
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
“Nano-garden cultivation” for electrocatalysis: controlled synthesis of Nature-inspired hierarchical nanostructures†
Xiaoyu
Yan
a,
Yang
Zhao
a,
Jasper
Biemolt
 b,
Kai
Zhao
a,
Petrus C. M.
Laan
b,
Xiaojuan
Cao
a and
Ning
Yan
*ab
b,
Kai
Zhao
a,
Petrus C. M.
Laan
b,
Xiaojuan
Cao
a and
Ning
Yan
*ab
aSchool of Physics and Technology, Wuhan University, Wuhan, China. E-mail: n.yan@uva.nl
bVan't Hoff Institute for Molecular Sciences (HIMS), University of Amsterdam, Amsterdam, The Netherlands
First published on 10th March 2020
Abstract
Three-dimensional intricate nanostructures hold great promise for real-life applications. Many of these hierarchical structures resemble shapes from Nature, demonstrating much improved physico-chemical properties. Yet, their rational design and controlled synthesis remain challenging. By simply manipulating (electro)chemical gradients using a combined hydrothermal and electrodeposition strategy, we herein show the controlled growth of Co(OH)2 nanostructures, mimicking the process of garden cultivation. The resulting “nano-garden” can selectively contain different patterns, all of which can be fully phosphidated into CoP without losing the structural integrity. Remarkably, these CoP nanostructures show distinct catalytic performance in oxygen evolution and hydrogen evolution reactions. Under pH-universal conditions, the CoP “soil + flower-with-stem” structure shows a much more “effective” surface area for gas-evolving reactions with lower activation and concentration overpotentials. This provides superior bifunctional catalytic activity for both reactions, outperforming noble metal counterparts.
Nature holds solutions to diverse scientific problems and serves as a major source of inspiration for human beings. The billions of years of evolution has led to the formation of numerous geographical and biological structures with stunning complexity. By studying these natural patterns and mimicking the topographic structure in the materials synthesis, researchers often find exciting solutions to optimize the physico-chemical properties of materials. In particular, with the bloom of nanotechnology, the rational synthesis of Nature-inspired hierarchical shapes at the nanometric scale becomes of great importance, showing potential for real-life applications in catalysis,1 electronics,2 optics3 and bio-medicine.4 A common approach for preparing nanostructures is top-down lithography.5 This subtractive method includes photo-,6 electron-beam-,7 and nanoimprint-lithography.8 It can indeed control the topology of the pattern accurately and is widely used in the nanofabrication of semiconductors.9–15 Nonetheless, this approach is rather time-consuming and cost-ineffective, hampering its wide applications in different fields.15,16
Therefore, the bottom-up synthesis is increasingly explored which promises to address the drawbacks of the top-down counterpart. This additive strategy is generally based on the self-assembly of molecules and/or nanoscale building blocks.17,18 Interesting nanostructures resembling natural shapes can be obtained. For instance, using solvothermal or hydrothermal strategies, flower-,19–23 leaf-,24,25 coral-26–30 and sea-urchin-like nanostructures have been reported recently,31,32 demonstrating much enhanced performances in different applications. However, the biomimetic appearance of these patterns during the synthesis, in many cases, is not based on predictive mechanisms.33 Besides, costly additives and surfactants are often used for geometry control, and large-scale material preparation is thus challenging.34–36 Electrodeposition is a simple alternative approach of nanofabrication which can be dated back to 1987 in the pioneering work of Martin et al.37 Yet, a template is often required for the synthesis and plating three-dimensional intricate structures at the nanometric scale remains problematic. Though the coprecipitation approach has recently allowed precise control of the formation of complex micrometer-scale shapes with amazing beauty, downsizing the shape to the nanometer scale is yet impossible.18,36,38,39
In this work, via coupling the hydrothermal and electrodeposition synthesis approaches under different reaction conditions, we managed to design and prepare a number of Co(OH)2-based nano-architectures that resemble various items in a garden (“soil”, “flake”, “sprout”, “grass”, “flower” and “leaf”). Such patterns can be fully phosphidated, forming CoP structures. The suitable combination of the nanometric items on the surface of carbon cloth provides superior bifunctional catalytic activity for overall water splitting, outperforming the state-of-the-art catalysts at high current densities.
Fig. 1a is the schematic cartoon of the step-by-step “nano-garden cultivation” on the carbon cloth (CC). The distinct topologies were sustained after phosphidation as shown in the corresponding scanning electron microscopy (SEM) images. The gardening started with “earthing” by encapsulating the fibers of CC with a dense layer of Co(OH)2via a modified hydrothermal approach. The detailed synthesis can be found in the ESI.† On this “soil” layer with a thickness of ∼100 nm (cf. the SEM image of the pristine fiber in Fig. S1†), many “sprouts” appeared after increasing the temperature of the hydrothermal synthesis. They were strongly “rooted” in the soil which is beneficial for the robust catalysis in gas-evolving reactions without suffering break-up or detachment (vide infra). The continuous growth finally led to the formation of “grass”, covering the entire soil land (see Fig. 1d and S2†). These grasses have a high aspect ratio with an average length of 1.5 μm and a thickness of 100 nm. By increasing the Co2+ concentration from 0.13 M to 0.3 M, they can progressively evolve into “lithic flakes” and “rocks” as shown in Fig. S3.†
 | ||
| Fig. 1 (a) The schematic steps of the nano-garden cultivation on carbon cloth; false color SEM images of various CoP nanostructures on carbon cloth after phosphidation for the (b) soil, (c) sprout, (d) grass, (e) flower and (f) leaf, and the true color SEM images are shown as the inset. | ||
The “blossom” of the “bush” was enabled by electrodeposition of Co(OH)2 following the reactions (1) and (2) below:40
| NO3− + H2O + 2e− → NO2− + 2OH−, E0 = 0.01 V | (1) |
| Co2+ + 2OH− → Co(OH)2(s) | (2) |
| Co2+ + 2e− → Co(s), E0 = −0.28 V | (3) |
| 2H+ + 2e− → H2(g), E0 = 0.00 V | (4) |
In 0.04 M Co2+-electrolyte, the electrodeposition dominantly proceeded from the tip of the grass stem, where the radius of curvature is smaller with a higher space charge density. At the lower parts of the stem, electrodeposition was suppressed as the competitive water reduction reaction (reaction (4)) occurred.41,42 The ramified growth of the “flower petals” was triggered by the electroconvection of the solution at the tip, resulting in the continuous splitting of the deposits into branches to all directions.43 This growth mechanism is detailed in the ESI in Fig. S4.†44,45 After 300 s deposition, a sphere-like flower, with a diameter of ca. 200 nm, was formed on the top of the stem (Fig. 1e). The SEM image in Fig. S5† with a lower magnification implies that nearly all the stems have “bloomed”.
On the contrary, electrodeposition can also initiate from the bottom of the stem when a 0.07 M Co(NO3)2 electrolyte was employed. In this more concentrated solution, the competing water reduction reaction was largely suppressed while reaction (1) prevailed. The formation of Co(OH)2 can therefore start from the bottom of the stem. This finally caused the deposition of the “leaf” pattern with the same orientation as shown in Fig. 1f. From the microscopic point of view, each leaf was in fact the interwoven dendritic structure of the deposits. The bottom side of the leaf spanned more than 0.5 μm and the inter-leaf distance was ca. 200 nm on average. A complete summary of all the nano-garden items via controlled synthesis is shown in Fig. S6.†
X-ray diffraction (XRD) patterns in Fig. S7a† compare the as-prepared crystal structures of all the nanostructures before and after phosphidation (denoted as soil/CC, sprout/CC, flake/CC, grass/CC, leaf/CC and flower/CC). The as-prepared precursor shows diffraction peaks at 26.0, 33.8, 35.5, 36.8, 39.6, 59.1, 61.1 and 62.3°, which can be indexed to the hexagonal phase Co(OH)0.44(CO3)0.78·0.29H2O.46 The incorporation of carbonate was due to the interaction with atmospheric CO2 before the XRD measurement. This is also evidenced by the Fourier-transform infrared spectra (FTIR) in Fig. S7b.† After phosphidation, all samples were fully converted to CoP. The patterns show four distinct peaks at 31.7, 36.2, 46.3, and 48.2°, which can be assigned to the (011), (111), (112) and (211) planes of the orthorhombic CoP phase (JCPDS no: 29-0497), respectively.47–50 Particularly, the corresponding SEM X-ray energy dispersive spectra (EDX) and the high-resolution transmission electron microscopy (HRTEM) micrograph in Fig. S8 and S9† also verified the formation of CoP. They also indicated that the flower petals and stems both uniformly consisted of CoP. We also performed X-ray photoelectron spectroscopy (XPS), confirming the formation of CoP. The Co 2p XPS spectrum in Fig. S7c† shows both 2p3/2 and 2p1/2 peaks in which the ones at 803.2 and 786.0 eV were the shake-up satellites.27,51 The peak at 781.8 eV was assigned to CoP and other oxidized forms of Co while the one at 778.1 eV was ascribed to the residual metallic Co.52–54 The reduced state of P in CoP was also seen in the P 2p spectrum at 129.0 eV in Fig. S7d.†43,55,56
We then examined the catalytic activity of all the CoP nanostructures in the hydrogen evolution reaction (HER) under acidic conditions. A standard three-electrode configuration was applied with a stationary working electrode to better simulate industrially relevant conditions. Fig. 2a compares the polarization curves (LSV, linear sweep voltammetry) of CC, soil/CC, sprout/CC, flake/CC, grass/CC, leaf/CC and flower/CC in 0.5 M H2SO4 aqueous solution. CC showed little activity for HER with an extremely high overpotential. The onset potential, defined as that at 1 mA cm−2 Faraday current, reached −325 mV (vs. RHE and hereafter other than specified). Interestingly, although other samples shared an identical chemical composition, their catalytic activity varied significantly. The flower/CC had the highest onset potential of −36 mV. The overpotentials at the benchmark 10, 20, 50 and 100 mA cm−2 current density were 68, 85, 103 and 112 mV, respectively (see Fig. 2b). These values are among the best of today's superior non-precious metal HER catalysts in an acidic environment. A detailed comparison can be found in Table S1.†
 | ||
| Fig. 2 The comparison of (a) LSVs, (b) overpotentials and (c) Tafel slopes for various nanomaterials in HER; (d) a schematic comparison showing the different reaction environments at the top and bottom positions of the nanostructure with a high aspect ratio; (e) CV cycles and chronoamperometric stability test at −0.094 V vs. RHE. H2-saturated 0.5 M H2SO4 aqueous electrolyte is used, the scan rate is 5 mV s−1, and post-iR compensation is applied. | ||
Fig. 2c shows the Tafel plots of all the catalysts. Remarkably, the Tafel slope of flower/CC was 68 mV dec−1, which was also the lowest among all the nanostructures. In addition, to evaluate the difference in intrinsic catalytic properties, the turnover frequency (TOF) was also plotted versus the overpotential (see Fig. S10†). The flower/CC catalysts illustrated significantly larger TOF values than those of leaf/CC in acidic environments, particularly at higher overpotentials when the mass transport limitation appeared. Specifically, the TOF of flower/CC reached 1.3 s−1 at an overpotential of 120 mV. This is in accordance with the electrochemical impedance spectra (EIS) in Fig. S11.† The Nyquist plot of the flower/CC exhibits a smaller charge-transfer resistance than that of the leaf/CC, suggesting faster kinetics at the interface.
The better performance of flower/CC mainly pertained to its physical geometry. Apparently, soil/CC and sprout/CC had lower surface areas and smaller numbers of active sites, which unsurprisingly showed limited activity. This was proven in the electrochemically active surface area (EASA) plots shown in Fig. S12.† A positive correlation between the HER performance and EASA can be drawn. Albeit that leaf/CC and flake/CC owned a larger number of active sites as reflected by the EASA, the interlayer distance between two leaves or flakes was less than 200 nm while the height of the layer was >2 μm (cf. the SEM images). When the hydrogen bubbles evolve at high current density, the reaction on the surface at the bottom side of the densely packed structure might suffer from slow mass transport,57 and the activity became increasingly lower than that of the flower/CC at higher current density. Conversely, the geometry of flower/CC was superior to the controls: the flowers on the top of the stems, comprising many nanometric petals, were all well exposed to the bulk electrolyte. Yet, the loosely packed stems, with much less CoP deposits, can also effectively catalyze HER (see the cartoon in Fig. 2d). Thus, such hierarchical structure provided complementary features in terms of abundant active sites and rapid mass transport. This set of experiments also implied that the physical geometry of nanomaterials can greatly affect the catalytic activity. Particularly for gas-evolving electrocatalytic reactions, the densely pack nanowires or nano-arrays must be fine-tuned to minimize the overpotentials arising from the lack of effective active sites and the limitation of mass transport.57
The stability test of flower/CC was carried out using both cyclic voltammetry (CV) and chronoamperometry. Fig. 2e demonstrates the comparison of the LSV curves of the catalyst after 0, 1000 and 2000 CV cycles in the voltage window from +0.2 to −0.3 V (vs. RHE). Little degradation was observed in comparison with the initial performance (the morphologies of the spent catalysts are shown in Fig. S13†). The overpotential showed a 2 mV increase at 100 mA cm−2. In the chronoamperometric analysis, the voltage was set at −0.094 V. The current density was maintained at ca. 31 mA cm−2 for more than 30 h. This suggested that the nanoflowers rooted in the “soil” have excellent structural robustness and this “nano-garden cultivation” approach has effectively increased the activity without any compromise of the stability.
The structural advantage of flower/CC was also proven in the HER performed under both neutral and alkaline conditions. Fig. 3 summarizes the electrocatalytic performance of both flower/CC and leaf/CC. In a 1.0 M phosphate buffer solution (PBS), flower/CC remained the best catalyst. The onset potential was −36 mV whereas the overpotentials at 10 and 50 mA cm−2 were 72 and 138 mV, respectively. In the stability test via the analogous approach used in acidic media, flower/CC also exhibited excellent robustness. Interestingly, it seemed that the catalyst was “activated” during the CV cycles or the chronoamperometric test, showing an activity jump. This phenomenon might be pertinent to the formation of low-valence Co complexes, such as Co(OH)x, on the surface of CoP under the cathodic potentials. In addition, the progressive incorporation of phosphonic acid pendant groups onto the surface increased the proton-accepting capability. Benefiting from these proven synergistic effects, the HER activity was enhanced after the longevity test.58,59
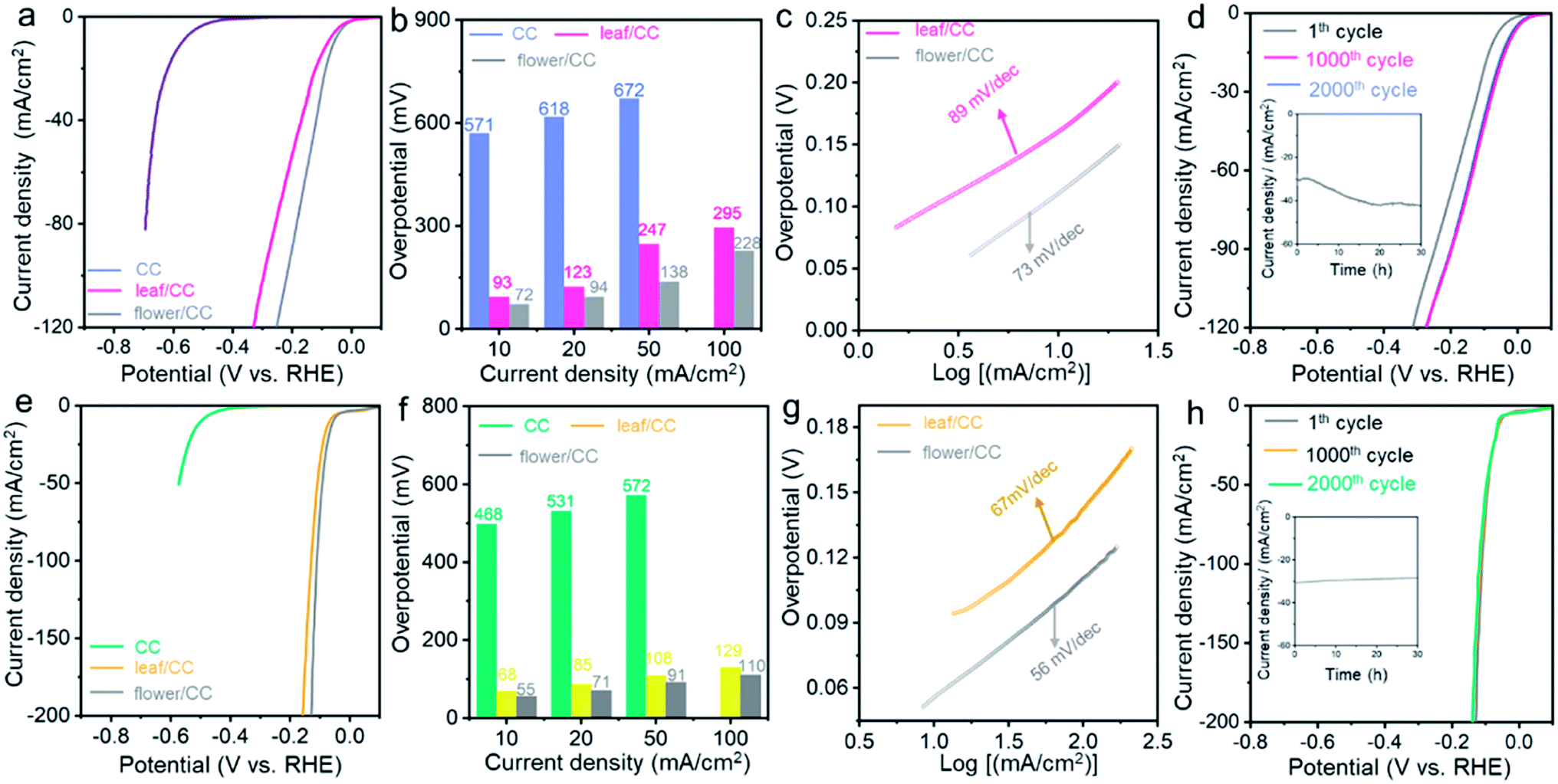 | ||
| Fig. 3 The comparison of (a) LSVs, (b) overpotentials, (c) Tafel slopes and (d) stability tests for two nanomaterials in HER under neutral conditions with H2-saturated phosphate buffer solution. The comparison of (e) LSVs, (f) overpotentials, (g) Tafel slopes and (h) stability tests for two nanomaterials in HER in H2-saturated 1.0 M KOH aqueous solution. The scan rate is 5 mV s−1, and post-iR compensation is applied. | ||
In 1.0 M KOH, flower/CC CoP still exhibited an ultra-low overpotential of 55 mV at the benchmark 10 mA cm−2, and the Tafel slope was 56 mV dec−1. Likewise, the overpotential difference relative to leaf/CC became increasingly large when the current density rose, indicating that concentration polarization was also suppressed in flower/CC compared with that of leaf/CC. In the longevity test, only trivial degradation (∼6%) was recorded during the chronoamperometric test biased at −0.082 V. We also compared the HER performance of flower/CC with the state-of-the-art non-noble metal catalysts in the literature (see Tables S2 and S3†). The activity was indeed among the best. This further supports our aforementioned structural advantages of flower/CC at high reaction rates. Therefore, we concluded that the CoP with a flower-with-stem structure rooted in a soil layer enabled superior HER activity under pH-universal conditions.
Apart from HER, the oxygen evolution reaction (OER) is another typical and important gas-evolving reaction for energy storage and conversion.60 We then evaluated the electrocatalytic performance of these CoP nanostructures in 1.0 M KOH electrolyte. Fig. 4a compares the LSV curves of flower/CC and leaf/CC with those of the state-of-the-art IrO2/CC OER catalysts. The onset potential of all the examined catalysts was essentially identical. However, at high current densities, both flower/CC and leaf/CC showed substantially improved activity. In particular, the overpotential of IrO2/CC at 50 mA cm−2 was 417 mV while that of flower/CC was only 324 mV. The outstanding OER performance of flower/CC was also supported by the low Tafel slope which was 73 mA dec−1. Both CV and chronoamperometric studies confirmed the stability of this material. A detailed comparison of the studied catalysts with those reported in the literature is summarized in Table S4.†
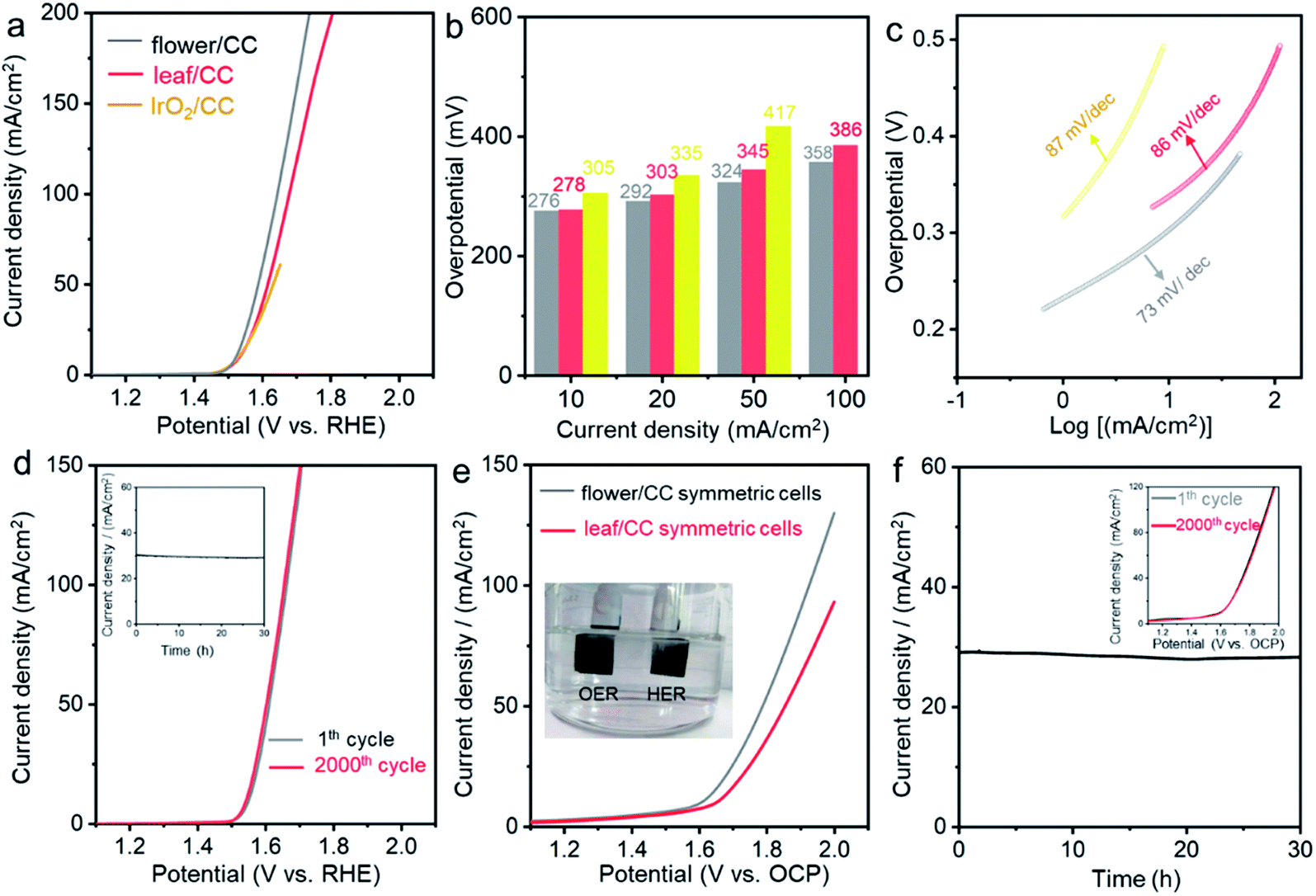 | ||
| Fig. 4 The comparison of (a) LSVs, (b) overpotentials, (c) Tafel slopes and (d) stability tests for three nanomaterials in OER in O2-saturated 1.0 M KOH aqueous solution; (e) LSV and (f) stability test of the symmetric cells in the overall water splitting reaction. The scan rate is 5 mV s−1, and post-iR compensation is applied. | ||
Finally, we carried out overall water splitting reactions using a symmetric two-electrode setup. The anode and cathode catalysts were completely identical to investigate their bifunctionality. Fig. 4e compares the LSV curves of flower/CC and leaf/CC cells. At the benchmark 10 mA cm−2, the overall overpotential was 370 mV for flower/CC which showed better performance. This potential was 49 mV higher than the sum of the two overpotentials obtained in the three-electrode setup (55 mV for HER and 276 mV for OER). We postulated that this difference might have originated from the additional polarization caused by the mass transport. In the stability test shown in Fig. 4f, we biased the cell at 1.78 V vs. open circuit potential (OCP) for 30 h, and the current density was stabilized at ∼29 mA cm−2 with little degradation. The redox stability was studied by cycling the cells from 1.0 to 1.6 V vs. OCP. No loss was observed after 2000 cycles (see the inset of Fig. 4e).
In conclusion, we developed a simple yet rational approach for accurately controlling the morphology of CoP catalysts at the nanoscale via the combined hydrothermal and electrodeposition approach. It enabled us to synthesize a number of Nature-inspired hierarchical nanostructures with distinct catalytic activity in water splitting reactions. Under pH-universal conditions, the “soil + flower-with-stem” is superior, showing a much more “effective” surface area for gas-evolving reactions with lower activation and concentration overpotentials. This work provides a simple approach for controllable synthesis, reveals the importance of fine-tuning the architecture of electrocatalysts and might open bona fide opportunities for the catalysis and materials science community in the context of sustainable energy research.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the funding through the Jiangsu Provincial Department of Science and Technology (BK20190216) and Wuhan University. J. Biemolt, P. Laan and N. Yan also acknowledge the financial support from the Netherlands Organization for Scientific Research (NWO) NWO-GDST Advanced Materials program (729.001.022) and VIDI program (VI.Vidi.192.045).References
- Z. Bao, M. R. Weatherspoon, S. Shian, Y. Cai, P. D. Graham, S. M. Allan, G. Ahmad, M. B. Dickerson, B. C. Church and Z. Kang, Nature, 2007, 446, 172–175 CrossRef CAS PubMed.
- G.-C. Yi, C. Wang and W. I. Park, Semicond. Sci. Technol., 2005, 20, S22–S34 CrossRef CAS.
- J. K. Gansel, M. Thiel, M. S. Rill, M. Decker, K. Bade, V. Saile, G. von Freymann, S. Linden and M. Wegener, Science, 2009, 325, 1513–1515 CrossRef CAS PubMed.
- J. Gao, Y. Feng, W. Guo and L. Jiang, Chem. Soc. Rev., 2017, 46, 5400–5424 RSC.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- J. H. Kim, M. Seo and S. Y. Kim, Adv. Mater., 2009, 21, 4130–4133 CrossRef CAS.
- V. R. Manfrinato, L. Zhang, D. Su, H. Duan, R. G. Hobbs, E. A. Stach and K. K. Berggren, Nano Lett., 2013, 13, 1555–1558 CrossRef CAS PubMed.
- H.-W. Sun, J.-Q. Liu, D. Chen and P. Gu, Microelectron. Eng., 2005, 82, 175–179 CrossRef CAS.
- H. Ding, Q. Zhang, H. Gu, X. Liu, L. Sun, M. Gu and Z. Gu, Adv. Funct. Mater., 2020, 30, 1901760 CrossRef CAS.
- S. Conrad, P. Kumar, F. Xue, L. Ren, S. Henning, C. Xiao, K. A. Mkhoyan and M. Tsapatsis, Angew. Chem., Int. Ed., 2018, 57, 13592–13597 CrossRef CAS PubMed.
- S. Fan, D. Wang, A. Kenaan, J. Cheng, D. Cui and J. Song, Small, 2019, 15, 1805554–1805566 CrossRef PubMed.
- J. Bae, K. Lee, S. Seo, J. G. Park, Q. Zhou and T. Kim, Nat. Commun., 2019, 10, 1–9 CrossRef PubMed.
- C.-Y. Wu, H.-P. Hsieh, S.-T. Chen, T.-Y. Liu and H.-Y. Chen, Langmuir, 2018, 34, 4651–4657 CrossRef CAS PubMed.
- M. Hwang, C. Kim, J. Kim, J. G. Son and B. Yeom, Adv. Funct. Mater., 2019, 29, 1808979 CrossRef.
- J. Biemolt, K. van der Veen, N. J. Geels, G. Rothenberg and N. Yan, Carbon, 2019, 155, 643–649 CrossRef CAS.
- A. Klinkova, R. M. Choueiri and E. Kumacheva, Chem. Soc. Rev., 2014, 43, 3976–3991 RSC.
- N. Nuraje, S. Mohammed, L. Yang and H. Matsui, Angew. Chem., Int. Ed., 2009, 48, 2546–2548 CrossRef CAS PubMed.
- F. Yu, D. Poole III, S. Mathew, N. Yan, J. Hessels, N. Orth, I. Ivanović-Burmazović and J. N. Reek, Angew. Chem., Int. Ed., 2018, 130, 11417–11421 CrossRef.
- B. Chang, Q. Liu, N. Chen and Y. Yang, ChemCatChem, 2019, 11, 1884–1888 CrossRef CAS.
- G. Li, X. Zhang, H. Zhang, C. Liao and G. Jiang, Appl. Catal., B, 2019, 249, 147–154 CrossRef CAS.
- L. Zhang, R. Abdullah, X. Hu, H. Bai, H. Fan, L. He, H. Liang, J. Zou, Y. Liu and Y. Sun, J. Am. Chem. Soc., 2019, 141, 4282–4290 CrossRef CAS PubMed.
- L. Shi, D. Li, J. Yu, H.-M. Zhang, S. Ullah, B. Yang, C. Li, C. Zhu and J. Xu, J. Power Sources, 2018, 387, 64–71 CrossRef.
- X. Li, T. Li, Y. Ma, Q. Wei, W. Qiu, H. Guo, X. Shi, P. Zhang, A. M. Asiri and L. Chen, Adv. Energy Mater., 2018, 8, 1801357 CrossRef.
- H. Du, W. Ai, Z. L. Zhao, Y. Chen, X. Xu, C. Zou, L. Wu, L. Su, K. Nan and T. Yu, Small, 2018, 14, 1801068–1801077 CrossRef PubMed.
- B. Chen, Z. Zhang, S. Kim, M. Baek, D. Kim and K. Yong, Appl. Catal., B, 2019, 259, 118017–118026 CrossRef CAS.
- S. Wang, P. He, L. Jia, M. He, T. Zhang, F. Dong, M. Liu, H. Liu, Y. Zhang and C. Li, Appl. Catal., B, 2019, 243, 463–469 CrossRef CAS.
- Y. Lin, Y. Gao and Z. Fan, Adv. Mater., 2017, 29, 1701736 CrossRef PubMed.
- Y. Lu, T. Chang, S.-H. Wu, C. Liu, K. Lai, Y. Chang, Y. Chang, H. Lu, C. Chu and K. Ho, Nano Energy, 2019, 58, 138–146 CrossRef CAS.
- J. Zhang, Y. Li, T. Zhu, Y. Wang, J. Cui, J. Wu, H. Xu, X. Shu, Y. Qin and H. Zheng, ACS Appl. Mater. Interfaces, 2018, 10, 31330–31339 CrossRef CAS PubMed.
- Y. Qu, Z. Li, N. Sun, X. Zhang, S. Chen and L. Jing, Catal. Today, 2019, 327, 288–294 CrossRef CAS.
- J. Rong, J. Xu, F. Qiu, Y. Zhu, Y. Fang, J. Xu and T. Zhang, Adv. Mater. Interfaces, 2019, 6, 1900502 CrossRef.
- T. Chen, B. Cheng, G. Zhu, R. Chen, Y. Hu, L. Ma, H. Lv, Y. Wang, J. Liang and Z. Tie, Nano Lett., 2016, 17, 437–444 CrossRef PubMed.
- W. L. Noorduin, A. Grinthal, L. Mahadevan and J. Aizenberg, Science, 2013, 340, 832–837 CrossRef CAS PubMed.
- A. Bhorde, A. Pawbake, P. Sharma, S. Nair, A. Funde, P. Bankar, M. More and S. Jadkar, Appl. Phys. A, 2018, 124, 133–141 CrossRef CAS.
- J. Pandey, B. Hua, W. Ng, Y. Yang, K. van der Veen, J. Chen, N. J. Geels, J.-L. Luo, G. Rothenberg and N. Yan, Green Chem., 2017, 19, 2793–2797 RSC.
- N. Yan, R. J. Detz, N. Govindarajan, J. M. Koelewijn, B. Hua, P. Li, E. J. Meijer and J. N. Reek, J. Mater. Chem. A, 2019, 7, 23098–23104 RSC.
- R. M. Penner and C. R. Martin, Anal. Chem., 1987, 59, 2625–2630 CrossRef CAS.
- T. Tang, W. J. Jiang, S. Niu, N. Liu, H. Luo, Q. Zhang, W. Wen, Y. Y. Chen, L. B. Huang and F. Gao, Adv. Funct. Mater., 2018, 28, 1704594 CrossRef.
- A. P. LaGrow, M. O. Besenhard, A. Hodzic, A. Sergides, L. K. Bogart, A. Gavriilidis and N. T. K. Thanh, Nanoscale, 2019, 11, 6620–6628 RSC.
- Z. Pu, Q. Liu, P. Jiang, A. M. Asiri, A. Y. Obaid and X. Sun, Chem. Mater., 2014, 26, 4326–4329 CrossRef CAS.
- J.-C. Bradley, H.-M. Chen, J. Crawford, J. Eckert, K. Ernazarova, T. Kurzeja, M. Lin, M. McGee, W. Nadler and S. G. Stephens, Nature, 1997, 389, 268–271 CrossRef CAS.
- L. Mirkova, J. Appl. Electrochem., 1994, 24, 420–425 CrossRef CAS.
- Y. Bai, H. Zhang, Y. Feng, L. Fang and Y. Wang, J. Mater. Chem. A, 2016, 4, 9072–9079 RSC.
- V. Fleury, J. Kaufman and D. Hibbert, Nature, 1994, 367, 435–438 CrossRef CAS.
- M. Wang, W. J. van Enckevort, N.-b. Ming and P. Bennema, Nature, 1994, 367, 438–441 CrossRef CAS.
- A. C. Kathalikkattil, R. Roshan, J. Tharun, H. G. Soek, H. S. Ryu and D. W. Park, ChemCatChem, 2014, 6, 284–292 CrossRef CAS.
- L. M. Cao, Y. W. Hu, S. F. Tang, A. Iljin, J. W. Wang, Z. M. Zhang and T. B. Lu, Adv. Sci., 2018, 5, 1800949 CrossRef PubMed.
- G. Zhang, B. Wang, J. Bi, D. Fang and S. Yang, J. Mater. Chem. A, 2019, 7, 5769–5778 RSC.
- K. Zhu, J. Liu, S. Li, L. Liu, L. Yang, S. Liu, H. Wang and T. Xie, Adv. Mater. Interfaces, 2017, 4, 1700377 CrossRef.
- H. Du, X. Zhang, Q. Tan, R. Kong and F. Qu, Chem. Commun., 2017, 53, 12012–12015 RSC.
- T. Liu, X. Yan, P. Xi, J. Chen, D. Qin, D. Shan, S. Devaramani and X. Lu, Int. J. Hydrogen Energy, 2017, 42, 14124–14132 CrossRef CAS.
- Y. Qian, Z. Liu, H. Zhang, P. Wu and C. Cai, ACS Appl. Mater. Interfaces, 2016, 8, 32875–32886 CrossRef CAS PubMed.
- P. D. Tran, S. Y. Chiam, P. P. Boix, Y. Ren, S. S. Pramana, J. Fize, V. Artero and J. Barber, Energy Environ. Sci., 2013, 6, 2452–2459 RSC.
- X. Yan, S. Devaramani, J. Chen, D. Shan, D. Qin, Q. Ma and X. Lu, New J. Chem., 2017, 41, 2436–2442 RSC.
- J. Tian, Q. Liu, A. M. Asiri and X. Sun, J. Am. Chem. Soc., 2014, 136, 7587–7590 CrossRef CAS PubMed.
- F. Yang, Y. Chen, G. Cheng, S. Chen and W. Luo, ACS Catal., 2017, 7, 3824–3831 CrossRef CAS.
- S. Flick, S. R. Dhanushkodi and W. Mérida, J. Power Sources, 2015, 280, 97–106 CrossRef CAS.
- L. Su, X. Cui, T. He, L. Zeng, H. Tian, Y. Song, K. Qi and B. Y. Xia, Chem. Sci., 2019, 10, 2019–2024 RSC.
- H. Huang, Y. Han, H. Lei, M. Chen and R. Cao, Chem. Commun., 2017, 53, 6195–6198 RSC.
- A. R. Zeradjanin, ChemSusChem, 2018, 11, 1278–1284 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta00870b |
| This journal is © The Royal Society of Chemistry 2020 |