
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImmobilisation and flow chemistry: tools for implementing biocatalysis
José
Coloma
ab,
Yann
Guiavarc’h
ac,
Peter-Leon
Hagedoorn
 a and
Ulf
Hanefeld
*a
a and
Ulf
Hanefeld
*a
aBiokatalyse, Afdeling Biotechnologie, Technische Universiteit Delft, Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: u.hanefeld@tudelft.nl
bUniversidad Laica Eloy Alfaro de Manabí, Avenida Circunvalación s/n, P. O. Box 13-05-2732, Manta, Ecuador
cLaboratory Reactions and Process Engineering, University of Lorraine, CNRS, LRGP, F-54000 Nancy, France
First published on 5th October 2021
Abstract
The merger of enzyme immobilisation and flow chemistry has attracted the attention of the scientific community during recent years. Immobilisation enhances enzyme stability and enables recycling, flow chemistry allows process intensification. Their combination is desirable for the development of more efficient and environmentally friendly biocatalytic processes. In this feature article, we aim to point out important metrics for successful enzyme immobilisation and for reporting flow biocatalytic processes. Relevant examples of immobilised enzymes used in flow systems in organic, biphasic and aqueous systems are discussed. Finally, we describe recent developments to address the cofactor recycling hurdle.
 José Coloma | José Coloma Hurel was born in 1981 in Guayaquil, Ecuador and received his master degree in food science from the Universidad Agraria del Ecuador in 2015. After working as an assistant lecturer at the Universidad Laica Eloy Alfaro de Manabí until 2018, he started his PhD studies with Prof. Ulf Hanefeld in the Biocatalysis section at Delft University of Technology, The Netherlands. He is working with hydroxynitrile lyases for the chiral synthesis of cyanohydrins in batch and continuous flow systems. |
 Yann Guiavarc*h | Yann Guiavarc*h is born in 1971 in Vannes, France. After a Master in Food Sciences at the University Blaise Pascal, he received his PhD degree in Applied Biological Sciences from the KULeuven (Belgium) where he developed enzymatic time temperature integrators with Prof. Marc E. Hendrickx. He then performed postdoctoral research in applied enzymology on UHPressure processed fruit juices (KULeuven) and bioremediation of effluents containing dyes (UCLouvain). In 2005, he was hired at the University of Lorraine, France, where his research mainly focuses on the study and use of enzymes in supercritical CO2 or water with special interest in their immobilisation. |
 Peter-Leon Hagedoorn | Peter-Leon Hagedoorn obtained his MSc degree Molecular Sciences in 1998 (Wageningen University, The Netherlands) after a short stay (1997–1998) in the laboratory of Prof. Michael Johnson (University of Georgia, Athens, USA). In 2002 he completed his PhD thesis on with Prof. Fred Hagen (Delft University of Technology in the Netherlands). He worked as a postdoctoral fellow (2002–2003) at Leiden University (The Netherlands) and subsequently (2003–2007) at Delft University of Technology. In 2007 he became an assistant professor in biocatalysis at the Delft University of Technology. His research is focussed on the elucidation of reaction mechanisms of (metallo-)enzymes and metalloproteomics. |
 Ulf Hanefeld | Ulf Hanefeld was born in 1966 in Köln, Germany, and grew up in then (West) Berlin and London. In 1993 he received his PhD from the Georg-August-Universität zu Göttingen, having performed the research both with Prof. H. Laatsch (Göttingen) and Prof. H. G. Floss (Seattle). After postdoctoral years with Prof. C. W. Rees (Imperial College London), Prof. J. Staunton (Cambridge) and Prof. J. J. Heijnen and Dr A. J. J. Straathof (TU Delft), he received a fellowship from the Royal Netherlands Academy of Arts and Sciences (KNAW). He rose through the ranks at the Technische Universiteit Delft and his research in Delft focuses on enzymes, their immobilisation and application in organic synthesis. |
Introduction
Most of the active pharmaceutical ingredients (API), natural products and fine chemicals are synthesised using (bio)chemical catalysts in large batch reactors. In recent years the utilisation of enzymes has facilitated the design of more environmentally friendly batch processes that fulfil 10 out of the 12 green chemistry principles.1,2 However, mass transfer limitation, the generation of significant amounts of waste and handling of large volumes of toxic reagents are still problems that have to be overcome. Flow chemistry solves most of these challenges. In a continuous reactor the substrates are pumped through the reactor and the product is collected continuously. This set up improves mass transfer thus increasing reaction rates and reducing reaction time. The reduced reactor volume in flow transformations minimizes energy requirements for heating and cooling (green chemistry – principle 6) and it is also of great benefit to the reduction of waste (green chemistry – principle 1).3–6 Indeed, there is an increasing interest in microreactor technology for the synthesis of high added-value products and for the development of high throughput methods at industrial scale and in academic research.7 In addition, the reduction in volume in continuous flow processes increases safety by avoiding handling of and thus potential exposure to large volumes of toxic compounds.8,9Soluble enzymes can be used for biotransformations in flow but reusability is difficult and the downstream processing needs to include a step for enzyme removal and its possible recycling. Immobilisation of enzymes allows straightforward reuse of the catalyst as it remains in the reactor. Moreover, in many cases increased operational stability is observed. This is an important contributor to the further development of flow chemistry.
With this feature article we aim to highlight important parameters to consider for a successful application of immobilised enzymes and for reporting continuous flow reactions. The latest applications in different reaction media will be discussed. In this context special attention will be paid to cofactors and their recycling in flow.
Challenges for biotransformations in flow with immobilised enzymes
Two main challenges have to be addressed in order to perform a successful biotransformation in flow: (i) immobilisation of the enzyme for recycling and straightforward downstream processing and (ii) suppressing the leaching of the enzyme and/or cofactor (if applicable) into the reaction medium during operation.Overall, an enzyme can be immobilised by adsorption/deposition, ionic binding, covalent attachment to solid carrier materials, chemical cross linking or encapsulation. All of these methods have advantages and disadvantages that have to be evaluated case by case.10–15
As mentioned above, enzyme and/or cofactor leaching are essential aspects that need to be addressed in flow systems. Enzymes themselves or organic cofactors that remain within the enzyme active site and are fully regenerated during the catalytic cycle such as pyridoxal 5′-phosphate (PLP) or thiamin diphosphate pose a relatively small problem. Conversely, organic cofactors that are transiently fixed to the enzyme (i.e. nicotinamide cofactors) need to be regenerated to their given oxidative state before re-entering the enzyme. Thus, the development of an efficient cofactor regeneration system that gives freedom to the cofactor to leave the active site without losing it from the reactor is essential to allow the economic feasibility of the process for industrial applications. Also, the system must be flexible, allowing the implementation of reactions in cascade with a rapid exchange of substrates and avoiding chemical modifications of the cofactor.16 Here we do not discuss metal containing enzymes among the cofactor containing enzymes. All the aspects discussed for organic cofactors (and metal containing organic cofactors) equally apply to these enzymes. In this feature for instance Granulicella tundricola hydroxynitrile lyase (GtHNL) is a Mn2+ cofactor containing enzyme.
A number of successful cofactor recycling systems in flow have been reported, for instance by immobilising onto different carriers.17–22 The performance of immobilised enzymes and the different cofactor regeneration systems in flow will be discussed for organic, biphasic and aqueous conditions.
Metrics
In a recent review, key developments of continuous flow biocatalysis from 2018 to September 2020 were discussed.23 It was found that the rise in the number of publications about this topic was not coupled to an increase in quality of reporting. Frequently, the productivity of the system as space-time-yield (STY) and the residence time were not given. This indicates that additional efforts must be made by the scientific community in order to ensure that the reproducibility and fair comparison between the results reported by different research groups is possible. This is actually a long-standing problem, and already more than a decade ago this was pointed out.24 We consider the following metrics important to achieve this goal.Immobilisation metrics
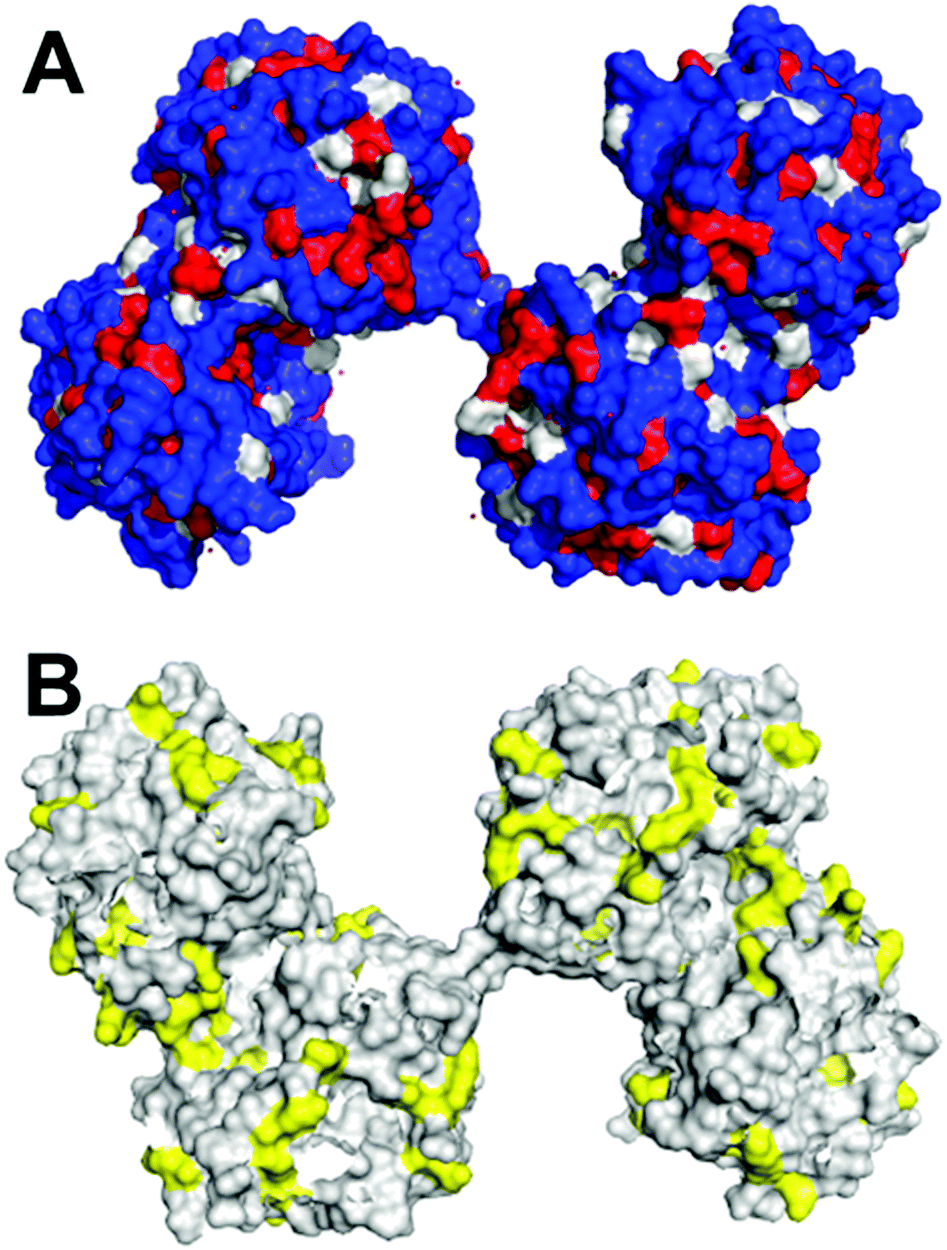 | ||
| Fig. 1 Surface visualisation of dimeric AtHNL (PDB code: 3dqz). (A) Residues in blue (arg, lys, his, glu, asp, asn, gln, thr, ser, cys) are hydrophilic, residues in grey (pro, tyr, typ) and residues in red (ala, gly, val, ile, leu, phe, met) are hydrophobic; B) residues in yellow are lysines. The images were created using PyMOL molecular Graphics System. | ||
Economic metrics
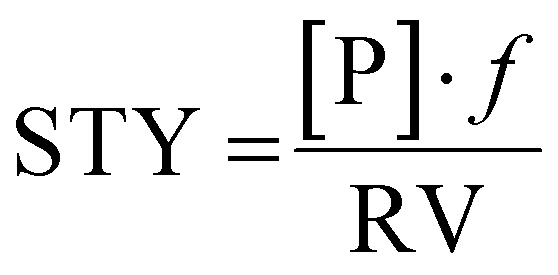 | (1) |
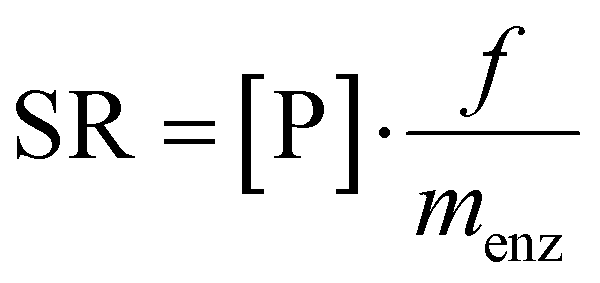 | (2) |
Additional important parameters related to reporting of biocatalytic reactions in flow processes are: (i) operational stability, (ii) biocatalyst loading, (iii) substrate concentration, (iv) reactor volume, (v) residence time. Details about these metrics have been extensively discussed in excellent reviews.33,34
Reaction medium
The reaction medium is an important aspect to consider for biocatalytic transformations, indeed for all transformations. For details about physical properties, environmental and health impacts, flammability/explosion limits and reactivity/stability of different solvents commonly used for biocatalytic transformations the GlaxoSmithKline (GSK) solvent selection guide can be consulted.35 In general the best solvent is no solvent, so reactions converting neat substrate into neat product would be the ideal. In biocatalysis this is often impossible due to inhibition effects. Overall, biocatalysis is usually performed under aqueous, biphasic or pure organic solvent conditions. Each of them has specific advantages and disadvantages. Water (buffered) is the natural reaction medium in which most bio-catalytically utilised enzymes display the highest activity. However, the separation of water from the product can be complicated and expensive due to its high boiling point. This might affect green metric indicators such as the E factor.36,37 In addition, apolar substrates dissolve poorly in water. This affects parameters such as STY and consequently, the economic performance of the process is often poor. Biphasic reactions, i.e. the addition of a water immiscible solvent is a straightforward method to improve economic and environmental metrics. Here, apolar substrates are soluble in the organic solvent layer, therefore high substrate loading is possible and the product is immediately extracted from the water layer and can be obtained from the organic phase by e.g. distillation. Moreover, product inhibition and hydrolysis of water sensitive compounds are avoided.38 However, the introduction of organic solvents as a second layer in a biphasic system might lead to mass transfer limitations and enzyme deactivation at the interphase.39 The utilisation of non-aqueous reaction media was introduced long ago and is today fully developed. Under these conditions, equilibria can be reverted and very high substrate loading can be achieved, enhancing economic parameters (indeed, no solvent is the best solvent).40–43 In order to perform a biotransformation in organic solvents, the enzyme must be immobilised on an appropriate carrier to avoid it lumping together. At the same time enzymes and cofactors are generally not soluble in organic solvents, thus this is an interesting approach to avoid leaching. For flow chemistry these are therefore good conditions.Only lipases have the ability to work in pure organic solvent medium.44 For all other enzymes, the water activity (aw) of the system must be carefully evaluated for optimal enzymatic performance. As a rule of thumb, enzymes work well in buffer saturated organic solvents with a log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P around 2, this provides the amount of water that the enzyme requires for conformational flexibility but still suppresses undesired side reactions. Overall, if different parameters such as type of solvent (logP), aw, immobilisation method and carrier are properly studied, an enzyme in an organic solvent medium is able to perform as well as in aqueous conditions.42,43
P around 2, this provides the amount of water that the enzyme requires for conformational flexibility but still suppresses undesired side reactions. Overall, if different parameters such as type of solvent (logP), aw, immobilisation method and carrier are properly studied, an enzyme in an organic solvent medium is able to perform as well as in aqueous conditions.42,43
To examine the influence of all these parameters in organic solvent, biphasic and aqueous systems, cofactor or cofactor free systems on the two challenges named-above, selected examples of biotransformations performed in flow systems are presented and will be discussed.
Biotransformations in organic solvents as reaction medium
Hydroxynitrile lyases (HNLs) comprise a diverse group of enzymes that catalyse the addition of cyanide to a prochiral aldehyde or ketone to produce chiral cyanohydrins, important building blocks for synthesis.45 They include metal containing cupins, α,β-hydrolase fold enzymes, FAD containing structures and many more. The metal containing cupins can equally well be viewed as cofactor dependent enzymes.11,25,26,45 A key challenge in every chiral cyanohydrin synthesis is the competing chemical, racemic background reaction. It can be suppressed by low pH values or, even better, by performing the reaction in organic solvents, as was already realised in the last century.11,46Recently, the immobilisation of Granulicella tundricola hydroxynitrile lyase (GtHNL; Mn2+ containing cupin) for the synthesis of (R)-mandelonitrile by using a packed bed reactor (Fig. 2) was reported.26
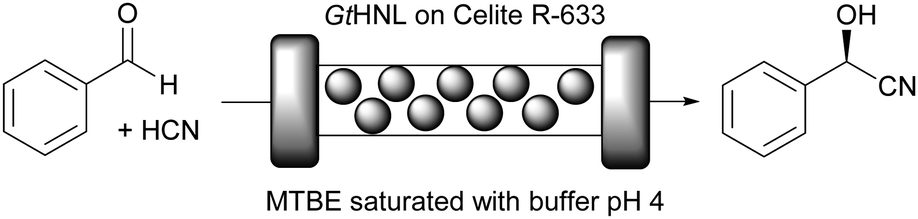 | ||
| Fig. 2 GtHNL catalysed synthesis of (R)-mandelonitrile. | ||
GtHNL was immobilised by adsorption on Celite R-633, also known as diatomaceous earth, a siliceous material obtained from diatoms, a type of microscopic algae. As described earlier, key aspects such as carrier pore volume and water absorption capacity were carefully evaluated.47 This carrier is: (i) environmentally friendly; (ii) the pore size is relatively large (6.5 μm), an important feature to minimise diffusion limitation; (iii) the immobilisation method is straightforward and no chemical treatment is required; (iv) it is a food grade material. All these important characteristics make Celite a green carrier for biocatalysis. However, as nothing is perfect all these advantageous features are accompanied by one main drawback: Celite also catalyses the racemic background reaction. This is suppressed by utilisation of organic solvents and by using continuous flow operation. Methyl tert-butyl ether (MTBE) was selected as reaction medium since other HNLs performed well in this organic solvent in batch systems47–49 and it is considered one of the ‘greenest’ organic solvents.35 It was used buffer saturated (pH 4) to ensure full enzyme activity.50 As expected no leaching of the enzyme was observed. Both, batch and flow produced enantiopure (R)-mandelonitrile. However, in batch, the STY was significantly lower than in the flow system even though otherwise identical reaction conditions were applied: 12 g h−1 L−1versus 784 g h−1 L−1. This represents a huge improvement in productivity (65 times), enabling an important reduction in waste generated due to the reduced volume, making the flow system a ‘greener’ process as compared to the batch approach. The potential of Celite was also demonstrated for the synthesis of cyanohydrins in batch and flow using organic solvents with another (R)-selective HNL. The enzyme from Arabidopsis thaliana (AtHNL; α,β-hydrolase fold) is structurally unrelated to GtHNL. Immobilisation on Celite improved the stability of the acid sensitive AtHNL.48 Conversion up to 96.8% and enantiomeric excess of 99.8% were reached after 45 minutes of reaction time in a batch system. Five years later, the successful synthesis of cyanohydrins with the same AtHNL preparation was compared in batch and flow systems.51 This time the safety limitation of this reaction (green chemistry – principles 3 and 12) related to the handling of toxic hydrogen cyanide (HCN) was addressed by performing the HCN generation in situ from the cheap and less toxic ethyl cyanoformate as well as the actual cyanohydrin synthesis in flow (Fig. 3).
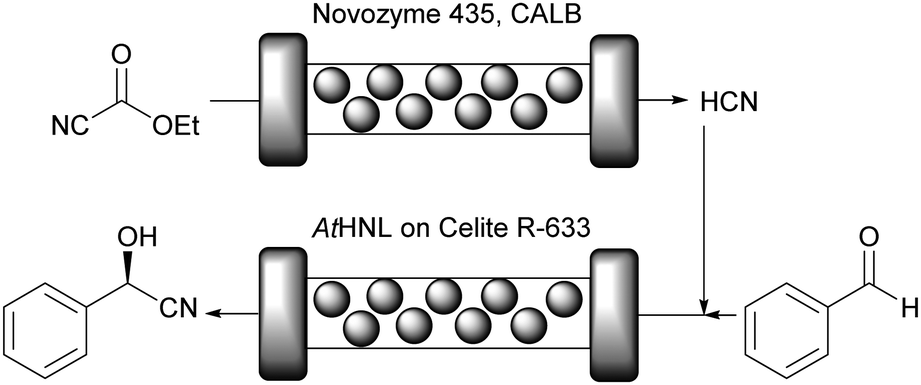 | ||
| Fig. 3 Two-step synthesis of (R)-mandelonitrile catalysed by CALB and AtHNL. | ||
The flow approach proved to be superior as compared to the batch system not only in terms of safety and waste reduction but also in terms of productivity: the reaction time was reduced from 345 min to 40 min by switching from batch to flow.
The same AtHNL was also immobilised via the his-tag on the carrier EziG Opal.52 This is a controlled porosity glass carrier bearing Fe3+ on its surface. The availability of metal ions for the enzyme binding was guaranteed by using a molar ratio of monomeric AtHNL:Fe3+ of 1:5. Again buffer saturated MTBE (here pH 5 rather than pH 4 for GtHNL) was used as reaction medium. After several steps of reaction engineering, near complete conversion and excellent enantioselectivity were achieved at low flow rate (0.1 mL min−1). No enzyme leached from the carrier. Although the racemic reaction was suppressed better in flow than in batch, an important decrease in enantioselectivity was observed at flow rates above 0.2 mL min−1. High flow rates reduce the contact time between enzyme and substrate allowing the racemic reaction to proceed. Again the flow system proved to be more efficient with a STY of 690 mol h−1 L−1 genzyme−1versus 187 mol h−1 L−1 genzyme−1 in batch.
GtHNL and AtHNL are both (R)-selective enzymes, however the multitude of different HNLs also offer access to the (S)-cyanohydrins. The (S)-selective Manihot esculenta HNL (MeHNL; α,β-hydrolase fold) and Hevea brasiliensis HNL (HbHNL; α,β-hydrolase fold) were therefore utilised to study siliceous monolithic micro-reactors (Fig. 4). The use of monolithic micro-reactors instead of packed bed reactors represents an interesting alternative in flow.25 These reactors further reduce reaction time as consequence of the large surface to volume ratio, high thermal efficiency and improved safety.53,54 In addition, mass transfer is enhanced due to its hierarchical/tortuous porous structure reducing diffusion limitation.55 Possible disadvantages are the higher costs compared to packed bed systems and the difficulty to scale up.7
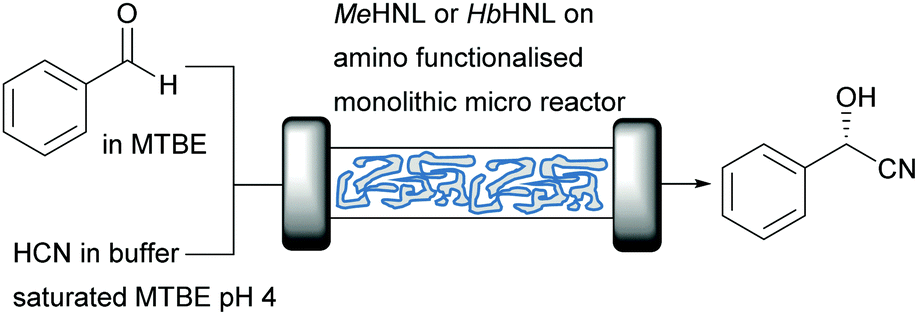 | ||
| Fig. 4 MeHNL or HbHNL catalysed synthesis of (S)-mandelonitrile. | ||
In this case, a careful examination of the surface characteristics of the enzymes and carriers enabled the successful covalent attachment. As explained above (Fig. 1), the most relevant parameters of MeHNL and HbHNL such as diameter, hydrophilicity, number of surface exposed lysine residues and their position related to the active site entrance were obtained by analysing the crystal structure. The pore size diameters of the carriers used for the immobilisation was estimated by transmission electron microscopy (TEM) images. Overall: (i) the carrier chosen had to be hydrophilic to avoid adverse folding effects, (ii) MeHNL and HbHNL had 36 lysine residues for covalent attachment without affecting the active site entrance and (iii) the pore size of the carriers was 2.6 to 3 times larger than the enzymes diameter even in the most unfavourable conformation which represent an internal pore volume 100 times larger as compared to the volume of either enzyme. The enzymes were covalently immobilised on carriers with the corresponding properties and probed in batch and flow reactions. In these cases the lysines were not close to the active site, independent of the carrier no extra actions needed to be taken. In other cases the lysines that obstructed the active site when covalently attached, were removed by mutations.13 Just as described above for the GtHNL and AtHNL catalysed cyanohydrin synthesis, the racemic background reaction was minimised in flow. Both immobilised enzymes showed very high productivity with STY of 1229 g L−1 h−1 and 613 g L−1 h−1 for MeHNL and HbHNL respectively.25
Overall, buffer saturated organic solvents have been successfully applied in flow systems for the synthesis of chiral cyanohydrins. Undesired reactions, here the racemic cyanohydrin formation, were efficiently suppressed, the enzymes did not leach from the carrier independent of the immobilisation method used, high substrate concentrations (500 mM) were employed and high STYs were achieved.
Biotransformations in biphasic systems as reaction medium
Biphasic systems are also widespread in biocatalysis as they enable high substrate loadings for apolar starting materials. Additionally they are ideal reaction conditions for lipases, that often display interfacial activation.44 On the downside additional diffusion limitation and partitioning occur which needs to be overcome by strong stirring in batch systems. In flow good mixing of the biphasic mixture before entering the reactor might be required, however with the monolith reactor mentioned above the mixing occurs in situ. The lipase mediated kinetic resolution of the rac-cyclopropanecarboxylate ester for the synthesis of (1R,2S)-2-(3,4-difluorophenyl) cyclopropan-1-amine generates the stereocentres for ticagrelor, one of the most important drugs for the treatment of acute coronary syndrome and stroke.56A comparison of batch and flow systems under biphasic conditions revealed the potential of the flow approach.57 Lipase from Thermomyces lanuginosus (TLL) was covalently attached to Immobead 100 and used as catalyst (Fig. 5). In batch, 53% conversion and enantioselectivity E = 52 were achieved after 23 hours of reaction time. Alternatively, when the reaction was performed in flow in a packed bed reactor (250 × 4 mm) at a total flow rate of 0.5 mL min−1 (0.25 mL min−1 of substrate in heptane + 0.25 mL min−1 0.1 M Glycine–NaOH buffer pH 9), 17% of conversion with slightly better enantioselectivity E = 58 were reached after only 5.5 minutes of residence time. In regard of productivity, the flow reactor displayed a STY of 28.2 mmol h−1 L−1 whereas the batch reactor yielded only 0.4 mmol h−1 L−1. Importantly, the maximum 50% of conversion would be attained (theoretically) with a residence time of 16.5 minutes by simply reducing the flow rate or coupling multiple reactors in series to extend the reactor length. This also highlights the flexibility of continuous flow systems to scale up processes.
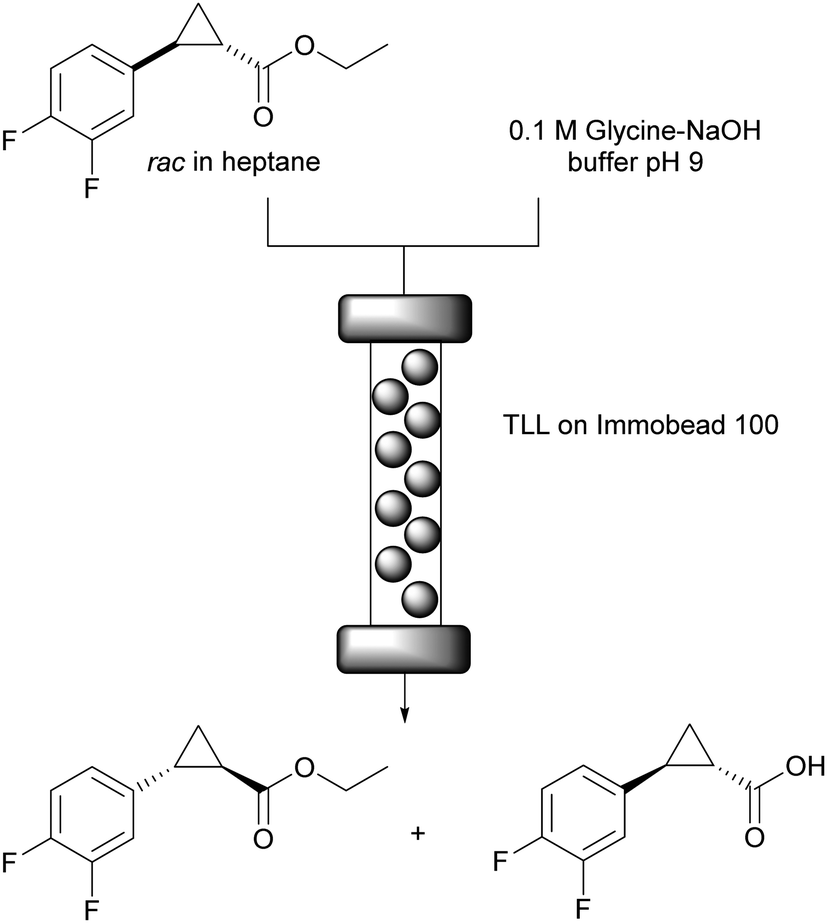 | ||
| Fig. 5 TLL catalysed kinetic resolution of rac-cyclopropanecarboxylate ester yielding (1S,2S)-2-(3,4-difluorophenyl) cyclopropan-1-acid. The unaltered (1R,2R) ester is converted into the desired amine. | ||
Transesterification reactions are commonly performed with lipases in anhydrous medium. Acyl transferase such as the one from Mycobacterium smegmatis (MsAcT) recently introduced the option of transesterification reactions under aqueous conditions.58 After a first report showed the potential of cell free extracts of MsAcT to perform acylation in water in a batch system,59 the system was transferred to flow and the acylating agent ethyl acetate was used as organic layer. The continuous transesterification of neopentylglycol (NPG) with immobilised MsAcT on siliceous monolith micro-reactors (see above) was performed in a biphasic 50/50% system and the enzyme was immobilised either by covalent bonding or his-tag interactions (Fig. 6).60
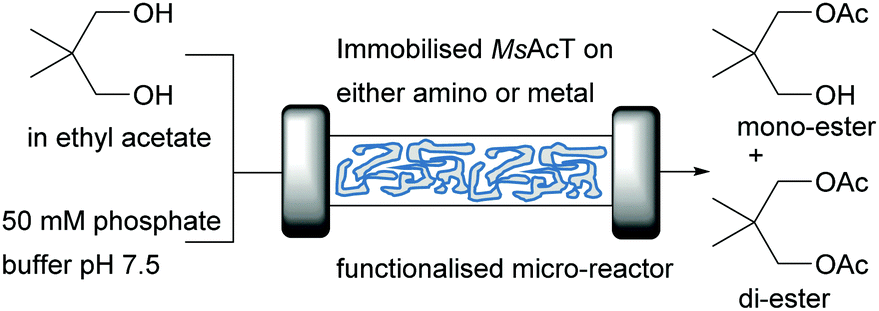 | ||
| Fig. 6 Continuous MsAcT catalysed transesterification of neopentylglycol in a biphasic system. | ||
An excess of either glutaraldehyde in the case of covalent binding or metal ions (Co2+ or Ni2+) when the his-tag was used, were available to ensure binding. The system displayed an exceptional performance for the synthesis of the mono-ester independent of the immobilisation method used: almost full conversion after just 45 seconds. The ratio of mono- to di-ester could be influenced with the flow rate, however full conversion to di-ester was not achieved. This represents a huge improvement over the batch system reported earlier, where full conversion of NPG was not even completed after 7 hours.59
The substrate scope of MsAcT for transesterification reactions in water revealed that different acyldonors such as vinyl acetate and phenyl acetate can be used and aliphatic and aromatic secondary alcohols are converted, while tert-alcohols are no substrates.61 This has opened up new possibilities for the synthesis of natural flavour compounds in a more sustainable fashion.
Recently an application of MsAcT for commercially relevant materials was reported.62 The successful immobilisation of MsAcT onto agarose (Fig. 7.) enabled improved STY in flow. Here, the goal was the synthesis of esters utilising exclusively natural substrates (obtained from nature or by biotechnological approaches). Thus, the natural but less reactive ethyl acetate was used as acyl donor instead of non-natural vinyl acetate. A drawback of performing the transesterification of alcohols and ethyl esters is the negative impact of ethanol on MsAcT. This was circumvented by the above mentioned immobilisation on agarose. The immobilised enzyme retained >75% of its activity after 24 hours of incubation in 500 mM ethanol whereas the free enzyme retained less than 60% of its original activity after only 2 hours of incubation. High conversions were reported for the acylation of 2-phenyl ethanol (75%), cinnamyl alcohol (76%) and n-hexanol (95%) with immobilised MsAcT (1 mg gagarose−1) in batch after 1, 2 and 0.5 hours respectively. By switching to a packed bed reactor and segmented flow (diameter = 6 mm and reactor volume = 1.4 mL) with immobilised MsAcT (1.9 g with enzyme loading of 1 mg gagarose−1) a drastic increase in productivity was observed. Five commercially relevant esters were synthesised with conversions ranging from 65% to 96% within 5 minutes of reaction time. The batch reaction achieved a STY of 23 g L−1 h−1 whereas the continuous flow system reached 318 g L−1 h−1.
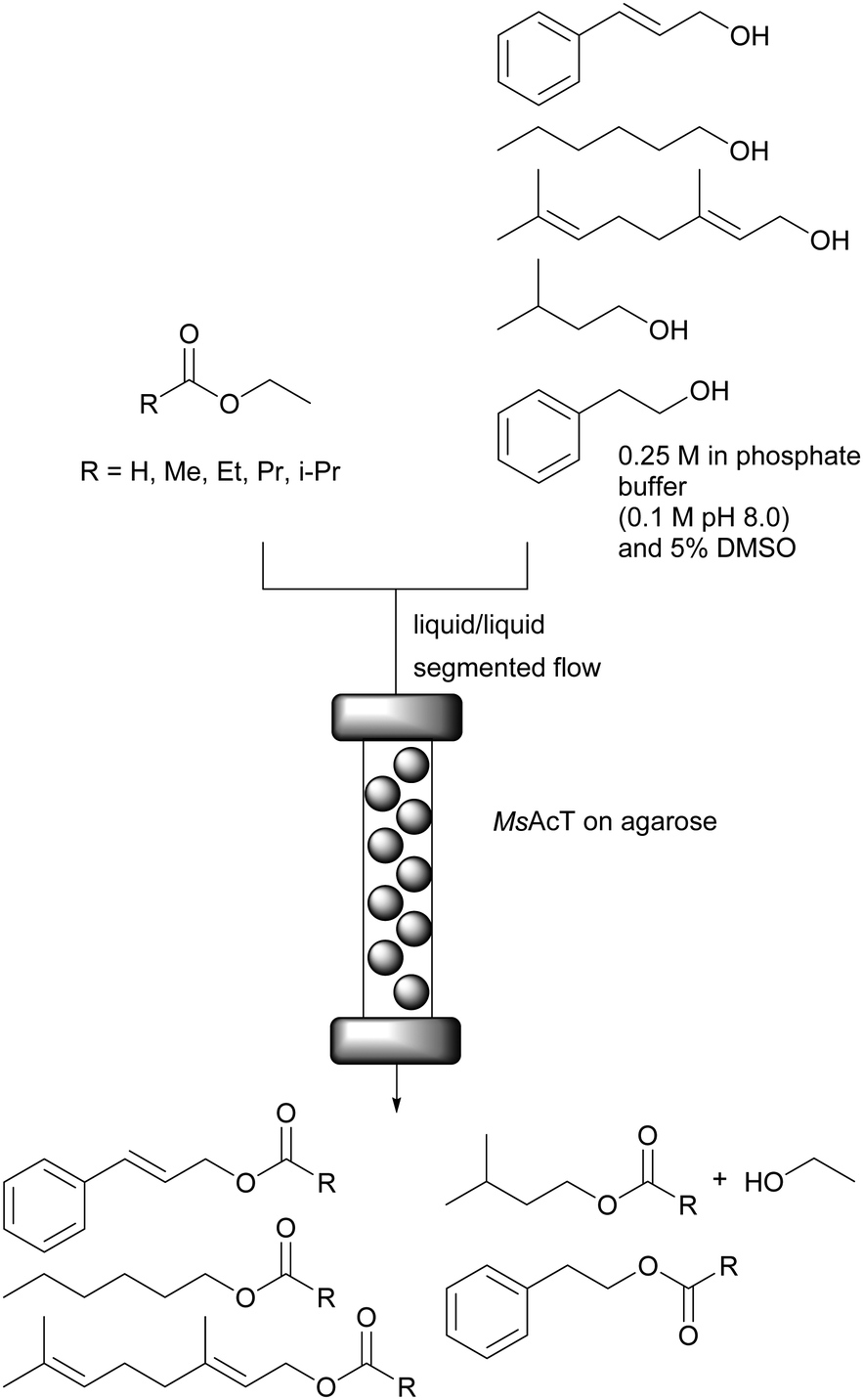 | ||
| Fig. 7 Continuous MsAcT catalysed transesterification of primary alcohols in a biphasic system with segmented flow. | ||
Overall, several successful examples of biotransformations in flow using biphasic systems have been reported. The enhanced mass transfer commonly observed in flow, including segmented flow, helps to circumvent the diffusion limitation of biphasic batch reactions and enables higher substrate loadings as compared to aqueous systems.
Biotransformations in aqueous systems as reaction medium
2-Deoxy-D-ribose-5-phosphate aldolase (DERA) is a very versatile enzyme for the synthesis of aldol products using acetaldehyde as donor. The sequential aldol condensation catalysed by DERA is one of the most efficient routes for the synthesis of the side chain of HMG-CoA reductase inhibitors called statins, important cholesterol lowering drugs (Scheme 1).63–65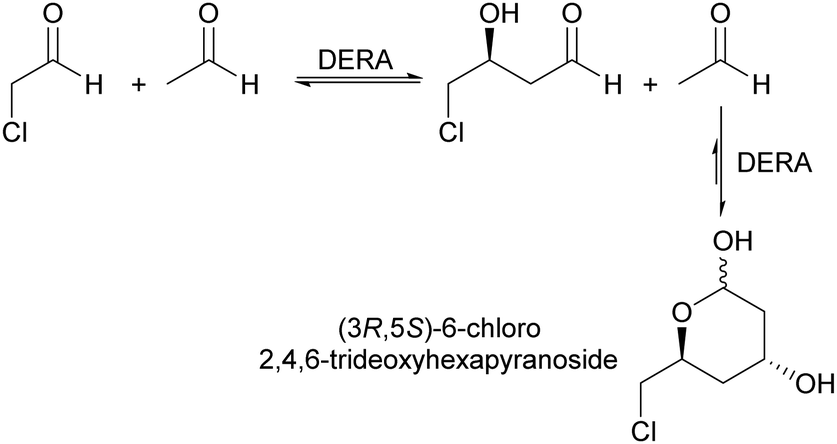 | ||
| Scheme 1 Sequential aldol condensation catalysed by DERA for the synthesis of a chiral statin precursor. | ||
However, the main limitation for an economically efficient industrial application is the enzymes sensitivity towards aldehydes, in particular acetaldehyde. Promising results with protein engineering techniques and reaction engineering were reported.66–70 The DERA from Lactobacillus brevis (LbDERA) already naturally displays high stability to acetaldehyde.71 The introduction of a single amino acid substitution, LbDERA-E78K, improved the enzyme stability even further. This made the synthesis of a chiral precursor of statins, (3R,5S)-6-chloro-2,4,6-trideoxyhexapyranoside, in a batch system possible, with an notable space-time-yield of 792.5 g L−1 d−1.
As demonstrated above for organic solvents and biphasic mixtures immobilisation and continuous flow are two important techniques to consider for improved enzyme stability for aqueous systems, too. Recently, DERA was utilised in a continuous flow approach in aqueous medium for the coupling of acetaldehyde and its chloro-derivative (Fig. 8).72
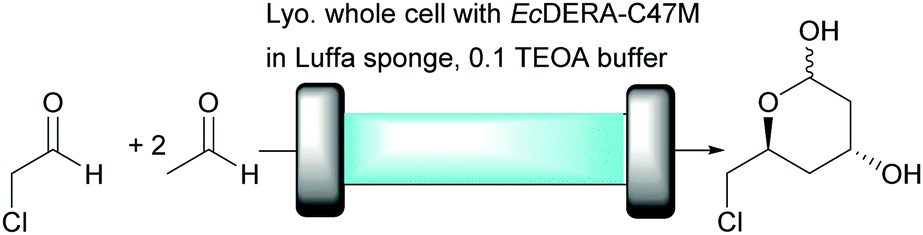 | ||
| Fig. 8 Continuous EcDERA-C47M catalysed aldol reaction for the synthesis of (3R,5S)-6-chloro-2,4,6-trideoxyhexapyranoside in aqueous medium. | ||
For this, lyophilised whole cells of E. coli BL21(DE3) expressing E. coli DERA-C47M, a variant more stable towards acetaldehyde,68 were immobilised inside an alginate matrix by encapsulation and fibrous material obtained from the fruit of the Egyptian Luffa plant, commonly known as the luffa bathroom sponge, was used as support to increase the surface area. From the green chemistry perspective, alginate and luffa sponge are excellent materials for biocatalysis. They are non-toxic, renewable and biodegradable. An enzyme loading of 700 mg led to 80% of conversion of chloro-acetaldehyde after circa 100 min at a flow rate of 0.1 mL min−1 and the enzyme was stable for more than 5 hours of continuous reaction. No enzyme leaching occurred. The productivity of the system was reported as 4.5 g of product per day but unfortunately different enzyme loadings and substrate concentrations were used for the continuous and batch systems making a reliable comparison of the two systems impossible. This once again emphasised the importance of reporting all metrics. On the other hand the DERA reactor is part of a plug-and-play system in which reactors with different catalysts are combined. The power of this is demonstrated in the next example.
Dihydroxyacetone phosphate (DHAP) dependent aldolases require a much more complex reaction system than DERA, as the unstable DHAP needs to be generated in situ. This multi-step procedure of phosphorylation, aldol reaction and dephosphorylation lends itself ideally to the plug-and-play approach. While the modules for phosphate chemistry can remain the same, different aldolases can be plugged in. The successful continuous flow synthesis of different carbohydrate analogues by immobilised Shigella flexneri acid phosphatase (Sf PhoN) and two aldolases (RAMA, rabbit muscle aldolase or RhuA from Thermotoga maritima) demonstrates this (Fig. 9).73 The three step cascade reaction starts with the SfPhoN to phosphorylate dihydroxyacetone (DHA). The resulting DHAP then, is converted by the desired aldolase, here either RAMA or RhuA, with different aldehydes and finally in the third step SfPhoN dephosphorylates the aldol product yielding the desired carbohydrate analogue.
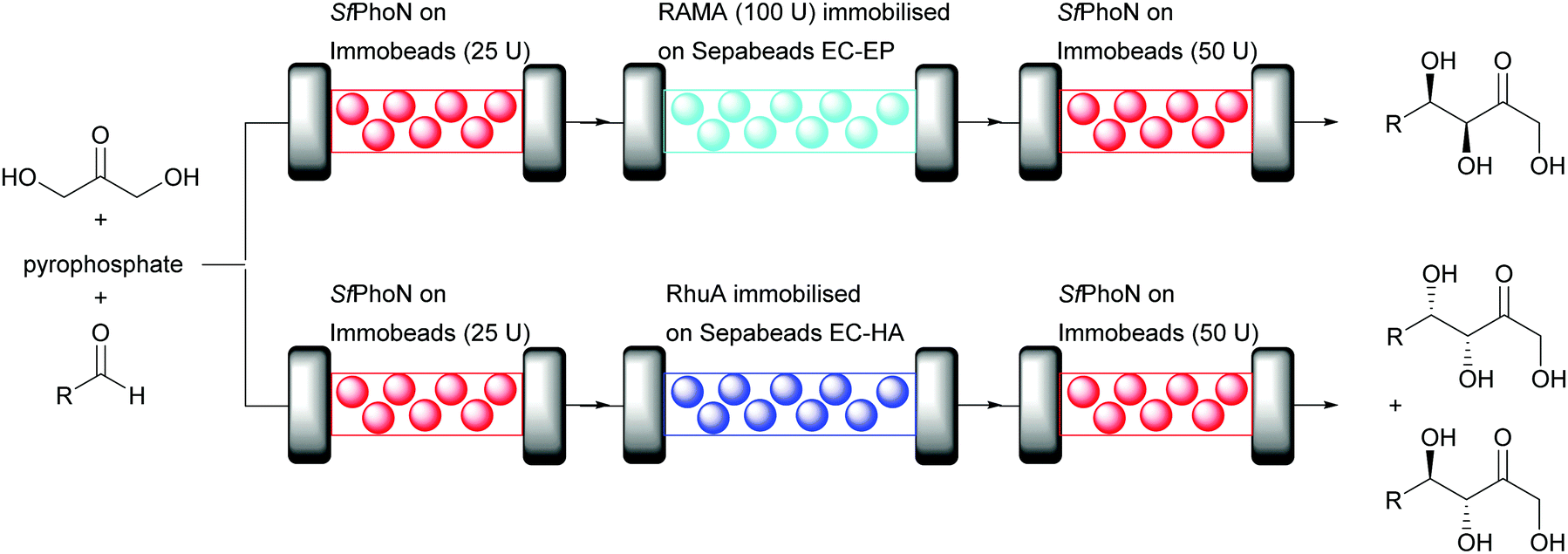 | ||
| Fig. 9 Aldol cascade synthesis catalysed by SfPhon and either RAMA or RhuA in a plug-and-play system in aqueous medium. | ||
SfPhoN was immobilised on methacrylate polymeric beads whereas the immobilisation of RAMA and RhuA was performed on different epoxy carriers. The stability of soluble and immobilised RAMA was evaluated after 24 hour cycles in batch under reaction conditions. Soluble RAMA was unstable with a 50% decrease of conversion after 3 cycles and the enzyme was completely inactive after 5 cycles. Immobilisation demonstrated to be a suitable technique to improve enzyme stability. The best results were observed when RAMA was immobilised on Sepabeads EC-EP or Relyzyme EP403 (rigid methacrylic polymeric beads). The enzyme was fully active after 6 cycles. Remarkably, immobilisation completely suppressed the retroaldol reaction. This might be explained by internal diffusion limitation or a modification of the equilibrium of the reaction. RhuA was also immobilised on epoxy carriers. Complete binding and high activity were observed when RhuA was immobilised on Sepabeads EC-HA. Having established suitable carriers for immobilisation, the cascade reaction was performed in flow with packed bed reactors (Fig. 9). The synthesis of various aldol products in good yield was possible, however higher conversion was observed with RAMA. 68% conversion was observed for the coupling of DHAP to propanal during the first day but this dropped to 51% after 5 days. Higher conversion (80%) was observed for 4-pentenal during the first day, unfortunately the conversion decreased to 7% after 5 days. Finally, 70% of conversion was observed for N-alloc-3-aminopropanal, an important starting material for the synthesis of D-fagomine (antidiabetic piperidine iminosugar drug) during the first day with a decrease to 10% after 5 days. Due to the covalent immobilisation methods chosen no leaching occurred.73
Aqueous systems are the most widely used reaction media in biotransformations. Here enzymes normally display their highest activity. However, the poor solubility of apolar substrates and unwanted side reactions are the main drawbacks for a wider industrial use. Special attention must be taken to avoid leaching of immobilised enzymes into the reaction media. Here this was achieved by covalent linking or the use of whole cells that do not leach easily.
Addressing the cofactor hurdle
Today, enzymes are used as biocatalysts with good success at an industrial scale. Conversely, cofactor recycling remains a hurdle for industrial scale application of cofactor dependent enzymes. The main differentiation in the type of cofactor is:• The cofactor is fully regenerated during the catalytic cycle and does not leave the active site; for example PLP or thiamin but also metal cofactors.
• The cofactor transiently binds to the enzyme, it has to leave the active site for regeneration. It requires a cofactor regeneration system very often involving a second enzyme and cofactor mobility is essential; NAD(P)H/NAD(P)+ are the prime examples.
The simplest system to approach the cofactor recycling hurdle is the use and immobilisation of whole cells as they have the metabolic pathways to regenerate their cofactors. Then, the problem is limited to avoid the leaching of the cofactor to the reaction medium. The use of organic solvents as reaction medium seems to be a good choice because of the often poor solubility of the cofactor in organic solvents.
Cofactors that are fully regenerated during the catalytic cycle
In line with the above the continuous flow synthesis of chiral amines by using a packed bed reactor and water saturated MTBE as reaction medium was reported.74 For this, E. coli cells containing both ω-transaminases (ω-TA) and PLP were immobilised on methacrylate beads, most probably via hydrogen bonding between the peptidoglycan layer of the E. coli cell wall (containing amide, alcohol and ether functional groups) and the polymeric carrier. PLP is fully regenerated during the catalytic cycle of the enzyme within the enzyme, therefore a regeneration system is not required. The conversion of several non-natural ketones (from 67% to 94%) with excellent enantioselectivity (>99%) and residence times between 30 and 60 min without leaching of E. coli cells, ω-TA and PLP was achieved (Fig. 10). Importantly, no quenching or purification was required and the system was operated for 10 days for the synthesis of mexiletine, a drug used to treat abnormal heart rhythms, chronic pain, and some causes of muscle stiffness.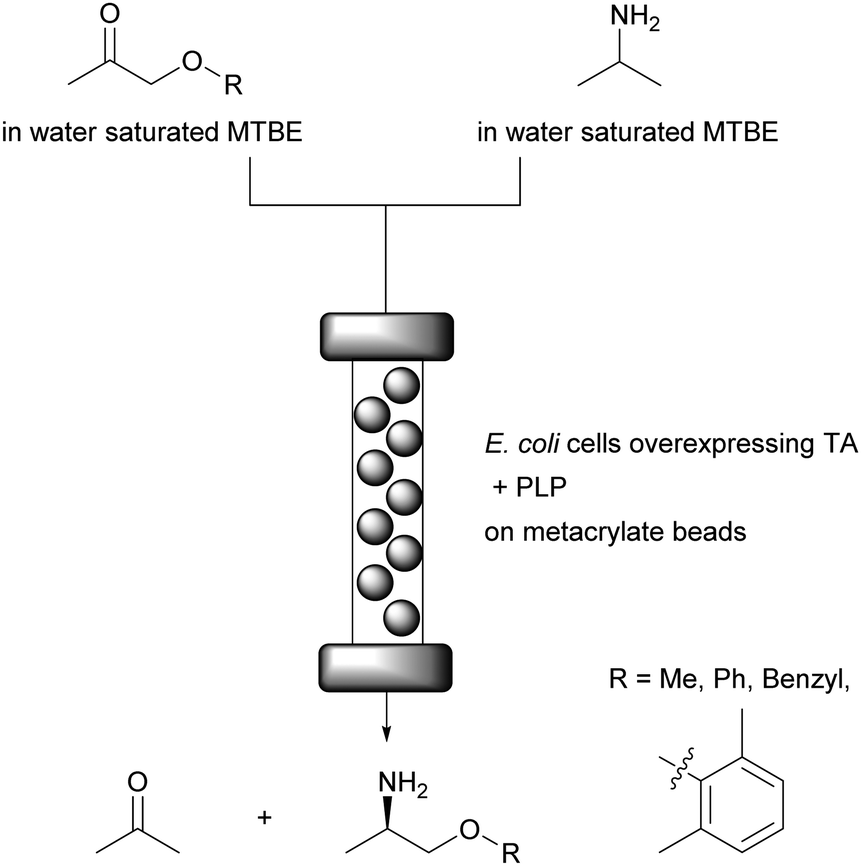 | ||
| Fig. 10 Stereoselective amine synthesis catalysed by immobilised E. coli cells and PLP on metacrylate beads. | ||
An essential breakthrough for cofactor application in flow was their attachment to ionic carriers.75 Any ionic material that can act as counter ion to phosphate moieties present in most cofactors will transiently bind the ionic cofactors. When a buffer of low ionic strength is used as reaction medium the cofactor will remain on the carrier and will be available for the enzyme, even when it needs to be recycled outside the enzyme active site. This principle was demonstrated with Flavin (FAD), PLP and also NAD+ (details discussed below).21 The enzymes were attached to porous carriers and polyethyleneimine (PEI), which is an amine, was the counter ion. For PLP and TA this was fully developed.76 Purified ω-TA from Halomonas elongata (HeTA) and PLP were successfully co-immobilised on porous methacrylate carriers for the continuous synthesis of optically pure amines in aqueous conditions. The beads were functionalised with different reactive groups (cobalt–chelates, epoxides and positively charged amines such as PEI) to allow the electrostatic interaction with his-tagged ω-TAs and PLP. The continuous enantioselective deamination of α-methylbenzyl amine gave >90% conversion for up to 50 column volumes at 1.45 mL min−1 without leaching of the cofactor to the reaction medium. The synthesis reaction of cinnamylamine required a doubling of the reaction time. After an initial decrease in activity it remained stable at 60% for at least 20 column volumes; equiv. to 40 min (Fig. 11). The initial loss of activity might be due to some PLP leaching, induced by the amine donor.
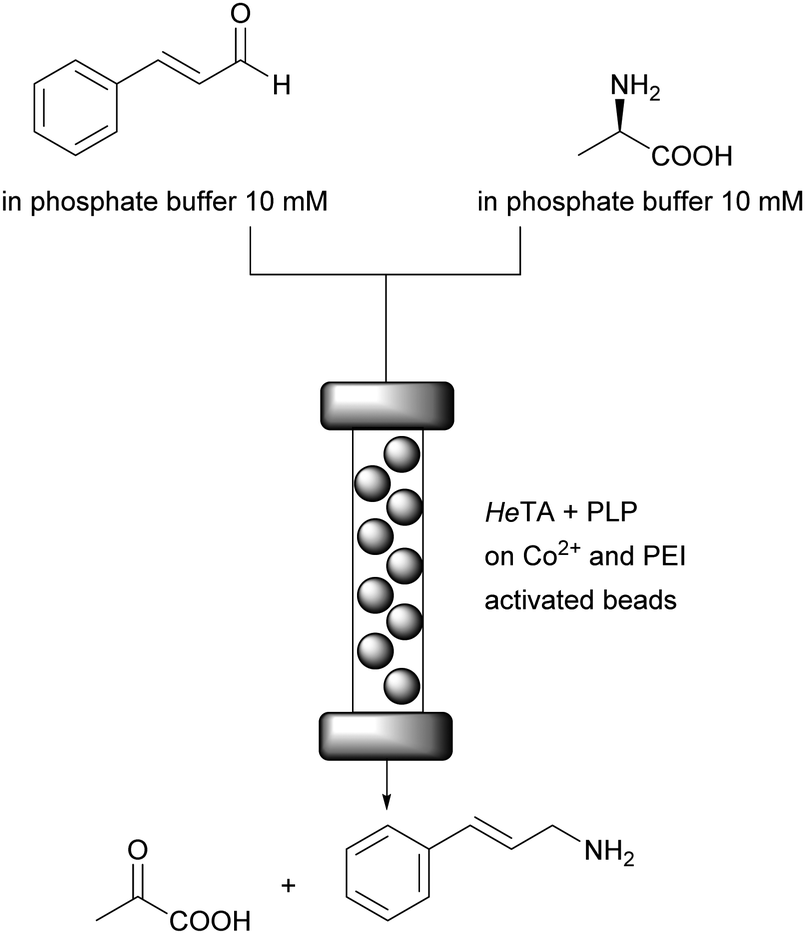 | ||
| Fig. 11 Synthesis of cinnamylamine in continuous flow. Enzyme and PLP are immobilised via ionic interactions. The HeTA via Co2+ on the carrier and a his-tag, the PLP via PEI attached to the carrier. | ||
An intriguing covalent immobilisation of the FAD containing phenylacetone monooxygenase (PAMO) via its cofactor was described. The cofactor was attached to agarose via a tether and then the apo enzyme from Thermobifida fusca was added.77 The immobilised enzyme displayed similar activity as compared to its free form but higher thermostability after 1 h of incubation at 60 °C. However, a recyclability study showed low enzyme stability with a decrease of circa 40% after 3 cycles.
Cofactors that do not require a recycling system and remain in the active site can readily be used in flow systems. Both organic solvents that suppress solubility or aqueous systems with ionic carriers at low buffer concentrations prevent leaching of enzyme and cofactor.
Cofactors that require recycling systems
The application of pure enzymes and cofactors reduces side reactions and it is therefore also preferred in systems which apply cofactors that need to be recycled. The above mentioned immobilisation via ionic interactions was of equal success here.21 Commercial porous carriers were coated with PEI to allow the co-immobilisation of enzymes and phosphorylated cofactors such as NAD+. The cofactor adsorption is dynamic and allows to establish an association–dissociation equilibrium without leaving the porous carrier. It thus is available for the enzyme performing the desired reaction, here alcohol dehydrogenase from Thermus thermophilus (TtADH2) and the enzyme required for cofactor recycling, here formate dehydrogenase from Candida boidinii (CbFDH). The two enzymes and the cofactor were co-immobilised on an anionic exchanger and tested in the continuous asymmetric reduction of 2,2,2-trifluoro-1-phenylethan-1-one (Scheme 2A). Full conversion with a productivity of 250 μM min−1 and a TTN of 85 for immobilised NAD+ after 107 hours on stream in continuous flow with less than 10% NAD+ loss were achieved.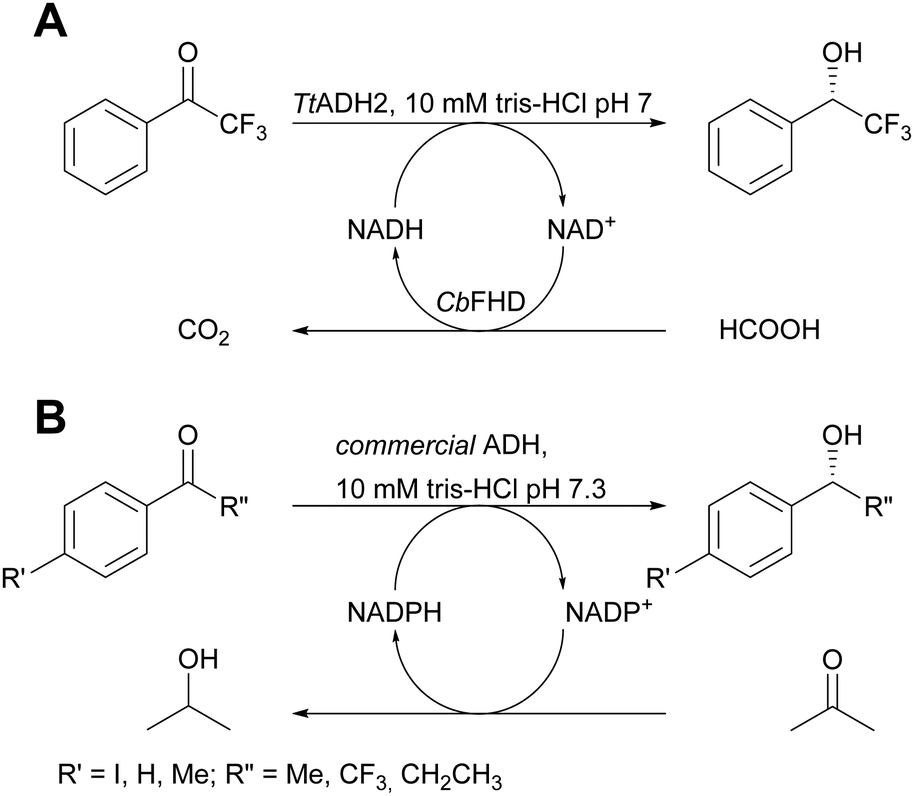 | ||
| Scheme 2 (A) Asymmetric reduction of 2,2,2-trifluoro-1-phenylethan-1-one catalysed by TtADH2 with external cofactor recycling by CbFDH; (B) asymmetric reduction of ketones with internal cofactor recycling. | ||
The system was further improved by applying a commercial ADH that can accept isopropanol as co-substrate. This makes the second enzyme redundant and the cofactor does not have to leave the active site.22 Enzyme and NADPH were co-immobilised on porous agarose beads coated with PEI. The system displayed STYs between 97 and 112 g L−1 day−1 for a range of ketones and the immobilised cofactor reached a TTN of 1076 for 120 hours. During this time, neither the enzyme nor the cofactor were inactivated or leached (Scheme 2B).
This can directly be compared to a recent,16 successful NADPH cofactor regeneration system for the synthesis of chiral alcohols based on a membrane liquid/liquid extractor for continuous flow. The cofactor remained in the aqueous layer and was recycled (Fig. 12).
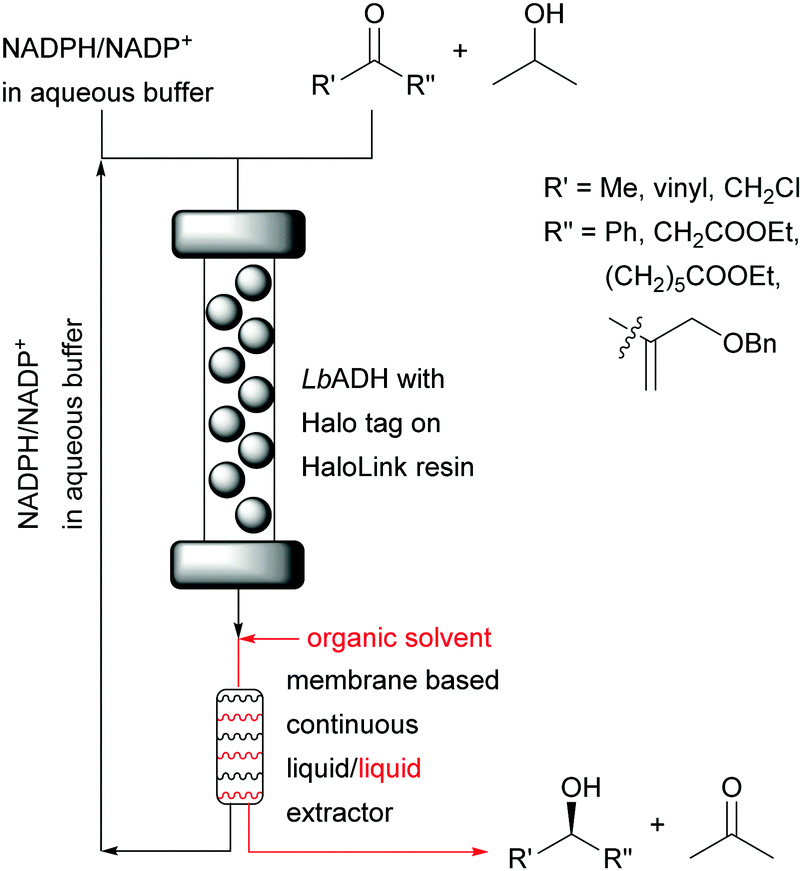 | ||
| Fig. 12 Synthesis of chiral alcohols catalysed by immobilised LbADH with cofactor recycling rather than immobilisation. | ||
The organic phase was added after the reaction mixture passed through the immobilised enzyme. This regeneration system without any chemical modification of the cofactor enabled the reduction of four different ketones with STYs from 14 g L−1 h−1 to 117 g L−1 h−1, cofactor turnover numbers ranging from 128 to 2023 mol mol−1 and excellent enantioselectivity (>99%). The reliability and robustness of the system was demonstrated with the continuous synthesis of ethyl (S)-4-chloro-3-hydroxybutanoate over 32 hours without any loss in performance displaying a STY of 121 g L−1 h−1. A longer run (123 h) exhibited an astonishing cofactor turnover number of 12855 mol mol−1 which represents a step forward compared to previous reports.17–19
Amine dehydrogenases (AmDH) enable the synthesis of chiral amines from cheap ammonium salts as amine donors. Unfortunately, this reaction is commonly hampered by substrate and product inhibition. Immobilisation and continuous flow helps to address these problems, as was recently shown.20 The asymmetric reductive amination of 5-methyl-2-hexanone was achieved by co-immobilisation of an amine dehydrogenase (AmDH) and FDH onto Nuvia IMAC (immobilised metal affinity chromatography) resin from BioRad (Fig. 13). By immobilising both his-tagged enzymes to Ni2+ on the surface of the carrier, the synthesis of (R)-5-methyl-2-aminohexane in a packed bed reactor became possible. This setup displayed a STY of 107 g L−1 day−1 at 48% of conversion moving up to 443 g L−1 day−1 at 24% of conversion. Remarkably the flow system remained operational for more than 120 hours.
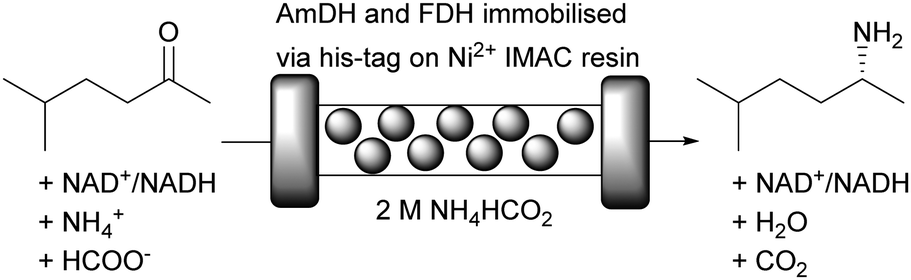 | ||
| Fig. 13 Reductive amination of 5-methyl-2-hexanone with cofactor regeneration catalysed by immobilised AmDH and immobilised FDH in aqueous ammonium formate buffer. Ratio ketone to NAD+ 10:1. | ||
Earlier the continuous synthesis of 4-fluoroamphetamine under aqueous conditions catalysed by co-immobilised AmDH and FDH was reported.78 EziG Amber, a controlled porosity glass carrier with Fe3+ on the surface for his-tag binding was used in a packed bed reactor. This system displayed a STY in the same range (300 g L−1 day−1), however a rapid enzyme deactivation was observed after 6 hours. In both examples low cofactor concentrations could be used and it was recycled during the reaction. Nonetheless, the cofactor was not immobilised and thus finally lost.
Recently, the cofactor regeneration problem in flow was beautifully addressed by applying an enzyme engineering approach.17 A three step cascade was employed for the conversion of glycerol to a precursor of D-fagomine (Fig. 14). In each step one enzyme reaction is performed by multi-enzyme modules. The modules consist of the catalytically active enzyme, if required the recycling enzyme and an esterase (Est) that reacts with the carrier to covalently immobilise the multi-enzyme modules. Thus, two three-enzyme and one two-enzyme modules are covalently attached to the carriers, each combination in a separate reactor. The two cofactors (ATP and NAD+) required were functionalised for the covalent attachment to the multi-enzymes modules with a long PEG-linker at the adenine. This linker allows the movement of the cofactor between the catalytic and cofactor recycling enzymes of the respective module. Subsequently, each cofactor was tethered to the protein linker between the catalytic and the recycling enzyme via an accessible thiol group. The key distinction of this set up is that the cofactor was immobilised to a specific amino acid close to catalytic and recycling enzymes whereas in previous reports the linkage was not specific.79,80
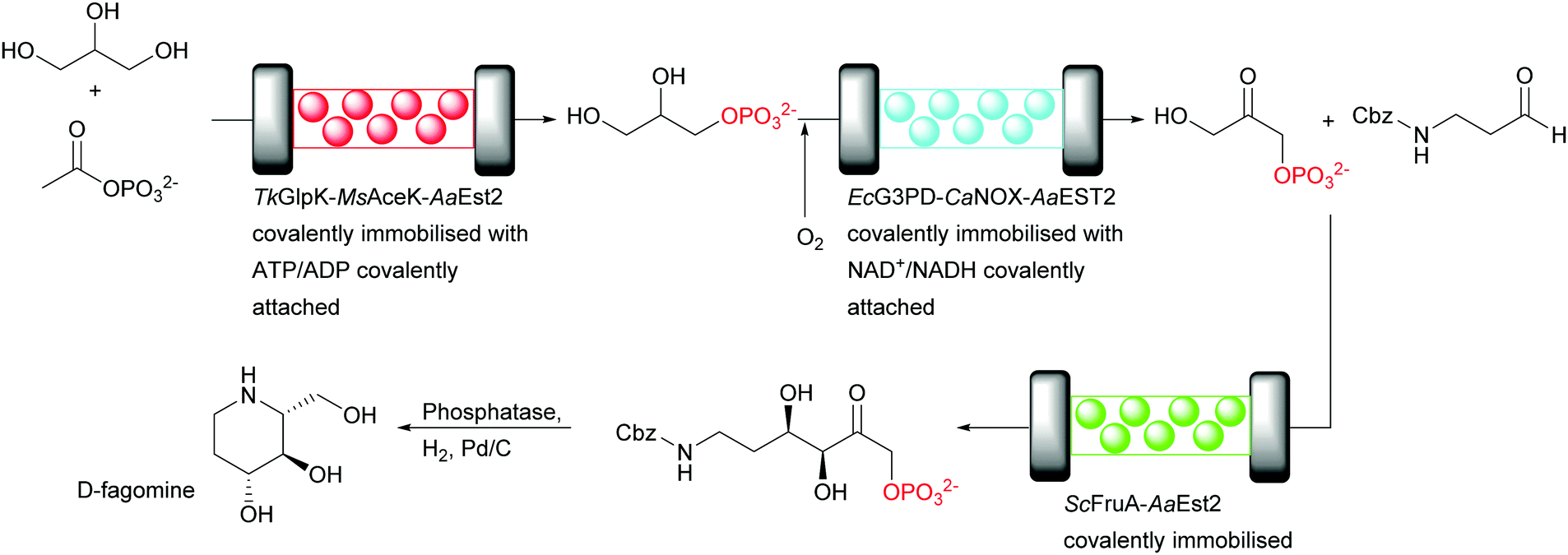 | ||
| Fig. 14 Cascade synthesis of D-fagomine in continuous flow in aqueous medium. The cofactors were covalently attached to the catalysts via a specific thiol group. The modules of two or three enzymes are attached to the carrier via a covalent linker between AaEST2 and the carrier. | ||
First, glycerol was phosphorylated to glycerol-3-phosphate by Thermococcus kodakarensis glycerol kinase (TkGlpK) and for the ATP regeneration Mycobacterium smegmatis acetate kinase (MsAceK) was used. Secondly, E. coli glycerol-3-phosphate dehydrogenase (EcG3PD) performed the oxidation of glycerol-3-phosphate to dihydroxyacetone phosphate (DHAP) and the cofactor NAD+ was regenerated by an NADH oxidase from Clostridium aminovalericum (CaNOX). Finally, an aldol reaction catalysed by a monomeric FruA from Staphylococcus carnosus produced the desired product, precursor of D-fagomine, with excellent STY (70 g L−1 h−1 g−1) and astonishing high cofactor turnovers (16848 for ATP and 10839 for NAD+).
Cofactor dependent enzymes are important biocatalysts for synthesis. When the cofactor is regenerated during the catalytic cycle such as PLP or FAD the main concern is to avoid leaching of the cofactor to the reaction media. The use of organic solvents has been reported as an important tool to circumvent this limitation. When the cofactor is not regenerated during the catalytic cycle, the implementation of a cofactor recycling system is required. For this co-immobilisation of enzymes and cofactor is an attractive approach. However, the efficiency of this recycling system is limited (TTNNADPH = 1076 and TTNNAD+ = 85).21,22 Higher cofactor recycling efficiency has been reported by using membrane technology (TTNNADPH = 12855).16 Nevertheless, the high cost of ultra or nanofiltration technologies impair their economic viability. Finally, the immobilisation of cofactors of either type using a protein engineering approach demonstrated to be as efficient as the use of membrane technology reaching a TTNATP = 16848 and TTNNAD+ = 10839.17
Conclusions
Immobilisation and flow chemistry are important tools for the further development of biocatalysis. Process intensification, better control of the processes, reduced reactor volumes and therefore increased safety are advantages commonly reported in organic, biphasic and aqueous systems. To fully appreciate the advantages and to also probe them rigorously solid metrics are essential.Similarly, large steps have been made to address the cofactor recycling challenge in flow. Co-immobilisation of enzymes and cofactors, membrane based separation and protein engineering techniques have allowed the development of efficient regeneration systems for challenging cascade reactions with cofactor dependent enzymes. Pronounced progress to answer the two challenges, enzyme immobilisation and prevention of leaching of enzyme or cofactor (including metals) during the flow process, have been made. Overall, the implementation of enzyme immobilisation and flow chemistry allow for more efficient, safe and thus green and environmentally friendly processes.
Author contributions
U. H. conceptualised, supervised and edited the manuscript; P.-L. H. and Y. G. edited the manuscript. J. C. wrote and edited the manuscript. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Secretary of Higher Education, Science, Technology and Innovation of Ecuador (Senescyt) and Universidad Laica Eloy Alfaro de Manabí (ULEAM).References
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- M. Santi, L. Sancineto, V. Nascimento, J. Braun Azeredo, E. V. M. Orozco, L. H. Andrade, H. Gröger and C. Santi, Int. J. Mol. Sci., 2021, 22, 990 CrossRef CAS PubMed.
- S. G. Newman and K. F. Jensen, Green Chem., 2013, 15, 1456–1472 RSC.
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef CAS PubMed.
- F. Ferlin, D. Lanari and L. Vaccaro, Green Chem., 2020, 22, 5937–5955 RSC.
- V. Sans, Curr. Opin. Green Sustain. Chem., 2020, 25, 100367 CrossRef.
- E. J. S. Brás, V. Chu, J. P. Conde and P. Fernandes, React. Chem. Eng., 2021, 6, 815–827 RSC.
- B. Bouchaut, L. Asveld, U. Hanefeld and A. Vlierboom, Int. J. Environ. Res. Public Health, 2021, 18, 1963 CrossRef PubMed.
- W. J. Yoo, H. Ishitani, Y. Saito, B. Laroche and S. Kobayashi, J. Org. Chem., 2020, 85, 5132–5145 CrossRef CAS PubMed.
- K. Faber, Biotransformations in Organic Chemistry, 2018, 7th edn, Springer Nature, Switzerland, pp. 224–233 Search PubMed.
- U. Hanefeld, Chem. Soc. Rev., 2013, 42, 6308–6321 RSC.
- M. T. de Martino, F. Tonin, V. R. L. J. Bloemendal, U. Hanefeld, F. P. J. T. Rutjes and J. C. M. van Hest, RSC Adv., 2021, 11, 21857–21861 RSC.
- U. Hanefeld, L. Gardossi and E. Magner, Chem. Soc. Rev., 2009, 38, 453–468 RSC.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- A. Basso and S. Serban, Mol. Catal., 2019, 479, 110607 CrossRef CAS.
- B. Baumer, T. Classen, M. Pohl and J. Pietruszka, Adv. Synth. Catal., 2020, 362, 2894–2901 CrossRef CAS.
- C. J. Hartley, C. C. Williams, J. A. Scoble, Q. I. Churches, A. North, N. G. French, T. Nebl, G. Coia, A. C. Warden, G. Simpson, A. R. Frazer, C. N. Jensen, N. J. Turner and C. Scott, Nat. Catal., 2019, 2, 1006–1015 CrossRef CAS.
- A. Ŝalić and B. Zelić, RSC Adv., 2014, 4, 41714–41721 RSC.
- M. L. Contente and F. Paradisi, Nat. Catal., 2018, 1, 452–459 CrossRef CAS.
- R. D. Franklin, J. A. Whitley, A. A. Caparco, B. R. Bommarius, J. A. Champion and A. S. Bommarius, Chem. Eng. J., 2021, 407, 127065 CrossRef CAS.
- S. Velasco-Lozano, A. I. Benítez-Mateos and F. López-Gallego, Angew. Chem., Int. Ed., 2017, 56, 771–775 CrossRef CAS PubMed.
- A. I. Benítez-Mateos, E. San Sebastian, N. Ríos-Lombardía, F. Morís, J. González-Sabín and F. López-Gallego, Chem. – Eur. J., 2017, 23, 16843–16852 CrossRef PubMed.
- P. De Santis, L. E. Meyer and S. Kara, React. Chem. Eng., 2020, 5, 2155–2184 RSC.
- L. Gardossi, P. B. Poulsen, A. Ballesteros, K. Hult, V. K. Svedas, Đ. Vasic-Racki, G. Carrea, A. Magnusson, A. Schmid, R. Wohlgemuth and P. J. Halling, Trends Biotechnol., 2010, 28, 171–180 CrossRef CAS PubMed.
- M. P. van der Helm, P. Bracco, H. Busch, K. Szymańska, A. B. Jarzębski and U. Hanefeld, Catal. Sci. Technol., 2019, 9, 1189–1200 RSC.
- J. Coloma, Y. Guiavarc'h, P. L. Hagedoorn and U. Hanefeld, Catal. Sci. Technol., 2020, 10, 3613–3621 RSC.
- R. A. Sheldon, M. Wallau, I. W. C. E. Arends and U. Schuchardt, Acc. Chem. Res., 1998, 31, 485–493 CrossRef CAS.
- L. Dettori, F. Vibert, Y. Guiavarc'h, S. Delaunay, C. Humeau, J. L. Blin and I. Chevalot, Microporous Mesoporous Mater., 2018, 267, 24–34 CrossRef CAS.
- M. C. Bourkaib, P. Gaudin, F. Vibert, Y. Guiavarc’h, S. Delaunay, X. Framboisier, C. Humeau, I. Chevalot and J.-L. Blin, Microporous Mesoporous Mater., 2021, 323, 111226 CrossRef CAS.
- H. H. P. Yiu and P. A. Wright, J. Mater. Chem., 2005, 15, 3690–3700 RSC.
- S. Hudson, J. Cooney and E. Magner, Angew. Chem., Int. Ed., 2008, 47, 8582–8594 CrossRef CAS PubMed.
- J. M. Bolivar, A. Hidalgo, L. Sanchez-Ruiloba, J. Berenguer, J. M. Guisan and F. Lopez-Gallego, J. Biotechnol., 2011, 155, 412–420 CrossRef CAS PubMed.
- L. Tamborini, P. Fernandes, F. Paradisi and F. Molinari, Trends Biotechnol., 2018, 36, 73–88 CrossRef CAS PubMed.
- M. P. Thompson, I. Peñafiel, S. C. Cosgrove and N. J. Turner, Org. Process Res. Dev., 2019, 23, 9–18 CrossRef CAS.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- Roger A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- P. Domínguez de María and F. Hollmann, Front. Microbiol., 2015, 6, 1257 Search PubMed.
- E. M. M. Abdelraheem, H. Busch, U. Hanefeld and F. Tonin, React. Chem. Eng., 2019, 4, 1878–1894 RSC.
- M. Villela Filho, T. Stillger, M. Müller, A. Liese and C. Wandrey, Angew. Chem., Int. Ed., 2003, 42, 2993–2996 CrossRef CAS PubMed.
- A. Zaks and A. M. Klibanov, Science, 1984, 224, 1249–1251 CrossRef CAS PubMed.
- E. Bourquelot and M. Bridel, Ann. Chim. Phys., 1913, 29, 145–218 Search PubMed.
- A. M. Klibanov, Nature, 2001, 409, 241–246 CrossRef CAS PubMed.
- M. M. C. H. van Schie, J. D. Spöring, M. Bocola, P. Domínguez de María and D. Rother, Green Chem., 2021, 23, 3191–3206 RSC.
- P. Bracco, N. van Midden, E. Arango, G. Torrelo, V. Ferrario, L. Gardossi and U. Hanefeld, Catalysts, 2020, 10, 308 CrossRef CAS.
- P. Bracco, H. Busch, J. von Langermann and U. Hanefeld, Org. Biomol. Chem., 2016, 14, 6375–6389 RSC.
- D. Costes, E. Wehtje and P. Adlercreutz, Enzyme Microb. Technol., 1999, 25, 384–391 CrossRef CAS.
- P. Bracco, G. Torrelo, S. Noordam, G. de Jong and U. Hanefeld, Catalysts, 2018, 8, 287 CrossRef.
- D. Okrob, M. Paravidino, R. V. A. Orru, W. Wiechert, U. Hanefeld and M. Pohl, Adv. Synth. Catal., 2011, 353, 2399–2408 CrossRef CAS.
- G. Torrelo, N. van Midden, R. Stloukal and U. Hanefeld, ChemCatChem, 2014, 6, 1096–1102 CrossRef CAS.
- M. Paravidino, M. J. Sorgedrager, R. V. A. Orru and U. Hanefeld, Chem. – Eur. J., 2010, 16, 7596–7604 CrossRef CAS PubMed.
- A. Brahma, B. Musio, U. Ismayilova, N. Nikbin, S. B. Kamptmann, P. Siegert, G. E. Jeromin, S. V. Ley and M. Pohl, Synlett, 2016, 27, 262–266 CAS.
- J. Coloma, T. Lugtenburg, M. Afendi, M. Lazzarotto, P. Bracco, P. L. Hagedoorn, L. Gardossi and U. Hanefeld, Catalysts, 2020, 10, 899 CrossRef CAS.
- T. Yu, Z. Ding, W. Nie, J. Jiao, H. Zhang, Q. Zhang, C. Xue, X. Duan, Y. M. A. Yamada and P. Li, Chem. – Eur. J., 2020, 26, 5729–5747 CrossRef CAS PubMed.
- Y. Zhu, Q. Chen, L. Shao, Y. Jia and X. Zhang, React. Chem. Eng., 2020, 5, 9–32 RSC.
- K. Szymańska, M. Pietrowska, J. Kocurek, K. Maresz, A. Koreniuk, J. Mrowiec-Białon, P. Widłak, E. Magner and A. Jarzębski, Chem. Eng. J., 2016, 287, 148–154 CrossRef.
- K. G. Hugentobler, H. Sharif, M. Rasparini, R. S. Heath and N. J. Turner, Org. Biomol. Chem., 2016, 14, 8064–8067 RSC.
- K. G. Hugentobler, M. Rasparini, L. A. Thompson, K. E. Jolley, A. J. Blacker and N. J. Turner, Org. Process Res. Dev., 2017, 21, 195–199 CrossRef CAS.
- I. Mathews, M. Soltis, M. Saldajeno, G. Ganshaw, R. Sala, W. Weyler, M. A. Cervin, G. Whited and R. Bott, Biochemistry, 2007, 46, 8969–8979 CrossRef CAS PubMed.
- L. Wiermans, S. Hofzumahaus, C. Schotten, L. Weigand, M. Schallmey, A. Schallmey and P. Dominguez de María, ChemCatChem, 2013, 5, 3719–3724 CrossRef CAS.
- K. Szymańska, K. Odrozek, A. Zniszczoł, G. Torrelo, V. Resch, U. Hanefeld and A. B. Jarzębski, Catal. Sci. Technol., 2016, 6, 4882–4888 RSC.
- N. de Leeuw, G. Torrelo, C. Bisterfeld, V. Resch, L. Mestrom, E. Straulino, L. van der Weel and U. Hanefeld, Adv. Synth. Catal., 2018, 360, 242–249 CrossRef CAS.
- M. L. Contente, L. Tamborini, F. Molinari and F. Paradisi, J. Flow Chem., 2020, 10, 235–240 CrossRef CAS.
- M. Haridas, E. M. M. Abdelraheem and U. Hanefeld, Appl. Microbiol. Biotechnol., 2018, 102, 9959–9971 CrossRef CAS PubMed.
- X. R. Wu, J. P. Jiang and Y. J. Chen, ACS Catal., 2011, 1, 1661–1664 CrossRef CAS.
- P. Hoyos, V. Pace and A. R. Alcántara, Catalysts, 2019, 9, 260 CrossRef CAS.
- M. Schürmann, Industrial Enzymes Applications, Wiley-VCH, Weinheim, Germany, 2019, pp. 385–403 Search PubMed.
- S. Jennewein, M. Schürmann, M. Wolberg, I. Hilker, R. Luiten, M. Wubbolts and D. Mink, Biotechnol. J., 2006, 1, 537–548 CrossRef CAS PubMed.
- M. Dick, R. Hartmann, O. H. Weiergräber, C. Bisterfeld, T. Classen, M. Schwarten, P. Neudecker, D. Willbold and J. Pietruszka, Chem. Sci., 2016, 7, 4492–4502 RSC.
- G. DeSantis, J. Liu, D. P. Clark, A. Heine, I. A. Wilson and C. H. Wong, Bioorg. Med. Chem., 2003, 11, 43–52 CrossRef CAS PubMed.
- J. Li, J. Yang, Y. Men, Y. Zeng, Y. Zhu, C. Dong, Y. Sun and Y. Ma, Appl. Microbiol. Biotechnol., 2015, 99, 7963–7972 CrossRef CAS PubMed.
- X. C. Jiao, J. Pan, G. C. Xu, X. D. Kong, Q. Chen, Z. J. Zhang and J. H. Xu, Catal. Sci. Technol., 2015, 5, 4048–4054 RSC.
- B. Grabner, Y. Pokhilchuk and H. Gruber-Woelfler, Catalysts, 2020, 10, 137 CrossRef CAS.
- L. Babich, A. F. Hartog, L. J. C. van Hemert, F. P. J. T. Rutjes and R. Wever, ChemSusChem, 2012, 5, 2348–2353 CrossRef CAS PubMed.
- L. H. Andrade, W. Kroutil and T. F. Jamison, Org. Lett., 2014, 16, 6092–6095 CrossRef CAS PubMed.
- G. E. Jeromin, Biotechnol. Lett., 2009, 31, 1717–1721 CrossRef CAS PubMed.
- A. I. Benítez-Mateos, M. L. Contente, S. Velasco-Lozano, F. Paradisi and F. López-Gallego, ACS Sustainable Chem. Eng., 2018, 6, 13151–13159 CrossRef.
- M. Krzek, H. L. van Beek, H. P. Permentier, R. Bischoff and M. W. Fraaije, Enzyme Microb. Technol., 2016, 82, 138–143 CrossRef CAS PubMed.
- M. P. Thompson, S. R. Derrington, R. S. Heath, J. L. Porter, J. Mangas-Sanchez, P. N. Devine, M. D. Truppo and N. J. Turner, Tetrahedron, 2019, 75, 327–334 CrossRef CAS.
- A. T. Keatinge-Clay, Nat. Prod. Rep., 2016, 33, 141–149 RSC.
- A. Nakamura, H. Minami, I. Urabe and H. Okada, J. Ferment. Technol., 1988, 66, 267–272 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |