
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbon supported noble metal nanoparticles as efficient catalysts for electrochemical water splitting†
Meng
Liu
a,
Ferdinand
Hof
 bc,
Miriam
Moro
a,
Giovanni
Valenti
a,
Francesco
Paolucci
a and
Alain
Pénicaud
*bc
bc,
Miriam
Moro
a,
Giovanni
Valenti
a,
Francesco
Paolucci
a and
Alain
Pénicaud
*bc
aDepartment of Chemistry “G. Ciamician”, University of Bologna, via Selmi 2, 40126 Bologna, Italy
bCNRS, Centre de Recherche Paul Pascal (CRPP), UMR 5031, F-33600 Pessac, France. E-mail: alain.penicaud@crpp.cnrs.fr
cUniversité Bordeaux, CRPP, UMR 5031, F-33600 Pessac, France
First published on 24th September 2020
Abstract
Due to an increasing requirement of clean and sustainable hydrogen energy economy, it is significant to develop new highly effective catalysts for electrochemical water splitting. In alkaline electrolyte, Platinum (Pt) shows a much slower hydrogen evolution reaction (HER) kinetics relative to acidic condition. Here, we show a versatile synthetic approach for combining different noble metals, such as Rhodium (Rh), RhPt and Pt nanoparticles, with carbon forming noble metal nanoparticles/nanocarbon composites, denoted as Rh(nP)/nC, RhPt(nP)/nC and Pt(nP)/nC, respectively. It was found that in alkaline media these composites exhibited higher performance for the HER than the commercial Pt/C. In particular, Rh(nP)/nC displayed a small overpotential of 44 mV at a current density of 5 mA cm−2 and a low Tafel slope of 50 mV dec−1. Meanwhile, it also showed a comparable activity for the oxygen evolution reaction (OER) to the benchmarking catalyst RuO2. The superior HER and OER performance benefits from the very small size of nanoparticles and synergy between carbon support and nanoparticles.
Introduction
Hydrogen has been considered as an ideal energy carrier due to its high energy density and potentially environmentally friendly production by the solar assisted splitting of water.1 Water electrolysis in alkaline media, which includes the hydrogen evolution reaction (HER) at the cathode and the oxygen evolution reaction (OER) at the anode, is an efficient approach to generate large volumes of hydrogen.2–4 The efficiency of electrocatalytic water splitting is directly related to the activity of the respective catalysts used in both electrochemical half cells. Noble metal-based materials, such as Pt/C and IrO2 (or RuO2), are used as benchmarking catalysts for the HER and OER, respectively.5 However, the catalytic activity of Pt for the HER in alkaline electrolyte is about two orders of magnitude lower than in acidic media.6 The OER, which ideally is a four-electron transfer process involving multiple intermediates, suffers from issues, such as high overpotential, product distribution, and activity loss over time.7 Therefore, it still remains a challenge to generate catalysts (beyond Pt, Ir and Ru) for both the HER and OER that can exhibit selectivity, keep stable over time and can be produced by low cost methods in large quantities.In order to reduce the cost for water electrolysis, substantial effort has been made to develop catalysts like non-noble metals and non-metallic materials.8 However, their catalytic performance largely falls behind noble metal catalysts.9 As a well-known noble metal, rhodium (Rh) has shown high catalytic property towards a couple of reactions including CO2 reduction, CO oxidation, N2O decomposition etc.10–14 For example, Huang et al. reported a route to synthesize a series of Rh nanocrystals, including tetrahedron, concave tetrahedron and nanosheet, which showed efficient HER and OER activity.15 Chen et al. explored ultrathin Rh2O3 nanosheet assemblies and used them as OER electrocatalyst.16 However, up to now the study of Rh as noble metal in electrocatalysis still lags much behind to the its Pt counterpart.
Generally, decreasing the size of metal nanostructures can increase the ratio of surface area to volume and enhance the exposure of surface atoms.17 Chen and his coworkers have reported many excellent works on electrocatalysis by means of in situ/operando studies specially for identifying real active sites.18–23 In one of their works, a series of operando measurements, including in situ X-ray absorption spectroscopy, liquid-phase transmission electron microscopy, and in situ Raman spectroscopy, were conducted to unravel in real time the structural and chemical stability of P-substituted CoSe2 electrocatalysts under both HER and OER.18 An attractive way to further improve the utilization of metal nanostructures is combining them with appropriate supports that possess both high electrical conductivity and high surface area. Carbon-based supports, such as carbon nanotubes and nitrogen-doped graphene, have already been employed to achieve increased surface loading and better dispersion of metal nanostructures.24–27 For instance, Guo et al. synthesized well-dispersed Rh nanocrystals on single-walled carbon nanotubes as a highly effective electrocatalyst for the HER both in acid and alkaline media.28
We have recently developed a novel synthetic approach for the preparation of carbon metal oxide/hydroxide composite materials using graphenide solutions, charge stabilized graphene layers in solution, directly as reducing agent.29,30 This approach allows to graft small and size calibrated nanoparticles randomly distributed onto the carbon frameworks by simultaneously controlling the grafting ratio and the respective size distribution.31,32 This control is achieved by the exploitation of the graphenide solution as the reduction agent as the redox process can take place only in the proximity of the carbon lattice and because the number of electrons on the carbon lattice is finite. This modular approach permits the accessibility of the catalytic centers by simultaneously maintaining the conductivity of the carbon lattice.
Herein, we utilized the graphitic nano carbon33,34 as precursor to generate firstly graphenide solution and secondly noble metal (Rh and Pt) nanoparticle/nanocarbon composites, Rh(nP)/nC and Pt(nP)/nC, by the reaction of graphenide solution with anhydrous RhCl3 or PtCl2. Considering that various combinations of metals, metal alloys, and metal oxides have been used to catalyze water splitting,35,36 we also prepared a mixed sample, RhPt(nP)/nC, by the reaction of graphenide solution with a mixture of equimolar RhCl3 and PtCl2. Electrocatalytic performance of the prepared materials were investigated for the OER and HER under alkaline condition. It was found that Rh(nP)/nC exhibited excellent OER activity with a low overpotential of 333 mV at a current density of 5 mA cm−2, which is comparable to that of RuO2. Meanwhile, all the three materials showed better HER property than commercial Pt/C. In order to achieve a current density of 5 mA cm−2 for Rh(nP)/nC towards the HER, an overpotential of 44 mV was observed and a Tafel slope of about 50 mV dec−1 was achieved. The superior catalytic performances might be traced back to the small particle size and the synergy of the two nanomaterials.
Characterization
As shown in Scheme 1, the synthesis of noble metal Rh and Pt nanoparticles/nanocarbon composites, denoted as Rh(nP)/nC, Pt(nP)/nC and RhPt(nP)/nC, respectively, involves the reduction of the synthetic, sustainable and almost defect free graphitic nano carbons by metallic potassium, followed by the dissolution to graphenide solution in absolute THF and reaction with equimolar amounts of anhydrous PtCl2 and/or RhCl3 salts. The achieved materials combined highly conductive carbon support with active metal nanoparticles and they were expected to display efficient performance for the OER and HER because of the synergy between carbon support and nanoparticles. Similar approaches have been utilized on transition metals (e.g. Cu, Fe, Mn, Ni and Co) and in all cases the formation of metal oxide/hydroxide nanoparticles grafted on the carbon sheets were observed.30–32 It was argued that the reduction generates in a first step metallic nanoparticles on the carbon framework that are oxidized during the work up-step due to the high oxophilicity of the transition metals. The electron transfer can occur only in close proximity of the carbon lattice because the charged carbon layers are the only reduction agent present. Presumably, the formation of the metallic nanoparticles is occurring due to a sequence of single electron transfer (SET) processes as argued for related reactions.37 By XRD measurements (CuKα, see Fig. 1a) this mechanistic interpretation can be corroborated because the obtained patterns for the Pt(nP)/nC and the RhPt(nP)/nC show broad peaks at 26.1°, 39.9° and 46.5° that can be attributed to the (002) pattern of the carbon material and the (111) and (200) patterns of metallic nanoparticles. In the case of the Rh(nP)/nC, only the peak at 26.1° is visible. The presence of the metallic nature of nanoparticles in the samples Pt(nP)/nC and RhPt(nP)/nC have been further corroborated by XPS measurements (see Fig. S1†) as in both samples the peaks for metallic platinum are clearly visible with only a minor contribution of oxidized species. In contrast, for the Rh(nP)/nC composite, peaks for Rh(III) are visible that can be attributed to Rh2O3 with a minor content of metallic rhodium. This observation may be explained by the less noble character of the rhodium metal and the oxidation of the rhodium metal to the respective oxide in the work-up step.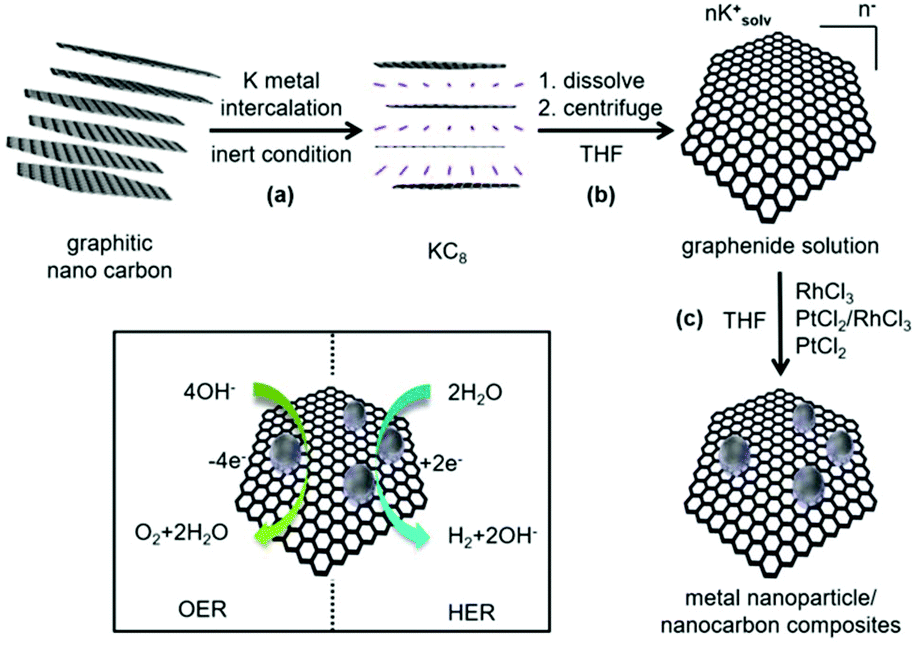 | ||
| Scheme 1 Reaction scheme of the synthesis of metal nanoparticles/nanocarbon composite materials. The respective steps are: (a) intercalation of carbon material with potassium metal (stoichiometry KC8); (b) dissolution and isolation of the graphenide solution in THF; (c) reaction with respective metal chlorides forming the final composites. The lower left scheme shows the combination between carbon support and nanoparticles, which may boost the exposure of active sites and the conductivity within the electrode for electrocatalysis of the OER and HER. | ||
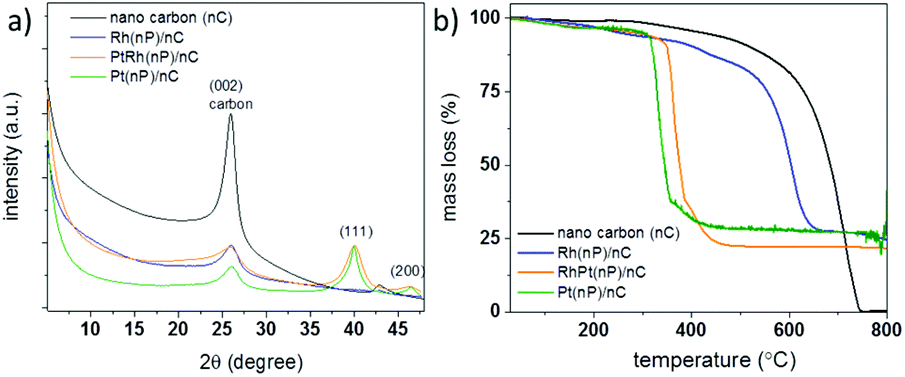 | ||
| Fig. 1 (a) XRD patterns (CuKα) of the three composite materials Rh(nP)/nC (blue trace), PtRh(nP)/nC (orange trace), and Pt(nP)/nC (green trace) in comparison to the carbon starting material (nC, black trace). (b) Thermogravimetric analysis (TGA) of the three composite materials and the starting carbon material between 50–800 °C under synthetic air (80% N2, 20% O2) with a heating ramp of 10 °C min−1. | ||
The metal contents of the samples were determined by TGA measurements (see Fig. 1b) and remaining mass of 27.2% for the Pt(nP)/nC composite material, 26.9% for the Rh(nP)/nC and 22.1% for the RhPt(nP)/nC could be found. The notable shift of the combustion temperature of the carbon framework to significantly lower temperatures can be traced back to the accessibility and the catalytic effect of the metallic nanoparticles. This observation is in line with soot combustion research performed for optimizing diesel engines.38 The respective atomic percentage (at%) of the noble metals in the different composites, taking into account the oxide nature for the Rh case, exhibit values between 2.1 and 2.3 at% and was corroborated by XPS analysis. The presence of oxidized species in the Rh(nP)/nC composite was further studied by measuring XRD patterns of the residue materials after the TGA measurements and in case of the Pt(nP)/nC sample peaks for metallic Pt were observed whereas for the Rh(nP)/nC peaks that can be attributed to Rh2O3 have been found (see Fig. S2†). A slight shift between the XRD patterns of Pt(nP)/nC and RhPt(nP)/nC can be seen in Fig. 1a, and is even more apparent in the enlarged diffractograms of Fig S3† of the composites and their respective residue materials (Fig. S3†). This shift has been observed in EXAFS studies of RhPt nanoalloys,39 and was explained by a compressing of the metallic lattice due to the simultaneous presence of Rh and Pt atoms in the same nanoparticle.
By means of HR-STEM/EDX mappings (Fig. S4 and S5†) it can be seen that the respective metal is present and is detectable only finely distributed on the carbon framework. This may be related to the grafting process of the nanoparticles that is occurring on the carbon framework during the synthesis. The distribution of the nanoparticles was studied in more detail by HR-TEM analysis and it can be seen that the nanoparticles are randomly and finely dispersed on the carbon framework (Fig. S6†). The nanoparticles are crystalline and the metallic nature in case of the Pt(nP)/nC can be once more observed in the diffraction pattern of the nanoparticles (Fig. S7†). The size distribution of the nanoparticles is in the range between 2 and 3 nm and was found to be 2.3 nm for the Rh(nP)/nC composite and 2.6 nm for the Pt(nP)/nC based on the analysis of about 20 HR-TEM images for each sample (Fig. S8†).
Electrochemical measurement
All the electrochemical measurement was performed with a SP-300 bipotentiostat (Biologic Instruments) workstation, using a three-electrode system composed of a saturated calomel electrode (SCE) as reference electrode, a Pt wire as counter electrode and a catalyst-modified glassy carbon electrode (GCE, 3 mm in diameter, geometric surface area 0.071 cm2) as working electrode. The obtained materials were dispersed in a mixed solvent of dimethylformamide (DMF) and 5% Nafion (v/v = 950/50) under sonication at least half an hour. Then, 30 microliters of the dispersion with a concentration of 1 mg mL−1 were drop cast onto a polished GCE surface and dried in air. Commercial RuO2 and Pt/C (20 wt%) were used as references for the OER and HER catalysis, and were dispersed in the same way. The electrocatalytic performance was evaluated in Ar-saturated 0.1 M KOH solution.The gas phase quantification was carried out by coupling a Gas Chromatograph (GC) with an electrochemical cell. The gas phase quantification was carried out during the electrolysis of least 1 hour at cathodic potentials, with sampling every 15 minutes. faradaic Efficiency (FE) for the HER was quantified following the procedure previously described by Baltrusaitis et al.:40 FE (%) = nFϕFm/I, where n is the number of electrons needed for HER, thus 2; F is the Faraday constant; ϕ is the volume fraction of the gas; I is the current and Fm is the molar Ar gas flow rate.
Results and discussion
The electrocatalytic activity of Rh(nP)/nC, RhPt(nP)/nC and Pt(nP)/nC for the OER was tested in Ar-saturated 0.1 M KOH solution. The LSV polarization curves in Fig. 2a showed that it followed an order of Pt(nP)/nC < RhPt(nP)/nC < Rh(nP)/nC. In detail, to achieve a current density of 5 mA cm−2, Rh(nP)/nC needed an overpotential of 333 mV, quite similar to that of commercial RuO2, which was 328 mV. However, for RhPt(nP)/nC and Pt(nP)/nC, much larger overpotentials were required, 414 mV and 555 mV, respectively. As shown in Fig. 2b, the Tafel slopes of commercial RuO2, Rh(nP)/nC, RhPt(nP)/nC and Pt(nP)/nC were 72, 123, 95 and 152 mV dec−1. An anodic peak at around 1.4 V (vs. RHE) of Rh(nP)/nC LSV polarization curve might be attributed to an oxidation process of Rh3+ to Rh4+ before oxygen evolution.41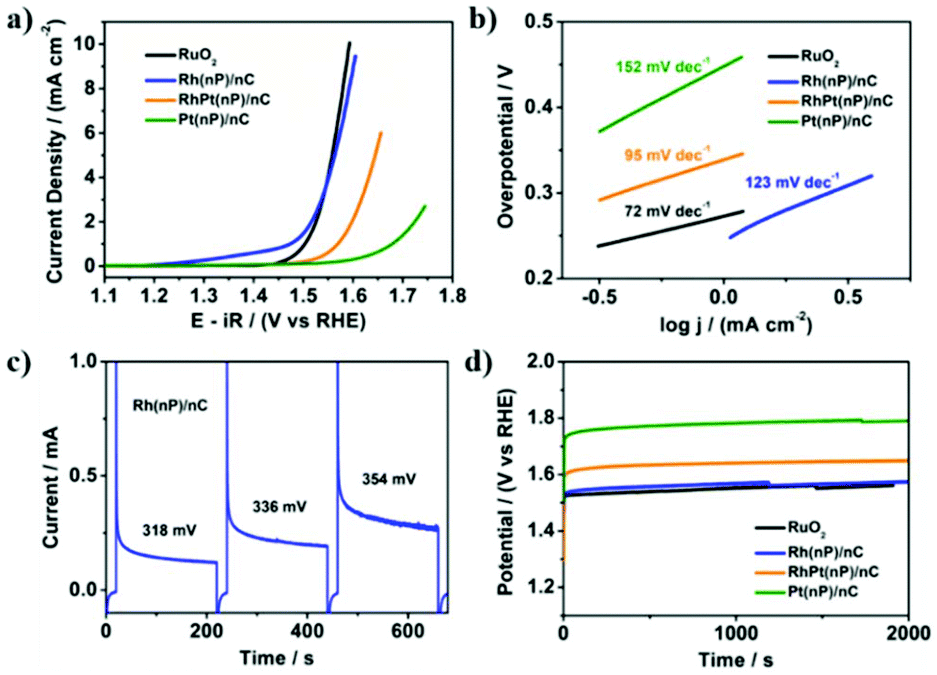 | ||
| Fig. 2 Electrocatalytic performance for the OER in 0.1 M KOH solution. (a) LSV polarization curves of commercial RuO2, Rh(nP)/nC, RhPt(nP)/nC and Pt(nP)/nC at a scan rate of 5 mV s−1. (b) Corresponding Tafel plots. (c) Representative 200 seconds potential steps of Rh(nP)/nC at overpotentials of 0.318 V, 0.336 V and 0.354 V, respectively. (d) Chronopotentiometry curves recorded at a current density of 2 mA cm−2. The potentials were corrected for iR drop and calibrated to RHE. | ||
Meanwhile, the electrochemical surface areas (ECSAs) were measured by means of CV in 0.1 M KOH solution as shown in Fig. S9.† By integrating the hydrogen underpotential deposition regions, the ECSAs of Rh(nP)/nC, RhPt(nP)/nC and Pt(nP)/nC were calculated to be 7.7, 17.5 and 22.2 m2 g−1, respectively, while the commercial Pt/C (20%) showed a value of 42.8 m2 g−1.
Meanwhile, a sequence of potential steps (200 seconds for each) under different overpotentials were carried out in order to achieve TOF for the OER (see Fig. 2c). Based on the charges transferred during potential steps, the TOF values of Rh(nP)/nC were 0.013 s−1, 0.019 s−1 and 0.026 s−1 at overpotentials of 318 mV, 336 mV and 354 mV, respectively, as shown in Fig. S10.† Here, we assumed that every single metal atom was catalytically active for the OER. Thus, the observed values are the lower limits of TOF, because likely not all particles are accessible. When compared with literatures, Rh(nP)/nC showed a high level of TOF. For example, at an overpotential of 350 mV, it is 2.5 times larger than that of the commercial RuO2 (see Fig. S11†).
Besides, the durability of RuO2, Rh(nP)/nC, RhPt(nP)/nC and Pt(nP)/nC for the OER was investigated by conducting chronopotentiometry measurement at a current density of 2 mA cm−2 as shown in Fig. 2d. After 2000 seconds only slight increase of potentials was observed indicating that the prepared nanocomposites were quite stable for the catalytic OER in alkaline media. This interpretation can be corroborated by SEM/EDX measurements of the surfaces before and after electrolysis, where the rhodium atomic content seems unchanged. Although the surface gets rougher and porous pointing towards limits in the preparation technique of the electrodes (Fig. S12†), the CV curves of Rh(nP)/nC before and after the OER durability test (Fig. S13†) did not change much, which means that the value of the ECSA of Rh(nP)/nC remained almost constant during catalysis. This confirmed that Rh(nP)/nC is stable for the OER in alkaline media. We compared the OER performance of Rh(nP)/nC in alkaline medium with previous works and it showed that Rh(nP)/nC displayed relatively lower overpotential at a current density of 10 mA cm−2 (see Table ST2†).
On the other side, the electrocatalytic activity of Rh(nP)/nC, RhPt(nP)/nC and Pt(nP)/nC for the HER was evaluated in Ar-saturated 0.1 M KOH solution. The HER polarization curves were recorded at a scan rate of 5 mV s−1. As shown in Fig. 3a, at a current density of 2 mA cm−2, overpotentials of the Rh(nP)/nC, RhPt(nP)/nC and Pt(nP)/nC are 44, 48 and 82 mV, respectively, better than commercial Pt/C with a higher overpotential of 90 mV, though in acidic media commercial Pt/C behaves better than Rh(nP)/nC (see Fig. S14†). The good performance of all here discussed composite materials can be traced back to the small size of the nanoparticles and the large amount of edges, shown to be beneficial for electrocatalytic performances.42 Noteworthy is the increased performance of the RhPt(nP)/nC over its Pt(nP)/nC counterpart for the HER. We attribute this fact to the nanoalloy structure of RhPt(nP)/nC.
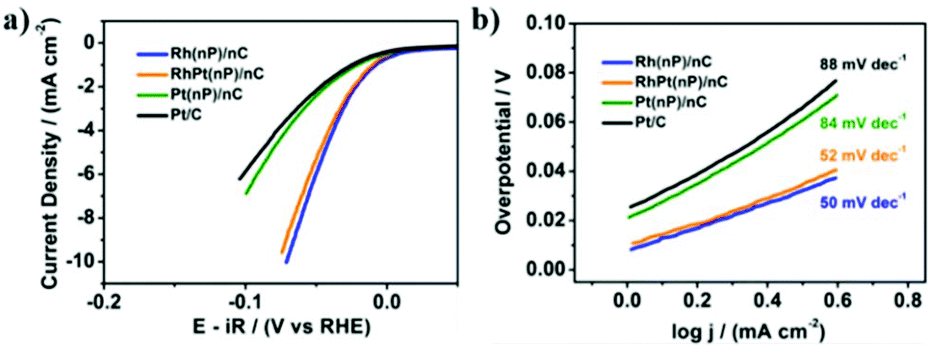 | ||
| Fig. 3 HER performance of Rh(nP)/nC, RhPt(nP)/nC, Pt(nP)/nC and commercial Pt/C in 0.1 M KOH solution. (a) LSV polarization curves at a scan rate 5 mV s−1. (b) Corresponding Tafel plots. The potentials were corrected for iR drop and calibrated to RHE. | ||
In order to study the HER reaction kinetics, Tafel slopes were calculated based on LSV polarization curves. We can see from Fig. 3b that Rh(nP)/nC, RhPt(nP)/nC and Pt(nP)/nC displayed low Tafel slopes, which were 50, 52 and 84 mV dec−1, respectively, while that of commercial Pt/C was 88 mV dec−1. It is known that lower Tafel slope means more rapid raise of product generation as overpotential increases. Therefore, the prepared carbon supported noble nanoparticles are supposed to have faster reaction kinetics than commercial Pt/C for the HER. Finally, faradaic Efficiency (FE) for the HER was quantified following the procedure previously described.43 After one hour of electrolysis, both electrocatalysts of Rh(nP)/nC and Pt(nP)/nC reached the 100% of FE.
We have physically mixed Rh(nP)/nC and Pt(nP)/nC with a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and investigated its electrocatalytic performances for the OER and HER. As can be seen from Fig. S15,† Rh–Pt mixture showed lower current densities at same overpotentials and larger Tafel slopes than the prepared RhPt(nP)/nC. This in some way confirmed that there might be a combination between Rh and Pt inside RhPt(nP)/nC sample, which contributes to its superior activities. When comparing the HER activity of Rh(nP)/nC with other reported catalysts (Table ST3†), we can see that Rh(nP)/nC is more efficient among them considering its low overpotential and Tafel slope. For practical use, a two-electrode electrolyzer using Rh(nP)/nC (loaded on two glassy carbon electrodes) as both anode and cathode was built for water splitting in 0.1 M KOH. As shown in Fig. S16,† a voltage of 1.68 V for Rh(nP)/nC||Rh(nP)/nC couple was required to achieve a current density of 10 mA cm−2, which corresponded to the potential difference between the HER and OER needed in the half cells when using Rh(nP)/nC as catalysts.
:1 and investigated its electrocatalytic performances for the OER and HER. As can be seen from Fig. S15,† Rh–Pt mixture showed lower current densities at same overpotentials and larger Tafel slopes than the prepared RhPt(nP)/nC. This in some way confirmed that there might be a combination between Rh and Pt inside RhPt(nP)/nC sample, which contributes to its superior activities. When comparing the HER activity of Rh(nP)/nC with other reported catalysts (Table ST3†), we can see that Rh(nP)/nC is more efficient among them considering its low overpotential and Tafel slope. For practical use, a two-electrode electrolyzer using Rh(nP)/nC (loaded on two glassy carbon electrodes) as both anode and cathode was built for water splitting in 0.1 M KOH. As shown in Fig. S16,† a voltage of 1.68 V for Rh(nP)/nC||Rh(nP)/nC couple was required to achieve a current density of 10 mA cm−2, which corresponded to the potential difference between the HER and OER needed in the half cells when using Rh(nP)/nC as catalysts.
Conclusions
In summary, well-dispersed noble metal nanoparticles (Rh and Pt) and mixed metal nanoparticles (RhPt) with an average size of about 2.5 nm on carbon were synthesized. Their catalytic activity for the HER and OER was investigated in alkaline media. It was found that all the prepared nanocomposites displayed superior HER performance than commercial Pt/C. Our results emphasis that the Rh(nP)/nC displays the lowest overpotential and smallest Tafel slope, and thus it is an excellent catalyst for this reaction. Furthermore, Rh(nP)/nC also exhibited a comparable activity for the OER to the commercial RuO2, which is one of the best catalytic materials known for this reaction. The superior performance of this catalyst can also be preserved when the active materials are mixed, either chemically or physically, with the respective platinum analogue. This is an important asset in a commercial context.The effective catalytic HER and OER performance can be attributed to a good combination between carbon support and nanoparticles The obtained carbon supported noble nanoparticles are excellent and promising electrocatalysts for alkaline water splitting, and further improvements on the preparation of the electrode will allow to exploit the full potential of these catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. H. and A. P. thank the French ANR (EdgeFiller project). F. H. thanks CNRS (Momentum grant). We thank Ahmed Bentaleb for XRD measurements and M. Gayot and C. Labrugère Sarroste, PLACAMAT, for HR-TEM and XPS measurements respectively. M. L. thanks Chinese Scholarship Council No. 201709370059 for funding support. M. L., M. M., G. V. and F. P. thank the Italian Ministero dell'Istruzione, Università e Ricerca, University of Bologna.References
- J. Wang, F. Xu, H. Jin, Y. Chen and Y. Wang, Adv. Mater., 2017, 29, 1605838 CrossRef
.
- M. E. G. Lyons and S. Floquet, Phys. Chem. Chem. Phys., 2011, 13, 5314–5335 RSC
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. L. Stephens, M. Risch, Z. J. Xu and Y. Shao-Horn, Adv. Mater., 2019, 31, 1806296 CrossRef
- B. You and Y. Sun, Acc. Chem. Res., 2018, 51, 1571–1580 CrossRef CAS
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC
- W. Sheng, H. A. Gasteiger and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1529–B1536 CrossRef CAS
- A. C. Garcia and M. T. M. Koper, ACS Catal., 2018, 8, 9359–9363 CrossRef CAS
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC
- G. Valenti, A. Boni, M. Melchionna, M. Cargnello, L. Nasi, G. Bertoni, R. J. Gorte, M. Marcaccio, S. Rapino, M. Bonchio, P. Fornasiero, M. Prato and F. Paolucci, Nat. Commun., 2016, 7, 13549 CrossRef CAS
- H. Duan, D. Li, Y. Tang, Y. He, S. Ji, R. Wang, H. Lv, P. P. Lopes, A. P. Paulikas, H. Li, S. X. Mao, C. Wang, N. M. Markovic, J. Li, V. R. Stamenkovic and Y. Li, J. Am. Chem. Soc., 2017, 139, 5494–5502 CrossRef CAS
- M. B. Chambers, X. Wang, N. Elgrishi, C. H. Hendon, A. Walsh, J. Bonnefoy, J. Canivet, E. A. Quadrelli, D. Farrusseng, C. Mellot-Draznieks and M. Fontecave, ChemSusChem, 2015, 8, 603–608 CrossRef CAS
- L. Zhu, H. Lin, Y. Li, F. Liao, Y. Lifshitz, M. Sheng, S.-T. Lee and M. Shao, Nat. Commun., 2016, 7, 12272 CrossRef CAS
- D. A. J. M. Ligthart, R. A. van Santen and E. J. M. Hensen, Angew. Chem., Int. Ed., 2011, 50, 5306–5310 CrossRef CAS
- V. Rico-Pérez, S. Parres-Esclapez, M. J. Illán-Gómez, C. S.-M. de Lecea and A. Bueno-López, Appl. Catal., B, 2011, 107, 18–25 CrossRef
- N. Zhang, Q. Shao, Y. Pi, J. Guo and X. Huang, Chem. Mater., 2017, 29, 5009–5015 CrossRef CAS
- J. Bai, S.-H. Han, R.-L. Peng, J.-H. Zeng, J.-X. Jiang and Y. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 17195–17200 CrossRef CAS
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K.-Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS
- Y. Zhu, H. C. Chen, C. S. Hsu, T. S. Lin, C. J. Chang, S. C. Chang, L. D. Tsai and H. M. Chen, ACS Energy Lett., 2019, 4, 987–994 CrossRef CAS
- S. F. Hung, Y. Chan, C. C. Chang, M. K. Tsai, Y. F. Liao, N. Hiraoka, C. S. Hsu and H. M. Chen, J. Am. Chem. Soc., 2018, 140, 17263–17270 CrossRef CAS
- Y. Zhu, J. Wang, H. Chu, Y. C. Chu and H. M. Chen, ACS Energy Lett., 2020, 5, 1281–1291 CrossRef CAS
- U. Narula, C. M. Tan and E. S. Tok, Adv. Mater. Interfaces, 2018, 5, 1800270 CrossRef
- S. F. Hung, Y. Zhu, G. Q. Tzeng, H. C. Chen, C. S. Hsu, Y. F. Liao, H. Ishii, N. Hiraoka and H. M. Chen, ACS Energy Lett., 2019, 4, 2813–2820 CrossRef CAS
- H. C. Chen, T. L. Chen, S. C. Lin, C. S. Hsu, T. S. Chan, M. Y. Liao and H. M. Chen, ChemCatChem, 2020, 12, 1926–1933 CrossRef CAS
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS
- N. Cheng, S. Stambula, D. Wang, M. N. Banis, J. Liu, A. Riese, B. Xiao, R. Li, T.-K. Sham, L.-M. Liu, G. A. Botton and X. Sun, Nat. Commun., 2016, 7, 13638 CrossRef CAS
- Q. Wang, M. Ming, S. Niu, Y. Zhang, G. Fan and J.-S. Hu, Adv. Energy Mater., 2018, 8, 1801698 CrossRef
- W. Zhang, X. Zhang, L. Chen, J. Dai, Y. Ding, L. Ji, J. Zhao, M. Yan, F. Yang, C.-R. Chang and S. Guo, ACS Catal., 2018, 8, 8092–8099 CrossRef CAS
- C. Vallés, C. Drummond, H. Saadaoui, C. A. Furtado, M. He, O. Roubeau, L. Ortolani, M. Monthioux and A. Pénicaud, J. Am. Chem. Soc., 2008, 130, 15802–15804 CrossRef
- F. Hof and A. Pénicaud, Chem. – Eur. J., 2018, 24, 16246–16250 CrossRef CAS
- F. Hof, A. Boni, G. Valenti, K. Huang, F. Paolucci and A. Pénicaud, Chem. – Eur. J., 2017, 23, 15283–15288 CrossRef CAS
- F. Hof, M. Liu, G. Valenti, E. Picheau, F. Paolucci and A. Pénicaud, J. Phys. Chem. C, 2019, 123, 20774–20780 CrossRef CAS
- F. Hof, K. Kampioti, K. Huang, C. Jaillet, A. Derré, P. Poulin, H. Yusof, T. White, K. Koziol, C. Paukner and A. Pénicaud, Carbon, 2017, 111, 142–149 CrossRef CAS
- L. Gustavo Cançado, M. Gomes da Silva, E. H. Martins Ferreira, F. Hof, K. Kampioti, K. Huang, A. Pénicaud, C. Alberto Achete, R. B. Capaz and A. Jorio, 2D Mater., 2017, 4, 025039 CrossRef
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K.-C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS
- J. Greeley, J. K. Nørskov, L. A. Kibler, A. M. El-Aziz and D. M. Kolb, ChemPhysChem, 2006, 7, 1032–1035 CrossRef CAS
- J. Chattopadhyay, S. Chakraborty, A. Mukherjee, R. Wang, P. S. Engel and W. E. Billups, J. Phys. Chem. C, 2007, 111, 17928–17932 CrossRef CAS
- D. Fino, S. Bensaid, M. Piumetti and N. Russo, Appl. Catal., A, 2016, 509, 75–96 CrossRef CAS
- T. J. P. Hersbach, R. Kortlever, M. Lehtimäki, P. Krtil and M. T. M. Koper, Phys. Chem. Chem. Phys., 2017, 19, 10301–10308 RSC
- R. Kas, R. Kortlever, A. Milbrat, M. T. M. Koper, G. Mul and J. Baltrusaitis, Phys. Chem. Chem. Phys., 2014, 16, 12194–12201 RSC
- J. Juodkazytė and K. Juodkazis, Electroanal. Int. J. Devoted Fundam. Pract. Asp. Electroanal., 2004, 16, 1622–1627 Search PubMed
- C. M. Zalitis, A. R. Kucernak, J. Sharman and E. Wright, J. Mater. Chem. A, 2017, 5, 23328–23338 RSC
- E. Verlato, S. Barison, Y. Einaga, S. Fasolin, M. Musiani, L. Nasi, K. Natsui, F. Paolucci and G. Valenti, J. Mater. Chem. A, 2019, 7, 17896–17905 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Scheme S1, Tables S1 and Fig. S1–S13. See DOI: 10.1039/d0nr05659f |
| This journal is © The Royal Society of Chemistry 2020 |