RSC.
- M. L. Jacobs, M. A. Boyd and N. P. Kamat, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4031–4036 CrossRef CAS.
- M. Lomora, M. Garni, F. Itel, P. Tanner, M. Spulber and C. G. Palivan, Biomaterials, 2015, 53, 406–414 CrossRef CAS.
- P. P. Ghoroghchian, P. R. Frail, K. Susumu, D. Blessington, A. K. Brannan, F. S. Bates, B. Chance, D. A. Hammer and M. J. Therien, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2922–2927 CrossRef CAS.
- S. Joshi, M. T. Hussain, C. B. Roces, G. Anderluzzi, E. Kastner, S. Salmaso, D. J. Kirby and Y. Perrie, Int. J. Pharm., 2016, 514, 160–168 CrossRef CAS.
- C. Sanson, C. Schatz, J.-F. Le Meins, A. Soum, J. Thévenot, E. Garanger and S. Lecommandoux, J. Controlled Release, 2010, 147, 428–435 CrossRef CAS.
- B. M. Geilich, I. Gelfat, S. Sridhar, A. L. van de Ven and T. J. Webster, Biomaterials, 2017, 119, 78–85 CrossRef CAS.
- Y. Chen, A. Bose and G. D. Bothun, ACS Nano, 2010, 4, 3215–3221 CrossRef CAS.
- G. Gaucher, R. H. Marchessault and J.-C. Leroux, J. Controlled Release, 2010, 143, 2–12 CrossRef CAS PubMed.
- D. C. Drummond, O. Meyer, K. Hong, D. B. Kirpotin and D. Papahadjopoulos, Pharmacol. Rev., 1999, 51, 691–743 CAS.
- S. Kaddah, N. Khreich, F. Kaddah, C. Charcosset and H. Greige-Gerges, Food Chem. Toxicol., 2018, 113, 40–48 CrossRef CAS.
- D. Richter, R. Zorn, B. Farago, B. Frick and L. J. Fetters, Phys. Rev. Lett., 1992, 68, 71–74 CrossRef CAS PubMed.
- A. R. Katritzky, S. Sild, V. Lobanov and M. Karelson, J. Chem. Inf. Comput. Sci., 1998, 38, 300–304 CrossRef CAS.
- N. Passerini and D. Q. Craig, J. Controlled Release, 2001, 73, 111–115 CrossRef CAS.
- J. S. Sharp and J. A. Forrest, Phys. Rev. Lett., 2003, 91, 235701 CrossRef CAS PubMed.
- D. Marsh, Handbook of Lipid Bilayers, CRC Press, 2013 Search PubMed.
- T. K. M. Nyholm, M. Nylund and J. Peter Slotte, Biophys. J., 2003, 84, 3138–3146 CrossRef CAS.
- J. Senior, C. Delgado, D. Fisher, C. Tilcock and G. Gregoriadis, Biochim. Biophys. Acta, Biomembr., 1991, 1062, 77–82 CrossRef CAS.
- N. Dos Santos, C. Allen, A.-M. Doppen, M. Anantha, K. A. K. Cox, R. C. Gallagher, G. Karlsson, K. Edwards, G. Kenner, L. Samuels, M. S. Webb and M. B. Bally, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 1367–1377 CrossRef CAS PubMed.
- L. M. Russell, M. Hultz and P. C. Searson, J. Controlled Release, 2018, 269, 171–176 CrossRef CAS PubMed.
- O. K. Nag and V. Awasthi, Pharmaceutics, 2013, 5, 542–569 CrossRef CAS PubMed.
- T. Ishida, R. Maeda, M. Ichihara, K. Irimura and H. Kiwada, J. Controlled Release, 2003, 88, 35–42 CrossRef CAS PubMed.
- P. Grenier, I. M. de, O. Viana, E. M. Lima and N. Bertrand, J. Controlled Release, 2018, 287, 121–131 CrossRef CAS PubMed.
- S. D. Allen, Y. G. Liu, S. Bobbala, L. Cai, P. I. Hecker, R. Temel and E. A. Scott, Nano Res., 2018, 11, 5689–5703 CrossRef CAS.
- R. Luxenhofer, Y. Han, A. Schulz, J. Tong, Z. He, A. V. Kabanov and R. Jordan, Macromol. Rapid Commun., 2012, 33, 1613–1631 CrossRef CAS.
- N. Maishi, T. Kawamoto, N. Ohga, K. Yamada, K. Akiyama, K. Yamamoto, T. Osawa, Y. Hida and K. Hida, Oncol. Rep., 2013, 30, 1695–1700 CrossRef CAS.
- T. Ishida, M. Ichihara, X. Wang, K. Yamamoto, J. Kimura, E. Majima and H. Kiwada, J. Controlled Release, 2006, 112, 15–25 CrossRef CAS.
- T. Siegal, A. Horowitz and A. Gabizon, J. Neurosurg., 1995, 83, 1029–1037 CAS.
- A. Gabizon, H. Shmeeda and Y. Barenholz, Clin. Pharmacokinet., 2003, 42, 419–436 CrossRef CAS PubMed.
- A. Bao, B. Goins, R. Klipper, G. Negrete, W. T. Phillips, N. van Rooijen, F. H. Corstens and G. Storm, J. Pharmacol. Exp. Ther., 2004, 308, 419–425 CrossRef CAS PubMed.
- T. M. Allen, C. Hansen, F. Martin, C. Redemann and A. Yau-Young, Biochim. Biophys. Acta, Biomembr., 1991, 1066, 29–36 CrossRef CAS.
- F. Ahmed, R. I. Pakunlu, A. Brannan, F. Bates, T. Minko and D. E. Discher, J. Controlled Release, 2006, 116, 150–158 CrossRef CAS.
- T. Ishida, K. Atobe, X. Wang and H. Kiwada, J. Controlled Release, 2006, 115, 251–258 CrossRef CAS PubMed.
- K. M. Laginha, S. Verwoert, G. J. R. Charrois and T. M. Allen, Clin. Cancer Res., 2005, 11, 6944–6949 CrossRef CAS PubMed.
- K. K. Upadhyay, A. N. Bhatt, A. K. Mishra, B. S. Dwarakanath, S. Jain, C. Schatz, J.-F. Le Meins, A. Farooque, G. Chandraiah, A. K. Jain, A. Misra and S. Lecommandoux, Biomaterials, 2010, 31, 2882–2892 CrossRef CAS PubMed.
- R. Ran, Y. Liu, H. Gao, Q. Kuang, Q. Zhang, J. Tang, H. Fu, Z. Zhang and Q. He, J. Pharm. Sci., 2015, 104, 476–484 CrossRef CAS PubMed.
- Q. G. van Hoesel, P. A. Steerenberg, D. J. Crommelin, A. van Dijk, W. van Oort, S. Klein, J. M. Douze, D. J. de Wildt and F. C. Hillen, Cancer Res., 1984, 44, 3698–3705 CAS.
- D. Shae, K. W. Becker, P. Christov, D. S. Yun, A. K. R. Lytton-Jean, S. Sevimli, M. Ascano, M. Kelley, D. B. Johnson, J. M. Balko and J. T. Wilson, Nat. Nanotechnol., 2019, 14, 269–278 CrossRef CAS PubMed.
- L. Brülisauer, G. Valentino, S. Morinaga, K. Cam, J. Thostrup Bukrinski, M. A. Gauthier and J.-C. Leroux, Angew. Chem., Int. Ed., 2014, 53, 8392–8396 CrossRef PubMed.
- L. Brülisauer, N. Kathriner, M. Prenrecaj, M. A. Gauthier and J.-C. Leroux, Angew. Chem., Int. Ed., 2012, 51, 12454–12458 CrossRef.
- L. Brülisauer, M. A. Gauthier and J.-C. Leroux, J. Controlled Release, 2014, 195, 147–154 CrossRef PubMed.
- X. Xu, W. Yang, Q. Liang, Y. Shi, W. Zhang, X. Wang, F. Meng, Z. Zhong and L. Yin, Nano Res., 2019, 12, 659–667 CrossRef CAS.
- Y. Xiao, H. Sun and J. Du, J. Am. Chem. Soc., 2017, 139, 7640–7647 CrossRef CAS PubMed.
- J. Cao, Y. Wei, Y. Zhang, G. Wang, X. Ji and Z. Zhong, ACS Appl. Mater. Interfaces, 2019, 11, 18953–18959 CrossRef CAS PubMed.
- T. X. Nguyen, L. Huang, M. Gauthier, G. Yang and Q. Wang, Nanomedicine, 2016, 11, 1169–1185 CrossRef CAS PubMed.
- M. A. Kisel, L. N. Kulik, I. S. Tsybovsky, A. P. Vlasov, M. S. Vorob’yov, E. A. Kholodova and Z. V. Zabarovskaya, Int. J. Pharm., 2001, 216, 105–114 CrossRef CAS PubMed.
- K. Iwanaga, S. Ono, K. Narioka, K. Morimoto, M. Kakemi, S. Yamashita, M. Nango and N. Oku, Int. J. Pharm., 1997, 157, 73–80 CrossRef CAS.
- H. Chen and R. Langer, Pharm. Res., 1997, 14, 537–540 CrossRef CAS PubMed.
- R. N. Rowland and J. F. Woodley, Biochim. Biophys. Acta, Lipids Lipid Metab., 1980, 620, 400–409 CrossRef CAS.
- R. Schubert, H. Jaroni, J. Schoelmerich and K. H. Schmidt, Digestion, 1983, 28, 181–190 CAS.
- K. Andrieux, L. Forte, S. Lesieur, M. Paternostre, M. Ollivon and C. Grabielle-Madelmont, Eur. J. Pharm. Biopharm., 2009, 71, 346–355 CrossRef CAS PubMed.
- C. J. O’Connor, R. G. Wallace, K. Iwamoto, T. Taguchi and J. Sunamoto, Biochim. Biophys. Acta, Biomembr., 1985, 817, 95–102 CrossRef.
- O. Zumbuehl and H. G. Weder, Biochim. Biophys. Acta, Biomembr., 1981, 640, 252–262 CrossRef CAS.
- M. Kokkona, P. Kallinteri, D. Fatouros and S. G. Antimisiaris, Eur. J. Pharm. Sci., 2000, 9, 245–252 CrossRef CAS PubMed.
- M. P. de la, C. Moreno, M. Oth, S. Deferme, F. Lammert, J. Tack, J. Dressman and P. Augustijns, J. Pharm. Pharmacol., 2006, 58, 1079–1089 CrossRef.
- L. Kalantzi, K. Goumas, V. Kalioras, B. Abrahamsson, J. B. Dressman and C. Reppas, Pharm. Res., 2006, 23, 165–176 CrossRef CAS.
- D.-A. Hallbäck, M. Jodal, M. Mannischeff and O. Lundgren, Acta Physiol. Scand., 1991, 143, 271–277 CrossRef.
- C. Ménager and V. Cabuil, J. Phys. Chem. B, 2002, 106, 7913–7918 CrossRef.
- S. Hupfeld, H. H. Moen, D. Ausbacher, H. Haas and M. Brandl, Chem. Phys. Lipids, 2010, 163, 141–147 CrossRef CAS.
- F. Ahmed and D. E. Discher, J. Controlled Release, 2004, 96, 37–53 CrossRef CAS.
- Y. Yun, Y. W. Cho and K. Park, Adv. Drug Delivery Rev., 2013, 65, 822–832 CrossRef CAS PubMed.
- V. Forster, R. D. Signorell, M. Roveri and J.-C. Leroux, Sci. Transl. Med., 2014, 6, 258ra141 CrossRef PubMed.
- G. Giacalone, S. Matoori, V. Agostoni, V. Forster, M. Kabbaj, S. Eggenschwiler, M. Lussi, A. De Gottardi, N. Zamboni and J.-C. Leroux, J. Controlled Release, 2018, 278, 57–65 CrossRef CAS PubMed.
- J. Parmentier, M. M. M. Becker, U. Heintz and G. Fricker, Int. J. Pharm., 2011, 405, 210–217 CrossRef CAS PubMed.
- S. Hu, M. Niu, F. Hu, Y. Lu, J. Qi, Z. Yin and W. Wu, Int. J. Pharm., 2013, 441, 693–700 CrossRef CAS.
- M. A. Khan, S. Ali, S. S. Venkatraman, M. F. Sohail, M. Ovais and A. Raza, Int. J. Pharm., 2018, 542, 196–204 CrossRef CAS.
- V. Pata, F. Ahmed, D. Discher and N. Dan, Langmuir, 2004, 20, 3888–3893 CrossRef CAS.
- X. Wang, J. Fan, Y. Liu, B. Zhao, Z. Jia and Q. Zhang, Int. J. Pharm., 2011, 419, 339–346 CrossRef CAS PubMed.
- C. Liu, Z. Chen, Y. Chen, J. Lu, Y. Li, S. Wang, G. Wu and F. Qian, Mol. Pharmaceutics, 2016, 13, 599–608 CrossRef CAS PubMed.
- M. Alibolandi, F. Alabdollah, F. Sadeghi, M. Mohammadi, K. Abnous, M. Ramezani and F. Hadizadeh, J. Controlled Release, 2016, 227, 58–70 CrossRef CAS PubMed.
- J. Gaitzsch, D. Appelhans, L. Wang, G. Battaglia and B. Voit, Angew. Chem., Int. Ed., 2012, 51, 4448–4451 CrossRef CAS PubMed.
- K. T. Kim, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Adv. Mater., 2009, 21, 2787–2791 CrossRef CAS.
- L. Klermund and K. Castiglione, Bioprocess Biosyst. Eng., 2018, 41, 1233–1246 CrossRef CAS.
- J. Gaitzsch, X. Huang and B. Voit, Chem. Rev., 2016, 116, 1053–1093 CrossRef CAS.
- M. El Idrissi, C. E. Meyer, L. Zartner and W. Meier, J. Nanobiotechnol., 2018, 16, 63 CrossRef.
- F. Wang, J. Xiao, S. Chen, H. Sun, B. Yang, J. Jiang, X. Zhou and J. Du, Adv. Mater., 2018, 30, 1705674 CrossRef.
- S. Matoori and J. C. Leroux, Adv. Drug. Delivery Rev., 2015, 90, 55–68 CrossRef CAS PubMed.
Footnote |
| † Current address: John A. Paulson School of Engineering and Applied Sciences, Harvard University, Cambridge, MA 02138, USA. |
| This journal is © The Royal Society of Chemistry 2020 |