
Enhanced charge separation and photocatalytic hydrogen evolution in carbonized-polymer-dot-coupled lead halide perovskites†
Yue
Zhao‡
a,
Qingsen
Zeng‡
a,
Yue
Yu
a,
Tanglue
Feng
a,
Yajie
Zhao
b,
Zidong
Wang
a,
Yi
Li
b,
Chongming
Liu
a,
Junjun
Liu
a,
Haotong
Wei
 a,
Shoujun
Zhu
a,
Zhenhui
Kang
*b,
Hao
Zhang
a and
Bai
Yang
*a
a,
Shoujun
Zhu
a,
Zhenhui
Kang
*b,
Hao
Zhang
a and
Bai
Yang
*a
aState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun 130012, China. E-mail: byangchem@jlu.edu.cn
bInstitute of Functional Nano and Soft Materials (FUNSOM), Jiangsu Key Laboratory for Carbon-based Functional Materials and Devices, Soochow University, Suzhou 215123, China. E-mail: zhkang@suda.edu.cn
First published on 28th July 2020
Abstract
Metal halide perovskites are promising candidates as photocatalysts due to their uniquely outstanding photophysical properties; however, the catalytic efficiency is limited by severe charge recombination. Herein, we show that carbonized polymer dots (CPDs) can act as an efficient charge modulator to stabilize photo-generated carriers in methylamine lead triiodide (MAPbI3) perovskites through ultra-fast hole transfer, and thus increase the rate of visible light-driven photocatalytic HI splitting 35-fold. The optimized CPD/MAPbI3/Pt hybrid photocatalytic system exhibits an impressive H2 evolution rate of 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 497 μmol h−1 g−1, a solar-to-hydrogen conversion efficiency of 2.15%, and an apparent quantum yield of 53.6% at 420 nm, which are among the highest values for metal halide perovskite photocatalysts. Moreover, the presented strategy of hole extraction via CPDs can be universally applied to improve the performance of previous electron-manipulated MAPbI3-based photocatalytic systems. The easy-to-prepare and bandedge-tunable CPDs with excellent charge-transfer ability may bring new insights in developing high-performance perovskite photocatalysts.
497 μmol h−1 g−1, a solar-to-hydrogen conversion efficiency of 2.15%, and an apparent quantum yield of 53.6% at 420 nm, which are among the highest values for metal halide perovskite photocatalysts. Moreover, the presented strategy of hole extraction via CPDs can be universally applied to improve the performance of previous electron-manipulated MAPbI3-based photocatalytic systems. The easy-to-prepare and bandedge-tunable CPDs with excellent charge-transfer ability may bring new insights in developing high-performance perovskite photocatalysts.
New conceptsMetal halide perovskite (MHP) photocatalysts have caught increasing attention because of their high intrinsic absorption coefficients and low surface recombination velocities. However, the performance is limited by the inefficient charge separation and utilization. In this work, we provide a completely new and highly efficient pathway to enhance charge separation in MHP through coupling with carbonized polymer dots (CPDs). CPDs can stabilize photo-generated carriers in methylamine lead triiodide (MAPbI3) perovskites through ultra-fast hole transfer. The long-lived photoeletrons in CPD-coupled MAPbI3 facilitate photocatalytic reduction reaction and thus increase the rate of visible light-driven HI splitting 35-fold. An H2 evolution rate of 11497 μmol h−1 g−1 and solar-to-hydrogen conversion efficiency of 2.15% are achieved in the optimized CPD/MAPbI3/Pt hybrid system, representing the state-of-the-art performance for MHP photocatalysts. This work highlights the excellent charge-transfer ability of CPDs. Moreover, the bandedge-tunable property makes CPDs promising candidates as co-catalysts to match with different-energy-alignment MHPs, indicating their great potential in developing efficient perovskite-based hybrid photocatalysts.
|
Introduction
Beyond photoelectronic devices,1–7 metal halide perovskites (MHP) have recently caught increasing attention in photocatalysis because of their wide high absorption coefficients, low surface recombination velocities and easily tunable energy levels.8–13 In addition to outstanding performance, the impressively labile perovskites exhibit unexpected photocatalytic stability,14–17 indicating bright prospects of MHP photocatalysts. Typical photocatalytic processes consist of light harvesting, charge transfer and surface reaction.18–22 Compared to ultrafast light absorption (10−12–10−9 s) and fast charge transfer (10−9–10−6 s), surface reactions are much slower (10−6–100 s). As this time scale mismatch intrinsically limits photocatalytic reaction efficiency, the enhancement of charge separation to increase separated charge lifetime favours improvement in surface reactions and thus efficiency. In 2017, Nam et al. discovered that methylammonium lead iodide (MAPbI3) could be stabilized in saturated aqueous hydroiodic acid (HI) solution, and pioneered perovskite photocatalysts on H2 evolution via HI splitting.23 Since this pioneering work, some groups have devoted efforts to regulating charge-transfer behaviors of perovskites in this photocatalytic model system. Gradient-level-alignment MHPs (MAPbBr3−xIx)24 or various electron-transfer materials, such as Pt–TiO2,10 reduced graphene oxide (rGO),16 Ni3C17 and black phosphorus (BP),25 have been used to increase charge separation and catalytic efficiencies in MAPbI3. So far, the highest H2 evolution rate and solar-to-hydrogen (STH) efficiency based on MHP photocatalysts for HI splitting are 3742 μmol h−1 g−1 and 1.42%,15,17 respectively, leaving much room for the further improvement of charge separation and utilization. In photocatalytic H2 evolution or other reduction reactions, one should develop ideal semiconductors with long-lifetime photoelectrons as well as co-catalysts to extract the photoholes, thus facilitating the reduction reaction instead of charge recombination.Carbonized polymer dots (CPDs), as an emerging but significant member of the carbon-based nanomaterials family,26 have a wide range of applications due to their color-tunable emission, high photo-chemical stability, excellent photophysical/chemical properties, low toxicity and facile preparation.27–30 CPDs are usually prepared through polymerization crosslinking and carbonization of small molecules and/or polymer precursors with multiple polar groups (carboxyl, amino or hydroxyl).31 The abundance of surface polar groups allows CPDs to easily bind to the surface of ionic lead halide perovskites with charged grain boundaries. Besides, the energy level of CPDs can be easily manipulated by the choice of precursors and reaction conditions.32,33 Recently, we observed enhanced charge separation and transfer in CPD/TiO2 photonic crystal hybrid photocatalysts and CPD-modified perovskite solar cells,34,35 witnessing outstanding charge-transfer ability of CPDs. Therefore, bandedge-tunable CPDs with excellent charge-transfer ability have great potential in developing efficient perovskite-based hybrid photocatalysts.
In this work, we demonstrate that CPDs can act as an efficient charge modulator to stabilize photo-generated carriers in MHPs, and make the first demonstration of efficient perovskite/CPD hybrid photocatalysts for H2 evolution. We find that the energy level of CPDs prepared from citric acid (CA) and p-aminosalicylic acid (PASA) well matches with that of MAPbI3, and show that CA-PASA CPDs can enhance the separation and stabilization of charges in MAPbI3 perovskites through ultra-fast hole extraction, resulting in a 35-fold increase in the rate of visible-light-driven photocatalytic splitting of HI compared to that observed for pure MAPbI3. Consequently, one of the highest STH conversion efficiencies of 2.15% is achieved for an optimized Pt/MAPbI3/CA-PASA CPD hybrid. Moreover, the novel strategy of CA-PASA CPD-assisted hole extraction can be universally applied to improve the performance of previously reported electron-manipulated MAPbI3-based photocatalytic systems. This work provides a new and efficient pathway to modulate carrier dynamics in MHP photocatalysts.
Results and discussion
MAPbI3 was hydrothermally synthesized according to a previously described procedure.23 The MAPbI3 shows extremely strong absorption in the visible and near-infrared regions (400–800 nm, Fig. 1a). The scanning electron microscopy (SEM) image of MAPbI3 powder shows the presence of microcrystals (inset of Fig. 1a, left), while the high-resolution transmission electron microscopy (HR-TEM) image indicates that these MAPbI3 microcrystals have a well-defined structure with a tetragonal lattice parameter of 0.31 nm along the [220] direction (inset of Fig. 1a, right), which is in good agreement with the corresponding X-ray diffraction pattern (Fig. S1, ESI†).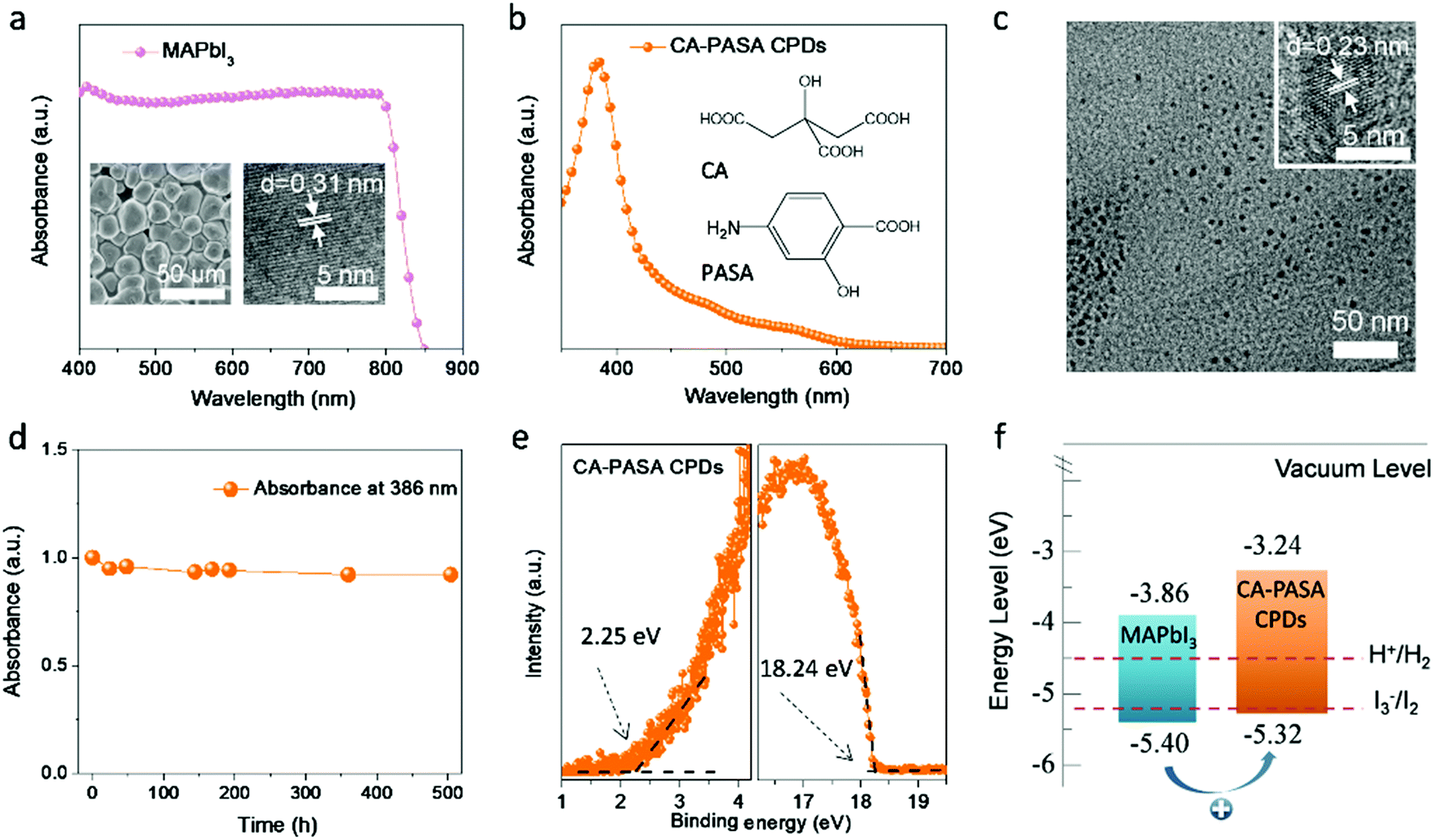 | ||
| Fig. 1 Characterization of MAPbI3 perovskites and CA-PASA CPDs. (a) Absorption spectrum, SEM and HR-TEM images of MAPbI3. (b) CA-PASA CPD absorption spectrum and their precursor molecular structures. (c) TEM and HR-TEM images of CA-PASA CPDs. (d) Time-dependent absorption intensity of CA-PASA CPDs at 386 nm in HI/H3PO2 solution. (e) UPS of CA-PASA CPDs: secondary electron cut-off region (left), magnified spectrum near the Femi edge (right). HOMO = −(21.21–18.24 + 2.25) eV = −5.32 eV. (f) Schematic energy level diagram of MAPbI3 and CA-PASA CPDs and the redox potentials for HI splitting. | ||
CA-PASA CPDs were hydrothermally prepared from CA and PASA (inset of Fig. 1b) under acidic conditions (pH 1.0), which were chosen to maximize the carbonization degree and crystallinity and thus improve light harvesting and charge transfer ability.36 Purified CA-PASA CPDs (Fig. 1b) exhibit strong absorption at 386 nm (with a tail extending to 600 nm) and a bandgap of 2.08 eV (estimated from the related Tauc plot shown in Fig. S2, ESI†). TEM and HR-TEM images show that CA-PASA CPDs are quasi-spherical particles with an average size of 4.9 nm and feature high crystallinity and a lattice pitch of 0.23 nm (Fig. 1c). CA-PASA CPDs exhibit strong fluorescence emission at 507 nm with negligible excitation dependency (Fig. S3, ESI†). Moreover, the stability of CA-PASA CPDs against HI was investigated by recording their time-dependent absorption spectra in HI solution (Fig. 1d and Fig. S4a, ESI†). The almost unchanged absorbance at 386 nm for >500 h, as well as intact chemical groups (Fig. S4b, ESI†), indicate that CA-PASA CPDs feature an excellent resistance to acids. Further characterizations illustrate that the multiple polar (carboxyl, amino and hydroxyl) groups of CA and PASA are partially preserved in CA-PASA CPDs (Fig. S4b and c, ESI†). As carboxyl and amino groups are efficient binding sites for stabilizing perovskite nanocrystals,3,37 the abundance of surface polar groups allows CA-PASA CPDs to easily bind to MAPbI3 perovskites for extracting charge carriers. Therefore, the CA-PASA CPDs/MAPbI3 hybrid can be obtained through self-assembling of CA-PASA CPDs and MAPbI3 in HI solution (Fig. S5a and b, ESI†). Studies on the hybrid catalysts indicate that CA-PASA CPDs bind with MAPbI3 mainly by COO–Pb coordination bond (Fig. S5c–e, ESI†).
Charge separation and transfer efficiencies depend on the alignment of energy levels between MAPbI3 perovskites and CA-PASA CPDs. Ultraviolet photoelectron spectroscopy (UPS) reveals that the CA-PASA CPD highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) lie at −5.32 and −3.24 eV, respectively (Fig. 1e). The valence band minimum and conduction band maximum of MAPbI3 perovskites are −5.40 and −3.86 eV, respectively.38 Thus, staggered energy levels are established between MAPbI3 and CA-PASA CPDs (Fig. 1f). This type-II energy level alignment provides a driving force for hole transfer from MAPbI3 to CA-PASA CPDs, also promoting electron transfer in the opposite direction. Because of the broad and strong absorption of MAPbI3 in hybrid systems, as demonstrated by similar absorption of CPDs/MAPbI3 as pure MAPbI3 (Fig. S6, ESI†), as well as in situ transient photovoltage (in situ TPV) measurement later. Therefore, the separation of photo-generated charges is concluded to be dominated by hole transfer from MAPbI3 to CA-PASA CPDs, while electron transfer is negligible from the opposite direction.
To study charge transfer between MAPbI3 perovskites and CA-PASA CPDs, we first conducted the steady-state PL and time-resolved PL (TRPL) decay measurements under 450 nm excitation. As shown in Fig. 2a, pure MAPbI3 features a strong interband emission peak at 746 nm, which is indicative of the severe photo-generated charge recombination. However, obvious fluorescence quenching is observed when MAPbI3 was coupled with 1 wt% CA-PASA CPDs. The distinctly suppressed radiative recombination in MAPbI3 suggests the occurrence of charge transfer from MAPbI3 to CA-PASA CPDs,10 as further demonstrated by TRPL measurements (Fig. 2b). The PL decay plots of MAPbI3 and MAPbI3/CA-PASA CPDs are fitted with a triexponential function (Table S1, ESI†), and the respective average PL lifetimes (56.5 and 35.0 ns) indicate that charge transfer is faster in the hybrid system. Thus, CA-PASA CPDs can effectively extract the holes of MAPbI3 because of the energy level alignment.10,13
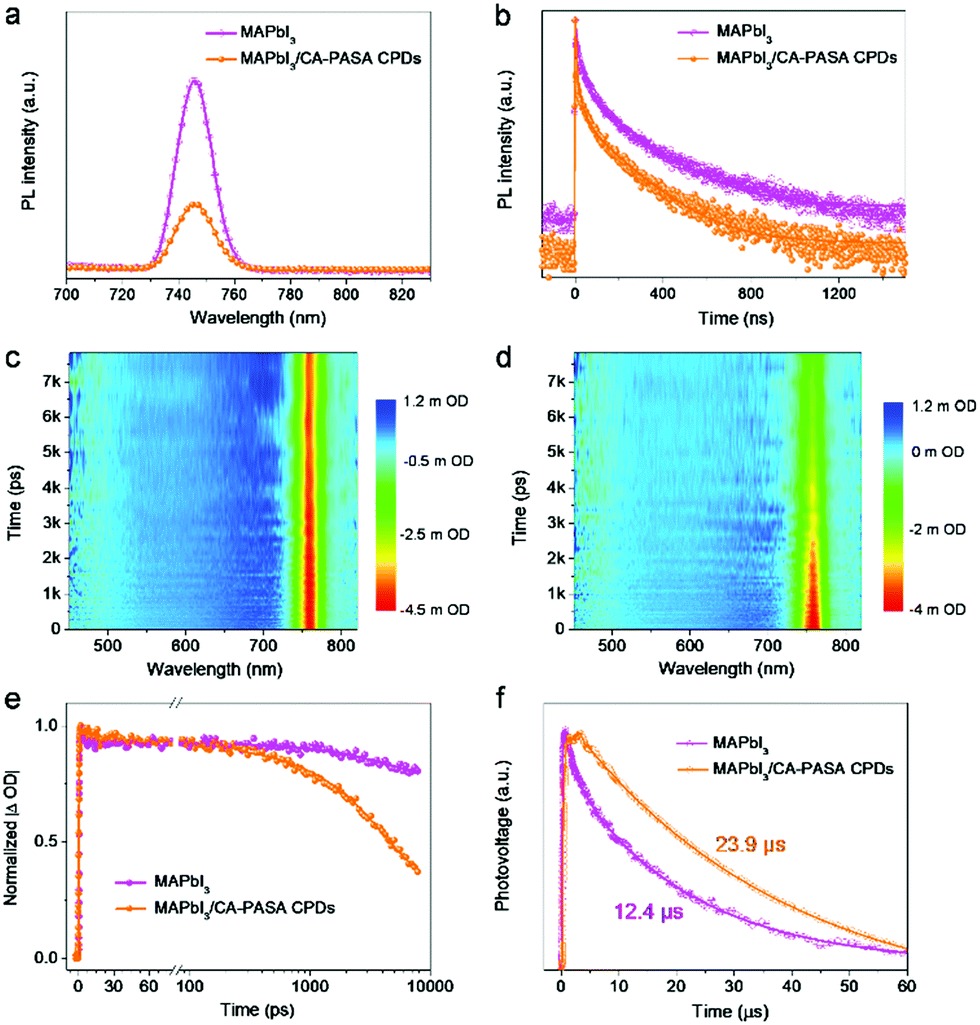 | ||
| Fig. 2 Enhanced charge separation and lifetime in MAPbI3/CA-PASA CPDs. (a) Steady-state PL and (b) TRPL spectra of MAPbI3 and MAPbI3/CPDs. (c) MAPbI3 and (d) MAPbI3/CPD spectro-temporal TA maps excited at 400 nm. (e) Characteristic dynamics for MAPbI3 and MAPbI3/CPDs probed at 760 nm. (f) TPV curves of MAPbI3 and MAPbI3/CPDs under 355 nm excitation. All hybrid samples contain 1 wt% CPDs. | ||
To gain further insights into ultrafast charge transfer, we investigated the spectral distribution of photo-generated carriers in MAPbI3 and MAPbI3/CA-PASA CPD samples by femtosecond transient absorption (fs-TA) spectroscopy. Two-dimensional fs-TA color maps (Fig. 2c and d) demonstrate an obvious photobleaching (PB) absorption peak at 760 nm for MAPbI3, in good agreement with the previous works.39 The absence of PB peak redshift indicates a flat energy landscape and negligible adverse bandtail states in the interband,3 suggesting that MAPbI3 has smooth charge transportation channels and low defect density, and is thus promising for high-performance photocatalytic reactions. After hybridization with CA-PASA CPDs, the fs-TA kinetics at 760 nm become much faster than that of sole MAPbI3 after 231 ps (Fig. 2e), implying that the CA-PASA CPDs can efficiently extract the holes of MAPbI3,39 consistent with the PL and TRPL results. Given the efficient charge transfer between MAPbI3 and CA-PASA CPDs, the photo-generated carrier lifetime of the MAPbI3/CA-PASA CPD hybrid is expected to be prolonged. To confirm this, we studied the carrier recombination dynamics by transient photovoltage (TPV) measurements, revealing that the half-lifetime of MAPbI3/CA-PASA CPDs (23.9 μs) exceeds that of MAPbI3 (12.4 μs) (Fig. 2f). This charge lifetime increase is ascribed to the enhanced photogenerated charge separation and suppressed recombination in the MAPbI3/CA-PASA CPD hybrid and benefits the surface reaction and, hence, catalytic efficiency.
Based on the above, CA-PASA CPDs are expected to improve the photocatalytic efficiency of MAPbI3 because of their outstanding hole-extraction properties and good stability in HI acid. Previous reports have shown that charge transfer materials can efficiently extract photo-generated carriers through dynamic interaction with perovskites,10,13 which greatly simplifies the preparation of high-performance perovskite-based hybrid photocatalysts. Therefore, we studied the photocatalytic hydrogen evolution performance of the hybrid prepared by the direct addition of CA-PASA CPDs into the MAPbI3 catalytic system. Photocatalytic reactions were conducted with 100 mg of MAPbI3 in 25 mL of MAPbI3-saturated HI solution under visible light irradiation (λ > 420 nm), and H2 evolution was monitored by gas chromatograph to probe the influence of CPD content on photocatalytic performance. After 4 h irradiation, 1.33 μmol of H2 is produced over bare MAPbI3, i.e., the H2 evolution rate is 3.33 μmol g−1 h−1 (Fig. 3a and Fig. S7, ESI†). H2 evolution is significantly enhanced by the introduction of CA-PASA CPDs, with the maximal H2 evolution rate (115 μmol g−1 h−1 for MAPbI3/1 wt% CA-PASA CPDs) exceeding that of pure MAPbI3 35-fold. Only traces of H2 are detected by gas chromatography when pure CA-PASA CPDs are used as a photocatalyst (Fig. S8, ESI†). Besides, CPD precursors (CA, PASA, and their mixture) have no positive effects on the photocatalytic performance of MAPbI3. All these control experiments indicate that the enhanced performance of MAPbI3/CA-PASA CPDs stems from the synergy between MAPbI3 and CPDs. Moreover, the photocatalytic experiments performed for three types of CPDs with different bandgaps and energy levels highlight the importance of matched energy alignment between CPDs and MAPbI3 (Fig. S9, ESI†). Therefore, the matched energy alignment and high charge transfer capacity of CA-PASA CPDs are concluded to greatly enhance hole extraction and thus improve the performance of MAPbI3-based perovskite photocatalysts.
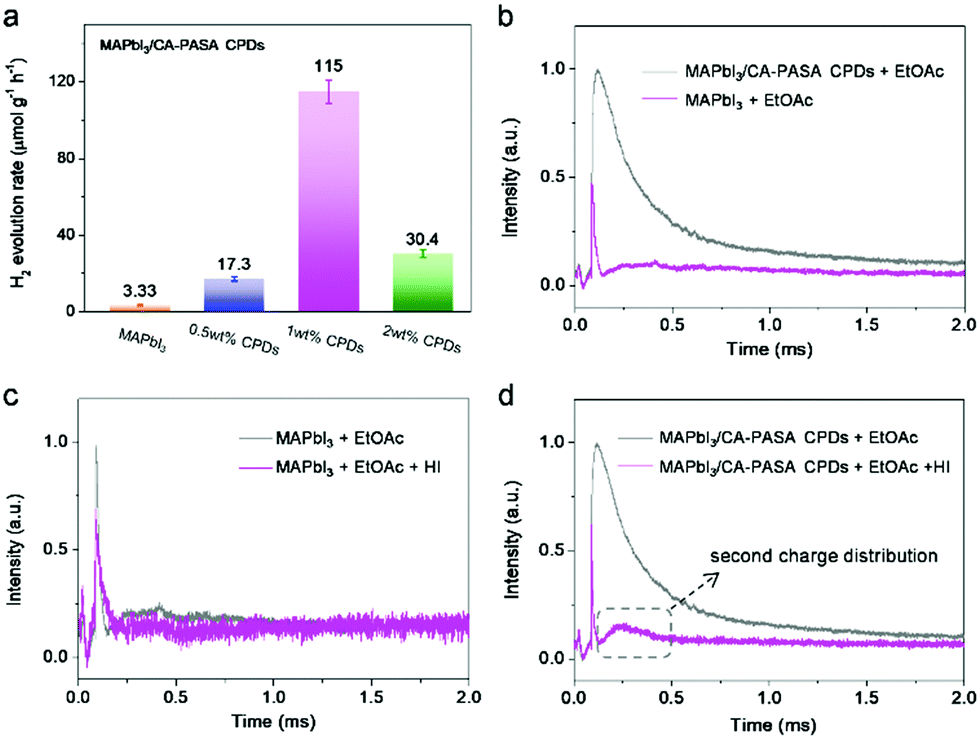 | ||
| Fig. 3 Photocatalytic performance and mechanism. (a) Rate of H2 evolution over MAPbI3 hybrids with different CPD contents. (b) In situ TPV curves of MAPbI3 and CA-PASA CPDs covered with EtOAc. In situ TPV curves recorded with (c) MAPbI3 and (d) CA-PASA CPD/MAPbI3 working electrodes in 1 vol% HI/EtOAc mixed solution (v/v). | ||
The mechanism of the photocatalytic reaction over MAPbI3/CA-PASA CPDs was further probed by in situ TPV measurements. The photocatalysts are deposited onto indium-tin oxide (ITO) as working electrodes, with details provided in the ESI.† During testing, the electrodes are covered with a N2-saturated inactive solvent (ethyl acetate, EtOAc) or reaction solution (1% HI/EtOAc, v/v), which allows one to directly observe the surface reactions of photo-generated charges. We first investigated the effects of CA-PASA CPDs on the photocurrent signals of MAPbI3 with no charge consumer (HI). As shown in Fig. 3b, the photocurrent intensity of MAPbI3/CA-PASA CPDs is much higher than that of MAPbI3, indicating a larger photo-generated charge concentration on the surface of the former and a higher electron–hole separation efficiency. Besides, the delayed attenuation of MAPbI3/CA-PASA CPDs suggests longer photo-generated charge lifetime. This observation is consistent with the conclusion that CA-PASA CPDs can enhance the charge separation and lifetime of MAPbI3 in the study of ultrafast photophysical processes (Fig. 2). Subsequently, the process of photocatalytic HI splitting was explored. For MAPbI3 as the working electrode (Fig. 3c), the photocurrent intensity decreases under 1% HI/EtOAc conditions, indicating that the photo-generated charges of MAPbI3 are consumed to split HI. When CA-PASA CPDs are introduced (Fig. 3d), the photocurrent of MAPbI3/CA-PASA CPDs in 1% HI/EtOAc presents a much sharper decay than that of MAPbI3, which implies a faster charge transfer from MAPbI3/CA-PASA CPDs to the catalyst/reactant interface. In addition, after the initial fast consumption of charges by HI, a broad shoulder peak at 0.2–0.5 ms appears again, which corresponds to the second distribution of photo-generated charges between the catalyst/solution interface. However, no second charge distribution is observed during HI splitting over pure MAPbI3 (Fig. 3c). Therefore, CA-PASA CPDs are concluded to induce a second charge migration from the interior of MAPbI3 to the catalyst/reactant interface, which increases the reaction efficiency of photo-generated charges. This phenomenon is ascribed to the excellent charge extraction and stabilization ability of CA-PASA CPDs.
Given the poor light response of CA-PASA CPDs (Fig. S10, ESI†), electrons and holes are concluded to be primarily generated from MAPbI3. These in situ TPV results confirm that MAPbI3 is the main photoactive component in this hybrid system, while CA-PASA CPDs are a superb charge transfer medium to stabilize photogenerated charges and increase their lifetime. The separated charges in MAPbI3/CA-PASA CPDs show a fast surface reaction rate and improved catalytic efficiency. The mechanism of catalytic HI splitting over MAPbI3/CA-PASA CPDs is provided in Scheme 1. When light strikes on the hybrid photocatalyts, MAPbI3 absorbs light energy to produce electrons and holes. Driven by the staggered energy alignment, CA-PASA CPDs extract holes and enhance charge separation to increase the lifetime of electrons in MAPbI3 and thus promote photocatalytic H2 evolution.
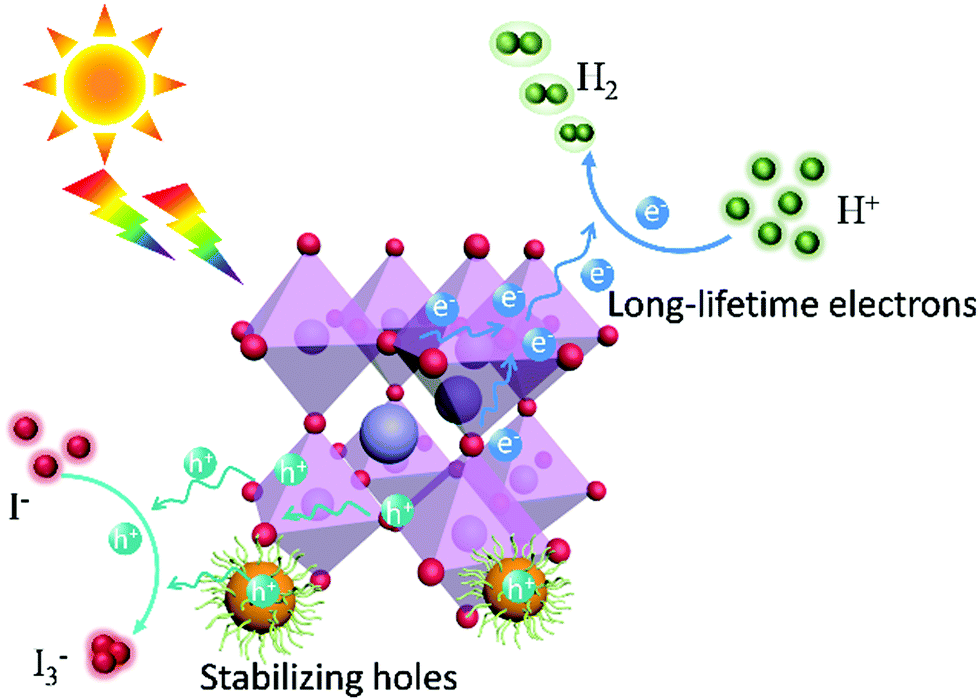 | ||
| Scheme 1 Schematic diagram of the photocatalytic mechanism for CA-PASA CPDs/MAPbI3. | ||
In MAPbI3/CPD photocatalysts, the long-lifetime photo-generated electrons in MAPbI3 reduce H+ to produce H2. Theoretical calculations indicate that both the MA+ and Pb2+ cations of MAPbI3 participate in reduction processes and act as active sites to produce H2via a two-step reaction.40 As H+ reduction on the perovskite surface is slow, we attempted to accelerate it by loading a Pt co-catalyst to further improve the catalytic efficiency of MAPbI3/CA-PASA CPDs.23,24 As shown in Fig. 4a, the H2 evolution rate of MAPbI3/1 wt% CA-PASA CPDs increases to 6594 μmol g−1 h−1 after the loading of 0.75 wt% Pt. Control experiments show that MAPbI3/Pt present a low H2 evolution rate of 25.4 μmol g−1 h−1, while the activity of CPDs/Pt is close to zero (Fig. S11, ESI†). These results suggest that the outstanding performance of Pt/MAPbI3/CA-PASA CPDs stems from the seamless cooperation between all constituents. The excellent ability of CA-PASA CPDs to extract holes from MAPbI3 promotes charge separation and prolongs the lifetime of photo-generated charges at the MAPbI3/CA-PASA CPD interface. Once Pt is loaded, the active sites of H2 reduction shift from MAPbI3 to Pt, which features a much higher H2 evolution activity, and the photo-generated electrons can be rapidly consumed. Therefore, one expects that photocatalytic efficiency can be appreciably improved by further enhancement of dynamic charge extraction and separation between MAPbI3 and CA-PASA CPDs through increasing the amount of CPDs. The H2 evolution rate of Pt/MAPbI3/7 wt% CPDs equals 11497 μmol g−1 h−1, and the corresponding STH conversion efficiency reaches 2.15%. Both of these values are among the highest reported for MHP-based photocatalysts (Fig. 4b and Table S2, ESI†). The above optimal photocatalyst presents a high apparent quantum yield (AQY) of 53.6% at 420 nm and shows good performance and structure stability during three reaction cycles (Fig. S13, ESI†).
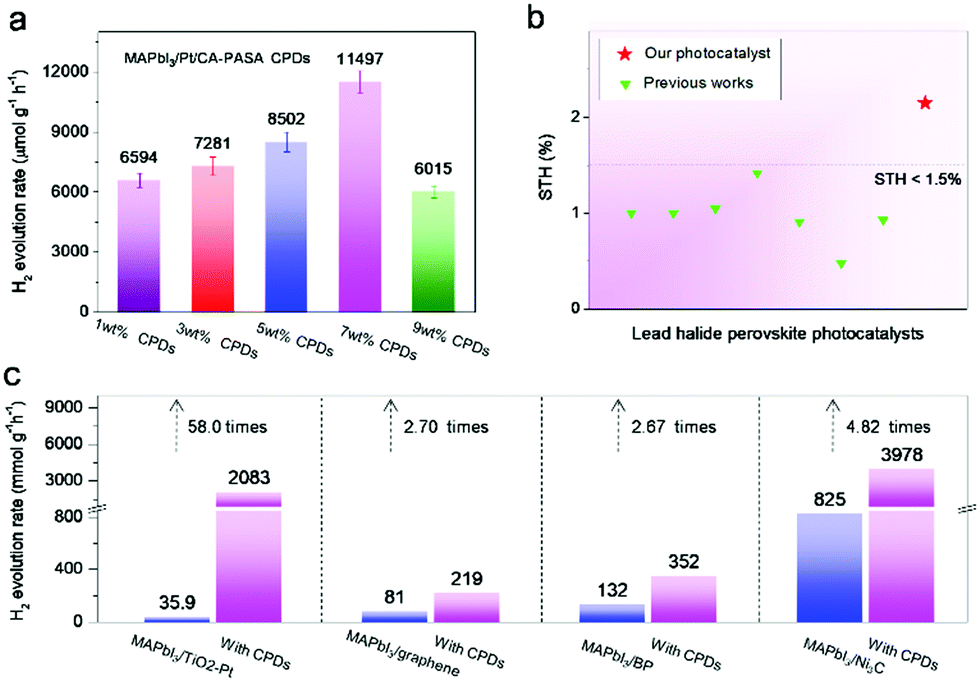 | ||
| Fig. 4 Efficiency optimization and performance comparison of CA-PASA CPDs with other electron-transfer materials. (a) Rate of H2 evolution on Pt/MAPbI3/CA-PASA CPDs with different CPD contents. (b) STH conversion efficiencies of previous MHP photocatalysts.10,13,15–17,23–25,37,38,41–44 (c) Rate of H2 evolution on MAPbI3/TiO2–Pt, MAPbI3/graphene, MAPbI3/BP, MAPbI3/Ni3C, and their CA-PASA CPD-hybrid systems. | ||
The above discussion indicates that hole extraction by CA-PASA CPDs is a novel and efficient method of enhancing charge separation and improving the lifetime of photo-generated electrons in the MAPbI3 perovskite. Therefore, this performance improvement strategy should be applicable to previously reported electron-manipulated MAPbI3 hybrid systems. To test this hypothesis, we prepared typical MAPbI3-based hybrid photocatalysts with four different electron-transfer materials, namely TiO2–Pt,10 graphene,16 black phosphorus (BP),25 and Ni3C17 (Fig. S14, ESI†), which were loaded at optimal contents determined in previous works. As shown in Fig. 4c, the above systems show higher H2 evolution rates than sole MAPbI3 (3.33 μmol g−1 h−1), consistent with the previous works. As expected, coupling with CA-PASA CPDs (Fig. 4c and Fig. S15, ESI†) improves the performance of all systems (by factors of 58.0, 2.70, 2.67, and 4.82 for MAPbI3/TiO2–Pt, MAPbI3/graphene, MAPbI3/BP, and MAPbI3/Ni3C, respectively). These results indicate that CA-PASA CPDs have a universally positive impact on electron-manipulated MAPbI3 photocatalytic systems.
Conclusions
In summary, we demonstrate that CPDs can act as an efficient charge modulator to stabilize photo-generated carriers in MHPs. The CA-PASA CPDs can efficiently enhance the charge separation and lifetime of MAPbI3 perovskite through ultra-fast hole transfer, thus leading to a 35-fold increase in the rate of visible-light-driven photocatalytic HI splitting. The optimal Pt/MAPbI3/CPD hybrid system exhibits a high H2 evolution rate of 11497 μmol h−1 g−1 and a record STH efficiency of 2.15%. Furthermore, CA-PASA CPDs can universally promote the performance of previous electron-manipulated MAPbI3-based photocatalysts. The bandedge-tunable property makes CPDs ideal candidates as co-catalysts to match with various MHP photocatalysts with different energy alignments. This work highlights the excellent charge-transfer ability of CPDs and their great potential in developing efficient perovskite-based hybrid photocatalysts.
Author contributions
B. Y. supervised the whole project. Y. Z., Q. Z. and B. Y. conceived the experiments. Y. Z. and Q. Z. contributed to all the experimental work. T. F., C. L. and J. L contributed to the synthesis and characterization of CPDs and perovskite. Y. Y. and Z. W. assisted the photocatalytic test. Y. Z., Y. L. and Z. K. carried out the in situ TPV and analysed the results. H. W. S. Z. and H. Z. helped to analyse the experimental results and modify English. Y. Z., Q. Z. and B. Y. wrote the manuscript. All the authors commented on the paper.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was financially supported by the National Science Foundation of China (NSFC) under Grant No. 51433003, the National Basic Research Program of China (973 Program) under Grant No. 2014CB643503, the China Postdoctoral Science Foundation under Grant No. 2019M661202, the Opening Funds of State Key Laboratory of Applied Optics, Changchun Institute of Optics, Fine Mechanics and Physics, Chinese Academy of Science, and the JLU Science and Technology Innovative Research Team 2017TD-06.References
- J. P. Correa-Baena, M. Saliba, T. Buonassisi, M. Gratzel, A. Abate, W. Tress and A. Hagfeldt, Science, 2017, 358, 739–744 CrossRef CAS PubMed.
- H. Wei, Y. Fang, P. Mulligan, W. Chuirazzi, H.-H. Fang, C. Wang, B. R. Ecker, Y. Gao, M. A. Loi, L. Cao and J. Huang, Nat. Photonics, 2016, 10, 333–339 CrossRef CAS.
- Q. Zeng, X. Zhang, X. Feng, S. Lu, Z. Chen, X. Yong, S. A. T. Redfern, H. Wei, H. Wang, H. Shen, W. Zhang, W. Zheng, H. Zhang, J. S. Tse and B. Yang, Adv. Mater., 2018, 30, 1705393 CrossRef PubMed.
- W. Zhu, W. Ma, Y. Su, Z. Chen, X. Chen, Y. Ma, L. Bai, W. Xiao, T. Liu, H. Zhu, X. Liu, H. Liu, X. Liu and Y. M. Yang, Light: Sci. Appl., 2020, 9, 112 CrossRef CAS PubMed.
- H. Sun, Z. Yang, M. Wei, W. Sun, X. Li, S. Ye, Y. Zhao, H. Tan, E. L. Kynaston, T. B. Schon, H. Yan, Z. H. Lu, G. A. Ozin, E. H. Sargent and D. S. Seferos, Adv. Mater., 2017, 29, 1701153 CrossRef PubMed.
- J. Xing, C. Zhao, Y. Zou, W. Kong, Z. Yu, Y. Shan, Q. Dong, D. Zhou, W. Yu and C. Guo, Light: Sci. Appl., 2020, 9, 111 CrossRef CAS PubMed.
- Q. Zeng, X. Zhang, C. Liu, T. Feng, Z. Chen, W. Zhang, W. Zheng, H. Zhang and B. Yang, Sol. RRL, 2019, 3, 1800239 CrossRef.
- M. Ou, W. Tu, S. Yin, W. Xing, S. Wu, H. Wang, S. Wan, Q. Zhong and R. Xu, Angew. Chem., 2018, 130, 13758–13762 CrossRef.
- J. Chen, C. Dong, H. Idriss, O. F. Mohammed and O. M. Bakr, Adv. Energy Mater., 2020, 10, 1902433 CrossRef CAS.
- X. Wang, H. Wang, H. Zhang, W. Yu, X. Wang, Y. Zhao, X. Zong and C. Li, ACS Energy Lett., 2018, 3, 1159–1164 CrossRef CAS.
- D. Yue, T. Zhang, T. Wang, X. Yan, C. Guo, X. Qian and Y. Zhao, EcoMat, 2020, 2, e12015 CrossRef CAS.
- Y. Zhao, T. Wang, T. Zhang and X. Li, Acta Chim. Sin., 2019, 77, 1075 CrossRef.
- H. Wang, X. Wang, R. Chen, H. Zhang, X. Wang, J. Wang, J. Zhang, L. Mu, K. Wu, F. Fan, X. Zong and C. Li, ACS Energy Lett., 2018, 4, 40–47 CrossRef.
- X. Zhu, Y. Lin, J. San Martin, Y. Sun, D. Zhu and Y. Yan, Nat. Commun., 2019, 10, 2843 CrossRef PubMed.
- Z. Zhao, J. Wu, Y.-Z. Zheng, N. Li, X. Li, Z. Ye, S. Lu, X. Tao and C. Chen, Appl. Catal., B, 2019, 253, 41–48 CrossRef CAS.
- Y. Wu, P. Wang, X. Zhu, Q. Zhang, Z. Wang, Y. Liu, G. Zou, Y. Dai, M. H. Whangbo and B. Huang, Adv. Mater., 2018, 30, 1704342 CrossRef PubMed.
- Z. Zhao, J. Wu, Y.-Z. Zheng, N. Li, X. Li and X. Tao, ACS Catal., 2019, 9, 8144–8152 CrossRef CAS.
- Z. Wang and L. Wang, Chin. J. Catal., 2018, 39, 369–378 CrossRef CAS.
- S. J. Phang and L.-L. Tan, Catal. Sci. Technol., 2019, 9, 5882–5905 RSC.
- X. C. Ma, Y. Dai, L. Yu and B. B. Huang, Light: Sci. Appl., 2016, 5, e16017 CrossRef CAS PubMed.
- Y. Wang, Y. Wu, K. Sun and Z. Mi, Mater. Horiz., 2019, 6, 1454–1462 RSC.
- F. Pincella, K. Isozaki and K. Miki, Light: Sci. Appl., 2014, 3, e133 CrossRef CAS.
- S. Park, W. J. Chang, C. W. Lee, S. Park, H.-Y. Ahn and K. T. Nam, Nat. Energy, 2017, 2, 16185 CrossRef CAS.
- Y. Wu, P. Wang, Z. Guan, J. Liu, Z. Wang, Z. Zheng, S. Jin, Y. Dai, M.-H. Whangbo and B. Huang, ACS Catal., 2018, 8, 10349–10357 CrossRef CAS.
- R. Li, X. Li, J. Wu, X. Lv, Y.-Z. Zheng, Z. Zhao, X. Ding, X. Tao and J.-F. Chen, Appl. Catal., B, 2019, 259, 118075 CrossRef CAS.
- S. Tao, T. Feng, C. Zheng, S. Zhu and B. Yang, J. Phys. Chem. Lett., 2019, 10, 5182–5188 CrossRef CAS PubMed.
- D. Li, D. Han, S. N. Qu, L. Liu, P. T. Jing, D. Zhou, W. Y. Ji, X. Y. Wang, T. F. Zhang and D. Z. Shen, Light: Sci. Appl., 2016, 5, e16120 CrossRef CAS PubMed.
- B. Wang, J. Li, Z. Tang, B. Yang and S. Lu, Sci. Bull., 2019, 64, 1285–1292 CrossRef CAS.
- Y. Liu, X. Li, Q. Zhang, W. Li, Y. Xie, H. Liu, L. Shang, Z. Liu, Z. Chen, L. Gu, Z. Tang, T. Zhang and S. Lu, Angew. Chem., Int. Ed., 2020, 59, 1718–1726 CrossRef CAS PubMed.
- X. Bao, Y. Yuan, J. Chen, B. Zhang, D. Li, D. Zhou, P. Jing, G. Xu, Y. Wang, K. Hola, D. Shen, C. Wu, L. Song, C. Liu, R. Zboril and S. Qu, Light: Sci. Appl., 2018, 7, 91 CrossRef PubMed.
- D. Qu and Z. Sun, Mater. Chem. Front., 2020, 4, 400–420 RSC.
- T. Ji, B. Guo, F. Liu, Q. Zeng, C. Yu, X. Du, G. Jin, T. Feng, S. Zhu, F. Li and B. Yang, Adv. Mater. Interfaces, 2018, 5, 1701519 CrossRef.
- J. Liu, D. Li, K. Zhang, M. Yang, H. Sun and B. Yang, Small, 2018, 14, e1703919 CrossRef PubMed.
- X. Zhang, Q. Zeng, Y. Xiong, T. Ji, C. Wang, X. Shen, M. Lu, H. Wang, S. Wen, Y. Zhang, X. Yang, X. Ge, W. Zhang, A. P. Litvin, A. V. Baranov, D. Yao, H. Zhang, B. Yang, A. L. Rogach and W. Zheng, Adv. Funct. Mater., 2020, 30, 1910530 CrossRef CAS.
- Y. Zhao, Q. Zeng, T. Feng, C. Xia, C. Liu, F. Yang, K. Zhang and B. Yang, Mater. Chem. Front., 2019, 3, 2659–2667 RSC.
- T. Feng, Q. Zeng, S. Lu, X. Yan, J. Liu, S. Tao, M. Yang and B. Yang, ACS Photonics, 2017, 5, 502–510 CrossRef.
- M. Xiao, M. Hao, M. Lyu, E. G. Moore, C. Zhang, B. Luo, J. Hou, J. Lipton-Duffin and L. Wang, Adv. Funct. Mater., 2019, 29, 1905683 CrossRef CAS.
- M. Wang, Y. Zuo, J. Wang, Y. Wang, X. Shen, B. Qiu, L. Cai, F. Zhou, S. P. Lau and Y. Chai, Adv. Energy Mater., 2019, 9, 1901801 CrossRef CAS.
- Z. Zhu, J. Ma, Z. Wang, C. Mu, Z. Fan, L. Du, Y. Bai, L. Fan, H. Yan, D. L. Phillips and S. Yang, J. Am. Chem. Soc., 2014, 136, 3760–3763 CrossRef CAS PubMed.
- L. Wang, H. Xiao, T. Cheng, Y. Li and W. A. Goddard 3rd, J. Am. Chem. Soc., 2018, 140, 1994–1997 CrossRef CAS PubMed.
- Z. Guan, Y. Wu, P. Wang, Q. Zhang, Z. Wang, Z. Zheng, Y. Liu, Y. Dai, M.-H. Whangbo and B. Huang, Appl. Catal., B, 2019, 245, 522–527 CrossRef CAS.
- Y. Guo, G. Liu, Z. Li, Y. Lou, J. Chen and Y. Zhao, ACS Sustainable Chem. Eng., 2019, 7, 15080–15085 CrossRef CAS.
- T. Wang, D. Yue, X. Li and Y. Zhao, Appl. Catal., B, 2020, 268, 118399 CrossRef.
- H. Zhao, Y. Li, B. Zhang, T. Xu and C. Wang, Nano Energy, 2018, 50, 665–674 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0mh00955e |
| ‡ Y. Z. and Q. Z. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |