DOI:
10.1039/D1NA00223F
(Review Article)
Nanoscale Adv., 2021,
3, 3353-3372
A review on bismuth oxyhalide based materials for photocatalysis
Received
24th March 2021
, Accepted 1st May 2021
First published on 3rd May 2021
Abstract
Photocatalytic solar energy transformation is the most encouraging solution to alleviate the environmental crisis and energy scarcity. Bismuth oxyhalide (BiOX) is an emerging class of materials that exhibits photocatalytic properties, such as resilient response to light, which causes enhanced energy conversion (solar energy) owing to their exceptional layered structure and attractive band structure. The present review presents a summary of results from the recent developments on the tuning and design of BiOX-based materials to improve the energy conversion. In particular, the preparation and tuning approaches that have the potential to enhance the photocatalytic behavior of BiOX and some other techniques, such as elemental doping, are addressed, which prevent the rapid recombination of charges, and formation of oxygen vacancies, facilitating an improvement in the photocatalytic reaction. Various frameworks are also presented, displaying the significance of BiOX-based nanocomposites. Finally, the main challenges and opportunities associated with the future progress of BiOX-based materials are presented. This review will provide an extended understanding and offer a preferred direction for the innovative design of BiOX-based materials for environmental and especially energy-based applications.
1. Introduction
Photocatalysis, which involves the successful use of solar energy, is a vital strategy for solving the energy crisis and environmental issues.1–4 The inexhaustible energy from the sun can be utilized for the generation of hydrogen and carbon-containing fuels via water splitting and CO2 photoreduction, respectively. Rational and novel photocatalyst designs are key factors for efficient photocatalytic activities.5–9 Hydrogen is the most important source of energy considering a sustainable and clean future. The high combustion yield of H2 energy generation (122 kJ mol−1) makes it superior to the commonly used fuels such as gasoline and petroleum, and other fossil fuels.4 Moreover, this method has the overwhelming advantage of environmental friendliness with no harmful and toxic by-products.10,11 However, one drawback is the production of CO2 during the process, which is extremely harmful to the environment.12,13 Thus, the development of novel H2 generation techniques is necessary to exclude the emission of harmful gases to the environment. Accordingly, one promising method to overcome this challenge involves the photocatalytic generation of H2 in the presence of a catalyst under solar light irradiation, which is commonly referred to as “photocatalysis”.14–18
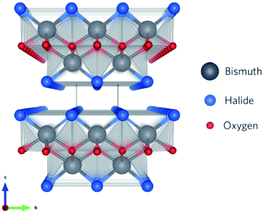
Recently, a new class of photocatalysts comprising ternary (V–VI–VII) semiconductors such as bismuth oxyhalides (BiOX, X = Cl, Br and I) and metal-free catalysts including g-carbon nitride has emerged.8,19–24 Specifically, slabs of [X–Bi–O–Bi–X] are combined via van der Waals forces to form a tetragonal matlockite crystal structure,25–27 as depicted in Fig. 1. A decahedral asymmetric structure is formed when four oxygen atoms and four halogen atoms surround each central bismuth ion in [X–Bi–O–Bi–X]. The outstanding properties (optical, electronic, and structural) of these layered materials are generated from the synergistic effect of strong covalent bonding and van der Waals interaction among the atomic layers.15
 |
| | Fig. 1 Framework of BiOX systems with stoichiometric X–Bi–O–Bi–X bilayers stacked along the c-axis. Reproduced with permission from ref. 28 Copyright 2016, the American Chemical Society. | |
In the past, BiOX materials were used as storage and ferroelectric materials, catalysts, and pigments. However, recently, they have found new applications in various fields including photocatalytic purification of wastewater and gases, oxidation of alcohols, water splitting, and organic synthesis.29–32 Extensive works have been reported on the use of BiOX for the photocatalytic degradation of organic dyes in wastewater, but no significant studies have been reported on their PEC properties.15,20,29,33,34 The high charge transfer ratio owing to their indirect band gaps together with their crystallinity makes these materials potential candidates for the production of H2. Besides, enhanced charge separation can be obtained along the (001) plane of BiOX via an induced perpendicular internal electric field.35 TiO2 has been abundantly used for photocatalytic applications, but only shows activity in the UV (ultraviolet) region. In contrast, BiOX materials have the advantage of a wide bandgap range, varying from 3.3 eV (BiOCl) to 1.8 eV (BiOI), allowing visible light photocatalysis.36,37 Since Zhang's group reported the fabrication of 3D-microspheres of BiOX on 2D-nanoplates via solvothermal synthesis for the degradation of methyl orange,21 researchers have focused on the photocatalytic activity of BiOX materials.38,39
Similar to BiOX, BixOyXz-based photocatalysts have a compact layered structure with weak interlayer interaction (nonbonding) and resilient interlayer bonding. A schematic illustration of the Bi4O5Br2 crystal is presented in Fig. 2a as an example of the BixOyXz-based structure. The layer structure of Bi4O5Br2 comprises Bi–O layers that are positioned among double slabs of bromine ions. The Bi–O layers with respect to the surrounding charge density are greater compared to the dual bromine slabs of Bi4O5Br2 (Fig. 2b).40 The non-uniform charge distribution among Bi–O layers and dual iodine slabs leads to polarization, resulting in a static internal electric field (IEF), which allows the splitting of excitons.41,42 The growth route and IEF of layered Bi4O5Br2 are in the [101] direction, which is altered to the [001] direction of BiOX. Experimental results and theoretical calculations (density functional theory (DFT) approach) have displayed the high photon absorption efficacy of BixOyXz, which has also been investigated in earlier studies.38,43 To enhance the activity of photocatalysts based on semiconductors, charge separation is considered an important factor. For BixOyXz-based photocatalysts, the carrier separation efficacy primarily results from the IEF among their layers.41 Usually, a higher value of IEF results in higher carrier separation efficiency for semiconductor photocatalysts. The electric field intensity is also based on the polarization force and space. The high dipole moment and interlayer spacing of BixOyXz result in an improved polarization force and space. Commonly, reducing the ratio of Bi to X results in a reduction in the optical absorption, which in turn reinforces the hybridization (for the conduction band to favor electron movement and support the splitting of photogenerated excitons).42 Consequently, BixOyXz are outstanding photocatalytic materials.
 |
| | Fig. 2 Structure models of Bi4O5Br. (a) IEF direction and (b) charge density contour plots of the (101) surface. Reproduced with permission from ref. 40 Copyright 2016, Elsevier B.V. | |
To date, many BiOX nanostructures (e.g., nanowires, nanobelts, nanocrystals, nanosheets, nanofibers, hollow microspheres, porous nanospheres, and flower-like structures) have been fabricated.26,44–47 However, a further improvement in the properties of BiOX is necessary for commercial applications, which is being attempted by boosting its photocatalytic potential via enhanced light absorption and photo-induced charge transfer efficiency. These improved BiOX photocatalysts show brilliant potential for wastewater treatment, H2 generation, N2 fixation, O2 evolution, CO2 reduction, disinfection, and organic synthesis.15,48–54
In the present review, we focus our discussion on the connection between the structural tailoring and enhancement of photocatalytic activity. This review is arranged as follows. Initially, we briefly discuss the synthesis protocols for BiOX, which is considered fundamental from an application point of view. Then, the photochemistry and schemes for solar energy harvesting such as hydrogen, oxygen production, and CO2 reduction are also explored. Subsequently, we combine the most promising and evolving strategies for the enhancement of the photocatalytic activity of BiOX-based photocatalysts. We aim to establish a bridge between modern strategies and structural engineering toward better photocatalytic reactivity combined with maximum solar energy harvesting opportunities, which is the focal point of the present review. All these findings and summaries are supplemented with the addition of theoretical studies. Finally, we provide the current challenges and opportunities.
2. Synthesis methods
2.1 Hydrolysis method
BiOCl can be prepared via the hydrolysis of different compounds including bismuth chlorides, nitrides, and oxides. Hydrolysis offers mild reaction conditions, but the products are not dimensionally uniform. Armelao et al. synthesized BiOCl nanostructures via the hydrolysis of BiCl3 for 6 h at 65 °C, which were stabilized using acetylacetone as an auxiliary solvent under an acidic environment.55 Similarly, Song et al. reported that thick nanosheets (21 nm to 85 nm) of BiOCl could be synthesized via the hydrolysis of Bi(NO3)3 with HCl and Na2CO3 by maintaining the pH (∼2) for 30 min at room temperature.56
2.2 Hydrothermal/solvothermal method
The hydrothermal method is the most common BiOCl preparation method, in which spontaneous pressure is produced to improve the reactivity and solubility of the precursors, causing chemical reactions to occur in the atmosphere.57,58 The size, crystal structure, exposed surface, and shape of BiOCl can be easily controlled by changing the thermodynamics and kinetics. Gao et al. prepared BiOCl nanospheres by adding Bi(NO3)3 in ethylene glycol and NaCl solvent for 12 h at 160 °C.59 Also, Liu et al. synthesized nanoplates by mixing Bi(NO3)3 in mannitol solution and NaCl was added to the resulting mixture.60 The reaction was carried out for 3 h to form a suspension at more than 150 °C. The (001) plane at the four-sided surface and (110) plane at the top and bottom of the prepared sample were exposed. Ye et al.61 fabricated Bi4O5I2 and Bi5O7I via different treatments of the molecular precursors (Fig. 3).
 |
| | Fig. 3 Schematic illustration of the synthetic procedure for the preparation of Bi4O5I2 and Bi5O7I via different methods. Reproduced with permission from ref. 61 Copyright 2016, Elsevier B.V. | |
2.3 Template method
A particular nano-sized material is used as a template, which enhances the growth in the template method. The symmetry (size and shape) of the prepared samples depends on the nature of the template used, which makes it a promising technique for the synthesis of BiOCl nanostructures. For instance, Cui et al. employed a template of carbonaceous microspheres and calcined Bi3+ with Cl− (absorbed on template surface) from raw HCl and Bi(NO3)3 at a very high temperature of 400 °C to prepare hollow microspheres of BiOCl. The shell thickness and diameter of the uniform BiOCl microspheres were about 40 nm and 200 nm, respectively.62 Yan et al. applied a biological template (butterfly wings) for the synthesis of hierarchical BiOCl.63 It was found that the subsequent seed deposition of BiOCl and animation of the used template resulted in the formation of 2D BiOCl nanosheets uniformly raised on the template surface, replicating the complex and original fine structure of butterfly wings.
2.4 Calcination
Modified BiOX-based photocatalysts can be produced via heat treatment of pure BiOX materials. The phase transformation can easily be achieved because the unstable halogen atoms can escape easily owing to the weak van der Waals forces. For instance, Yu, et al. synthesized plate-like Bi24O31Br10 through the heat treatment of BiOBr flakes at 750 °C and investigated its photocatalytic activity for the decomposition of Acid Orange II.64 Besides, instead of the plate-like morphology, there various morphologies have also been observed. Bi24O31Cl10 hollow microspheres were obtained via the thermal treatment (600 °C) of BiOCl in the presence of carbonaceous materials as sacrificial templates.62 The mechanism occurring is shown in Fig. 4.
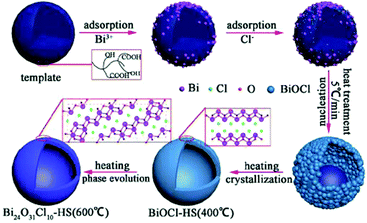 |
| | Fig. 4 Formation mechanism of bismuth oxychloride hollow microspheres. Reproduced with permission from ref. 62 Copyright 2016, Springer Nature. | |
2.5 Molecular precursor method
Bismuth-rich BixOyXz photocatalysts are usually synthesized via the hydrothermal method under alkaline conditions. However, it has been found that a small change in pH leads to a phase transition in the product, and thus obtaining high purity single-phase samples is still challenging. Recently, Ye et al. reported the preparation of single-phase Bi4O5X2 (X = Br and I) samples via the molecular precursor method, where Bi4O5X2 was obtained by hydrolyzing the precursor.40,65 Two main products, namely Bi4O5Br2 microspheres (diameter: 1 μm) and Bi4O5Br2 nanosheets (thickness: 1 μm) were obtained. Interestingly, the photocatalytic data shows that the bismuth-rich and thickness-ultrathin strategies play quite distinct roles in the reaction. The bismuth-rich strategy only increases the efficiency of CH4 generation for the photoreduction of CO2, whereas efficient CO generation is only associated with the thickness-ultrathin strategies.40 Presently, hierarchical bismuth-rich Bi4O5BrxI2−x can also be obtained via the molecular precursor method, and enhanced activity was found in the photocatalytic reaction of CO2 conversion under visible light.66 BiOCl with a nanosheet morphology can be acquired via the hydrolysis of Bin(Tu)xCl3n (Tu = thiourea). Importantly, the percentage of {001} plane was determined by adjusting the BiCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Tu feed ratio, and it was found that the percentage of {001} plane was positively proportional to the photoactivity of BiOCl.
:Tu feed ratio, and it was found that the percentage of {001} plane was positively proportional to the photoactivity of BiOCl.
3. Fundamentals of photocatalysis
3.1 H2 evolution
The sun is the most remarkable energy source in nature because of its clean nature and diversity, though it is not easy to harvest, convert and store. Therefore, researchers have focused their attention on using sunshine and turning it into a green energy supply that is effective and readily available. The difficulties regarding the capture and storage of energy can be addressed by using hydrogen as an alternative clean energy source, which has many future applications such as stationary power generation, domestic heating, and environmental-friendly vehicles with zero pollution. The energy shortage problem can be resolved if we can use scattered sunlight for hydrogen generation via electrochemical, photochemical, and photoelectrochemical methods. Several limited methods have been used for the conversion of solar energy, but the production of hydrogen by water splitting using sunlight has attracted more interest due to its many advantages as follows.67–72 (i) This process is based on clean water, which is a source of energy together with photon energy. (ii) It produces no pollutants and by-products (environmentally friendly). (iii) Hydrogen production via photochemical reaction efficiently deals with solar influx seasonal variation.73
Two main processes are involved in hydrogen generation via photocatalysis, i.e., photo-physical (PP) and electrochemical (EC) (see Fig. 5). In the photophysical process, the absorption of photons by electrons present in the valence band (VB) produces electron–hole pairs and transports electrons to the reaction sites at the conduction band (CB). In contrast, the photochemical process defines water splitting by redox reactions. Generally, a photocatalyst absorbs only photons whose energy corresponds to its bandgap energy. A photon with the same or greater energy than the bandgap energy of the photocatalyst will have enough potential to eject an electron from the VB of the photocatalyst to its CB (leaving holes behind). Photon absorption results in the creation of excited electrons and holes, while the electrons in the CB may recombine with holes radiatively/nonradiative. This is an inevitable process regardless of the semiconductor used for photocatalysis.4,75–77 The recombination rate is a vital element for low quantum efficiency for any photocatalyst.75,78–80 The photo-excited carriers that survive recombination then migrate to the surface of the photocatalyst, where photocatalytic reactions (hydrogen evolution reaction (HER) and oxygen evolution reaction (OER)) occur via diffusion/electric fields, which are related to the electrolyte or semiconductor interfaces.81,82
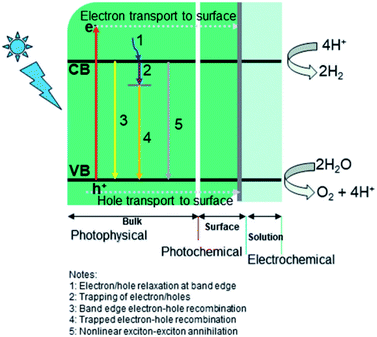 |
| | Fig. 5 Key dynamic processes involved in water-splitting: (i) photophysical, (ii) photochemical, and (iii) electrochemical. Reproduced with permission from ref. 74 Copyright 2019, the American Chemical Society. | |
The generation of excitons is typically limited below 100 fs (femtosecond) and the average lifetime of excitons is around a few hundred ps (picoseconds). The diffusion of an electron lasts for a few ps, while the corresponding time for the transition of holes, at most, may be less than 100–300 fs.75,83 Trap sites in semiconductors also play a role in restricting the recombination of photo-excited carriers. The greater the number of trap sites present in a catalyst (semiconductor), the lower the diffusion coefficients of electron/holes.84,85 The trapping of electrons has a shorter lifetime (a few microseconds) than holes, whose trapping, recombination, and surface transfer processes occur significantly faster (in pico- and nano-second range).
3.2 O2 evolution
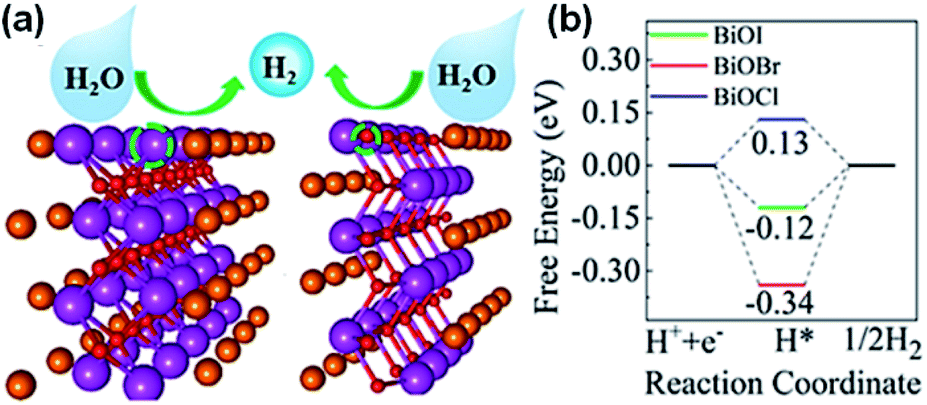
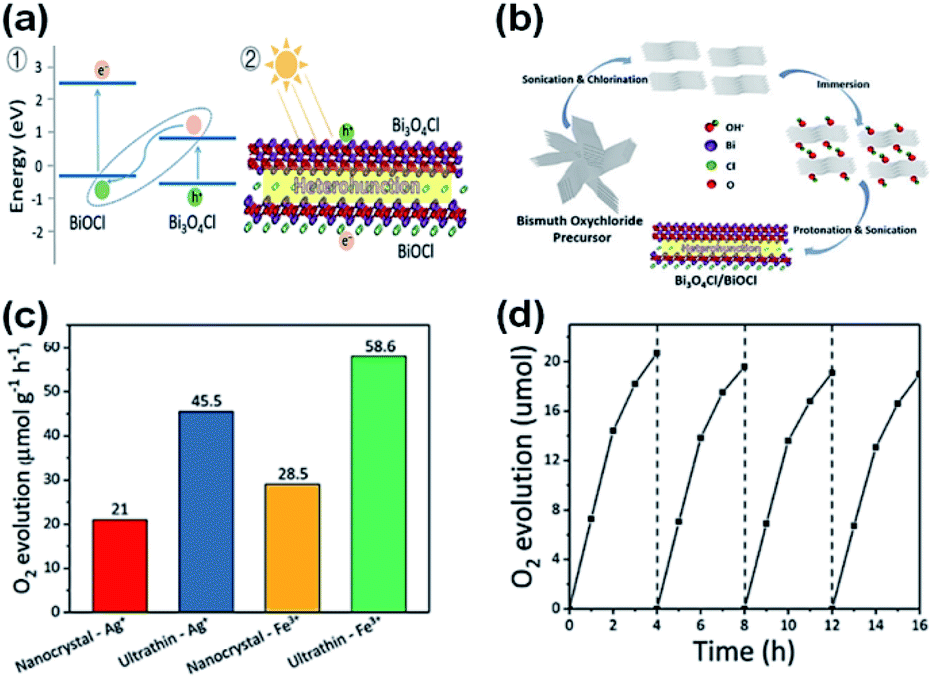
The photocatalytic production of O2 is considered the rate-determining phase for splitting water to produce H2. Compared to the transformation process, which involves two electrons from H+ to H2, the production of O2 requires four holes to participate in the oxidation of OH− and H2O to obtain O2 to overcome the large kinetic barrier. Thus, it is necessary to boost the water splitting oxidation half-reaction for the more efficient generation of H2. Previously, studies have been reported on the O2 production capability of BiOBr and BiOCl.87,88 For the currently established BixOyXz materials, improved activity for the production of O2 can be achieved. Lin et al.89 established a heterostructure of Bi3O4Cl/BiOCl and employed it via a Z-scheme for O2 production. Bi3O4Cl/BiOCl exhibited a formation rate 58.6 μmol g−1 h−1 O2 with FeCl3 and AgNO3 as electron scavengers. They established that the oxygen vacancy adjusted the Bi7O9I3 flowered microspheres through the ionic liquid-assisted method.90 These Bi7O9I3 microspheres with excessive oxygen could deliver O2 with a generation rate of 199.26 μmol g−1 h−1 with AgNO3 as an electron sacrificial agent. Hu et al.91 used polyvinylpyrrolidone (PVP) and cetyltrimethylammonium bromide (CTAB) to tune the morphology of Bi3O4Br to nanorings. Due to the ring structures, appropriate band potentials, and exposure of the (001) plane, the improved adsorption/migration of carriers and mass could be achieved over the nanorings of Bi3O4Br. Therefore, the Bi3O4Br nanorings delivered a production rate of 725.4 μmol g−1 h−1 O2 with Fe(NO3)3 as an electron capture agent under sunlight.
Besides surface defect engineering and morphological control, Li et al.86 employed C doping to boost the intrinsic electrical field in Bi3O4Cl (Fig. 6). Due to the substitution of Cl atoms in the crystal lattice by C doping, the electrostatic potential between [Cl] and [Bi3O4] could be enhanced up to 7.36. Thus, the internal electrical field in Bi3O4Cl can be maximized, which can considerably reduce the possibility of carrier recombination. Upon irradiation, photo-generated electrons will transfer along the [Bi3O4] layer to the [010] plane and holes will transfer along the [CxCl1−x] layer to the [110] plane, as verified by the photo-deposition experiment and calculated density of states (DOS). Due to these features, C–Bi3O4Cl delivered a production rate of ∼860 μmol g−1 h−1 O2 under sunlight irradiation. This O2 production rate could be maintained for up to 73 h in the cycle experiment.
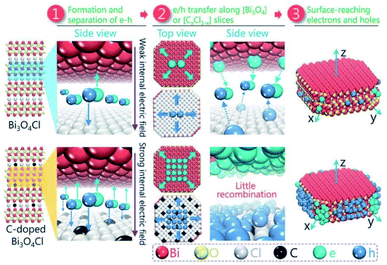 |
| | Fig. 6 Schematic design of the separation and the migration of holes and electrons in the bulk of pure and C-doped Bi3O4Cl. Reproduced with permission from ref. 86 Copyright 2016, Wiley-VCH. | |
3.3 CO2 reduction
In the last two decades, photocatalytic activity has been considered a green method for converting CO2 into useful hydrocarbon fuels and eradicating the greenhouse effect.92–94 In comparison with water splitting ability to obtain H2, BixOyXz displays encouraging potential in the photoreduction of CO2 into solar fuel (CH3OH, CH4, CO, etc.). BixOyXz exhibits an enhanced photocatalytic reduction performance compared to BiOX due to its lower CBM potential. For example, Bi4O5Br2 microspheres were synthesized and used for the photoreduction of CO2. It was demonstrated that Bi4O5Br2 could effectively photoreduce CO2 under sunlight because based on the bismuth-rich and thickness-ultrathin strategies.40 As illustrated in Fig. 7, the generation of CH4 and CO by Bi4O5Br2 was 2.04 and 2.73 μmol g−1 h−1, which was higher than that by ultrathin BiOBr (CH4: 0.16 μmol g−1 h−1: CO: 2.67 μmol g−1 h−1) and bulk BiOBr (CH4: μmol g−1 h−1; CO: 1.68 μmol g−1 h−1), respectively. Moreover, it was also perceived that the bismuth-rich approach and the thick-ultrathin strategy induced the selective generation of CH4 and CO, respectively. The apparent quantum yield (AQY) for the photoreduction CO2 into solar fuels at different wavelengths was calculated based on the following equation:22
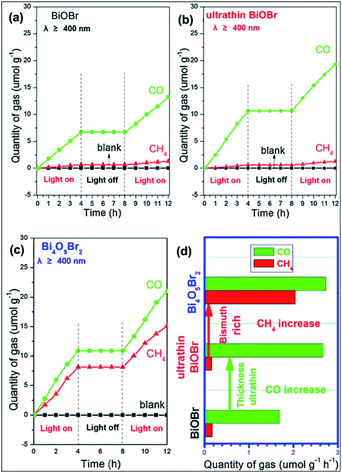 |
| | Fig. 7 Photocatalytic reduction performance for the conversion of CO2 on (a) BiOBr; (b) ultrathin BiOBr; and (c) Bi4O5Br2 and (d) comparison of solar fuel generation. Reproduced with permission from ref. 40 Copyright 2016, Elsevier B.V. | |
Bi4O5I2, Bi4O5BrI, Bi4O5Br2, and Bi5O7I have been developed for the photoreduction of CO2 into CH4 and CO upon exposure to sunlight. The AQY for solar fuels (CO and CH4) manufactured by Bi4O5I2 approached a maximum of 0.37% at the wavelength of 420 nm under monochromatic light irradiation.
3.4 N2 fixation
The photocatalytic fixation of N2 is very complex compared to other applications given that the photocatalytic reduction of N2 to NH3 is severely impaired by the weak nitrogen binding force on the surface of catalytic materials and the intermediate-high energy levels involved in the reaction.38 Thus, to resolve this problem, efficient interface charge transfer from the catalyst to N2 requires catalysts having strong electron donor ability and excess catalytic activation centers. The development of BiOBr nanosheets with oxygen vacancies on (001) facets is a typical example, which can fix N2 to NH3 upon exposure to visible light (Fig. 8).50,95 For the manufacture of fertilizers and chemicals, the synthesis of ammonia (NH3) is very important in agriculture and industry. The Haber–Bosch approach dominates commercial ammonia synthesis, which involves stringent reaction conditions (over 300 °C and 100 atm).96 Thus, low-cost approaches for synthesizing artificial NH3 need to be investigated under ambient conditions, among which, the sustainable reduction of photocatalytic N2 is an excellent solution. A series of N2 reduction experiments, particularly for bismuth-rich bismuth oxyhalides, was carried by Zhang's group,50,97 demonstrating that BiOBr has suitable properties for the photoreduction of N2 to yield ammonia. Ye et al.98 reported that the N2 photoreduction efficiency of Bi5O7I is facet dependent. Nanosheets of Bi5O7I with exposed (001) facets showed 2.3 times enhanced activity (111.5 vs. 47.6 μmol L−1 h−1). However, Bi5O7I with exposed (100) facets displayed a higher NO3-generation average than Bi5O7I with exposed (001) facets. It is important to note that 20% CH3OH was used in this method as a sacrificial reagent.
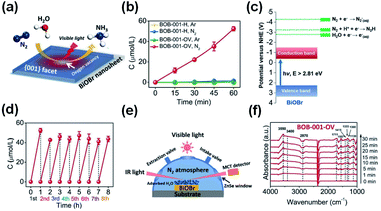 |
| | Fig. 8 (a) Scheme of N2 fixation with water over BiOBr nanosheets. (b) Quantitative measurement of NH3 generated under visible light (λ > 420 nm). (c) Energy-level diagram and (d) Multi-cycle N2 fixation on BOB-001-OV. (e) Reaction cell for the in situ IR signal measurement. (f) In situ IR signals of the photocatalytic N2 fixation over BOB-001-OV. Reprinted with permission from ref. 50 Copyright 2015, the American Chemical Society. | |
For N2 photoreduction in distilled water, researchers used defect-rich atomic-layer Bi3O4Br to prevent the use of a sacrificial reagent (Fig. 9).99 The charges generated in the bulk will diffuse easily to the surface, benefiting from the single-unit-cell thickness. To facilitate surface-charge separation, surface defects also act as a center of charge separation (see Fig. 9a and b). In addition, the defective structure allows improved adsorption and N2 activity (Fig. 9c), as shown by the reduction in adsorption energy on the perfect Bi3O4Br surface from −0.087 eV to −0.241 eV on a surface having defects. Consequently, the NH3 formation rate if 50.8 μmol g−1 h−1 in water could be produced on a defect-rich Bi3O4Br layer between 9.2 and 30.9 times higher than the Bi3O4Br atomic layer (defect deficient) and bulk Bi3O4Br. In addition to the chemisorbed mode N2 and N–H bending (Fig. 9e), the adsorption, activation and conversion of N2 into NH3 were further confirmed via in situ FTIR (Fig. 9d).
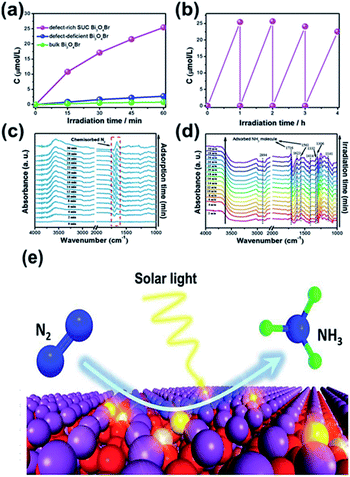 |
| | Fig. 9 (a) Quantitative evaluation of NH3 generated and (b) stability of defect-rich SUC Bi3O4Br under light irradiation. In situ FTIR spectra of SUC Bi3O4Br catalysts during N2 adsorption without irradiation (c) and under irradiation (d). (e) Scheme of the photocatalytic N2 fixation process. Reprinted with permission from ref. 99 Copyright 2019 Wiley-VCH. | |
In 2021, Tiana et al.100 prepared 1T phase WS2 nanosheet (NS)-decorated Bi5O7Br NSs (1T-WS2@Bi5O7Br) have a narrow band gap, effective carrier transport efficiency and good light absorption ability. After testing, the 1T-WS2@Bi5O7Br-5 composite presented the best photocatalytic nitrogen fixation performance (8.43 mmol L−1 h−1 g−1) and excellent stability. The probable photocatalytic mechanism of the 1T-WS2@Bi5O7Br composites was proposed. The Bi5O7Br NSs were prepared via a self-assembly water-induced process (Scheme 1a). Firstly, ammonia solution and bismuth(III) nitrate pentahydrate were added to a beaker, and then potassium bromide was added. Finally, the mixture was heated at 40 °C to obtain Bi5O7Br NSs with rich oxygen vacancies. As shown in Scheme 1b, the metallic 1T-WS2 NSs were prepared via a solvothermal process. Tungsten(VI) chloride and TAA were dissolved in dimethylformamide (DMF), and then the mixed solution was hydrothermally heated at 200 °C for 24 h to obtain 1T-WS2 NSs. The 1T-WS2@Bi5O7Br composites were synthesized via an ordinary grinding process (Scheme 1c). The Bi5O7Br NSs and 1T-WS2 NSs were added to a mortar with ethanol and hexane to fully grind. The mixed powder was dried to obtain 1T-WS2@Bi5O7Br composites.
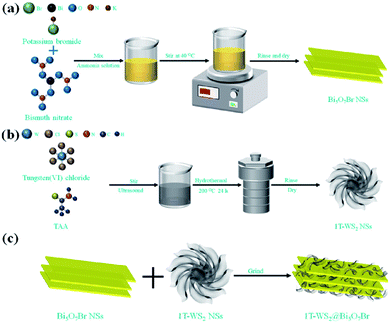 |
| | Scheme 1 Schematic route for the synthesis of (a) pure Bi5O7Br NSs, (b) 1T-WS2 NSs and (c) 1T-WS2@Bi5O7Br composites. Reprinted with permission from ref. 101 Copyright 2015, the American Chemical Society. | |
3.5 Pollutant removal
Pollutant removal is the most widely active photocatalytic application of BixOyXz. Upon irradiation, electron and hole pairs can be generated in BixOyXz, yielding various oxygen reactive species, such as singlet oxygen, ˙O2−, and ˙OH. By virtue of direct hole oxidation or reactive oxygen, different types of pollutants can be effectively removed, such as 4-tert-butylphenol, Rhodamine B (RhB), bisphenol-A (BPA), sodium pentachlorophenate, phenol, ciprofloxacin (CIP), resorcinol, acetaldehyde, tetracycline hydrochloride (TC), NO, and resorcinol.102–109 Using the Bi-rich technique of Bi4O5Br2 for upshifting the CB edge, more active oxygen species could be produced over Bi4O5Br2 relative to BiOBr. Consequently, 75% CIP was degraded by Bi4O5Br2 after 120 min and 53.3% was mineralized after 180 min. In another report, our group synthesized Bi4O5I2 and Bi4O5I2 hollow nanotubes through a PVP-supported solvothermal process.110 In comparison with Bi4O5I2 nanosheets, the Bi4O5I2 hollow nanotubes displayed an enhanced photocatalytic performance towards the degradation of BPA and RhB. The enhanced activity can be attributed to the confined highly concentrated active species induced by the tubular structure and the excellent electric conductivity. Xiao et al. prepared hierarchical microsheets of Bi7O9I3via a microwave-assisted method, which were employed for the photodegradation of BPA.111 According to the complete organic carbon measurement, 89.1% BPA was eliminated by the Bi7O9I3 microsheets almost after 1 h of irradiation, indicating that Bi7O9I3 degrades BPA effectively. Conversely, the BPA degradation intermediates were measured by LC-MS chromatography. Besides water body pollutants, gas pollutants such as acetaldehyde and NO could be effectively removed by BixOyXz.105,112 For instance, Dong et al.108 revealed that Bi4O5Br2 nanosheets exhibited stable and high activity for the removal of NO. The NO removal efficiency over Bi4O5Br2 reached 41.8% after 30 min irradiation and ˙OH was found to be the main active species.
3.6 Organic syntheses
Currently, BiOX-based photocatalysts show potential under mild conditions for selective organic synthesis.113 For example, surface-chlorinated BiOBr/TiO2 transformed alkanes into an oxygenated product via the activation of C–H bonds under sunlight irradiation.114 During the reaction, BiOBr with a narrow bandgap is excited, generating electron–hole pairs, and holes are transferred to the VB of TiO2 which results in a positive VB position for TiO2. The chlorine groups, which are chemisorbed on the catalyst surface, trap the holes, producing chlorine radicals, and then control the activation of the C–H bond. In a recent report, ultrathin sheets of BiOCl were synthesized with hydrophobic properties, which was assisted by the in situ formation of water in an octadecylene solution via the reaction between oleic acid and oleylamine.48 The fabricated ultrathin sheets had abundant oxygen vacancies, resulting in an improved photocatalytic performance for the aerobic oxidation of secondary amines to the corresponding imines under sunlight at room temperature. Compared to the BiOCl ultrathin nanosheets prepared through the hydrothermal process, the colloidal ultrathin sheets showed higher selectivity, conversion, and cycling stability. This improved photocatalytic performance was based on the strong absorption in the visible range and strong hydrophobic surface of the BiOCl colloidal sheets.
Recently, Xie et al.115 revealed that significant electron–hole interactions can be produced in BiOBr owing to its restrained layered structure, which result in robust excitonic properties (Fig. 10). Their investigation showed that singlet oxygen can be easily generated for (001) plane-exposed BiOBr owing to the robust excitonic effects in BiBr (001), whereas H2O2 is the main product for BiOBr (010). The singlet oxygen produced by BiOBr (001) could assist the mild oxidation of the sulfides to the corresponding sulfoxides with an improved photocatalytic performance and high selectivity for five consecutive cycles.
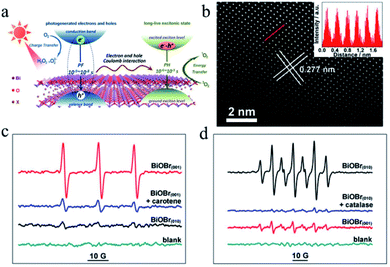 |
| | Fig. 10 (a) Schematic illustration of optical relaxation excitation processes presented in confined layered structure systems, where PH and PF are phosphorescence and prompt fluorescence, respectively. (b) Atomic-resolution scanning transmission electron microscopy (STEM) image of BiOBr(001). (c and d) Electron spin resonance (ESR) spectra. Reproduced with permission from ref. 115 Copyright 2017, the American Chemical Society. | |
4. Techniques for enhancing the photocatalytic activity of bismuth halides
4.1 Elemental doping
4.1.1 Transition metal doping.
As dopants, transition metal ions may produce lattice defects in semiconductors or alter their crystal structure to avoid electron–hole pair combination, and thereby create long-lasting carriers. Doping with unique metal ions will increase the region of light absorption, and the 3D subshells and variable valence configuration have a significant impact on the photoelectrochemical behavior of semiconductors. After doping with certain transition metal ions such as Fe3+ and Mn3+, a redshift in the energy band may be observed. The incorporation of these metal ions in the crystal lattice acts as an impurity in the forbidden band of semiconductors and the observed redshift is ascribed to the transition of an electron from these impurity bands to the CB or VB. Generally, very effective doped photocatalysts are found to be transition metal ion-based semiconductors, which should meet the following criteria: (a) the dopant can trap the electron–hole pairs, besides permitting powerful local separation and (b) the captured electrons and holes can be released and migrate to the interface.7,15,116,117
Numerous transition metal ions such as Fe,116,119–122 Cu,123 Zn,124,125 Ti,118 Al,126 Sn,127 and Mn128 have been used as dopants in BiOX materials. For instance, Mn doping in BiOCl with oxygen vacancies could reduce its bandgap and expand its optical absorption to the visible and IR regions.129 Both theoretical first-principle calculations and experimental observations proved the reduction in bandgap and the formation of an impurity band in the forbidden region in BiOCl after doping with tungsten.130 For example, Fe and Cu could be incorporated into the BiOCl system through the reaction between the self-doped reactive ionic liquids 1-octyl-3-methylimidazolium tetrachloroferrate ([Omim]FeCl4) and [Omim]CuCl3.119 The latter reactant, i.e. [Omim] CuCl3, in this process not only served as a source of Cl and Cu, but also behaved as a model for Cu/BiOCl microspheres. Most importantly, Jiang et al. prepared a BiOBr photocatalyst doped with Ti and Ag, resulting in the successful enhancement of dye degradation, as illustrated in Fig. 11.118
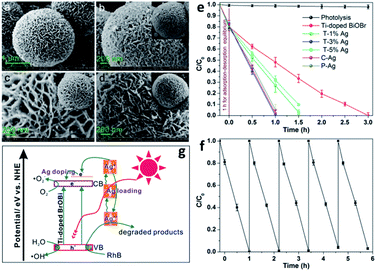 |
| | Fig. 11 FESEM images of (a) as-prepared Ti-doped BiOBr microspheres, (b) C–Ag, (c) P–Ag, and (d) T–3% Ag. (e) Photocatalytic degradation of RhB over Ti-doped BiOBr. (f) Cycling runs for the photodegradation of RhB over the as-prepared T–3% Ag composites. (g) Schematic illustration of the photocatalytic mechanism of RhB degradation over T–Ag photocatalyst. Reproduced with permission from ref. 118 Copyright 2012, the American Chemical Society. | |
4.1.2 Rare earth ion doping.
The special electronic configurations of the inner transition metals impart spectral properties to these elements, which are not characteristic in other elements in the periodic table. This is due to the presence of 4f orbitals with a multi-electron system. Oxides of the rare earth metals possess various crystal structures, which result in highly selective adsorption, good conductivity, and thermal stability. Various energy levels are present in these rare earth metals, where photogenerated electrons and holes can be captured easily.131 Thus, doping of these elements enhances the electron–hole pair separation, which in turn boosts the photocatalytic ability. Y-doped microspheres of BiOBr were synthesized via an IL-assisted solvothermal scheme, and a shift in the X-ray diffraction (XRD) peaks was observed, demonstrating the substitution of Bi by Y in the BiOBr lattice.132 The photocatalytic activity for the degradation of RhB and ciprofloxacin under visible-light irradiation was improved in the presence of the Y-doped sample. The enhancement in the photocatalytic properties is due to the formation of a sub-band under the conduction band, where electrons are trapped, thus reducing the recombination rate of electron–hole pairs. Other rare-earth elements such as europium and erbium were also used as dopants for the BiOX system recently.131 Dash et al. synthesized a photocatalyst composed of poly(vinyl alcohol)-functionalized Eu-doped BiOX nanoflakes, which demonstrates significantly enhanced RhB degradation activity.133
4.1.3 Non-metal doping.
The optical properties of any substance are attributed to its basic electronic configuration, i.e., due to the transition of an electron from a filled orbital to an empty energy level. There is also a transition of electrons from the valence to conduction bands in the case of conductors and semiconductors.134,135 Additional extrinsic energy levels are created between the energy bandgap upon the addition of non-metal dopants. This is supported by two hypotheses: the valence band is shifted upwards to its maximum level as a result of (i) the hybridization of the valence orbitals of the non-metal elements and (ii) the localized states from the valence orbits of the non-metal elements.
Basically, in the former case, the band to band absorption occurs as a result of the transition of electrons from the recently created valence band (VB) to the conduction band (CB), whereas according to the latter hypotheses, a pristine absorption edge remains, and small absorption peaks appear in the form of a shoulder or tail.71,80,136 Considering the case of C-doped BiOCl materials that are formed by adsorbing dopants via heat treatment (Fig. 12), the mechanism for the homogeneous adsorption of carbon in the crystal surface was interpreted by utilizing nanosheets of BiOCl with high {001} or {010} facet exposure.137 Efficient homogeneous doping of hydrothermally grafted carbonaceous nanoclusters into the crystal lattice essentially depended on the facet-related surface atomic structure.138,139
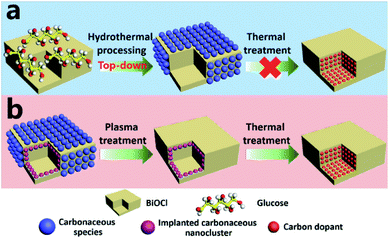 |
| | Fig. 12 (a) Exploration of the contribution of surface-modified carbonaceous species to carbon doping. (b) Elucidation of the role of implanted carbonaceous nanoclusters during carbon doping. Reproduced with permission from ref. 137 Copyright 2015, Wiley-VCH. | |
4.2 Creation of oxygen vacancies
Surface modification via the creation of O2 vacancies is a fruitful method to improve the photocatalytic potential of BiOCl.46,140,141 The electronic properties can be tuned via the incorporation of O2 vacancies, which consequently improve the electron transfer and light absorption in BiOCl. For example, Guan et al. employed the solvothermal route to fabricate a highly thin (thickness ∼2.7 nm) sheet of BiOCl and reported the associated triple vacancy as the major defect in the prepared nanosheet (Fig. 13a). Besides an enhancement in the absorption range, a reduction in the bandgap energy and exciton recombination rate was observed owing to the triple vacancy defect in BiOCl. These alterations in the electronic properties of the BiOCl nanosheet resulted in enhanced activity for the degradation of Rhodamine B dye (Fig. 13b).142
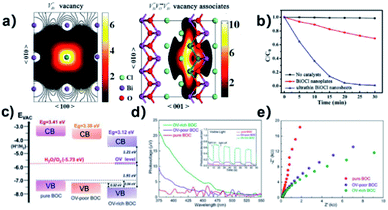 |
| | Fig. 13 Schematic representation of the trapped positrons of (a) 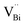 defect and defect and 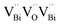 vacancy associates, respectively. (b) Comparison of the photodecomposition of Rhodamine B with ultrathin BiOCl nanosheets and BiOCl nanoplates under simulated solar irradiation. (c) Schematic illustration of the band structure of OV-rich/poor BOC and pure BOC. (d) Surface photovoltage spectrum of OV-rich/poor BOC and pure BOC. Current density transients of OV-rich/poor BOC and pure BOC under visible-light irradiation are shown in the inset. (e) Nyquist impedance plots of OV-rich/poor BOC and pure BOC. Reproduced with permission from ref. 142 Copyright 2013, the American Chemical Society, and ref. 143 Copyright 2018, The Royal Society of Chemistry. vacancy associates, respectively. (b) Comparison of the photodecomposition of Rhodamine B with ultrathin BiOCl nanosheets and BiOCl nanoplates under simulated solar irradiation. (c) Schematic illustration of the band structure of OV-rich/poor BOC and pure BOC. (d) Surface photovoltage spectrum of OV-rich/poor BOC and pure BOC. Current density transients of OV-rich/poor BOC and pure BOC under visible-light irradiation are shown in the inset. (e) Nyquist impedance plots of OV-rich/poor BOC and pure BOC. Reproduced with permission from ref. 142 Copyright 2013, the American Chemical Society, and ref. 143 Copyright 2018, The Royal Society of Chemistry. | |
Another similar study was reported by Cui et al., where they prepared nanosheets of BiOCl enriched with abundant O2 vacancies via solvothermal synthesis.143 The generation of a new energy level (defective) upon the incorporation of O2 vacancies was confirmed both theoretically and experimentally. This enabled photocatalytic oxygen evolution under visible light irradiation by enhancing the absorption range of BiOCl films from the UV to visible region. Moreover, the increased number of O2 vacancies offered a large photocurrent and superior charge transfer compared to BiOCl with fewer O2 vacancies (Fig. 13d and e), respectively.
4.3 Heterojunction construction
The construction of a heterojunction is considered to be an effective way to enhance the photocatalytic activity of bismuth oxyhalides. Constructing a heterojunction at the interface of different semiconductor materials can promote the separation of photogenerated charges in the photocatalytic system.144–147 There are various types of heterojunctions such as the conventional, direct Z-scheme, 2D/3D, and P–N heterojunctions.97 The conventional semiconductor heterojunction (type II) is the most studied type due to its appropriate band edge location, which generates effective charge separation.148–153 Peng et al.154 prepared Bi12O17Br2/Bi24O31Br10 type-II heterojunctions via the calcination of BiOBr/Bi(OHC2O4)·2H2O. Due to the rapid electron–hole separation in the type-II heterojunction, Bi12O17Br2/Bi24O31Br10 exhibited excellent photocatalytic activity for the degradation of RhB and phenol. Generally, bismuth oxyhalide nanomaterials may play three important roles in semiconductor heterojunction systems. Initially, bismuth oxyhalides have a wide range of band redox potentials, and thus they can easily match various semiconductors in energy level, which provides a driving force for the directional separation of photogenerated charges.
Next, narrow band gap bismuth oxyhalide semiconductors (BiOI and BiOBr) can be used to photosensitize other semiconductors to effectively utilize solar energy. For example, BiOI with TiO2 and ZnO was coupled to construct p–n heterostructures via a simple chemical bath method at low temperature.155,156 By adjusting the molar ratio of Bi to Ti/Zn, the morphology, chemical composition, and photoresponse range of the TiO2/BiOI and ZnO/BiOI heterostructures can be carefully controlled. Compared with TiO2 and ZnO, the more negative conduction band level of BiOI will result in the effective transfer of photogenerated electrons between the catalysts, which may prolong the lifetime of photogenerated charges and reduce the probability of photogenerated charge recombination (Fig. 14). Finally, due to the structural diversity of bismuth oxyhalides, carbon-based nanomaterials can be easily coupled with bismuth oxyhalides, which can partly increase the charge separation and transfer.157,158 For instance, BiOCl/graphene, BiOBr/graphene, and BiOI/graphene have been utilized due to the enhanced bond between bismuth oxyhalide and graphene.159,160 Besides, graphene can be used as a co-catalyst to extract photogenerated electrons. Therefore, the electron transfer from bismuth oxyhalides to graphene can effectively inhibit the electron–hole recombination, prolong the carrier lifetime and improve the photocatalytic efficiency. Above all, the heterojunction strategy is often used to construct binary or multicomponent composite photocatalyst systems, which is one of the universal strategies in solar photocatalysis and photoelectrocatalysis.117
 |
| | Fig. 14 (a) UV-vis diffuse reflectance spectra and (b) corresponding colors of pure BiOI, pure ZnO, and ZnO/BiOI heterostructures. Plots of (αhv)1/2versus energy (hv) for the band-gap energies of (c) BiOI and (d) ZnO. (e) Schematic illustration of ZnO/BiOI heterostructures with different Bi/Zn molar ratios. Reproduced with permission from ref. 155 Copyright 2011, the American Chemical Society. | |
4.4 Strain modulation
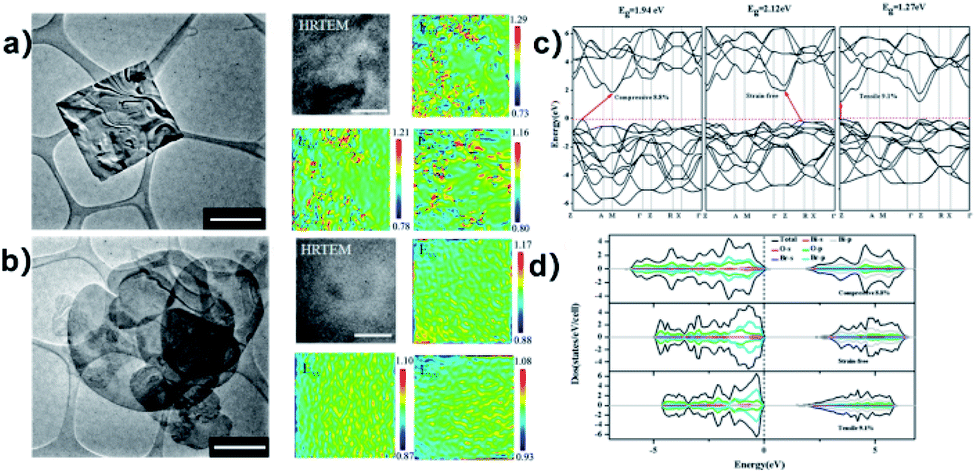
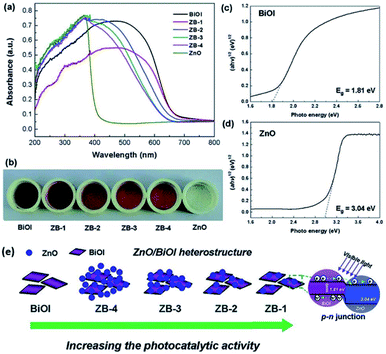
The synthesis of bismuth oxyhalides is very sensitive to strain perturbation due to their specific 2D layered structure.162 Also, the electronic structure of bismuth oxyhalides may change greatly even with the existence of subtle inner strain variation. This was investigated by Du and co-workers, who found how strain affects the photocatalytic performance over a BiOBr-square and BiOBr-circle.161 Based on HRTEM images, they initially observed some in-plane wrinkles in the BiOBr-square, and then obtained an inhomogeneous strain distribution from GPA simulation (Fig. 15a).161,163 This type of local lattice distortion was proven to be certainly located on the BiOBr-square. On the contrary, the strain distribution was found to be homogeneous on the surface of the BiOBr-circle (Fig. 15b), suggesting much less lattice distortion than that in the BiOBr-square. Overall, it was clear that the strain in the BiOBr-square was much stronger than that in the BiOBr-circle. Meanwhile, the strain effect on the electronic structures was carefully studied through DFT calculations.164 As shown in Fig. 15c and d, two types of stains (tensile strain and compressive strain) could promote the solar-light harvesting abilities in a wide range by narrowing the band gap of the photocatalysts.
 |
| | Fig. 15 (a) TEM image and simulated strain distribution based on HRTEM over a BiOBr square. The scale bar is 10 nm. (b) TEM image and simulated strain distribution based on HRTEM over a BiOBr square. The scale bar is 10 nm. (c) Band structure of BiOBr with biaxial strain calculated by DFT. (d) Calculated DOS of BiOBr with different types of strain. Reproduced with permission from ref. 161 Copyright 2015, the American Chemical Society. | |
4.5 Co-catalyst
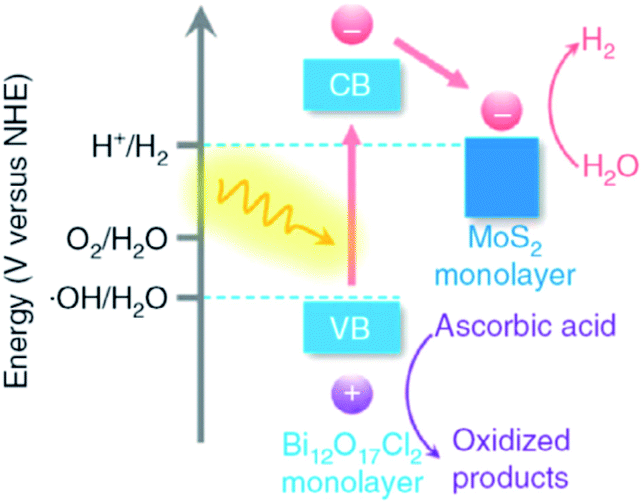
In semiconductor photocatalysts, loading of co-catalysts plays a crucial role, which behave as reaction active sites and also facilitate the trapping of photogenerated charges, and hence the photocatalytic response can be significantly enhanced.165,166 Co-catalysts formally include the deriving-electron-type (Ag, Pt, Au, MoS2, and Ni) and deriving-hole-type (PbO2, MnOx, and NiO). It was reported that Bi24O31Br10 loaded with silver nanoparticles exhibited increased photocatalytic activity in contrast with bare Bi24O31Br10 and P25.167 More recently, Zhang and co-workers demonstrated Bi12O17Cl2 monolayers with MoS2 nanosheets with the aid of noble metal co-catalysts (Fig. 16).167 Notably, monolayers (MoS2/Bi12O17Cl2) result in a higher rate of H2 production. These outcomes suggest that the use of co-catalysts serves as a suitable technique for the development of highly proficient BixOyXz photocatalysts. However, the relevant literature regarding this strategy is scarce.
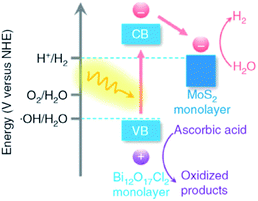 |
| | Fig. 16 Band alignments in 1L-BOC and 1L-MS. Reproduced with permission from ref. 167 Copyright 2016, Springer Nature. | |
4.6 Solid solution
Constructing solid solutions is an effective way to tune the band gap structure, crystal structure, and local electronic structure of materials. Due to the varying proportional range of the components, it is promising to synthesize solid-solution photocatalysts with enhanced photocatalytic activity.168 For example, it has been widely reported that BiOCl1−xBrx, BiOBr1−xIx, and BiOCl1−xIx solid solutions have much higher photocatalytic activity than their individual compounds.169,170 However, due to the non-uniform synthesis conditions, little work has been performed on bismuth-rich BixOyXz solid solutions. In 2015, Liu et al. reported the synthesis of Bi24O31ClxBr10−x solid solutions using a solvothermal method for the first time,171 where the Bi4O5BrI solid solution with the optimal ratio (x = 1) displayed the highest photocatalytic activity for the photocatalytic reduction of CO2 and Cr(VI) under visible light irradiation (Fig. 17).66 This may be attributed to its higher CB edges than that of the other Bi4O5BrxI2−x photocatalysts. These results clearly demonstrate that the formation of solid solutions is an effective method to modify the energy structure and enhance the photocatalytic performance of BixOyXz.
 |
| | Fig. 17 Photocatalytic activity for the reduction of CO2 over hierarchical bismuth-rich Bi4O5BrxI2−x solid solutions. Reproduced with permission from ref. 66 Copyright 2017, Elsevier B.V. | |
4.7 Composite framework
4.7.1 Bismuth oxyhalide-based binary composites.
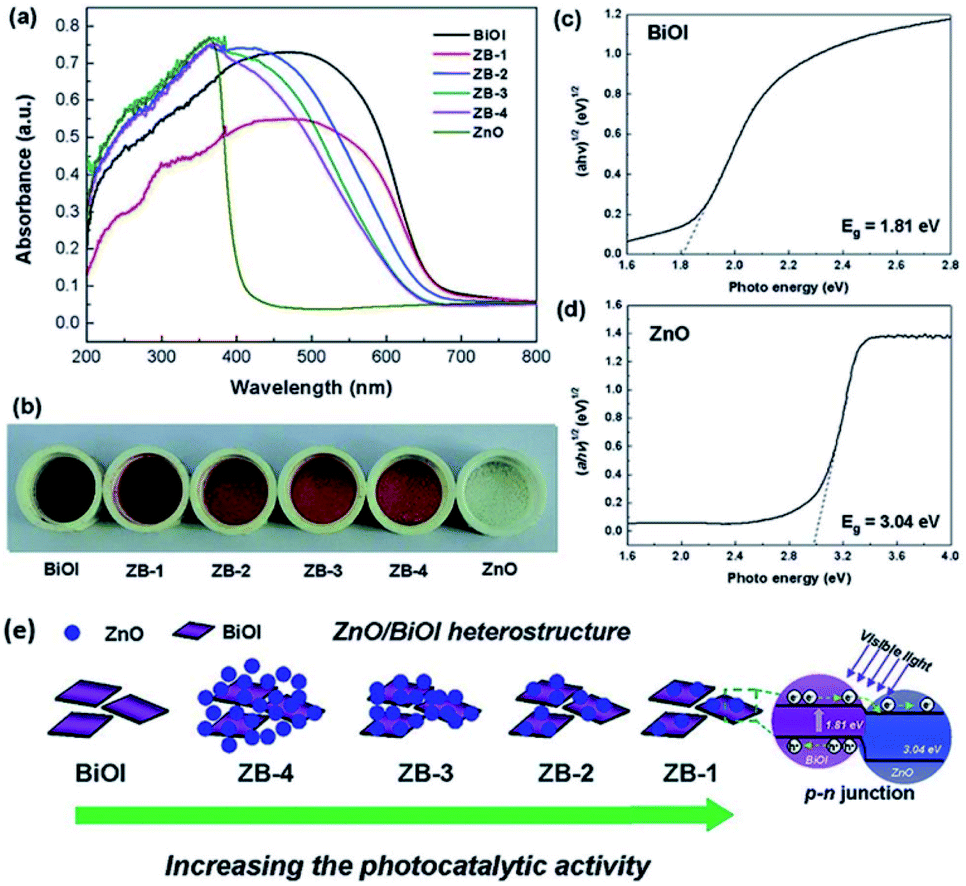
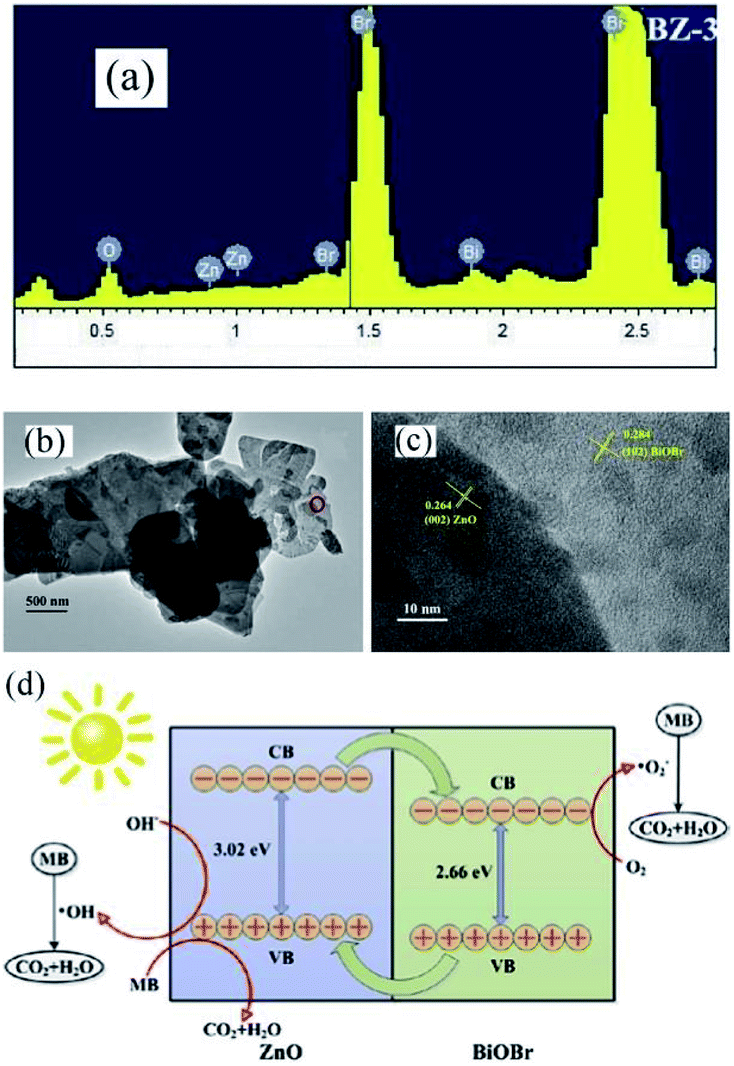
Recently, the impressive photocatalytic activity and remarkable structures of BiOX-based photocatalysts have gained increasing attention.20,146,172–175 BiOX possess a [Bi2O2]2+ coating assembly of crystals surrounded by halogen atom slabs. Additionally, the internal electric field (static) present between two layers filled with separated photo-induced electrons and holes improves its photocatalytic performance.176,177 ZnO is considered an effective photocatalyst for the purification of water.178–186 However, its wide bandgap energy (3.2 eV) restricts its photocatalytic performance to the UV region.187–191 Thus, to enhance its photocatalytic performance and ability to absorb sunlight, different photocatalyst composites (ZnO/BiOX) have been synthesized.155,192–194 Geng et al. prepared various BiOBr/ZnO materials through the hydrothermal method for the removal of methylene blue (MB) in the aqueous phase.177 Suspended Zn(OH)2 was prepared by adding Zn (NO3)2·6H2O to a KOH solution, and in a separate beaker, a specific concentration of KBr with Bi(NO3)3·5H2O was dripped in water (deionized), and the resulting mixture was added dropwise to the above solution at pH 7 with continuous stirring. Then, the obtained solution was autoclaved for 12 h at 120 °C. The EDS spectrum of BiOBr/ZnO confirmed that the sample contained Br, O, Bi, and Zn and the construction of a heterojunction (Fig. 18a). Due to the interaction among BiOBr and ZnO, the symmetry of heterojunction did not change during the preparation, as revealed by the TEM images (Fig. 18b). HR-TEM (Fig. 18c) indicated a lattice spacing of 0.284 nm {102} and 0.264 nm {002} for ZnO and BiOBr, respectively, and the formation of heterojunction BiOBr. The photocatalytic activity improved due to the formed heterojunction given that it reduced the recombination rate of light-generated excitons. The synthesized heterojunction of BiOBr/ZnO mirrored the Z-scheme process for dye (MB) degradation (Fig. 18d).177 Consequently, the formed O2− decomposed the dye (MB) molecules into CO2 and H2O.
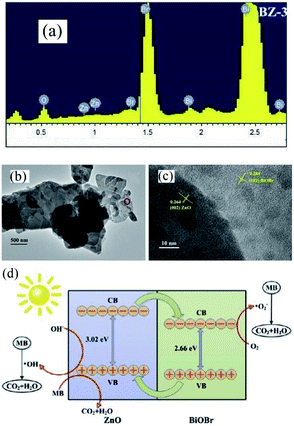 |
| | Fig. 18 (a) EDS pattern, (b) TEM image, (c) HRTEM image, and (d) photocatalytic mechanism of BiOBr/ZnO heterojunction. Reproduced with permission from ref. 177 Copyright 2017 Elsevier B.V. | |
4.7.2 Bismuth oxyhalide-based ternary composites.
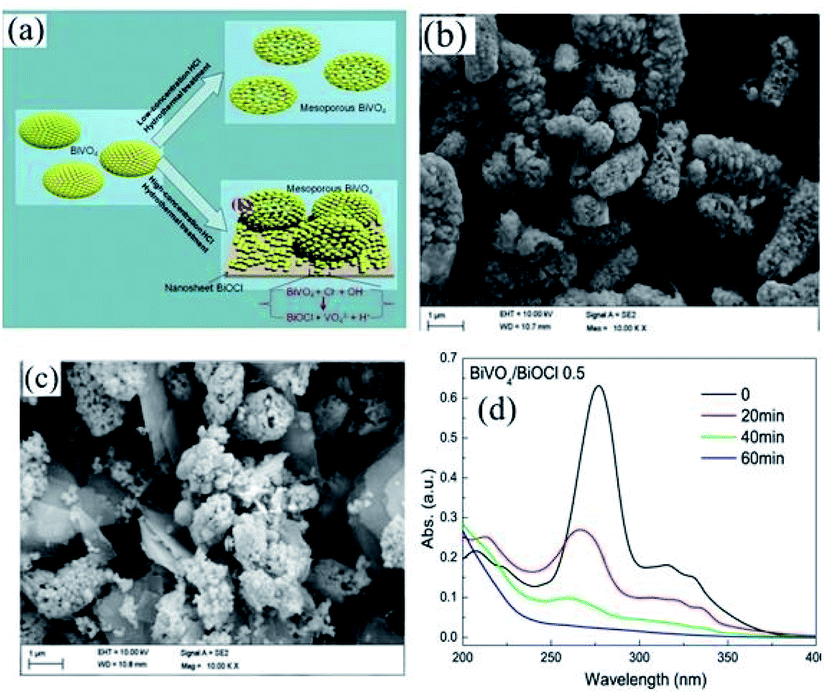
Nowadays, metal oxides with multiple components have attracted significant attention for water purification via photocatalysis.195 Recently, CdWO4,196 BiWO4,197 and ZnWO4,198 which are ternary metal oxides, have been utilized as photocatalysts under visible light. Cao et al. fabricated a CdWO4/BiOBr photocatalyst based on visible light through a hydrothermal and chemical precipitation process for the degradation of RhB dye.196 During the synthesis of CdWO4/BiOBr, a certain amount of Bi(NO3)3·5H2O and KBr amount was dissolved in solution, while CdWO4 was observed as a suspended component. Further, the obtained solution was kept for 5 h at 80 °C on a water bath. CdWO4/BiOBr composites were synthesized with 5%, 15%, and 25% molar ratios of CdWO4 in CdWO4/BiOBr. The sample that showed the maximum degradation of RhB under visible light in 8 min was the 15% molar ratio. This sample exhibited the maximum performance for degradation compared to the individual BiOBr and CdWO4 photocatalysts. The p–n junction promoted electron–hole pair separation, which improved the catalytic activity of the composite. A BiVO4/BiOCl composite was synthesized by Ma et al. via a hydrothermal process for the degradation of norfloxacin. BiVO4 and PVP were stirred for homogeneous mixing together with concentrated HCl (0.7, 0.5, 0.3, and 0.1).199 The mixture was further heated in an autoclave at 180 °C for 10 h (Fig. 19a). The structure obtained was mesoporous at a concentration of 0.1 M HCl. The TEM images displayed nanosheets of the BiVO4/BiOCl composite at different concentrations of HCl. However, although nanosheets were obtained for 0.1 M HCl concentration (Fig. 19b and c), the nanosheets formed for 0.5 M HCl concentration showed the maximum photocatalytic activity under visible light for the degradation of the norfloxacin antibiotic, which was caused by the formation of a p–n junction in 1 h between the composite (Fig. 19d). As mentioned above, the formation of a p–n junction restricted the recombination of charge carriers, which resulted in an enhancement in photocatalytic activity.
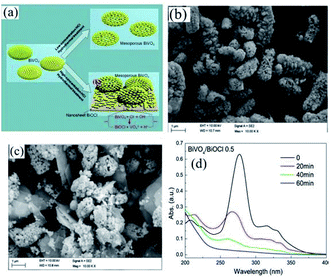 |
| | Fig. 19 (a) Formation of BiOCl nanosheets over BiVO4/BiOCl heterojunction. TEM image of BiVO4/BiOCl heterojunction at (b) 0.1 HCl and (c) 0.5 HCl concentration. (d) Photocatalytic activity of BiVO4/BiOCl-0.5 heterojunction for the degradation of norfloxacin. Reproduced with permission from ref. 199 Copyright 2017, Elsevier B.V. | |
4.7.3 CNT-based bismuth oxyhalide composites.
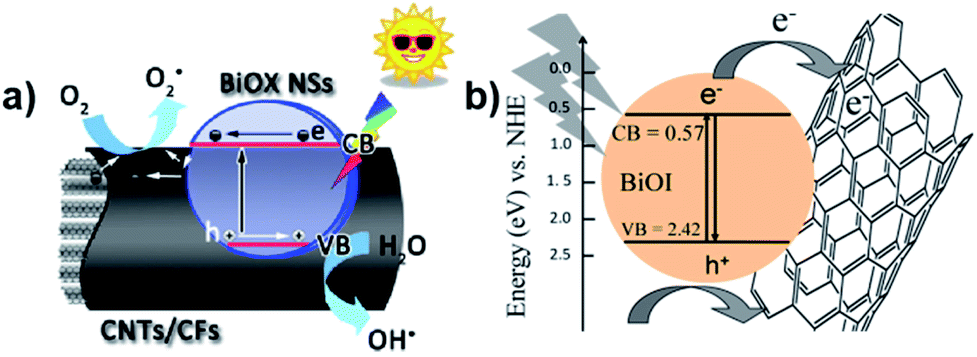
Carbon nanotubes (CNTs) have been well developed due to their effective conductivity, capture capability, and high electron affinity. Many researchers reported an enhancement in photocatalytic efficiency caused by the addition of CNTs in nanocomposites. The use of carbon-based materials can also be an efficient strategy to enhance photocatalytic efficiency. For instance, Xu et al. reported the synthesis of hierarchical structure BiOX (X = Cl/I) NSs via an ionic layer adsorption and reaction method, which showed notable photocatalytic activity for the degradation of Methyl Orange (MO).19 Improved stability and recycling ability under simulated sunlight were observed. As presented in Fig. 20a, the CNTs act as a promising material to tailor the energy gaps of semiconductors via covalent bonds due to their excellent mechanical properties and high pollutant absorption ability. A similar result was described by Xiong et al., where BiOI–MWCNT composites synthesized via an EG-assisted solvothermal method exhibited higher visible-light photocatalytic activity than that of BiOI, which can be attributed to the interfacial charge-transfer, as shown in Fig. 20b.200 Due to its extensive specific surface area and the promising charge transfer properties, the CNT–BiOI composite can be used as a highly efficient and delicate system for the degradation of organic pollutants.
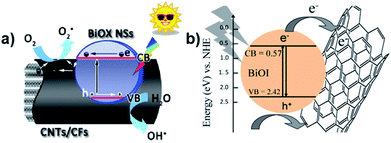 |
| | Fig. 20 (a) Photocatalytic mechanism of the hierarchical CNT/CF-BiOX NS structures. (b) Photoinduced electron transfer between an excited BiOI particle and MWCNT and charge equilibration. Reproduced with permission from ref. 19 Copyright 2014, Springer Nature and ref. 200 Copyright 2012, Elsevier B.V. | |
4.7.4 Graphene-based bismuth oxyhalide composite.
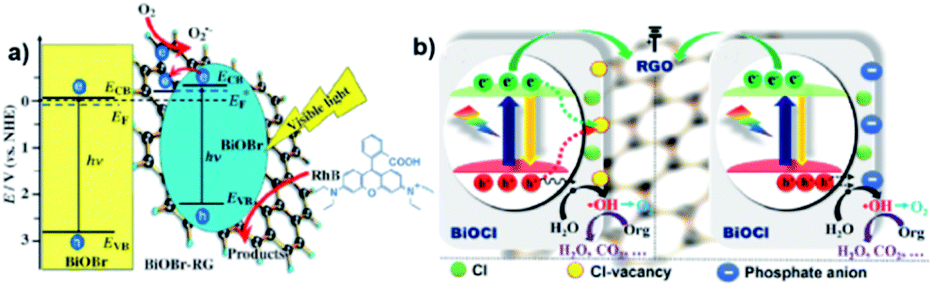
Graphene displayed an array of advantages in photocatalytic composite systems in recent decades as a conventional layered material. Li et al., for example, reported the one-pot synthesis of a composite of BiOBr–graphene with significantly enhanced photocatalytic activity for the degradation of Rhodamine B (RhB), which simultaneously exhibited excellent dye absorption, intense charge-separation, and transport properties.129 In this work, graphene oxide could be reduced to graphene via the solvothermal method, and the BiOBr particles were found to grow on the graphene surface simultaneously, as illustrated in Fig. 21a. Moreover, the functional graphene–BiOBr composites exhibited novel properties owing to the combination of the advantages of graphene nanosheets and BiOBr. Zhang et al. found that modified RGO/BiOCl nanocomposites showed 8.4-times and 3.8-times greater activity enhancement for the degradation of MO and oxidation of water than that of BiOCl, respectively, which was attributed to the enhanced charge separation and extended carrier lifetime of BiOCl by chemically bonding with RGO.201 Interestingly, the results in this study indicated that chloride vacancies act as recombination centers, which may impede photogenerated charge separation, as shown in Fig. 21b. In addition, it has also been reported that Er-doped BiOBr–graphene hollow microspheres exhibited a major improvement in photocatalytic activity for the degradation of RhB under simulated sunlight irradiation, which may be due to the strong synergistic effect between charge separation and solar energy harvesting.202 In summary, compared with other layered materials, graphene is much more appealing to exactly regulate the behaviors of photogenerated electrons and holes in bismuth oxyhalide-based composite photocatalysis systems, largely due to its flexible atom-thin 2D nature, unique electronic properties and excellent absorptivity, which are promising candidates in photocatalysis.203
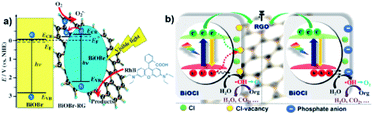 |
| | Fig. 21 (a) Schematic illustration of energy-band diagrams of BiOBr and BiOBr–RG, schematic structure of BiOBr–RG, and tentative processes of the photodegradation of RhB over BiOBr–RG. (b) Proposed mechanism for the photogenerated charge transfer and separation in RGO/BOC photoanodes before and after phosphate modification. Reproduced with permission from ref. 204 Copyright 2012 Wiley-VCH and ref. 201 Copyright 2012, Elsevier B.V. | |
4.8 Plasmonic material-supported photocatalysts
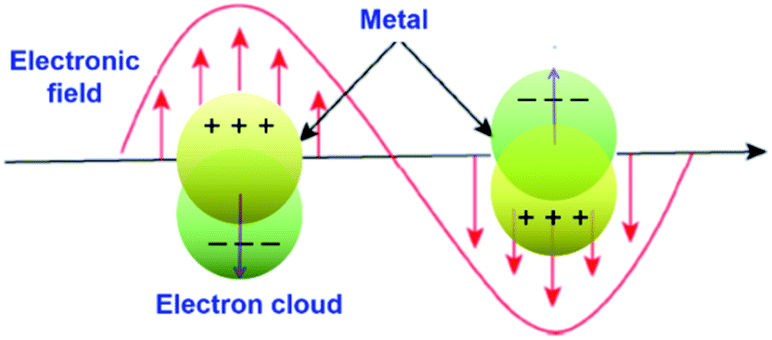
Surface plasmon resonance (SPR) is a phenomenon involving electromagnetic oscillation produced on the surface of a metal via the interaction of free electrons and photons. Surface plasmons have many distinctive properties due to the association between surface charge oscillation and lightwave electromagnetic field. As the light pulse (electromagnetic wave) enters the metal–dielectric interface, the free electrons collectively oscillate on the metal surface. Then, the electromagnetic wave is coupled with the free electrons on the metal surface to form a type of near-field electromagnetic wave propagating along the metal surface. When the oscillation frequency of the electron is close to or consistent with the frequency of the incident light wave, resonance will occur, which makes the metal absorb and scatter the incident light strongly and causes resonance extinction. In the resonance state, the energy of the electromagnetic field is effectively transformed into the collective vibration energy of the free electrons on the metal surface. Meanwhile, a special electromagnetic mode is formed, where the electromagnetic field is confined to a small area of the metal surface and is significantly enhanced (that is, the intensity of the local electric field generated near the metal is much stronger than that of the incident light), which is defined as surface plasmon resonance (Fig. 22).205,206
 |
| | Fig. 22 Schematic illustration of surface plasmon resonance (SPR) on the metal surface. | |
5. Theoretical studies
5.1 H2 production
Recently, Pan et al.207 used DFT calculations discover the source of catalysis on single-layer BiOX (Fig. 23a). The core possible calculations proposed that the single-layer BiOX-Bi and BiO-terminations are constant in O-poor and O-rich conditions, respectively. The Bi and BiO-terminations of single-layer BiOX were observed to have distinct HER active sites, while the (001) basal planes are inert. The Gibbs free energies (GFE) are similar to the optimum values of 0 eV for the H-atoms adsorption on Bi- and BiO-terminations, signifying that there are favorable HER performances for single-layer BiOX (Fig. 23b). These termination-enhanced HER behaviors are attributed to the clustered edge states around the Fermi stage induced by the Bi 6p fringe bismuth atom orbital density and O 2p fringe oxygen atom orbital density, respectively.
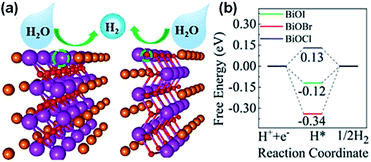 |
| | Fig. 23 (a) H2 evolution mechanism and (b) free energy diagram for HER occurring on the (001) basal plane of single-layer BiOX. Reproduced with permission from ref. 207 Copyright 2019, the American Chemical Society. | |
5.2 O2 production
Further, oxygen can be generated by doping BiOCl with elemental sulfur and the sacrificial reagent AgNO3 under UV-vis irradiation.208 Thus, by doping BiOCl with sulfur, the photogenerated charge carrier separation efficiency boosts the evolution of oxygen by five times than in BiOCl. For improving strength of IEF, allowing 80% charge splitting efficiency by directing it, BiOCl and homogeneous carbon-doped Bi3O4Cl nanosheets were fabricated.209 Femtosecond-resolved transient absorption spectroscopy showed that the strong IEF separated the electron–hole pairs restricted within the [Bi3O4] and [Cl] slices when they drifted from the bulk to surface. Finally, water could be split to give out oxygen with an efficiency of about 90 μmol h−1 when 0.1 g of Bi3O4Cl nanosheets doped with carbon was dispersed in 200 mL water in the absence of a co-catalyst and electron-scavenger under visible light (150 W Xe arc lamp equipped with a 420 nm cutoff filter). However, the Bi3O4Cl nanosheets doped with carbon were not able to generate hydrogen given that their conduction band is more positive than the activation potential of H2. O2 was generated by heterostructure photocatalysts (mechanism shown in Fig. 24a with DFT calculations) of Bi3O4Cl/BiOCl with a Z-scheme by Lin et al.210 (synthesis procedure shown in Fig. 24b). They suggested an operative method to promote the photocatalytic O2 progress through the dual-induced {0 0 1} plane of BiOCl nanosheet heterostructures. Using an electron scavenger (AgNO3 and FeCl3), O2 was produced at a rate of 58.6 μmol g−1 h−1 by Bi3O4Cl/BiOCl (Fig. 24c and d).
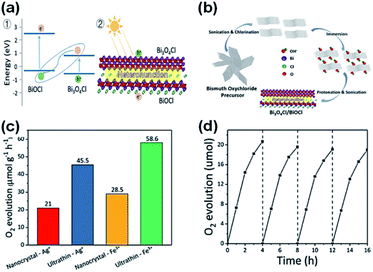 |
| | Fig. 24 (a) Band alignment for ultrathin Bi3O4Cl/BiOCl heterostructures (a type-II heterojunction) by theoretical calculation. (b) Synthetic route for the fabrication of the Bi3O4Cl/BiOCl heterostructures. (c) Demonstration of an ultrathin Bi3O4Cl/BiOCl with a Bi3O4Cl monolayer lying on top of a BiOCl monolayer. Photo-generated charge carriers separated into different layers. (c) O2 evolution rates of pure BiOCl, nanocrystal Bi3O4Cl/BiOCl, and ultrathin Bi3O4Cl/BiOCl. (d) O2 rate with respect to time. Reproduced with permission from ref. 210 Copyright 2019, Wiley-VCH. | |
5.3 CO2 production
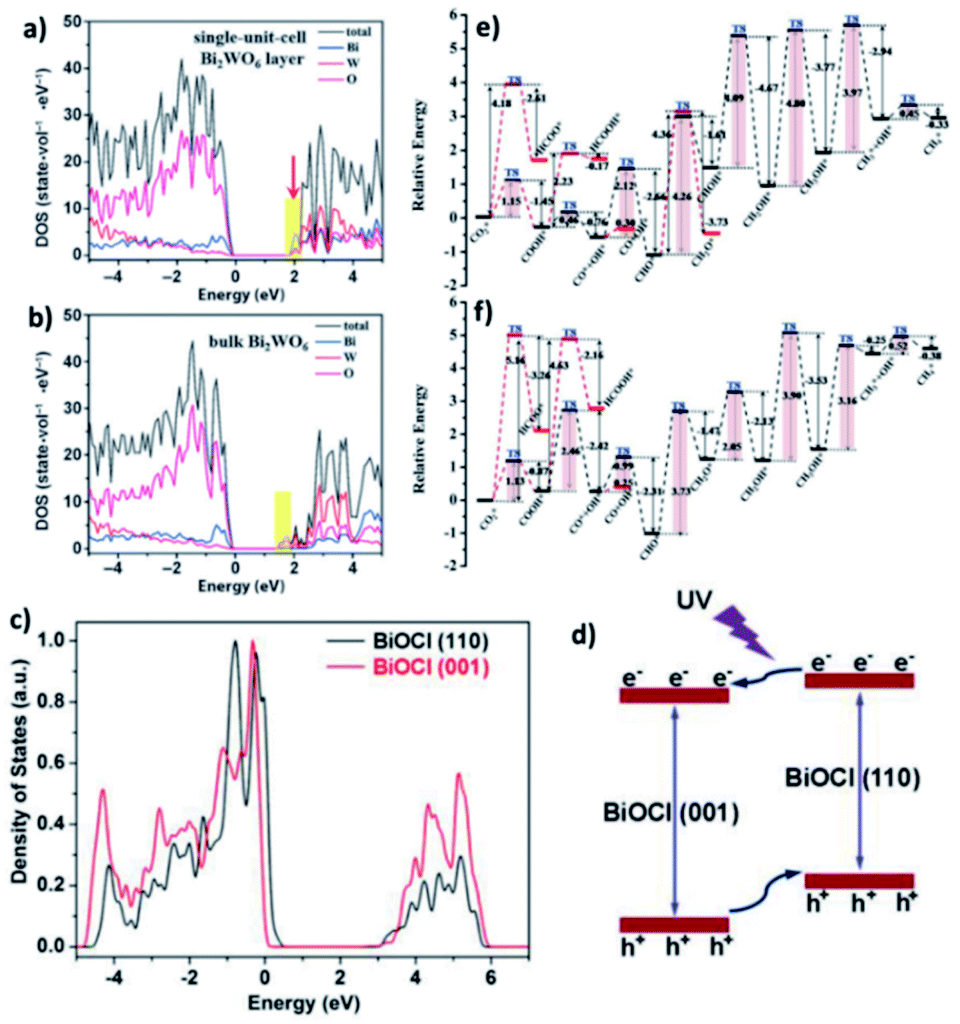
DFT calculations verified that for the band structure, oxygen vacancies introduce impurity levels among the valence and conduction band of BiOCl. The introduction of impurity levels is the intention for the absorption of visible and infrared light for BiOC with oxygen vacancies (BOC-OV).211 Moreover, DFT calculations evidenced that for the case of BiOV4, the vanadium vacancies introduce impurity levels similar to that of BOC-OV.5 Additionally, density of states (DOS) studies showed that the CO2 conversion performance of photocatalysts based upon Bi is merely based on the thickness of the catalyst. Similar to its bulk counterpart, the few-layered Bi2WO6 structure possess an amplified DOS at the CBM, as presented in Fig. 25a and b.212 The thickness of the catalyst also causes the DOS of BiOI to fluctuate, given that the CBM and VBM of bulk BiOI are lower than that of few-layered BiOI, which is quite higher in previous reports. Bismuth-rich Bi–O–X revealed a similar relation, given that the CBM of compounds based on Bi generally contains 6p orbitals for Bi. According to the DOS calculations, the order of the CBM is BixOyBr ≫ BiOBr, which reveals the greater capacity of BixOyBr for the photoreduction (using visible light) of CO2 to hydrocarbon fuel.104,212,213
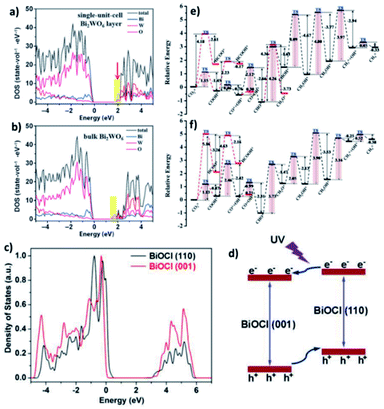 |
| | Fig. 25 (a) DFT calculations of Bi2WO6 single-unit-cell layer slab and (b) bulk Bi2WO6 slab. Reproduced with permission from ref. 213 Copyright 2015, Wiley-VCH. (c) PDOS diagrams from first-principles simulations for BiOCl (001) and (110) facets and (d) schematic showing the facet-dependent charge migration propensities in BiOCl. Reproduced with permission from ref. 87 Copyright 2015, Wiley-VCH. (e and f) Reaction pathways for CO2 reduction to CH4 on BOC-OV and BOC, respectively, where “*” represents adsorption on the substrate. The red route is the alternative pathway. Reproduced with permission from ref. 211 Copyright 2017, The Royal Society of Chemistry. | |
Additionally, the change in the performances for the photocatalytic reduction of CO2 with different planes was shown by DOS calculation. It exhibited that the (001) plane for BiOI has a more positive CBM capacity compared to the (100) plane of BiOI for the reduction of CO2.214 Further, the (100) plane of BiOIO3 exhibits a more negative VB compared to the (010) plane of BiOIO3, which displays a more positive CB. Hence, excitons are transferred to the (010) and (100) planes, respectively.215 The transfer of electrons among dissimilar planes of BiOCl can boost the charge migration, resulting in higher activity, as displayed in Fig. 25c and d.87 For a better understanding of the possible reaction paths of CO2 conversion, an investigation on the transient state of CO2 using a photocatalyst based on Bi was performed. As an example, the photocatalytic dissociation path of CO2 between various crystalline planes of Bi2WO6 was reported. Thermodynamically greater stability was achieved for CO2 adsorbed on the Bi2WO6 planes compared to that of non-adsorbed CO2. However, thermodynamic instability was observed for CO–O adsorption on the (001) plane. Meanwhile, the energy barrier of the Bi2WO6 (101) facets is higher compared to that of the Bi2WO6 (001) planes and spontaneous CO2 dissociation without photocatalysis. Therefore, the most thermodynamically favoured planes for photocatalytic CO2 reduction are the Bi2WO6 (001) planes.216 A higher energy barrier was observed for the direct dissociation of CO2 compared to CO2 hydrogenation dissociation. Additionally, the energy barrier of COOH is lower compared to HCOO with respect to the (001) planes on both BOC and BOC-OV. Hence, CO2 hydrogenation to COOH offers the best thermodynamically favored mechanism of CO2 reduction. Besides, the low energy barrier clarifies that CO is the main product of CO2 reduction on BOC and BOC-OV, as shown in Fig. 25e and f, respectively. Likewise, the hydrogenation of CH2OH* is the rate-determining step in the CH4*-generation path. Therefore, the formation of CH4 is not energetically favored. Furthermore, the rate-determining step of CO formation is the hydrogenation of CO2.211
5.4 N2 fixation
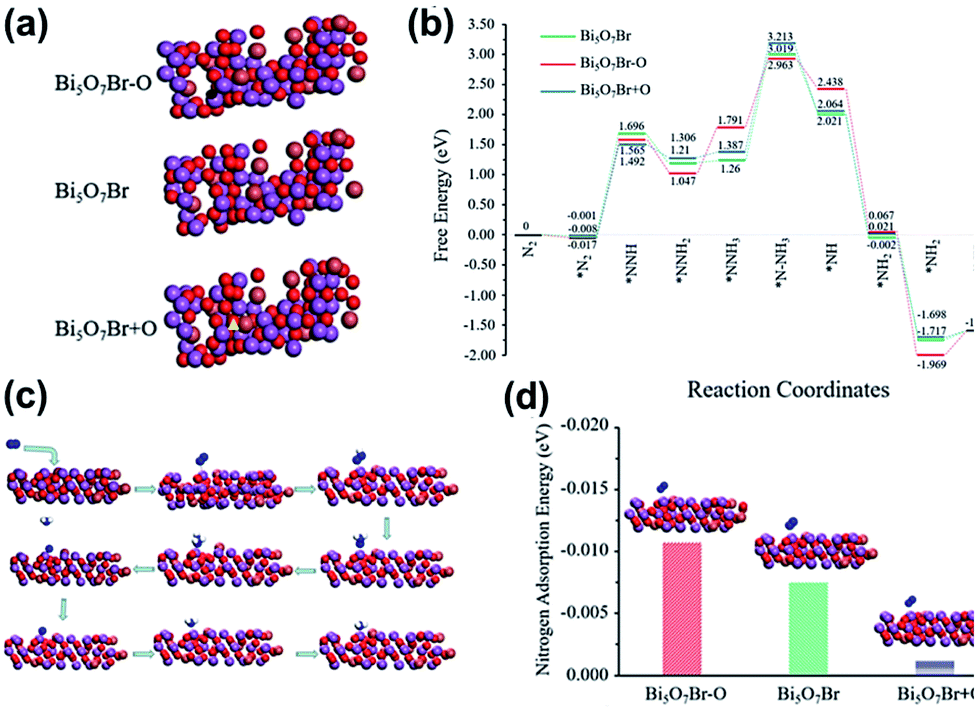
Ye et al.217 developed 5 nm-diameter nanotubes of Bi5O7Br via a self-assembly approach. In the Bi5O7Br nanotubes, oxygen vacancies could be generated to give plenty of localized electrons to activate N2 under the light irradiation. These nanotubes effectively reduced N2 to NH3 in water at a of rate up to ∼1.38 mmol g−1 h−1 under visible light. The apparent quantum efficiency at 420 nm exceeded 2.3%. Also, no obvious loss in activity was observed during four-fold cycle tests. Recently, Xiong et al.218 showed that the NH3 generation rates could be extended to 12.72 mmol g−1 h−1 under visible light (xenon bulb, 300 W) through Bi5O7Br nanostructures. This is highest ammonia generation rate recorded to date over a BiaObXc material (Fig. 26).
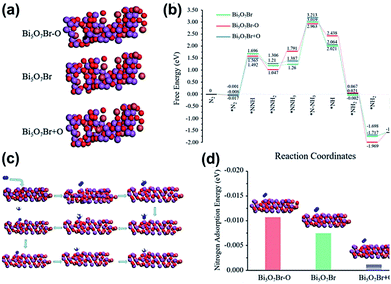 |
| | Fig. 26 (a) Structures of anoxic Bi5O7Br–O, pristine Bi5O7Br, and oxygen-enriched Bi5O7Br + O. Black circle and yellow triangle represent oxygen vacancy sites and additional oxygen sites, respectively. (b) Reaction energy diagram of N2 fixation catalyzed by Bi5O7Br–O, Bi5O7Br and Bi5O7Br + O. (c) N2 fixation process. (d) N2 adsorption energy on the surfaces of Bi5O7Br–O, Bi5O7Br and Bi5O7Br + O. Reprinted with permission from ref. 218 Copyright 2020, the American Chemical Society. | |
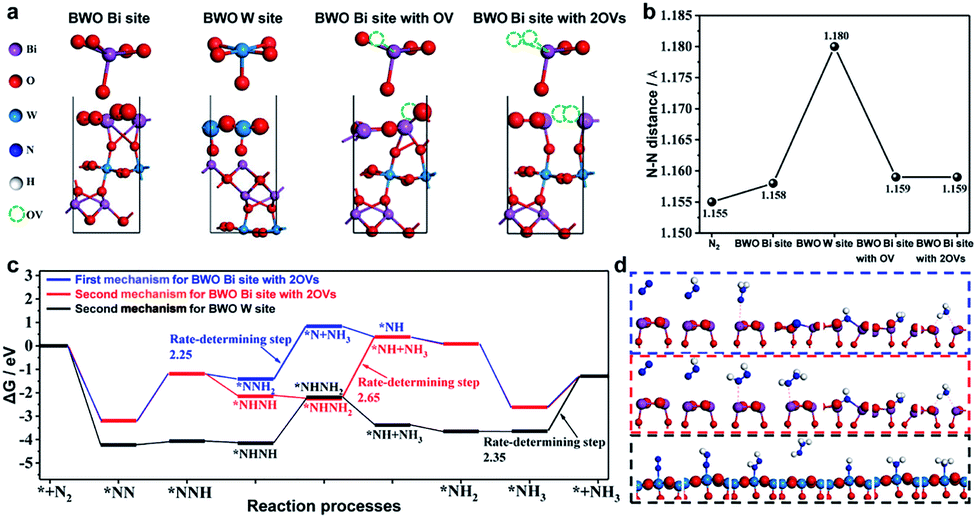
Recently, in 2021 Xue et al.100 used a solvothermal template-free approach to synthesize Bi2WO6 hollow microspheres with oxygen vacancies (OVs-BWO). Ethylene glycol has the ability to scavenge terminal oxygen atoms, resulting in the formation of oxygen vacancies in the form of W5+ and Bi(3−x)+ species with low oxidation states. The experimental results combined with DFT calculations showed that the optimal N2 fixation with an output of 80.5 m2 g−1 exhibited the ammonia yield of 106.4 μmol gcat−1 under simulated sunlight for 2 h, which was 18 times higher than equal-area nest-like Bi2WO6 without OVs. The enhanced activity is mainly attributed to the transfer of metastable electrons within the sub-band (i.e., defect energy level) induced by oxygen vacancies to the π-anti-bonding orbitals of N2via non-radiative transfer, leading to the activation of the nitrogen molecules. The activation and hydrogenation of nitrogen at the active sites of OVs-BWO surface were investigated by DFT calculation (Fig. 27). Subsequently, three possible nitrogen reduction reaction (NNR) processes on the active sites together with their corresponding rate-determining steps were proposed. The material is durable and stable, and there is almost no loss in activity after continuous repeated experiments in the nitrogen reduction process. Therefore, the OVs-BWO material provides an alternative for efficient N2 fixation under ambient conditions using atmosphere and water as the feedstock and sunlight as the driving force.
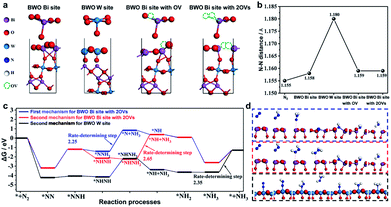 |
| | Fig. 27 (a) Models of the four possible active sites for Bi2WO6 (010) surface, W site, Bi site, Bi site with OV, and Bi site with 2OVs. (b) N–N distance of free N2 and N2 on the four possible active sites. (c) Gibbs free energy during the reaction processes via two different mechanisms (the first mechanism: * + N2 → *NN → *NNH → *NNH2 → *N + NH3 → *NH → *NH2 → *NH3 → * + NH3 and the second mechanism: * + N2 → *NN → *NNH → *NHNH → *NHNH2 → *NH + NH3 → *NH2 → *NH3 → * + NH3, where * represents the active site) and (d) the free-energy profile of three possible processes for BWO W site and BWO Bi site with 2OVs. Reprinted with permission from ref. 100 Copyright 2021, the American Chemical Society. | |
6. Conclusion, challenges, and future prospects
Encouraging photocatalysts for environmental and especially energy-associated applications correspond to a special class of materials that includes BiOX. The advantage of this class is its innovative structure comprised of a layered arrangement, tremendous physicochemical properties, and tunable electronic features. However, bare BiOX demonstrates some inherent drawbacks, including low light-harvesting efficiency, huge probability for the recombination of photogenerated excitons, lack of active sites for catalytic features, and lastly low specific surface area. Thus, great efforts have been dedicated to minimizing or eliminating these drawbacks, resulting in the optimization of BiOX materials for an enhancement in photocatalytic activity. The present review presented the recent developments in the design of new approaches for the preparation of BiOX materials. Additionally, we also discussed numerous schemes for the enhancement in photocatalytic activity, and finally the introduction of several BiOX-based nanocomposite frameworks with a minor focus on plasmonic-based photocatalysts. The tailoring of porous crystal structures and fabrication of ultrathin assemblies can enhance the photocatalytic activity of BiOX materials. Numerous routes have been utilized for the additional enhancement of photocatalytic activity, including elemental doping with rare earth metals, metals, and non-metals, and the generation of oxygen vacancies. BiOX-based materials are exceptional candidates for a wide range of applications involving photocatalysis. These applications include organic synthesis, oxygen and hydrogen evolution, disinfection, nitrogen fixation, pollutant removal, and reduction of CO2. Therefore, significant efforts have been devoted to developing BiOX-based materials and enhancing their photocatalytic activity; however, their, potential has not been completely exploited.
This review on 2D materials disclosed that the reduction of the thickness of these materials to the atomic level can result in intrinsic properties related to the bulk material and unique properties not visualized in the bulk material. The layered assembly of BiOX allows its facile transformation into a mono/few-layer assembly. However, additional consideration is necessary for the controlled fabrication of atomically thick BiOX. To optimize the photocatalysis process, the incorporation of defects in BiOX nanostructures has been studied. Generally, the type of defects observed in BiOX-based materials is oxygen vacancies. Various supplementary defects are not formed in nanostructures of BiOX such as dislocations, halogen vacancies, and dual vacancies. However, they may promote the transfer rate of electron and induce extra electronic states in BiOX. An enhancement in the density of states is typically aimed to enhance the photocatalytic activity of these materials. However, more effective approaches must be developed to construct different defects in BiOX and gain an in depth understanding of the relation between the type of defect and enhancement in photocatalytic activity.
Additionally, varying the content of Bi content in BiOX-based materials can be employed for the reduction of CO2 using a photocatalytic scheme. Presently, the main products from the photoreduction CO2 by BiOX are CH4 and CO. A manageable Bi-rich approach can practically tune the band edge positions and bandgap energy of BiOX-based materials. Thus, it is extremely necessary to achieve extraordinary selectivity for the photo-reduction products of CO2via the additional engineering of BiOX nanosheet materials. Despite the significant development in photocatalysts based on BiOX materials, the nature of the active catalytic sites present in these materials is still uncertain. Thus, studies on the mechanism of BiOX-based photocatalysts in numerous applications at the atomic level are desirable. Extra consideration should be dedicated to defining and calculating the active catalytic sites through the help of direct experimental measurements and calculation.
Supported co-catalysts can promote the redox reaction, especially the synergistic effect of two catalysts (deriving-electron-type (Ag, Pt, Au, MoS2, and Ni) and derived hole-type (PbO2, MnOx, IrOx, CoOx, and NiO)). It has been reported that BixOyXz photocatalysts have high space charge separation efficiency, which can accelerate the oxidation reaction and reduction reaction rate, and subsequently result in a higher total reaction rate. However, the synthesis and application of BixOyXz photocatalysts are still challenging. Also, it is very difficult to synthesize pure bismuth-rich bismuth oxyhalides. Currently, two synthetic methods, i.e., hydrothermal alkalization and calcination, are usually applied for the preparation of bismuth-rich oxyhalide. However, a slight change in pH or temperature can lead to differences in the physicochemical properties of the BixOyXz photocatalyst. Moreover, only Bi4O5X2 (X = Br and I) was obtained by the molecular precursor method. Therefore, it is urgent to explore new synthetic routes for the application of BixOyXz photocatalysts. Currently, photocatalysts are mainly applied in environmental remediation and solar power generation, but Bi4O5X2 can be used in other applications such as the photocatalytic inactivation of bacteria and photocatalytic organic synthesis, which will be of great importance in the future. Meanwhile, many advanced characteristic technologies have been developed to investigate the photocatalytic mechanism of BixOyXz, such as in situ infrared spectroscopy and synchrotron radiation technology, which have greatly promoted the research on BixOyXz photocatalysts.
BiOX-based materials can be further explored as nanocomposite catalysts, such as supported IB metal particles, for photocatalytic conversions to yield high-value chemicals. Notably, IB metal particles exhibit good surface plasmon resonance properties and can emerge as excellent electron acceptors for the adsorption and activation of oxygen, thus remarkably promoting photocatalytic conversions. However, more in situ characterization routes and measurements that are close to realistic situations are desirable to improve the atomic level understanding of the relationship between active catalytic sites and photocatalysis, which will be advantageous to enhance the tailored design of BiOX materials for photocatalytic applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the financial support by the Funds of National Natural Science Foundation of China (21701168 and 21802008), Liaoning Natural Science Foundation (2020-MS-024), and Liaoning Revitalization Talents Program (XLYC1807121).
Notes and references
- A. A. Ismail and D. W. Bahnemann, Sol. Energy Mater. Sol. Cells, 2014, 128, 85–101 CrossRef CAS
 .
.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS .
- T. Hisatomi and K. Domen, Nat. Catal., 2019, 2, 387–399 CrossRef CAS .
- P. Ganguly, M. Harb, Z. Cao, L. Cavallo, A. Breen, S. Dervin, D. D. Dionysiou and S. C. Pillai, ACS Energy Lett., 2019, 4, 1687–1709 CrossRef CAS .
- S. Gao, B. Gu, X. Jiao, Y. Sun, X. Zu, F. Yang, W. Zhu, C. Wang, Z. Feng, B. Ye and Y. Xie, J. Am. Chem. Soc., 2017, 139, 3438–3445 CrossRef CAS PubMed .
- L. Liang, F. Lei, S. Gao, Y. Sun, X. Jiao, J. Wu, S. Qamar and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 13971–13974 CrossRef CAS PubMed .
- J. Xiong, P. Song, J. Di and H. Li, J. Mater. Chem. A, 2020, 8, 21434–21454 RSC .
- D. S. Bhachu, S. J. A. Moniz, S. Sathasivam, D. O. Scanlon, A. Walsh, S. M. Bawaked, M. Mokhtar, A. Y. Obaid, I. P. Parkin, J. Tang and C. J. Carmalt, Chem. Sci., 2016, 7, 4832–4841 RSC .
- M. Ikram, A. Raza, M. Imran, A. Ul-Hamid, A. Shahbaz and S. Ali, Nanoscale Res. Lett., 2020, 15, 95 CrossRef PubMed .
- S. Cao, L. Piao and X. Chen, Trends Chem., 2020, 2, 57–70 CrossRef CAS .
- Z. Wei, J. Liu and W. Shangguan, Chin. J. Catal., 2020, 41, 1440–1450 CrossRef CAS .
- P. Chiesa, S. Consonni, T. Kreutz and W. Robert, Int. J. Hydrogen Energy, 2005, 30, 747–767 CrossRef CAS .
- N. Fajrina and M. Tahir, Int. J. Hydrogen Energy, 2019, 44, 540–577 CrossRef CAS .
- U. Gupta and C. N. R. Rao, Nano Energy, 2017, 41, 49–65 CrossRef CAS .
- J. Di, J. Xia, H. Li, S. Guo and S. Dai, Nano Energy, 2017, 41, 172–192 CrossRef CAS .
- J. Di, J. Xia, M. Ji, L. Xu, S. Yin, Q. Zhang, Z. Chen and H. Li, Carbon, 2016, 98, 613–623 CrossRef CAS .
- Y. Zhou, W. Wang, C. Zhang, D. Huang, C. Lai, M. Cheng, L. Qin, Y. Yang, C. Zhou, B. Li, H. Luo and D. He, Adv. Colloid Interface Sci., 2020, 279, 102144 CrossRef CAS PubMed .
- A. Raza, M. Ikram, M. Aqeel, M. Imran, A. Ul-Hamid, K. N. Riaz and S. Ali, Appl. Nanosci., 2020, 10, 1535–1544 CrossRef CAS .
- B. Weng, F. Xu and J. Xu, J. Nanopart. Res., 2014, 16, 2766 CrossRef .
- K. Sharma, V. Dutta, S. Sharma, P. Raizada, A. Hosseini-Bandegharaei, P. Thakur and P. Singh, J. Ind. Eng. Chem., 2019, 78, 1–20 CrossRef CAS .
- X. Zhang, Z. Ai, F. Jia and L. Zhang, J. Phys. Chem. C, 2008, 112, 747–753 CrossRef CAS .
- X. Jin, L. Ye, H. Xie and G. Chen, Coord. Chem. Rev., 2017, 349, 84–101 CrossRef CAS .
- G.-J. Lee, Y.-C. Zheng and J. J. Wu, Catal. Today, 2018, 307, 197–204 CrossRef CAS .
- X. Wang, C. Zhou, L. Yin, R. Zhang and G. Liu, ACS Sustainable Chem. Eng., 2019, 7, 7900–7907 CrossRef CAS .
- J. Li, H. Li, G. Zhan and L. Zhang, Acc. Chem. Res., 2017, 50, 112–121 CrossRef CAS PubMed .
- Y. Wang, Y. Long and D. Zhang, ACS Sustainable Chem. Eng., 2017, 5, 2454–2462 CrossRef CAS .
- D. Kato, K. Hongo, R. Maezono, M. Higashi, H. Kunioku, M. Yabuuchi, H. Suzuki, H. Okajima, C. Zhong, K. Nakano, R. Abe and H. Kageyama, J. Am. Chem. Soc., 2017, 139, 18725–18731 CrossRef CAS PubMed .
- A. M. Ganose, M. Cuff, K. T. Butler, A. Walsh and D. O. Scanlon, Chem. Mater., 2016, 28, 1980–1984 CrossRef CAS PubMed .
- D. Kandi, S. Martha, A. Thirumurugan and K. M. Parida, J. Phys. Chem. C, 2017, 121, 4834–4849 CrossRef CAS .
- R. Dong, Y. Hu, Y. Wu, W. Gao, B. Ren, Q. Wang and Y. Cai, J. Am. Chem. Soc., 2017, 139, 1722–1725 CrossRef CAS PubMed .
- C. Lai, M. Zhang, B. Li, D. Huang, G. Zeng, L. Qin, X. Liu, H. Yi, M. Cheng, L. Li, Z. Chen and L. Chen, Chem. Eng. J., 2019, 358, 891–902 CrossRef CAS .
- Z. Qin, D. Zhao, L. Zhao, Q. Xiao, T. Wu, J. Zhang, C. Wan and G. Li, Nanoscale Adv., 2019, 1, 2529–2536 RSC .
- Q. Shi, G. Ping, X. Wang, H. Xu, J. Li, J. Cui, H. Abroshan, H. Ding and G. Li, J. Mater. Chem. A, 2019, 7, 2253–2260 RSC .
- S. Zhang, X. Gong, Q. Shi, G. Ping, H. Xu, A. Waleed and G. Li, ACS Omega, 2020, 5, 15942–15948 CrossRef CAS PubMed .
- J. Hu, W. Fan, W. Ye, C. Huang and X. Qiu, Appl. Catal., B, 2014, 158–159, 182–189 CrossRef CAS .
- A. Sudhaik, P. Raizada, P. Shandilya, D.-Y. Jeong, J.-H. Lim and P. Singh, J. Ind. Eng. Chem., 2018, 67, 28–51 CrossRef CAS .
- S. O. Ganiyu, C. A. Martínez-Huitle and M. A. Rodrigo, Appl. Catal., B, 2020, 270, 118857 CrossRef CAS .
- J. Li, H. Li, G. Zhan and L. Zhang, Acc. Chem. Res., 2017, 50, 112–121 CrossRef CAS PubMed .
- B. Li, S. Liu, C. Lai, G. Zeng, M. Zhang, M. Zhou, D. Huang, L. Qin, X. Liu, Z. Li, N. An, F. Xu, H. Yi, Y. Zhang and L. Chen, Appl. Catal., B, 2020, 266, 118650 CrossRef .
- L. Ye, X. Jin, C. Liu, C. Ding, H. Xie, K. H. Chu and P. K. Wong, Appl. Catal., B, 2016, 187, 281–290 CrossRef CAS .
- J. Jiang, K. Zhao, X. Xiao and L. Zhang, J. Am. Chem. Soc., 2012, 134, 4473–4476 CrossRef CAS PubMed .
- Z. Fan, Y. Zhao, W. Zhai, L. Qiu, H. Li and M. R. Hoffmann, RSC Adv., 2016, 6, 2028–2031 RSC .
- L. Wang, J. Shang, W. Hao, S. Jiang, S. Huang, T. Wang, Z. Sun, Y. Du, S. Dou, T. Xie, D. Wang and J. Wang, Sci. Rep., 2014, 4, 7384 CrossRef PubMed .
- G. Zhang, L. Zhang, Y. Liu, L. Liu, C.-P. Huang, H. Liu and J. Li, ACS Appl. Mater. Interfaces, 2016, 8, 26783–26793 CrossRef CAS PubMed .
- D. Mao, S. Ding, L. Meng, Y. Dai, C. Sun, S. Yang and H. He, Appl. Catal., B, 2017, 207, 153–165 CrossRef CAS .
- Y. Huang, H. Li, W. Fan, F. Zhao, W. Qiu, H. Ji and Y. Tong, ACS Appl. Mater. Interfaces, 2016, 8, 27859–27867 CrossRef CAS PubMed .
- Q. Mu, Q. Zhang, H. Wang and Y. Li, J. Mater. Chem., 2012, 22, 16851–16857 RSC .
- Y. Wu, B. Yuan, M. Li, W.-H. Zhang, Y. Liu and C. Li, Chem. Sci., 2015, 6, 1873–1878 RSC .
- Y. Shi, X. Xiong, S. Ding, X. Liu, Q. Jiang and J. Hu, Appl. Catal., B, 2018, 220, 570–580 CrossRef CAS .
- H. Li, J. Shang, Z. Ai and L. Zhang, J. Am. Chem. Soc., 2015, 137, 6393–6399 CrossRef CAS PubMed .
- H. Cheng, B. Huang, P. Wang, Z. Wang, Z. Lou, J. Wang, X. Qin, X. Zhang and Y. Dai, Chem. Commun., 2011, 47, 7054–7056 RSC .
- X. Zhang, X. Liu, C. Fan, Y. Wang, Y. Wang and Z. Liang, Appl. Catal., B, 2013, 132–133, 332–341 CrossRef CAS .
- G. He, C. Xing, X. Xiao, R. Hu, X. Zuo and J. Nan, Appl. Catal., B, 2015, 170–171, 1–9 CAS .
- A. Waheed, Q. Shi, N. Maeda, D. M. Meier, Z. Qin, G. Li and A. Baiker, Catalysts, 2020, 10, 933 CrossRef CAS .
- L. Armelao, G. Bottaro, C. Maccato and E. Tondello, Dalton Trans., 2012, 41, 5480–5485 RSC .
- Z. Song, X. Dong, N. Wang, L. Zhu, Z. Luo, J. Fang and C. Xiong, Chem. Eng. J., 2017, 317, 925–934 CrossRef CAS .
- Z. Zhuang, Q. Peng and Y. Li, Chem. Soc. Rev., 2011, 40, 5492–5513 RSC .
- M. Ikram, R. Tabassum, U. Qumar, S. Ali, A. Ul-Hamid, A. Haider, A. Raza, M. Imran and S. Ali, RSC Adv., 2020, 10, 20559–20571 RSC .
- X. Gao, W. Peng, G. Tang, Q. Guo and Y. Luo, J. Alloys Compd., 2018, 757, 455–465 CrossRef CAS .
- W. Liu, Y. Shang, A. Zhu, P. Tan, Y. Liu, L. Qiao, D. Chu, X. Xiong and J. Pan, J. Mater. Chem. A, 2017, 5, 12542–12549 RSC .
- C. Ding, L. Ye, Q. Zhao, Z. Zhong, K. Liu, H. Xie, K. Bao, X. Zhang and Z. Huang, J. CO2 Util., 2016, 14, 135–142 CrossRef CAS .
- P. Cui, J. Wang, Z. Wang, J. Chen, X. Xing, L. Wang and R. Yu, Nano Res., 2016, 9, 593–601 CrossRef CAS .
- X. Yan, H. Zhao, T. Li, W. Zhang, Q. Liu, Y. Yuan, L. Huang, L. Yao, J. Yao, H. Su, Y. Su, J. Gu and D. Zhang, Nanoscale, 2019, 11, 10203–10208 RSC .
- C. Yu, W. Zhou, J. Yu, F. Cao and X. Li, Chin. J. Chem., 2012, 30, 721–726 CrossRef CAS .
- Y. Bai, T. Chen, P. Wang, L. Wang and L. Ye, Chem. Eng. J., 2016, 304, 454–460 CrossRef CAS .
- Y. Bai, L. Ye, T. Chen, P. Wang, L. Wang, X. Shi and P. K. Wong, Appl. Catal., B, 2017, 203, 633–640 CrossRef CAS .
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed .
- C. Huang, Y. Wen, J. Ma, D. Dong, Y. Shen, S. Liu, H. Ma and Y. Zhang, Nat. Commun., 2021, 12, 320 CrossRef CAS PubMed .
- G. Zhou, Y. Shan, Y. Hu, X. Xu, L. Long, J. Zhang, J. Dai, J. Guo, J. Shen, S. Li, L. Liu and X. Wu, Nat. Commun., 2018, 9, 3366 CrossRef PubMed .
- J. Kosco, M. Bidwell, H. Cha, T. Martin, C. T. Howells, M. Sachs, D. H. Anjum, S. Gonzalez Lopez, L. Zou, A. Wadsworth, W. Zhang, L. Zhang, J. Tellam, R. Sougrat, F. Laquai, D. M. DeLongchamp, J. R. Durrant and I. McCulloch, Nat. Mater., 2020, 19, 559–565 CrossRef CAS PubMed .
- M. Ikram, E. Umar, A. Raza, A. Haider, S. Naz, A. Ul-Hamid, J. Haider, I. Shahzadi, J. Hassan and S. Ali, RSC Adv., 2020, 10, 24215–24233 RSC .
- A. Raza, U. Qumar, J. Hassan, M. Ikram, A. Ul-Hamid, J. Haider, M. Imran and S. Ali, Appl. Nanosci., 2020, 10, 3875–3899 CrossRef CAS .
-
F. Deng, J.-P. Zou, L.-N. Zhao, G. Zhou, X.-B. Luo and S.-L. Luo, in Nanomaterials for the Removal of Pollutants and Resource Reutilization, ed. X. Luo and F. Deng, Elsevier, 2019, pp. 59–82, DOI:10.1016/B978-0-12-814837-2.00003-2 .
- M. Z. Rahman and C. B. Mullins, Acc. Chem. Res., 2019, 52, 248–257 CrossRef CAS PubMed .
- M. Z. Rahman, M. G. Kibria and C. B. Mullins, Chem. Soc. Rev., 2020, 49, 1887–1931 RSC .
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed .
- D. Ravelli, D. Dondi, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2009, 38, 1999–2011 RSC .
- B. Ohtani, J. Photochem. Photobiol., C, 2010, 11, 157–178 CrossRef CAS .
- M. Ikram, J. Hassan, M. Imran, J. Haider, A. Ul-Hamid, I. Shahzadi, M. Ikram, A. Raza, U. Qumar and S. Ali, Appl. Nanosci., 2020, 10, 3525–3528 CrossRef CAS .
- M. Ikram, J. Hassan, A. Raza, A. Haider, S. Naz, A. Ul-Hamid, J. Haider, I. Shahzadi, U. Qamar and S. Ali, RSC Adv., 2020, 10, 30007–30024 RSC .
- T. Hisatomi, K. Takanabe and K. Domen, Catal. Lett., 2015, 145, 95–108 CrossRef CAS .
- Q. Usman, H. Jahanzeb, N. Sadia, H. Ali, R. Ali, U.-H. Anwar, H. Junaid, S. Iram, A. Iqbal and I. Muhammad, Nanotechnology, 2021, 32, 255704 CrossRef PubMed .
- J. E. Kroeze, T. J. Savenije and J. M. Warman, J. Am. Chem. Soc., 2004, 126, 7608–7618 CrossRef CAS PubMed .
- J. Bisquert, Phys. Chem. Chem. Phys., 2008, 10, 3175–3194 RSC .
- M. Shen and M. A. Henderson, J. Phys. Chem. Lett., 2011, 2, 2707–2710 CrossRef CAS .
- J. Li, L. Cai, J. Shang, Y. Yu and L. Zhang, Adv. Mater., 2016, 28, 4059–4064 CrossRef CAS PubMed .
- S. Bai, X. Li, Q. Kong, R. Long, C. Wang, J. Jiang and Y. Xiong, Adv. Mater., 2015, 27, 3444–3452 CrossRef CAS PubMed .
- W. Wu, Z. Zhang, J. Di and W. Zhao, ChemNanoMat, 2019, 5, 215–223 CrossRef CAS .
- S. Ning, X. Shi, H. Zhang, H. Lin, Z. Zhang, J. Long, Y. Li and X. Wang, Sol. RRL, 2019, 3, 1900059 CrossRef .
- M. Ji, R. Chen, J. Di, Y. Liu, K. Li, Z. Chen, J. Xia and H. Li, J. Colloid Interface Sci., 2019, 533, 612–620 CrossRef CAS PubMed .
- X. Xiong, T. Zhou, X. Liu, S. Ding and J. Hu, J. Mater. Chem. A, 2017, 5, 15706–15713 RSC .
- N. Du, H. B. Park, G. P. Robertson, M. M. Dal-Cin, T. Visser, L. Scoles and M. D. Guiver, Nat. Mater., 2011, 10, 372–375 CrossRef CAS PubMed .
- B. Kumar, M. Asadi, D. Pisasale, S. Sinha-Ray, B. A. Rosen, R. Haasch, J. Abiade, A. L. Yarin and A. Salehi-Khojin, Nat. Commun., 2013, 4, 2819 CrossRef .
- H. Zhou, J. Guo, P. Li, T. Fan, D. Zhang and J. Ye, Sci. Rep., 2013, 3, 1667 CrossRef PubMed .
- L. Wang, S.-q. Guo, Y. Chen, M. Pan, E. H. Ang and Z.-h. Yuan, ChemPhotoChem, 2020, 4, 110–119 CrossRef CAS .
- J. Di, J. Xia, M. Ji, B. Wang, X. Li, Q. Zhang, Z. Chen and H. Li, ACS Sustainable Chem. Eng., 2016, 4, 136–146 CrossRef CAS .
- J. Liu, R. Li, X. Zu, X. Zhang, Y. Wang, Y. Wang and C. Fan, Chem. Eng. J., 2019, 371, 796–803 CrossRef CAS .
- Y. Bai, L. Ye, T. Chen, L. Wang, X. Shi, X. Zhang and D. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 27661–27668 CrossRef CAS PubMed .
- J. Di, J. Xia, M. F. Chisholm, J. Zhong, C. Chen, X. Cao, F. Dong, Z. Chi, H. Chen, Y.-X. Weng, J. Xiong, S.-Z. Yang, H. Li, Z. Liu and S. Dai, Adv. Mater., 2019, 31, 1807576 CrossRef PubMed .
- T. Wang, C. Feng, J. Liu, D. Wang, H. Hu, J. Hu, Z. Chen and G. Xue, Chem. Eng. J., 2021, 414, 128827 CrossRef CAS .
- P. Qiu, J. Wang, Z. Liang, Y. Xue, Y. Zhou, X. Zhang, H. Cui, G. Cheng and J. Tian, Chin. Chem. Lett., 2021 DOI:10.1016/j.cclet.2021.03.077 .
- J. Wang, Y. Yu and L. Zhang, Appl. Catal., B, 2013, 136–137, 112–121 CrossRef CAS .
- X. Xiao, C. Xing, G. He, X. Zuo, J. Nan and L. Wang, Appl. Catal., B, 2014, 148–149, 154–163 CrossRef CAS .
- Y. Su, C. Ding, Y. Dang, H. Wang, L. Ye, X. Jin, H. Xie and C. Liu, Appl. Surf. Sci., 2015, 346, 311–316 CrossRef CAS .
- S. Sun, W. Wang, L. Zhang, L. Zhou, W. Yin and M. Shang, Environ. Sci. Technol., 2009, 43, 2005–2010 CrossRef CAS PubMed .
- J. Yang, L. Xu, C. Liu and T. Xie, Appl. Surf. Sci., 2014, 319, 265–271 CrossRef CAS .
- J. Xia, Y. Ge, J. Di, L. Xu, S. Yin, Z. Chen, P. Liu and H. Li, J. Colloid Interface Sci., 2016, 473, 112–119 CrossRef CAS PubMed .
- W. Zhang, X. Liu, X. a. Dong, F. Dong and Y. Zhang, Chin. J. Catal., 2017, 38, 2030–2038 CrossRef CAS .
- M. Ikram, M. I. Khan, A. Raza, M. Imran, A. Ul-Hamid and S. Ali, Phys. E, 2020, 124, 114246 CrossRef CAS .
- M. Ji, J. Di, Y. Liu, R. Chen, K. Li, Z. Chen, J. Xia and H. Li, Appl. Catal., B, 2020, 268, 118403 CrossRef CAS .
- X. Xiao, R. Hao, X. Zuo, J. Nan, L. Li and W. Zhang, Chem. Eng. J., 2012, 209, 293–300 CrossRef CAS .
- W. Zhang, X. a. Dong, B. Jia, J. Zhong, Y. Sun and F. Dong, Appl. Surf. Sci., 2018, 430, 571–577 CrossRef CAS .
- L. Ding, H. Chen, Q. Wang, T. Zhou, Q. Jiang, Y. Yuan, J. Li and J. Hu, Chem. Commun., 2016, 52, 994–997 RSC .
- R. Yuan, S. Fan, H. Zhou, Z. Ding, S. Lin, Z. Li, Z. Zhang, C. Xu, L. Wu, X. Wang and X. Fu, Angew. Chem., Int. Ed., 2013, 52, 1035–1039 CrossRef CAS PubMed .
- H. Wang, S. Chen, D. Yong, X. Zhang, S. Li, W. Shao, X. Sun, B. Pan and Y. Xie, J. Am. Chem. Soc., 2017, 139, 4737–4742 CrossRef CAS PubMed .
- C. Huang, J. Hu, S. Cong, Z. Zhao and X. Qiu, Appl. Catal., B, 2015, 174–175, 105–112 CrossRef CAS .
- J. Li, Y. Yu and L. Zhang, Nanoscale, 2014, 6, 8473–8488 RSC .
- G. Jiang, R. Wang, X. Wang, X. Xi, R. Hu, Y. Zhou, S. Wang, T. Wang and W. Chen, ACS Appl. Mater. Interfaces, 2012, 4, 4440–4444 CrossRef CAS PubMed .
- J. Xia, L. Xu, J. Zhang, S. Yin, H. Li, H. Xu and J. Di, CrystEngComm, 2013, 15, 10132–10141 RSC .
- G. Jiang, X. Wang, Z. Wei, X. Li, X. Xi, R. Hu, B. Tang, R. Wang, S. Wang, T. Wang and W. Chen, J. Mater. Chem. A, 2013, 1, 2406–2410 RSC .
- M. Z. Shahid, R. Mehmood, M. Athar, J. Hussain, Y. Wei and A. Khaliq, ACS Appl. Nano Mater., 2021, 4, 746–758 CrossRef CAS .
- M. Gao, D. Zhang, X. Pu, H. Li, W. Li, X. Shao, D. Lv, B. Zhang and J. Dou, Sep. Purif. Technol., 2016, 162, 114–119 CrossRef CAS .
- J. Di, J. Xia, S. Yin, H. Xu, L. Xu, Y. Xu, M. He and H. Li, RSC Adv., 2014, 4, 14281–14290 RSC .
- W. T. Li, W. Z. Huang, H. Zhou, H. Y. Yin, Y. F. Zheng and X. C. Song, J. Alloys Compd., 2015, 638, 148–154 CrossRef CAS .
- J. Zhou, J. Zhou, Z. Hu and L. Wang, Mater. Sci. Semicond. Process., 2019, 90, 112–119 CrossRef CAS .
- Z. Liu, B. Wu, Y. Zhao, J. Niu and Y. Zhu, Ceram. Int., 2014, 40, 5597–5603 CrossRef CAS .
- X. Tu, S. Qian, L. Chen and L. Qu, J. Mater. Sci., 2015, 50, 4312–4323 CrossRef CAS .
- B. Pare, B. Sarwan and S. B. Jonnalagadda, Appl. Surf. Sci., 2011, 258, 247–253 CrossRef CAS .
- W. Wang, R. Dai, L. Zhang, Q. Wu, X. Wang, S. Zhang, T. Shao, F. Zhang, J. Yan and W. Zhang, J. Mater. Sci., 2020, 55, 11226–11240 CrossRef CAS .
- W. Yang, Y. Wen, R. Chen, D. Zeng and B. Shan, Phys. Chem. Chem. Phys., 2014, 16, 21349–21355 RSC .
- J. Xia, M. Ji, W. Li, J. Di, H. Xu, M. He, Q. Zhang and H. Li, Colloids Surf., A, 2016, 489, 343–350 CrossRef CAS .
- W. Zhao, C. Li, A. Wang, C. Lv, W. Zhu, S. Dou, Q. Wang and Q. Zhong, Phys. Chem. Chem. Phys., 2017, 19, 28696–28709 RSC .
- A. Dash, S. Sarkar, V. N. K. B. Adusumalli and V. Mahalingam, Langmuir, 2014, 30, 1401–1409 CrossRef CAS PubMed .
- M. Arumugam and M. Y. Choi, J. Ind. Eng. Chem., 2020, 81, 237–268 CrossRef CAS .
- X. Chen, L. Liu and F. Huang, Chem. Soc. Rev., 2015, 44, 1861–1885 RSC .
- G. Liu, C. Sun, L. Cheng, Y. Jin, H. Lu, L. Wang, S. C. Smith, G. Q. Lu and H.-M. Cheng, J. Phys. Chem. C, 2009, 113, 12317–12324 CrossRef CAS .
- J. Li, K. Zhao, Y. Yu and L. Zhang, Adv. Funct. Mater., 2015, 25, 2189–2201 CrossRef CAS .
- X. Zhang and L. Zhang, J. Phys. Chem. C, 2010, 114, 18198–18206 CrossRef CAS .
- L. Zeng, F. Zhe, Y. Wang, Q. Zhang, X. Zhao, X. Hu, Y. Wu and Y. He, J. Colloid Interface Sci., 2019, 539, 563–574 CrossRef CAS PubMed .
- H. Li, J. Shang, H. Zhu, Z. Yang, Z. Ai and L. Zhang, ACS Catal., 2016, 6, 8276–8285 CrossRef CAS .
- H. Li, J. Li, Z. Ai, F. Jia and L. Zhang, Angew. Chem., Int. Ed., 2018, 57, 122–138 CrossRef CAS PubMed .
- M. Guan, C. Xiao, J. Zhang, S. Fan, R. An, Q. Cheng, J. Xie, M. Zhou, B. Ye and Y. Xie, J. Am. Chem. Soc., 2013, 135, 10411–10417 CrossRef CAS PubMed .
- D. Cui, L. Wang, K. Xu, L. Ren, L. Wang, Y. Yu, Y. Du and W. Hao, J. Mater. Chem. A, 2018, 6, 2193–2199 RSC .
- B. Gao, J.-R. Zhang, L. Chen, J. Guo, S. Shen, C.-T. Au, S.-F. Yin and M.-Q. Cai, Appl. Surf. Sci., 2019, 492, 157–165 CrossRef CAS .
- J. Cao, B. Xu, H. Lin, B. Luo and S. Chen, Chem. Eng. J., 2012, 185–186, 91–99 CrossRef CAS .
- X. Jia, J. Cao, H. Lin, M. Zhang, X. Guo and S. Chen, Appl. Catal., B, 2017, 204, 505–514 CrossRef CAS .
- Y. Wang, Y. Long, Z. Yang and D. Zhang, J. Hazard. Mater., 2018, 351, 11–19 CrossRef CAS PubMed .
- M. Sun, Q. Wei, Y. Shao, B. Du, T. Yan, L. Yan and D. Li, Appl. Catal., B, 2018, 233, 250–259 CrossRef CAS .
- Y. Xu, Y. You, H. Huang, Y. Guo and Y. Zhang, J. Hazard. Mater., 2020, 381, 121159 CrossRef CAS PubMed .
- X. Yan, Z. Wu, P. Yang, Y. Mao, B. Liu, Y. Zhao, H. Shen and D. Wang, Catal. Commun., 2019, 123, 91–95 CrossRef CAS .
- H. Peng, L. Wang, J. Xu, S. Jiang, X. Xu and Q. Zhang, Mater. Lett., 2019, 256, 126694 CrossRef CAS .
- J. Xu, Y.-G. Mao, T. Liu and Y. Peng, CrystEngComm, 2018, 20, 2292–2298 RSC .
- D. Hou, F. Tang, B. Ma, M. Deng, X.-q. Qiao, Y.-L. Liu and D.-S. Li, CrystEngComm, 2019, 21, 4158–4168 RSC .
- Y. Peng, P.-P. Yu, Q.-G. Chen, H.-Y. Zhou and A.-W. Xu, J. Phys. Chem. C, 2015, 119, 13032–13040 CrossRef CAS .
- J. Jiang, X. Zhang, P. Sun and L. Zhang, J. Phys. Chem. C, 2011, 115, 20555–20564 CrossRef CAS .
- X. Zhang, L. Zhang, T. Xie and D. Wang, J. Phys. Chem. C, 2009, 113, 7371–7378 CrossRef CAS .
- Q. Xiang, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2012, 41, 782–796 RSC .
- W. Tu, Y. Zhou, Q. Liu, S. Yan, S. Bao, X. Wang, M. Xiao and Z. Zou, Adv. Funct. Mater., 2013, 23, 1743–1749 CrossRef CAS .
- F. Gao, D. Zeng, Q. Huang, S. Tian and C. Xie, Phys. Chem. Chem. Phys., 2012, 14, 10572–10578 RSC .
- H. Liu, Y. Su, Z. Chen, Z. Jin and Y. Wang, J. Hazard. Mater., 2014, 266, 75–83 CrossRef CAS PubMed .
- H. Feng, Z. Xu, L. Wang, Y. Yu, D. Mitchell, D. Cui, X. Xu, J. Shi, T. Sannomiya, Y. Du, W. Hao and S. X. Dou, ACS Appl. Mater. Interfaces, 2015, 7, 27592–27596 CrossRef CAS PubMed .
- J. Feng, X. Qian, C.-W. Huang and J. Li, Nat. Photonics, 2012, 6, 866–872 CrossRef CAS .
- L. Wang, L. Wang, Y. Du, X. Xu and S. X. Dou, Mater. Today Phys., 2021, 16, 100294 CrossRef .
- D. Shiri, Y. Kong, A. Buin and M. P. Anantram, Appl. Phys. Lett., 2008, 93, 073114 CrossRef .
- R. Li, F. Zhang, D. Wang, J. Yang, M. Li, J. Zhu, X. Zhou, H. Han and C. Li, Nat. Commun., 2013, 4, 1432 CrossRef PubMed .
- L. Ye, X. Liu, Q. Zhao, H. Xie and L. Zan, J. Mater. Chem. A, 2013, 1, 8978–8983 RSC .
- J. Shang, W. Hao, X. Lv, T. Wang, X. Wang, Y. Du, S. Dou, T. Xie, D. Wang and J. Wang, ACS Catal., 2014, 4, 954–961 CrossRef CAS .
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed .
- Y. Liu, W.-J. Son, J. Lu, B. Huang, Y. Dai and M.-H. Whangbo, Chem.–Eur. J., 2011, 17, 9342–9349 CrossRef CAS PubMed .
- K. Ren, J. Liu, J. Liang, K. Zhang, X. Zheng, H. Luo, Y. Huang, P. Liu and X. Yu, Dalton Trans., 2013, 42, 9706–9712 RSC .
- Z. Liu, J. Niu, P. Feng and Y. Zhu, Ceram. Int., 2015, 41, 4608–4615 CrossRef CAS .
- H. Cheng, B. Huang and Y. Dai, Nanoscale, 2014, 6, 2009–2026 RSC .
- Y. Yang, C. Zhang, C. Lai, G. Zeng, D. Huang, M. Cheng, J. Wang, F. Chen, C. Zhou and W. Xiong, Adv. Colloid Interface Sci., 2018, 254, 76–93 CrossRef CAS PubMed .
- Z. Wang, M. Chen, D. Huang, G. Zeng, P. Xu, C. Zhou, C. Lai, H. Wang, M. Cheng and W. Wang, Chem. Eng. J., 2019, 374, 1025–1045 CrossRef CAS .
- H. Cheng, B. Huang, Y. Dai, X. Qin and X. Zhang, Langmuir, 2010, 26, 6618–6624 CrossRef CAS PubMed .
- C. Xue, J. Xia, T. Wang, S. Zhao, G. Yang, B. Yang, Y. Dai and G. Yang, Mater. Lett., 2014, 133, 274–277 CrossRef CAS .
- Y. Geng, N. Li, J. Ma and Z. Sun, J. Energy Chem., 2017, 26, 416–421 CrossRef .
- M. Rashid, M. Ikram, A. Haider, S. Naz, J. Haider, A. Ul-Hamid, A. Shahzadi and M. Aqeel, Dalton Trans., 2020, 49, 8314–8330 RSC .
- M. Ali, S. Sharif, S. Anjum, M. Imran, M. Ikram, M. Naz and S. Ali, Mater. Res. Express, 2020, 6, 1250–1255 Search PubMed .
- J. Liu, Z. Luo, W. Han, Y. Zhao and P. Li, Mater. Sci. Semicond. Process., 2020, 106, 104761 CrossRef CAS .
- L. Wang, Z. Li, J. Chen, Y. Huang, H. Zhang and H. Qiu, Environ. Pollut., 2019, 249, 801–811 CrossRef CAS PubMed .
- L. Zhu, H. Li, P. Xia, Z. Liu and D. Xiong, ACS Appl. Mater. Interfaces, 2018, 10, 39679–39687 CrossRef CAS PubMed .
- L. Zhu, H. Li, Z. Liu, P. Xia, Y. Xie and D. Xiong, J. Phys. Chem. C, 2018, 122, 9531–9539 CrossRef CAS .
- M. Laurenti, N. Garino, G. Canavese, S. Hernandéz and V. Cauda, ACS Appl. Mater. Interfaces, 2020, 12, 25798–25808 CrossRef CAS PubMed .
- Y. Chen, Y. Wang, J. Fang, B. Dai, J. Kou, C. Lu and Y. Zhao, Chin. J. Catal., 2021, 42, 184–192 CrossRef CAS .
- R. Beura, R. Pachaiappan and T. Paramasivam, J. Phys. Chem. Solids, 2021, 148, 109689 CrossRef CAS .
- H. J. Biswal, A. Yadav, P. R. Vundavilli and A. Gupta, RSC Adv., 2021, 11, 1623–1634 RSC .
-
O. Yanushevska, T. Dontsova, S. Nahirniak and V. Alisova, TiO2–ZnO Nanocomposites for Photodegradation of Dyes in Water Bodies, in Nanomaterials and Nanocomposites, Nanostructure Surfaces, and Their Applications, ed. O. Fesenko and L. Yatsenko, Springer International Publishing, Cham, 2021, pp. 719–731 Search PubMed .
- L. Zhou, Z. Han, G.-D. Li and Z. Zhao, J. Phys. Chem. Solids, 2021, 148, 109719 CrossRef CAS .
- A. H. Zyoud, M. Dwikat, S. Anabtawi, R. Alkowni, N. Qamhieh, A. Hajamohideen, S. H. Zyoud, M. H. S. Helal, S. H. Zyoud, H. Nassar and H. S. Hilal, JOM, 2021, 73, 420–431 CrossRef CAS .
- S. Sharon Tamil Selvi, G. Hannah Priya, R. Ragu, D. Ancelia, L. Allwin Joseph and J. Mary Linet, J. Cluster Sci., 2021 DOI:10.1007/s10876-020-01963-9 .
- H. J. Yoon, Y. I. Choi, E.-S. Jang and Y. Sohn, J. Ind. Eng. Chem., 2015, 32, 137–152 CrossRef CAS .
- Z. Zhang, C. Shao, X. Li, L. Zhang, H. Xue, C. Wang and Y. Liu, J. Phys. Chem. C, 2010, 114, 7920–7925 CrossRef CAS .
- L. Kong, Z. Jiang, T. Xiao, L. Lu, M. O. Jones and P. P. Edwards, Chem. Commun., 2011, 47, 5512–5514 RSC .
- M. M. Khan, S. F. Adil and A. Al-Mayouf, J. Saudi Chem. Soc., 2015, 19, 462–464 CrossRef .
- Q. W. Cao, X. Cui, Y. F. Zheng and X. C. Song, J. Alloys Compd., 2016, 670, 12–17 CrossRef CAS .
- Y. Zhao, R. Li, L. Mu and C. Li, Cryst. Growth Des., 2017, 17, 2923–2928 CrossRef CAS .
- G. Huang and Y. Zhu, Mater. Sci. Eng., B, 2007, 139, 201–208 CrossRef CAS .
- X. Ma, Z. Ma, T. Liao, X. Liu, Y. Zhang, L. Li, W. Li and B. Hou, J. Alloys Compd., 2017, 702, 68–74 CrossRef CAS .
- M. Su, C. He, L. Zhu, Z. Sun, C. Shan, Q. Zhang, D. Shu, R. Qiu and Y. Xiong, J. Hazard. Mater., 2012, 229–230, 72–82 CrossRef CAS PubMed .
- Z. Li, Y. Qu, K. Hu, M. Humayun, S. Chen and L. Jing, Appl. Catal., B, 2017, 203, 355–362 CrossRef CAS .
- C. Liu, X. Dong, Y. Hao, X. Wang, H. Ma and X. Zhang, RSC Adv., 2017, 7, 22415–22423 RSC .
- W. Liu, J. Cai and Z. Li, ACS Sustainable Chem. Eng., 2015, 3, 277–282 CrossRef CAS .
- X. Tu, S. Luo, G. Chen and J. Li, Chem.–Eur. J., 2012, 18, 14359–14366 CrossRef CAS PubMed .
- M. J. Kale, T. Avanesian and P. Christopher, ACS Catal., 2014, 4, 116–128 CrossRef CAS .
- Z. Zhang, Y. Zhou, S. Yu, M. Chen and F. Wang, Mater. Lett., 2015, 150, 97–100 CrossRef CAS .
- H.-x. Pan, L.-p. Feng, W. Zeng, Q.-c. Zhang, X.-d. Zhang and Z.-t. Liu, Inorg. Chem., 2019, 58, 13195–13202 CrossRef CAS PubMed .
- Z. Jiang, Y. Liu, T. Jing, B. Huang, Z. Wang, X. Zhang, X. Qin and Y. Dai, RSC Adv., 2015, 5, 47261–47264 RSC .
- J. Li, L. Cai, J. Shang, Y. Yu and L. Zhang, Adv. Mater., 2016, 28, 4059–4064 CrossRef CAS PubMed .
- S. Ning, X. Shi, H. Zhang, H. Lin, Z. Zhang, J. Long, Y. Li and X. Wang, Sol. RRL, 2019, 3, 1900059 CrossRef .
- Z. Ma, P. Li, L. Ye, Y. Zhou, F. Su, C. Ding, H. Xie, Y. Bai and P. K. Wong, J. Mater. Chem. A, 2017, 5, 24995–25004 RSC .
- L. Liang, F. Lei, S. Gao, Y. Sun, X. Jiao, J. Wu, S. Qamar and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 13971–13974 CrossRef CAS PubMed .
- L. Ye, H. Wang, X. Jin, Y. Su, D. Wang, H. Xie, X. Liu and X. Liu, Sol. Energy Mater. Sol. Cells, 2016, 144, 732–739 CrossRef CAS .
- L. Ye, X. Jin, X. Ji, C. Liu, Y. Su, H. Xie and C. Liu, Chem. Eng. J., 2016, 291, 39–46 CrossRef CAS .
- F. Chen, H. Huang, L. Ye, T. Zhang, Y. Zhang, X. Han and T. Ma, Adv. Funct. Mater., 2018, 28, 1804284 CrossRef .
- Y. Zhou, Z. Tian, Z. Zhao, Q. Liu, J. Kou, X. Chen, J. Gao, S. Yan and Z. Zou, ACS Appl. Mater. Interfaces, 2011, 3, 3594–3601 CrossRef CAS PubMed .
- S. Wang, X. Hai, X. Ding, K. Chang, Y. Xiang, X. Meng, Z. Yang, H. Chen and J. Ye, Adv. Mater., 2017, 29, 1701774 CrossRef PubMed .
- P. Li, Z. Zhou, Q. Wang, M. Guo, S. Chen, J. Low, R. Long, W. Liu, P. Ding, Y. Wu and Y. Xiong, J. Am. Chem. Soc., 2020, 142, 12430–12439 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c and
Gao
Li
*c and
Gao
Li
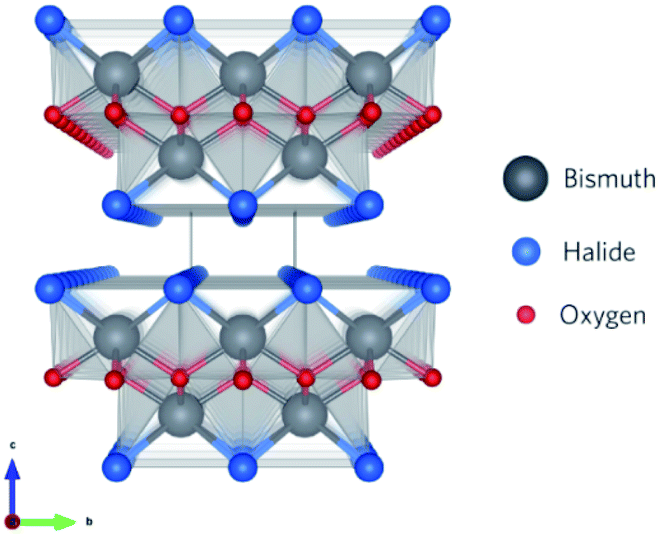
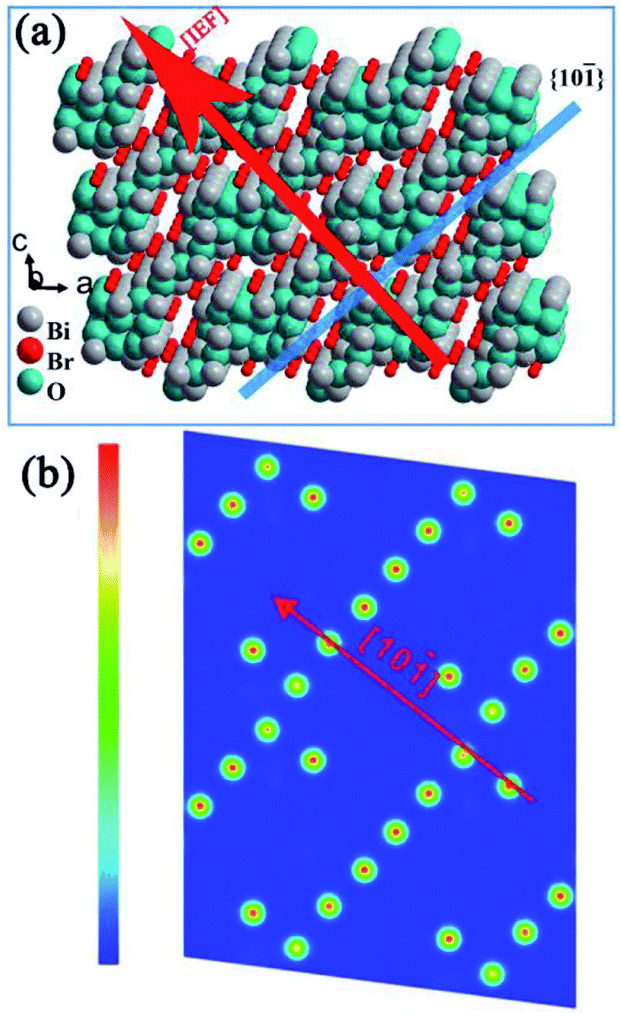
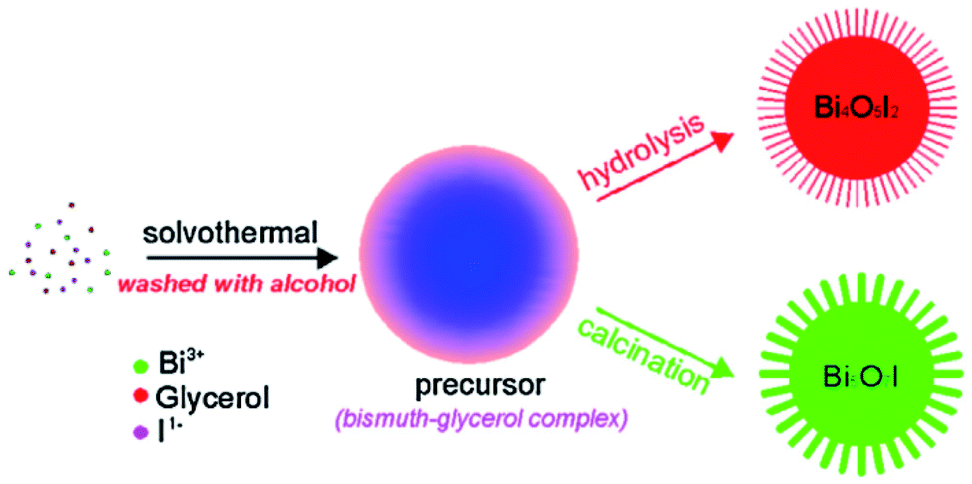
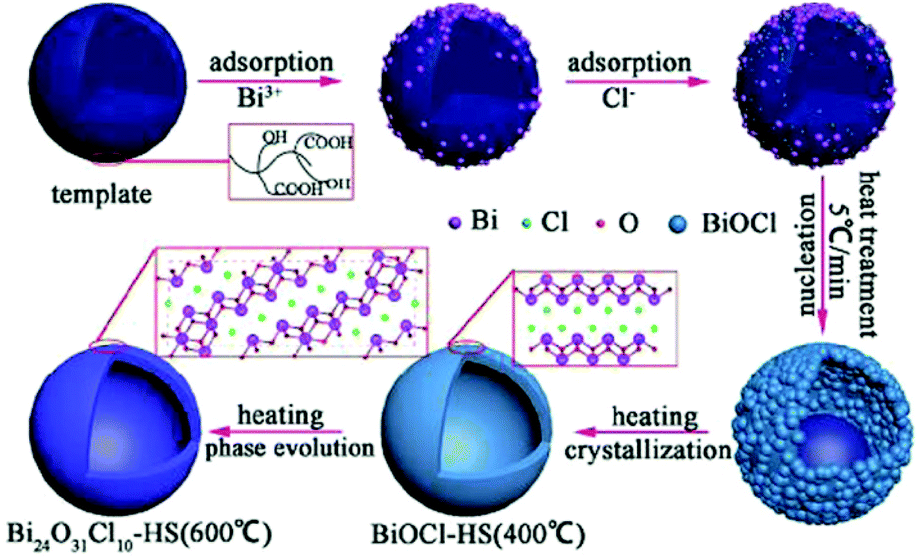
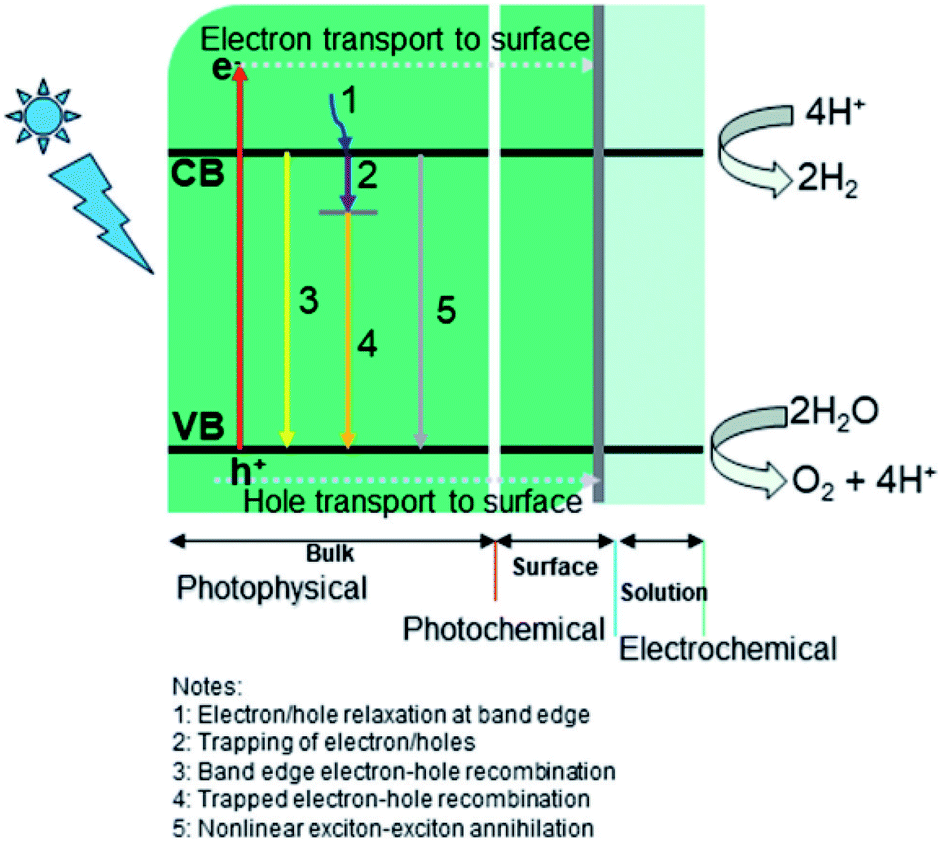
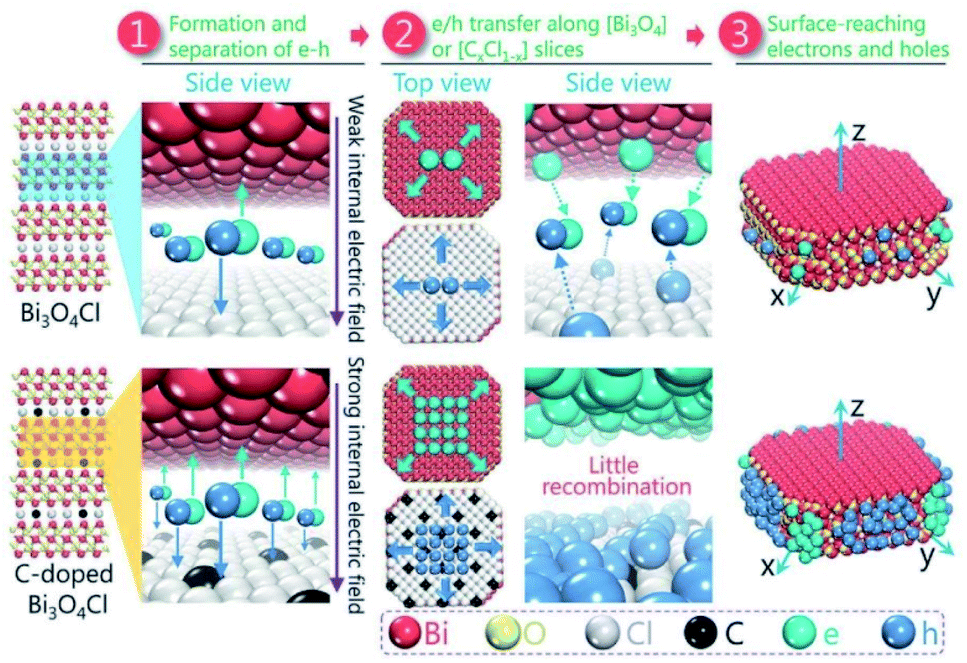
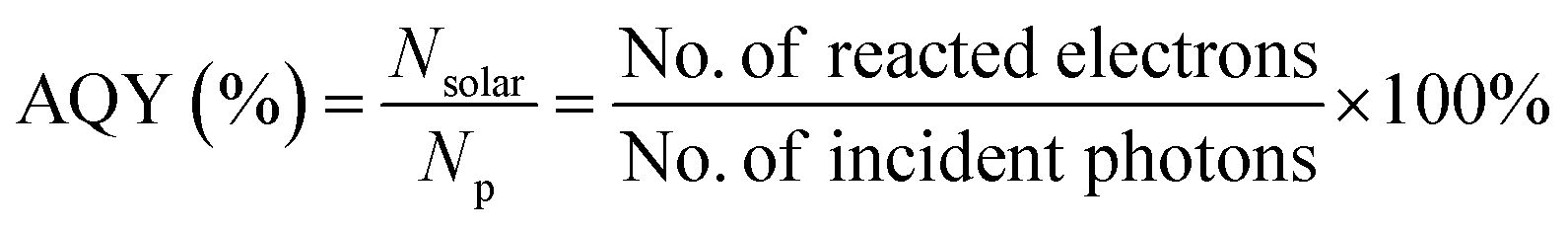
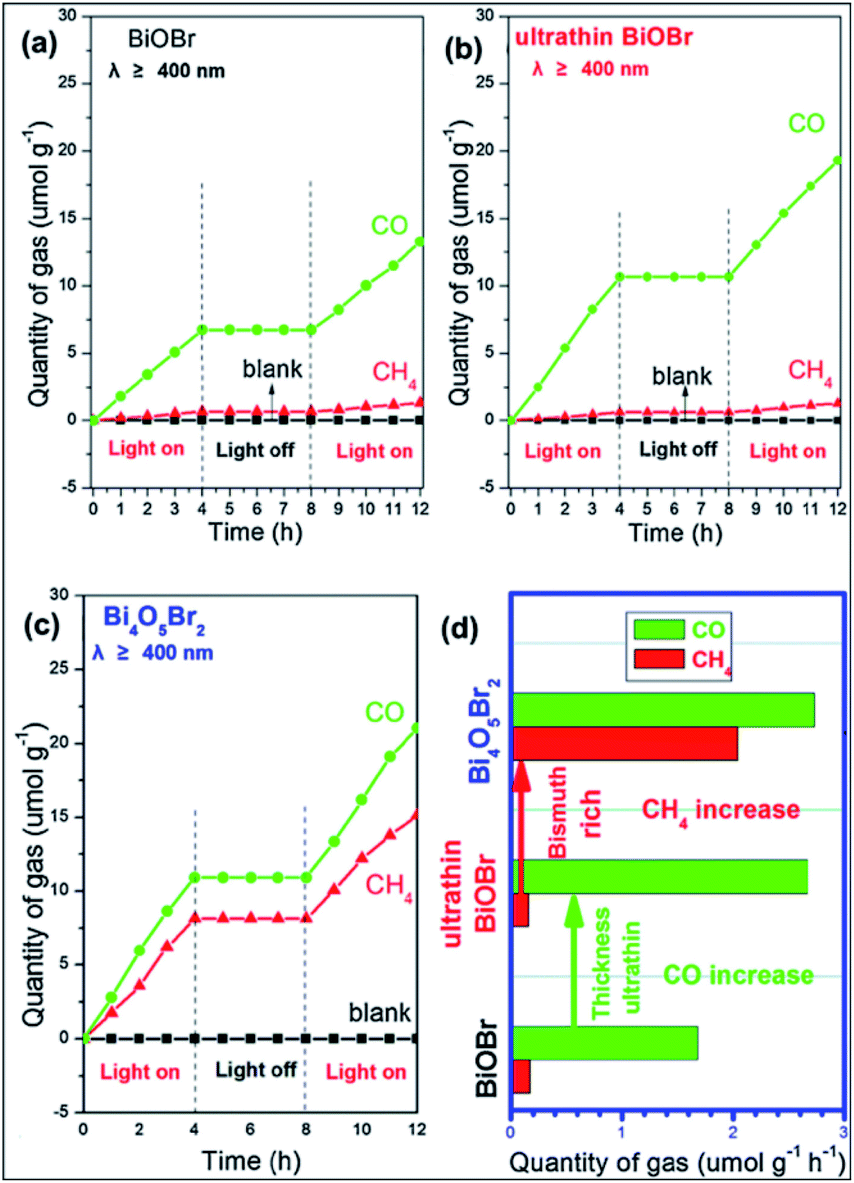
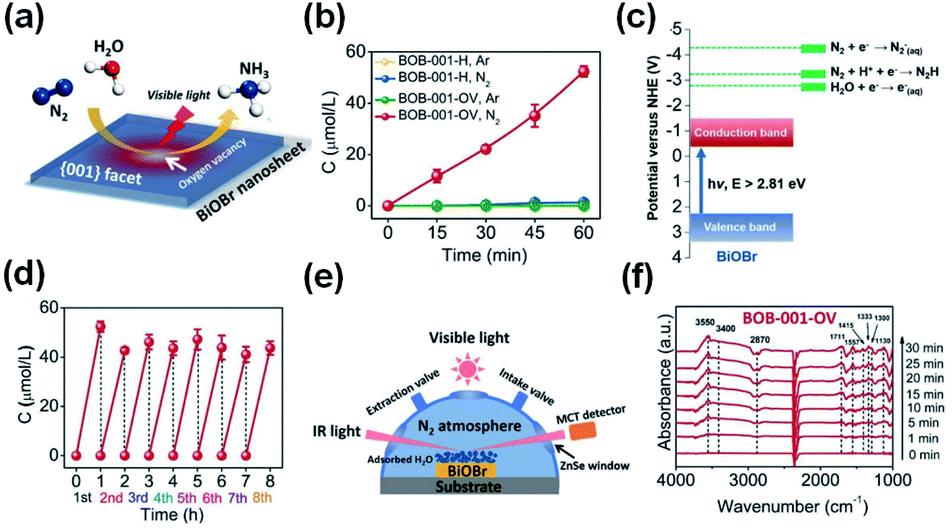
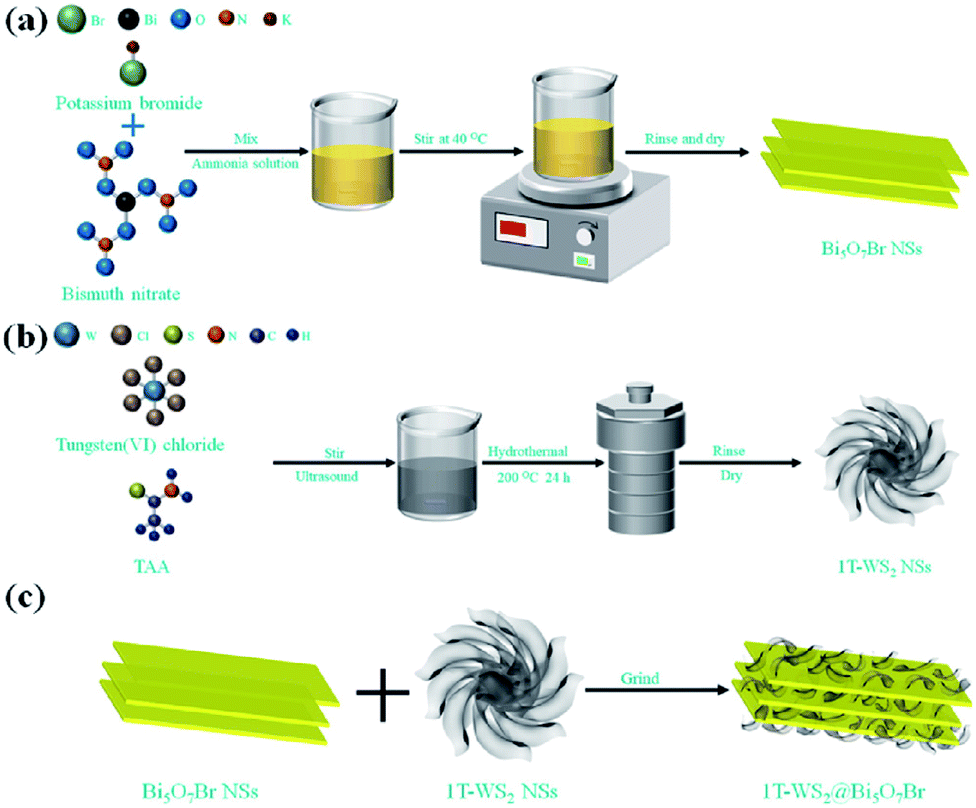
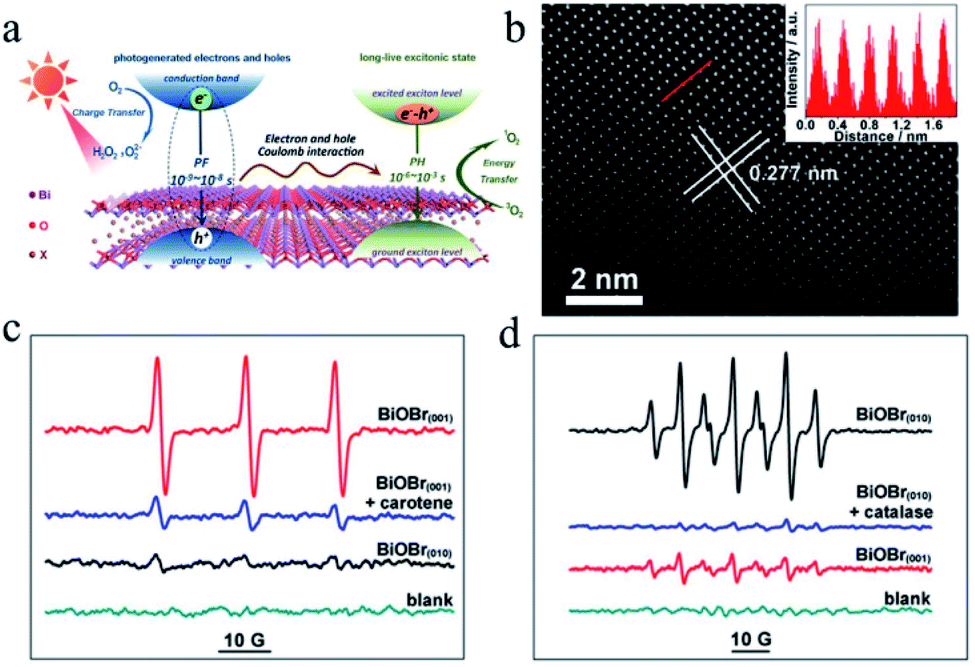
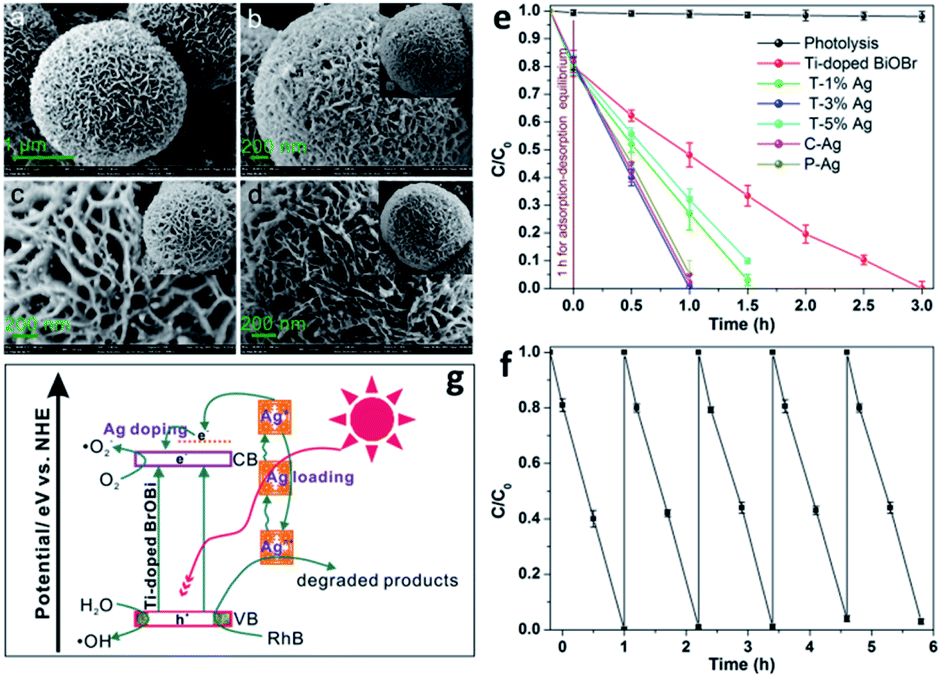
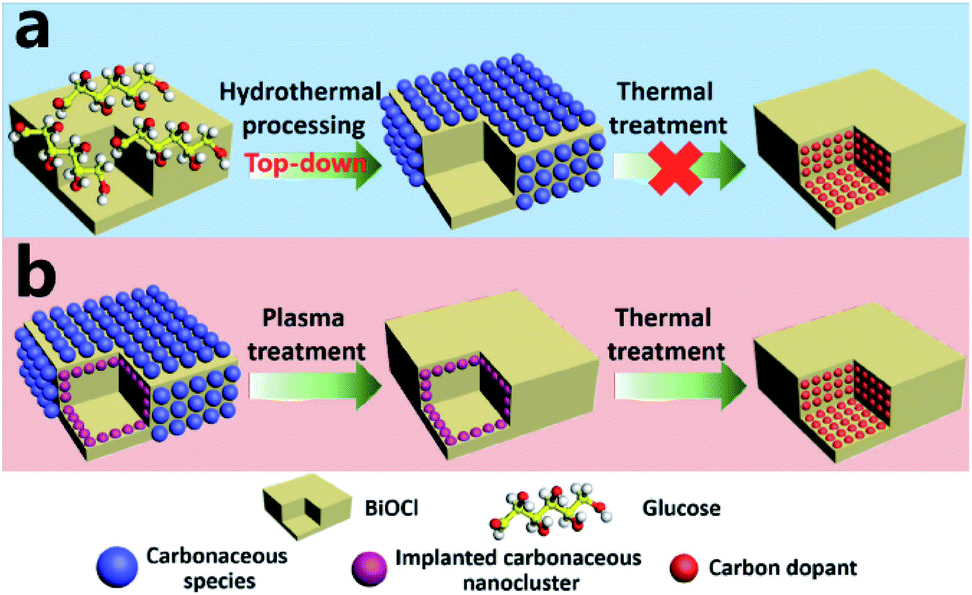
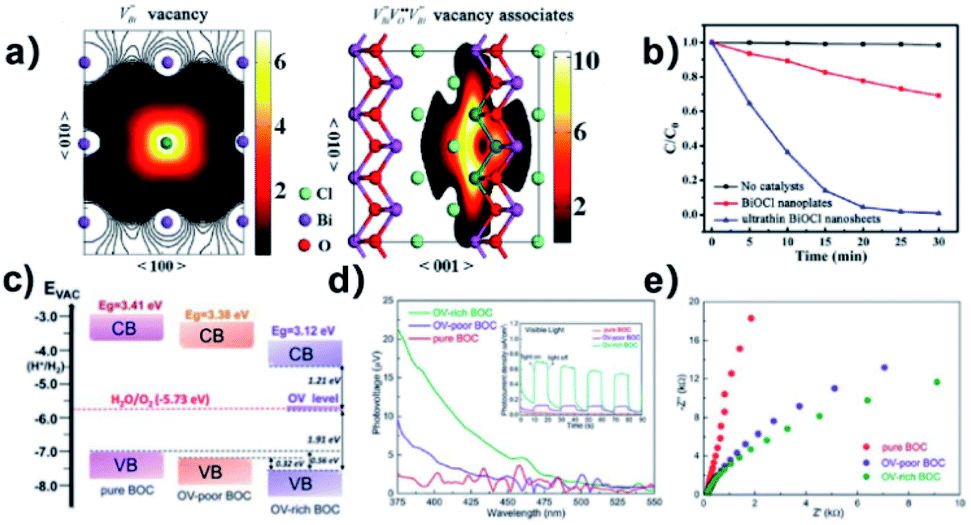
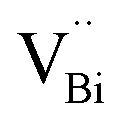 defect and
defect and 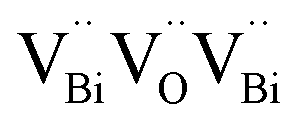 vacancy associates, respectively. (b) Comparison of the photodecomposition of Rhodamine B with ultrathin BiOCl nanosheets and BiOCl nanoplates under simulated solar irradiation. (c) Schematic illustration of the band structure of OV-rich/poor BOC and pure BOC. (d) Surface photovoltage spectrum of OV-rich/poor BOC and pure BOC. Current density transients of OV-rich/poor BOC and pure BOC under visible-light irradiation are shown in the inset. (e) Nyquist impedance plots of OV-rich/poor BOC and pure BOC. Reproduced with permission from ref. 142 Copyright 2013, the American Chemical Society, and ref. 143 Copyright 2018, The Royal Society of Chemistry.
vacancy associates, respectively. (b) Comparison of the photodecomposition of Rhodamine B with ultrathin BiOCl nanosheets and BiOCl nanoplates under simulated solar irradiation. (c) Schematic illustration of the band structure of OV-rich/poor BOC and pure BOC. (d) Surface photovoltage spectrum of OV-rich/poor BOC and pure BOC. Current density transients of OV-rich/poor BOC and pure BOC under visible-light irradiation are shown in the inset. (e) Nyquist impedance plots of OV-rich/poor BOC and pure BOC. Reproduced with permission from ref. 142 Copyright 2013, the American Chemical Society, and ref. 143 Copyright 2018, The Royal Society of Chemistry.