
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isothiourea-catalysed enantioselective Michael addition of N-heterocyclic pronucleophiles to α,β-unsaturated aryl esters†
Chang
Shu‡
,
Honglei
Liu‡
,
Alexandra M. Z.
Slawin
 ,
Cameron
Carpenter-Warren
and
Andrew D.
Smith
*
,
Cameron
Carpenter-Warren
and
Andrew D.
Smith
*
EaStCHEM, School of Chemistry, University of St Andrews, North Haugh, St Andrews, Fife, KY16 9ST, UK. E-mail: ads10@st-andrews.ac.uk
First published on 23rd October 2019
Abstract
The isothiourea-catalysed enantioselective Michael addition of 3-aryloxindole and 4-substituted-dihydropyrazol-3-one pronucleophiles to α,β-unsaturated p-nitrophenyl esters is reported. This process generates products containing two contiguous stereocentres, one quaternary, in good yields and excellent enantioselectivities (>30 examples, up to > 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 dr and 99:1 er). This protocol harnesses the multifunctional ability of p-nitrophenoxide to promote effective catalysis. In contrast to previous methodologies using tertiary amine Lewis bases, in which the pronucleophile was used as the solvent, this work allows bespoke pronucleophiles to be used in stoichiometric quantities.
:5 dr and 99:1 er). This protocol harnesses the multifunctional ability of p-nitrophenoxide to promote effective catalysis. In contrast to previous methodologies using tertiary amine Lewis bases, in which the pronucleophile was used as the solvent, this work allows bespoke pronucleophiles to be used in stoichiometric quantities.
Introduction
Catalytic enantioselective Michael addition of enolate equivalents to α,β-unsaturated carbonyl compounds represents an efficient methodology for stereoselective C–C bond formation.1 Within this field, considerable advances in catalytic enantioselective Michael additions to enals and enones have been reported.2 Typical strategies involve activation of the Michael acceptor through iminium ion formation,3 H-bonding organocatalysis,4 or Lewis acid catalysis.5 In comparison to enals and enones, the intrinsic recalcitrance of α,β-unsaturated esters6 represents a significant challenge in enantioselective catalysis (Scheme 1a).7 Established metal-based catalytic systems allow, for example, conjugate additions of aryl boronic acids and Grignard reagents to α,β-unsaturated esters.8 Broader reactivity has been targeted through developing the use of ester surrogates such as N-acylpyrroles,9 2-acyl imidazoles,10 activated imides11 and β,γ-unsaturated acyl phosphonates.12 Catalytic strategies using these motifs typically rely upon two-point binding between the enoyl substrate and either a Lewis acidic metal catalyst13 or a H-bond donor organocatalyst.14 Despite these advances, only limited organocatalytic strategies have been developed that allow activation of α,β-unsaturated ester substrates, with the current state-of-the-art strategies having been showcased by List (silylium catalysis)15 and Dixon (BIMP catalysis).16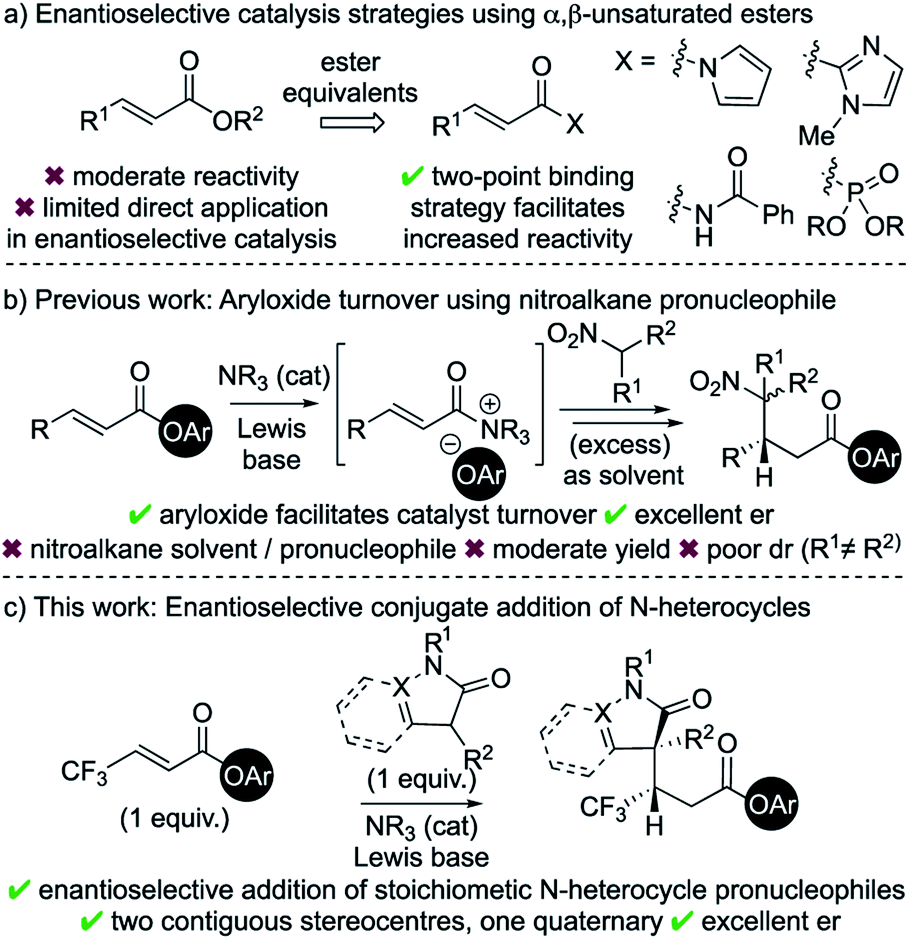 | ||
| Scheme 1 Enantioselective Michael addition to α,β-unsaturated esters. | ||
Chiral α,β-unsaturated acyl ammonium intermediates are readily prepared in situ from α,β-unsaturated acyl halides and anhydrides using tertiary amine Lewis base catalysts.17 They have been utilised as convenient and powerful synthons in a number of organocascade reactions,18 yet the Lewis base catalysed activation of α,β-unsaturated esters are rare. One major limitation within this field is the requirement of the reactive partner to contain two distinct nucleophilic functionalities to facilitate conjugate addition and subsequent catalyst turnover.18a In previous work we exploited aryloxide- facilitated catalyst turnover to show the compatibility of monofunctional nucleophilic reaction partners in this reaction manifold.19 This allowed the conjugate addition of nitronate anions to in situ generated α,β-unsaturated acyl ammonium intermediates. However, this transformation was inherently limited to the use of excess nitroalkane as both solvent and pronucleophile and was not applicable for the formation of quaternary stereogenic centres.
In the area of medicinal chemistry, the incorporation of N-heterocycles and fluorinated substituents into substrates are common strategies to improve physiochemical properties.20 In this context, the application of this aryloxide turnover strategy to incorporate these valuable motifs was targeted.21 The major challenges were to identify suitable pronucleophiles containing N-heterocycles that could (i) be used as stoichiometric reagents rather than solvent; (ii) lead to the formation of a quaternary stereocentre; and (iii) be compatible with a range of acyl ammonium precursors. In this manuscript, these challenges are met through the enantioselective conjugate addition of enolates derived from dihydropyrazol-3-ones and 3-substituted oxindoles to a range of α,β-unsaturated aryl esters, particularly those bearing a β-trifluoromethyl substituent.
Results and discussion
Initial studies probed the addition of a range of model N-heterocycle containing pronucleophiles 2–9 (1 equiv.) to β-trifluoromethyl α,β-unsaturated p-nitrophenyl (PNP) ester 1 to assess the feasibility of this process (Fig. 1). Screening showed that oxazolones and thiazolones 2–5, pyrrolidinone 6 and 3-benzyl oxindole 7 gave < 10% conversion to product using HyperBTM as the Lewis base catalyst (see ESI† for full details).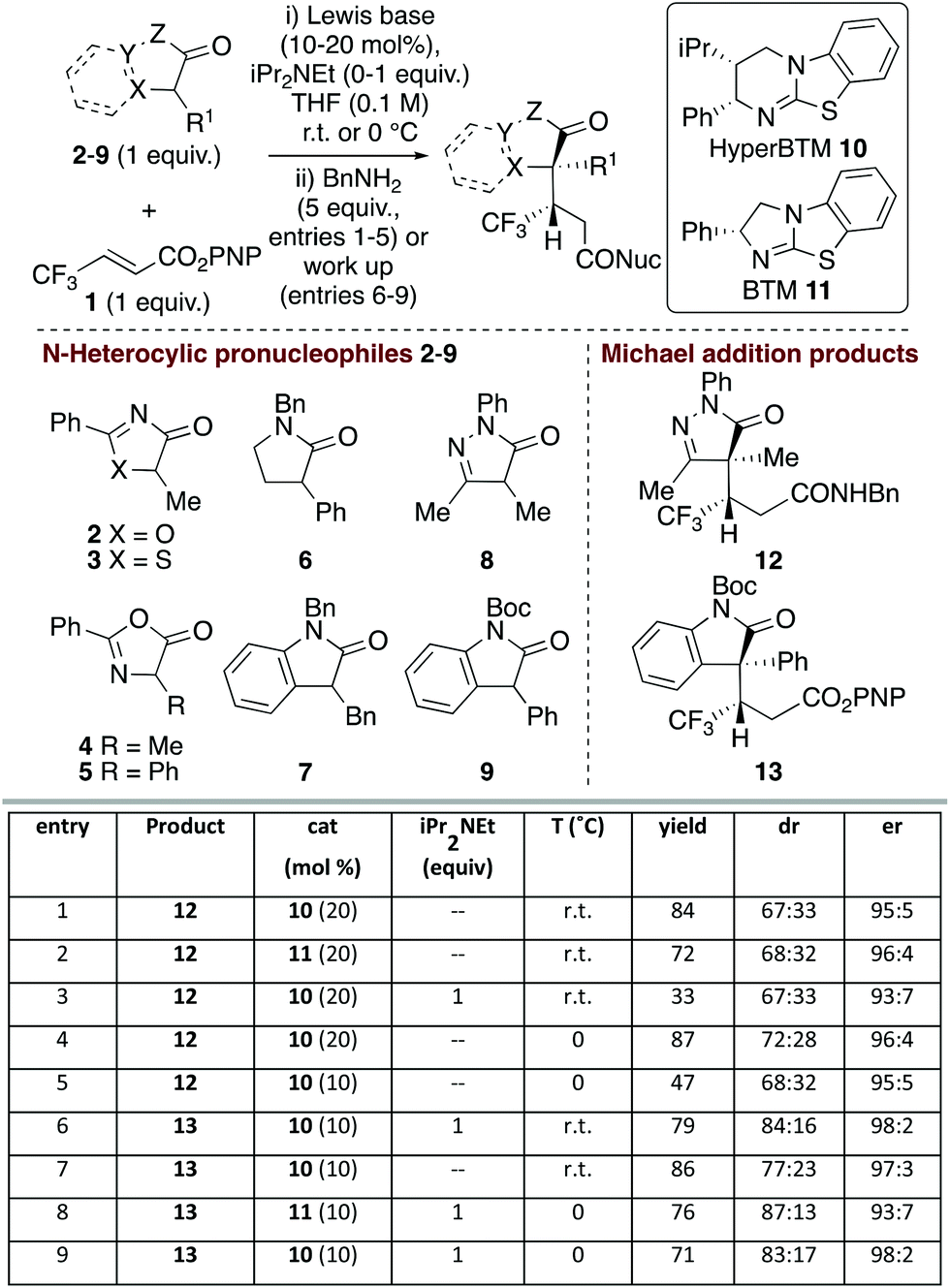 | ||
| Fig. 1 Screening of pronucleophiles. Isolated overall yields given; dr determined by 1H NMR spectroscopic analysis of crude mixture; er determined by chiral HPLC analysis of purified products and refers to er of major diastereoisomer. | ||
However dihydropyrazol-3-one 8 and 3-phenyl oxindole 9 pronucleophiles22 gave good reactivity, consistent with their expected lower pKa and associated ease of enolate formation. Using dihydropyrazol-3-one 8, followed by addition of benzylamine, gave isolable amide 12,23 with HyperBTM 10 giving marginally improved yield over BTM 11 (entries 1 and 2). Consistent with the oxidative susceptibility of dihydropyrazol-3-ones in the presence of base,24 only low product yield was observed with iPr2NEt (entry 3). Variation of solvent and temperature showed that THF at 0 °C was optimal, giving the desired amide in 72:28 dr.25 Purification of the separable diastereoisomers provided 87% overall yield, with each diastereoisomer obtained with high enantioselectivity (96:4 er) (entry 4). Attempted reduction of the catalyst loading to 10 mol% gave 12 with high enantioselectivity but markedly reduced yield (entry 5). Moving to the oxindole series, N-Boc-3-phenyl-oxindole 9 gave isolable ester 13, with iPr2NEt required for optimal diastereoselectivity (entries 6 and 7). Variation of Lewis base catalysts 10 and 11 at 0 °C showed that HyperBTM 10 was optimal, giving 13 in 71% yield, 83:17 dr and 98:2 er (entry 9).
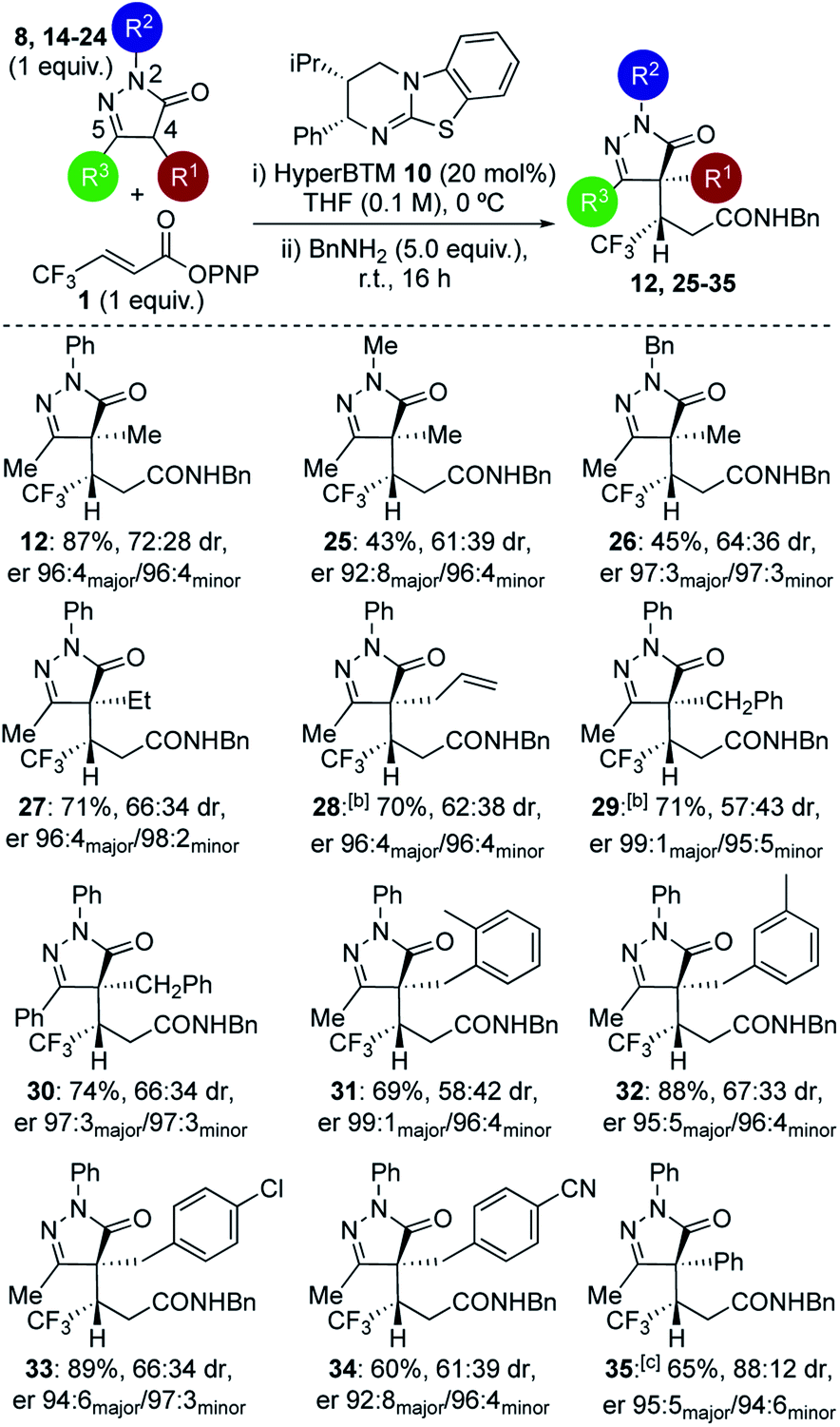
Further studies focused on structural variations of the pronucleophile. In the dihydropyrazol-3-one series, variation of the N(2)-, C(4)- or C(5)-substituents was investigated (Table 1). Variation of the N(2)-substituent showed that N-Ph gave higher product yield than N-Me or N-Bn substitution. Although moderate diastereoselectivities were observed, in each case purification allowed separation of the diastereoisomers, which were all obtained with excellent enantioselectivity (products 12, 25–26). C(4)-Ethyl and allyl variants, as well as a range of substituted C(4)-benzyl26 derivatives, were also tolerated, giving good product yields with high enantioselectivity (products 27–34). Using a C(4)-phenyl substituted pronucleophile with HyperBTM 10 gave 35 in moderate yield, while the use of (R)-BTM 11 gave improved yield and stereocontrol.
Within the 3-aryloxindole class, N-methyl substitution led to improved stereoselectivity over the N-Boc variant, giving amide 53 in 80% yield, >95:5 dr and 98:2 er (Fig. 2A). Using N-benzyl 3-phenyl oxindole led to similarly excellent stereoselectivity, giving ester 54 directly in 74% yield, >95:5 dr and 98:2 er,26 or the corresponding amides 55–57 (following addition of the appropriate amine) in excellent yield, >95:5 dr and >98:2 er in each case. Further investigation probed the scope and limitations of this transformation through subjecting a variety of substituted 3-aryloxindoles to this protocol. Notably, studies showed that 5-, 6-, and 7-substituents within the oxindole were readily tolerated (Fig. 2B). For example, 5-MeO substituted 3-phenyl oxindole gave 58 in 72% yield, 90:10 dr and 98:2 er, while 5-Cl-, 6-Cl and 7-Cl derivatives gave 59–61 respectively in excellent yield, >95:5 dr and up to 95:5 er. This protocol was extended to a range of 3-aryloxindole pronucleophiles bearing either extended π-systems, fluorinated substituents, electron-donating or electron-withdrawing substituents, as well as the heteroaryl 3-(2-thienyl) motif (Fig. 2C). Products 62–69 were isolated in good to excellent yields with 91:9 to >95:5 dr and 92:8 to >99:1 er. Either 2-substitution of the oxindole 3-aryl group (2-MeOC6H4, 2-FC6H4), or the incorporation of a strongly electron-donating 4-Me2NC6H4 substituent, led to no reactivity, presumably due to either additional steric encumberance or increased pKa of the substrate disfavouring enolate formation (Fig. 2D). Extension of this procedure to a range of β-substituted α,β-unsaturated esters was also probed, using N-benzyl-3-phenyloxindole 37 as a representative pronucleophile. Electron-withdrawing groups at the β-position were readily tolerated, with C(3)-difluoromethyl-, C(3)-chlorodifluoromethyl-, and C(3)-bromodifluoromethyl-substituted esters giving the corresponding derivatives 76–78 in good to excellent yields and high diastereo- and enantiocontrol (Fig. 2E). Significantly, both crotonic and cinnamic PNP esters also proved compatible with this methodology. In our previous work crotonic esters proved low yielding (∼20%) while cinnamic derivatives gave no reaction,19 highlighting the significant potential of this approach. For example, addition of 37 to the crotonic PNP ester gave 79 in 85% yield with promising stereocontrol (86:14 dr and 86:14 er). Addition of 37 to the cinnamate PNP ester derivative gave 80 with poor diastereocontrol but high enantioselectivity in excellent yield.27 The incorporation of a β-ester substituent gave product 81 in moderate dr but excellent overall yield and enantioenrichment, indicating that the hybridisation of the β-substituent may be significant in determining diastereocontrol.
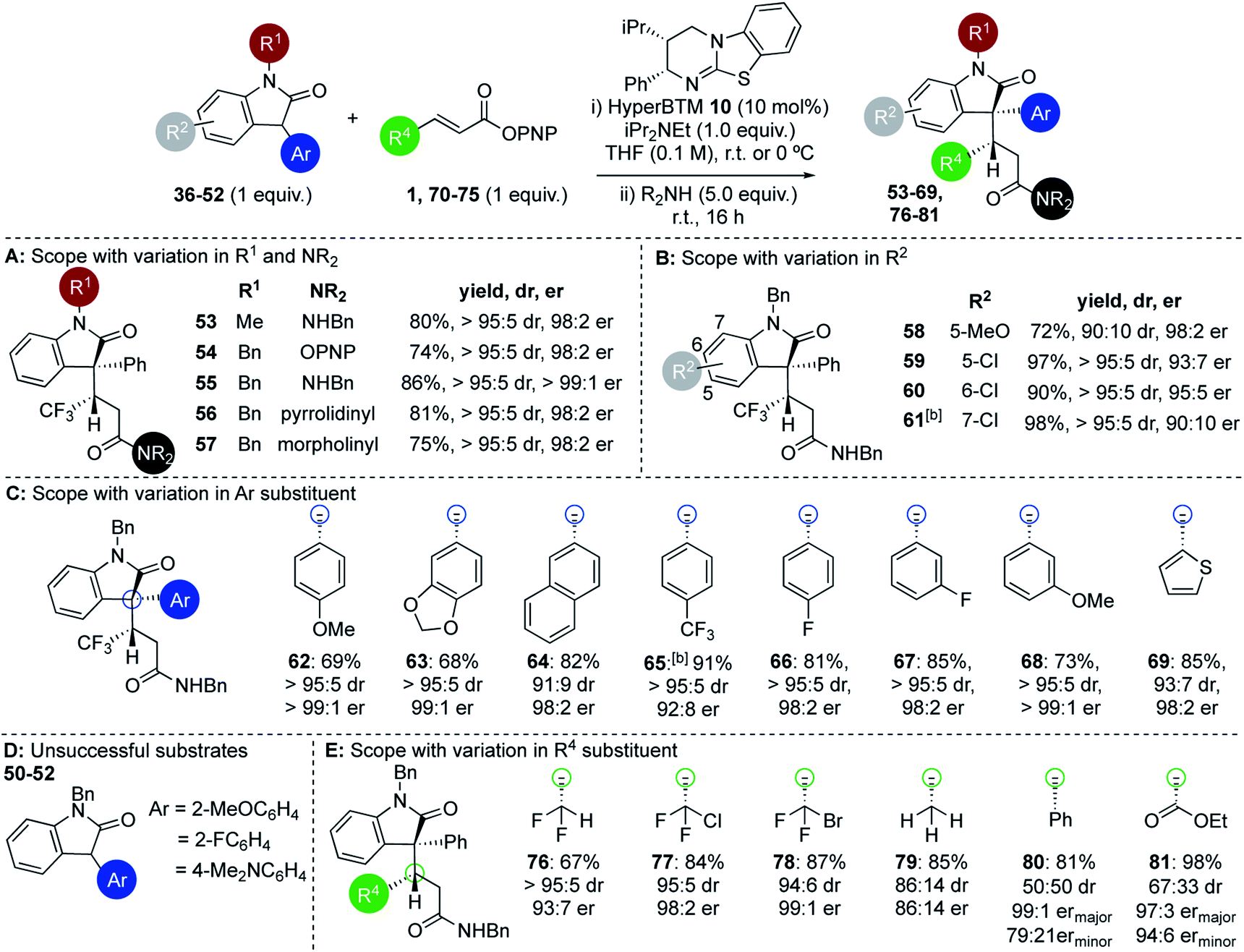 | ||
| Fig. 2 (A–E) Scope and limitations: 3-aryloxindole pronucleophiles and α,β-unsaturated aryl esters.a Isolated overall yields given; dr determined by 1H NMR spectroscopic analysis of crude mixture; er determined by chiral HPLC analysis.b Reaction carried out at −40 °C. | ||
To demonstrate the utility of this process a gram scale reaction was carried out (Scheme 2). At this practical scale the catalyst loading of HyperBTM 10 could be readily reduced to 5 mol%, giving product 55 in 76% yield, >95:5 dr and 98:2 er.
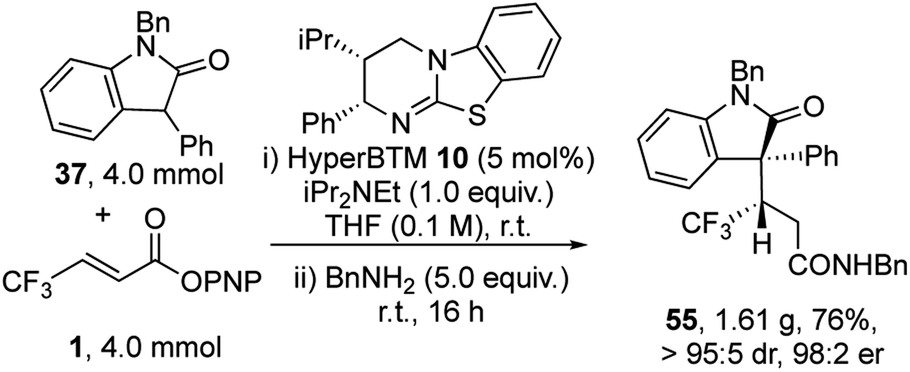 | ||
| Scheme 2 Gram-scale experiment. | ||
Following our previous mechanistic investigations,19 a proposed catalytic cycle is outlined in Scheme 3. Catalysis is initiated through rapid and reversible catalyst acylation by the α,β-unsaturated PNP ester 1 to give α,β-unsaturated acyl isothiouronium ion pair 82. Deprotonation of the pronucleophile by the released p-nitrophenoxide,28 followed by Michael addition of the resultant enolate to the α,β-unsaturated acyl isothiouronium 82, in the assumed stereo-determining step, will generate isothiouronium enolate 83. Subsequent protonation, presumably by the generated p-nitrophenol, gives acyl isothiouronium ion pair 84. Finally, catalyst turnover either directly by p-nitrophenoxide, or by intramolecular participation from the oxindole C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O to give 86, followed by addition of p-nitrophenoxide, gives the Michael addition product 87 and regenerates the isothiourea. The stereochemical outcome of the reaction can be rationalised by the α,β-unsaturated acyl isothiouronium 82 adopting an s-cis conformation, with a 1,5 S⋯O interaction between the acyl O and catalyst S providing a conformational lock.17p,29 Enantioselective conjugate addition of the N-heterocycle-derived enolate to the Si-face of the α,β-unsaturated acyl isothiouronium 82 takes place anti- to the stereodirecting pseudo-axial phenyl substituent of the acylated HyperBTM isothiourea catalyst.
O to give 86, followed by addition of p-nitrophenoxide, gives the Michael addition product 87 and regenerates the isothiourea. The stereochemical outcome of the reaction can be rationalised by the α,β-unsaturated acyl isothiouronium 82 adopting an s-cis conformation, with a 1,5 S⋯O interaction between the acyl O and catalyst S providing a conformational lock.17p,29 Enantioselective conjugate addition of the N-heterocycle-derived enolate to the Si-face of the α,β-unsaturated acyl isothiouronium 82 takes place anti- to the stereodirecting pseudo-axial phenyl substituent of the acylated HyperBTM isothiourea catalyst.
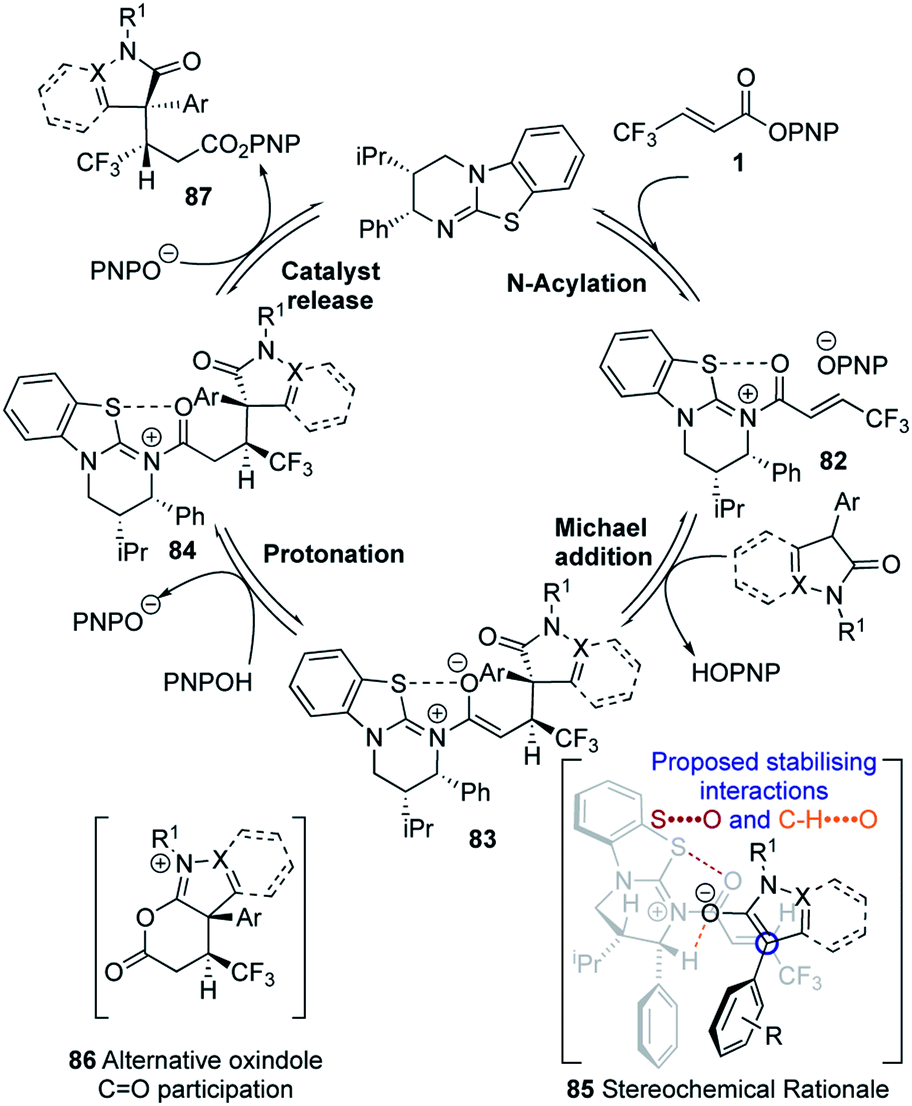 | ||
| Scheme 3 Proposed mechanism for the reaction. | ||
The observed diastereoselectivity is consistent with the reaction proceeding through pre-transition state assembly 85, in which non-bonding interactions around the prostereogenic centres are minimised while allowing for a potentially-stabilising C–H⋯O interaction30 between the enolate oxygen and acidic α-ammonium C–H of the acylated catalyst.
Conclusions
In summary, we have developed an isothiourea-catalysed enantioselective protocol for the Michael addition of 3-aryloxindole and 4-substituted-dihydropyrazol-3-one pronucleophiles to a range of α,β-unsaturated p-nitrophenyl esters. This protocol allows the use of pronucleophiles as stoichiometric reagents rather than solvent, affording products containing N-heterocycles and fluorinated substituents bearing contiguous quaternary and tertiary stereocenters in moderate to high yield and with generally excellent diastereo- and enantioselectivity (>30 examples, up to >95:5 dr and 98:2 er). A broad range of substitution patterns within the heterocyclic pronucleophiles is tolerated, with 3-aryloxindoles leading to optimal diastereo- and enantiocontrol. Variation of the β-substituent within the α,β-unsaturated ester showed that electron-withdrawing β-substituents provided optimal stereocontrol. Notably, in contrast to our previous work, both crotonic and cinnamic esters gave high product yields, further demonstrating the generality of this process. This protocol enhances the utility of α,β-unsaturated acyl ammonium catalysis and uses the multifunctional ability of the aryloxide to act as a leaving group, a proton shuttle (through acting as a Brønsted base, then Brønsted acid as p-nitrophenol) and as a Lewis base to promote catalyst turnover.31
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to these results (C. S., H. L.) has received funding from the Royal Society Newton Fellowship Programme. A. D. S. thanks the Royal Society for a Wolfson Research Merit Award.Notes and references
- For selected reviews see: (a) J. d'Angelo, D. Desmaële, F. Dumas and A. Guingant, Tetrahedron: Asymmetry, 1992, 3, 459 CrossRef; (b) S. C. Jha and N. N. Joshi, ARKIVOC, 2002, 167 Search PubMed; (c) C. Bhanja, S. Jena, S. Nayak and S. Mohapatra, Beilstein J. Org. Chem., 2012, 8, 1668 Search PubMed; (d) Y. Zhang and W. Wang, Catal. Sci. Technol., 2012, 2, 42 RSC; (e) M. M. Heravi, P. Hajiabbasi and H. Hamidi, Curr. Org. Chem., 2014, 18, 489 CrossRef CAS; (f) S. Nayak, S. Chakroborty, S. Bhakta, P. Panda and S. Mohapatra, Res. Chem. Intermed., 2016, 42, 2731 CrossRef CAS; (g) C. Hui, F. Pu and J. Xu, Chem.–Eur. J., 2017, 23, 4023 CrossRef CAS; (h) S. Yumiko, Mini-Rev. Org. Chem., 2018, 15, 236 CrossRef.
- For select reviews see: (a) N. Krause and A. Hoffmann-Röder, Synthesis, 2001, 171 CrossRef CAS; (b) J. Christoffers, G. Koripelly, A. Rosiak and M. Rössle, Synthesis, 2007, 1279 CrossRef CAS.
- For selected examples see: (a) J.-W. Xie, L. Yue, D. Xue, X.-L. Ma, Y.-C. Chen, Y. Wu, J. Zhu and J.-G. Deng, Chem. Commun., 2006, 1563 RSC; (b) M. P. Patil and R. B. Sunoj, Chem. Asian J., 2009, 4, 714 CrossRef CAS PubMed; (c) X. Huang, Y.-H. Wen, F.-T. Zhou, C. Chen, D.-C. Xu and J.-W. Xie, Tetrahedron Lett., 2010, 51, 6637 CrossRef CAS; (d) I. Mager and K. Zeitler, Org. Lett., 2010, 12, 1480 CrossRef CAS PubMed; (e) Z. Mao, Y. Jia, W. Li and R. Wang, J. Org. Chem., 2010, 75, 7428 CrossRef CAS PubMed; (f) J. F. Wang, C. Qi, Z. M. Ge, T. M. Cheng and R. T. Li, Chem. Commun., 2010, 46, 2124 RSC; (g) J. F. Wang, X. Wang, Z. M. Ge, T. M. Cheng and R. T. Li, Chem. Commun., 2010, 46, 1751 RSC; (h) K. Patora-Komisarska, M. Benohoud, H. Ishikawa, D. Seebach and Y. Hayashi, Helv. Chim. Acta, 2011, 94, 719 CrossRef CAS; (i) A. Quintard and A. Alexakis, Chem. Commun., 2011, 47, 7212 RSC; (j) S. Vera, Y. K. Liu, M. Marigo, E. C. Escudero-Adan and P. Melchiorre, Synlett, 2011, 489 CAS; (k) N. Lu, L. Meng, D. Chen and G. Zhang, J. Phys. Chem. A, 2012, 116, 670 CrossRef CAS PubMed; (l) H. S. Yoon, X. H. Ho, J. Jang, H. J. Lee, S. J. Kim and H. Y. Jang, Org. Lett., 2012, 14, 3272 CrossRef CAS PubMed; (m) Y. Gu, Y. Wang, T. Y. Yu, Y. M. Liang and P. F. Xu, Angew. Chem., Int. Ed., 2014, 53, 14128 CrossRef CAS PubMed; (n) R. Lagoutte, C. Besnard and A. Alexakis, Eur. J. Org. Chem., 2016, 4372 CrossRef CAS; (o) A. Ueda, T. Umeno, M. Doi, K. Akagawa, K. Kudo and M. Tanaka, J. Org. Chem., 2016, 81, 6343 CrossRef CAS PubMed; (p) Z. L. Jia, Y. Wang, C. G. Zhao, X. H. Zhang and P. F. Xu, Org. Lett., 2017, 19, 2130 CrossRef CAS; (q) F. Larnaud, A. Sulpizi, N. O. Haggman, J. M. E. Hughes, D. F. Dewez and J. L. Gleason, Eur. J. Org. Chem., 2017, 2637 CrossRef CAS; (r) Z.-X. Wang, Z. Zhou, W. Xiao, Q. Ouyang, W. Du and Y.-C. Chen, Chem.–Eur. J., 2017, 23, 10678 CrossRef CAS PubMed; (s) B. Mondal, M. Balha and S. C. Pan, Asian J. Org. Chem., 2018, 7, 1788 CrossRef CAS; (t) K. Swiderek, A. R. Nodling, Y. H. Tsai, L. Y. P. Luk and V. Moliner, J. Phys. Chem. A, 2018, 122, 451 CrossRef CAS.
- For an excellent overview see: (a) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS PubMed. For select examples see: (b) Y. Kobayashi, Y. Taniguchi, N. Hayama, T. Inokuma and Y. Takemoto, Angew. Chem., Int. Ed., 2013, 52, 11114 CrossRef CAS; (c) T. Inokuma, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2006, 128, 9413 CrossRef CAS PubMed.
- For selected seminal examples see: (a) S. Kobayashi, S. Suda, M. Yamada and T. Mukaiyama, Chem. Lett., 1994, 97 CrossRef CAS; (b) A. Bernardi, K. Karamfilova, S. Sanguinetti and C. Scolastico, Tetrahedron, 1997, 53, 13009 CrossRef CAS; (c) H. Kitajima and T. Katsuki, Synlett, 1997, 568 CrossRef CAS; (d) D. A. Evans, T. Rovis, M. C. Kozlowski and J. S. Tedrow, J. Am. Chem. Soc., 1999, 121, 1994 CrossRef CAS; (e) D. A. Evans, M. C. Willis and J. N. Johnston, Org. Lett., 1999, 1, 865 CrossRef CAS; (f) D. A. Evans, K. Scheidt, J. N. Johnston and M. C. Willis, J. Am. Chem. Soc., 2001, 123, 4480 CrossRef CAS PubMed.
- D. S. Allgäuer, H. Jangra, H. Asahara, Z. Li, Q. Chen, H. Zipse, A. R. Ofial and H. Mayr, J. Am. Chem. Soc., 2017, 139, 13318 CrossRef PubMed.
- E. Badiola, B. Fiser, E. Gomez-Bengoa, A. Mielgo, I. Olaizola, I. Urruzuno, J. M. Garcia, J. M. Odriozola, J. Razkin, M. Oiarbide and C. Palomo, J. Am. Chem. Soc., 2014, 136, 17869 CrossRef CAS.
- (a) S. R. Harutyunyan, F. Lopez, W. R. Browne, A. Correa, D. Peña, R. Badorrey, A. Meetsma, A. J. Minnaard and B. L. Feringa, J. Am. Chem. Soc., 2006, 128, 9103 CrossRef CAS PubMed; (b) S. R. Harutyunyan, T. den Hartog, K. Geurts, A. J. Minnaard and B. L. Feringa, Chem. Rev., 2008, 108, 2824 CrossRef CAS PubMed; (c) Y. Takaya, T. Senda, H. Kurushima, M. Ogasawara and T. Hayashi, Tetrahedron: Asymmetry, 1999, 10, 4047 CrossRef CAS; (d) T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829 CrossRef CAS PubMed.
- S. Matsunaga, T. Kinoshita, S. Okada, S. Harada and M. Shibasaki, J. Am. Chem. Soc., 2004, 126, 7559 CrossRef CAS PubMed.
- (a) D. A. Evans, K. R. Fandrick and H.-J. Song, J. Am. Chem. Soc., 2005, 127, 8942 CrossRef CAS PubMed; (b) J. Mansot, J.-J. Vasseur, S. Arseniyadis and M. Smietana, ChemCatChem, 2019 DOI:10.1002/cctc.201900743.
- C. D. Vanderwal and E. N. Jacobsen, J. Am. Chem. Soc., 2004, 126, 14724 CrossRef CAS PubMed.
- (a) D. A. Evans, K. A. Scheidt, K. R. Fandrick, H. W. Lam and J. Wu, J. Am. Chem. Soc., 2003, 125, 10780 CrossRef CAS PubMed; (b) D. A. Evans and J. S. Johnson, J. Am. Chem. Soc., 1998, 120, 4895 CrossRef CAS; (c) H. Jiang, M. W. Paixão, D. Monge and K. A. Jørgensen, J. Am. Chem. Soc., 2010, 132, 2775 CrossRef CAS PubMed.
- For select examples see: (a) J. S. Johnson and D. A. Evans, Acc. Chem. Res., 2000, 33, 325 CrossRef CAS; (b) M. P. Sibi, S. Manyem and J. Zimmerman, Chem. Rev., 2003, 103, 3263 CrossRef CAS PubMed; (c) A. W. Hird and A. M. Hoveyda, Angew. Chem., Int. Ed., 2003, 42, 1276 CrossRef CAS PubMed; (d) K. Itoh and S. Kanemasa, J. Am. Chem. Soc., 2002, 124, 13394 CrossRef CAS PubMed.
- For select examples see: (a) J. Zu, J. Wang, H. Li, H. Xe, W. Jiang and W. Wang, J. Am. Chem. Soc., 2007, 129, 1036 CrossRef PubMed; (b) M. P. Sibi and K. Itoh, J. Am. Chem. Soc., 2007, 129, 8064 CrossRef CAS; (c) B. Tan, X. Zeng, W. W. Y. Leong, Z. Shi, C. F. Barbas III and G. Zhong, Chem.–Eur. J., 2012, 18, 63 CrossRef CAS; (d) D. Uraguchi, Y. Vek and T. Ooi, Science, 2009, 326, 120 CrossRef CAS PubMed; (e) T. Liu, Y. Wang, G. Wu, H. Song, Z. Zhou and C. Tang, J. Org. Chem., 2011, 76, 4119 CrossRef CAS PubMed; (f) A. Baschieri, L. Bernardi, A. Ricci, S. Suresh and M. F. A. Adamo, Angew. Chem., Int. Ed., 2009, 48, 9342 CrossRef CAS PubMed; (g) J. Zhang, X. Liu, X. Ma and R. Wang, Chem. Commun., 2013, 49, 9329 RSC; (h) D.-Q. Xu, Y.-F. Wang, W. Zhang, S.-P. Luo, A. G. Zhong, A.-B. Xia and Z.-Y. Xu, Chem.–Eur. J., 2010, 16, 4177 CrossRef CAS PubMed.
- T. Gatzenmeier, P. S. J. Kaib, J. B. Lingnau, R. Goddard and B. List, Angew. Chem., Int. Ed., 2018, 57, 2464 CrossRef CAS PubMed.
- J. Yang, A. J. M. Farley and D. J. Dixon, Chem. Sci., 2017, 8, 606 RSC.
- For selected examples see: (a) E. Bappert, P. Muller and G. C. Fu, Chem. Commun., 2006, 2604 RSC; (b) S. Pandiancherri, S. J. Ryan and D. W. Lupton, Org. Biomol. Chem., 2012, 10, 7903 RSC; (c) J. Cheng, Z. Huang and Y. R. Chi, Angew. Chem., Int. Ed., 2013, 52, 8592 CrossRef CAS PubMed; (d) G. Liu, M. E. Shirley, K. N. Van, R. L. McFarlin and D. Romo, Nat. Chem., 2013, 5, 1049 CrossRef CAS PubMed; (e) E. R. T. Robinson, C. Fallan, C. Simal, A. M. Z. Slawin and A. D. Smith, Chem. Sci., 2013, 4, 2193 RSC; (f) S. Vellalath, K. N. Van and D. Romo, Angew. Chem., Int. Ed., 2013, 52, 13688 CrossRef CAS PubMed; (g) J. Xu, Z. Jin and Y. R. Chi, Org. Lett., 2013, 15, 5028 CrossRef CAS PubMed; (h) M. E. Abbasov, B. M. Hudson, D. J. Tantillo and D. Romo, J. Am. Chem. Soc., 2014, 136, 4492 CrossRef CAS PubMed; (i) P. Chauhan and D. Enders, Angew. Chem., Int. Ed., 2014, 53, 1485 CrossRef CAS PubMed; (j) Y. Fukata, T. Okamura, K. Asano and S. Matsubara, Org. Lett., 2014, 16, 2184 CrossRef CAS PubMed; (k) Y. Fukata, K. Asano and S. Matsubara, J. Am. Chem. Soc., 2015, 137, 5320 CrossRef CAS PubMed; (l) S. V. Vellalath, N. Khoi and D. Romo, Tetrahedron Lett., 2015, 56, 3647 CrossRef CAS; (m) N. A. Ahlemeyer and V. B. Birman, Org. Lett., 2016, 18, 3454 CrossRef CAS PubMed; (n) Z. Fu, X. Wu and Y. R. Chi, Org. Chem. Front., 2016, 3, 145 RSC; (o) A. Matviitsuk, J. E. Taylor, D. B. Cordes, A. M. Slawin and A. D. Smith, Chem.–Eur. J., 2016, 22, 17748 CrossRef CAS PubMed; (p) E. R. T. Robinson, D. M. Walden, C. Fallan, M. D. Greenhalgh, P. H.-Y. Cheong and A. D. Smith, Chem. Sci., 2016, 7, 6919 RSC; (q) M. E. Abbasov, B. M. Hudson, W. Kong, D. J. Tantillo and D. Romo, Org. Biomol. Chem., 2017, 15, 3179 RSC; (r) M. E. Abbasov, B. M. Hudson, D. J. Tantillo and D. Romo, Chem. Sci., 2017, 8, 1511 RSC; (s) E. R. T. Robinson, A. B. Frost, P. Elías-Rodríguez and A. D. Smith, Synthesis, 2017, 49, 409 CAS; (t) G. Kang, M. Yamagami, S. Vellalath and D. Romo, Angew. Chem., Int. Ed., 2018, 57, 6527 CrossRef CAS PubMed; (u) K. N. Van and D. Romo, J. Org. Chem., 2018, 83, 632 CrossRef CAS; (v) M. D. Greenhalgh, S. Qu, A. M. Z. Slawin and A. D. Smith, Chem. Sci., 2018, 9, 4909 RSC.
- For selected reviews see: (a) S. Vellalath and D. Romo, Angew. Chem., Int. Ed., 2016, 55, 13934 CrossRef CAS; (b) J. Merad, J.-M. Pons, O. Chuzel and C. Bressy, Eur. J. Org. Chem., 2016, 5589 CrossRef CAS.
- A. Matviitsuk, M. D. Greenhalgh, D.-J.B. Antúnez, A. M. Z. Slawin and A. D. Smith, Angew. Chem., Int. Ed., 2017, 56, 12282 CrossRef CAS.
- For selected examples that emphasise the importance of products containing nitrogen heterocycles see: (a) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed; (b) R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845 CrossRef CAS; (c) D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383 CrossRef CAS PubMed. For selected examples that showcase the widespread interest in the formation fluorinated substituents at stereogenic centres see: (d) J. Nie, H.-C. Guo, D. Cahard and J.-A. Ma, Chem. Rev., 2011, 111, 455 CrossRef CAS; (e) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826 CrossRef CAS; (f) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed.
- For selected examples of aryloxide-assisted catalyst turnover in ammonium enolate catalysis see: (a) T. H. West, D. S. B. Daniels, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2014, 136, 4476 CrossRef CAS PubMed; (b) K. J. Schwarz, J. L. Amos, J. C. Klein, D. T. Do and T. N. Snaddon, J. Am. Chem. Soc., 2016, 138, 5214 CrossRef CAS PubMed; (c) X. Jiang, J. J. Beiger and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 87 CrossRef CAS; (d) T. H. West, D. M. Walden, J. E. Taylor, A. C. Brueckner, R. C. Johnston, P. H.-Y. Cheong, G. C. Lloyd-Jones and A. D. Smith, J. Am. Chem. Soc., 2017, 139, 4366 CrossRef CAS PubMed. For an overview see: (e) W. C. Hartley, T. J. C. O'Riordan and A. D. Smith, Synthesis, 2017, 49, 3303 CrossRef CAS.
- For select examples of pyrazolone pronucleophiles in Michael addition processes see: (a) Z. Wang, Z. Yang, D. Chen, X. Liu, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2011, 50, 4928 CrossRef CAS PubMed; (b) J.-H. Li and D.-M. Du, Org. Biomol. Chem., 2013, 11, 6215 RSC. For select examples of 3-aryloxindole pronucleophiles in Michael addition processes see: (c) T. Bui, S. Syed and C. F. Barbas III, J. Am. Chem. Soc., 2009, 131, 8758 CrossRef CAS PubMed; (d) K. Zhao, Y. Zhi, A. Wang and D. Enders, ACS Catal., 2016, 6, 657 CrossRef CAS; (e) Y.-H. Liao, X.-L. Liu, Z.-J. Wu, L.-F. Cun, X.-M. Zhang and W.-C. Yuan, Org. Lett., 2010, 12, 2896 CrossRef CAS PubMed.
- The PNP esters are prone to hydrolysis upon extended purification resulting in overall moderate yields; furthermore the product diastereoisomers are difficult to separate. Formation of the corresponding amide was found to both increase product stability and facilitate separation by chromatographic purification.
- X. Sheng, J. Zhang, H. Yang and G. Jiang, Org. Lett., 2017, 19, 2618 CrossRef CAS PubMed.
- See ESI† for more details on reaction optimisation.
- The relative and absolute configuration within both diastereoisomers of 29, as well as that within 54, was confirmed by X-ray crystallographic analysis followed by HPLC analysis. CCDC 1882825 (29), 1882826 (29′, minor diastereoisomer) and 1882818 (54) contain the supplementary crystallographic data for this paper.†.
- Following a suggestion from a referee the poor diastereoselectivity observed upon addition of 37 to cinnamate derivative 70 was further probed through electronic variation of the cinnamate substitution pattern. Addition of 37 to a 4-methyl substituted cinnamate PNP ester under standard conditions gave only moderate product yield (26%) with poor 59:41 dr. Addition of 37 to a 4-nitro substituted cinnamate PNP ester gave improved 57% product but again with poor 52:48 dr. See ESI† for full details.
- For a review on the use of ammonium aryloxide salts as Brønsted and Lewis base catalysts, see: J. Godemert, S. Oudeyer and V. Levacher, ChemCatChem, 2016, 8, 74 CrossRef CAS.
- Non-covalent S⋯O interactions have recognised importance in medicinal chemistry: (a) B. R. Beno, K.-S. Yeung, M. D. Bartberger, L. D. Pennington and N. A. Meanwell, J. Med. Chem., 2015, 58, 4383 CrossRef CAS; (b) X. Zhang, Z. Gong, J. Li and T. Lu, J. Chem. Inf. Model., 2015, 55, 2138 CrossRef CAS PubMed. For discussions of S⋯O interactions in isothiourea-catalysed reactions: (c) V. B. Birman, X. Li and Z. Han, Org. Lett., 2007, 9, 37 CrossRef CAS PubMed; (d) P. Liu, X. Yang, V. B. Birman and K. N. Houk, Org. Lett., 2012, 14, 3288 CrossRef CAS PubMed; (e) M. D. Greenhalgh, S. M. Smith, D. M. Walden, J. E. Taylor, Z. Brice, E. R. T. Robinson, C. Fallan, D. B. Cordes, A. M. Z. Slawin, H. C. Richardson, M. A. Grove, P. H.-Y. Cheong and A. D. Smith, Angew. Chem., Int. Ed., 2018, 57, 3200 CrossRef CAS. For an excellent discussion on the origin of chalcogen-bonding interactions see: (f) D. J. Pascoe, K. B. Ling and S. L. Cockcroft, J. Am. Chem. Soc., 2017, 139, 15160 CrossRef CAS.
- Ammonium N+−C(sp3)–H⋯O interactions are expected to be strong and significant, see: (a) C. E. Cannizzaro and K. N. Houk, J. Am. Chem. Soc., 2002, 124, 7163 CrossRef CAS PubMed. For a general discussion of C–H⋯O interactions see: (b) R. C. Johnston and P. H.-Y. Cheong, Org. Biomol. Chem., 2013, 11, 5057 RSC. For selected discussions of the importance of C–H⋯O interactions in isothiourea-catalysed reactions see: ref. 17p, 29e and (c) D.-J. B. Antúnez, M. D. Greenhalgh, A. C. Brueckner, D. M. Walden, P. Elías-Rodríguez, P. Roberts, B. G. Young, T. H. West, A. M. Z. Slawin, P. H.-Y. Cheong and A. D. Smith, Chem. Sci., 2019, 10, 6162 RSC.
- The research data underpinning this publication can be found at, DOI:10.17630/aa042eff-5b55-4096-991f-e92cf4f51375.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, product characterisation data (mpt, NMR, IR, HRMS, [α]D, HPLC), X-ray crystallographic data. CCDC 1882825, 1882826 and 1882818. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04303a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |