
Zeolite-confined carbon dots: tuning thermally activated delayed fluorescence emission via energy transfer†
Hongyue
Zhang
 ab,
Jiancong
Liu
c,
Bolun
Wang
a,
Kaikai
Liu
a,
Guangrui
Chen
a,
Xiaowei
Yu
a,
Jiyang
Li
*a and
Jihong
Yu
*ab
ab,
Jiancong
Liu
c,
Bolun
Wang
a,
Kaikai
Liu
a,
Guangrui
Chen
a,
Xiaowei
Yu
a,
Jiyang
Li
*a and
Jihong
Yu
*ab
aState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, Changchun 130012, P. R. China. E-mail: lijiyang@jlu.edu.cn; jihong@jlu.edu.cn
bInternational Center of Future Science, Jilin University, 2699 Qianjin Street, Changchun 130012, P. R. China
cKey Laboratory of Functional Inorganic Material Chemistry, Ministry of Education of the People's Republic of China, Heilongjiang University, Harbin 150080, China
First published on 2nd October 2019
Abstract
Confining carbon dots (CDs) in zeolites may boost the thermally activated delayed fluorescence (TADF) emission. However, so far only blue-emissive CD-based TADF materials are reported, and rationally tuning the TADF emission of these materials remains challenging. Herein, by in situ embedding different emissive CDs into a zeolite matrix, we have successfully prepared a series of CD@zeolite composites with tunable TADF emission from blue to green by utilizing energy transfer (ET) between confined CDs. The as-prepared CD@zeolite composites exhibit lifetimes varying from 271 ms to 860 ms and quantum yields ranging from 20% to 42%. The ET process occurs from the singlet excited state of the blue emissive CD donor to the singlet excited state of green emissive CD acceptors with different polymerization degrees, promoting tunable TADF with cyan, mint green and olive green emission. The design concept proposed in this work may open a way to judicious tuning of the emission of TADF materials, and thus may broaden the applications of CD-based TADF materials.
Introduction
Thermally activated delayed fluorescence (TADF) materials have the ability to harvest triplet excitons and facilitate the reverse intersystem crossing (T1 → S1) process, and have been inspiring intensive research for their potential applications in the optoelectronics, molecular imaging, and security fields.1–5 To date, the reported TADF materials are mainly based on metal organic complexes (e.g., Cu(I), Ag(I), and Au(I) complexes)6,7 and organic molecules (e.g., single-component organics, fullerenes and organic donor–acceptor molecules).8,9 Most of these materials have limited TADF lifetimes within microseconds to milliseconds, except one organic host–guest system was reported to possess the longest TADF lifetime over 1 h through photo-induced charge separation and recombination.4 However, the oxygen and temperature sensitivity, as well as complex fabrication processes, may restrict the practical application of these organic TADF materials. Therefore, it is highly desirable to develop environmentally stable, easy to synthesize and long-lasting TADF materials. In recent years, a new class of carbon dot (CD)-based afterglow materials have been prepared by embedding CDs into various matrices such as polyvinyl alcohol,10,11 potash alum,12 porous materials,13–15 and other matrices.16–19 In such composite materials, the host matrices can form hydrogen bonds with confined CDs, thus stabilizing the triplet states of CDs to achieve long-lifetime afterglow emission. Based on this design principle, several CD-based composites with room temperature phosphorescence (RTP) emission have been prepared. Compared with CD-based RTP materials, CD-based TADF materials usually show a shorter lifetime but higher quantum efficiency due to the reduction of thermal dissipation caused by non-radiative transitions during the long afterglow emission. However, so far CD-based TADF materials remain rarely reported. Our group developed a “dots-in-zeolite” strategy to prepare a series of blue emissive CD-based TADF materials with ultra-long lifetimes.14 In addition, we found that through varying the carbon precursors in the synthesis, the energy gap between S1 and T1 of the CDs can be regulated, thus achieving the modulation of the TADF and RTP behaviors of the composites.20 However, the fabrication of CD-based composite materials with tunable TADF emission remains a challenge. Notice that energy transfer (ET) from the donor to the acceptor has been generally utilized to improve the energy storage capacity and lower the energy gap of the acceptor in constructing organic afterglow materials and metal organic TADF complexes.21–23 For instance, Yan's group reported a TADF-assisted ET route between two small organic molecules to prolong the RTP lifetime to 2 s.24 More recently, our group prepared a red RTP CD-based composite based on ET between CDs and a Mn-doped zeolite matrix.25 Inspired by these studies, we hypothesize that the ET process might be also feasible between two kinds of CDs with matched energy levels in a zeolite matrix, thus affording the tuning of the photoluminescence properties.Herein, we demonstrate an ET-assisted strategy to prepare CD-based TADF materials with tunable emission by confining different emitting CDs serving as donor–acceptor into a zeolite by a one-step hydrothermal synthesis. The as-made CD@zeolite composites exhibit tunable TADF emission colors from blue to green, variable lifetimes from 271 ms to 860 ms and quantum yields from 20% to 42%. Such composites show potential applications in multicolour display and time-dependent security protection.
Results and discussion
Synthesis and characterization of CD@MgAPO-5 composites
The synthetic process of the CD@MgAPO-5 composite is illustrated in Scheme 1. Two different kinds of CD precursors were simultaneously carbonized into CDs and embedded into a zeolite matrix, giving CD-based composites with modulated TADF emission. The blue emissive TADF composite CD1@MgAPO-5 was synthesized by using 4,7,10-trioxa-1,13-tridecanediamine (TTDDA) as the template and carbon precursor of CD1 according to our previous work.14 Meanwhile, another kind of green emissive CD, CD2, was synthesized by adding m-phenylenediamine (mPD)26 into the CD1@MgAPO-5 composite. The composites were prepared in the hydrothermal reaction system of Al(OiPr)3–MgHPO4·3H2O–H3PO4–TTDDA–xmPD–H2O at 180 °C for 3 days, during which TTDDA and mPD would carbonize into different kinds of CDs and were in situ confined into the zeolite matrix. By changing the initial molar ratio of mPD and TTDDA to 0, 0.007, 0.014 and 0.042, respectively, a series of CD-based composites named CD1@MgAPO-5, CD1CD2@MgAPO-5-a, CD1CD2@MgAPO-5-b and CD1CD2@MgAPO-5-c were prepared. The scanning electron microscopy (SEM) images of CD1@MgAPO-5, CD1CD2@MgAPO-5-a, CD1CD2@MgAPO-5-b and CD1CD2@MgAPO-5-c all display a similar polyhedral morphology (Fig. 1a, d and Fig. S1, ESI†), and their powder X-ray diffraction (PXRD) patterns reveal that they all have the AFI zeolite framework structure (Fig. S2, ESI†). The fluorescence microscopy images show that CD1@MgAPO-5 emits blue light, while CD1CD2@MgAPO-5-c emits green light under excitation with UV light (Fig. 1b and e). The transmission electron microscopy (TEM) images reveal that the CDs are all well-dispersed in the crystalline MgAPO-5 matrices (Fig. 1c, f and Fig. S3, ESI†). The TEM images of the CD1@MgAPO-5 and CD1CD2@MgAPO-5-c mother liquids show that the CDs have the same lattice spacing of 0.21 nm (Fig. 1c and f inset), but different average particle diameters, in which the CDs in CD1@MgAPO-5 (3.33 nm) have a larger diameter than that of CD1CD2@MgAPO-5-c (2.57 nm) (Fig. S4, ESI†).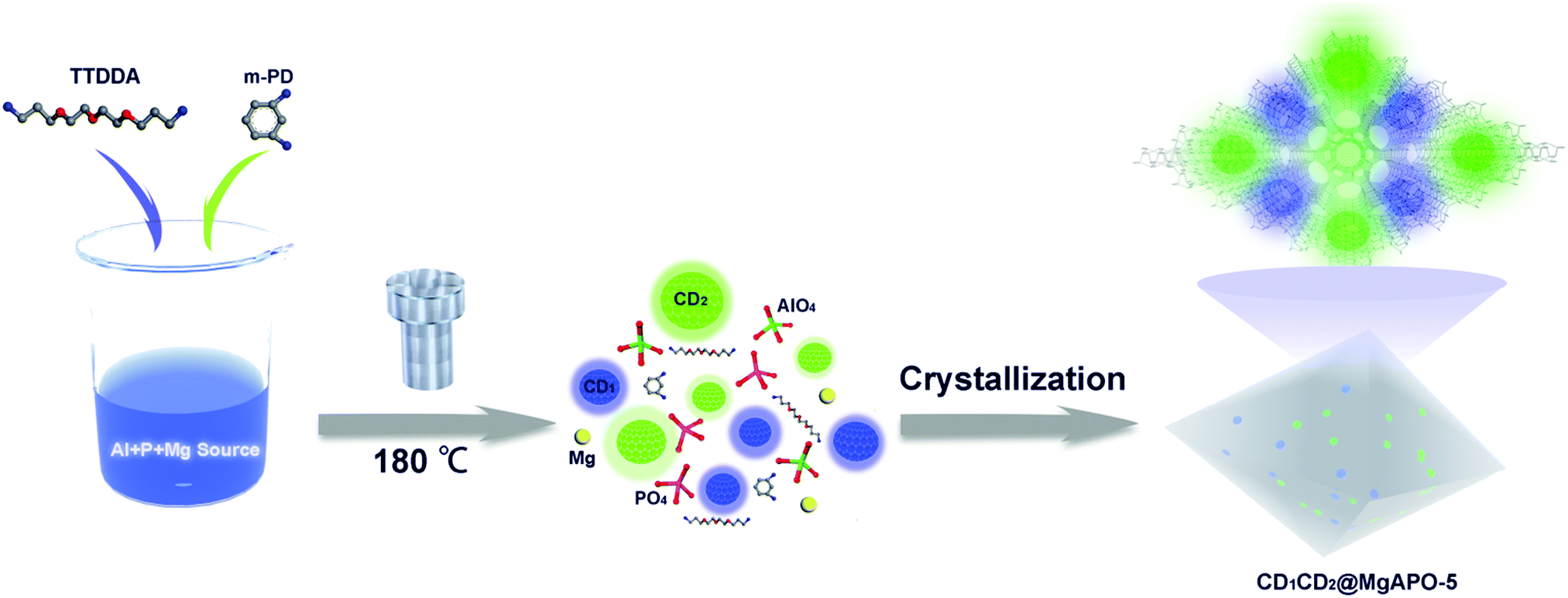 | ||
| Scheme 1 Formation process of the CD1CD2@MgAPO-5 composites. The reaction mixture is composed of an Al source, P source and Mg source for synthetic MgAPO-5; TTDDA acted as a template as well as the precursor of CD1, and mPD as the precursor of CD2. Under hydrothermal conditions, TTDDA and mPD are carbonized into blue emissive CD1 and green emissive CD2, respectively; meanwhile, the MgAPO-5 zeolite framework is crystallized. As a result, the two kinds of CDs are in situ embedded inside the zeolite matrix. | ||
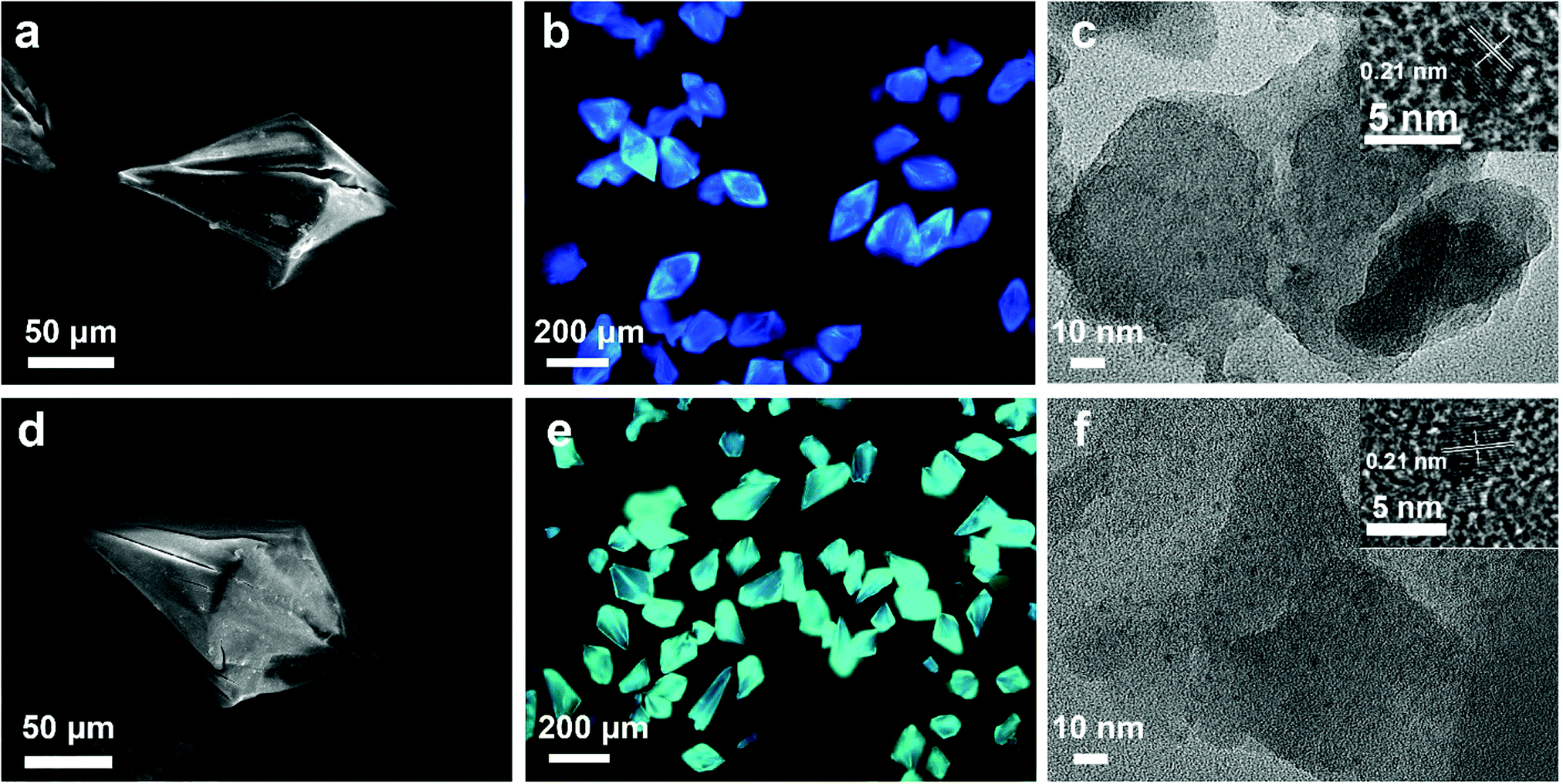 | ||
| Fig. 1 (a and d) SEM images of the CD1@MgAPO-5 and CD1CD2@MgAPO-5-c composites, respectively. (b and e) Fluorescence microscopy images of CD1@MgAPO-5 and CD1CD2@MgAPO-5-c upon UV light excitation, respectively. (c and f) TEM images of the CD1@MgAPO-5 and CD1CD2@MgAPO-5-c composites, respectively (inset, HRTEM images of CDs from the mother liquid with a lattice spacing of 0.21 nm). | ||
Photoluminescence properties
The as-synthesized composites show different photoluminescence behaviors. As shown in Fig. 2a, CD1@MgAPO-5 shows blue emission at around 425 nm when excited by 360 nm UV light. For the series of CD1CD2@MgAPO-5 composites, with the increasing addition of mPD, they present fluorescence emission varying from cyan and mint green to olive green (i.e., CD1CD2@MgAPO-5-a: 484 nm, CD1CD2@MgAPO-5-b: 498 nm, and CD1CD2@MgAPO-5-c: 515 nm). The corresponding CIE coordinates of these CD-based composites vary from (0.17, 0.12) to (0.29, 0.48) (Fig. S5, ESI†).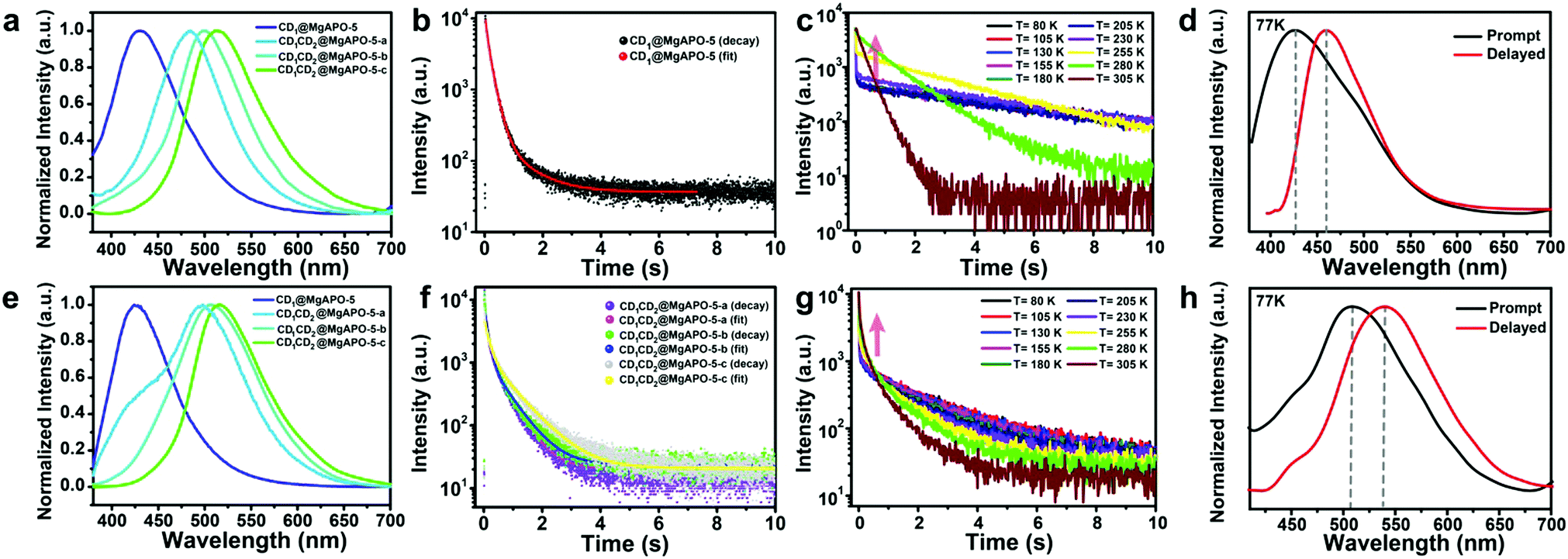 | ||
| Fig. 2 Photoluminescence (PL) spectra of the CD-based composites. (a) The normalized steady-state PL spectra of the CD-based composites excited by 360 nm UV light. (b) The time-resolved decay spectra of CD1@MgAPO-5 at room temperature. (c) Temperature-dependent time-resolved decay spectra of CD1@MgAPO-5 monitored at 425 nm emission. (d) The normalized steady-state (black line) and delayed (red line) PL spectra of CD1@MgAPO-5 excited by λ = 360 nm at 77 K. (e) The normalized delayed PL spectra of the CD-based composites at room temperature and in an ambient atmosphere. (f) The time-resolved decay spectra of the CD-based composites at room temperature. (g) Temperature-dependent time-resolved decay spectra of CD1CD2@MgAPO-5-c monitored at 515 nm emission. (h) The normalized steady-state (black line) and delayed (red line) photoluminescence spectra of CD1CD2@MgAPO-5-c excited by λ = 360 nm at 77 K. | ||
Remarkably, these CD-based composites all show long-lifetime photoluminescence when the excitation source is turned off (Fig. 2b and f). The emission bands of the delayed fluorescence (Fig. 2e) are almost consistent with those of the prompt fluorescence (Fig. 2a). In detail, the emission bands of the delayed fluorescence are centered at 425 nm for CD1@MgAPO-5, 497 nm for CD1CD2@MgAPO-5-a, 505 nm for CD1CD2@MgAPO-5-b and 515 nm for CD1CD2@MgAPO-5-c. According to the previous report, the CD1@MgAPO-5 composite is a typical TADF material.14 In order to investigate the species of delayed fluorescence in the composites after adding CD2, the lifetime of delayed fluorescence at 515 nm for the CD1CD2@MgAPO-5-c composite was monitored when the temperature was increased from 80 K to 305 K (Fig. 2g). As the temperature rises, the long-lifetime components increase (Table S1, ESI†), being similar to that of the CD1@MgAPO-5 composite (Fig. 2c and Table S2, ESI†), which is characteristic of thermally activated delayed fluorescence.27,28 In addition, CD1CD2@MgAPO-5-a and CD1CD2@MgAPO-5-b also show similar phenomena of increased long-lifetime components with increased temperature (Fig. S6, ESI†). Considering a slight red shift in the delayed spectra compared with the prompt spectra of CD1CD2@MgAPO-5-a and CD1CD2@MgAPO-5-b, we could not exclude the co-existence of a minor component of phosphorescence emission with the TADF emission because of the broad emission peak of the delayed spectra. Therefore, we suppose that these CD1CD2@MgAPO-5 composite materials should be categorized as TADF materials. Furthermore, the energy gap (ΔEST) between the excited singlet state (S1) and the excited triplet state (T1) is also an important criterion for TADF materials. The S1 and T1 energy levels of CD1CD2@MgAPO-5-c are determined using the prompt and delayed spectra at 77 K. ΔEST of CD1CD2@MgAPO-5-c is 0.14 eV, which is lower than that of CD1@MgAPO-5 (0.20 eV) (Fig. 2h and d). This suggests that the CD1CD2@MgAPO-5-c composite has a small energy gap for facilitating the electron flow through intersystem crossing (ISC) and reverse intersystem crossing (RISC) between S1 and T1 to achieve the TADF phenomenon.29,30 Moreover, through varying the addition amount of mPD, the quantum yields and TADF lifetimes of the composites can be modulated from 20% to 42%, and 271 ms to 860 ms at λex = 360 nm, respectively (Tables S3 and S4, ESI†).
To gain insight into the photoluminescence mechanism of these CD-based composites, the structures of the CDs in the composites were studied. The mother liquid of CD1CD2@MgAPO-5-c was separated by thin layer silica chromatography. Unexpectedly, several components of the CDs with multi-emissive properties were obtained, named CD1 (λem = 435 nm), CD2-a (λem = 487 nm), CD2-b (λem = 503 nm) and CD2-c (λem = 515 nm) (Fig. S7, ESI†). According to the previous reports, CD1 originates from the precursor of TTDDA, while CD2 with three components originates from organic amine mPD. X-ray photoelectron spectroscopy (XPS) spectra show that the three kinds of CD2 all have more pyrrolic N and pyridinic N, but a little amino N compared with CD1 (Fig. S8, ESI†). Specifically, the C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/CN components increase and the C–O component decreases from CD2-a to CD2-c. It may be caused by the different polymerization degrees resulting from different amounts of the added mPD in the synthesis, which gives different functional groups on the resulting CD2. The different surface groups of CD2-a to CD2-c lead to the distinct fluorescence emission. Note that the fluorescence spectra of CD2-a, CD2-b and CD2-c are almost consistent with those of the CD1CD2@MgAPO-5-a, CD1CD2@MgAPO-5-b and CD1CD2@MgAPO-5-c composites, respectively, suggesting CD2-a, CD2-b and CD2-c are the respective dominant CD2 components in the corresponding composites, although they may co-exist. The XPS results of the composites show that the C–N (especially for pyrrolic N and pyridinic N) and CO/CN components increase and the C–O component decreases from CD1CD2@MgAPO-5-a to CD1CD2@MgAPO-5-c, being consistent with the above XPS analysis results of separated CD2-a, CD2-b and CD2-c (Fig. S9, ESI†).
O/CN components increase and the C–O component decreases from CD2-a to CD2-c. It may be caused by the different polymerization degrees resulting from different amounts of the added mPD in the synthesis, which gives different functional groups on the resulting CD2. The different surface groups of CD2-a to CD2-c lead to the distinct fluorescence emission. Note that the fluorescence spectra of CD2-a, CD2-b and CD2-c are almost consistent with those of the CD1CD2@MgAPO-5-a, CD1CD2@MgAPO-5-b and CD1CD2@MgAPO-5-c composites, respectively, suggesting CD2-a, CD2-b and CD2-c are the respective dominant CD2 components in the corresponding composites, although they may co-exist. The XPS results of the composites show that the C–N (especially for pyrrolic N and pyridinic N) and CO/CN components increase and the C–O component decreases from CD1CD2@MgAPO-5-a to CD1CD2@MgAPO-5-c, being consistent with the above XPS analysis results of separated CD2-a, CD2-b and CD2-c (Fig. S9, ESI†).
The unique confinement effect of the zeolite matrix was further confirmed by the spectra of CD solutions. By taking CD1 and CD2-c as examples, Fig. S10 (ESI†) shows their emission spectra under the excitation of 360 nm UV light. When changing the excitation wavelength from 320 nm to 480 nm, CD1 emits excitation-dependent luminescence varying from 400 to 500 nm, with the strongest emission centered at around 435 nm (excited at 360 nm), while CD2-c emits excitation-independent luminescence centered at around 515 nm and the strongest emission is achieved upon excitation at 460 nm. The lifetimes of CD1 and CD2 aqueous solution are 6.68 ns and 9.45 ns, respectively (Fig. S11 and Table S5, ESI†). Long-lifetime emission was not observed for both CD1 and CD2-c when the UV light was turned off. This phenomenon implies that the zeolitic matrix can effectively stabilize the triplet excited state of the CDs and restrict the non-radiation dissipated by rotation and vibration to realize long-lifetime emission. The CD1CD2@MgAPO-5-c composite also shows strong photoluminescence (PL) stability under UV radiation or in air. When being irradiated under UV light or ground in air for 8 hours, it retains a PL intensity of 80% (Fig. S12, ESI†). Moreover, the CD@MgAlPO-5 composites are not sensitive to oxygen, and their TADF lifetimes are similar under air and N2 conditions (Fig. S13, ESI†). This high photo-stability and air-stability of the CD1CD2@MgAPO-5-c composite are superior to most organic TADF materials,31–33 which may benefit from the stabilization effect of the zeolite matrix.
Energy transfer process
Comparing the fluorescence spectra and the delayed fluorescence spectra of these four composites, we found that with the increasing addition of mPD in the synthesis, the blue emission from CD1 becomes weak and almost vanishes in the CD1CD2@MgAPO-5-c composite (Fig. S14, ESI†). Considering the existence of multiple CD species in the composites, we suppose that there may exist an energy transfer (ET) process between CD1 (from template TTDDA) and CD2 (from introduced mPD) in these composites. The absorption bands of the three types of CD2 all have large spectral overlap integrals with the emission band of CD1 at 400–470 nm (Fig. 3a), suggesting that ET may occur between the donor CD1 and the acceptor CD2 with three different components. The increasing overlap indicates a more efficient ET process from CD1CD2@MgAPO-5-a to CD1CD2@MgAPO-5-c. Moreover, the zeolite matrix provides a suitable space that makes the D(CD1)–A(CD2) pairs near enough to achieve the ET process according to Dexter's theory of energy transfer.34–36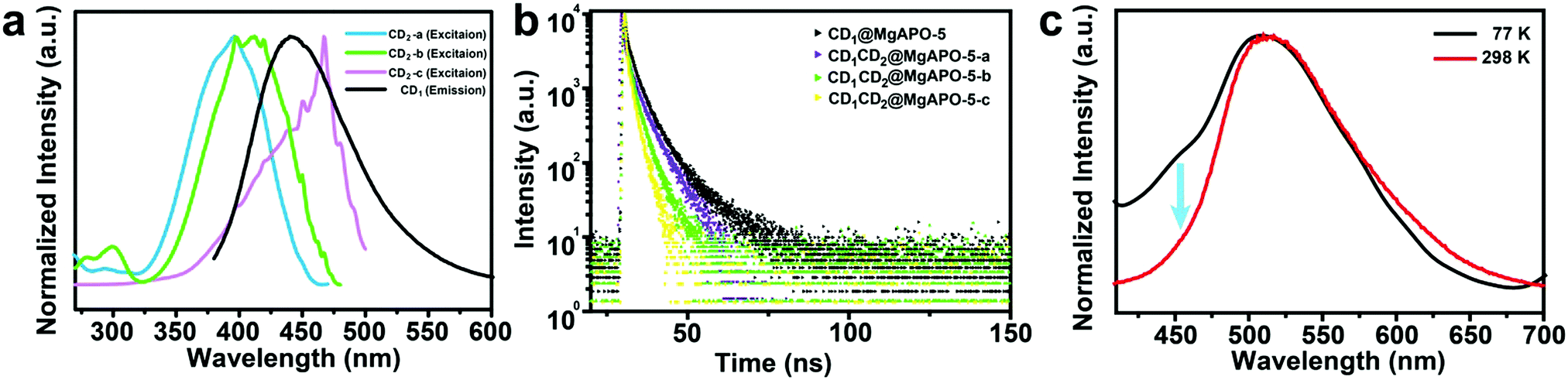 | ||
| Fig. 3 (a) The excitation spectra of CD2 and emission spectrum of CD1. (b) Time-resolved decay spectra of the CD@MgAPO-5 composites monitored at λem = 425 nm. (c) The photoluminescence spectra of the CD1CD2@MgAPO-5-c composite measured at 298 K (red line) and 77 K (black line). | ||
Furthermore, the prompt fluorescence lifetimes of these CD-based composites were also detected (Fig. 3b). Compared with CD1@MgAPO-5, the lifetime values of the emission band at 425 nm for CD1CD2@MgAPO-5-a, CD1CD2@MgAPO-5-b and CD1CD2@MgAPO-5-c decrease greatly at λex = 360 nm, which indicates that the effective ET originated from the singlet states of CD1 and CD2. Thus, the possible ET mechanism for these composites is proposed as follows: CD2 acceptors with different emission and energy levels were produced when different amounts of mPD were added in the synthesis. CD2 could efficiently harvest singlet excitons of the donor CD1, while the stabilized triplet states and small energy gap (ΔEST) of CD2 make possible ISC and RISC between the S1 and T1 states, leading to enhanced TADF emission in CD2 (Fig. 4a). In addition, the photoluminescence spectrum of CD1CD2@MgAPO-5-c at 77 K shows an additional blue emission peak located around 450 nm, which is quite weak at room temperature (Fig. 3c). This indicates that the ET process between different CDs is suppressed at low temperature, which is a typical feature for energy transfer.37,38
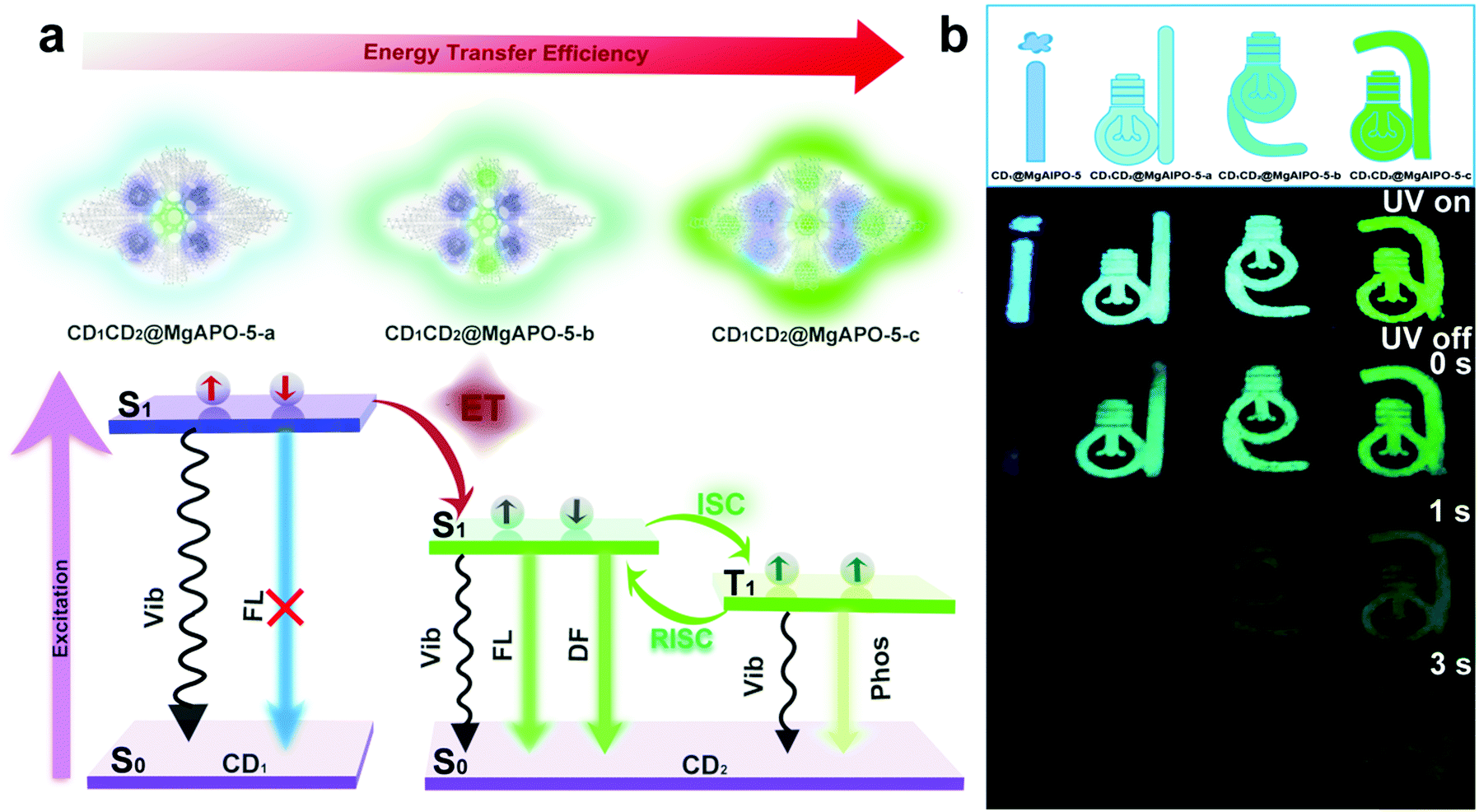 | ||
| Fig. 4 (a) Proposed ET mechanism of different CDs in the composite. (b) Multicolour display and time-dependent security protection of the CD-based composites. | ||
The ET efficiency of these CD-based composites can be quantified according to the equation η = 1 − τDA/τD, in which τDA and τD represent the PL lifetimes of the energy donor in the presence and absence of the energy acceptor, respectively.39–42 Based on this, the ET efficiencies from CD1CD2@MgAPO-5-a to CD1CD2@MgAPO-5-c are calculated as 43.72%, 53.10% and 73.0%, respectively. This result manifests that the ET efficiency increases through enhancing the polymerization degree of the introduced CD2 in the zeolite matrix, which is consistent with the above-mentioned increasing overlap between the absorption peaks of CD2 and emission peak of CD1 from CD1CD2@MgAPO-5-a to CD1CD2@MgAPO-5-c (Fig. 3a). The high ET efficiency may facilitate the increasing TADF lifetimes and QYs of these composites. It should be pointed out that although our previous work has demonstrated ET between CDs and a Mn-doped zeolite matrix,25 ET between different types of CDs in a matrix is presented for the first time in this CD-based system.
Application: multicolour display and time-dependent security protection
On the basis of the modulated TADF lifetimes and emission colors of the as-synthesized CD-based composites, we demonstrate their potential applications in multicolour display and time-dependent security protection. An encryption algorithm was designed with three modes of operation: “UV on”, “UV off (instantly)”, and “UV off (for 1 s and 3 s)”. As shown in Fig. 4b, the pattern “idea” with a size of 4 cm was fabricated with the above-mentioned four kinds of CD-based composites (CD1@MgAPO-5, CD1CD2@MgAPO-5-a, CD1CD2@MgAPO-5-b and CD1CD2@MgAPO-5-c). Under excitation with 365 nm UV light, the four letters “i”, “d”, “e” and “a” are blue, cyan, mint green and olive green, respectively. When switching off the excitation source, these four letters all can be seen by the naked eye and their TADF colors are similar to their fluorescence emission under UV on. As time extends, these four letters fade away gradually due to different TADF lifetimes. In detail, after turning off the UV light for 1 s, the letters “i” and “d” are invisible, and gradually, extending to 3 s, only the letter “e” can be captured. Besides the application in security protection, it is believed that the CD@zeolite composites with long-lived TADF can serve as ideal contrast agents for future high-sensitivity bioimaging to efficiently avoid short-lived background fluorescence.Conclusion
A series of CD@zeolite TADF materials with modulated emission colors, lifetimes and QYs have been successfully prepared by embedding different kinds of CDs into a zeolite matrix via in situ hydrothermal synthesis. CD1 with blue emission has a relative high energy level, while CD2 composed of three components with emission from cyan to green has relatively low energy levels; their co-existence in the nanospace of the zeolite matrix enables energy transfer between them. The energy transfer efficiency increases with the enhanced polymerization degree of CD2 in the composite. By varying the composition of the CDs, four kinds of CD-based composites are achieved with multicolour emission from blue, cyan, and mint green to olive green, TADF lifetimes from 271 ms, 578 ms, and 801 ms to 860 ms, and QYs from 20%, 25%, and 37% to 42%. Based on the unique TADF properties, these CD-based composites can be potentially used in multicolour display and time-dependent security protection. This work provides a feasible strategy to modulate the afterglow emission of CD-based composites in terms of the ET route, which will be helpful for the design and fabrication of novel CD-based composites with desired photoluminescence properties.Experimental
Materials
All reagents were used as received without purification. Aluminum isopropoxide (98% purity) and TTDDA (97% purity) were purchased from Sigma-Aldrich. MgHPO4·3H2O and H3PO4 (85 weight% (wt%) in water) were obtained from Tianjin Fuchen Chemical Co. Ltd. M-Phenylenediamine was supplied by Beijing Chemical Reagent Factory. Deionized (DI) water was obtained from Milli-Q purification equipment.Instruments
Powder X-ray diffraction (PXRD) data were measured with a RigakuD/Max 2500 diffractometer with Cu Kα radiation (λ = 1.5418 Å). The transmission electron microscopy (TEM) images were taken on a Tecnai F20 electron microscope. Scanning electron microscopy (SEM) images were taken on a JSM-6510 (JEOL) electron microscope. Fluorescence microscopy images were taken by using an Olympus BX51 microscope under UV excitation. The XPS measurements were collected on a ThermoESCALAB250 spectrometer with monochromatized Al Kα excitation.Synthesis of the CD-based composites
The series of CD-based composites were synthesized by using TTDDA as the SDA under hydrothermal conditions. In the synthesis of CD1@MgAPO-5, aluminum isopropoxide was dispersed in H3PO4 aqueous solution. Then, MgHPO4·3H2O and TTDDA were added into the mixture with stirring. A homogeneous gel was formed with a molar composition of 1.0Al2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.1MgO:2.0P2O5:2.1TTDDA:139H2O. After crystallization at 180 °C for 3 days, the crystalline product was washed with distilled water, and dried at 80 °C in an oven. Similarly, CD1CD2@MgAPO-5 composites were synthesized by adding m-PD into the above-mentioned homogeneous gel with stirring. Through changing the molar ratio of mPD and TTDDA in the reaction gel to 0.007, 0.014 and 0.042, respectively, a series of CD1CD2@MgAPO-5 composites were produced, which were named CD1CD2@MgAPO-5-a, CD1CD2@MgAPO-5-b, and CD1CD2@MgAPO-5-c. The subsequent steps were the same as the above mentioned for the synthesis of CD1@MgAPO-5.
:0.1MgO:2.0P2O5:2.1TTDDA:139H2O. After crystallization at 180 °C for 3 days, the crystalline product was washed with distilled water, and dried at 80 °C in an oven. Similarly, CD1CD2@MgAPO-5 composites were synthesized by adding m-PD into the above-mentioned homogeneous gel with stirring. Through changing the molar ratio of mPD and TTDDA in the reaction gel to 0.007, 0.014 and 0.042, respectively, a series of CD1CD2@MgAPO-5 composites were produced, which were named CD1CD2@MgAPO-5-a, CD1CD2@MgAPO-5-b, and CD1CD2@MgAPO-5-c. The subsequent steps were the same as the above mentioned for the synthesis of CD1@MgAPO-5.
Photoluminescence measurements of the CD-based composites
Steady-state and delayed spectra were measured on a HORIBA Scientific FluoroMax-4 spectrofluorometer. The fluorescence lifetime spectra were measured using a FluoroHub-B fluorescence lifetime system equipped with a photomultiplier tube detector in a HORIBA Scientific FluoroMax-4. Ludox was selected to rectify the instrument response factor (IRF). The lifetime data were fitted by using DAS6 software with a multi-exponential model. The average lifetimes (τavg) are shown as τavg = ΣAiτi2/ΣAiτi, in which Ai is the pre-exponential for lifetime τi. Temperature-dependent transient photoluminescence decay was collected on an Edinburgh FLS920 fluorescence spectrophotometer. The fluorescence QY was collected on an Edinburgh FLS920 with an integrating sphere. To prove the accuracy of the as-measured QY, sodium salicylate solid was used as a reference. The measured QY (52.33%) was close to its standard QY of 52.0%.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful for financial support from the National Natural Science Foundation of China (Grant No. 21835002, 21621001 and 21671075), and the 111 Project of China (B17020).Notes and references
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS.
- S. Xu, R. Chen, C. Zheng and W. Huang, Adv. Mater., 2016, 28, 9920–9940 CrossRef CAS.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- R. Kabe and C. Adachi, Nature, 2017, 550, 384–387 CrossRef CAS.
- K. Y. Zhang, Q. Yu, H. Wei, S. Liu, Q. Zhao and W. Huang, Chem. Rev., 2018, 118, 1770–1839 CrossRef CAS.
- A. Endo, M. Ogasawara, A. Takahashi, D. Yokoyama, Y. Kato and C. Adachi, Adv. Mater., 2009, 21, 4802–4806 CrossRef CAS.
- Q. Zhang, Q. Zhou, Y. Cheng, L. Wang, D. Ma, X. Jing and F. Wang, Adv. Funct. Mater., 2006, 16, 1203–1208 CrossRef CAS.
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326 CrossRef CAS.
- Y. Deng, D. Zhao, X. Chen, F. Wang, H. Song and D. Shen, Chem. Commun., 2013, 49, 5751–5753 RSC.
- K. Jiang, L. Zhang, J. Lu, C. Xu, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2016, 55, 7231–7235 CrossRef CAS.
- X. Dong, L. Wei, Y. Su, Z. Li, H. Geng, C. Yang and Y. Zhang, J. Mater. Chem. C, 2015, 3, 2798–2801 RSC.
- K. Jiang, Y. Wang, C. Cai and H. Lin, Chem. Mater., 2017, 29, 4866–4873 CrossRef CAS.
- J. Liu, N. Wang, Y. Yu, Y. Yan, H. Zhang, J. Li and J. Yu, Sci. Adv., 2017, 3, e1603171 CrossRef.
- Y. Mu, H. Shi, Y. Wang, H. Ding and J. Li, J. Mater. Chem. C, 2017, 5, 10894–10899 RSC.
- Q. Li, M. Zhou, M. Yang, Q. Yang, Z. Zhang and J. Shi, Nat. Commun., 2018, 9, 734 CrossRef.
- Q. Li, M. Zhou, Q. Yang, Q. Wu, J. Shi, A. Gong and M. Yang, Chem. Mater., 2016, 28, 8221–8227 CrossRef CAS.
- J. Tan, R. Zou, J. Zhang, W. Li, L. Zhang and D. Yue, Nanoscale, 2016, 8, 4742–4747 RSC.
- Y. Chen, J. He, C. Hu, H. Zhang, B. Lei and Y. Liu, J. Mater. Chem. C, 2017, 5, 6243–6250 RSC.
- J. Liu, H. Zhang, N. Wang, Y. Yu, Y. Cui, J. Li and J. Yu, ACS Mater. Lett., 2019, 1, 58–63 CrossRef CAS.
- Q. Wei, N. Fei, A. Islam, T. Lei, L. Hong, R. Peng, X. Fan, L. Chen, P. Gao and Z. Ge, Adv. Opt. Mater., 2018, 6, 1800512 CrossRef.
- H. Tanaka, K. Shizu, H. Miyazaki and C. Adachi, Chem. Commun., 2012, 48, 11392–11394 RSC.
- Y. Gong, L. Zhao, Q. Peng, D. Fan, W. Z. Yuan, Y. Zhang and B. Z. Tang, Chem. Sci., 2015, 6, 4438–4444 RSC.
- B. Zhou and D. Yan, Adv. Funct. Mater., 2019, 29, 1807599 CrossRef.
- B. Wang, Y. Mu, H. Zhang, H. Shi, G. Chen, Y. Yu, Z. Yang, J. Li and J. Yu, ACS Cent. Sci., 2019, 5, 349–356 CrossRef CAS.
- K. Jiang, S. Sun, L. Zhang, Y. Lu, A. Wu, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2015, 54, 5360–5363 CrossRef CAS.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS.
- S. Wang, X. Yan, Z. Cheng, H. Zhang, Y. Liu and Y. Wang, Angew. Chem., Int. Ed., 2015, 54, 13068–13072 CrossRef CAS.
- Q. Zhang, H. Kuwabara, W. J. Potscavage, S. Huang, Y. Hatae, T. Shibata and C. Adachi, J. Am. Chem. Soc., 2014, 136, 18070–18081 CrossRef CAS.
- T. Hofbeck, U. Monkowius and H. Yersin, J. Am. Chem. Soc., 2015, 137, 399–404 CrossRef CAS.
- S. Hirata, Y. Sakai, K. Masui, H. Tanaka, S. Y. Lee, H. Nomura, N. Nakamura, M. Yasumatsu, H. Nakanotani, Q. Zhang, K. Shizu, H. Miyazaki and C. Adachi, Nat. Mater., 2015, 14, 330–336 CrossRef CAS.
- D. Zhang, L. Duan, Y. Zhang, M. Cai, D. Zhang and Y. Qiu, Light: Sci. Appl., 2015, 4, e232 CrossRef CAS.
- L. Bergmann, G. J. Hedley, T. Baumann, S. Bräse and I. D. W. Samuel, Sci. Adv., 2016, 2, e1500889 CrossRef.
- R. Gao and D. Yan, Chem. Sci., 2017, 8, 590–599 RSC.
- T. Ha, Methods, 2001, 25, 78–86 CrossRef CAS.
- P. Lutsyk, R. Arif, J. Hruby, A. Bukivskyi, O. Vinijchuk, M. Shandura, V. Yakubovskyi, Y. Kovtun, G. A. Rance, M. Fay, Y. Piryatinski, O. Kachkovsky, A. Verbitsky and A. Rozhin, Light: Sci. Appl., 2016, 5, e16028 CrossRef CAS.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS.
- G. D. Scholes, G. R. Fleming, A. Olaya-Castro and R. van Grondelle, Nat. Chem., 2011, 3, 763 CrossRef CAS PubMed.
- C. Vijayakumar, V. K. Praveen, K. K. Kartha and A. Ajayaghosh, Phys. Chem. Chem. Phys., 2011, 13, 4942–4949 RSC.
- P. Parkinson, C. E. I. Knappke, N. Kamonsutthipaijit, K. Sirithip, J. D. Matichak, H. L. Anderson and L. M. Herz, J. Am. Chem. Soc., 2014, 136, 8217–8220 CrossRef CAS.
- T. Senden, R. J. A. van Dijk-Moes and A. Meijerink, Light: Sci. Appl., 2018, 7, 8 CrossRef.
- P.-Z. Chen, Y.-X. Weng, L.-Y. Niu, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Angew. Chem., Int. Ed., 2016, 55, 2759–2763 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qm00549h |
| This journal is © the Partner Organisations 2020 |