
Interfaces in rechargeable magnesium batteries
Jiayan
Shi
 ab,
Jian
Zhang
c,
Juchen
Guo
*ac and
Jun
Lu
*b
ab,
Jian
Zhang
c,
Juchen
Guo
*ac and
Jun
Lu
*b
aDepartment of Chemical and Environmental Engineering, University of California-Riverside, Riverside, California 92521, USA. E-mail: jguo@engr.ucr.edu
bChemical Sciences and Engineering Division, Argonne National Laboratory, 9700 Cass Ave, Lemont, IL 60439, USA. E-mail: junlu@anl.gov
cProgram of Materials Science and Engineering, University of California-Riverside, Riverside, California 92521, USA
First published on 2nd September 2020
Abstract
This minireview provides a concise overview on the development of electrolytes for rechargeable magnesium (Mg) batteries. It elucidates the intrinsic driving force of the evolution from Grignard-based electrolytes to electrolytes based on simple Mg salts. Additional discussion includes the key electrochemical processes at the interfaces in Mg electrolytes, with a focus on unaddressed issues and future research directions.
 Jiayan Shi | Jiayan Shi earned her Bachelor degree of Chemical Engineering from Nanjing Tech University in 2017. She is currently a PhD candidate at the University of California, Riverside working at Argonne National Laboratory as a visiting student. Her research interest lies in fundamental electrochemistry and energy storage technologies including Li-ion, Li-sulfur, and multivalent ion batteries. She is the 2020 distinguished alumni of Nanjing Tech University. |
 Jian Zhang | Jian Zhang received his BS in Materials Science and Engineering in 2016 from Zhejiang University of Technology. He is currently a PhD candidate at the University of California, Riverside. His research focuses on lithium-ion, lithium metal, and multivalent ion batteries. His research interests also include developing electrochemical techniques for battery characterizations. |
 Juchen Guo | Juchen Guo is an Associate Professor in the Department of Chemical and Environmental Engineering and Materials Science and Engineering Program at University of California – Riverside. He earned his Bachelor's degree from Zhejiang University in 1999 and his PhD degree from University of Maryland in 2007, both in chemical engineering. He performed postdoctoral research at University of Maryland from 2007 to 2011 and Cornell University from 2011 to 2012. He was the recipient of 2014 Hellman Fellowship and 2018 NSF CAREER Award. His research interests are interfacial phenomena and material properties in electrochemical energy storage systems. |
 Jun Lu | Dr Jun Lu is a chemist at Argonne National Laboratory. His research interests focus on the electrochemical energy storage and conversion technology, with main focus on beyond Li-ion battery technology. Dr Lu earned his bachelor degree in Chemistry Physics from University of Science and Technology of China (USTC) in 2000. He completed his PhD from the Department of Metallurgical Engineering at University of Utah in 2009. He is the awardee of the first DOE-EERE postdoctoral fellow under Vehicles Technology Program from 2011–2013. He serves as the associate editor of ACS Applied Materials and Interfaces. He was elected as associate president and board committee member of the International Academy of Electrochemical Energy Science (IAOEES). He is also the first awardee of IAOEES Award for Research Excellence in Electrochemistry Energy in 2016. Dr Lu has authored/co-authored more than 400 peer-reviewed research articles, including Nature; Science; Nature Energy; Nature Nanotechnology; Chem. Rev.; Nat. Commun.; J. Am. Chem. Soc.; etc., and has filed over 20 patents and patent applications. |
Historical overview of Mg electrolytes
Rechargeable magnesium (Mg) batteries have received tremendous attention as a new electrochemical energy storage technology with the theoretic advantages of lower cost, better safety, and even higher energy density than lithium-ion batteries. However, rechargeable Mg batteries are encountering significant challenges on all fronts, including electrolytes, anodes, and cathodes. The research community has yet to reach the stage of evaluating the technical and commercial feasibility of rechargeable Mg batteries. Instead, current studies should continue to explore new chemistry and emphasize fundamental understandings.An intrinsic requirement for rechargeable Mg batteries is the use of Mg metal anode. Although one can argue that Mg alloys formed electrochemically may be used, such anodes would defeat a main purpose of Mg batteries, i.e., high energy density. The first attempt at Mg electrodeposition can be traced back to the report by Gaddum and French in 1927 using Grignard reagent solutions in ethers.1 In the next half century, Mg electrolytes and Mg electrodeposition were scarcely reported in literature. Notably, Connor and coworkers in 1957 reported Mg deposition from an ethereal solution of magnesium borohydride (Mg(BH4)2) among a number of potential electrolytes.2 Brenner in 1971 reported Mg deposition from an ethereal solution of a Mg–boron complex derived from the reaction between magnesium chloride (MgCl2) and a Li derivative of decaborane. The same electrolyte could also be obtained from the reaction between a Grignard reagent and decaborane with MgCl2 as the additive in tetrahydrofuran (THF).3 These early studies preluded the burst of Mg battery research starting in the 1990s, of which the work of Gregory and coworkers in 1990 was representative.4 Despite the considerable citations received by this work, its significant implications may still be undervalued and deserve further discussion. The authors elucidated two types of electrolytes, both of which strongly influenced later studies: (1) electrolytes composed of Grignard reagents and aluminum chloride (AlCl3) and (2) Mg salts with bulky organoborate anions with alkyl and/or phenyl substituents including tetrabutylborate (B(C4H9)4− or BBu4−), tributylphenylborate (BBu3Ph−), and dibutyldiphenylborate (BBu2Ph2−). More importantly, they studied the chemical and electrochemical magnesiation of a number of materials listed in Table 1. The successful chemical magnesiation using dibutyl-magnesium indicated that these materials could potentially host Mg via either intercalation or conversion (the mechanisms were not clear from the paper). However, the electrochemical magnesiation of these materials failed in the Grignard-based electrolytes (RMgCl + AlCl3) and the Mg(BBu4)2 electrolyte.
| Material | Open-circuit potential vs. Mg0 | Capacity | |
|---|---|---|---|
| Moles Mg/mole host | mA h g−1 | ||
| Co3O4 | 2.28 | 0.80 | 222 |
| Mn2O3 | 2.40 | 0.66 | 224 |
| Mn3O4 | 2.40 | 0.66 | 154 |
| MoO3 | 2.28 | 0.50 | 143 |
| PbO2 | 3.10 | 0.25 | 56 |
| Pb3O4 | 3.10 | 0.25 | 20 |
| RuO2 | 2.55 | 0.66 | 266 |
| V2O5 | 2.66 | 0.66 | 194 |
| WO3 | 2.16 | 0.50 | 116 |
| TiS2 | 1.63 | 0.15 | 157 |
| VS2 | 1.71 | 0.34 | 154 |
| ZrS2 | 2.60 | 0.66 | 228 |
| MoB2 | 1.15 | 0.66 | 301 |
| TiB2 | 1.25 | 0.42 | 324 |
| ZrB2 | 1.20 | 0.66 | 313 |
The authors attributed the failure to the chemical magnesiation by the MgR2 species, which were from the Schlenk equilibrium in the Grignard-based electrolytes and the decomposition of tetrabutylborate in Mg(BBu4)2, respectively. On the other hand, Mg(BBu2Ph2)2 electrolyte in THF/1,2-dimethoxyethane (DME) mixture was found to be stable against decomposition. Despite the failed magnesiation in Grignard and Mg(BBu4)2 electrolytes, some of the materials in Table 1 (evidenced by ruthenium oxide (RuO2) shown in Fig. 1a) were successfully electrochemically magnesiated in the magnesium perchloride (Mg(ClO4)2) electrolyte as primary cells; Mg(ClO4)2 electrolyte could only enable Mg stripping. Based on the results above, the authors demonstrated the first rechargeable Mg battery (Fig. 1b) composed of Mg anode, cobalt oxide (Co3O4) cathode, and Mg(BBu2Ph2)2 electrolyte in THF/DME. This work is regarded as the first successful demonstration of rechargeable Mg batteries. Inadvertently, it also served as the first hint at the importance and complexity of the interfaces in rechargeable Mg batteries.
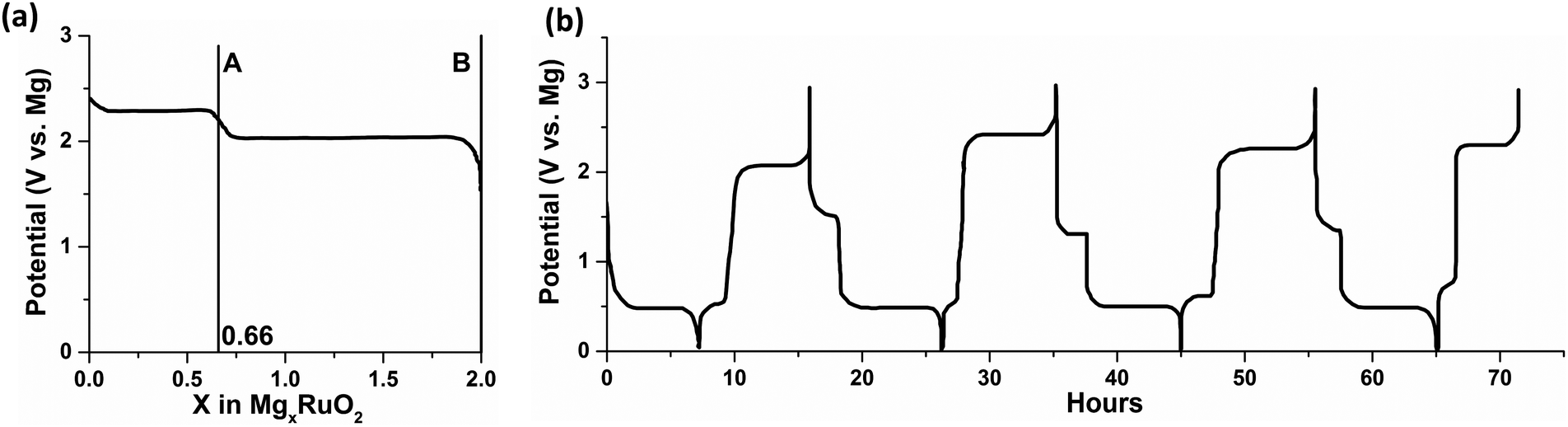 | ||
| Fig. 1 (a) Magnesiation potential curve of RuO2 in 1 M Mg(ClO4)2 in THF versus Mg at 1 mA cm−2. The first plateau (point A) is reversible and the full magnesiation (point B) is irreversible due to the complete reduction of Ru4+ to Ru0; (b) cycling curve of Co3O4versus Mg in 0.25 M Mg(BBu2Ph2)2 in THF/DME at 24 mA g−1 with respect to Co3O4. The plots are reproduced from ref. 4. | ||
A great thrust of rechargeable Mg battery research came from the work by Aurbach and coworkers in 2000.5 This work made two important contributions by (1) demonstrating Chevrel phase molybdenum sulfide (Mo6S8) as a new cathode material and (2) inventing a new type of electrolytes, namely dichloro-complex (DCC), composed of dialkyl Grignard (MgR2) and aluminum Lewis acids (AlCl3 or alkylAlCl2). Mo6S8 remains as the “benchmark” Mg cathode mainly because to date it is the only cathode for which electrochemical intercalation is unambiguous and the mechanism is relatively well understood. Separately, the DCC electrolytes showed improved anodic stability but were not found to be fundamentally different from the Grignard electrolytes in Gregory's study. The active species in these electrolytes are THF-solvated MgCl+ or Mg2Cl3+ cations and the anions are chloroaluminates (AlCl4−nRn−).6–10 Aurbach and coworkers further improved the anodic stability by replacing the alkyl groups in DCC with phenyl moieties, resulting in the so-called all phenyl complex (APC) electrolytes.10–12 However, these Grignard-based Mg electrolytes are problematic in Mg batteries containing electrophilic cathodes due to the nucleophilicity of the Grignard reagents. Therefore, researchers shifted their focus to Grignard-free electrolytes based on Mg-containing Lewis bases such as Mg alkoxides,13,14 Mg fluorinated alkoxides,15 and Mg amides such as hexamethyldisilazide magnesium chloride (HMDSMgCl)16 and magnesium bis(hexamethyldisilazide) (Mg(HMDS)2).17–19 These compounds are not completely non-nucleophilic, and they still need to be combined with Lewis acids such as AlCl3 to generate the active Mg-containing cations via transmetalation. The active halides (mainly chloride) render these electrolytes corrosive and limit the anodic stability through chlorine generation. Therefore, the latest trend in Mg electrolyte research is to look to the “simple salt” electrolytes composed of solvated divalent Mg2+ cations (Mg(solvent)n2+) and weakly coordinating anions. This type of Mg electrolyte was proven feasible for Mg deposition–stripping, as Connor reported Mg(BH4)2 in 1957![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2 and Gregory reported Mg organobrates in 1990.4 To date, Mg simple salts that have been studied for Mg electrolytes, in addition to Mg(BH4)2 and Mg organobrates, include Mg(ClO4)2,11 magnesium hexafluorophosphate (Mg(PF6)2),20 magnesium bis(trifluoromethanesulfonyl)imide (Mg(TFSI)2),21 magnesium fluorinated alkoxyborate,22 magnesium fluorinated alkoxyaluminate,23–25 and magnesium carboranes.26,27 The electrochemical properties of simple salt electrolytes vastly vary, and are clearly affected by the solvents, cation–anion interactions, and anionic stability. In the following sections, we will discuss the interfaces in the aforementioned Mg electrolytes in the hope of shedding some light on the important issues of rechargeable Mg batteries.
2 and Gregory reported Mg organobrates in 1990.4 To date, Mg simple salts that have been studied for Mg electrolytes, in addition to Mg(BH4)2 and Mg organobrates, include Mg(ClO4)2,11 magnesium hexafluorophosphate (Mg(PF6)2),20 magnesium bis(trifluoromethanesulfonyl)imide (Mg(TFSI)2),21 magnesium fluorinated alkoxyborate,22 magnesium fluorinated alkoxyaluminate,23–25 and magnesium carboranes.26,27 The electrochemical properties of simple salt electrolytes vastly vary, and are clearly affected by the solvents, cation–anion interactions, and anionic stability. In the following sections, we will discuss the interfaces in the aforementioned Mg electrolytes in the hope of shedding some light on the important issues of rechargeable Mg batteries.
Anode interfaces (Mg deposition–stripping)
Here, we focus on the interfacial phenomena during Mg deposition–stripping determined by the molecular structure and properties of the species in the electrolyte. For most of the Mg electrolytes from the combination of a Lewis base (Mg-containing species such as MgCl2, RMgCl, MgR2, ROMgCl, Mg(OR)2, etc., where R is an organic ligand) and a Lewis acid (typically AlCl3 or AlCl2R), active cations are produced via transmetalation represented by the simplified reactions in Scheme 1a. The [MgCl]+ monomer cation in Scheme 1a is solvated by solvent molecules, and the resultant solvation structures may vary depending on the solvents as in the examples shown in the left panel of Scheme 1b: [MgCl(THF)5]+28versus [MgCl(DME)2]+ (Mg prefers four or six-coordination environment, thus this five-coordination is metastable).29,30 Mg–Cl dimmer cations (right panel of Scheme 1b) can coexist under equilibrium with the monomers, along with other species in the electrolytes such as neutral species MgCl2(solvent)n, and AlCl4− or RAlCl3− anions. The equilibrium is affected by the Lewis base/acid ratio and the relative stability of the solvation structures.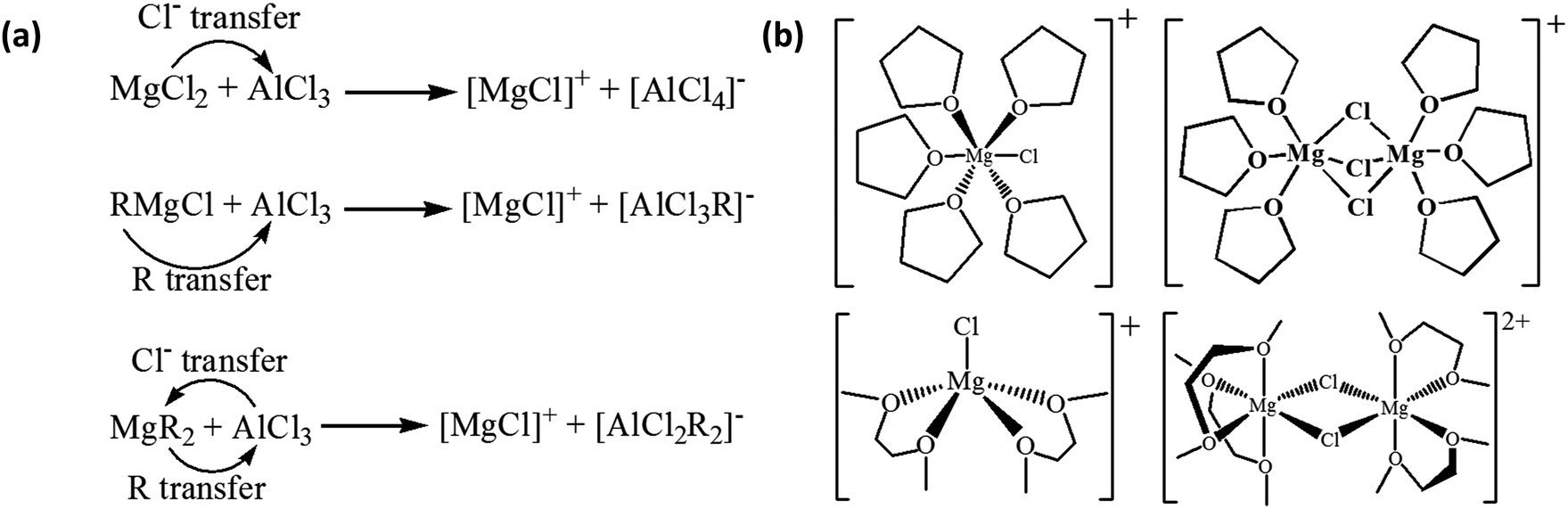 | ||
| Scheme 1 (a) Simplified transmetalation reactions between Lewis base and acid to produce active cations in the Mg electrolytes; (b) representative solvation structures of [MgCl(THF)5]+ (top left),28 [MgCl(DME)2]+ (bottom left)29 monomer cations and [Mg2Cl3(THF)6]+ (top right),28 [Mg2Cl2(DME)4]2+ (bottom right)29 dimer cations. | ||
With the [MgCl(THF)5]+ cation as an example, the simplified Mg deposition process is illustrated in Scheme 2 (modified from ref. 28). The properties of the cations and their interactions with the Mg surface, including the configuration of the adsorption, the binding energy of the solvation, the cathodic stability of the solvent molecules, and the fate of the deslovated solvents and chloride, all affect the Mg deposition–stripping process. In principle, the solvents used in Mg electrolytes should be inherently resistant to electrochemical reduction and oxidization. It is widely recognized that only ether solvents are suitable for Mg electrolytes due to their good cathodic stability. On the other hand, organic carbonate solvents are considered unsuitable for Mg electrolytes due to their proneness to cathodic decomposition to form a passivation layer, although the composition of such a layer has not been studied to date. The effect of the deslovated chloride (Cl−) at the interface on the efficiency of Mg deposition–stripping is also not clear. However, it is reasonable to speculate that free Cl− can interact with the surface species in a number of routes. For instance, free Cl− can combine with surface-adsorbed Mg–Cl cations to form a MgCl2 rich surface layer, which may facilitate cation transport and prevent anion decomposition. On the other hand, in the presence of a trace amount of water, which is inevitable in ethereal electrolytes, free Cl− can attack Mg metal to reduce the deposition efficiency.
 | ||
| Scheme 2 Illustration of the possible processes of Mg deposition from [MgCl(THF)5]+ cation. This illustration is modified from ref. 28. | ||
The recent development of Mg electrolytes has shifted significantly to focus on the ones based on simple Mg salts with weakly coordinating anions. The composition of these electrolytes has the beauty of simplicity, containing only solvent solvated [Mg(solvent)n]2+ cations and the weakly coordinating anions. Thus, the mechanisms of Mg deposition–stripping and cathode reactions can be elucidated with fewer intertwined processes. The simple salt Mg electrolytes also have the potential benefits of higher Mg deposition efficiency and higher anodic stability due to the absence of active Cl−. However, the simple salts of Mg still face steep challenges dictated by the properties of their anions. Mg(BH4)2 was the first simple salt to demonstrate reversible Mg deposition–stripping in the work of Connor and coworkers, albeit with co-deposition of boron.2 Mohtadi and coworkers in 2012 demonstrated improved Mg deposition from Mg(BH4)2 with LiBH4 as the additive in DME.31 However, the same study also revealed the low anodic stable window of Mg(BH4)2 at 1.5 V on platinum and 2 V on stainless steel versus Mg, which seriously limits its realistic application in batteries. It is also worthwhile to note that the deposition of Mg from the “dual ion” (Mg2+ + Li+) electrolytes may take an alternative route from the direct electrochemical reduction of [Mg(solvent)n]2+: under high overpotential built at the interface, metallic Li can be deposited first, after which Mg will be deposited via displacement reaction. This mechanism may work in favor of Mg deposition, but its long-term effect on the interface cannot be overlooked. The displacement reaction at the Mg anode/electrolyte interface was also observed in the electrolytes containing Mg–Cl cations and chloroaluminate anions, particularly in the magnesium aluminum chloride complex (MACC) system.32,33 Mg was deposited first, but metallic Al was subsequently deposited from the chloroaluminate anions by the oxidization of Mg. Such displacement at the Mg interface is clearly undesirable.
Mg(ClO4)2 is another simple salt that was studied during the early stages of Mg battery research. An interesting aspect of Mg(ClO4)2 electrolytes is that although Mg deposition is infeasible, Mg can be stripped despite an high overpotential of around 1.5 V in its ethereal solutions.11 This suggests that the difficulty of Mg deposition originates from the properties of the (ClO4)− anion instead of the [Mg(solvent)n]2+ cation. Indeed, (ClO4)− anion can be both chemically reduced by Mg metal and electrochemically reduced, resulting in a passivation layer on the surface of the Mg anode. Although the composition of this layer has not been investigated, one can speculate that it may contain Mg oxide. It is also believed that magnesium tetrafluoroborate (Mg(BF4)2), magnesium trifluoromethanesulfonate ((Mg(CF3SO3)2), Mg(PF6)2, and Mg(TFSI)2 are all chemically and/or electrochemically unstable at the Mg surface in a similar fashion. As reported by Jay et al., the passivation layer from Mg(TFSI)2 was composed of Mg fluoride, sulfide, oxide, and sulfite/dithionite compounds from the decomposition of TFSI− anions.34 The study by Yu et al. also suggested that the undercoordinated Mg (kinks on MgO or Mg(OH)2) on the Mg surface and the water impurity in the electrolyte synergistically contributed to the chemical decomposition of the Mg(TFSI)2 electrolyte in DME.35 A number of studies have reported that adding MgCl2 into the Mg(TFSI)2 electrolytes can significantly reduce the overpotentials of Mg deposition–stripping and improve roundtrip efficiency.36,37 However, one must understand that the electrolytes containing both Mg(TFSI)2 and MgCl2 are not simple salt electrolytes, because the active cations are Mg–Cl monomers or dimers generated from the comproportionation simplified as the following reaction:
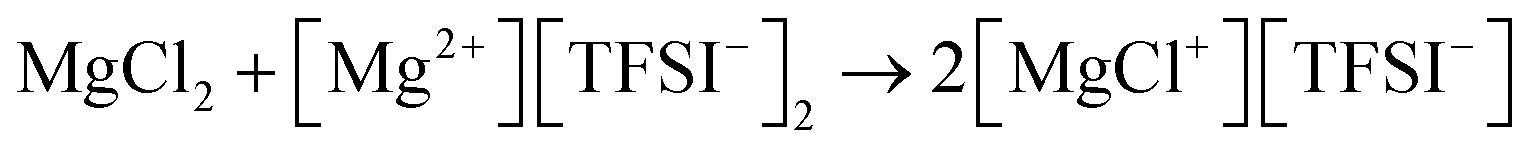
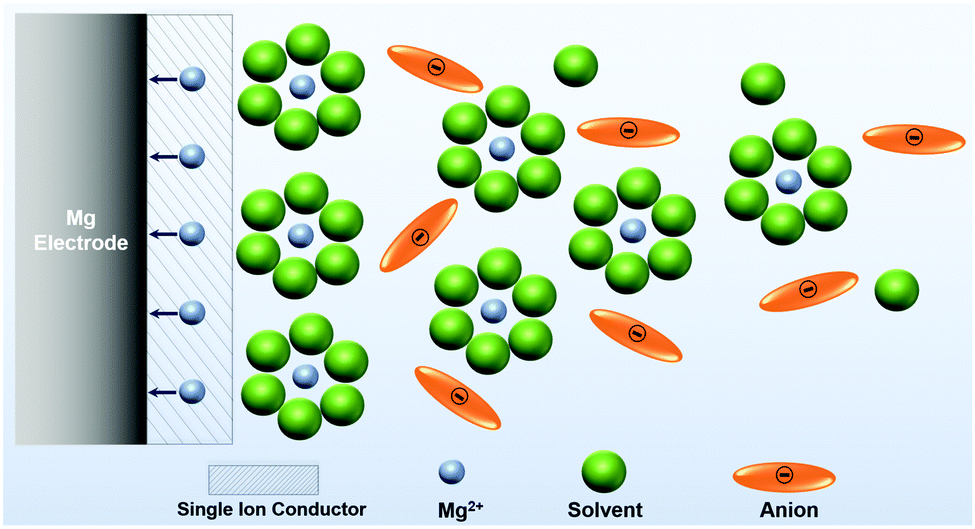 | ||
| Scheme 3 Illustration of an idealized single ion conducting (artificial) SEI on the Mg anode enable desolvation and screening out anions. | ||
Recently, more simple salt Mg electrolytes using unconventional weakly coordinating anions including tetrakis(hexafluoroisopropyloxy)aluminate ([Al(hfip)4]−),23 tetrakis(hexafluoroisopropyloxy)borate ([B(hfip)4]−),24 icosahedral carborane ((HCB11H11)−),26,27 and 10-vertex carborane ((HCB9H9)−)42 were studied. These electrolytes unambiguously demonstrated facile Mg deposition–stripping with low overpotential and high efficiency. Various cathode materials such as Mo6S8, TiS2, alpha manganese dioxide (α-MnO2), and sulfur (S) were demonstrated in these electrolytes, though with certain discrepancies and ambiguities to be discussed in a later section.
Dendrite formation and growth is the universal fear for all rechargeable batteries using metal anodes. Mg has been cited in many publications as a “dendrite-free” anode,43 but a number of recent studies have clearly demonstrated that this is not true.44,45 Simply, the key parameter for distinguishing dendritic and non-dendritic metal deposition is the surface current density, which determines whether the deposition is under mass transfer limitation for the Mg-containing cations. When mass transfer is not the rate-limiting step, the morphology of electrochemically deposited metal is closely related to thermodynamic and kinetic properties such as adsorption energy of ions, charge transfer activation energy, overpotential, bond energy of the metal atoms, surface diffusion coefficient, etc. Computational modeling indicates that the deposition of Li prefers one-dimensional growth, forming wires and whiskers, while Mg is in favor of two-dimensional and three-dimensional growth as observed in most Mg deposition studies.46 Dendritic deposition of Mg occurs under the mass transfer limitation, i.e., the current is high enough to result in zero concentration of Mg-containing cations on the anode surface.47 Nevertheless, studies on the morphology of Mg deposition have to consider the different governing deposition mechanisms under different current densities. Future Mg batteries required to operate under high current conditions need to overcome the challenge raised by Mg dendrite formation.
Cathode interface
The interfacial processes on the Mg cathode surface may be more complex than those on the anode side. However, there are few studies that have discussed the interfacial effects on the electrochemical reactions of the cathodes. In fact, most of the publications on Mg cathode materials heavily emphasize the demonstration of “performance”, despite the frequent failure of apparent discharge–charge performance to fully reflect the underlying mechanisms. In the electrolytes from the combination of Lewis bases and acids, one must attend to the cathode–electrolyte chemical compatibility. It is known that Grignard reagents are not compatible with certain transition metal oxides and sulfides due to their nucleophilicity, as demonstrated by Gregory and coworkers in 1990.4 The transition metals at high oxidation state in the oxides and sulfides can be reduced by MgR2 (where R is alkyl or phenol) species generated from the Schlenk equilibrium in the electrolytes. The same concern also applies to electrolytes using Mg-containing Lewis bases (such as ROMgCl, Mg(OR)2, HMDSMgCl, and Mg(HMDS)2) and Al halides Lewis acids. These electrolytes are still nucleophilic, so the chemical compatibility with the (relatively) electrophilic cathode materials must be validated. A recent study revealed the chemical reaction between vanadium pentoxide (V2O5) and chloroaluminate anions including AlCl4−, which is a common anion in Lewis base/acid electrolytes.48 Iron disulfide was also reported to possibly react with chloroaluminate anions.49 These studies have brought the chemical stability of the metal oxides and sulfides in Mg electrolytes containing AlCl4− anions into question.The reaction mechanisms of the cathodes, i.e., intercalation and conversion, are also affected by the structures of the Mg-containing cations. Although it is outside the focus of this paper, it is worth mentioning that there are discrepancies among reports of the intercalation-type of Mg cathodes in the literature. The diffusion of divalent Mg2+ in metal oxide crystal structures can be extremely difficult due to the strong Coulombic attraction with the oxide anionic framework. Metal sulfides in principle are easier to intercalate due to the better polarizability of sulfide, but they suffer from lower intercalation potential.50,51 Intercalation reactions are characterized by distinct changes in the crystal structures of the hosts; however, with the exception of Mo6S8, reports on Mg intercalation with unambiguous crystallographic evidence and mechanism elucidation are rare.52–59 To date, Mo6S8 is the only Mg cathode material with a well-elucidated intercalation mechanism and high cycling stability, and it has remained as the benchmark cathode since it was first reported in 2000.5,60 Recent studies on Mo6S8 as the host material for electrochemically intercalated Al further demonstrate the extraordinary ability of the Chevrel phase structure to accommodate multivalent foreign cations.61–63 In contrast with the well-elucidated Mg2+ transport process inside the crystal structure of Mo6S8, the interface processes on the surface of Mo6S8 is much less known. The original report of Mo6S8 was in the DCC electrolytes containing [MgnCl2n−1(solvent)m]+ monomer or dimer cations. The Cl ligand in the cations played a critical role in facilitating Mg intercalation in Mo6S8via a surface mechanism, revealed by Wan et al. using a cluster model to study the properties of Mo6S8(100) surface. It was found that the Mo6S8(100) surface can reduce the Mg–Cl dissociation energy from ∼3 eV to as low as ∼0.2 eV due to the surface catalytical property granted by Mo, which could bond Cl− anions.64 In addition to the mechanism of Mg2+ intercalation via Cl desolvation, intercalation of monovalent MgCl+ was also reported by Hyun et al. in expanded layered TiS2 in 0.25 M APC electrolyte with 0.2 M 1-butyl-1-methylpyrrolidinium chloride additive. Both first-principles calculations and experimental results indicated favorable diffusion of MgCl+ over Mg2+ in the expanded TiS2 crystal structure.65
On the other hand, Mg cations in simple salt electrolytes, i.e., solvated divalent [Mg(solvent)n]2+ cations, may have distinct interfacial process from those cations containing Cl. To our best knowledge, there have been no studies devoted to comparing the interface processes between solvated simple Mg2+ cations and Mg–Cl complex cations. A literature survey unambiguously indicates that the magnesiation–demagnesiation behaviors of the benchmark Mo6S8 in Lewis base/acid electrolytes and simple salt electrolytes are very different.31,66,67 In general, the magnesiation-demagnesiation curve in simple salt electrolytes has a slope or single plateau (compared to two flat plateaus in Lewis base/acid electrolytes) and lower magnesiation potential. The cycle stability in Mg simple salt electrolytes is also generally worse. This observation indicates that the Cl ligand helps Mg intercalation on the Mo6S8 surface, which is consistent with the simulation work from Wan et al., while the solvation of divalent Mg2+ cations has a negative impact on Mg intercalation. Understanding the potentially different desolvation processes of these two types of cations during Mg intercalation is worth pursuing.
The great advantage of simple salt Mg electrolytes is that they enable sulfur cathodes, which are not compatible with nucleophilic electrolytes. Despite the numerous reports on Mg–S batteries, the detailed reaction mechanism at the S cathode is not well understood. The typical magnesiation curve of sulfur has two stepwise plateaus, which reassemble the lithiation curve of sulfur, although with lower potential. It is widely believed that polysulfides are produced as the intermediate products from the Mg–S reaction and that the polysulfide shuttling occurs during the charge process. This is indicated by the longer charge curve compared to the discharge curve observed in a number of studies. The work from Robba and coworkers showed differing structures between electrochemically formed MgS and chemically formed MgS, which is clear evidence for the complex nature of the reaction taking place on the sulfur cathode.68 In addition to the effects on the cathode interface, Mg anode passivation could also shut down the reversible reaction in Mg–S batteries. Wang and coworkers demonstrated that the sluggish magnesiation of sulfur in 0.5 M MgTFSI2-DME electrolyte due to the Mg surface passivation could be alleviated by adding MgCl2 in the electrolyte.69,70 This observation is consistent with the speculation discussed in the Anode Interfaces Section that MgCl2 can remove the Mg surface passivation layer and/or facilitate the interfacial cation transport.
To overcome the issue of polysulfides, conventional strategies succeed, to some extent, in improving cycle stability by encapsulating sulfur with porous carbon structures.71 A systematic study by Salama and coworkers pointed out that a trace amount of polysulfide in Mg(TFSI)2/MgCl2/DME electrolyte could poison Mg anode, but reversible Mg deposition-stripping persisted until a considerable amount of polysulfide dissolved in the electrolyte.72 These studies imply complex interfaces in Mg–S batteries that requires thorough understanding for future development.
Interfaces in hybrid electrolytes
To avoid the aforementioned difficulties at the Mg cathodes, hybrid batteries composed of Mg anode and Li+ ion cathodes have been proposed and studied. The first Mg–Li hybrid system can be traced back to 2006, when Gofer et al. studied tetrabutylammonium (TBA+) chloride and lithium chloride (LiCl) as additives in DCC electrolytes to improve the ionic conductivity.73 The solubilities of TBACl and LiCl were higher in DCC electrolyte than in pure THF, indicating the formation of new species with DCC. More interestingly, co-intercalation of Li+ in Mo6S8 cathode was observed in the 0.25 M DCC electrolyte with a very small amount of LiCl (0.01 M). The intercalation of both Mg2+ and Li+ has opened up opportunities for hybrid batteries in which Li-hosting cathodes may be used. The mechanism of co-intercalation was later verified by several independent studies. It was found that the feasibility of Li+ intercalation from a Mg–Li hybrid electrolyte is closely related to the Li+ activity in the electrolyte. Cheng et al. studied the intercalation of Mo6S8 in Mg electrolytes with a much higher Li+ ion concentration (1.0 M LiCl in 0.4 M APC) than that of Gofer's study.74 Instead of co-intercalation of Li+ and Mg2+, the authors found that only Li+ was intercalated into Mo6S8. They concluded that to ensure exclusive intercalation of Li, the amount of Li+ in the electrolyte must be much greater than that required to fully lithiate Mo6S8. Cho and coworkers reached a similar conclusion by calculating the ion activity using methods based on density functional theory.75 Based on their calculation, a threshold of Li+ activity must be achieved for Mo6S8 to prefer lithiation instead of magnesiation. In addition to Mo6S8, TiS2 has also been studied as the cathode in hybrid systems. Yao and coworkers observed sole intercalation of Li in TiS2 in 0.25 M APC electrolyte with 0.5 M LiCl as the additive.76 Similar results were also obtained by Wang and coworkers using 0.4 M APC electrolyte with 0.4 M LiCl.77 Conventional Li transition metal oxide cathodes would be ideal for Mg–Li hybrid systems in terms of energy density, but the intrinsically low anodic stability of Mg electrolyte (due to the ethereal solvents) prohibits the use of high potential cathodes such as LiCoO2 and LiNixMnyCo(1−x−y)O2. Yagi et al. demonstrated a Mg–Li hybrid cell using LiFePO4 cathode in a dual-salt THF electrolyte containing 1.0 M PhMgCl, 0.20 M AlCl3, and 0.20 M LiBF4. However, due to the incompatibility between PhMgCl and LiBF4, the full cell performance suffered from low coulombic efficiency.78 The idea of a hybrid Mg–Li system was also studied using sulfur cathodes. Gao et al. demonstrated improvement of the electrochemical reduction kinetics of sulfur by adding LiTFSI into the Mg(HMDS)2-based Mg electrolyte.79Based on the research discussed above, a Mg–Li hybrid battery is theoretically feasible through the use of Mg anode and Li cathodes. However, the disadvantage and challenges of hybrid cells should be taken into consideration. The complex nature of the hybrid electrolytes makes the mechanism of reactions and interfacial behaviors even more complicated.
Conclusions
Ultimately, there are no benchmark electrolytes for rechargeable Mg batteries to date. Interfacial processes vary significantly in different electrolytes and are still in need of more thorough understanding. The electrolytes containing both Mg–Cl complex cations and chloroaluminate anions are clearly problematic. This is mainly due to the chloroaluminate anions, which are chemically corrosive in the presence of water impurity. Moreover, their corrosiveness can be anodically enhanced, thus limiting the electrochemical window of these electrolytes and undermining battery performance. In comparison, electrolytes containing Mg–Cl complex cations and weakly coordinating anions such as the Mg(TFSI)2/MgCl2 system can be advantageous due to the absence of anions prone to oxidation and the improved Mg anode interface alleviating TFSI− anion reduction. The electrolytes based on simple Mg salts are conceptually superior, but the (electro)chemical instability of their anions under reducing conditions is a challenge. The stable weakly coordinating anions are often associated with sophisticated synthetic processes. In addition, the effects of the solvated divalent Mg2+ cations at the interface of cathodes, particularly intercalation-type materials represented by Mo6S8, may be disadvantageous in comparison to those Mg–Cl complex cations. Simple salt Mg electrolytes with weakly coordinating anions are also feasible for Mg–S batteries in theory, but the electrochemical characterizations in the literature suggest a lack of fundamental understanding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Argonne National Laboratory is operated for DOE Office of Science by UChicago Argonne, LLC, under contract No. DE-AC02-06CH11357. The authors gratefully acknowledge the support from the U. S. Department of Energy (DOE), Vehicle Technologies Office. J. G. gratefully acknowledges the support from the U. S. National Science Foundation though grant DMR-2004497.Notes and references
- L. W. Gaddum and H. E. French, J. Am. Chem. Soc., 1927, 49, 1295–1299 CrossRef CAS
.
- J. H. Connor, W. E. Reid and G. B. Wood, J. Electrochem. Soc., 1957, 104, 38–41 CrossRef CAS
- A. Brenner, J. Electrochem. Soc., 1971, 118, 99–100 CrossRef
- T. D. Gregory, R. J. Hoffman and R. C. Winterton, J. Electrochem. Soc., 1990, 137, 775–780 CrossRef CAS
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 CrossRef CAS
- D. Aurbach, H. Gizbar, A. Schechter, O. Chusid, H. E. Gottlieb, Y. Gofer and I. Goldberg, J. Electrochem. Soc., 2002, 149, 115–121 CrossRef
- L. W. F. Wan and D. Prendergast, J. Am. Chem. Soc., 2014, 136, 14456–14464 CrossRef CAS
- H. Gizbar, Y. Vestfrid, O. Chusid, Y. Gofer, H. E. Gottlieb, V. Marks and D. Aurbach, Organometallics, 2004, 23, 3826–3831 CrossRef CAS
- Y. Vestfried, O. Chusid, Y. Goffer, P. Aped and D. Aurbach, Organometallics, 2007, 26, 3130–3137 CrossRef CAS
- N. Pour, Y. Gofer, D. T. Major and D. Aurbach, J. Am. Chem. Soc., 2011, 133, 6270–6278 CrossRef CAS
- Z. Lu, A. Schechter, M. Moshkovich and D. Aurbach, J. Electroanal. Chem., 1999, 466, 203–217 CrossRef CAS
- Y. Gofer, O. Chusid, H. Gizbar, Y. Viestfrid, H. E. Gottlieb, V. Marks and D. Aurbach, Electrochem. Solid-State Lett., 2006, 9, 257–260 CrossRef
- F. F. Wang, Y. S. Guo, J. Yang, Y. Nuli and S. Hirano, Chem. Commun., 2012, 48, 10763–10765 RSC
- E. G. Nelson, J. W. Kampf and B. M. Bartlett, Chem. Commun., 2014, 50, 5193–5195 RSC
- A. J. Crowe, K. K. Stringham and B. M. Bartlett, ACS Appl. Mater. Interfaces, 2016, 8, 23060–23065 CrossRef CAS
- C. Liao, N. Sa, B. Key, A. K. Burrell, L. Cheng, L. A. Curtiss, J. T. Vaughey, J. J. Woo, L. B. Hu, B. F. Pan and Z. C. Zhang, J. Mater. Chem. A, 2015, 3, 6082–6087 RSC
- H. S. Kim, T. S. Arthur, G. D. Allred, J. Zajicek, J. G. Newman, A. E. Rodnyansky, A. G. Oliver, W. C. Boggess and J. Muldoon, Nat. Commun., 2011, 2, 427 CrossRef
- Z. Zhao-Karger, X. Y. Zhao, O. Fuhr and M. Fichtner, RSC Adv., 2013, 3, 16330–16335 RSC
- L. C. Merrill and J. L. Schaefer, Chem. Mater., 2018, 30, 3971–3974 CrossRef CAS
- E. N. Keyzer, H. F. J. Glass, Z. G. Liu, P. M. Bayley, S. E. Dutton, C. P. Grey and D. S. Wright, J. Am. Chem. Soc., 2016, 138, 8682–8685 CrossRef CAS
- T. T. Tran, W. M. Lamanna and M. N. Obrovac, J. Electrochem. Soc., 2012, 159, A2005–A2009 CrossRef CAS
- Z. Zhao-Karger, M. E. G. Bardaji, O. Fuhr and M. Fichtner, J. Mater. Chem. A, 2017, 5, 10815–10820 RSC
- J. T. Herb, C. A. Nist-Lund and C. B. Arnold, ACS Energy Lett., 2016, 1, 1227–1232 CrossRef CAS
- J. T. Herb, C. A. Nist-Lund and C. B. Arnold, J. Mater. Chem. A, 2017, 5, 7801–7805 RSC
- E. Keyzer, J. Lee, Z. G. Liu, A. D. Bond, D. S. Wright and C. P. Grey, J. Mater. Chem. A, 2019, 7, 2677–2685 RSC
- O. Tutusaus, R. Mohtadi, T. S. Arthur, F. Mizuno, E. G. Nelson and Y. V. Sevryugina, Angew. Chem., Int. Ed., 2015, 54, 7900–7904 CrossRef CAS
- S. G. McArthur, L. X. Geng, J. C. Guo and V. Lavallo, Inorg. Chem. Front., 2015, 2, 1101–1104 RSC
- H. D. Yoo, I. Shterenberg, Y. Gofer, G. Gershinsky, N. Pour and D. Aurbach, Energy Environ. Sci., 2013, 6, 2265–2279 RSC
- Y. W. Cheng, R. M. Stolley, K. S. Han, Y. Y. Shao, B. W. Arey, N. M. Washton, K. T. Mueller, M. L. Helm, V. L. Sprenkle, J. Liu and G. S. Li, Phys. Chem. Chem. Phys., 2015, 17, 13307–13314 RSC
- Y. Nakayama, Y. Kudo, H. Oki, K. Yamamoto, Y. Kitajima and K. Noda, J. Electrochem. Soc., 2008, 155, A754–A759 CrossRef CAS
- R. Mohtadi, M. Matsui, T. S. Arthur and S. J. Hwang, Angew. Chem., Int. Ed., 2012, 51, 9780–9783 CrossRef CAS
- R. E. Doe, R. Han, J. Hwang, A. J. Gmitter, I. Shterenberg, H. D. Yoo, N. Pour and D. Aurbach, Chem. Commun., 2014, 50, 243–245 RSC
- T. B. Liu, Y. Y. Shao, G. S. Li, M. Gu, J. Z. Hu, S. C. Xu, Z. M. Nie, X. L. Chen, C. M. Wang and J. Liu, J. Mater. Chem. A, 2014, 2, 3430–3438 RSC
- R. Jay, A. W. Tomich, J. Zhang, Y. F. Zhao, A. De Gorostiza, V. Lavallo and J. C. Guo, ACS Appl. Mater. Interfaces, 2019, 11, 11414–11420 CrossRef CAS
- Y. Yu, A. Baskin, C. Valero-Vidal, N. T. Hahn, Q. Liu, K. R. Zavadil, B. W. Eichhorn, D. Prendergast and E. J. Crumlin, Chem. Mater., 2017, 29, 8504–8512 CrossRef CAS
- T. T. Tran, W. M. Lamanna and M. N. Obrovac, J. Electrochem. Soc., 2012, 159, 2005–2009 CrossRef
- N. Y. Sa, B. F. Pan, A. Saha-Shah, A. A. Hubaud, J. T. Vaughey, L. A. Baker, C. Liao and A. K. Burrell, ACS Appl. Mater. Interfaces, 2016, 8, 16002–16008 CrossRef CAS
- E. Peled and H. Straze, J. Electrochem. Soc., 1977, 124, 1030–1035 CrossRef CAS
- E. Peled, J. Electrochem. Soc., 1979, 126, 2047–2051 CrossRef CAS
- A. Meitav and E. Peled, J. Electrochem. Soc., 1981, 128, 825–831 CrossRef CAS
- S. B. Son, T. Gao, S. P. Harvey, K. X. Steirer, A. Stokes, A. Norman, C. S. Wang, A. Cresce, K. Xu and C. M. Ban, Nat. Chem., 2018, 10, 532–539 CrossRef CAS
- S. G. McArthur, R. Jay, L. X. Geng, J. C. Guo and V. Lavallo, Chem. Commun., 2017, 53, 4453–4456 RSC
- M. Matsui, J. Power Sources, 2011, 196, 7048–7055 CrossRef CAS
- M. S. Ding, T. Diemant, R. J. Behm, S. Passerini and G. A. Giffin, J. Electrochem. Soc., 2018, 165, 1983–1990 CrossRef
- R. Davidson, A. Verma, D. Santos, F. Hao, C. Fincher, S. S. Xiang, J. Van Buskirk, K. Xie, M. Pharr, P. P. Mukherjee and S. Banerjee, ACS Energy Lett., 2019, 4, 375–376 CrossRef CAS
- C. Ling, D. Banerjee and M. Matsui, Electrochim. Acta, 2012, 76, 270–274 CrossRef CAS
- C. Brissot, M. Rosso, J. N. Chazalviel and S. Lascaud, J. Power Sources, 1999, 81, 925–929 CrossRef
- X. Y. Wen, Y. H. Liu, A. Jadhav, J. Zhang, D. Borchardt, J. Y. Shi, B. W. Wong, B. Sanyal, R. J. Messinger and J. C. Guo, Chem. Mater., 2019, 31, 7238–7247 CrossRef CAS
- T. Mori, Y. Orikasa, K. Nakanishi, K. Z. Chen, M. Hattori, T. Ohta and Y. Uchimoto, J. Power Sources, 2016, 313, 9–14 CrossRef CAS
- E. Levi, M. D. Levi, O. Chasid and D. Aurbach, J. Electroceram., 2009, 22, 13–19 CrossRef CAS
- Z. Q. Rong, R. Malik, P. Canepa, G. S. Gautam, M. Liu, A. Jain, K. Persson and G. Ceder, Chem. Mater., 2015, 27, 6016–6021 CrossRef CAS
- E. Levi, E. Lancry, A. Mitelman, D. Aurbach, G. Ceder, D. Morgan and O. Isnard, Chem. Mater., 2006, 18, 5492–5503 CrossRef CAS
- E. Levi, E. Lancry, A. Mitelman, D. Aurbach, O. Isnard and D. Djurado, Chem. Mater., 2006, 18, 3705–3714 CrossRef CAS
- E. Levi, A. Mitelman, D. Aurbach and M. Brunelli, Chem. Mater., 2007, 19, 5131–5142 CrossRef CAS
- E. Levi, A. Mitelman, O. Isnard, M. Brunelli and D. Aurbach, Inorg. Chem., 2008, 47, 1975–1983 CrossRef CAS
- P. Bonnick, L. Blanc, S. H. Vajargah, C. W. Lee, X. Q. Sun, M. Balasubramanian and L. F. Nazar, Chem. Mater., 2018, 30, 4683–4693 CrossRef CAS
- X. Q. Sun, P. Bonnick and L. F. Nazar, ACS Energy Lett., 2016, 1, 297–301 CrossRef CAS
- A. Mukherjee, N. Sa, P. J. Phillips, A. Burrell, J. Vaughey and R. F. Klie, Chem. Mater., 2017, 29, 2218–2226 CrossRef CAS
- J. L. Andrews, A. Mukherjee, H. D. Yoo, A. Parija, P. M. Marley, S. Fakra, D. Prendergast, J. Cabana, R. F. Klie and S. Banerjee, Chem, 2018, 4, 564–585 CAS
- C. Ling and K. Suto, Chem. Mater., 2017, 29, 3731–3739 CrossRef CAS
- L. X. Geng, G. C. Lv, X. B. Xing and J. C. Guo, Chem. Mater., 2015, 27, 4926–4929 CrossRef CAS
- L. X. Geng, J. P. Scheifers, J. Zhang, K. N. Bozhilov, B. P. T. Fokwa and J. C. Guo, Chem. Mater., 2018, 30, 8420–8425 CrossRef CAS
- B. Lee, H. R. Lee, T. Yim, J. H. Kim, J. G. Lee, K. Y. Chung, B. W. Cho and S. H. Oh, J. Electrochem. Soc., 2016, 163, 1070–1076 CrossRef
- L. W. F. Wan, B. R. Perdue, C. A. Apblett and D. Prendergast, Chem. Mater., 2015, 27, 5932–5940 CrossRef CAS
- H. D. Yoo, Y. L. Liang, H. Dong, J. H. Lin, H. Wang, Y. S. Liu, L. Ma, T. P. Wu, Y. F. Li, Q. Ru, Y. Jing, Q. Y. An, W. Zhou, J. H. Guo, J. Lu, S. T. Pantelides, X. F. Qian and Y. Yao, Nat. Commun., 2017, 8, 339 CrossRef
- R. Attias, M. S. Chae, B. Dlugatch, M. Oliel, Y. Gofer and D. Aurbach, ACS Catal., 2020, 10, 7773–7784 CrossRef CAS
- F. Tuerxun, Y. Abulizi, Y. N. NuLi, S. J. Su, J. Yang and J. L. Wang, J. Power Sources, 2015, 276, 255–261 CrossRef CAS
- A. Robba, A. Vizintin, J. Bitenc, G. Mali, I. Arcon, M. Kavcic, M. Zitnik, K. Bucar, G. Aquilanti, C. Martineau-Corcos, A. Randon-Vitanova and R. Dominko, Chem. Mater., 2017, 29, 9555–9564 CrossRef CAS
- T. Gao, X. Ji, S. Hou, X. L. Fan, X. G. Li, C. Y. Yang, F. D. Han, F. Wang, J. J. Jiang, K. Xu and C. S. Wang, Adv. Mater., 2018, 30, 1704313 CrossRef
- T. Gao, S. Hou, F. Wang, Z. H. Ma, X. G. Li, K. Xu and C. S. Wang, Angew. Chem., Int. Ed., 2017, 56, 13526–13530 CrossRef CAS
- X. W. Yu and A. Manthiram, ACS Energy Lett., 2016, 1, 431–437 CrossRef CAS
- M. Salama, R. Attias, B. Hirsch, R. Yemini, Y. Gofer, M. Noked and D. Aurbach, ACS Appl. Mater. Interfaces, 2018, 10, 36910–36917 CrossRef CAS
- Y. Gofer, O. Chusid, H. Gizbar, Y. Viestfrid, H. E. Gottlieb, V. Marks and D. Aurbach, Electrochem. Solid-State Lett., 2006, 9, A257–A260 CrossRef CAS
- Y. W. Cheng, Y. Y. Shao, J. G. Zhang, V. L. Sprenkle, J. Liu and G. S. Li, Chem. Commun., 2014, 50, 9644–9646 RSC
- J. H. Cho, M. Aykol, S. Kim, J. H. Ha, C. Wolverton, K. Y. Chung, K. B. Kim and B. W. Cho, J. Am. Chem. Soc., 2014, 136, 16116–16119 CrossRef CAS
- H. D. Yoo, Y. L. Liang, Y. F. Li and Y. Yao, ACS Appl. Mater. Interfaces, 2015, 7, 7001–7007 CrossRef CAS
- T. Gao, F. D. Han, Y. J. Zhu, L. M. Suo, C. Luo, K. Xu and C. S. Wang, Adv. Energy Mater., 2015, 5, 1401507 CrossRef
- S. Yagi, T. Ichitsubo, Y. Shirai, S. Yanai, T. Doi, K. Murase and E. Matsubara, J. Mater. Chem. A, 2014, 2, 1144–1149 RSC
- T. Gao, M. Noked, A. J. Pearse, E. Gillette, X. L. Fan, Y. J. Zhu, C. Luo, L. M. Suo, M. A. Schroeder, K. Xu, S. B. Lee, G. W. Rubloff and C. S. Wang, J. Am. Chem. Soc., 2015, 137, 12388–12393 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2020 |