
Advances and challenges in electrochemical CO2 reduction processes: an engineering and design perspective looking beyond new catalyst materials
Sahil
Garg
a,
Mengran
Li
 *a,
Adam Z.
Weber
b,
Lei
Ge
ac,
Liye
Li
d,
Victor
Rudolph
a,
Guoxiong
Wang
a and
Thomas E.
Rufford
*a
*a,
Adam Z.
Weber
b,
Lei
Ge
ac,
Liye
Li
d,
Victor
Rudolph
a,
Guoxiong
Wang
a and
Thomas E.
Rufford
*a
aSchool of Chemical Engineering, The University of Queensland, St Lucia 4072, Australia. E-mail: m.li6@uq.edu.au; t.rufford@uq.edu.au
bJoint Center for Artificial Photosynthesis, Lawrence Berkeley National Laboratory, 1 Cyclotron Rd, Berkeley, CA 94720, USA
cCenter for Future Materials, University of Southern Queensland, Springfield 4300, Australia
dHBIS Group Technology Research Institute, Shijiazhuang, 050023, China
First published on 12th December 2019
Abstract
Electrochemical CO2 reduction (CO2R) is one of several promising strategies to mitigate CO2 emissions. Electrochemical processes operate at mild conditions, can be tuned to selective products, allow modular design, and provide opportunities to integrate renewable electricity with CO2 reduction in carbon-intensive manufacturing industries such as iron and steel making. In recent years, significant advances have been achieved in the development of highly efficient and selective electrocatalysts for CO2R. However, to realize fully the potential benefits of new electrocatalysts in low cost, large scale CO2R electrolyzers requires advances in design and engineering of the CO2R process. In this review, we examine the state-of-the-art in electrochemical CO2R technologies, and highlight how the efficiency of CO2R processes can be improved through (i) electrolyzer configuration, (ii) electrode structure, (iii) electrolyte selection, (iv) pH control, and (v) the electrolyzer's operating pressure and temperature. Although a comprehensive review of catalytic materials is beyond this review's scope, we illustrate how other engineering and design decisions may also influence CO2R reaction pathways because of effects on mass transfer rates, the electrode surface chemistry, interactions with intermediate reaction species, and rates of charge transfer.
 Sahil Garg | Sahil Garg is a PhD student in the School of Chemical Engineering at the University of Queensland, Australia investigating the interactions between electrolytes and catalysts for CO2 reduction. Sahil received his B. Technology degree in Chemical Engineering from the Indian Institute of Technology Gandhinagar (IITGN), India in 2012, and a Masters in in Chemical Engineering from Universiti Teknologi PETRONAS, Malaysia in 2016. In September 2019, Sahil was named a Rising Star in the field of chemical engineering and materials by The Australian newspaper. |
 Mengran Li | Mengran Li is a postdoctoral research fellow in the School of Chemical Engineering and UQ-HBIS Innovation Centre for Sustainable Steel at the University of Queensland, Australia. Mengran completed his Bachelor of Engineering in Chemical Engineering at Tianjin University, China (2008), and PhD from the University of Queensland in Australia (2012) for a thesis on cathode materials for solid oxide fuel cells. His current research focuses on development of transition metal-based material and electrochemical process for energy conversion and carbon mitigation. |
 Adam Z. Weber | Adam Z. Weber is a staff scientist and Group Leader of the Energy Conversion Group at Lawrence Berkeley National Laboratory, USA. Adam holds B.S. and M.S. degrees from Tufts University, and a Ph.D. in chemical engineering from the University of California, Berkeley. He has authored over 100 peer-reviewed articles and 10 book chapters on fuel cells, flow batteries, and related electrochemical devices. Adam's work has been recognized by a number of awards including a a 2012 Presidential Early Career Award for Scientists and Engineers (PECASE), the 2014 Charles W. Tobias Young Investigator Award of the Electrochemical Society, and the 2016 Sir William Grove Award of the International Association for Hydrogen Energy. |
 Lei Ge | Lei Ge is a research fellow in the Centre for Future Materials at the University of Southern Queensland, and an adjunct fellow in the School of Chemical Engineering at the University of Queensland, Australia. He completed his PhD in 2013 at the University of Queensland and ME in 2008 at the Nanjing University of Technology, China. His research interests includes materials for membrane separation and selective gas adsorption, metal organic frameworks related materials for electrolysis, and energy devices (e.g. fuel cells). |
 Geoff Wang | Guoxiong (Geoff) Wang is a Professor in Chemical Engineering at the University of Queensland, Australia. Geoff received his BEng in 1983, MEng in 1986, and PhD in 1990 from Northeastern University, China and specialized in process metallurgy focusing on iron- and steel-making. He joined the University of Queensland in 1996 and leads research on modeling and simulation of the Chemical and Metallurgical Engineering processes, coalbed methane (CBM) extraction and carbon dioxide geo-sequestration with enhanced coalbed methane (CO2-ECBM) recovery. His research activity and interests are directed towards developing energy and environmental technologies. |
 Thomas E. Rufford | Thomas (Tom) Rufford is an Associate Professor in the School of Chemical Engineering at the University of Queensland, Australia. Tom completed his Bachelor and PhD degrees in Chemical Engineering at the University of Queensland in 2000 and 2009, respectively, with four years engineering practice in the oil industry between these degrees. His research interests include electrode materials for supercapacitors and electrocatalysts for CO2 reduction, industrial gas separation and purification technologies, and fluid–solid interactions in coal bed methane production systems. Tom has published more than 65 journal articles, 2 book chapters, and an edited book on carbon materials. |
1 Introduction
Utilization of CO2 from industrial waste gases is considered a complementary route to other CO2 emission reduction strategies such as renewable energy sources, CO2 capture and storage (CCS), and other low carbon emission technologies.1 The main pathways to utilize CO2 include reuse of CO2 without conversion (e.g. enhanced oil recovery, supercritical CO2)2,3 or to convert the CO2 to a valuable fuel, energy storage vector, or chemical feedstock. Among CO2 conversion technologies such as biochemical, photosynthetic, thermo-catalytic, and photocatalytic processes,4–7 electrochemical CO2 reduction (CO2R) is one of the most promising CO2 utilization strategies because of the mild electrolyzer operating conditions, opportunities to tune the process towards desired products, the potential to use industrial or municipal wastewaters as electrolytes, and modular reactor designs.8 In addition, CO2R technologies could be integrated with renewable electricity generation from solar or wind9–11 (as shown in Fig. 1) to reduce carbon footprints in carbon intensive manufacturing industries such as ammonia production or iron and steel production through CO recycling,12 for example.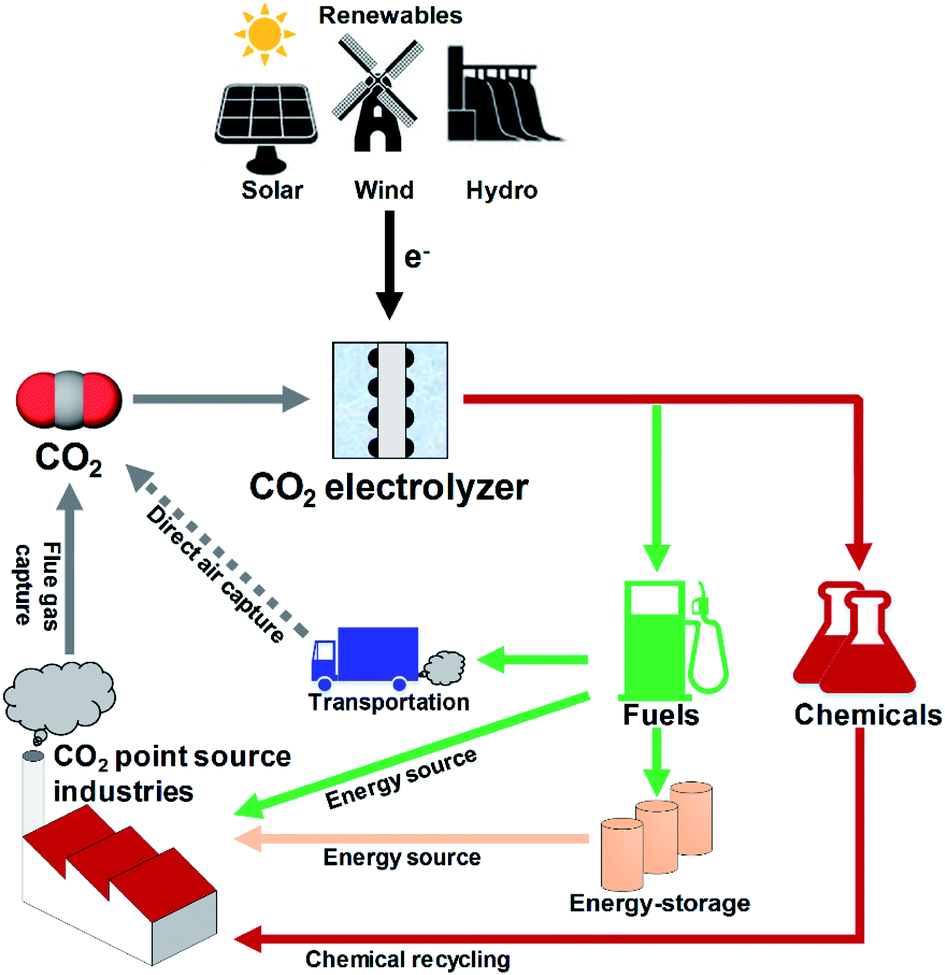 | ||
| Fig. 1 Closing the carbon cycle. CO2 electrolyzer utilizing renewable energy can convert the captured CO2 into chemicals or fuels for direct usage or energy storage. | ||
Table 1 shows several organisations report electrochemical CO2R technologies to operate at pilot-scale with current densities in the range j = 100–200 mA cm−2. These technologies could lead to commercially viable processes to convert CO2 to CO,13 light hydrocarbons including CH4 and C2H4,14 alcohols,15 and chemical feedstocks like formic acid (HCOOH).16 However, most of these technologies are currently too costly for practical applications and market penetration. The first challenge to low cost CO2R is the high energy requirement to break bonds in the CO2 molecule.17 The second challenge is to achieve a high selectivity of CO2 to desired products to minimize costs and complexity of product separation processes. Achieving high selectivity is difficult because a large number of CO2R reactions and the competing hydrogen evolution reaction (HER) all have standard potentials (Eo) in a narrow range (−0.25 V to 0.17 V vs. standard hydrogen electrode (SHE)) as shown in Fig. 2. The third challenge is to ensure the overall rate of reaction is not limited by rates of CO2 mass transfer from the gas phase to electrolyte and to active sites on the cathode catalyst. The fourth practical challenge is to maintain stable electrocatalyst performance over extended operating periods because the catalyst can be poisoned by impurities in the electrolytes18,19 or CO2 feed gas (e.g. sulphur compounds), or by products stemming from corrosion of the electrolyzer components.20–26
| Technology (location) | Throughput/scale | Reactor configuration | Catalysts | Electrolyte | Target Products | Notes | Reference |
|---|---|---|---|---|---|---|---|
| a The information presented in above table is mostly collected from relevant papers or websites. Some companies have not publicly disclosed any data on CO2 electrolysis amid high competition. | |||||||
| Opus-12 (Berkeley, USA) | Lab-scale | Proton exchange membrane (PEM) type electrolyzer | Anode: IrO2, cathode: Ag nanoparticles supported on carbon foam42 | Water | CO and O2 | Announced plans to develop renewable electricity operated CO2R to ethylene, ethanol | 13 |
| Dioxide materials (Boca Raton, USA) | Lab-scale | Sandwich-type CO2 electrolyzer where cathode and anode catalysts are painted on either side of the membrane | Anode: IrO2 or RuO2, cathode: carbon paper coated with silver/ionomer mixture43 | 10 mM KHCO3 as anolyte and humidified CO2 as catholyte43 | >95% selectivity to CO43 | Report 6 months stable catalyst operation | 44 |
| Carbon electrocatalytic recycling Toronto (CERT) (Toronto, Canada) | Lab-scale/pilot-scale cell | Flow cell modified from state-of-the-art fuel cells | Anode: made from cheap and conventional abundant earth metals, cathode: nanostructured metals based on copper45 | 7 M KOH45 | 70% selectivity for C2H4 (ref. 45) | Uniform selectivity for the initial 150 hours | 14 |
| Mantra energy alternatives Ltd (Vancouver, Canada) | 100 kg per day pilot plant | Fuel-cell type CO2 electrolyzer | Not disclosed publicly | Water or wastewater | Formate/formic acid | Successfully demonstrated CO2RR for greater than 2500 hours | 16 |
| Skyre or sustainable innovations (East Hartford, USA) | Pilot plant | Not disclosed publicly | Not disclosed publicly | Not disclosed publicly | Hydrocarbon fuels | — | 46 |
| Siemens and Evonik (Germany) | Lab-scale | CO2R electrolyzer | Anode: IrO2 coated titanium, cathode: silver gas diffusion electrode based on oxygen depolarization cathode (ODC used in industrial chlorine–alkaline electrolysis)47 | 0.1 M K2SO4/1.5 M KHCO3 as both anolyte and catholyte47 | ∼70% selectivity for CO, then CO fermentation to higher alcohols47 | Stable CO selectivity for almost 1200 hours | 15 |
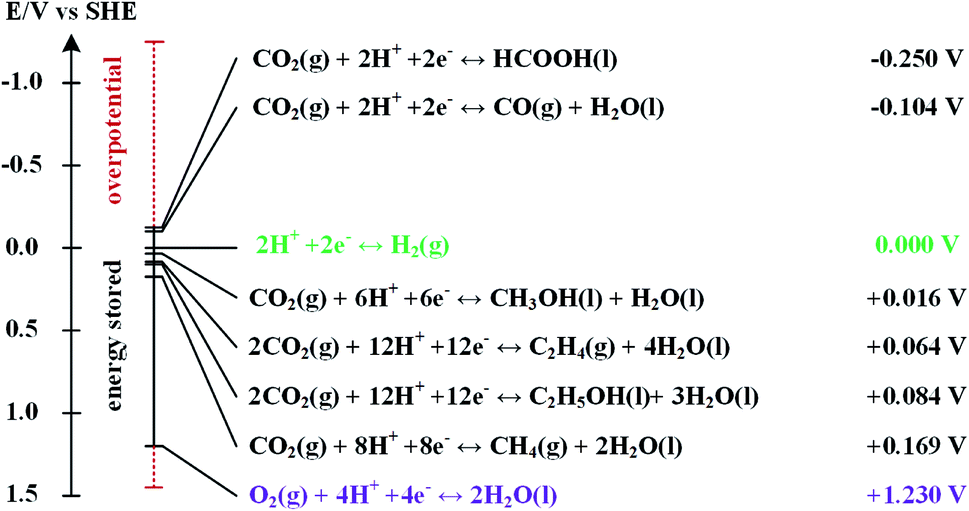 | ||
| Fig. 2 Standard equilibrium potentials for hydrogen evolution half-cell reaction and several other half-cell reactions to reduce CO2 into various products at 1 atm and 25 °C. Data presented was taken from Qiao et al.27 (2014). | ||
To circumvent these challenges, a significant amount of research effort aims to develop highly efficient, stable, and selective CO2R electrocatalysts. Many comprehensive reviews on CO2R catalysts are available,28–38 and these reviews cover advances in transition metals, alloys, metal–organic complexes, metal chalcogenides, metal–nitrogen–carbon materials, and carbon materials. Further review of electrocatalysts is beyond the scope of this article. Instead, we complement existing catalyst reviews with a critical analysis of engineering factors that affect the performance of CO2R electrolyzers. These factors include the reactor configuration, electrode structures, electrolyte selection, and the choice of reaction conditions such as pH, pressure, and temperature. These engineering factors not only predetermine the CO2R mass-transport characteristics but can also have significant impacts on the catalytic reaction pathways.39–41 The review concludes with a discussion of the priorities for future research to understand better the fundamental mechanisms of CO2R and improve CO2R performance.
2 Working principles of electrochemical CO2 reduction
A typical CO2 electrolyzer (see for example Fig. 3a showing a H-cell type reactor used in research laboratories) consists of a cathode to reduce CO2 to products such as CO or HCOOH/HCOO− and produce hydroxyl ions (OH−); an anode to oxidize water via the oxygen evolution reaction (OER) that consumes OH− or generates protons (H+) and electrons (e−); an electrolyte to conduct ions and to dissolve and transport CO2 to the cathode active sites; an ion-exchange membrane or porous diaphragm to separate the cathode and anode electrodes; and a voltage source with sufficient potential (E) to transfer electrons from anode to cathode. In such a system, there are several key steps involved in a CO2R process, including (1) mass transfer of CO2 from the gas phase to the bulk electrolyte, (2) transport of dissolved CO2 from the bulk electrolyte to cathode/electrolyte interface, (3) absorption of CO2 at the cathode surface, (4) dissociation of adsorbed CO2 species into adsorbed intermediates such as *COOH, *CO, *CHO, and *COH, (5) electron transfer from the cathode catalyst to intermediates, (6) desorption of products from the electrode, and (7) migration of products away from the cathode/electrolyte interface to the bulk gas or liquid phases.48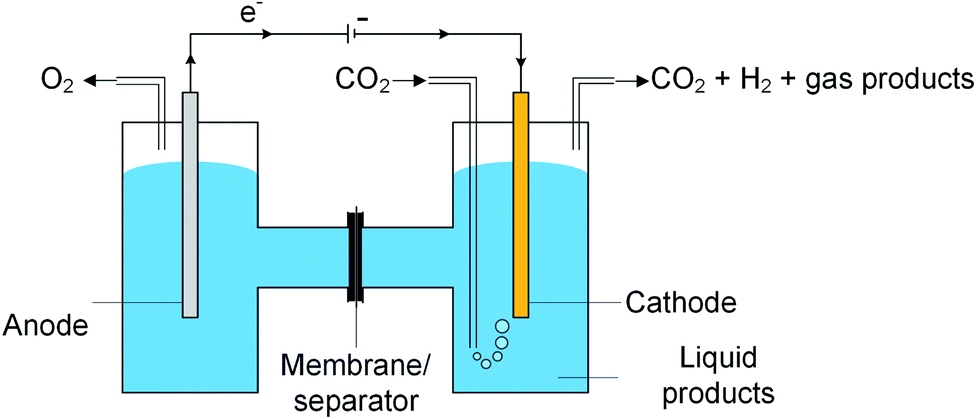 | ||
| Fig. 3 Schematic of a laboratory electrochemical H-cell (reactor) for CO2 reduction at the cathode to gas products such as CO and liquid products such as formic acid and methanol with water being oxidized at the anode. | ||
We start our discussion at the electron transfer step (5) to examine the minimum theoretical energy requirement for CO2R. The minimum potential required for a CO2R reaction is the half-cell standard potential described by Eo = −ΔG0/nF, where ΔG0 is the Gibbs free energy at 1 atm and 298 K, n is the number of moles of electrons transferred in the half-cell reaction, and F is Faraday constant (96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485C mol−1). For example, the half-cell reaction CO2(g) + 2H+(aq.) + 2e− → CO(g) + H2O(l) with ΔG0 = 20.09 kJ mol−1 has E° = −0.104 V vs. SHE.49 Other half-cell standard potentials at 1 atm and 298 K are shown in Fig. 2. To drive a sufficient CO2R rate, an excess voltage or overpotential to E° must be applied to overcome the sum (Rtotal) of several energy barriers or resistances as described in eqn (1):
485C mol−1). For example, the half-cell reaction CO2(g) + 2H+(aq.) + 2e− → CO(g) + H2O(l) with ΔG0 = 20.09 kJ mol−1 has E° = −0.104 V vs. SHE.49 Other half-cell standard potentials at 1 atm and 298 K are shown in Fig. 2. To drive a sufficient CO2R rate, an excess voltage or overpotential to E° must be applied to overcome the sum (Rtotal) of several energy barriers or resistances as described in eqn (1):
| Rtotal = Rcathode + Ranode + Rions + Rmembrane + Rbubble,cathode + Rbubble,anode + R | (1) |
The resistances include (1) the activation barriers or activation overpotentials (ηs)50 for CO2R at cathode (Rcathode) and OER at the anode (Ranode); (2) ohmic losses from conduction of ions (Rions) in the bulk electrolytes, ion transport across the membrane (Rmembrane); (3) loss of active electrode area from the bubbles formation at the electrodes (e.g. Rbubble,cathode for CO and H2 at the cathode, and Rbubble,anode for O2 at the anode);51,52 and (4) the sum (R) of electrical resistances in other cell components and contact resistances between components.
Because the CO2R reaction depletes CO2 concentrations at the electrode surface, at high current densities the overall reaction rate can be limited by the rates of CO2 mass-transfer to the electrode surface.53 In addition according to the Nernst equation, the change in concentration affects the equilibrium potential and this effect can be approximated by the concentration overpotential.49 In large scale industrial electrolyzers operating at high temperature concentration overpotential becomes important because at higher temperatures the activation overpotential is lower and the improved electrolyte conductivity leads to smaller ohmic losses.54 Therefore, now that we have established the minimum energy requirements for CO2R, we will look next at the steps involved in transferring CO2 from the gas phase to the electrode surface.
In most CO2R electrolyzers, gaseous CO2 is first dissolved in the liquid electrolyte, then transferred through the liquid to the cathode–electrolyte interface. This process is driven by CO2 concentration gradients as illustrated in Fig. 4a, and the rate of CO2 transfer depends on the interfacial contact area, film and overall mass transfer coefficients, and the overall concentration driving force. The concentration gradients in the system are dependent on the solubility of CO2 in the electrolyte and the selected operating pressures and temperature of the electrolyzer cell. However, prediction of these gradients is complex during CO2R because acid/base reactions (for example CO2 consumption by OH−) in the electrolyte can lead to non-linear deviation of the concentration away from Fick's law behavior,55 which reduces the concentrations of CO2 available to react at the electrode's surface.56 This effect can be partially controlled with buffering electrolytes such as potassium carbonate (KHCO3) to maintain pH at the cathode.55 The interfacial contact area can maximized by reducing the size of gas bubbles injected to the electrolyte57 or using a 3D-structured electrode such as a gas diffusion electrode (GDE).58,59 The magnitudes of CO2 and CO2R product mass transfer coefficients generally increase with temperature, pressure, and the velocities of gas and liquid in the electrolyzer, but also are effected by electrolyte density, viscosity and solubility relationships.
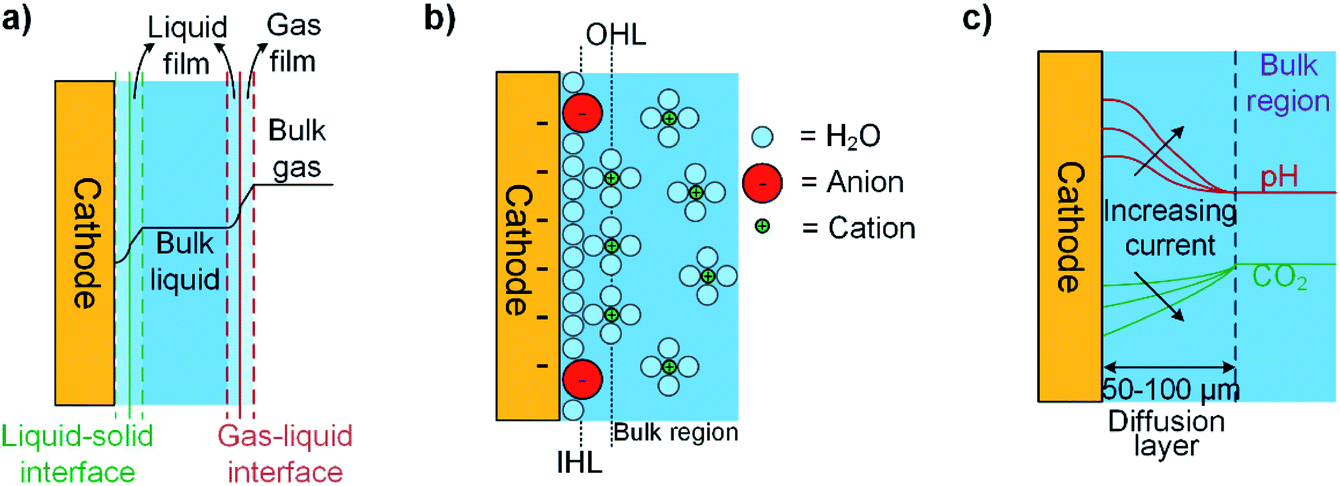 | ||
| Fig. 4 Schematic illustrations of (a) the concentration profile of CO2 across gas, liquid, and solid interfaces; (b) the electrochemical double layer with specifically adsorbed anions at the inner Helmholtz layer and solvated cations at outer Helmholtz layer; and (c) changes in CO2 and pH next to the diffusion layer at increasing current density in a system where CO2 is supplied from the bulk electrolyte. | ||
Next we examine the cathode–electrolyte interface where CO2R occurs in a typical aqueous electrolyzer. Cations in the electrolyte migrate towards the negatively charged cathode surface to form an electrochemical double layer (DL), as shown in Fig. 4b. This DL is formed by the outer Helmholtz layer (OHL) of fully-solvated cations, and the inner Helmholtz layer (IHL) of less-solvated halide ions or CO2-related adsorbed species directly adsorbed at the electrode surface.49 The presence of this DL can effect CO2R through several mechanisms. For example, the local electrical field between the negatively charged cathode and the positively-charged adsorbed cations has been reported to stabilize CO2R-related intermediates such as *CO2 and *COOH.60,61 On the other hand, in the OHL solvated cations like Li+, Na+, and K+ act as a source of protons for the HER and disrupt the local pH within the DL.62,63 Another effect relates to interactions between anions and the electrode surface, which have been reported in some cases like I− and a Cu surface to be strong enough to allow anion absorption within the IHL.64 In most cases, anions with a pKa close to the local pH may help buffer the pH and adsorbed ions may be directly involved in CO2R pathways by affecting the binding strength or adsorption geometry of CO2R intermediates such as *COOH.65,66Fig. 4c illustrates that the concentrations and rates of consumption of protons, CO2, and other species in the DL are directly proportional to the current density and product selectivity during a CO2R reaction and these changes in the local reaction environment could occur even at low currents, and thus limit the overall reaction rate before all available CO2 becomes depleted at the electrode surface.53,67
This background discussion of the principles of CO2R highlights that even though the electrocatalyst determines the underlying reaction kinetics, other factors such as reactor configuration, electrode structure, and conditions including the type of electrolyte, pH, pressure, and temperature can affect the overall rate of CO2R reactions.
2.1 Figures of merit to describe electrochemical CO2R
We briefly describe here the figures of merit commonly used to evaluate and compare electrochemical CO2R processes. These are faradaic efficiency, current density, and energy efficiency.• Faradaic efficiency (FE) is the ratio of the amount of charge used to form a product species (e.g. CO) calculated from Faraday's law to the total charge (Q) supplied:68
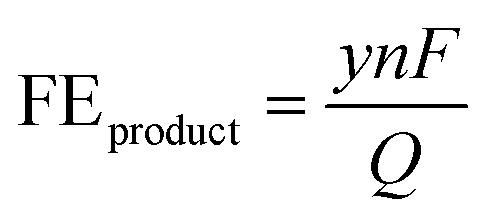 | (2) |
• Current density (j or CD) is the total current (I, in Amps) per unit area of the cathode (A, m2 or commonly cm2) calculated by eqn (3), and describes the total rate of reaction so is an important input to estimate electrolyzer size and capital cost for a CO2R process.69
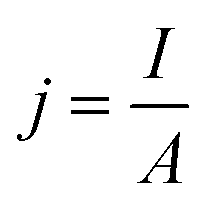 | (3) |
Partial current density (jproduct) for a specific product can be obtained by:
| jproduct = FEproduct × j | (4) |
• Energy efficiency (EE) is a measure of net energy consumption toward a specific product expressed in eqn (5) as a ratio of amount of energy used to produce the specific product to the net electrical energy supplied to the system.
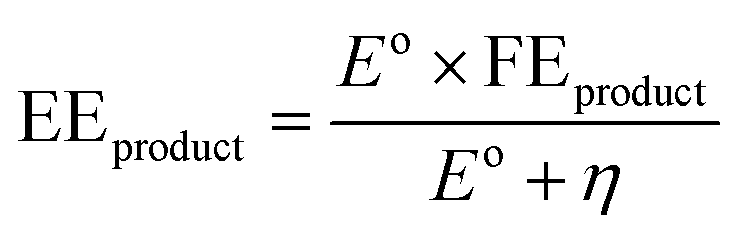 | (5) |
3 Electrolyzer configurations
The background discussion in Section 2 highlights how, in addition to catalyst materials, the configuration of the electrolyzer impacts the overall efficiency of a CO2R process by effecting the limiting rates of CO2 mass transfer to the catalyst, controlling resistances of cell components, and determining the reaction distribution across electrodes. The two general categories of electrochemical reactors are (i) batch or semi-batch cell and (ii) continuous flow-cell configurations. Batch and semi-batch electrolyzers like the H-cell in Fig. 3a are commonly used in laboratory CO2R studies with electrodes simply immersed in liquid electrolytes with CO2 gas bubbled to saturate the catholyte.70 This configuration is simple, low cost, and allows rapid screening of novel electrocatalysts and electrolytes, but is not practical for treatment of large volumes of CO2 gases40,71 because rates of CO2 mass transfer to the electrode surface (even if the catholyte is vigorously stirred) are too slow and limit CDs to less than 100 mA cm−2.39 In addition, in a semi-batch cell any cations (e.g. K+) that pass through the separator accumulate in the catholyte, and this accumulation can degrade the electrode kinetics and CO2R selectivity over extended cell operation or result in high ohmic losses due to electrodialysis.72,73Industrial-scale CO2R processes require continuous processes to achieve sufficient reaction rates and be economically viable. Table 1 presented examples of continuous flow-cell electrolyzers reported for pilot-plant and scale-up studies, and most of these follow scale-up and engineering strategies that were developed for polymer electrolyte (PE) electrolyzers. In this section we will discuss the separator, which is a critical component in all batch cells and continuous flow-cells, and then describe the liquid-fed, vapor-fed, and microfluidic electrolyzers illustrated in Fig. 5. Our review does not cover flow-field patterns (e.g. straight, parallel, serpentine) that can be manufactured on current collectors to optimize CO2 and electrolyte contact with the catalyst, and to minimize pressure drop, and to manage heat transfer in the cell.74 We refer readers interested on flow-field patterns to reviews of PE electrolyzers.74–76
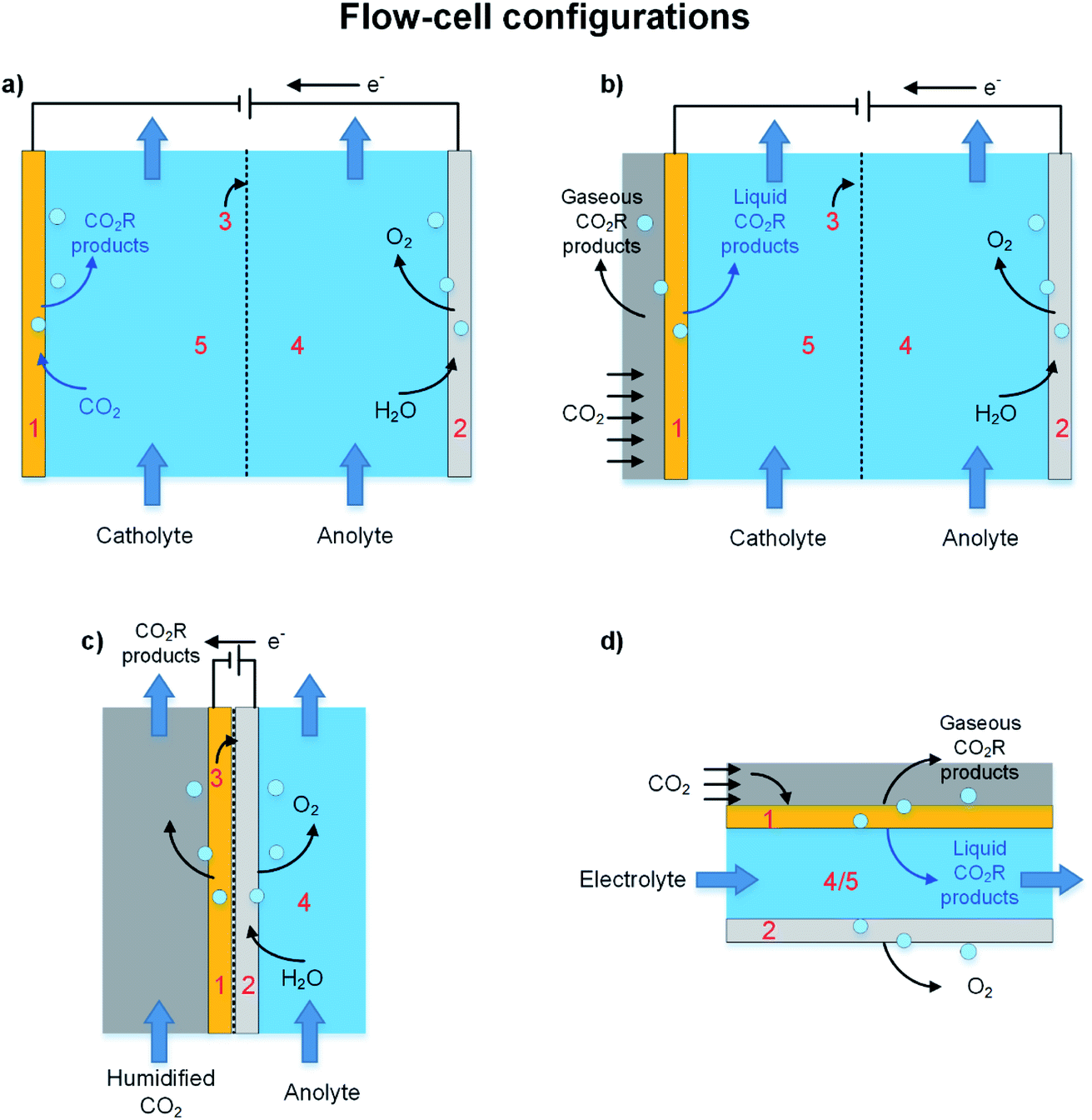 | ||
| Fig. 5 Schematic of CO2 electrolyzer in flow-cell configurations (a and b) liquid-fed electrolyzer, (c) vapor-fed electrolyzer and (d) microfluidic reactor. The main cell components are labelled as (1) cathode, (2) anode, (3) separator or ion-exchange membrane which could be a cation exchange membrane, anion exchange membrane, or a bipolar membrane, (4) anolyte, and (5) catholyte. | ||
3.1 Separator considerations
In a CO2R electrolyzer the separator between cathode and anode chambers, just like separators in water splitting electrolyzers,73,77 is critical to safe and efficient operation of the cell. An effective separator (1) minimizes the risk of a short circuit between electrodes; (2) prevents exchange of reactants and CO2R products between cell chambers to reduce the risk of forming unsafe gas mixtures (e.g. H2 and O2) and prevent oxidation of products at the anode; (3) maintains desired local conditions in the anode and cathode reactions, (4) provides mechanical support to withstand any pressure differences between chambers, and (5) must have a good conductivity for certain ions. The two classes of materials that can provide the required selective ion transport and low permeability to other species are porous separators and ion exchange membranes (IEM). Porous separators include plastic mesh (e.g. polyolefin or Netlon) with large pores (0.5–12 mm widths) and microporous diaphragms with pore sizes 0.1–50 μm (e.g. glass fibres, polytetrafluoroethylene (PTFE)).77Semi-permeable ion exchange membranes selectively transport certain dissolved ions but are not permeable to other ions or non-charged species. Most CO2R electrolyzers use monopolar IEMs that are either cation exchange membranes (CEM) such as Nafion® or anion exchange membranes (AEM) such as Sustainion®. Bipolar membranes (BPM) with electrocatalyst sandwiched between CEM and AEM layers are also available,78 and are reported to achieve more stable pH levels between two electrodes at steady state.79–81 A further advantage of the BPM is that this design may allow lower, cost abundant metals to be used as catalysts instead of precious and noble metals.82,83 Further detailed descriptions of IEM working principles and recent advances in IEMs are provided in reviews by Kusoglu and Weber,84 Kaczur et al.,85 Luo and Wessling,86 and Kimberly et al.87
The selection of a cation exchange, anion exchange, or bipolar membrane must be considered together with catalyst selection, electrolyte selection, and the targeted CO2R products. For example, a CEM is commonly used for CO2R to formate because this IEM blocks formate anions from crossing over to the anode chamber. If the CEM is also proton exchange membrane like Nafion then an acidic anolyte must also be selected to manage proton concentrations,88 and these decisions limit the choice of OER anode catalysts to expensive precious noble metals such as Ru and Ir. Another consideration for proton exchange membranes is that excess protons will promote HER at the cathode.89
Anion exchange membranes can transport anions such as HCO3−, OH− and CO32− ions from alkaline catholytes to the anode chamber.90 Hori et al.91 reported a FECO up to 92% at 20 mA cm−2 which shows the improved performance of AEM-based reactor over CEM. However, there are some important considerations in the use of AEM. For example, CO32− and HCO3− transported to the anode chamber are expected to produce CO2, and this reduces the overall efficiency of the CO2R process. Further, the extended exposure of AEM to alkaline catholytes can lead to blockage of the membrane with less mobile HCO3− and CO32− anions, which degrades the AEM's ionic conductivity and increases the membrane's ohmic resistance.87,92–94 In addition, AEM may be susceptible to degradation by excessive OH−, especially if the membrane is insufficiently hydrated. A recent report by Sun et al. revealed CO2R to methanol and ethanol can also accelerate such degradation.95
3.2 Continuous liquid-fed electrolyzers
Fig. 5a depicts a two-chamber liquid-fed electrolyzer in which a CO2-saturated catholyte and an anolyte are pumped through the separate cell chambers. In this configuration, the catholyte is saturated with CO2 outside the electrolyzer which requires additional CO2 capture process units. A more recent integrated design for a liquid-fed electrolyzer is the three chamber cell shown in Fig. 5b that uses a gas diffusion electrode to enhance transfer of CO2 from the gas phase to the electrolyte–cathode interface. We provide further discussion of GDEs in Section 4.Obviously, one control on the overall mass transfer rates and thus reaction rate in liquid-fed electrolyzers is the flow-rate of the catholyte because it directly effects superficial liquid velocities in the cell.96–98 For example, Alvarez-Guerra et al.97 reported that at low current density (CD = 2.5 mA cm−2) the overall rate of CO2R to formate over a lead-based cathode was insensitive to catholyte flow rates. However, they reported that at higher current densities (12.25–22 mA cm−2) increasing the catholyte flow-rate from 0.57 mL min−1 cm−2 to 1.44 mL min−1 cm−2 enhanced formate production because of an improved supply of dissolved CO2 at the electrode interface for CO2R.
Both liquid-fed configurations provide larger active electrode area to electrolyte volume ratios than semi-batch cells, and thus can achieve higher overall reaction rates and lower ohmic losses than batch operation.99–102 An additional advantage of the high electrode area to electrolyte volume ratio in liquid-fed electrolyzers in laboratory studies is that this allows detection of low concentration products and accurate voltage measurement. For example, Kuhl et al.99 used a liquid-fed flow-cell electrolyzer like that in Fig. 5a to detect for the first time acetone, glycolaldehyde, ethylene glycol, glyoxal, and hydroxyacetone as CO2R products at low concentrations. The expected H2, CH4, CO, HCOOH, and C2H4 were also reported by Kuhl et al. Other groups report also sandwich type compression flow-cells for detection of low concentration products in CO2R reactions.57,63,101,103,104
Fig. 6a shows an example of a liquid-fed electrolyzer with a BPM separator that due to the better control of pH imparted by the BPM achieved a more stable cell voltage during a 12 hour CO2R experiment than an electrolyzer with a CEM (Fig. 6b and c).71 However, a potential issue with BPMs in liquid-fed electrolyzers is ensuring the rate of water flux across the BPM matches the rate of water dissociation to prevent dry-out of the membrane, which leads to significant increases in ohmic resistance.105 Another potential issue in liquid-fed electrolyzers is that the ionic conductivity of the BPM depends on the concentration-gradients of salts in the electrolyte,106 and because a BPM inherently leads to depletion of charge at the CEM/AEM interface this can create significant junction gradients. Therefore, operation of liquid-fed electrolyzers with BPMs requires ion concentrations in both the catholyte and anolyte to be controlled for the reaction kinetics and for the material and thickness of the membrane(s).
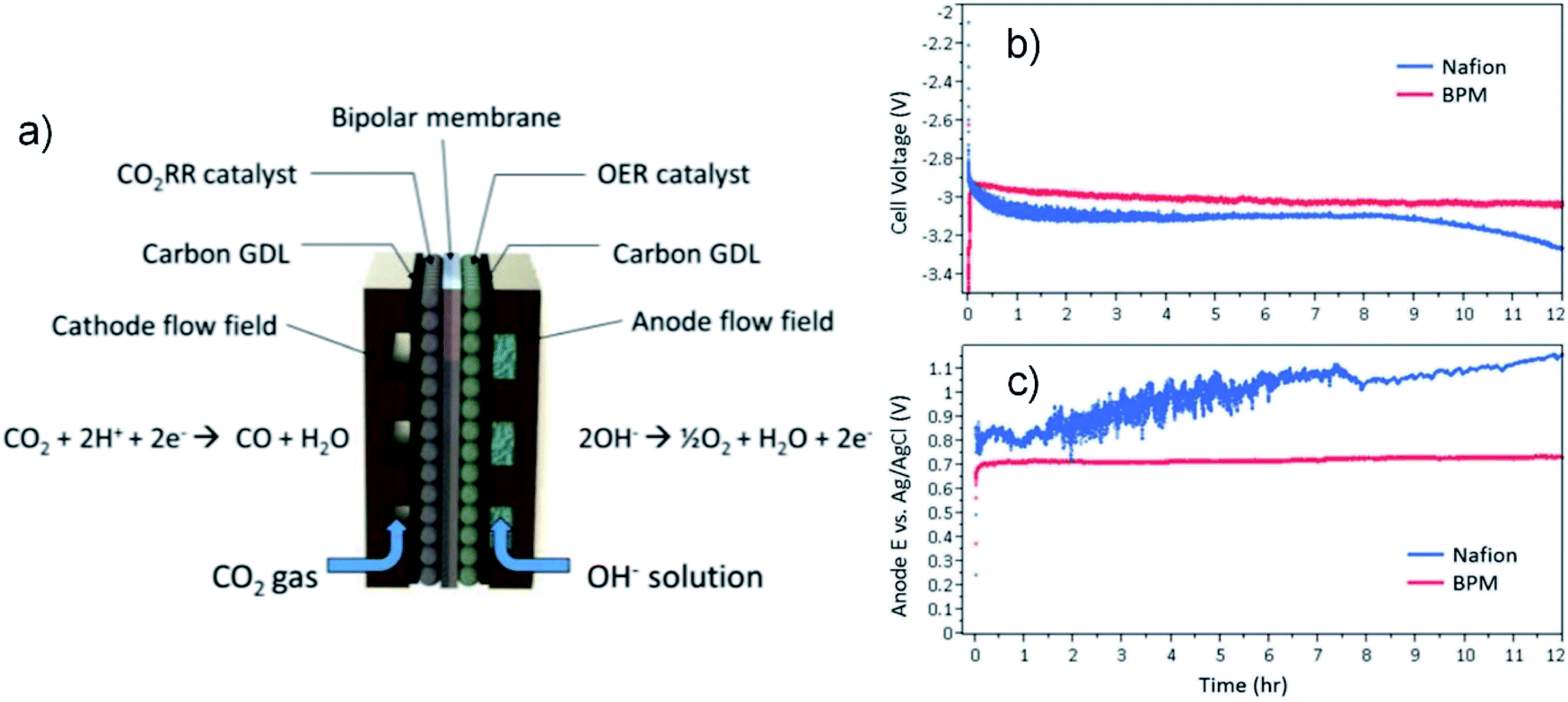 | ||
| Fig. 6 (a) Schematic diagram of a CO2 electrolyzer employing a BPM; comparison of (b) overall cell voltage and (c) anode potential vs. Ag|AgCl between BPM and Nafion membrane. 0.1 M KOH was used as anolyte and 0.5 M KHCO3 as catholyte. Reprinted with permission from Li et al.,71 Copyright 2016, American Chemical Society. | ||
In liquid-fed electrolyzers with a GDE, unstable CO2R operation can result from liquid flooding of the GDE. Several researcher groups suggest suppling the CO2 gas at higher pressures to prevent liquid ingress into the GDE and in this case any gaseous CO2R products leave the electrolyzer with the catholyte.54,107–109 For example, Haas et al.47 operated a liquid-fed electrolyzer at 50 mA cm2 for more than 1000 h, but report that to achieve this current density they had to sacrifice CO selectivity. Jeanty et al.110 ran a CO2 electrolyzer at 150 mA cm2 with a CO FE close to 60% for more than 200 h over a 100 cm2 electrode area. A potential adverse effect of operating with a high gas overpressure across the GDE in a liquid-fed electrolyzer is that the higher partial pressure of CO2 leads to precipitation from the bicarbonate/carbonates catholyte which can reduce electrolyte conductivity, block GDE pores or modulate pH. All such effect increase the overall ohmic losses in the cell.
3.3 Vapor-fed electrolyzers
Fig. 5c shows a vapor-fed flow-cell electrolyzer111 with an IEM coated on one-side with a cathode catalyst and on the other side with an anode catalyst to create a zero-gap cell. In this configuration, the catholyte is supplied via humidified CO2-containing gas to maintain membrane hydration during CO2R. Compared to liquid-fed electrolyzers, the key advantages of vapor-fed electrolyzers are lower ohmic losses and reduced risk of catalyst poisoning from impurities in the catholyte. In addition because the vapor-fed cell does not require pumps to feed the electrolyzer the equipment and operating costs may be lower than a liquid-fed cell; however, this comparison depends on the cost of the processes required to vaporize catholyte into the CO2 feed gas. One potential drawback of vapor-fed electrolyzers is liquid CO2R products like alcohols can flood back into GDE pores and thereby hinder CO2 access to the active sites.Most vapor-fed electrolyzers reported in the literature use a CEM to transport protons from the anode chamber to the cathode for CO2R. For example, Lee et al.112 reported more stable formate production over a tin nanoparticle cathode catalyst in a vapor-fed electrolyzer than a liquid-fed electrolyzer, and they attributed the improved performance to a shorter CO2 diffusion pathway to the catalyst. However extended CO2R operation in CEM vapor-fed electrolyzers is reported to lead to acidification at the cathode, which promotes unwanted HER.113 In an alternative design, Kutz et al. used AEM like a methylimidazolium-based styrene polymer in a vapor-fed electrolyzers with Ag-based catalyst and reported stable operation for 6 months at CD = 50 mA cm−2 with a FECO = 90%.43 Mallouk's laboratory71 reported a vapor-fed electrolyzer with a BPM separator and ionic liquid catholyte that achieved CDs two times larger than a liquid-fed cell and relatively stable cell voltage close to 3 V during operation at 80 mA cm−2 for 14 h. However, they did report that the FECO began to degrade after 1 h, which may have been due to de-wetting of the ionic liquid IL from the surface of the catalyst.71 Salvatore et al.114 enhanced the stability of this vapor-fed with BPM configuration using a solid support layer of aqueous NaHCO3 between the Ag-decorated GDE and the BPM, and demonstrated a steady FECO = 65% at 100 mA cm−2 and 3.4 V for 24 h.114
3.4 Microfluidic electrolyzer
A microfluidic electrolyzer (Fig. 5d), such as the cells described by Kenis and co-workers,115,116 does not use a membrane separator but instead uses a thin (less than 1 mm) electrolyte flow-field channel to separate electrodes. In this configuration gaseous CO2 diffuses to the electrode–electrolyte interface through a gas diffusion layer (GDL), and crossover of reactants and products is controlled at laminar flow conditions because diffusion of the products is slow. Due to its compact design and high surface area to volume ratio, a microfluidic electrolyzer design could allow fast rates of CO2 mass transfer to the cathode surface and thus high CDs for CO2R.115,116 Additionally, microfluidic electrolyzers may provide new opportunities for fundamental studies into the effects of temperature, pH, catalyst deposition methods, electrolyte composition, GDL composition, and channel length on CO2R processes and thus help provide new insights for technology improvement.115,1173.5 Separation of CO2R products downstream of the electrolyzer
Efforts towards improving the selectivity of CO2R through catalyst innovations and optimization the electrolysis process are aimed partly at increasing the concentration of products in the electrolyzer effluents so as to the cost of product separation processes.118,119 Greenblatt et al.120 showed just how energy intensive separation of liquid products from a photo-electrochemical CO2R could be, with 4.7 to 45 MJ kg−1 of product required using distillation to recovery products from 10 wt% to 1 wt% in the catholyte stream from the electrolyzer.120 Greenblatt et al. reported more energy efficient technologies such as membranes and solvent extraction could potentially reduce the energy requirements to 0.1–8.3 MJ kg−1 product, but these technologies cannot always compete with distillation in terms of higher throughput and desired purity.120One strategy to avoid separation costs could be direct use of effluent streams from the CO2R reactor. For example, potential opportunities could include tuning the CO2R performance tuned to produce CO + H2 mixtures to feed a Fischer–Tropsch process, CH4 + C2H4 mixtures for synthesis of C3H6,121 or alcohol and/or hydrocarbon mixtures for liquid fuels.122,123 However, that strategy is likely only viable in a small number of circumstances where the CO2 source and the potential use of CO2R products are closely located and integrated. Therefore, consideration must be made for separation of CO2R product streams. One of the challenges with CO2R processes compared to many other industrial conversion processes is that the concentration of products, especially liquid products, leaving the electrolyzer are low. For example, in the conventional formic acid route of hydrolysis of methyl formate produces formic acid + methanol mixtures with more than 10% formic acid that are relatively easy to separate by distillation or liquid/liquid extraction.124,125 However, the concentration of formic acid leaving a CO2R electrolyzer is typically less than 1% in a mixture of water and the electrolyte salts. Recovery of the formic acid from this CO2R effluent stream requires an acidification process then azeotropic separation to obtain a pure formic acid product. Furthermore, formic acid separation processes are sensitive to pH like acidification and potential inorganic salts separation (e.g. crystallization) so conditions in the electrolyzer may affect downstream process.126
Control of the engineering factors described in the article (e.g. reactor design, electrode structure, electrolyte, pH, pressure, and temperature) may ultimately help to design efficient downstream separation processes. For example, the choices of conducting salts, solvents, IEMs, and flow rates can all effect the concentration and types of liquid CO2R products that must be recovered from the electrolyte. Yang et al.127 provide a clear demonstration of the relationships between reactor design and product distribution in their report of electrolyzer with a Sustainion™ AEM to obtain streams with up to 20 wt% formic acid from the reactor. In summary, the overall reactor design is a key factor that governs the properties of the CO2R product streams.
4 Electrode structure
The cathode in a CO2R electrolyzer must provide active catalyst sites, facilitate sufficient contact between CO2, electrolyte and catalysts, and conduct electrons. Fig. 7 depicts the three types of cathode architectures: (a) planar electrodes (e.g. a metal foil or glassy carbon plate),128–133 (b) simple porous electrodes (e.g. carbon paper or mesh),134–138 and (c, d) gas-diffusion electrodes (GDEs).139–141 Most CO2R electrocatalyst screening and fundamental catalysis studies30,31,47,142–144 use planar electrodes or simple porous electrodes because these types are relatively simple to construct and immerse in a CO2 saturated electrolyte (for example, in a electrochemical cell like that shown in Fig. 3a).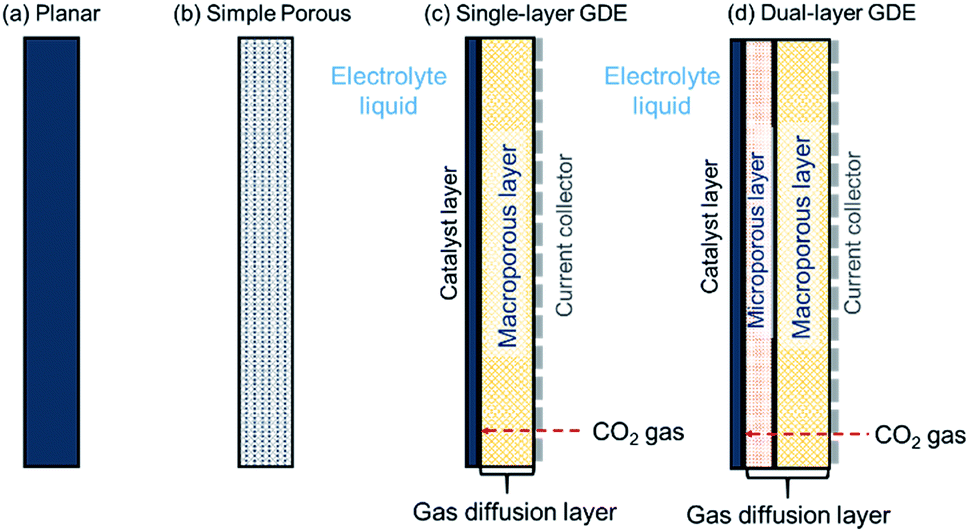 | ||
| Fig. 7 Schematic diagrams of (a) planar electrode, (b) simple porous electrode, (c) single-layer gas diffusion electrode (GDE), and (d) dual-layer GDE. | ||
The planar electrode is useful to screen catalyst materials in a laboratory scale because of its relatively simple geometry that rules out impacts induced by complex factors such as structures of the electrodes. In addition, placement of a reference electrode is straightforward, thereby enabling single electrode overpotential measurements. However, the CO2R half-cell reaction rates achieved with a planar electrode or porous electrode are often limited by the rate of CO2 transfer across the hydrodynamic layer from the bulk electrolyte to the electrode surface especially at a high CD.58 In such systems, mass-transfer rates could be improved by operating the electrolyzer at high pressure, low temperature, or selecting organic electrolytes to increase the solubility of CO2 in the electrolyte. But those options add costs and complexity to the CO2R process.
For high CD electrolyzers, one prefers the application of the last three electrode types (Fig. 7b–d) that are 3D-structured catalytically-active materials or electron conducting material with coverage of catalysts. A 3D-structure increases the active electrode area and decreases the transport resistance of gaseous and liquid reactants and products. Increasing the active electrode area reduces overall cell voltage and increase the rate of charge and mass-transport. Fast charge and reactant transport also accelerate the electrode kinetics. However, challenge arises in understanding the property-performance relationship and optimisation of the 3D-structure for efficient CO2R conversion. This results from the complexity of the 3D-structured electrodes that involves multiphase flow in the pores, interactions at interfaces and multiscale kinetics at the catalysts. Though this area still remains underexplored in CO2R application, a lot can be drawn from the studies in other electrochemical conversion applications such as PEM fuel cells and redox flow batteries. Recently, Shojaeefard et al.,145 Weber et al.,146 and Fadzillah et al.147 reviewed the electrode microstructure restructuring and pore-scale simulations. Moreover, Lai et al.148 and Walsh et al.149 reviewed the design and fabrication of general 3D-structured electrodes, including the 3D-electrode architecture and decoration of catalysts.
Overall, the ultimate goal of developing 3D-structured electrode is to minimise the various cell resistances due to electron and ion transport, multiphase flow (e.g. bubble), transport-related, and electrochemical reaction. The ohmic resistance of 3D-structured electrode is mainly influenced by both the 3D-skeleton and the interfacial conductivity. The 3D-electrode structure is a composite made of conductive matrix (carbon or metals) and less-conductive binders (e.g. PTFE or ionomers) and/or pores containing electrolyte or gas, thus requiring multiple percolations pathways for reactant molecules, ions and electrons.150–152 Simply put, increasing porosity decreases the overall conductivity of the matrix and therefore increases the overall ohmic resistance. The interfacial conductivity, governed by the interfacial contacts and heterogeneous phases, contributes more to the ohmic resistance. This is valid especially for a pure metal matrix as prepared through sintering, where sintering temperature, interfacial contact and sizes of metal particles and pore formers are important factors to consider.151 3D-structured electrodes decorated with heterogeneous catalyst materials also have extensive interfaces between the conductive backbone and less conductive catalysts, thus facing an increase of ohmic resistance. Therefore, the size and shapes of the catalysts and pore structures of the matrix also matter for the overall electrical conductivity of the structure.153 Compared with the ohmic resistance, the resistance related to transport is more critical, particularly for mass-transport-controlled CO2R electrolysis.111 In a 3D-porous electrode, transport of liquid and/or gas reactants and products is dominated by either molecular or Knudsen diffusion, depending on the pore size and electrode structures. Similarly, wettability and pore size dominate the intrinsic saturation/capillary-pressure relationship that is critical for optimal multiphase performance.146
The electrochemical reaction resistance is related to the electrode kinetics. In addition to the catalyst materials (where compositions, surface orientations, morphology, and sizes are important factors) that directly affect the kinetics, catalyst support, multiphase flows within the electrode and local environments are all crucial for CO2R surface reaction rate and selectivity. For the effects of catalyst support, one could refer to a recent review published by Li, MacFarlane, and Zhang,30 as well as herein. However, there are still gaps remaining, especially regarding the exact interfacial structure of the catalysts (especially with ionomer),146 and how these structures affect the electrode kinetics. Answering these questions is essential to guide where and how to deposit catalysts in the electrode structure. It is also important to note that an optimal balance has to be achieved among these resistances. For example, oxide-derived catalysts are active for CO2R154,155 but may not be very electrically conductive, leading to a decrease of electrode kinetic resistance but an increase of ohmic resistance. In the case where liquid products are targeted products, a high reaction rate consumes quickly the CO2 gas and lowers the local gas pressure, which may lead to local flooding that blocks gas transport and in turn degrades CO2R selectivity. Such a balance appears more crucial in the GDEs that include the transport of gases in the electrode structure. Because recent works have demonstrated a superior CO2R performance of GDEs compared to planar and simple porous electrodes, in the following subsections we mainly focus the review of recent GDE development for CO2R.
4.1 Gas diffusion electrodes
GDEs offer an alternative approach to improve mass-transfer and overall CO2R rates.156,157 The key difference between a GDE and a simple porous electrode is that in a GDE, the CO2 gas diffuses through a gas-diffusion layer (GDL) to the electrode–electrolyte interface inside the catalyst layer (CL). This type of GDE electrolysis has been reported to achieve reaction rates up to an order of magnitude faster than porous electrodes that require CO2 transfer from the bulk electrolyte to the electrode surface.58 A typical GDE, as shown in Fig. 8, consists of a GDL, a CL, and a current collector as shown in the two examples in Fig. 7c and d. The porous metal mesh or foam current collector serves to distribute current and maybe engineered with gas flow channels.142,158–160 The porous GDL should enable fast transport of gaseous CO2 to the CL, where the electrochemical reactions occur. A critical property of the GDL is that this layer must be hydrophobic (or more generally, the GDL must be gas wet relative to the wetting with electrolyte) to prevent liquid electrolyte from seeping to the gas flow channel. The GDL can be designed with a single-layer of porous materials (Fig. 7c) or dual-layers with different porosities and wettabilities (Fig. 7d). In a single-layer GDL, the hydrophobic macroporous layer might be a metal mesh or metal foam, or a hydrophobically-treated porous carbon such as polytetrafluoroethylene (PTFE) treated carbon paper. A dual layer GDL (Fig. 7d) includes a macroporous layer and a microporous layer (MPL).161 The MPL is normally a hydrophobic layer with small pores that is composed of carbon powder and PTFE.162 The catalyst layer (CL) is commonly prepared by depositing the catalytically active phase, including catalysts and an ionic binder on the GDL or membrane.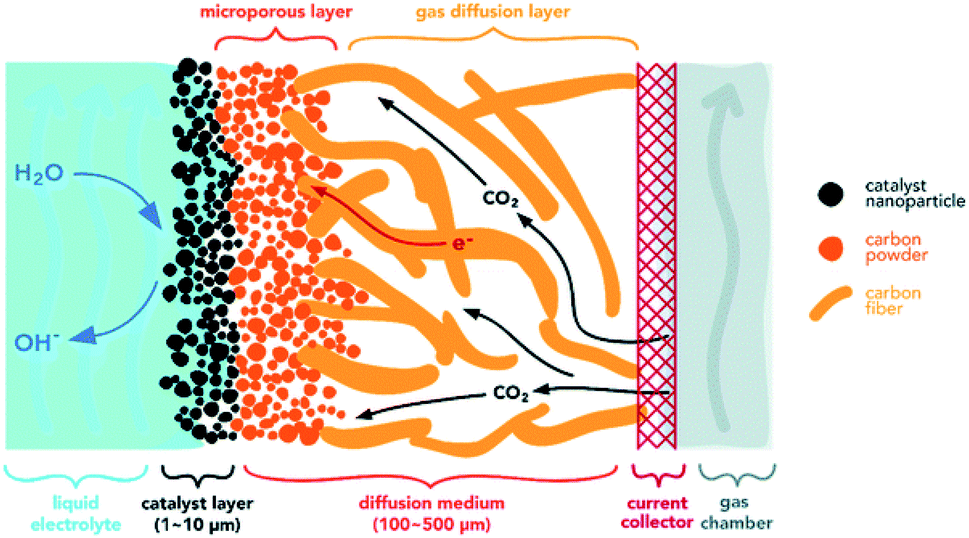 | ||
| Fig. 8 Schematic diagrams of a gas diffusion electrode.58 | ||
In a recent publication, Weng, Bell and Weber predicted with a mathematical model that a Ag-based GDE could potentially achieve a partial CD (PCD) for CO one order of magnitude higher than that achieved with a planar Ag electrode.58 That study concluded that good GDE performance is achieved by (1) a high density of active sites per geometric electrode area, and (2) a low mass-transfer resistance in the GDE, especially at more negative potentials. The predictions of the Weng, Bell and Weber modelling study are consistent with various experimental studies.58 For example, Castillo et al. reported that a Sn-based GDE with 1.5 mg Sn cm−2 achieved a maximum FE of ∼70% in producing formate at a current density of 40 mA cm−2, which was more efficient than the planar Sn electrode with a maximum formate FE of 67% at 12 mA cm−2.163 In another example, Hass et al.47 demonstrated a commercial Ag-based GDE, which was developed by Covestro as an oxygen depolarized electrode (ODE) for chlor-alkali applications, as a cathode in a CO2 flow cell electrolyzer with stable operation at a CD of 300 mA cm−2 and CO FE close to 70% for over 1200 h. A brief summary of other recent reports of GDEs as cathode for CO2R is provided in Table 2.
| Catalyst material | Catalyst loading | GDE configuration | Current collector | Remarks | Reference |
|---|---|---|---|---|---|
| Ag | NA | Dual layer | NA | Oxygen depolarization cathode (from Covestro) | 47 |
| Ag | 0.8 mg cm−2 | Dual layer | Carbon | 20 wt% PTFE MPL, 10 wt% carbon fiber substrate 190 μm | 162 |
| Cu | 20 nm | Dual layer | Carbon | (20 nm thick catalyst layer) | 45 |
| Cu (−350 mesh, 5N purity)/carbon | NA | Single layer | Cu gauze | Cu mixed with carbon black (CB, hydrophilic) and CBhydrophobic as the catalyst layer, Cu/(CBhydrophilic + CBhydrophobic) = 1.2 | 164 |
| Cu | 7 mg cm−2 | Single layer | Cu grid | 157 | |
| Cu2O/ZnO | 1 mg cm−2 | Single layer | Carbon | Air brushed on porous carbon paper | 165 |
| In/C | 1 ± 0.05 mg cm−2 | Single layer | Carbon | 140 | |
| La1.8Sr0.2CuO4 | NA | Single layer | Stainless steel mesh | Carbon/Teflon | 141 |
| Pt | 0.56 mg cm−2 | Single layer | Stainless steel mesh | 159 | |
| Pt/CNTs | NA | Dual layer | Carbon | Sigracet 25 BC GDL with imidazolate-based SIM-1 | 166 |
| Sn | NA | Single layer | Carbon | Sn electrodeposited on carbon fibers | 167 |
| Sn | 1.5 mg cm−2 | Single layer | Carbon | Sn on Toray carbon paper | 163 |
| Sn | 1.9 mg cm−2 | Dual layer | Carbon | Sn electrodeposited on dual layer GDL | 168 |
| Sn | 5 mg cm−2 | Single layer | Carbon | 11.1 wt% PTFE in catalyst layer | 169 |
| Sn nanoparticles (10–15 nm) | 0.75 mg cm−2 | Dual layer | Carbon | 144 | |
| Sn@Cu | NA | Dual layer | Cu mesh | Sn loaded on a Cu mesh through electroless deposition, and subsequently rolled on the GDL | 160 |
| Sn | NA | Single layer | Carbon | Sn electrodeposited on carbon fiber paper | 170 |
Although only a relative small number of studies report use of GDEs for CO2R,141,157,159,171 GDEs have been extensively developed and optimized for fuel-cell applications.161,172,173 The knowledge from this field can be leveraged to develop more efficient GDEs for CO2R, but the requirements for CO2-electrolyte contacts in a CO2R electrolyzer are more challenging than in a fuel cell. We describe in the following sections recent advances to optimize GDEs for use as the cathode in a CO2R electrolyzer.
4.2 Engineering the gas diffusion layer
The thickness and hydrophobicity of the macroporous layer predetermine the mass-transfer resistance of CO2 in the GDEs, and thus have an impact on the CO2R reaction rate. For example, Kim et al. compared the CO2 gas permeability and CO2R performance over Ag-based GDEs with carbon-fiber substrates (i.e. macroporous layer) in different thicknesses ranging from 170 to 380 μm.162 With the reduction of substrate thickness from 370 to 190 μm, they found that the CO2 gas permeability increases from 69.25 ± 0.69 to 72.42 ± 0.72 mL min−1, and that the PCD of CO also improves from ∼180 to ∼220 mA cm−2 at −2.05 V vs. Ag|AgCl. The enhanced PCD of CO2R for thinner substrates is attributed to the improved CO2 gas permeability. However, a too thin substrate (Toray Carbon Paper 30 with a thickness of 110 μm) leads to electrolyte flooding (i.e. the electrolyte fully occupies the pores) in the GDE during the CO2R operation, in essence turning into a simple electrode case. Therefore, an optimal thickness of the microporous layer is essential to ensure a balanced gas permeability and effective electrolyte management in the GDE.
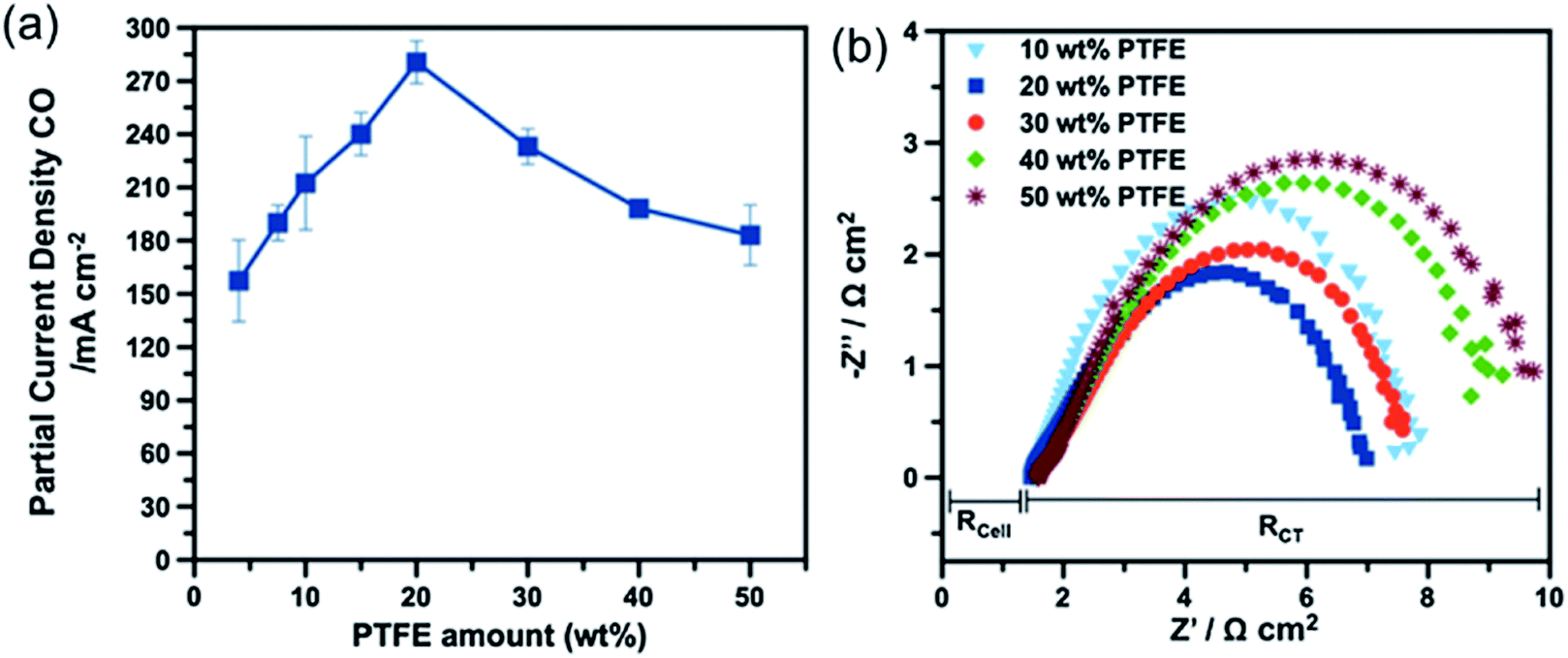
Another key factor is the wettability of the macroporous layer: too high a hydrophobicity may cause poor electronic conduction due to the high amount of non-conductive hydrophobic agents, while too high of a hydrophilicity may limit the diffusion of CO2 due to flooding propensity and promote unwanted HER.40,58 The wettability can be adjusted by controlling the content of hydrophobic agents (e.g. PTFE),161,177,178 and hydrophilic treatments (such as plasma treatments, addition of inorganic oxides or carbon black, etc.).161,179,180 Ikeda et al. observed similar CO2R reaction rates for the macroporous layer with PTFE contents between 10 and 30 wt%, but a degraded performance for the one over 30 wt%.164 A recent detailed investigation on Ag-based GDE showed that the macroporous layer with 10 wt% of PTFE has a higher CO PCD of 224 mA cm−2 than those with PTFE content of 30 wt% (PCD = 190 mA cm−2) and 50 wt% (PCD = 158.41 mA cm−2) at −2.05 V vs. Ag|AgCl.162 (Fig. 9a) The better performance for the GDEs with lower PTFE content in macroporous layer is a consequence of the better electronic conduction than those with higher PTFE content, as evidenced by the observed lower charge-transfer resistance from the electrochemical impedance spectra (Fig. 9b). In the studied PTFE range (10 to 50 wt%), additionally, the authors observed negligible effects of the PTFE content in the macroporous layer on the CO2 gas permeability and durability of the GDEs.
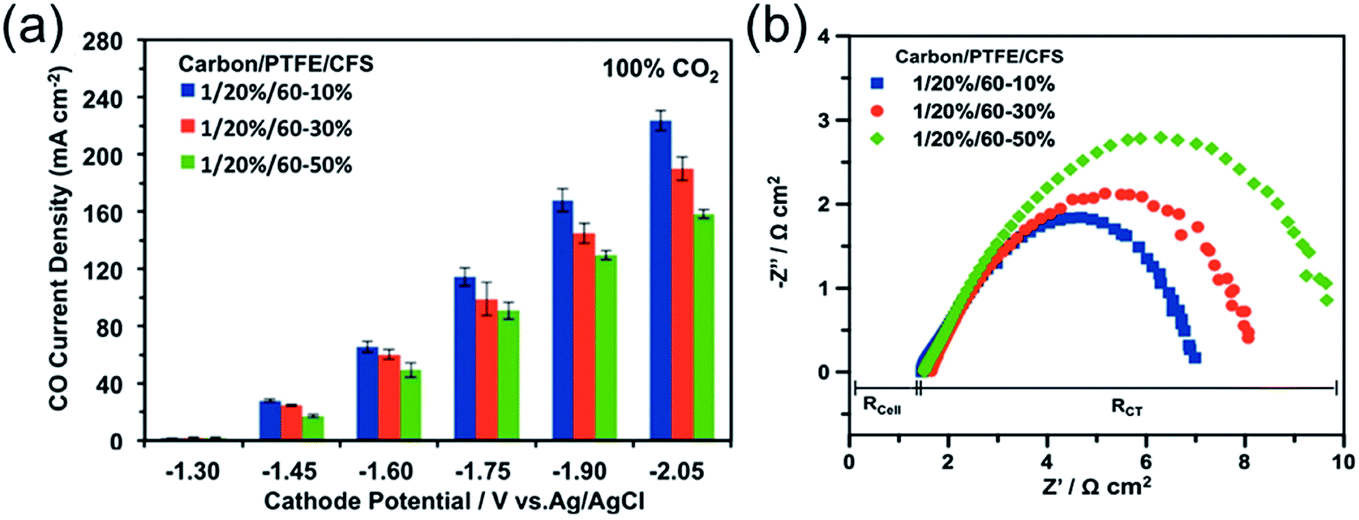 | ||
| Fig. 9 (a) A comparison of GDE with 10, 30, and 50 wt% PTFE in the macroporous layer as a function of potential. (b) The Nyquist plot of electrochemical impedance spectra of the corresponding GDEs at −2.0 V vs. Ag|AgCl. CFS means the carbon fiber substrate, which is the macroporous layer of the GDE. Rcell represents the ohmic resistance of the cell, and RCT is the polarization resistance related to charge transfer. Reprinted with permission from Kim et al.,162 Copyright 2016, Elsevier Ltd. | ||
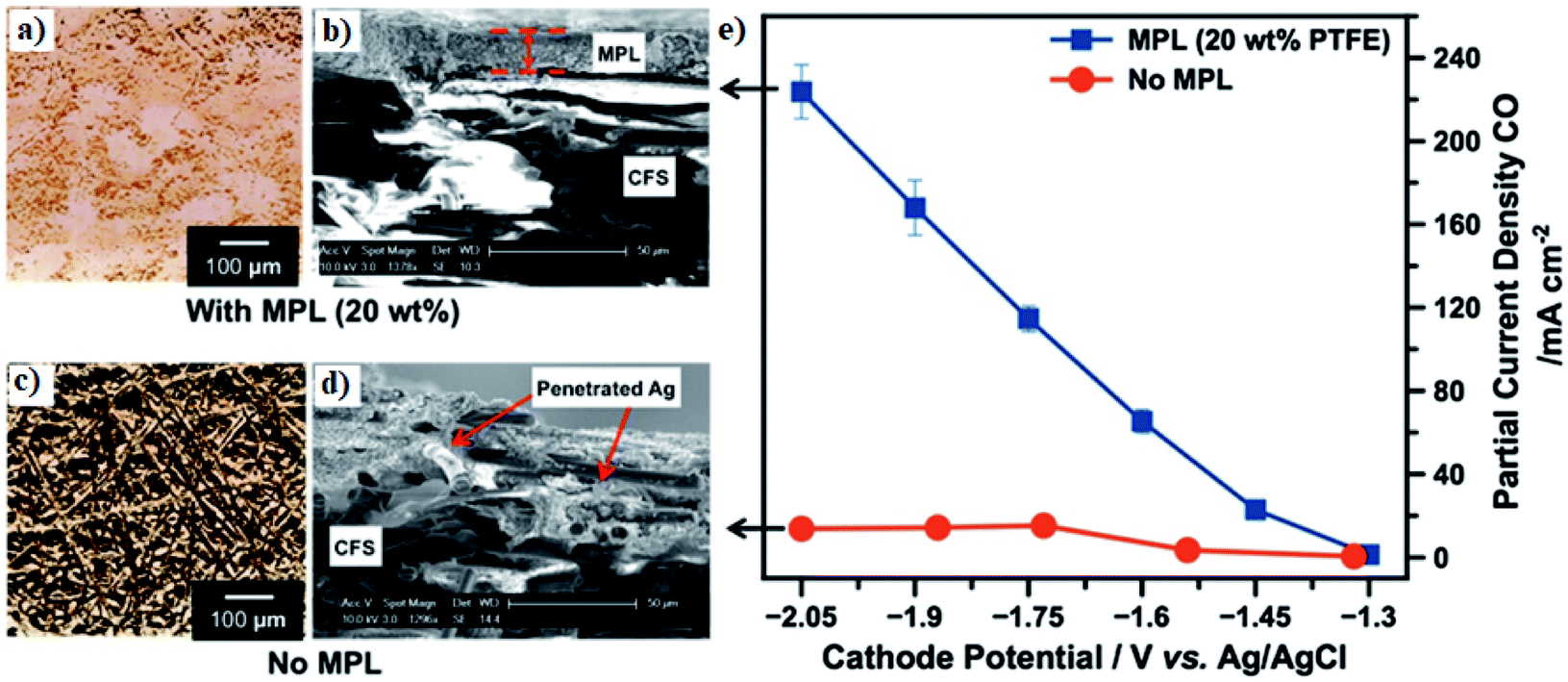 | ||
| Fig. 10 (a and c) Micro-computed tomography and (b and d) SEM images of Ag-based GDE with and without the microporous layer. (e) A comparison of CO partial CD of Ag-based GDE with and without MPL as a function of potential. Reprinted with permission from Kim et al.,162 Copyright 2016, Elsevier Ltd. | ||
Additionally, in the same work, the authors also studied the effects of PTFE content of the MPLs on CO2R reaction rate. As shown in Fig. 11a, the CO PCD increases with PTFE at low PTFE contents ≤20 wt%, but decreases with PTFE at contents >20 wt%.162 Too little PTFE content (i.e. 4.5–10 wt%) in the MPL is insufficient to prevent flooding of the layer by electrolyte and does not provide strong binding between the carbon and catalysts in the CLs, which resulted in the higher HER level and poorer cathode durability. A higher content of PTFE (>20 wt%) in the MPL degrades the electronic conductivity of the GDE (Fig. 11b) and also reduces the MPL porosity.185 Correspondingly, both the resistances of GDE for electron transfer and CO2 mass transfer become higher, thus leading to a degraded CO2R performance.
 | ||
| Fig. 11 (a) A comparison of PCD of CO for Ag-based GDE as a function of PTFE content in the MPLs. (b) The Nyquist plot of electrochemical impedance spectra for the corresponding GDEs at cathode potential of −2.2 V vs. Ag|AgCl. Reprinted with permission from Kim et al.,162 Copyright 2016, Elsevier Ltd. | ||
4.3 Optimization of the catalyst layer
The CL is mainly a layer of the electroactive cathode materials normally immobilized on one side of the GDL (typically on the MPL) facing the electrolyte or membrane through various deposition methods (e.g. electrodeposition,168 drop casting,45 thermal evaporation,45 ionic displacement, or pressing139), though the position of the CL can be also extended inside the GDL structure to further increase the electroactive interface boundaries.186 There are several factors that could contribute to the activity and product distribution of the CO2 electrolysis, such as the catalyst materials, catalysts loading (i.e. the mass of catalyst per electrode geometric area), ionic binders, and CL deposition methodology.Immense research efforts have been and continue to be devoted to the study and development of catalyst materials suitable for CO2R, and the recent advances in the catalyst development have been extensively reviewed recently.28,30,31,187,188 Most of these studies mainly focused on catalysts supported by a planar electrode or simple porous electrode, but not by a GDE, where the CL must deal with complex transport and reaction processes simultaneously and there is an inherent need for porosity.
The processes taking place at the CLs of GDE cathodes are even more complex than those in the GDL, since all of those processes occur as well as the electrochemical reactions and ionic transport. Weng et al. recently discussed in detail regarding the processes involved in the CL of an Ag-based GDE.58,189 In addition to local environment change close to the electrode surface in the electrolyte during CO2R operation, as discussed in the electrolyte selection and pH effects sections, the CL of the GDE also needs to manage the transport of gas and ions to achieve an optimal cathode performance. Too dry of a CL results in catalytically active sites becoming inactive due to the absence of supporting electrolyte or ionic pathways, but flooded pores increase the mass-transfer resistance of gaseous CO2. An ideal CL should facilitate a sufficient contact of liquid electrolyte and gas with the catalysts. Furthermore, the reaction kinetics are intimately connected with the transport phenomena. What also matters is the pore size in the CL. Small hydrophilic pores will be flooded when the liquid/gas pressure difference is low. When the pressure difference between gas and electrolyte increases, the susceptibility to electrolyte flooding of the pores in CL follows the trend: large hydrophilic pores > large hydrophobic pores > small hydrophobic pores.58
The amount of catalysts loading in the CL also alters the CD and product distributions. For example, through varying the Sn catalysts loading content ranging from 0 to 15 mg cm−2, Kopljar and co-workers found that a higher content of Sn increases the CO2R activity as evidenced by the observed higher CD and lower Tafel slope, especially at the higher potential range as shown in Fig. 12a.139 Such enhancement could be a result of the increased concentration of catalytically active sites as imparted by the higher Sn loading. Moreover, the product distribution was also found to be dependent on the catalyst loading: a Sn loading of less than 5 mg cm−2 could lead to ∼90% of FE for HCOO− formation, <10% for CO and 3% for H2, and further increase of the loading decreases the selectivity of the HCOO− production but promotes CO and H2 evolution (see Fig. 12b). The authors considered the effects of catalyst loading on product distribution analogous to the effects of cathodic potentials, where a low cathode potential leads to the promotion of CO and H2. Alternatively, we explain such phenomena by two possible reasons. First, a higher Sn loading may hamper the diffusion of CO2 to the active regions and therefore lead to the promotion of HER. Second, similar to the effects of interparticle interactions on Cu nano-particles for CO2R catalysis,190 a higher catalyst loading could increase the availability of neighboring active sites and therefore promote re-adsorption of HCOO− for further reduction to become CO as the final product.
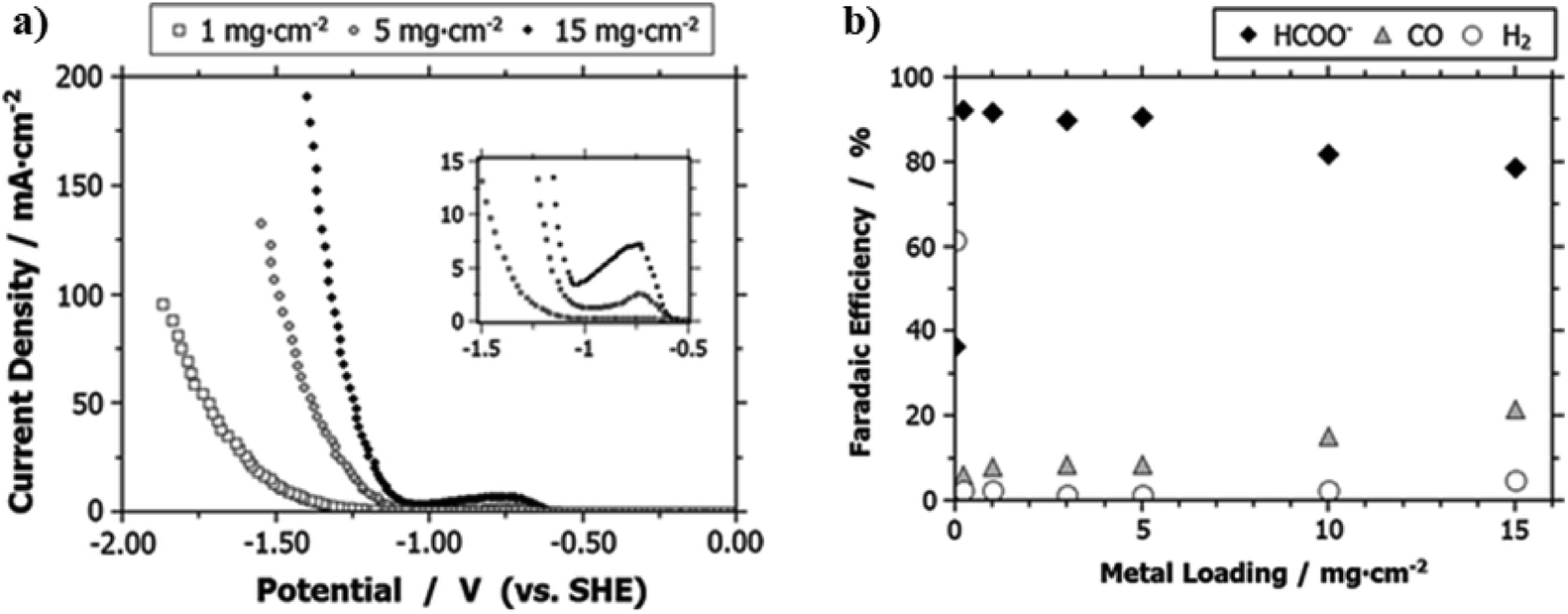 | ||
| Fig. 12 (a) A comparison of CD for GDEs with various Sn loading as a function of potential. (b) The FE for HCOO−, CO and H2 production for GDEs with different Sn loading at 50 mA cm−2. All the experiment were conducted in the 0.1 M KHCO3 aqueous electrolyte. Reprinted with permission from Kopljar et al.,139 Copyright 2014, Springer Nature. | ||
Sargent et al. recently used Cu-based GDEs with a very thin Cu CL (i.e. 10 or 25 nm thick) as the CO2R cathode to achieve a higher CD and higher FE for C2H4 formation than the ones with thicker Cu CLs (i.e. 1000 nm thick and ∼1000 μg cm−2 CL) at a higher cathodic potential (i.e. <−0.54 V vs. RHE).45 At lower cathodic potential (>−0.4 V vs. RHE), in contrast, the GDEs with thick CLs exhibited higher CDs than the thin CLs. They explained the higher FE for GDE with the thin Cu CLs by the catalyst-mediated abrupt interface as imparted by the thin layer of Cu, which could accelerate the rate-determining CO dimerization step for C2H4 formation.45 It could be thought that the thin layers and high flowrates of electrolyte also did not provide sufficient time for the homogenous acid/base reactions of CO2 to occur, thus resulting in higher CO2 local concentrations compared to the thicker CLs, where the CO2 and electrolyte residence time was higher. The multi-physics model of Ag-GDE developed recently for CO2R showed that a thinner CL could enhance the mass transfer of the CO2 in the GDE, which normally dominates the overall reaction rate at high cathodic potential.58,189 Consequently, the negative effect on CD due to the low density of active sites of thin CL could be less significant at high cathodic potential. This mechanism could also partially explain the higher CD of Cu-based GDEs with thin CLs at higher cathodic potentials, as experimentally observed by Sargent et al.45
In addition to the catalyst loading, the ionic binder also influences the cathode performance. Taking Nafion ionomer for example,84 it not only forms a continuous matrix in the CL that promotes cation conduction, but also has an impact on the microstructure of the CL that governs CO2 diffusion. For efficient CO2 electrocatalysis, therefore, an optimal balance needs to be achieved among the pores for CO2 diffusion, catalyst particles for electron conduction and catalysis, and ionomer for ion conduction.179 Through constraining the Sn loading, Zhou and co-workers found that the electrode performance is dependent on the content of the Nafion ionomer, and reported an optimal Nafion loading of 20 wt% in terms of CD and FE.179 Moreover, the ionomer in the CL can also serve as a co-catalyst promoting the CO2R reaction. An example is the incorporation of the imidazole-based ionomers to an Sn-based CL could stabilize the *CO2− intermediates and therefore enable the electroreduction of CO2 to HCOOH.127,191,192
The dispersion of the catalysts in the CL is another crucial factor for the electrode performance towards CO2R. Kenis's and coworkers studied the effects of catalyst dispersion by comparing the performance of Ag-based GDEs prepared using hand painting and air-brushing CL deposition methods.193 They found that the automated air-brushing technique renders a more uniform catalyst distribution and reduced particle agglomeration in the CL, as shown in Fig. 13a–c, thereby suppressing the HER and promoting the evolution of CO, though the overall CD was not significantly affected by the microstructural difference. They considered such product yield difference to be a result of exposed carbon from MPL that promotes HER, which is evidenced by the observed high HER PCD for bare GDL (Fig. 13d and e); a uniformly dispersed CL could reduce such carbon exposure and therefore suppress the unwanted HER.193
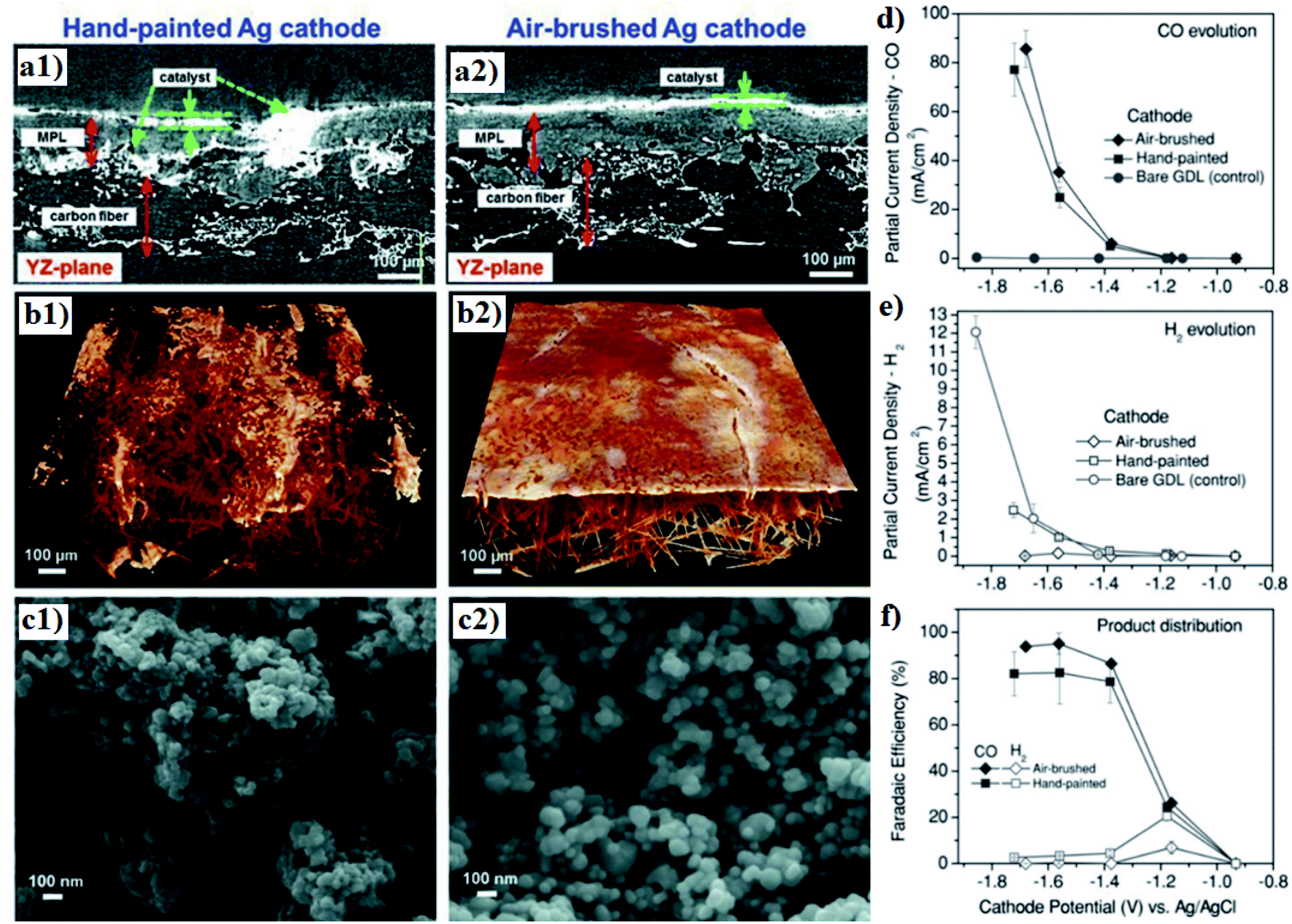 | ||
| Fig. 13 (a) SEM image of the cross-section (b) MicroCT 3D tomographic virtual models and (c) SEM topography of the Ag GDEs with hand-painted and air-brushed CLs. A comparison of the partial CD of (d) CO and (e) H2 as well as (f) product distribution for Ag-based GDEs fabricated through hand painting and air brushing. Reprinted with permission from Jhong et al.,193 Copyright 2013, John Wiley and Sons. | ||
5 Electrolyte selection
The primary function of the electrolyte in a CO2R electrolyzer is to conduct ionic charge between electrodes. An electrolyte generally consists of three components: an inert electrolyte or salt, the solvent (e.g. water), and the electroactive species. A good solvent should have (1) a high solubility for the reactant (CO2) and desired electrolyte to provide conduction; (2) electrochemically stable; (3) be chemically compatible with the electrode materials including the active catalysts; (4) low viscosity if liquid at the cell operating temperature to ensure good rates of CO2 mass transfer from the bulk electrolyte solution to the electrode surfaces194,195 and (5) easy to handle, storage, and safe. Water is the most common solvent for CO2R electrolytes because water satisfies the properties listed above, and can acts as both a proton donor and proton acceptor to facilitate production of different electroactive species.A key requirement of inert electrolytes commonly used in CO2R processes is that the electrolyte easily dissociates into cations and anions so as to provide a high ionic conductivity.196 However, the effect of the electrolyte ions on CO2R is far more complex than a simple charge carrier relationship.197,198 Even if the ions of inert electrolytes do not participate directly in redox reactions, an inert electrolyte can affect CO2R, for example, through interactions of electrolyte ions with radicals and ions produced in the CO2R reaction as described by Setterfield-Price and Dryfe.199
An operational issue common for all types of electrolytes is that impurities in the electrolyte can poison cathode catalysts. For example, trace metal impurities electrodeposited at the cathode during the CO2R process can lead to loss of CO2R selectivity with increased relative rates of the HER.18 Therefore, usually high purity inert electrolytes or electrolyte purification by pre-electrolysis is required. Alternatively, chelating agent such as ethylenediaminetetraacetic acid (EDTA)19 or a solid-supported iminodiacetate resin (Chelex)200 could be used to mitigate effects of impurities on CO2R.
5.1 Aqueous solutions of inorganic salts
The most commonly reported electrolytes for CO2R are simple aqueous, inorganic salt solutions such as potassium bicarbonate (KHCO3), and some common examples are summarized in Table 3. Although CO2 solubility in aqueous salt solutions201,202 may be low as compared to other CO2 capture solvents such as aqueous amine solutions;203,204 aqueous salt solutions are widely available at large scale and low cost; are relatively easy to prepare, handle, and store safely; and exhibit stable ionic conductivity.154,205–207 Since the 1950s the natural gas and ammonia industries have captured CO2 using the Benfield Process with hot solutions (100–116 °C) of potassium carbonate (K2CO3) to capture CO2 to form KHCO3.208 It's not surprising that given this long industrial history, KHCO3 solutions have become the most commonly reported aqueous electrolytes for CO2R.196,209–217 Carbonates are also chemically compatible with most electrode materials (relative to other conducting salts such as sulfides,218 sulphates,219 and halides190,220,221). Importantly, bicarbonate solutions provide capacity to buffer the local pH at the electrode surface during CO2R.56 The KHCO3 cases described here highlight the complexity of the effects of electrolyte choice in a CO2 electrolyzer, and to understand these effects more clearly, we discuss separately the roles of cations and anions in aqueous salt electrolytes.| Electrolyte | Catalyst | Major CO2R products (faradaic efficiency, %) | Applied potential (V vs. RHE) | Current density (mA cm−2)/mass activity (A g−1) | Reactor-typeref. |
|---|---|---|---|---|---|
| a GDE means gas diffusion electrodes. | |||||
| 0.1 M KClO4 | Cu nanowires | C2H6 (20.3%) | −1.10 | — | H-cell222 |
| 0.1 M K2HPO4 | C2H6 (10%) | ||||
| 0.1 M KHCO3 | C2H6 (17.6%) | ||||
| 0.1 M LiHCO3 | Cu foil | CH4 (32.2%), C2H4 (5.2%), C2H5OH (1.6%), HCOO− (4.7%) | −1.45 (vs. SHE) | 5 | H-cell223 |
| 0.1 M NaHCO3 | CH4 (55.1%), C2H4 (12.9%), C2H5OH (4.2%), HCOO− (7%) | −1.45 (vs. SHE) | |||
| 0.1 M KHCO3 | CH4 (32%), C2H4 (30.3%), C2H5OH (10.9%), HCOO− (8.3%) | −1.39 (vs. SHE) | |||
| 0.1 M CsHCO3 | CH4 (16.3%), C2H4 (30.5%), C2H5OH (2.4%), HCOO− (15.8%) | −1.38 (vs. SHE) | |||
| 1 M NaCl | Ag-GDE | CO (75%) | −1.87 (vs. Ag|AgCl) | 72.7 A g−1 | Flow-cell224 |
| 1 M KCl | CO (95.6%) | −1.84 (vs. Ag|AgCl) | |||
| 1 M RbCl | CO (93.6%) | −1.83 (vs. Ag|AgCl) | |||
| 1 M CsCl | CO (87%) | −1.81 (vs. Ag|AgCl) | |||
| 1 M NaBr | CO (60.8%) | −2.33 (vs. Ag|AgCl) | |||
| 1 M KBr | CO (96.6%) | −1.76 (vs. Ag|AgCl) | |||
| 1 M RbBr | CO (95.8%) | −1.80 (vs. Ag|AgCl) | |||
| 1 M CsBr | CO (93.6%) | −1.64 (vs. Ag|AgCl) | |||
| 1 M NaI | CO (80.8%) | −1.81 (vs. Ag|AgCl) | |||
| 1 M KI | CO (96.6%) | −1.64 (vs. Ag|AgCl) | |||
| 1 M RbI | CO (96.5%) | −1.59 (vs. Ag|AgCl) | |||
| 1 M CsI | CO (101.7%) | −1.56 (vs. Ag|AgCl) | |||
| 1 M NaOH | CO (83.0%) | −1.86 (vs. Ag|AgCl) | |||
| 1 M KOH | CO (96.7%) | −1.70 (vs. Ag|AgCl) | |||
| 1 M RbOH | CO (91.6%) | −1.63 (vs. Ag|AgCl) | |||
| 1 M CsOH | CO (89.8%) | −1.60 (vs. Ag|AgCl) | |||
| 3 M KHCO3 | Ag-GDEa | CO (82.5%) | −0.74 | 23.1 | Flow-cell225 |
| 3 M KOH | CO (101.5%) | −0.80 | 234.8 | ||
| 3 M KCl | CO (73.6%) | −0.81 | 10.7 | ||
| 0.1 M LiHCO3 | Ag foil | CO (59.1%) | −1.0 | 1.97 | H-cell62 |
| 0.1 M NaHCO3 | CO (68.4%) | 2.75 | |||
| 0.1 M KHCO3 | CO (82.9%) | 4.06 | |||
| 0.1 M RbHCO3 | CO (82.2%) | 4.65 | |||
| 0.1 M CsHCO3 | CO (80.3%) | 5.54 | |||
| 0.1 M LiHCO3 | Cu foil | CH4 (6.2%) | 2.40 | ||
| 0.1 M NaHCO3 | CH4 (17.7%), C2H4 (5.5%) | 2.57 | |||
| 0.1 M KHCO3 | CH4 (15.3%), C2H4 (10.2%), HCOO− (4.7%) | 3.03 | |||
| 0.1 M RbHCO3 | CH4 (13.2%), C2H4 (24.4%), C2H5OH (9.6%) | 4.03 | |||
| 0.1 M CsHCO3 | CH4 (9.4%), C2H4 (31.1%), C2H5OH (11.4%) | 4.80 | |||
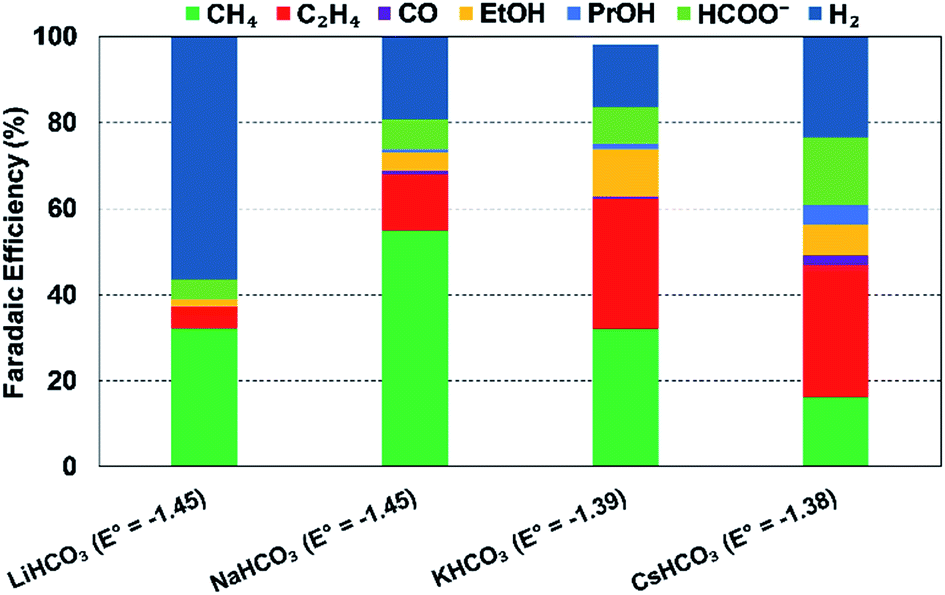 | ||
| Fig. 14 Effect of cationic species (different 0.1 M bicarbonate solutions) on FEs of various products at a Cu electrode and at 5 mA cm−2; Eo values are vs. SHE, reproduced from the data in Akira and Hori (1991).223 | ||
Thorson et al.224 proposed that adsorbed cations at the electrode surface could stabilize the intermediate *CO2− and thus promote the CO2R. This theory could explain the observed enhancement in total CO2R efficiencies with cation size from Li+ to K+ (Fig. 14). Recently, Kim et al.232 provided further evidence to support this proposed mechanism by observing an efficient CO production over a Au electrode in K+-based electrolyte (K+ is prone to adsorb at the Au electrode) than in a Na+-based one. Further DFT calculations reported by Liu et al.233 revealed that higher electron-density of adsorbed K+ close to the carbon atom could facilitate in the stabilization of intermediates such as *COOH and *CO.
However, there remains some debate around the conclusions of Thorson et al. For example, Mills et al.234 and Strmcnik et al.235 argued that the specific adsorption of cations seems impossible under the operating conditions for CO2R (usually for potentials larger than −1.4 V vs. a normal hydrogen electrode (NHE, potential defined at 1 M H+ concentration and 1 atm pressure)). Bell and co-workers proposed that the observed cationic effect originates from the hydrolysis of the solvated cations close to the cathode surface.62 At the cathode surface, pKa of hydrolysis decreases significantly due to an increasing electrostatic force between the water molecules in the hydration sphere of cation and negatively charged cathode. These interactions can further polarize O–H bonds of water molecules, and consequently facilitate water dissociation to increase the local proton concentration as illustrated in Fig. 15a. This effect decreases the local pH, which permits an increase of dissolved CO2 concentration locally that can approach the CO2 concentration in the bulk electrolyte (Fig. 15b and c). The net effect reported by Singh et al.62 was that switching from Li+ to Cs+ in an aqueous electrolyte suppressed HER and significantly promoted CO2R over Ag cathodes (Fig. 15d) or Cu cathodes (Fig. 15e).
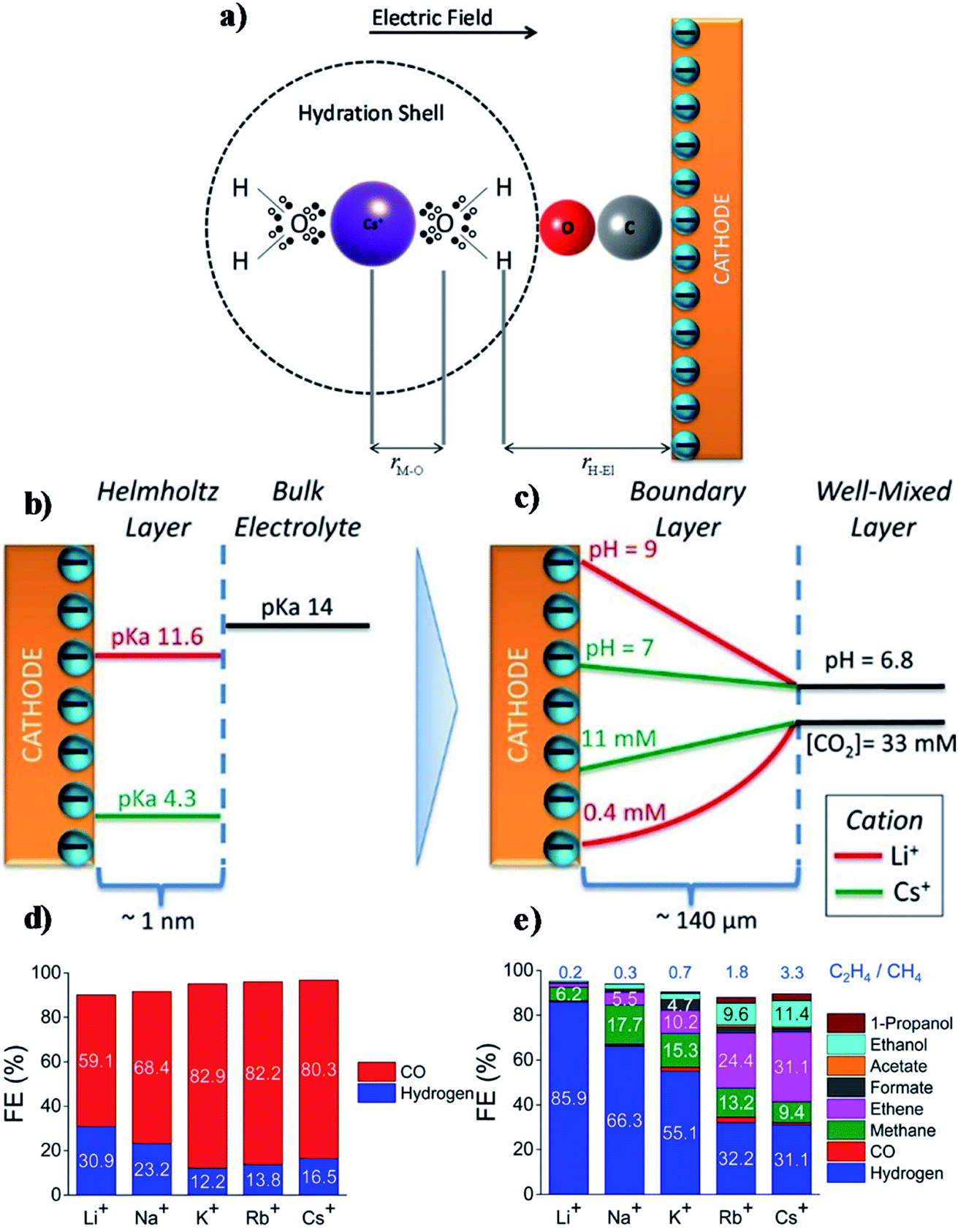 | ||
| Fig. 15 (a) Interaction of hydrated cation with the negatively charged cathode surface. The additional electrostatic force between the H atom of the H2O in the primary hydration shell and the cathode causes a decrease in the pKa of hydrolysis; (b) pKa of hydrolysis of hydrated Li+ and Cs+ inside the Helmholtz layer and in the bulk electrolyte; (c) concentration of CO2 and pH distribution in the boundary layer; (d and e) FE of different products over Ag and Cu electrodes respectively at −1.0 V vs. RHE. The electrolyte was saturated and the concentration was kept at 0.1 M XHCO3 (X = Li, Na, K, Rb, and Cs). Reprinted (adapted) with permission from Singh et al.,62 Copyright 2016, American Chemical Society. | ||
Note that these interpretations of the effects of cation hydrolysis only explain experimental observations at potentials less than −1.1 V vs. RHE. The phenomenon of hydrolysis of solvated cations is only applicable when (1) the pH of the electrolyte is close to 7; and (2) the pKa for cation hydrolysis is close to local pH at the electrode; and (3) the reactant concentration is pH dependent.62 The hydrolysis of the solvated cations close to the cathode surface does not adequately explain the same cationic effects in CO reduction,223 because the CO concentration is not pH dependent. Koper et al.236 used density functional theory (DFT) calculations to predict the effect of cations on CO reduction to C2H2 at pH close to 13, and found that cations could stabilize intermediates, especially dimers *OCCO and *OCCOH, via interactions of oxygen atoms in these intermediates.236 Moreover, the shift in average reaction energies (for Li+, Na+, and Cs+) of CO to C1 products is close to that reported by Nørskov and co-workers,60,61 who showed that the cation-induced local electric effect could alter the free-energy landscape of CO2R to CO by stabilizing the key reaction intermediates.236
Bell along with other JCAP researchers63 conducted experimental measurements and DFT calculations at low cell potentials, and reported electrostatic interactions between the hydrated cations located at the OHL and the adsorbed reaction intermediates that have high dipole moments (such as *CO, *CO2, *OCCO) at the cathode surface. Such electrostatic interactions lower the energy required for *CO2 adsorption (an intermediate for HCOO−) and for C–C dimerization to form *OCCHO or *OCCO (which are the key intermediates for C2H4 and C2H5OH, respectively).63 As a consequence, the PCD for H2 and CH4 formation are uninfluenced by the cation size, while the PCD for HCOO−, C2H4, and C2H5OH enhanced with cation size.
In future research, the investigation of monovalent cations for CO2R should be expanded to multivalent cations. Schizodimou and Kyriacou237 showed that rates of CO2R can be accelerated by increasing the size of cations and the surface charge of cations. They demonstrated that the CO2R rate is almost two-fold higher in electrolyte containing La3+ than in electrolytes containing Na+ at similar operating conditions.
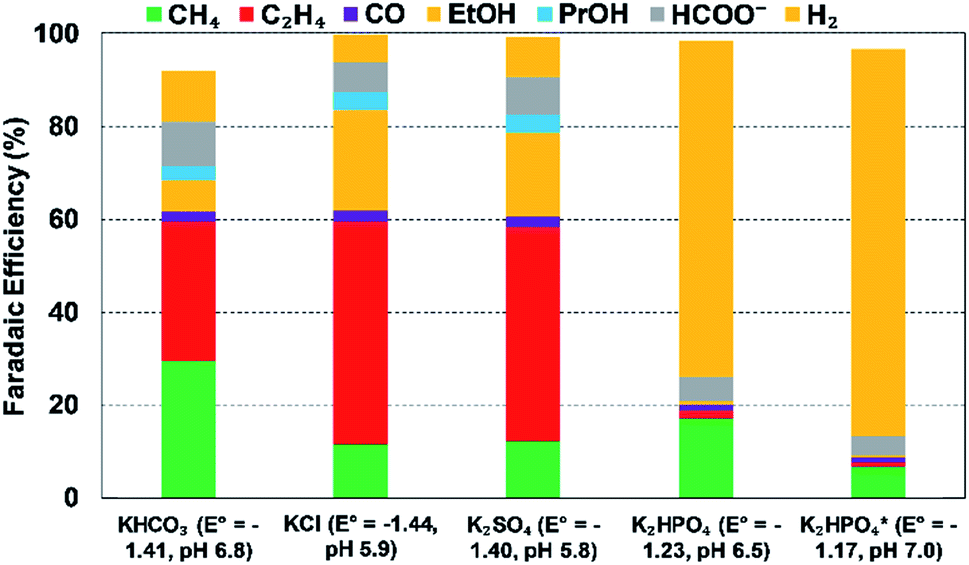 | ||
| Fig. 16 Effect of anionic species (different 0.1 M bicarbonate solutions) on FEs of various products at a Cu electrode and at 5 mA cm−2; Eo values are vs. SHE; *refer to 0.5 M, reproduced from the data in Hori et al.238 (1989). | ||
The specific adsorption of anions on cathode can also alter the CO2R activity and selectivity.190,225,245,246 In particular, the adsorption of halide ions on the electrode surface can alter the electronic structure of the catalysts, and thereby influence the interactions between the electrode surface and intermediates.190 For example, Varela et al. showed that Br− and Cl− enhanced CO formation, but I− facilitated formation of CH4 instead of CO.190 Moreover, specifically adsorbed halide anions can suppress proton adsorption and thus prefer CO2R to HER for a Cu-metal electrode.246 This can be attributed to the presence of covalent interaction between the halides and Cu electrodes, which facilitates the transfer of electrons from Cu-surface to CO2.246 In addition, several studies have reported the changes of morphology of a catalyst's surface in the presence of halide anions, especially via oxidation–reduction cycles to promote C2+ products over copper catalysts.66,68,247–249 However, it still remains unclear which anion factor influences CO2R reaction the most.
5.2 Organic solvent electrolytes
Some typical examples of organic solvent electrolytes used for CO2R are shown in Table 4 with dimethyl sulfoxide (DMSO), dimethylformamide (DMF), propylene carbonate (PC), and acetonitrile (ACN)250,251 the most commonly reported organic solvents used in CO2R. The solubility of CO2 in organic solvents could be much higher than in water (as presented in Fig. 17). As a result of their higher CO2 solubility and absence of proton, organic solvents can achieve higher CD and better product distribution for CO2R than in aqueous electrolytes.27,197,198,229 Vassilev et al.252 reported that DMSO, DMF, PC, and ACN assist in dimerization of *CO2 with adsorbed CO2 molecules to form oxalate (C2O42−) over Pb, In, Sn, and Hg cathodes, which tend to produce HCOO−/HCOOH in aqueous electrolytes.229 Moreover, the overpotential required to convert CO2 into products is lower than that in aqueous solution, making the process energy-efficient.253 Mixed electrolytes containing both organic solvents and water are reported, which allows tuning of proton concentrations.254 Hori et al.254 found that upon increasing the concentration of water in ACN, CO2R favors the formation of formic acid rather than oxalic acid.| Electrolyte | Catalyst | Major CO2R products (faradaic efficiency, %) | Applied potential (V vs. RHE) | Current density (mA cm−2) | Reactor-typeref. |
|---|---|---|---|---|---|
| a At high pressure (10 atm). b At low temperature (−30 °C); PC – propylene carbonate; TBAP – tetrabutylammonium perchlorate; TEAP – tetraethylammonium perchlorate; DMF – dimethylformamide; LiCl – lithium chloride; CsOH – cesium hydroxide. | |||||
| 0.1 M TBAP/PC | Au foil | CO (91.8%) | −3.02 (vs. Fc/Fc+) | 2.8 | H-cell260 |
| 0.1 M TEAP/PC | Pb foil | H2C2O4 (73.3%) | −2.4 (vs. SHE) | — | H-cell250 |
| 0.1 M TEAP/PC | In foil | CO (85.3%) | |||
| DMF | Pb foil | CO (n.a.) | — | — | H-cell251 |
| 0.5 M LiCl/methanol | Cu foila | CO (∼65%), CH4 (20%) | −2.8 (vs. SHE) | 15 | H-cell261 |
| 0.08 M CsOH/methanol | Cu foil | CH4 (8.3%), C2H4 (23.7%) | −4.0 (vs. Ag|AgCl) | — | H-cell262 |
| 0.08 M benzalkonium/methanol | Cu foilb | CO (∼7%), CH4 (42.5%), C2H4 (2.1%) | −2.0 (vs. SCE) | — | H-cell263 |
 | ||
| Fig. 17 The solubility of CO2 in various organic solvents at 298 K and 1 atm.255–258 *Note: CO2 solubility in 5 wt% K2CO3 was calculated from the CO2 loading data (0.830 mol CO2/mol K2CO3)257 and since it includes both chemical and physical CO2 solubility, therefore, 5 wt% K2CO3 has higher CO2 solubility than organic solvents. Moreover, 5 wt% K2CO3 has higher CO2 loading than 30 wt% aqueous monoethanolamine solution, which is 0.540 (mol CO2/mol amine) at 298 K and 2.80 kPa (PCO2).259 | ||
Although organic solvents offer higher CO2 solubility than aqueous electrolytes, some critical disadvantages of organic solvent electrolytes are their high cost, their volatility and flammability, and possible toxicity. As the organic solvents are not typically consumed in the CO2R reaction it could be possible to recycle solvents to reduce the operational costs of CO2R.198 However, recycling of organic solvents from the electrolyzer liquid product may be complicated and costly due to the volatility and toxicity of the solvent and potential miscibility with the desired products.260
5.3 Ionic liquids
Ionic liquids (IL) have been proposed as alternatives to aqueous amines for CO2 capture process264 because ILs exhibit high CO2 solubility265 and high selectivity for CO2 over other gases such as N2, O2, and CH4.266 Ionic liquids also exhibit other desirable properties for CO2R electrolytes such as excellent thermal stability,267,268 stability across a wide electrochemical potential window,268,269 high ionic conductivity,268 and low vapor pressure.268,270 In addition, some ILs can form a complex with the intermediates during CO2R and thus lower the energy barrier of the reaction.191Table 5 provides a summary of some ILs reported as CO2R electrolytes. The most extensively studied ILs for use in CO2R electrolyzers are two commercially available ILs: 1-ethyl-3-methylimidazolium tetrafluoroborate ([EMIM][BF4]), and 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIM][BF4]).191,271,272| Electrolyte | Catalyst | Major CO2R products (faradaic efficiency, %) | Applied potential (V vs. RHE) | Current density (mA cm−2) | Reactor-typeref. |
|---|---|---|---|---|---|
| a [EMIM] – 1-ethyl-3-methylimidazolium; [BMIM] – 1-butyl-3-methylimidazolium; [TBA] – tetrabutylammonium; [BMMIM] – 1-butyl-2,3-dimethylimidazolium; [DMPIM] – 1,3-dimethyl-2-phenyl-imidazolium; [BF4] – tetrafluoroborate; [PF6] – hexafluorophosphate; [NTF2] – bis(trifluoromethylsulfonyl)imide; [CN2] – dicyanamide; ACN – acetonitrile. | |||||
| 18 mol% [EMIM][BF4]/water | Ag-GDE | CO (∼96%) | 1.5 to 2.5 V (cell potential) | — | Flow-cell191 |
| [EMIM][BF4] | Ag NPs (40–200 nm) onto Sigracet carbon paper (5 mg cm−2) | CO (−) | 3.25 V (cell potential) | 4 | Flow-cell |
| 0.02 M [EMIM][BF4]/0.1 M [TBA][PF6]/ACN | Bi electrodeposited onto a glassy carbon electrode | CO (93 ± 7%) | −1.95 (V vs. SCE) | ∼3.77 | H-cell279 |
| 0.02 M [BMIM][BF4]/0.1 M [TBA][PF6]/ACN | CO (95 ± 6%) | ∼5.51 | |||
| 0.02 M [BMIM][PF6]/0.1 M [TBA][PF6]/ACN | CO (90 ± 9%) | ∼4.82 | |||
| 0.02 M [BMMIM][BF4]/0.1 M [TBA][PF6]/ACN | CO (76 ± 6%) | ∼0.67 | |||
| 80 wt% [BMIM][Cl]/water | Ag foil | CO (>99%) | −1.5 (V vs. SCE) | — | H-cell271 |
| 4 mol% [EMIM][BF4]/water | Bulk MoS2 | CO (∼98%) | −0.764 | 65 | H-cell275 |
| 0.1 M[EMIM][NTf2]/ACN | |||||
| 0.02 M [DMPIM][BF4]/0.1 M [TBA][PF6]/ACN | Ag foil | CO (∼100%) | −1.48 (V vs. Fc/Fc+) | 4.2 | H-cell282 |
| 0.5 M [EMIM][N(CN2)] | Sn NPS/glassy carbon | HCOO− (81.9%) | −1.2 | ∼4.18 | H-cell283 |
| 0.1 M [TBA][PF6]/ACN | Ag foil | CO (74%) | −2.1 (V vs. SCE) | 17 | H-cell284 |
| 0.5 M [BMIM][PF6]/0.1 M [TBA][PF6]/ACN | CO (97%) | 50 | |||
| [BMIM][PF6] | Pb foil | CO (∼100%) | −2.2 (V vs. Ag|Ag+) | 0.33 | H-cell285 |
| 30 wt% [BMIM][PF6]/ACN | HCOOH (46.3%), CO (40.2%) | 2.63 | |||
| 30 wt% [BMIM][PF6]/5 wt% water/ACN | Pb foil | HCOOH (91.6%) | −2.3 (V vs. Ag|Ag+) | 37.6 | |
| 30 wt% [BMIM][PF6]/5 wt% water/ACN | Sn foil | HCOOH (92%) | 32.1 | ||
In 2011, Rosen et al.191 first reported the catalytic effect of an 18 mol% aqueous solution of ([EMIM][BF4]) in the CO2R to CO over an Ag electrode at low overpotentials to achieve FECO of over 96%. Fig. 18 illustrates Rosen et al.'s191 hypothesis that an IL cation can stabilize the *CO2 intermediate, and therefore substantially lower the energy barrier for the formation of *CO2 and its subsequent reduction to CO.191,273,274 Subsequently, the Rosen's team investigated the effect of water on CO2R in ([EMIM][BF4]) solutions and found that addition of water in the IL enhanced the CO selectivity, with an optimal CO selectivity close to 100% in a solution of 10.5 mol% IL in water. Such performance enhancement was attributed to a synergistic effect of mass-transfer improvement due to lower the electrolyte's viscosity by dilution with water and an optimal pH (pH around 3.2 at 89.5% water).274
 | ||
| Fig. 18 A change in free energy of CO2 to CO in water (solid line) to CO2 to CO in EMIM-BF4 (dashed line). Reprinted with permission from Rosen et al.,191 Copyright 2011, American Association for the Advancement of Science. | ||
Following Rosen et al.'s work,191 others have attempted to understand the catalytic effect of the ILs in CO2R, and although there is still more to understand about the reaction mechanism most reports suggest that the cation is primarily responsible for the observed catalytic effects.271,275–278 For example, Rosenthal and co-workers proposed that ILs serve as a proton source by investigating the catalytic performance of the Bi-based catalyst in AN solutions of [BMIM]+-based ILs.279,280 Deprotonation from the C2 position (refer to Fig. 19) of the [EMIM]+ or [BMIM]+ cation would result in the conversion of CO2 to CO via the 2e−/2H+ reduction pathway, which has a lower energy barrier.279,281,282
 | ||
| Fig. 19 Structure of imidazolium cation; white is H2, grey is C and blue is N2. Reprinted (adapted) with permission from Lau et al.,282 Copyright 2016, American Chemical Society. | ||
Despite the many promising properties of ILs for CO2R electrolytes, the high viscosity of ILs can severely limit the rates of CO2 diffusion, and thus ILs achieve very low CDs272 and, as shown in Table 5, most CO2R studies dilute ILs in aqueous solutions or organic solvents such as AN, DMF, or CH3OH to reduce the electrolyte's viscosity.191,271,275,277 Progress in development and application of ILs has progressed rapidly in the last 30 years to provide a large amount of fundamental understanding of IL properties and physicochemical data, and to lower IL production costs significantly. Thus, ILs are becoming more available and could soon be viable options for use in CO2R electrolyzers.
6 pH effects
The pH of the electrolyte affects the phase stability of CO2 in aqueous solutions as illustrated by the Pourbaix diagram in Fig. 20.229 The Pourbaix diagram highlights the acid/base homogeneous interactions of CO2 into different forms, such as carbonic acid (H2CO3), bicarbonate (HCO3−), carbonate (CO32−) and methanol (CH3OH) in water as a function of pH and potential. reaction (6) shows that the introduction of CO2 into an aqueous solution entails a complex series of reversible reactions between CO2, water, carboxylic acid, and its deprotonated species. In solutions with a pH up to about 6, CO2 is in the form of a weak carboxylic acid; at pH between 6 and 10.3, HCO3− anions are formed; and at pH above 10.3 HCO3− deprotonates further to CO32− (reaction (7)). Due to the various reactions and the feed CO2, most aqueous solutions are normally in the HCO3− form and around pH 9 unless specific measures are used to counteract them (e.g., high flow rates).45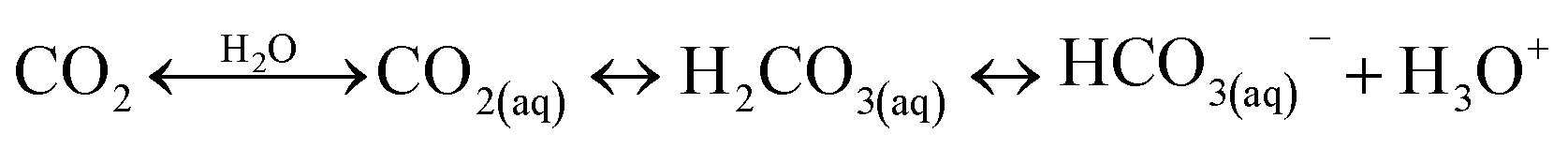 | (6) |
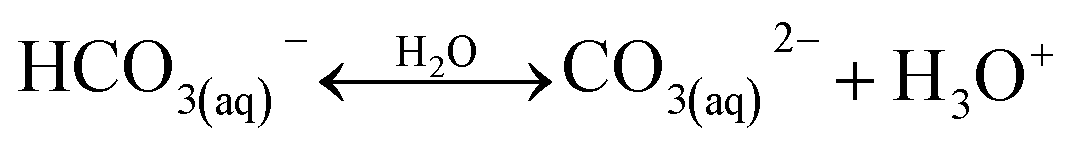 | (7) |
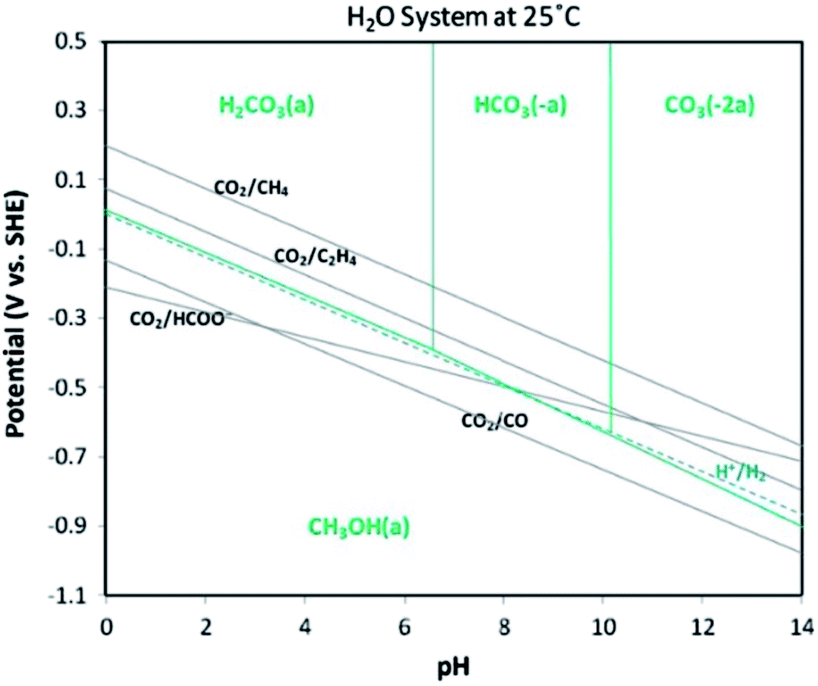 | ||
| Fig. 20 Pourbaix diagram for electrochemical CO2R at 25 °C. Reprinted with permission from Ganesh,286 Copyright 2014, Elsevier Ltd. | ||
Note that the local pH near the cathode surface is usually different to that in the bulk electrolyte due to the catalytic reactions generating OH− or consuming H+, and diffusion limitations.287 Despite the significance of local pH on CO2R as discussed in Hori's works,229,238,239 only recently has the influence of local pH been re-re-examined.55,287–292 Additionally, some electrolytes, such as bicarbonates provide pH buffering effects (see Section 5.1.2 for more details).240
As the major competing side reaction, HER needs to be minimized for an optimal CO2R reaction. Therefore, pH is crucial in determining the HER activity, as it characterizes the availability of the protons. Although CO2R produces OH−, its equilibrium potential is not much influenced by the pH in comparison to HER.286 A rise in pH moves the equilibrium potential of HER to a more negative value, thus significantly slowing down HER.293 As shown in Fig. 21, the equilibrium potential denoted by Erev reflects the potential of the HER derived from the Nernst equation. Moreover, the mechanism of HER switches from protons-to-H2 in acidic media to water-to-H2 in alkaline media, where the kinetics of latter is significantly slower and does not have a pH dependence, although the concentration is typically much greater.294–297
 | ||
| Fig. 21 Hydrogen evolution polarization data at copper (Cu) with varying solution pHs. Reprinted with permission from Gattrell et al.,293 Copyright 2006, Elsevier Ltd. | ||
The pH is also an essential factor in determining the products of CO2R. Taking Cu as an example, an increase of pH shifts the product selectivity from H2 and CH4 to higher carbon products such as C2H4.45,238,241,298,299 The HER suppression at higher pH can be easily described by the reduced availability of H+/Hads. However, how pH affects CO2R products (i.e. CH4 preferred at low pH and C2H4 at higher pH) is very complex, and is likely related to the multiple proton-coupled-electron steps involved in their various reaction pathways, and the rate-determining steps in those various microkinetics.294,298,300–302 The different onset potentials238,299,303 and Tafel slopes304–306 for CH4 and C2H4 evolution suggest that they follow different reaction pathways. Hori and co-workers proposed that C2H4 evolution is pH independent, while the pathway for CH4 evolution is pH-dependent, during or prior to the rate-determining step (RDS).306 By using online electrochemical mass spectrometry (OLEMS), however, Schouten's group found that the onset potential for forming CH4 and C2H4 are both pH-dependent particularly over Cu (111) electrode.307,308 Schouten and co-workers proposed two possible reaction pathways for the formation of C2H4 as illustrated in Fig. 22a. One pathway is pH-independent, involving the formation of CO dimer where proton transfer is not the RDS, and preferentially takes place at Cu (100) facets. This pathway has also been theoretically confirmed by Calle-Vallejo and Koper.309 Another pathway is pH dependent, sharing the same *CHO intermediate, which is also critical in evolving CH4, and takes place both at Cu (100) and (111) facets. More interestingly, the overpotential for both CH4 and H2 evolution is lowered at a very high pH (e.g. pH = 13, see Fig. 22b).308,310 The reduced overpotential of HER at higher pH could be a result of the possible reaction pathway shift from pH dependent (due to proton availability) to pH independent (due to water discharge).293,311 Likewise, it could be understood that a similar shift likely takes place during the formation of the CH4 at higher pH. Based on this understanding, electrolytes with higher pH can favor the formation of higher hydrocarbon products (especially over copper catalysts). Recently, Sargent and co-workers45 showed that strong alkaline media (7 M KOH) accelerates the kinetics of CO2R by lowering the C–C coupling energy barrier over Cu catalyst and could achieve a C2H4 FE of ∼70% at −0.55 V vs. RHE over 150 h of continuous CO2R operation. Therefore, more researchers are investigating KOH as electrolyte to further enhance the efficiency and selectivity of >C2 products.45,312
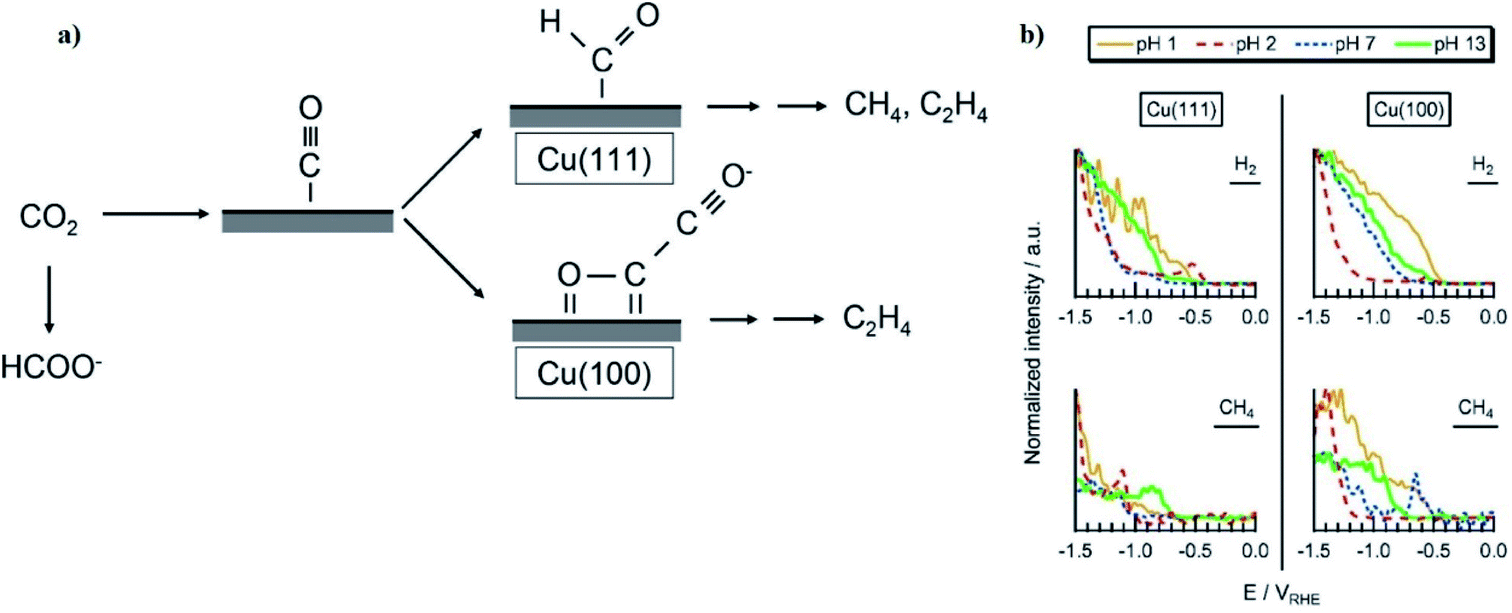 | ||
| Fig. 22 (a) The proposed reaction mechanism for the reduction of CO2 on Cu single crystal electrodes and (b) the reduction of CO in 0.1 M HClO4 (pH 1), 0.2 M NaClO4 (pH: 2 and 7) and 0.1 M NaOH (pH: 13) on Cu (111) left and Cu (100) right. Reprinted with permission from Schouten et al.,308 Copyright 2014, Elsevier Ltd. | ||
Additionally, it could be understood that a rise in the local pH could be beneficial for >C2 products; however, with a rise in local pH, the equilibrium of CO2 and water neutralization reaction shifts more towards bicarbonate formation, which eventually depletes the local CO2 concentration and thus reduces the CO2R selectivity by promoting HER.62,290 This conflicting statement really begs a question on whether a high local pH is really desirable or deleterious for the CO2R reaction. At higher pH, CO2 may still exist in low quantity (if ample CO2 is transported into the solution) due to its limited hydration kinetics.242,313 Therefore, an optimal pH may exist for an efficient reaction and sufficient CO2 supply. Recent modeling studies found a relationship between pH and selectivity of CO2R with a maximum CO2 selectivity over an optimal local pH range of 9 to 10 (especially for C2 products).154,314 Interestingly, at higher pH, the selectivity and activity of >C2 products reach a maximum and finally start decreasing, whereas, CH4 evolution starts increasing.63 A possible reason for this behavior could be the increased coverage of H* caused by the depletion of CO* in the limited mass-transport regime. Such adsorbed protons promote CH4 evolution (by CO hydrogenation) and reduce the chances of CO* coupling to make >C2 products.315,316
Furthermore, in a relatively alkaline environment, the Cannizzaro reaction (Fig. 23a) could take place during CO2 electrolysis. In a Cannizzaro reaction, an aldehyde can disproportionate to the corresponding carboxylic acid and alcohol.317 Through investigating the reduction of formaldehyde in various electrolytes, such as perchloric acid (HClO4), sodium perchlorate (NaClO4), and phosphate buffers, Koper and co-workers observed that the disproportionation reactions are strongly influenced by the electrolyte pH and buffering strength.317 As OH− ions are essential for the Cannizzaro reaction, HCOOH is easier to form in the HClO4-based electrolyte (pH = 3) than the one with pH = 1 (see Fig. 23b). In phosphate electrolyte with a high buffering capacity and thereby a relatively low local pH, Cannizzaro reaction was significantly hindered, leading to a negligible formation of HCOOH.317 Therefore, researchers should be careful in distinguishing the products such as acids and alcohols formed from the disproportionation reactions and those formed by direct CO2R, especially for the liquid products such as methanol and ethanol, where the corresponding aldehyde is often proposed as a reaction intermediate.318 This is especially true since interrogation of the just produced products is hard to measure compared to the ones that further react and transport and are detected in the bulk solution or gas flows.
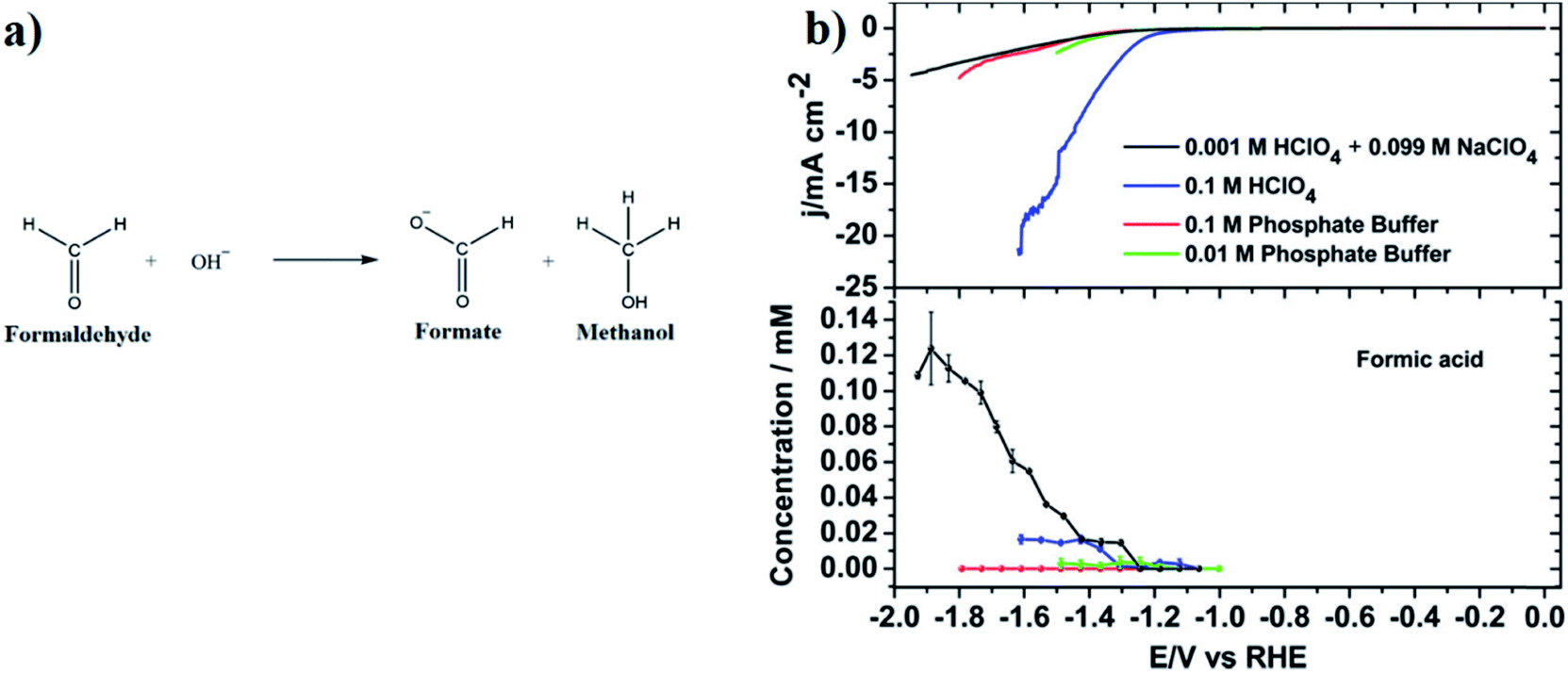 | ||
| Fig. 23 (a) Cannizzaro disproportionation reactions showing formaldehyde disproportionate to formate and methanol, adapted from Birdja and Koper317 (2017), (b) formic acid formation during reduction of formaldehyde in perchloric acid (pH: 1 and 3), 0.1 M phosphate buffer pH 6.6, and 0.01 M phosphate buffer pH 6.8. Scan rate: 1 mV s−1. Reprinted with permission from Birdja and Koper,317 Copyright 2017, American Chemical Society. | ||
Enhanced mass transport makes the value of local pH closer to the bulk pH, thus influencing the activity and selectivity of cathodic reactions.295,319 Through rotating a Cu cylinder electrode to enhance the mass transfer, Marshall and co-workers found that the CO2R activity decreased with the increasing rotation speed, along with a change in product preference from CH4 to CO, while HER was promoted.319 Such degradation of CO2R is attributed to the formation of graphitic carbon on electrode surface due to low local pH at higher rotation speed, resulting in the deactivation of the active sites for CO2R.319 This evidence is also supported by Mul and co-workers, who presented a pH-dependent pathway for the deactivation of Cu electrode due to carbon formation.288 In addition, the change in product selectivity from CH4 to CO at higher rotation speed could be due to lower coverage of adsorbed *CO, which is a precursor for CH4 formation.319
7 Pressure and temperature effects
7.1 Pressure
The partial pressure of CO2 in the gas fed to the electrolyzer directly impacts the rate of CO2 mass transfer to the electrode surface due CO2-solvent solubility relationship. For example, at a given temperature CO2 solubility in aqueous electrolytes and physical organic solvents typically increases linearly with pressure according to Henry's law.320 Other solvent-electrolytes like amines321 and ILs322 exhibit non-linear CO2 solubility relationships that may reach a plateau at a critical pressure (e.g. around 10 MPa for imidazolium-based ionic liquids322 and ∼1 MPa for amines323) beyond which further increase of pressure does not improve solubility. Raising the operating pressure increases complexity and cost of electrolyzer construction and operation, so optimization of the pressure must consider costs and the effect of pressure on CO2R selectivity.A large number of studies report the effects of pressure on product selectivity and FE of CO2R processes.67,108,109,159,210,212,288,324–330 Hara et al.330 reported that on a Cu wire (0.16 cm2) in 0.1 M KHCO3 the CO2R products shifted from H2 at 1 atm to hydrocarbons then to CO and/or HCOOH as the pressure was increased to 30 atm, with PCDCO up to 523 mA cm−2 observed. Table 6 summarizes a typical example reported by Hori et al.210 of the effect of CO2 pressure on product selectivity over different metal electrodes. Hori et al. observed that over group B catalysts, which include Fischer–Tropsch catalysts like Ni, Co and Fe, product selectivity shifted from H2 at ambient pressures to HCOOH and CO at high pressure.210 Kudo et al.331 reported similar shifts from H2 at low pressures to CO, HCOOH, and hydrocarbons at 60 atm over Ni catalysts as shown in Fig. 24a. They observed that the hydrocarbons from CO2R over Ni followed a Schulz–Flory probability distribution like that obtained by the Fischer–Tropsch reaction, and proposed that like the conventional Fischer–Tropsch process electrochemical CO2R at high pressures may involve hydrogenation of metal carbonyl intermediates to facilitate production of carbene groups (–CH2–) that polymerize to longer-chain hydrocarbons. In addition, high CO2 pressure increases the surface coverage of CO2R intermediates on the electrode surface, which promotes CO2R and suppresses of HER as illustrated in Fig. 24b and c.210,331
| Group | Cathode catalyst | Effect of pressure | |
|---|---|---|---|
| Major products at 1 atm | Major products at 30 atm | ||
| A | Ti, Nb, Ta, Mo, Mn, and Al | H2 | H2 |
| B | Zr, Cr, W, Fe, co, Rh, ir, Ni, Pd, Pt, C and Si | H2 | CO and HCOOH |
| C | Ag, Au, Zn, In, Sn, Pb, and Bi | CO and HCOOH | CO and HCOOH |
| D | Cu | CH4 and C2H6 | CO and HCOOH |
 | ||
| Fig. 24 Electrochemical reduction of CO2 on Ni electrodes, (a) effect of pressure on the FE of various products; reaction mechanism (b) at high pressure (60 atm), and (c) at atmospheric pressure. Adapted with permission from Kudo et al.331 Copyright 1993, The Electrochemical Society. | ||
For group C catalysts like Ag and Sn, Hara et al.210 did not report a major shift in the type of CO2R products at higher pressure but the FE towards CO and HCOOH did increase (relative to H2), and this was attributed to increased CO2 solubility at higher pressures. Over Cu catalysts (group D), Hara et al.210 reported a shift from hydrocarbon production at 1 atm to CO and HCOOH at 30 atm. Jesús-Cardona et al.326 proposed that at concentrations of CO2 in the electrolyte at elevated pressure, CO2 molecules displace may displace at the electrode's surface some of the CO* that are a key intermediate in the CO2R to hydrocarbon pathway.
High-pressure CO2R requires balancing the pressure in the anode and cathode chambers to prevent damage to the separator.98,212,262 Ramdin et al.98 compared the effects of BPM and CEM on CO2R to formic acid/formate at high CO2 pressure, and after the experiment observed delamination of BPM layers. Note that even in electrolyzer operating at near ambient pressures there can be a pressure imbalance across the separator, especially in CO2R processes where the anode produces O2 and the cathode reduces CO2 to liquid products. This pressure imbalance can deform the separator, so to improve mechanical strength thicker membranes, fabric-reinforced membranes, or additional porous substrates may be require. These approaches may improve strength and durability, but also increase the resistances across the separator.332
7.2 Temperature
Temperature effects CO2R electrolyzer performance because of both CO2 solubility (i.e. the Van't Hoff equation)333,334 and minimum half-cell potentials are temperature dependent, and temperature influences CO2R kinetics because of its effect on mass transfer rates, reaction pathways, viscosity and conductivity of electrolytes, and the conductivity of IEM separators. The stability of cell components like membranes, electrodes, and gaskets may also be effected by temperature extremes or by temperature cycles. The range of temperatures reported for electrochemical CO2R processes is commonly between 20–40 °C, although a few studies have explored CO2R at temperatures as low as 0 °C (ref. 209) and up to 125 °C.54 Within this operating range the temperature dependency of electrical conductivity of cell components like current collectors is unlikely to have a significant effect on CO2R electrolyzer performance.The main CO2R reactions and the HER have a positive entropy change so that lower overall thermodynamic cell voltages are required for reactions at higher temperatures. Additionally, lower activation overpotential are required at higher temperatures than lower temperatures to drive a sufficient overall CO2R rate, which can be quantitatively determined through the Butler–Volmer equation.335 The effect of temperature on overpotential appears to be more significant than the effect on thermodynamic potentials. For example, Dufek et al. reported a significant reduction of cathode potentials from −2.19 V to −1.87 V to achieve 70 mA cm−2 over an Ag-based GDEs when operating temperature was elevated from 18 to 70 °C, while less than 0.1 V decrement in thermodynamic voltage was observed when the temperature was increased from 25 to 125 °C.54 Ryu et al.336 reported a similar trend for CO2R over Hg electrodes.
Temperature has a significant effect on hydrodynamic properties (viscosity and density) and concentration gradients (due to solubility relationships) that control rates of CO2 mass transfer. For example, faster CO2 mass transfer coefficients from the gas to electrolyte is observed at higher temperatures because diffusivity of CO2 in gas and liquid phases increases, and the viscosity of the catholyte decreases.337,338 But, a reduced solubility at higher temperatures lowers the overall driving force for the mass-transport across the gas/liquid interface. The relative changes in solubility with temperature for CO2 compared to CO2R products such as CH4 and C2H4 that are sparingly soluble in the catholyte,339,340 could be advantageous to promote quick desorption of products from the electrode surface. However, proton transfer rates may also increase at high temperatures and so increasing temperature can potentially promote HER at the cathode.
The enhanced diffusivity and thinner static laminar layer at the electrode exhibited at higher temperatures reduces ohmic resistances in the electrolyte. In GDEs, temperature can also change the phase-change induced flow and result in dryers CLs.189 However, increased temperature means higher water entering into the electrolyzer at same relative humidity and so that can help get to higher CDs, especially if the system is water limited.189
Temperature is known to affect the product distribution of the CO2R reaction over different catalysts.341–344 for example, Hori et al.209 reported a decrease of FECH4 and increase of FEC2H4 over Cu foil at 5 mA cm−2 when the temperature was raised from 0 to 40 °C. Ahn et al.343 reported a similar trend when increasing the temperature from 2 to 22 °C. A close proximity of *CO intermediates is important for the formation of C–C bond, meaning that a high *CO coverage should make it easier for the C2H4 evolution. However, the reported suppression of C2H4 at high temperature, where the coverage of *CO is low, suggests that *CO coverage may not play a dominating role at least within the studied range. Alternatively, several recent theoretical studies proposed that *COH and *CHO intermediates are crucial to determine whether the product is CH4 or C2H4, respectively with a Cu electrode.318,345,346 According to the work reported by Luo et al.,346 the availability of absorbed protons is essential to form *CHO, while the absorbed H2O is important to serve as a shuttle (H3Oδ+) to transfer the proton to the *CO intermediate to form *COH. Therefore, one possible explanation of the reported temperature effect is that low temperature can stabilize the formation of (H3Oδ+) proton shuttle, making it easier to form *COH intermediate, thus facilitating the production of CH4. On the other hand, higher temperatures increase the availability of adsorbed protons at the electrode surface, resulting in an easier formation of *CHO intermediates that promote C2H4. Moreover, the adsorbed protons could also partially contribute towards promoting HER. When temperature further increased from 22 °C, a degraded FE(C2H4) over Cu electrode was observed by Ahn et al.343 This suggests that the aforementioned coverage of *CO starts to dominate instead of the coverage of adsorbed protons.343 Interestingly, potential vs. RHE showed an opposite trend when comparing the temperature-effect results reported by Hori et al. and Ahn et al. Nevertheless, both studies report similar observations towards FE of CH4 and C2H4, suggesting that the potential effect as imparted by the temperature change on the product distribution could be negligible in the studied temperature range.
Considering the effects of the potential on the reactivity and selectivity as mentioned, we recommend the design of experiment to keep the potential vs. RHE constant when investigating temperature effects, as this potential is corrected by both temperature and pH. Overall, the temperature has significant impacts on the surface coverage of CO2R-related species (*COOH and *CO) through affecting their mass-transfer characteristics (e.g. solubility), as well as the coverage of proton-related adsorbates (e.g. H* and H3Oδ+) that also are important for the formation of key intermediates (e.g. *CHO and *COH). Such effects likely contribute the observed temperature-dependent CO2R selectivity.
A small increase in temperature can lower the ohmic resistance of the separator.94,347 However, too high of a temperature degrades the ionic conductivity and mechanical structure of the membranes. Taking perfluorosulfonic acid (PFSA) polymer-based membrane as an example, its clusters of the hydrophilic sulfonic-acid group uptake sufficient water and allow protons to move easily in the membrane. In a fuel-cell configuration, a too high temperature (>90 °C) lowers the water content in the membrane, leading to a decreased ionic conductivity.348 Common AEMs, such as quaternary ammonia polysulfone, limit the operation to a much lower temperature than CEMs due to chemical stability concerns.349 The Sustainion® membrane, developed by Dioxide Materials, has an improved ionic conductivity (∼140 mS cm−1 in 1 M KOH aqueous solution at 80 °C) and can stably be operated at temperature up to 80 °C.347 Ceramic based separators such as ZrO2 diaphragm can withstand higher operating temperature, but again they also pose other key risks such as product cross-over that lose conversion efficiency of the cells.
8 Summary and outlook
We have examined in this review the impact of design and operating decision related to electrolyzer configurations, electrode structure, electrolyte selection, pH, pressure, and temperature affect the reaction conditions at catalyst sites in a CO2R electrolyzer, and thus impact on the overall efficiency of the process. The key challenges for low cost CO2R that can be addressed by these factors together with catalyst materials are reducing the required cell voltages, improving selectivity, and lowering product separation costs.The most promising CO2R electrolyzers under development are based on designs and understanding translated from polymer electrolyte fuel-cells. Of the types of continuous CO2R electrolyzers under development, vapor-fed electrolyzers may be the most promising for the large-scale CO2R processes because this configuration provides opportunity to feed wet CO2-rich flue gas directly to the electrolyzer without CO2 capture process units.350 This technology if coupled with renewable electricity generation could also provide opportunities for passive CO2 capture from the atmosphere.351 Vapor-fed electrolyzers may also reduce risks of catalyst poisoning from trace impurities in liquid electrolytes. For large-area electrodes in industrial scale electrolyzers, achieving uniform current and voltage distribution throughout the electrode surface and managing pressure gradients at the gas–electrolyte interface are critical, thus further work is required to optimize the electrode and flow-field design. In this regard, advanced manufacturing technologies like 3D printing352 combined with computational modelling may help to optimize electrode and electrolyzer geometries.
There are fundamental aspects of electrolyte interactions in CO2R processes that are not fully understood. For example, although the charge carrying capacity of electrolytes is the primary function, researchers have discovered that electrolyte cations contribute in other ways to effect to the availability of protons and to stabilize CO2R intermediates. Likewise, electrolyte anions can provide local pH buffering effects due to specific adsorption of anions at the cathode surface. Low CO2 solubility and competitive HER remain major challenges in inorganic salts-based aqueous electrolytes. Other new solvent-electrolytes such as ionic liquids and deep eutectic solvents may become more attractive if their cost reduces in the future, but to be effective CO2R electrolytes the viscosity of these solvents will also need to be managed, for example by mixing with a co-solvent such as water.
While our review has focused on electrochemical CO2R at the cathode, we acknowledge that the anode catalyst is also critical and especially because the state-of-the-art Ru and Ir based anode materials are a major contributor to the cost of electrochemical CO2R. Therefore, any advances in anode technologies achieved in other applications such as the oxygen evolution reaction in water splitting should help inform development of electrochemical CO2R processes. Water purification from treating wastewater through anodic oxidation of organic pollutants could be another approach to be considered for coupling with CO2R.353 Moreover, since the oxidation potential of chloride is similar to that of water, producing chlorine at anode354,355 (similar to chlor-alkali cells) without the expense of extra energy could be beneficial for optimizing the overall process.
A final consideration in research needs for CO2R relates to experimental methods used to test novel catalysts and electrolyes. Most laboratory studies still use H-type batch setups40 in which the solubility of CO2 in electrolyte and gas–liquid mass transfer limits can control the overall rate of CO2R reactions, plus the transport of ions across separators in H-type cells can limit applicability of long term stability tests. Lab scale continuous flow-cell electrolyzers offer opportunities to detect minor CO2R products99 and to better control reaction conditions than in H-cells. The cost of flow-cell apparatus is no longer significantly higher than a batch cells, so we expect the trend in reporting continuous CO2R measurements in catalyst studies may become more common. Clark et al.200 have also highlighted the urgent need for standardized protocols, such as those developed for battery testing and fuel cell performance, to benchmark performance of catalyst and electrolyzers for electrochemical CO2.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the HBIS Group, China, and Australian Research Council (ARC) Linkage Project No. LP160101729. S. Garg received scholarship support from the University of Queensland Graduate School and the HBIS-UQ Innovation Centre for Sustainable Steel. A. Z. Weber acknowledges support for his effort on this review from the Joint Center for Artificial Photosynthesis, a DOE Energy Innovation Hub, supported through the Office of Science of the U.S. Department of Energy under Award No. DE-SC0004993.References
- H. Naims, Environ. Sci. Pollut. Res., 2016, 23, 22226–22241 CrossRef CAS PubMed.
- Global CCS Institute, The Global Status of CCS: 2015, Summary Report, Melbourne, Australia, 2015 Search PubMed.
- M. M. Halmann, Chemical Fixation of Carbon DioxideMethods for Recycling CO2 into Useful Products, CRC press, 1993 Search PubMed.
- A. Al-Mamoori, A. Krishnamurthy, A. A. Rownaghi and F. Rezaei, Energy Technol., 2017, 5, 834–849 CrossRef.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294 CrossRef CAS PubMed.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazabal and J. Perez-Ramirez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC.
- H.-R. M. Jhong, S. Ma and P. J. A. Kenis, Curr. Opin. Chem. Eng., 2013, 2, 191–199 CrossRef.
- A. S. Agarwal, Y. Zhai, D. Hill and N. Sridhar, ChemSusChem, 2011, 4, 1301–1310 CrossRef CAS PubMed.
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS.
- D. R. Kauffman, J. Thakkar, R. Siva, C. Matranga, P. R. Ohodnicki, C. Zeng and R. Jin, ACS Appl. Mater. Interfaces, 2015, 7, 15626–15632 CrossRef CAS PubMed.
- R. J. Lim, M. Xie, M. A. Sk, J.-M. Lee, A. Fisher, X. Wang and K. H. Lim, Catal. Today, 2014, 233, 169–180 CrossRef CAS.
- N. Pardo and J. A. Moya, Energy, 2013, 54, 113–128 CrossRef CAS.
- Opus-12, We have developed a device that recycles CO2 into chemicals and fuels, https://www.opus-12.com/technology.
- C. E. R. Toronto, Technology,https://co2cert.com/technology/.
- Evonik, Evonik and siemens to generate high-value specialty chemicals from carbon dioxide and eco-electricity, https://corporate.evonik.com/en/media/search/pages/news-details.aspx?NewsId=72457.
- The Powerhaus Pavilion, http://greenangelenergy.ca/ph/mantraenergy.html, accessed at 1st December 2018.
- E. Alper and O. Yuksel Orhan, Petroleum, 2017, 3, 109–126 CrossRef.
- Y. Hori, H. Konishi, T. Futamura, A. Murata, O. Koga, H. Sakurai and K. Oguma, Electrochim. Acta, 2005, 50, 5354–5369 CrossRef CAS.
- A. Wuttig and Y. Surendranath, ACS Catal., 2015, 5, 4479–4484 CrossRef CAS.
- B. Jermann and J. Augustynski, Electrochim. Acta, 1994, 39, 1891–1896 CrossRef CAS.
- G. Nogami, H. Itagaki and R. Shiratsuchi, J. Electrochem. Soc., 1994, 141, 1138–1142 CrossRef CAS.
- J. Yano, T. Morita, K. Shimano, Y. Nagami and S. Yamasaki, J. Solid State Electrochem., 2007, 11, 554–557 CrossRef CAS.
- J. Yano and S. Yamasaki, J. Appl. Electrochem., 2008, 38, 1721 CrossRef CAS.
- P. Kedzierzawski and J. Augustynski, J. Electrochem. Soc., 1994, 141, L58–L60 CrossRef CAS.
- B. Kumar, J. P. Brian, V. Atla, S. Kumari, K. A. Bertram, R. T. White and J. M. Spurgeon, ACS Catal., 2016, 6, 4739–4745 CrossRef CAS.
- L. M. Chiacchiarelli, Y. Zhai, G. S. Frankel, A. S. Agarwal and N. Sridhar, J. Appl. Electrochem., 2012, 42, 21–29 CrossRef CAS.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- D. Voiry, H. S. Shin, K. P. Loh and M. Chhowalla, Nat. Rev. Chem., 2018, 2, 0105 CrossRef CAS.
- Z. D. Dong, L. J. Long and Q. S. Zhang, Adv. Mater. (Weinheim, Ger.), 2016, 28, 3423–3452 CrossRef PubMed.
- F. Li, D. R. MacFarlane and J. Zhang, Nanoscale, 2018, 10, 6235–6260 RSC.
- W. Yang, K. Dastafkan, C. Jia and C. Zhao, Adv. Mater. Technol., 2018, 1700377 CrossRef.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS PubMed.
- T. Ziqi, P. Chad and C. Liang, Adv. Theory Simul., 2018, 1, 1800004 CrossRef.
- C. W. Lee, K. D. Yang, D. H. Nam, J. H. Jang, N. H. Cho, S. W. Im and K. T. Nam, Adv. Mater. (Weinheim, Ger.), 2018, 1704717 CrossRef.
- K. A. Grice, Coord. Chem. Rev., 2017, 336, 78–95 CrossRef CAS.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- N. Kornienko, Y. Zhao, C. S. Kley, C. Zhu, D. Kim, S. Lin, C. J. Chang, O. M. Yaghi and P. Yang, J. Am. Chem. Soc., 2015, 137, 14129–14135 CrossRef CAS PubMed.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS.
- B. Endrődi, G. Bencsik, F. Darvas, R. Jones, K. Rajeshwar and C. Janáky, Prog. Energy Combust. Sci., 2017, 62, 133–154 CrossRef.
- M. Bevilacqua, J. Filippi, H. A. Miller and F. Vizza, Energy Technol., 2015, 3, 197–210 CrossRef CAS.
- S. Ma, J. Liu, K. Sasaki, S. M. Lyth and P. J. A. Kenis, Energy Technol., 2017, 5, 861–863 CrossRef CAS.
- R. B. Kutz, Q. Chen, H. Yang, S. D. Sajjad, Z. Liu and I. R. Masel, Energy Technol., 2017, 5, 929–936 CrossRef CAS.
- D. Materials, CO2 Electrolyzers, https://dioxidematerials.com/technology/co2-electrolysis/.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS.
- Skyre, Products: CO2RENEWTM, https://www.skyre-inc.com/products/co2renew/.
- T. Haas, R. Krause, R. Weber, M. Demler and G. Schmid, Nat. Catal., 2018, 1, 32–39 CrossRef CAS.
- J. Qiao, Y. Liu and J. Zhang, Electrochemical Reduction of Carbon Dioxide: Fundamentals and Technologies, CRC Press, Boca Raton, 2016 Search PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical methods: fundamentals and applications, Wiley, 1980 Search PubMed.
- K. Scott, in Sustainable and Green Electrochemical Science and Technology, 2017, pp. 27–85, DOI:10.1002/9781118698075.ch2.
- M. S. Faber, R. Dziedzic, M. A. Lukowski, N. S. Kaiser, Q. Ding and S. Jin, J. Am. Chem. Soc., 2014, 136, 10053–10061 CrossRef CAS.
- K. Zeng and D. Zhang, Prog. Energy Combust. Sci., 2010, 36, 307–326 CrossRef CAS.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- E. J. Dufek, T. E. Lister and M. E. McIlwain, J. Appl. Electrochem., 2011, 41, 623–631 CrossRef CAS.
- H. Hashiba, L.-C. Weng, Y. Chen, H. K. Sato, S. Yotsuhashi, C. Xiang and A. Z. Weber, J. Phys. Chem. C, 2018, 122, 3719–3726 CrossRef CAS.
- N. Gupta, M. Gattrell and B. MacDougall, J. Appl. Electrochem., 2006, 36, 161–172 CrossRef CAS.
- P. Lobaccaro, M. R. Singh, E. L. Clark, Y. Kwon, A. T. Bell and J. W. Ager, Phys. Chem. Chem. Phys., 2016, 18, 26777–26785 RSC.
- L.-C. Weng, A. T. Bell and A. Z. Weber, Phys. Chem. Chem. Phys., 2018, 20, 16973–16984 RSC.
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019, 18, 1222–1227 CrossRef CAS PubMed.
- L. D. Chen, M. Urushihara, K. Chan and J. K. Nørskov, ACS Catal., 2016, 6, 7133–7139 CrossRef CAS.
- R. B. Sandberg, J. H. Montoya, K. Chan and J. K. Nørskov, Surf. Sci., 2016, 654, 56–62 CrossRef CAS.
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager and A. T. Bell, J. Am. Chem. Soc., 2016, 138, 13006–13012 CrossRef CAS PubMed.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS PubMed.
- A. S. Varela, W. Ju, T. Reier and P. Strasser, ACS Catal., 2016, 6, 2136–2144 CrossRef CAS.
- M. S. Xie, B. Y. Xia, Y. Li, Y. Yan, Y. Yang, Q. Sun, S. H. Chan, A. Fisher and X. Wang, Energy Environ. Sci., 2016, 9, 1687–1695 RSC.
- D. Gao, F. Scholten and B. Roldan Cuenya, ACS Catal., 2017, 7, 5112–5120 CrossRef CAS.
- M. R. Singh, J. D. Goodpaster, A. Z. Weber, M. Head-Gordon and A. T. Bell, Proc. Natl. Acad. Sci., 2017, 114, E8812–E8821 CrossRef CAS PubMed.
- C. S. Chen, A. D. Handoko, J. H. Wan, L. Ma, D. Ren and B. S. Yeo, Catal. Sci. Technol., 2015, 5, 161–168 RSC.
- D. T. Whipple and P. J. A. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451–3458 CrossRef CAS.
- C. Delacourt, P. L. Ridgway and J. Newman, J. Electrochem. Soc., 2010, 157, B1902–B1910 CrossRef CAS.
- Y. C. Li, D. Zhou, Z. Yan, R. H. Gonçalves, D. A. Salvatore, C. P. Berlinguette and T. E. Mallouk, ACS Energy Lett., 2016, 1, 1149–1153 CrossRef CAS.
- M. R. Singh, K. Papadantonakis, C. Xiang and N. S. Lewis, Energy Environ. Sci., 2015, 8, 2760–2767 RSC.
- C. Xiang, K. M. Papadantonakis and N. S. Lewis, Mater. Horiz., 2016, 3, 169–173 RSC.
- X. Li and I. Sabir, Int. J. Hydrogen Energy, 2005, 30, 359–371 CrossRef CAS.
- J. Wang and H. Wang, Fuel Cells, 2012, 12, 989–1003 CrossRef CAS.
- P. J. Hamilton and B. G. Pollet, Fuel Cells, 2010, 10, 489–509 CrossRef CAS.
- D. M. F. Santos, C. A. C. Sequeira and J. L. Figueiredo, Quim. Nova, 2013, 36, 1176–1193 CrossRef CAS.
- F. Harnisch, U. Schröder and F. Scholz, Environ. Sci. Technol., 2008, 42, 1740–1746 CrossRef CAS PubMed.
- N. M. Vargas-Barbosa, G. M. Geise, M. A. Hickner and T. E. Mallouk, ChemSusChem, 2014, 7, 3017–3020 CrossRef CAS PubMed.
- M. B. McDonald, J. P. Bruce, K. McEleney and M. S. Freund, ChemSusChem, 2015, 8, 2645–2654 CrossRef CAS PubMed.
- J. Luo, D. A. Vermaas, D. Bi, A. Hagfeldt, W. A. Smith and M. Grätzel, Adv. Energy Mater., 2016, 6, 1600100 CrossRef.
- M. B. McDonald, S. Ardo, N. S. Lewis and M. S. Freund, ChemSusChem, 2014, 7, 3021–3027 CrossRef CAS PubMed.
- R. S. Reiter, W. White and S. Ardo, J. Electrochem. Soc., 2016, 163, H3132–H3134 CrossRef CAS.
- A. Kusoglu and A. Z. Weber, Chem. Rev., 2017, 117, 987–1104 CrossRef CAS PubMed.
- J. J. Kaczur, H. Yang, Z. Liu, S. D. Sajjad and R. I. Masel, Front. Chem., 2018, 6, 263 CrossRef PubMed.
- T. Luo, S. Abdu and M. Wessling, J. Membr. Sci., 2018, 555, 429–454 CrossRef CAS.
- K. F. L. Hagesteijn, S. Jiang and B. P. Ladewig, J. Mater. Sci., 2018, 53, 11131–11150 CrossRef CAS.
- D. Y. Leung and J. Xuan, Micro & nano-engineering of fuel cells, Crc Press, 2015 Search PubMed.
- D. W. Dewulf and A. J. Bard, Catal. Lett., 1988, 1, 73–79 CrossRef CAS.
- C. Delacourt, P. L. Ridgway, J. B. Kerr and J. Newman, J. Electrochem. Soc., 2008, 155, B42–B49 CrossRef CAS.
- Y. Hori, H. Ito, K. Okano, K. Nagasu and S. Sato, Electrochim. Acta, 2003, 48, 2651–2657 CrossRef CAS.
- N. Ziv and D. R. Dekel, Electrochem. Commun., 2018, 88, 109–113 CrossRef CAS.
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135–3191 RSC.
- K. N. Grew, X. Ren and D. Chu, Electrochem. Solid-State Lett., 2011, 14, B127–B131 CrossRef CAS.
- Z. Sun, J. Pan, J. Guo and F. Yan, Adv. Sci., 2018, 5, 1800065 CrossRef PubMed.
- J. Albo, A. Sáez, J. Solla-Gullón, V. Montiel and A. Irabien, Appl. Catal., B, 2015, 176, 709–717 CrossRef.
- M. Alvarez-Guerra, S. Quintanilla and A. Irabien, Chem. Eng. J. (Lausanne), 2012, 207, 278–284 CrossRef.
- M. Ramdin, A. R. T. Morrison, M. de Groen, R. van Haperen, R. de Kler, L. J. P. van den Broeke, J. P. M. Trusler, W. de Jong and T. J. H. Vlugt, Ind. Eng. Chem. Res., 2019, 58, 1834–1847 CrossRef CAS PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- T. Hatsukade, K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Phys. Chem. Chem. Phys., 2014, 16, 13814–13819 RSC.
- J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Nørskov and T. F. Jaramillo, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS.
- E. R. Cave, J. H. Montoya, K. P. Kuhl, D. N. Abram, T. Hatsukade, C. Shi, C. Hahn, J. K. Norskov and T. F. Jaramillo, Phys. Chem. Chem. Phys., 2017, 19, 15856–15863 RSC.
- K. Manthiram, B. J. Beberwyck and A. P. Alivisatos, J. Am. Chem. Soc., 2014, 136, 13319–13325 CrossRef CAS PubMed.
- E. L. Clark, M. R. Singh, Y. Kwon and A. T. Bell, Anal. Chem., 2015, 87, 8013–8020 CrossRef CAS PubMed.
- J. Balster, D. F. Stamatialis and M. Wessling, Chem. Eng. Process., 2004, 43, 1115–1127 CrossRef CAS.
- J. Kamcev, R. Sujanani, E.-S. Jang, N. Yan, N. Moe, D. R. Paul and B. D. Freeman, J. Membr. Sci., 2018, 547, 123–133 CrossRef CAS.
- E. J. Dufek, T. E. Lister and M. E. McIlwain, Electrochem. Solid-State Lett., 2012, 15, B48–B50 CrossRef CAS.
- E. J. Dufek, T. E. Lister, S. G. Stone and M. E. McIlwain, J. Electrochem. Soc., 2012, 159, F514–F517 CrossRef CAS.
- K. Hara and T. Sakata, Bull. Chem. Soc. Jpn., 1997, 70, 571–576 CrossRef CAS.
- P. Jeanty, C. Scherer, E. Magori, K. Wiesner-Fleischer, O. Hinrichsen and M. Fleischer, J. CO2 Util., 2018, 24, 454–462 CrossRef CAS.
- D. Higgins, C. Hahn, C. Xiang, T. F. Jaramillo and A. Z. Weber, ACS Energy Lett., 2019, 4, 317–324 CrossRef CAS.
- W. Lee, Y. E. Kim, M. H. Youn, S. K. Jeong and K. T. Park, Angew. Chem., 2018, 130, 6999–7003 CrossRef.
- I. Merino-Garcia, J. Albo and A. Irabien, Nanotechnology, 2017, 29, 014001 CrossRef PubMed.
- D. A. Salvatore, D. M. Weekes, J. He, K. E. Dettelbach, Y. C. Li, T. E. Mallouk and C. P. Berlinguette, ACS Energy Lett., 2018, 3, 149–154 CrossRef CAS.
- D. T. Whipple, E. C. Finke and P. J. A. Kenis, Electrochem. Solid-State Lett., 2010, 13, B109–B111 CrossRef CAS.
- R. S. Jayashree, S. K. Yoon, F. R. Brushett, P. O. Lopez-Montesinos, D. Natarajan, L. J. Markoski and P. J. A. Kenis, J. Power Sources, 2010, 195, 3569–3578 CrossRef CAS.
- M. M. Morgan, L. Peter, L. Yanwei and W. A. Joel, J. Phys. D: Appl. Phys., 2017, 50, 154006 CrossRef.
- S. Verma, B. Kim, H.-R. M. Jhong, S. Ma and P. J. A. Kenis, ChemSusChem, 2016, 9, 1972–1979 CrossRef CAS PubMed.
- J. Spurgeon and B. Kumar, Energy Environ. Sci., 2018, 11, 1536–1551 RSC.
- J. B. Greenblatt, D. J. Miller, J. W. Ager, F. A. Houle and I. D. Sharp, Joule, 2018, 2, 381–420 CrossRef CAS.
- J. H. Lunsford, P. Qiu, M. P. Rosynek and Z. Yu, J. Phys. Chem. B, 1998, 102, 167–173 CrossRef CAS.
- A. Elfasakhany, Eng. Sci. Technol. Int J., 2015, 18, 713–719 CrossRef.
- P. Tian, Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS.
- S. Şahin, Ş. S. Bayazit, M. Bilgin and İ. İnci, J. Chem. Eng. Data, 2010, 55, 1519–1522 CrossRef.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, J. Org. Chem., 2009, 74, 487–498 CrossRef CAS PubMed.
- H. S. Abdelkader, L. Touhami, B. M. Lakhdar, K. Abdellah, G. Toufik and B. B. Fouzi, presented in part at the International Conference on Environmental, Biomedical and Biotechnology Singapore, 2012 Search PubMed.
- H. Yang, J. J. Kaczur, S. D. Sajjad and R. I. Masel, J. CO2 Util., 2017, 20, 208–217 CrossRef CAS.
- H. S. Jeon, S. Kunze, F. Scholten and B. Roldan Cuenya, ACS Catal., 2018, 8, 531–535 CrossRef CAS.
- C. W. Lee, J. S. Hong, K. D. Yang, K. Jin, J. H. Lee, H.-Y. Ahn, H. Seo, N.-E. Sung and K. T. Nam, ACS Catal., 2018, 8, 931–937 CrossRef CAS.
- F. Li, L. Chen, M. Xue, T. Williams, Y. Zhang, D. R. MacFarlane and J. Zhang, Nano Energy, 2017, 31, 270–277 CrossRef CAS.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 3242 CrossRef PubMed.
- S. Liu, J. Xiao, X. F. Lu, J. Wang, X. Wang and X. W. Lou, Angew. Chem., Int. Ed., 2019, 58, 8499–8503 CrossRef CAS PubMed.
- M. Rahaman, A. Dutta, A. Zanetti and P. Broekmann, ACS Catal., 2017, 7, 7946–7956 CrossRef CAS.
- Q. Li, J. Fu, W. Zhu, Z. Chen, B. Shen, L. Wu, Z. Xi, T. Wang, G. Lu, J.-j. Zhu and S. Sun, J. Am. Chem. Soc., 2017, 139, 4290–4293 CrossRef CAS PubMed.
- L. Dai, Q. Qin, P. Wang, X. Zhao, C. Hu, P. Liu, R. Qin, M. Chen, D. Ou, C. Xu, S. Mo, B. Wu, G. Fu, P. Zhang and N. Zheng, Sci. Adv., 2017, 3, e1701069 CrossRef PubMed.
- P. Su, W. Xu, Y. Qiu, T. Zhang, X. Li and H. Zhang, ChemSusChem, 2018, 11, 848–853 CrossRef CAS PubMed.
- H. Li and C. Oloman, J. Appl. Electrochem., 2005, 35, 955–965 CrossRef CAS.
- D. Kopljar, A. Inan, P. Vindayer, N. Wagner and E. Klemm, J. Appl. Electrochem., 2014, 44, 1107–1116 CrossRef CAS.
- Z. Bitar, A. Fecant, E. Trela-Baudot, S. Chardon-Noblat and D. Pasquier, Appl. Catal., B, 2016, 189, 172–180 CrossRef CAS.
- M. Schwartz, R. L. Cook, V. M. Kehoe, R. C. MacDuff, J. Patel and A. F. Sammells, J. Electrochem. Soc., 1993, 140, 614–618 CrossRef CAS.
- M. N. Mahmood, D. Masheder and C. J. Harty, J. Appl. Electrochem., 1987, 17, 1159–1170 CrossRef CAS.
- G. K. S. Prakash, F. A. Viva and G. A. Olah, J. Power Sources, 2013, 223, 68–73 CrossRef CAS.
- A. Del Castillo, M. Alvarez-Guerra, J. Solla-Gullón, A. Sáez, V. Montiel and A. Irabien, J. CO2 Util., 2017, 18, 222–228 CrossRef CAS.
- M. H. Shojaeefard, G. R. Molaeimanesh, M. Nazemian and M. R. Moqaddari, Int. J. Hydrogen Energy, 2016, 41, 20276–20293 CrossRef CAS.
- A. Z. Weber, R. L. Borup, R. M. Darling, P. K. Das, T. J. Dursch, W. Gu, D. Harvey, A. Kusoglu, S. Litster, M. M. Mench, R. Mukundan, J. P. Owejan, J. G. Pharoah, M. Secanell and I. V. Zenyuk, J. Electrochem. Soc., 2014, 161, F1254–F1299 CrossRef.
- D. M. Fadzillah, M. I. Rosli, M. Z. M. Talib, S. K. Kamarudin and W. R. W. Daud, Renewable Sustainable Energy Rev., 2017, 77, 1001–1009 CrossRef CAS.
- J. Lai, A. Nsabimana, R. Luque and G. Xu, Joule, 2018, 2, 76–93 CrossRef CAS.
- F. C. Walsh, L. F. Arenas and C. Ponce de León, J. Chem. Technol. Biotechnol., 2018, 93, 3073–3090 CrossRef CAS.
- F. Lux, J. Mater. Sci., 1993, 28, 285–301 CrossRef CAS.
- L. I. Chernyshev and V. V. Skorokhod, Powder Metall. Met. Ceram., 2003, 42, 88–93 CrossRef CAS.
- B. I. Shklovskiĭ and A. L. Éfros, Phys.-Usp., 1975, 18, 845–862 CrossRef.
- R. Lipton, Proc. R. Soc. London, Ser. A, 1998, 454, 1371–1382 CrossRef.
- Y. Lum, B. Yue, P. Lobaccaro, A. T. Bell and J. W. Ager, J. Phys. Chem. C, 2017, 121, 14191–14203 CrossRef CAS.
- M. Ma, B. J. Trześniewski, J. Xie and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 9748–9752 CrossRef CAS PubMed.
- H. Yano, F. Shirai, M. Nakayama and K. Ogura, J. Electroanal. Chem., 2002, 519, 93–100 CrossRef CAS.
- R. L. Cook, R. C. MacDuff and A. F. Sammells, J. Electrochem. Soc., 1990, 137, 607–608 CrossRef CAS.
- K. Hara and T. Sakata, J. Electrochem. Soc., 1997, 144, 539–545 CrossRef CAS.
- K. Hara, A. Kudo, T. Sakata and M. Watanabe, J. Electrochem. Soc., 1995, 142, L57–L59 CrossRef CAS.
- Q. Wang, H. Dong and H. Yu, J. Power Sources, 2014, 271, 278–284 CrossRef CAS.
- S. Park, J.-W. Lee and B. N. Popov, Int. J. Hydrogen Energy, 2012, 37, 5850–5865 CrossRef CAS.
- B. Kim, F. Hillman, M. Ariyoshi, S. Fujikawa and P. J. A. Kenis, J. Power Sources, 2016, 312, 192–198 CrossRef CAS.
- A. Del Castillo, M. Alvarez-Guerra and A. Irabien, AIChE J., 2014, 60, 3557–3564 CrossRef CAS.
- S. Ikeda, T. Ito, K. Azuma, K. Ito and H. Noda, Denki Kagaku, 1995, 63, 303–309 CAS.
- J. Albo and A. Irabien, J. Catal., 2016, 343, 232–239 CrossRef CAS.
- B. C. Marepally, C. Ampelli, C. Genovese, T. Saboo, S. Perathoner, F. M. Wisser, L. Veyre, J. Canivet, E. A. Quadrelli and G. Centi, ChemSusChem, 2017, 10, 4442–4446 CrossRef CAS PubMed.
- E. Irtem, T. Andreu, A. Parra, M. D. Hernández-Alonso, S. García-Rodríguez, J. M. Riesco-García, G. Penelas-Pérez and J. R. Morante, J. Mater. Chem. A, 2016, 4, 13582–13588 RSC.
- Q. Wang, X. Wang, C. Wu, Y. Cheng, Q. Sun, H. Dong and H. Yu, Sci. Rep., 2017, 7, 13711 CrossRef PubMed.
- Q. Wang, H. Dong, H. Yu and H. Yu, J. Power Sources, 2015, 279, 1–5 CrossRef CAS.
- S. Guan, A. Agarwal, E. Rode, D. Hill and N. Sridhar, Advances in Materials Science for Environmental and Energy Technologies II: Ceramic Transactions, 2013, vol. 241, pp. 231–243 Search PubMed.
- D. Masheder and K. P. J. Williams, J. Raman Spectrosc., 1987, 18, 387–390 CrossRef CAS.
- L. Cindrella, A. M. Kannan, J. F. Lin, K. Saminathan, Y. Ho, C. W. Lin and J. Wertz, J. Power Sources, 2009, 194, 146–160 CrossRef CAS.
- J. Park, H. Oh, T. Ha, Y. I. Lee and K. Min, Appl. Energy, 2015, 155, 866–880 CrossRef CAS.
- L. Han, W. Zhou and C. Xiang, ACS Energy Lett., 2018, 3, 855–860 CrossRef CAS.
- D. Kopljar, N. Wagner and E. Klemm, Chem. Eng. Technol., 2016, 39, 2042–2050 CrossRef CAS.
- Q. Wang, H. Dong and H. Yu, RSC Adv., 2014, 4, 59970–59976 RSC.
- T. Van Nguyen, A. Ahosseini, X. Wang, V. Yarlagadda, A. Kwong, A. Z. Weber, P. Deevanhxay, S. Tsushima and S. Hirai, J. Electrochem. Soc., 2015, 162, F1451–F1460 CrossRef.
- R. Omrani and B. Shabani, Int. J. Hydrogen Energy, 2017, 42, 28515–28536 CrossRef CAS.
- J. Wu, F. G. Risalvato, S. Ma and X.-D. Zhou, J. Mater. Chem. A, 2014, 2, 1647–1651 RSC.
- A. Li, H. Wang, J. Han and L. Liu, Front. Chem. Sci. Eng., 2012, 6, 381–388 CrossRef CAS.
- T.-J. Park, S. Banerjee, T. Hemraj-Benny and S. S. Wong, J. Mater. Chem., 2006, 16, 141–154 RSC.
- M. Pumera, A. Ambrosi and E. L. K. Chng, Chem. Sci., 2012, 3, 3347–3355 RSC.
- C. M. Long, M. A. Nascarella and P. A. Valberg, Environ. Pollut., 2013, 181, 271–286 CrossRef CAS PubMed.
- A. Z. Weber and J. Newman, J. Electrochem. Soc., 2005, 152, A677–A688 CrossRef CAS.
- M. Prasanna, H. Y. Ha, E. A. Cho, S. A. Hong and I. H. Oh, J. Power Sources, 2004, 131, 147–154 CrossRef CAS.
- T. Turek, I. Moussallem, A. Bulan, N. Schmitz and P. Weuta, European Patent Office, EP2397578A2, 2016.
- Y. Wang, J. Liu, Y. Wang, A. M. Al-Enizi and G. Zheng, Small, 2017, 13, 1701809 CrossRef PubMed.
- J. He, N. J. J. Johnson, A. Huang and C. P. Berlinguette, ChemSusChem, 2018, 11, 48–57 CrossRef CAS PubMed.
- L.-C. Weng, A. T. Bell and A. Z. Weber, Energy Environ. Sci., 2019, 12, 1950–1968 RSC.
- H. Mistry, F. Behafarid, R. Reske, A. S. Varela, P. Strasser and B. Roldan Cuenya, ACS Catal., 2016, 6, 1075–1080 CrossRef CAS.
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. A. Kenis and R. I. Masel, Science, 2011, 334, 643–644 CrossRef CAS PubMed.
- H. Yang, J. J. Kaczur, S. D. Sajjad and R. I. Masel, ECS Trans., 2017, 77, 1425–1431 CrossRef CAS.
- H. R. M. Jhong, F. R. Brushett and P. J. A. Kenis, Adv. Energy Mater., 2013, 3, 589–599 CrossRef CAS.
- K. Nakata, T. Ozaki, C. Terashima, A. Fujishima and Y. Einaga, Angew. Chem., 2014, 126, 890–893 CrossRef.
- P. P. Sharma and X. D. Zhou, Wiley Interdiscip. Rev.: Energy Environ., 2017, 6, e239 Search PubMed.
- W. Plieth, in Electrochemistry for Materials Science, Elsevier, Amsterdam, 2008, pp. 1–26, DOI:10.1016/B978-044452792-9.50003-8.
- X. Lu, D. Y. C. Leung, H. Wang, M. K. H. Leung and J. Xuan, ChemElectroChem, 2014, 1, 836–849 CrossRef CAS.
- J.-P. Jones, G. K. S. Prakash and G. A. Olah, Isr. J. Chem., 2014, 54, 1451–1466 CrossRef CAS.
- B. M. Setterfield-Price and R. A. W. Dryfe, J. Electroanal. Chem., 2014, 730, 48–58 CrossRef CAS.
- E. L. Clark, J. Resasco, A. Landers, J. Lin, L.-T. Chung, A. Walton, C. Hahn, T. F. Jaramillo and A. T. Bell, ACS Catal., 2018, 8, 6560–6570 CrossRef CAS.
- Á. Pérez-Salado Kamps, E. Meyer, B. Rumpf and G. Maurer, J. Chem. Eng. Data, 2007, 52, 817–832 CrossRef.
- Y. Liu, M. Hou, G. Yang and B. Han, J. Supercrit. Fluids, 2011, 56, 125–129 CrossRef CAS.
- D. Tong, J. P. M. Trusler, G. C. Maitland, J. Gibbins and P. S. Fennell, Int. J. Greenhouse Gas Control, 2012, 6, 37–47 CrossRef CAS.
- M. Wagner, I. von Harbou, J. Kim, I. Ermatchkova, G. Maurer and H. Hasse, J. Chem. Eng. Data, 2013, 58, 883–895 CrossRef CAS.
- W. Lv, R. Zhang, P. Gao and L. Lei, J. Power Sources, 2014, 253, 276–281 CrossRef CAS.
- W. Lv, J. Zhou, F. Kong, H. Fang and W. Wang, Int. J. Hydrogen Energy, 2016, 41, 1585–1591 CrossRef CAS.
- F. Cai, D. Gao, H. Zhou, G. Wang, T. He, H. Gong, S. Miao, F. Yang, J. Wang and X. Bao, Chem. Sci., 2017, 8, 2569–2573 RSC.
- A. L. Kohl, R. Nielsen and A. L. Kohl, Gas purification, Gulf Pub., Houston, Tex, 5th edn, 1997 Search PubMed.
- Y. Hori, K. Katsuhei, A. Murata and S. Suzuki, Chem. Lett., 1986, 15, 897–898 CrossRef.
- K. Hara, A. Kudo and T. Sakata, J. Electroanal. Chem., 1995, 391, 141–147 CrossRef.
- M. Jitaru, D. A. Lowy, M. Toma, B. C. Toma and L. Oniciu, J. Appl. Electrochem., 1997, 27, 875–889 CrossRef CAS.
- M. Todoroki, K. Hara, A. Kudo and T. Sakata, J. Electroanal. Chem., 1995, 394, 199–203 CrossRef.
- D. Ren, Y. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, ACS Catal., 2015, 5, 2814–2821 CrossRef CAS.
- D. Raciti, K. J. Livi and C. Wang, Nano Lett., 2015, 15, 6829–6835 CrossRef CAS PubMed.
- A. Bandi, M. Specht, T. Weimer and K. Schaber, Energy Convers. Manage., 1995, 36, 899–902 CrossRef CAS.
- M. Gattrell, N. Gupta and A. Co, Energy Convers. Manage., 2007, 48, 1255–1265 CrossRef CAS.
- S. Stucki, A. Schuler and M. Constantinescu, Int. J. Hydrogen Energy, 1995, 20, 653–663 CrossRef CAS.
- K. Hara, A. Tsuneto, A. Kudo and T. Sakata, J. Electroanal. Chem., 1997, 434, 239–243 CrossRef CAS.
- P. Dubé and G. M. Brisard, J. Electroanal. Chem., 2005, 582, 230–240 CrossRef.
- K. Ogura, J. CO2 Util., 2013, 1, 43–49 CrossRef CAS.
- K. Ogura and M. D. Salazar-Villalpando, JOM, 2011, 63, 35–38 CrossRef CAS.
- M. Ma, K. Djanashvili and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 6680–6684 CrossRef CAS PubMed.
- A. Murata and Y. Hori, Bull. Chem. Soc. Jpn., 1991, 64, 123–127 CrossRef CAS.
- M. R. Thorson, K. I. Siil and P. J. A. Kenis, J. Electrochem. Soc., 2013, 160, F69–F74 CrossRef CAS.
- S. Verma, X. Lu, S. Ma, R. I. Masel and P. J. A. Kenis, Phys. Chem. Chem. Phys., 2016, 18, 7075–7084 RSC.
- W. Paik, T. N. Andersen and H. Eyring, Electrochim. Acta, 1969, 14, 1217–1232 CrossRef CAS.
- H. Yoshio and S. Shin, Bull. Chem. Soc. Jpn., 1982, 55, 660–665 CrossRef.
- S. Ringe, E. L. Clark, J. Resasco, A. Walton, B. Seger, A. T. Bell and K. Chan, Energy Environ. Sci., 2019, 12, 3001–3014 RSC.
- Y. Hori, in Modern Aspects of Electrochemistry, ed. C. G. Vayenas, R. E. White and M. E. Gamboa-Aldeco, Springer, New York, New York, NY, 2008, pp. 89–189, DOI:10.1007/978-0-387-49489-0_3.
- M. E. Essington, Soil and water chemistry: an integrative approach, CRC Press, 2015 Search PubMed.
- A. N. Frumkin, Trans. Faraday Soc., 1959, 55, 156–167 RSC.
- H. Kim, H. S. Park, Y. J. Hwang and B. K. Min, J. Phys. Chem. C, 2017, 121, 22637–22643 CrossRef CAS.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. G. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382 CrossRef CAS PubMed.
- J. N. Mills, I. T. McCrum and M. J. Janik, Phys. Chem. Chem. Phys., 2014, 16, 13699–13707 RSC.
- D. Strmcnik, K. Kodama, D. van der Vliet, J. Greeley, V. R. Stamenkovic and N. M. Marković, Nature Chem., 2009, 1, 466 CrossRef CAS PubMed.
- E. Pérez-Gallent, G. Marcandalli, M. C. Figueiredo, F. Calle-Vallejo and M. T. M. Koper, J. Am. Chem. Soc., 2017, 139, 16412–16419 CrossRef PubMed.
- A. Schizodimou and G. Kyriacou, Electrochim. Acta, 2012, 78, 171–176 CrossRef CAS.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- Y. Hori, in Handbook of Fuel Cells, John Wiley & Sons, Ltd, 2010, DOI:10.1002/9780470974001.f207055.
- J. Resasco, Y. Lum, E. Clark, J. Z. Zeledon and A. T. Bell, ChemElectroChem, 2018, 5, 1064 CrossRef CAS.
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, J. Chem. Soc., Chem. Commun., 1988, 17–19, 10.1039/C39880000017.
- A. Wuttig, Y. Yoon, J. Ryu and Y. Surendranath, J. Am. Chem. Soc., 2017, 139, 17109–17113 CrossRef CAS PubMed.
- T. Cheng, H. Xiao and W. A. Goddard, J. Am. Chem. Soc., 2016, 138, 13802–13805 CrossRef CAS.
- A. Wuttig, M. Yaguchi, K. Motobayashi, M. Osawa and Y. Surendranath, Proc. Natl. Acad. Sci., 2016, 113, E4585–E4593 CrossRef CAS.
- H. Yano, T. Tanaka, M. Nakayama and K. Ogura, J. Electroanal. Chem., 2004, 565, 287–293 CrossRef CAS.
- K. Ogura, J. R. Ferrell, A. V. Cugini, E. S. Smotkin and M. D. Salazar-Villalpando, Electrochim. Acta, 2010, 56, 381–386 CrossRef CAS.
- F. S. Roberts, K. P. Kuhl and A. Nilsson, Angew. Chem., Int. Ed., 2015, 54, 5179–5182 CrossRef CAS PubMed.
- Y. Kwon, Y. Lum, E. L. Clark, J. W. Ager and A. T. Bell, ChemElectroChem, 2016, 3, 1012–1019 CrossRef CAS.
- S. Lee, D. Kim and J. Lee, Angew. Chem., 2015, 127, 14914–14918 CrossRef.
- I. Shoichiro, T. Takehiko and I. Kaname, Bull. Chem. Soc. Jpn., 1987, 60, 2517–2522 CrossRef.
- C. Amatore and J. M. Saveant, J. Am. Chem. Soc., 1981, 103, 5021–5023 CrossRef CAS.
- Y. B. Vassiliev, V. S. Bagotsky, N. V. Osetrova, O. A. Khazova and N. A. Mayorova, J. Electroanal. Chem. Interfacial Electrochem., 1985, 189, 271–294 CrossRef.
- Y. Matsubara, ACS Energy Lett., 2017, 2, 1886–1891 CrossRef CAS.
- Y. Tomita, S. Teruya, O. Koga and Y. Hori, J. Electrochem. Soc., 2000, 147, 4164–4167 CrossRef CAS.
- O. Aschenbrenner and P. Styring, Energy Environ. Sci., 2010, 3, 1106–1113 RSC.
- B. P. Sullivan, K. Krist and H. Guard, Electrochemical and electrocatalytic reactions of carbon dioxide, Elsevier, 2012 Search PubMed.
- S.-B. Park, C.-S. Shim, H. Lee and K.-H. Lee, Fluid Phase Equilib., 1997, 134, 141–149 CrossRef CAS.
- Y. Qiu, H. Zhong, W. Xu, T. Zhang, X. Li and H. Zhang, J. Mater. Chem. A, 2019, 7, 5453–5462 RSC.
- F.-Y. Jou, A. E. Mather and F. D. Otto, Can. J. Chem. Eng., 1995, 73, 140–147 CrossRef CAS.
- L. L. Snuffin, L. W. Whaley and L. Yu, J. Electrochem. Soc., 2011, 158, F155–F158 CrossRef CAS.
- S. Kaneco, K. Iiba, H. Katsumata, T. Suzuki and K. Ohta, Electrochim. Acta, 2006, 51, 4880–4885 CrossRef CAS.
- S. Kaneco, K. Iiba, N.-h. Hiei, K. Ohta, T. Mizuno and T. Suzuki, Electrochim. Acta, 1999, 44, 4701–4706 CrossRef CAS.
- T. Mizuno, A. Naitoh and K. Ohta, J. Electroanal. Chem., 1995, 391, 199–201 CrossRef.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS PubMed.
- J. L. Anderson, J. K. Dixon and J. F. Brennecke, Acc. Chem. Res., 2007, 40, 1208–1216 CrossRef CAS PubMed.
- J. F. Brennecke and B. E. Gurkan, J. Phys. Chem. Lett., 2010, 1, 3459–3464 CrossRef CAS.
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 RSC.
- M. V. Fedorov and A. A. Kornyshev, Chem. Rev., 2014, 114, 2978–3036 CrossRef CAS.
- M. Smiglak, J. Pringle, X. Lu, L. Han, S. Zhang, H. Gao, D. MacFarlane and R. Rogers, Chem. Commun., 2014, 50, 9228–9250 RSC.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621 CrossRef CAS PubMed.
- F. Zhou, S. Liu, B. Yang, P. Wang, A. S. Alshammari and Y. Deng, Electrochem. Commun., 2014, 46, 103–106 CrossRef CAS.
- M. Alvarez-Guerra, J. Albo, E. Alvarez-Guerra and A. Irabien, Energy Environ. Sci., 2015, 8, 2574–2599 RSC.
- B. A. Rosen, J. L. Haan, P. Mukherjee, B. Braunschweig, W. Zhu, A. Salehi-Khojin, D. D. Dlott and R. I. Masel, J. Phys. Chem. C, 2012, 116, 15307–15312 CrossRef CAS.
- B. A. Rosen, W. Zhu, G. Kaul, A. Salehi-Khojin and R. I. Masel, J. Electrochem. Soc., 2013, 160, H138–H141 CrossRef CAS.
- M. Asadi, B. Kumar, A. Behranginia, B. A. Rosen, A. Baskin, N. Repnin, D. Pisasale, P. Phillips, W. Zhu, R. Haasch, R. F. Klie, P. Král, J. Abiade and A. Salehi-Khojin, Nat. Commun., 2014, 5, 4470 CrossRef CAS PubMed.
- D. Niu, H. Wang, H. Li, Z. Wu and X. Zhang, Electrochim. Acta, 2015, 158, 138–142 CrossRef CAS.
- L. Sun, G. K. Ramesha, P. V. Kamat and J. F. Brennecke, Langmuir, 2014, 30, 6302–6308 CrossRef CAS PubMed.
- A. Salehi-Khojin, H.-R. M. Jhong, B. A. Rosen, W. Zhu, S. Ma, P. J. A. Kenis and R. I. Masel, J. Phys. Chem. C, 2013, 117, 1627–1632 CrossRef CAS.
- J. L. DiMeglio and J. Rosenthal, J. Am. Chem. Soc., 2013, 135, 8798–8801 CrossRef CAS PubMed.
- J. Medina-Ramos, J. L. DiMeglio and J. Rosenthal, J. Am. Chem. Soc., 2014, 136, 8361–8367 CrossRef CAS.
- C. Costentin, M. Robert and J.-M. Saveant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- G. P. S. Lau, M. Schreier, D. Vasilyev, R. Scopelliti, M. Grätzel and P. J. Dyson, J. Am. Chem. Soc., 2016, 138, 7820–7823 CrossRef CAS PubMed.
- X. Zhang, Y. Zhao, S. Hu, M. E. Gliege, Y. Liu, R. Liu, L. Scudiero, Y. Hu and S. Ha, Electrochim. Acta, 2017, 247, 281–287 CrossRef CAS.
- Q. Wang, C. Chen, J. Zhong, B. Zhang and Z. Cheng, Aust. J. Chem., 2017, 70, 293–300 CrossRef CAS.
- Q. Zhu, J. Ma, X. Kang, X. Sun, H. Liu, J. Hu, Z. Liu and B. Han, Angew. Chem., Int. Ed., 2016, 55, 9012–9016 CrossRef CAS PubMed.
- I. Ganesh, Renewable Sustainable Energy Rev., 2014, 31, 221–257 CrossRef CAS.
- A. S. Varela, M. Kroschel, T. Reier and P. Strasser, Catal. Today, 2016, 260, 8–13 CrossRef CAS.
- R. Kas, R. Kortlever, H. Yılmaz, M. T. M. Koper and G. Mul, ChemElectroChem, 2015, 2, 354–358 CrossRef CAS.
- I. Katsounaros, J. C. Meier, S. O. Klemm, A. A. Topalov, P. U. Biedermann, M. Auinger and K. J. J. Mayrhofer, Electrochem. Commun., 2011, 13, 634–637 CrossRef CAS.
- M. R. Singh, E. L. Clark and A. T. Bell, Phys. Chem. Chem. Phys., 2015, 17, 18924–18936 RSC.
- A. S. Hall, Y. Yoon, A. Wuttig and Y. Surendranath, J. Am. Chem. Soc., 2015, 137, 14834–14837 CrossRef CAS PubMed.
- H. Hashiba, H. K. Sato, S. Yotsuhashi, K. Fujii, M. Sugiyama and Y. Nakano, Sustainable Energy Fuels, 2017, 1, 1734–1739 RSC.
- M. Gattrell, N. Gupta and A. Co, J. Electroanal. Chem., 2006, 594, 1–19 CrossRef CAS.
- L. Wang, S. A. Nitopi, E. Bertheussen, M. Orazov, C. G. Morales-Guio, X. Liu, D. C. Higgins, K. Chan, J. K. Nørskov, C. Hahn and T. F. Jaramillo, ACS Catal., 2018, 8, 7445–7454 CrossRef CAS.
- H. Ooka, M. C. Figueiredo and M. T. M. Koper, Langmuir, 2017, 33, 9307–9313 CrossRef CAS.
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. van der Vliet, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Nat. Chem., 2013, 5, 300 CrossRef CAS PubMed.
- J. Zheng, W. Sheng, Z. Zhuang, B. Xu and Y. Yan, Sci. Adv., 2016, 2, e1501602 CrossRef.
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, J. Am. Chem. Soc., 1987, 109, 5022–5023 CrossRef CAS.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Phys. Chem. B, 2002, 106, 15–17 CrossRef CAS.
- J. H. Montoya, C. Shi, K. Chan and J. K. Nørskov, J. Phys. Chem. Lett., 2015, 6, 2032–2037 CrossRef CAS PubMed.
- K. Jiang, R. B. Sandberg, A. J. Akey, X. Liu, D. C. Bell, J. K. Nørskov, K. Chan and H. Wang, Nat. Catal., 2018, 1, 111–119 CrossRef CAS.
- J. D. Goodpaster, A. T. Bell and M. Head-Gordon, J. Phys. Chem. Lett., 2016, 7, 1471–1477 CrossRef CAS PubMed.
- N. Hidetomo, I. Shoichiro, O. Yoshiyuki and I. Kaname, Chem. Lett., 1989, 18, 289–292 CrossRef.
- K. W. Frese Jr, in Electrochemical and Electrocatalytic Reactions of Carbon Dioxide, ed. B. P. Sullivan, K. Krist and H. E. Guard, Elsevier, Amsterdam, 1993, pp. 145–216, DOI:10.1016/B978-0-444-88316-2.50010-3.
- J. J. Kim, D. P. Summers and K. W. Frese, J. Electroanal. Chem. Interfacial Electrochem., 1988, 245, 223–244 CrossRef CAS.
- Y. Hori, R. Takahashi, Y. Yoshinami and A. Murata, J. Phys. Chem. B, 1997, 101, 7075–7081 CrossRef CAS.
- K. J. P. Schouten, Y. Kwon, C. J. M. van der Ham, Z. Qin and M. T. M. Koper, Chem. Sci., 2011, 2, 1902–1909 RSC.
- K. J. P. Schouten, E. Pérez Gallent and M. T. M. Koper, J. Electroanal. Chem., 2014, 716, 53–57 CrossRef CAS.
- F. Calle-Vallejo and M. T. M. Koper, Angew. Chem., Int. Ed., 2013, 52, 7282–7285 CrossRef CAS PubMed.
- K. J. P. Schouten, Z. Qin, E. Pérez Gallent and M. T. M. Koper, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS PubMed.
- J. O. M. Bockris and N. Pentland, Trans. Faraday Soc., 1952, 48, 833–839 RSC.
- M. Jouny, W. Luc and F. Jiao, Nat. Catal., 2018, 1, 748–755 CrossRef CAS.
- M. Dunwell, X. Yang, B. P. Setzler, J. Anibal, Y. Yan and B. Xu, ACS Catal., 2018, 8, 3999–4008 CrossRef CAS.
- D. Raciti, M. Mao and C. Wang, Nanotechnology, 2017, 29, 044001 CrossRef PubMed.
- A. D. Handoko, C. W. Ong, Y. Huang, Z. G. Lee, L. Lin, G. B. Panetti and B. S. Yeo, J. Phys. Chem. C, 2016, 120, 20058–20067 CrossRef CAS.
- X. Liu, P. Schlexer, J. Xiao, Y. Ji, L. Wang, R. B. Sandberg, M. Tang, K. S. Brown, H. Peng, S. Ringe, C. Hahn, T. F. Jaramillo, J. K. Nørskov and K. Chan, Nat. Commun., 2019, 10, 32 CrossRef PubMed.
- Y. Y. Birdja and M. T. M. Koper, J. Am. Chem. Soc., 2017, 139, 2030–2034 CrossRef CAS.
- A. J. Garza, A. T. Bell and M. Head-Gordon, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS.
- C. F. C. Lim, D. A. Harrington and A. T. Marshall, Electrochim. Acta, 2017, 238, 56–63 CrossRef CAS.
- J. F. Richardson, J. H. Harker and J. R. Backhurst, Coulson & Richardson's Chemical Engineering, Butterworth-Heinemann London, 2002 Search PubMed.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- F. Karadas, B. Köz, J. Jacquemin, E. Deniz, D. Rooney, J. Thompson, C. T. Yavuz, M. Khraisheh, S. Aparicio and M. Atihan, Fluid Phase Equilib., 2013, 351, 74–86 CrossRef CAS.
- P. Singh, D. W. F. Brilman and M. J. Groeneveld, Int. J. Greenhouse Gas Control, 2011, 5, 61–68 CrossRef CAS.
- T. Mizuno, K. Ohta, A. Sasaki, T. Akai, M. Hirano and A. Kawabe, Energy Sources, 1995, 17, 503–508 CrossRef CAS.
- S. Nakagawa, A. Kudo, M. Azuma and T. Sakata, J. Electroanal. Chem. Interfacial Electrochem., 1991, 308, 339–343 CrossRef CAS.
- H. De Jesús-Cardona, C. del Moral and C. R. Cabrera, J. Electroanal. Chem., 2001, 513, 45–51 CrossRef.
- K. Hara, A. Kudo and T. Sakata, J. Electroanal. Chem., 1997, 421, 1–4 CrossRef CAS.
- K. Hara, A. Kudo and T. Sakata, J. Electroanal. Chem., 1995, 386, 257–260 CrossRef.
- N. Sonoyama, M. Kirii and T. Sakata, Electrochem. Commun., 1999, 1, 213–216 CrossRef CAS.
- K. Hara, A. Tsuneto, A. Kudo and T. Sakata, J. Electrochem. Soc., 1994, 141, 2097–2103 CrossRef CAS.
- A. Kudo, S. Nakagawa, A. Tsuneto and T. Sakata, J. Electrochem. Soc., 1993, 140, 1541–1545 CrossRef CAS.
- E. Stránská and D. Neděla, J. Ind. Text., 2018, 48, 432–447 CrossRef.
- F. L. Smith and A. H. Harvey, Chem. Eng. Prog., 2007, 103, 33–39 CAS.
- J. H. Perry, Chemical engineers' handbook, ACS Publications, 1950 Search PubMed.
- M. J. Moran, H. N. Shapiro, D. D. Boettner and M. B. Bailey, Fundamentals of engineering thermodynamics, John Wiley & Sons, 2010 Search PubMed.
- J. Ryu, T. N. Andersen and H. Eyring, J. Phys. Chem., 1972, 76, 3278–3286 CrossRef CAS.
- J. Kestin, M. Sokolov and W. A. Wakeham, J. Phys. Chem. Ref. Data, 1978, 7, 941–948 CrossRef CAS.
- E. N. Fuller, P. D. Schettler and J. C. Giddings, Ind. Eng. Chem., 1966, 58, 18–27 CrossRef CAS.
- J. J. Carroll, F.-Y. Jou, A. E. Mather and F. D. Otto, Can. J. Chem. Eng., 1998, 76, 945–951 CrossRef CAS.
- J. E. Davis and J. J. McKetta, J. Chem. Eng. Data, 1960, 5, 374–375 CrossRef CAS.
- M. Azuma, K. Hashimoto, M. Hiramoto, M. Watanabe and T. Sakata, J. Electroanal. Chem. Interfacial Electrochem., 1989, 260, 441–445 CrossRef CAS.
- M. Azuma, K. Hashimoto, M. Hiramoto, M. Watanabe and T. Sakata, J. Electrochem. Soc., 1990, 137, 1772–1778 CrossRef CAS.
- S. T. Ahn, I. Abu-Baker and G. T. R. Palmore, Catal. Today, 2017, 288, 24–29 CrossRef CAS.
- S. Kaneco, N.-h. Hiei, Y. Xing, H. Katsumata, H. Ohnishi, T. Suzuki and K. Ohta, Electrochim. Acta, 2002, 48, 51–55 CrossRef CAS.
- J. Hussain, H. Jónsson and E. Skúlason, ACS Catal., 2018, 8, 5240–5249 CrossRef CAS.
- W. Luo, X. Nie, M. J. Janik and A. Asthagiri, ACS Catal., 2016, 6, 219–229 CrossRef CAS.
- Sustainion®Anion Exchange Membranes, https://dioxidematerials.com/technology/sustainion-membranes/, accessed 15/08/2019, 2019.
- K. Scott, C. Xu and X. Wu, Wiley Interdiscip. Rev.: Energy Environ., 2014, 3, 24–41 CAS.
- I. Vincent and D. Bessarabov, Renewable Sustainable Energy Rev., 2018, 81, 1690–1704 CrossRef CAS.
- F. Proietto, B. Schiavo, A. Galia and O. Scialdone, Electrochim. Acta, 2018, 277, 30–40 CrossRef CAS.
- S. J. Davis, N. S. Lewis, M. Shaner, S. Aggarwal, D. Arent, I. L. Azevedo, S. M. Benson, T. Bradley, J. Brouwer, Y.-M. Chiang, C. T. M. Clack, A. Cohen, S. Doig, J. Edmonds, P. Fennell, C. B. Field, B. Hannegan, B.-M. Hodge, M. I. Hoffert, E. Ingersoll, P. Jaramillo, K. S. Lackner, K. J. Mach, M. Mastrandrea, J. Ogden, P. F. Peterson, D. L. Sanchez, D. Sperling, J. Stagner, J. E. Trancik, C.-J. Yang and K. Caldeira, Science, 2018, 360 Search PubMed.
- J. R. Hudkins, D. G. Wheeler, B. Pena and C. P. Berlinguette, Energy Environ. Sci., 2016, 9, 3417–3423 RSC.
- S. Sabatino, A. Galia, G. Saracco and O. Scialdone, ChemElectroChem, 2017, 4, 150–159 CrossRef CAS.
- T. E. Lister and E. J. Dufek, Energy Fuels, 2013, 27, 4244–4249 CrossRef CAS.
- I. Moussallem, J. Jörissen, U. Kunz, S. Pinnow and T. Turek, J. Appl. Electrochem., 2008, 38, 1177–1194 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |