
Physicochemical implications of surface alkylation of high-valent, Lindqvist-type polyoxovanadate-alkoxide clusters†
Alex A.
Fertig
a,
S. M. Gulam
Rabbani
b,
Melissa D.
Koch
 a,
William W.
Brennessel
a,
Pere
Miró
*b and
Ellen M.
Matson
*a
a,
William W.
Brennessel
a,
Pere
Miró
*b and
Ellen M.
Matson
*a
aDepartment of Chemistry, University of Rochester, Rochester, NY 14627, USA. E-mail: matson@chem.rochester.edu
bDepartment of Chemistry, University of South Dakota, Vermillion, SD 57069, USA. E-mail: pere.miro@usd.edu
First published on 11th March 2021
Abstract
We report a rare example of the direct alkylation of the surface of a plenary polyoxometalate cluster by leveraging the increased nucleophilicity of vanadium oxide assemblies. Addition of methyl trifluoromethylsulfonate (MeOTf) to the parent polyoxovanadate cluster, [V6O13(TRIOLR)2]2− (TRIOL = tris(hydroxymethyl)methane; R = Me, NO2) results in functionalisation of one or two bridging oxide ligands of the cluster core to generate [V6O12(OMe)(TRIOLR)2]1− and [V6O11(OMe)2(TRIOLR)2]2−, respectively. Comparison of the electronic absorption spectra of the functionalised and unfunctionalised derivatives indicates the decreased overall charge of the complex results in a decrease in the energy required for ligand to metal charge transfer events to occur, while simultaneously mitigating the inductive effects imposed by the capping TRIOL ligand. Electrochemical analysis of the family of organofunctionalised polyoxovanadate clusters reveals the relationship of ligand environment and the redox properties of the cluster core: increased organofunctionalisation of the surface of the vanadium oxide assembly translates to anodic shifts in the reduction events of the Lindqvist ion. Overall, this work provides insight into the electronic effects induced upon atomically precise modifications to the surface structure of nanoscopic, redox-active metal oxide assemblies.
Introduction
Nanocrystalline metal oxides have been used in a wide variety of applications in material sciences, such as catalysis,1–6 energy storage,7–9 and optoelectronics.10–12 One strategy that has been invoked to optimise the physicochemical properties of these systems involves manipulation of their surface structure through ligand substitution/modification. Indeed, functionalisation of metal oxide nanocrystals with organic ligands allows for the control of many of their characteristics, including solubility, size, stability, and reactivity.6,12,14–16 While there has been a significant amount of studies focused on the addition of ligands that possess the ability to perform redox reactions or, alternatively, ligands that can bind active components to the surface of metal oxide nanoparticles,3,12,17–19 little work has focused on the development of descriptions of the direct electronic consequences of surface functionalisation on the metal oxide core. This is largely due to the complicated nature of ligand-core interactions at the surface of nanoparticles, as well as the inhomogeneity of substitution patterns and surface ligand densities in a sample of organofunctionalised nanoscale materials.15,20,21Efforts to elucidate the physicochemical consequences of organic ligation to the surface of metal oxide materials has led researchers to the development of atomically precise, monodisperse models of these assemblies, using a class of molecular metal oxide clusters called polyoxometalates (POMs). POMs possess surface structures containing alternating terminal and bridging oxide moieties, resembling the surface of bare metal oxide nanocrystals.22 This attribute has translated to their use as molecular models for the surface reactivity of their nanoscale congeners.23–25 With interest in elucidating the consequences of surface ligation, researchers have developed synthetic methods in order to establish organic–inorganic POM hybrid systems.26–28 In a similar fashion to strategies invoked for ligation of metal oxide nanoparticles, an organic functional group is grafted to the surface of a POM through carboxylate, phosphonate, siloxide or alkoxide linkages.
Most often, synthetic approaches to generate these hybrid cluster complexes feature post-synthetic modification of basic sites at the surface of a metal oxide assembly. This renders the organofunctionalisation of plenary polyoxotungstate and polyoxomolybdate clusters challenging, as surface oxygen atoms of these metal oxide assemblies possess limited nucleophilicity as a result of charge distribution across the cluster core.29,30 To generate reactive oxygen atoms at the surface of the cluster, researchers have invoked functionalisation of Lacunary POMs. In this class of cluster, one or more [MO]n− fragments are removed from the POM core via exposure of the assembly to basic conditions, resulting in the formation of a well-defined pocket of nucleophilic oxygen atoms at the surface of the cluster.27,29–31 The increased basicity of the newly formed surface sites has translated to a variety of organofunctionalised POM clusters, all featuring grafted organic moieties bound to the oxygen atoms flanking the vacant site(s) of the Lacunary metal oxide surface. This strategy allows for the amount of surface bound species of organofunctionalised POMs to be tuned, where the number of organic species bound to the surface of the clusters can be altered based on the number of vacant sites present.29,30
The requirement of basic sites at the surface of the metal oxide assembly for successful organofunctionalisation renders polyoxovanadates (POVs) particularly disposed to surface modification. In contrast to their Group(VI) counterparts, POVs typically adopt highly charged cores, as the lower-valent vanadium centres are unable to charge compensate the oxido (O2−) ligands that compose the metal oxide cluster.30,32–34 These effects have been observed experimentally through the site-selective addition of protons to a vanadium-doped Keggin cluster, [γ-XV2W10O39(μ-OR)] (X = Ge, Si; R = Me, Et, Pr).35 Upon addition of an acid to a solution containing the heterometallic metal oxide assembly, proton binding was observed exclusively at the μ2-O2− ligand bridging two vanadium centres (e.g. V–O–V site). The increased basicity of bridging oxide ligands bound to vanadium centres renders these sites particularly susceptible to condensation reactions with organic ligands, affording straightforward methodologies for post-synthetic surface functionalisation of POVs.13,28,30,33 Alternatively, formation of organofunctionalised POVs has been demonstrated possible through self-assembly; addition of vanadium-containing precursors to the desired organic ligand affords formation of the hybrid POV architectures.28,30,32,36 Due in part to the variety of organic ligands available for association to the cluster, POVs contain a high degree of structural diversity, demonstrating the control with which selection of organic ligands can dictate self-assembly reactions.26–28
Although there have been many examples of organofunctionalised POVs reported to date, limited work has focused on the consequences of ligand substitution on the electronic properties of the vanadium oxide core. A seminal contribution from Zubieta and coworkers showed that the redox properties of the metal centres of the Lindqvist-type polyoxovanadate-alkoxide (POV-alkoxide) cluster, [V6O13(TRIOLR)2]2− (TRIOL = tris(hydroxymethyl)methane; R = Me, Et, CH2OH, NO2, NMe2, and Bn), can be tuned as a function of the electron withdrawing/donating properties of the functional group bound to the periphery of the hexavanadate core (first reduction potentials of the aforementioned clusters range from −1.2 to −0.67 V vs. Fc+/0).13 However, recent work from our research group has shown that the incorporation of similar functional groups at the surface of Lindqvist-type POV-alkoxide clusters fully saturated with bridging alkoxide ligands has a limited influence on the electrochemical profile of the assembly.37 This result is surprising, given the similar structure of the vanadium oxide core of the two classes of cluster complexes. Nominally, these assemblies differ only by the number of bridging alkoxide ligands substituted at their surfaces. These contrasting results demonstrate the complex relationship between the electronic structure of the cluster and the surface chemistry surrounding the metal oxide core.
Here we present the synthesis and characterisation of a series of POV-alkoxide clusters, sequentially functionalised at the bridging oxido ligands with methyl substituents (Fig. 1). Exploitation of the nucleophilic properties of the parent POV-alkoxide cluster allows for the selective addition of alkoxide ligands directly to the surface of the cluster via post-synthetic functionalisation. Structural effects imposed by increased surface ligation are described. Analysis of the functionalised clusters via cyclic voltammetry (CV) reveals substantial changes in the electrochemical profile of the clusters, relating to the ability for bridging alkoxide ligands to allow for electrochemical communication between metal sites. Computational methods are employed in order to complement the results established experimentally, and to analyse the localisation of electrons as each cluster is reduced. Collectively, these results provide an insight into the consequences modification of the surface of metal oxide clusters impart on their structural, electrochemical, and nucleophilic characteristics.
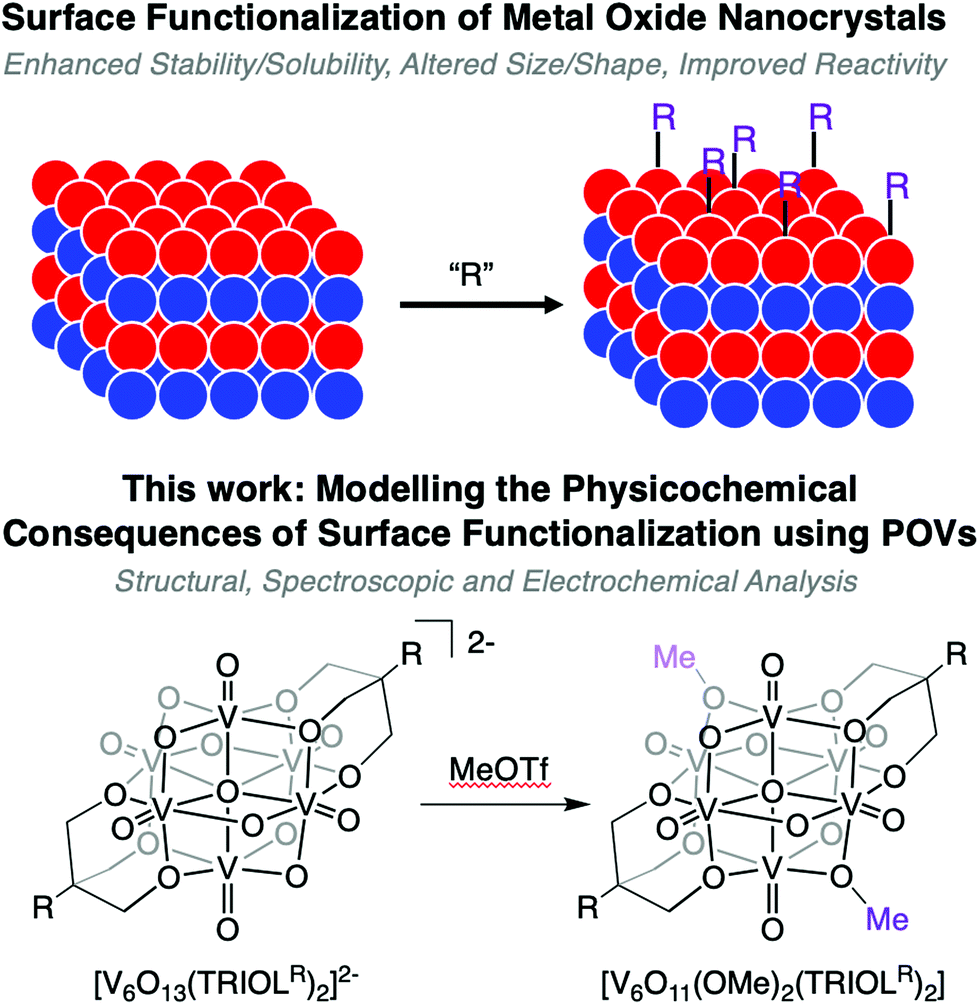 | ||
| Fig. 1 Schematic representation of surface functionalisation of metal oxide nanomatierals (top); here, we present the sequential alkylation of high-valent POV-alkoxide clusters as homogeneous model complexes for the physicochemical implications of organofunctionalisation of metal oxide surfaces. | ||
Results and discussion
Synthesis of [V6O12(OMe)(TRIOLR)2]1− and [V6O11(OMe)2(TRIOLR)2]2−
The established nucleophilicity of high-valent POV clusters prompted further investigation into synthetic routes for direct functionalisation of the cluster surface. Indeed, previous reports have described bridging oxido atoms on the Lindqvist-type POV-alkoxide cluster, [V6O13(TRIOLMe)2]2− (1) as nucleophilic sites capable of binding to protons, resulting in the isolation of a series of clusters containing bridging hydroxide ligands.13 Additionally, Zubieta describes the reported synthesis of trimethylsilyl substituted forms of this POV-alkoxide cluster through addition of trimethylsilylchloride to the parent cluster (structural and electronic characterisation of this substituted assembly was not reported). Given the established reactivity of the vanadium oxide assembly, we hypothesised that addition of organic cations, such as alkyl triflates, to complex 1 would result in the installation of new bridging alkoxide ligands at the cluster surface, creating opportunities to study the physicochemical consequences of organofunctionalisation of vanadium oxide assemblies with atomic precision.To accomplish this goal, we adapted established synthetic protocols for the formation of clusters bearing bridging hydroxide and siloxide moieties; direct alkylation of high-valent POV-alkoxide clusters using methyl trifluoromethylsulfonate (MeOTf) was performed. Addition of 2.5 equiv. of MeOTf to a slurry of 1 in dichloromethane (45 °C) resulted in complete dissolution of the starting material to give a red solution (Scheme 1). Purification of the crude reaction mixture was accomplished via recrystallisation of the product (vapor diffusion of diethyl ether into a concentrated DCM solution). Analysis of the product via1H NMR spectroscopy revealed substantial changes in comparison to the parent cluster, 1: the signal assigned to methylene group of the tridentate capping ligand has shifted downfield, and is split into three inequivalent signals as a result of the loss of symmetry imparted to the assembly relative to the parent cluster by formation of an alkoxide ligand (Fig. 2). An additional resonance integrating to three protons is observed at 4.48 ppm, assigned to the new bridging methoxide ligand. Further support for the formation of the monoalkylated product was obtained via electrospray ionization mass spectrometry (ESI-MS; Fig. S2†); a single prominent signal was observed at m/z = 763, corresponding to the predicted molecular weight of the product, complex [V6O12(OMe)(TRIOLMe)2]1− (1-OMe).
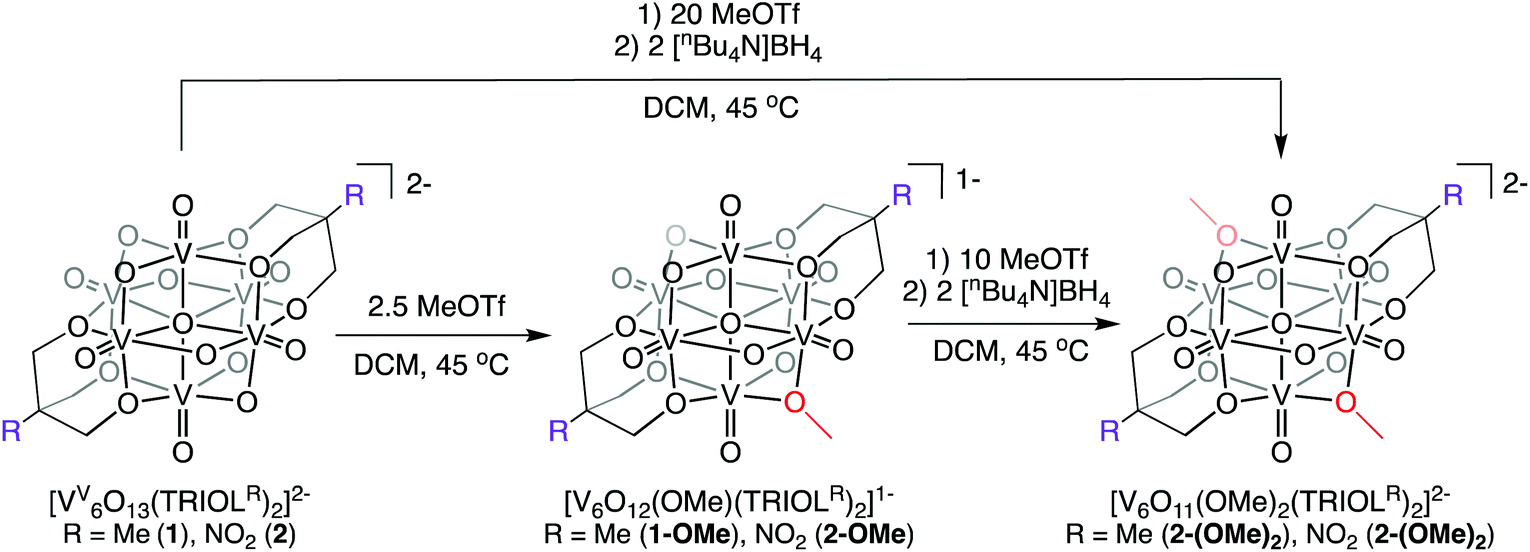 | ||
| Scheme 1 Synthesis of complexes [V6O12(OMe)(TRIOLR)2]1− (X-OMe) and [V6O11(OMe)2(TRIOLR)2]2− (X-(OMe)2) (X = 1; R = Me; X = 2; R = NO2) via alkylation of [V6O13(TRIOLR)2]2− (R = Me (1), NO2 (2)). | ||
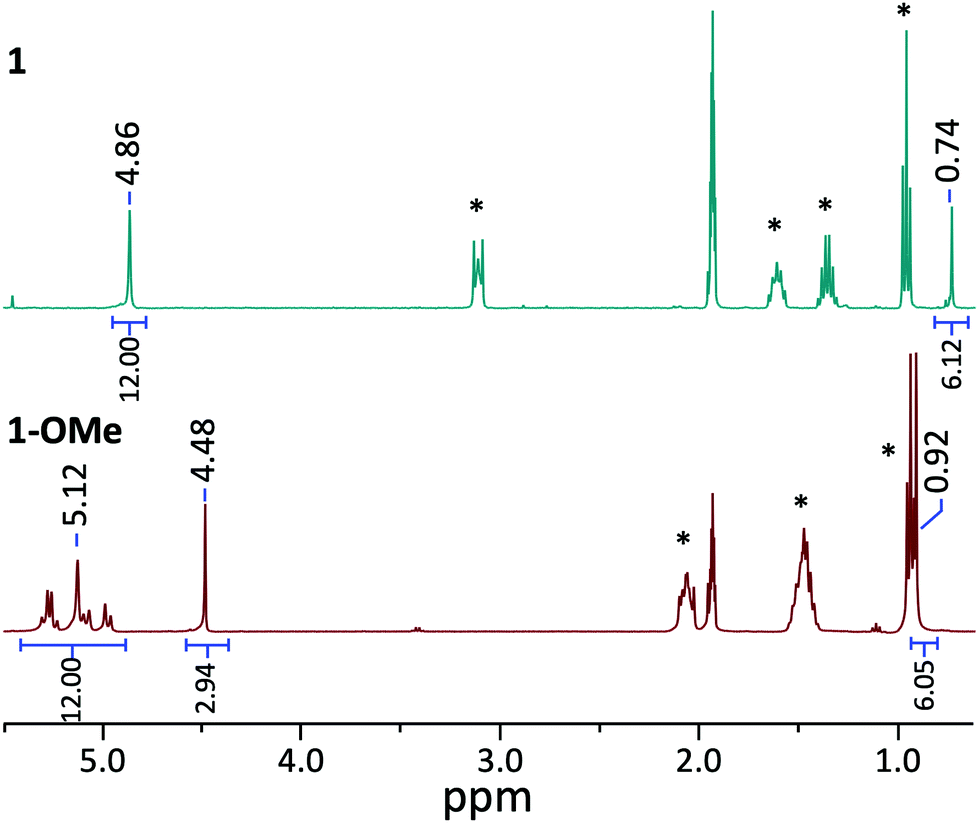 | ||
| Fig. 2 1H NMR spectra of complexes 1 (top, blue) and 1-OMe (bottom, red) collected in CD3CN at 21 °C. Formation of the alkylated species 1-OMe is indicated by the appearance of a singlet peak belonging to the bridging methoxide ligand at 4.48 ppm, along with a downfield shift of the signal corresponding to the terminal methyl group of the capping triol ligand (0.74 and 0.92 ppm for 1 and 1-OMe respectively). Additionally, the singlet peak corresponding to the methylene group of the parent cluster 1 (4.86 ppm) splits into a multi-peak pattern upon formation of the alkylated species 1-OMe (peaks centred at 5.12 ppm). Peaks labelled with * indicate signals belonging to the cations [nBu4N]+ and [nBu4P]+ for 1 and 1-OMe respectively. Peak assignments for alkylated species can be found in Fig. S1 and S7† for 1-OMe and 2-OMe respectively. | ||
To unambiguously confirm the molecular structure of [V6O12(OMe)(TRIOLMe)2]1−, crystals suitable for single crystal X-ray diffraction (SCXRD) were grown from vapor diffusion of diethyl ether into a concentrated solution of dichloromethane. Refinement of the data reveals formation of the desired product, a hexavanadate Lindqvist cluster similar to that of complex 1, with one bridging oxide moiety transformed to a methoxide ligand (Fig. 3, Table 1, additional crystallographic parameters can be found in Table S1†). The new alkoxide ligand is disordered across an inversion centre, inhibiting our ability to ascertain precise changes in bond metrics following surface functionalisation (V–Obvs. V–OMe distances appear averaged in refinement due to resolution limitations). That being said, it is clear that this V–Ob distance is slightly elongated in comparison to other μ2-oxido ligands in the structure that are not disordered with the bridging alkoxide ligand (V–Ob/OMe = 1.894 Å vs. V–Ob = 1.819 Å), suggesting, as expected, that alkylation of a surface oxide ligand translates to the reduction in bond order of V–Ob. Broadly, comparison of V–O bond distances (e.g. V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ot, V–Oc, V–Ob) in complex 1-OMe to that of the parent POV-alkoxide clusters reveals that the dimensions of the Lindqvist core have not changed upon surface functionalisation (Table 1).
Ot, V–Oc, V–Ob) in complex 1-OMe to that of the parent POV-alkoxide clusters reveals that the dimensions of the Lindqvist core have not changed upon surface functionalisation (Table 1).
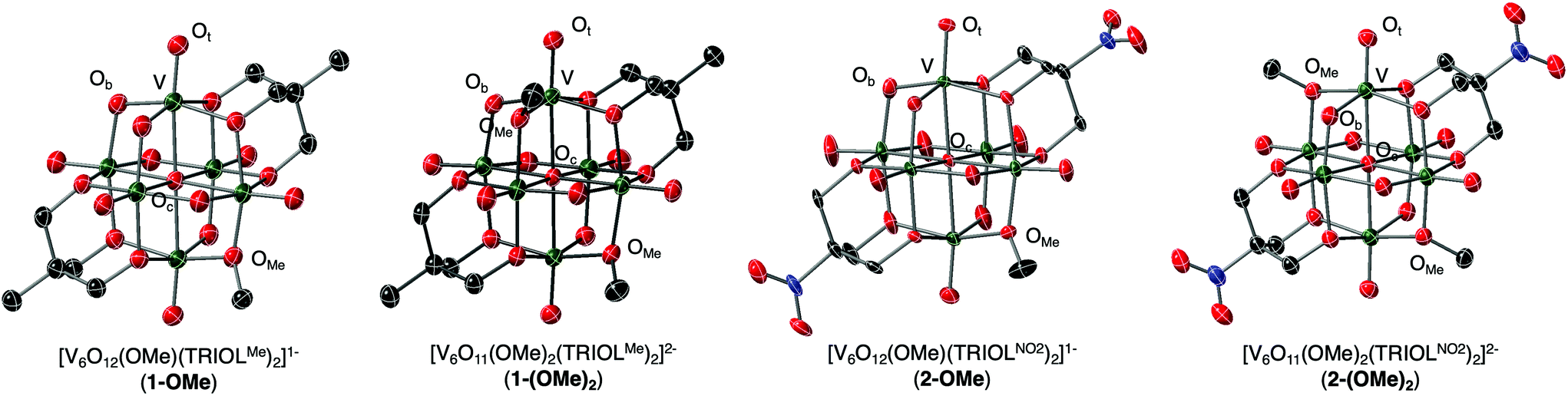 | ||
| Fig. 3 Molecular structures of [V6O12(OMe)(TRIOLMe)2]1− (1-OMe), [V6O11(OMe)2(TRIOLMe)2]2− (1-(OMe)2), [V6O12(OMe)(TRIOLNO2)2]1− (2-OMe), and [V6O11(OMe)2(TRIOLNO2)2]2− (2-(OMe)2) shown with 40% probability ellipsoids. Counter cations, solvent molecules and hydrogen atoms have been removed for clarity. Dark green ellipsoids: V; red ellipsoids: O; dark grey ellipsoids: C; blue ellipsoids: N. | ||
| Bond | 1 | 1-OMe | 1-(OMe)2 | 2 13 | 2-OMe | 2-(OMe)2 |
|---|---|---|---|---|---|---|
| a Density functional theory structures for 1, 1-OMe, 1-(OMe)2, 2, 2-OMe, and 2-(OMe)2 are in agreement with the ones determined experimentally (Tables S2–S4†). Confirmation of retention of Lindqvist structure upon alkylation is observed in the root mean square deviation (RMSD) of the 1-OMe and 1-(OMe)2 vanadium cores being only 0.05 and 0.07 with respect to 1. | ||||||
| VOt (avg) |
1.6079 Å | 1.599 Å | 1.610 Å | 1.597 Å | 1.596 Å | 1.602 Å |
| V–Ob (avg) | 1.8307 Å | 1.819 Å | 1.840 Å | 1.822 Å | 1.826 Å | 1.835 Å |
| V–OMe (avg) | — | 1.894 Å | 1.983 Å | — | 1.879 Å | 1.976 Å |
| V–Oc (avg) | 2.2402 Å | 2.2395 Å | 2.264 Å | 2.241 Å | 2.236 Å | 2.2631 Å |
| V–Ob–V (avg) | 111.65° | 112.36° | 112.88° | 112.3° | 111.93° | 112.53° |
| V-OMe-V (avg) | — | 110.39° | 108.17° | — | 111.02° | 107.44° |
Previous work from a variety of research teams has described the synthesis and molecular structures of a series of POV-alkoxide clusters with varying degree of bridging alkoxide ligands (general formula [V6O19−x(OR)x]; x = 6,38 7,39 8,40 11,41 and 1232,41,42). In all examples, V–Ob bond lengths associated with bridging alkoxide ligands (1.974(7)–2.034(3) Å) are elongated in comparison to the V–Ob bonds of bridging oxido moieties (1.752(4)–1.897(3) Å). This parallels our observations of changes in bond lengths associated with V–O bonds at the surface of the assembly following alkylation. Notably, in all structural examples of the Lindqvist-type POV-alkoxide clusters, V–O bond lengths (VOt, V–Ob, V–OMe, and V–Oc) in each assembly are almost identical, indicating that structurally, the metal oxide core is not affected by the degree of alkoxide ligand incorporation at the surface of the vanadium oxide assembly.
The isolation of the mono-functionalised POV-alkoxide cluster, 1-OMe, is intriguing. Previous work involving surface modification of high-valent POV-alkoxide clusters describe pair-wise addition of cationic substituents to the cluster core.13 For example, Zubieta reports that addition of 2.5 equiv. of HBF4OEt2 to [V6O13(TRIOLMe)2]2− results in formation of the bis-protonated species, [V6O11(OH)2(TRIOLMe)2]. Sole formation of the di-protonated species was confirmed by 17O NMR spectroscopy; a resonance at 331 ppm appears in the spectrum, assigned to the two new hydroxide moieties (confirmed by integration). Similarly, addition of 2 equiv. of Me3SiCl results in the formation of the disilylated species, [V6O11(OSiMe3)2(TRIOLMe)2]. The observation of the formation of the monosubstituted variants in either case are not mentioned in this work, despite the similar reaction conditions invoked for the synthesis of each.
Intrigued by isolation of complex 1-OMe, we sought to develop reaction conditions that would lead to isolation of further functionalised variants of the assembly. We hypothesised that we could push the reaction toward formation of the bis-alkylated cluster, [V6O11(OMe)2(TRIOLMe)2] by addition of excess MeOTf (Scheme 1). Addition of 20 equiv. of MeOTf to a slurry of complex 1 in dichloromethane (45 °C for 3 hours) resulted in the formation of a dark red precipitate. The solid was collected by filtration through filter paper and washed with a small amount of dichloromethane and diethyl ether. The insolubility of the resultant product in organic solvent is consistent with the formation of a neutral assembly; retention of the Lindqvist structure was confirmed by two distinct absorption bands in the IR spectrum (Fig. S3†), corresponding to ν(VOt) (968 cm−1) and ν(Ob–CR3) (981 cm−1).
To increase the solubility of the cluster, allowing for characterisation of the assembly via techniques reserved of homogeneous samples, reduction of the purported bis-alkoxide species, [V6O11(OMe)2(TRIOLMe)2], was performed. Addition of 2 equiv. of tetrabutylammonium borohydride, [nBu4N][BH4], to a slurry of the red product in acetonitrile at room temperature results in a gradual colour change to dark green. Characterisation of the product by 1H NMR spectroscopy is difficult due to the paramagnetic nature of the reduced cluster (Fig. S4†). However, ESI-MS proved useful for confirming the formation of the desired product; a signal observed at m/z = 389 (Fig. S5†) indicates formation of the dianionic form of the bis-methylated cluster [V6O11(OMe)2(TRIOLMe)2]2− (1-(OMe)2). Purification of the cluster was performed by vapor diffusion of diethyl ether into a concentrated solution of 1-(OMe)2 in acetonitrile.
The purification procedure described above for complex 1-(OMe)2 afforded single crystals suitable for X-ray analysis (Fig. 3, Table 1, additional crystallographic parameters can be found in Table S1;† note that extremely concentrated acetonitrile solutions of 1-(OMe)2 were required to grow crystals of sufficient size for SCXRD). The asymmetric unit cell contains one cluster molecule, two tetrabutylammonium cations, and one co-crystallised acetonitrile solvent molecule, all located in general positions. Broadly speaking, the structure resembles that of the parent cluster, 1; however, two new alkoxide ligands are observed in complex 1-(OMe)2, consistent with successful alkylation of the surface of the vanadium oxide assembly. The lack of disorder of alkoxide ligands in the structure of 1-(OMe)2 allows for rigorous bond metric analysis. Substantial elongation of V–Ob bond lengths are observed upon alkylation of bridging oxide ligands (V–Ob (avg) = 1.840 Å vs. V–OMe (avg) = 1.983 Å). V–Ob–V bond angles become more acute upon alkylation, likely due to the displacement of the oxygen atom of the alkoxide ligand from the Lindqvist lattice.
The general positions and lack of disorder in the unit cell of complex 1-(OMe)2 allows for relative positions of the new alkoxide functionalities to be ascertained. The bridging oxide ligands make up a belt of six possible positions for surface functionalisation to occur across the cluster (Fig. 4); three possible relative positions of alkyl functionalisation can be proposed (Fig. 4c). In the case of complex 1-(OMe)2, the two μ2-O2− ligands functionalised by methyl cations are separated by a single oxo ligand, corresponding to the “meta” configuration designated in Fig. 4c. This result is surprising, given that in their original report, Zubieta and coworkers describe exclusive formation of the “para” product upon functionalisation of the cluster surface with two siloxide moieties.13 In the original report, selective formation of the single product was confirmed by 17O NMR; unfortunately, the paramagnetic nature of complex 1-(OMe)2 precludes this form of analysis (recall reduction was required to increase solubility).
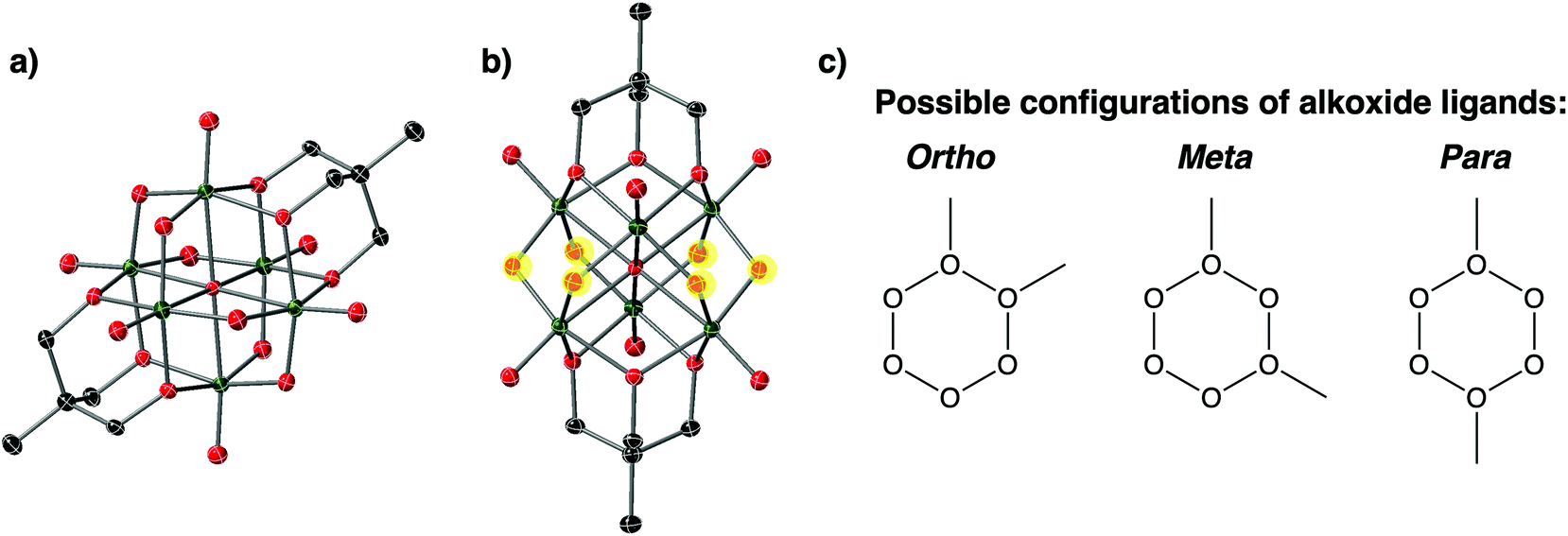 | ||
| Fig. 4 (a) Molecular structure of POV-alkoxide cluster shown in typical view, red ellipsoids: O, dark green ellipsoids: V, dark grey ellipsoids: C; (b) rotation of POV-alkoxide cluster to highlight the belt of six bridging oxido ligands; note that μ2-O2− moieties available for surface functionalisation are highlighted in yellow; (c) possible relative positions of new alkoxide ligands in bis-functionalised POV-alkoxide clusters. | ||
Density functional theory calculations were performed at the M06/def2-TZVP//M06-L/def2-TZVP,def2-SVP level of theory to study the sequential alkylation of complex 1. Gibbs free energy of activation was 24.9 kcal mol−1 for the first alkylation of the vanadium oxide surface. Entropic contributions associated with bringing the reactants together contribute significantly to the observed barrier since the enthalpy of activation in each alkylation event is only 13.0 kcal mol−1. The second alkylation of 1-OMe has a barrier 4.8 kcal mol−1 higher on average than the first alkylation, which is in agreement with the more facile carbocation addition to the more negatively charged POV-alkoxide surface as observed in mapping the comparative electrostatic potentials of complexes 1 and 1-OMe (Fig. S6†). The most favourable alkylation for 1-OMe occurs in the meta position with a Gibbs free energy barrier of 28.4 kcal mol−1; however, the para- and ortho-alkylations are only 2.0 kcal mol−1 higher in energy, on average. The formation of the dialkylated ortho, meta, and para species with respect to 1-OMe is always exergonic (−9.7, −10.8, and −9.1 kcal mol−1, respectively), further indicating a slight thermodynamic preference for the meta-alkylated products over the two other isomers. This observation is consistent with the observed distribution of methoxide ligands in the structure of 1-(OMe)2.
The sequential alkylation of 1 reveals a significant reduction of the favourability of the alkylation product with each alkylation (10.8 kcal mol−1 per alkyl group on average; Fig. S6†). The first alkylation is exergonic by −22.3 kcal mol−1 with respect to free reactants. The second alkylation to form the meta-1-(OMe)2 product is still exergonic, but only by −10.8 kcal mol−1. The addition of a third methyl substituent to the surface of the neutral, bis-alkylated POV-alkoxide cluster, [V6O11(OMe)2(TRIOLMe)2], is predicted to be a thermoneutral process (−0.7 to 2.0 kcal mol−1). This progression is a clear indication of a significant decrease of the thermodynamic stability of the alkylated products upon the increase of the number of alkyl groups at the surface of the Lindqvist assembly. In consequence, the combination of a relatively high activation energy and the slightly thermodynamically unfavourable trisalkylated product is expected to make it difficult to form species beyond dialkylated clusters.
In their original report, Zubieta and coworkers describe significant discrepancies in the electronic properties of high-valent POV-alkoxide clusters supported by disparate variants of the tris(hydroxymethyl)methane ligand, [V6O13(TRIOLR)2]2− (R = Me, NO2).13 The authors credit the inductive character of the peripheral functional groups (e.g. methyl vs. nitro) as the source of these differences, manifesting in dramatic shifts in the electrochemical properties of the hexavanadate assembly (vide infra). Interested in understanding how these electron withdrawing/donating functional groups influence the propensity of the surface of the POV-alkoxide cluster to undergo alkylation reactions, we next explored the reactivity of [V6O13(TRIOLNO2)2]2− (2) with MeOTf. We hypothesised that the electron withdrawing character of the nitro functional group might translate to deactivation of the surface of the assembly, rendering installation of methyl substituents at bridging oxygen atoms more challenging.
Identical reaction conditions invoked for the formation of 1-OMe and 1-(OMe)2 were investigated for the synthesis of the surface functionalised derivatives of complex 2 (Scheme 1). Following work-up, [V6O12(OMe)(TRIOLNO2)2]1− (2-OMe) and [V6O11(OMe)2(TRIOLNO2)2]2− (2-(OMe)2) were isolated in good yield (65% and 68%, respectively; see ESI† for details of characterisation of these complexes, Fig. S7–S10†). In the case of complex 2-OMe, analysis of the product by 1H NMR spectroscopy revealed similar changes in spectrum to that observed upon formation of 1-OMe; most notably, the reduction in symmetry imparted to the cluster surface as a result of alkylation of a bridging oxido ligand results in the splitting of resonances corresponding to the methylene moieties of the tris(hydroxymethyl)nitromethane ligands (Fig. S7†). Both complexes were additionally characterised by ESI-MS (m/z (2-OMe) = 824, Fig. S8;†m/z (2-(OMe)2) = 419.5, Fig. S10†), confirming installation of the methyl cation(s) at the surface of the assembly.
It is worth mentioning that the yield of both nitro-backed POV-alkoxide clusters is significantly diminished from that reported for the methyl-backed derivatives (1-OMe: 89%; 1-(OMe)2: 88%). We hypothesise this is due to the decreased nucleophilicity of the bridging oxide ligands of complex 2. This was also confirmed computationally; the decrease in nucleophilicity of the bridging oxide ligands of 2 as compared to 1 is clearly observed in the map of the surface electrostatic potentials of these complexes (Fig. S11†). This translates to an increase in the Gibbs free energy barrier for methylation of complex 2 by 2.0 kcal mol−1 with respect to 1 (26.9 kcal mol−1cf. 24.9 kcal mol−1). A similar increase in the activation energy is noted for the alkylation of 2-OMe (Fig. S12†).
Structural characterisation of complexes 2-OMe and 2-(OMe)2 was performed via SCXRD. Broadly speaking, the structures of the surface functionalised derivatives of the POV-alkoxide cluster containing nitro substituents closely resembled those of 1-OMe and 1-(OMe)2 (Fig. 3, Table 1, crystallographic parameters details in Table S1†). In the case of complex 2-OMe, while each vanadium centre crystallised in a unique position within the unit cell, the single alkoxide ligand was disordered across three bridging positions, rendering elucidation of changes in V–Ob bond length as a consequence of surface functionalisation challenging.
In the case of the bis-alkoxide derivative, complex 2-(OMe)2, the asymmetric unit contains one-half of the vanadium cluster positioned on a crystallographic inversion centre that coincides with the central oxygen atom. Notably, the alkoxide moieties are not disordered in this structure, allowing for assignment of relative positions to be ascertained. Unlike complex 1-(OMe)2, the two new alkoxide ligands in complex 2-(OMe)2 are positioned directly across the cluster core from one another, adopting the “para” configuration (see Fig. 4). Justification for the difference in relative positions of alkoxide moieties in complexes 1-(OMe)2 and 2-(OMe)2 is unclear. From Density Functional Theory, it is predicted that all isomers of the bis-alkylated, methyl- and nitro-backed POV-alkoxide clusters are similar in energy, with a slight preference for the formation of the meta-product as discussed above (fig. S12†). This information, coupled with the crystallisation of the two distinct isomers (complex 1-(OMe)2vs.2-(OMe)2) suggest multiple species might be present in solution, and that a single isomer has crystalised in each sample. However, the inherent differences in the electronic properties of the cluster core as a result of the disparate inductive properties of the tris(hydroxymethyl)methane ligands might also govern selectivity in surface functionalisation.
Consequences of surface functionalisation on the electron structure of POV-alkoxides: Changes in electronic absorption spectra
The absorption spectra for the parent POV-alkoxide clusters, complexes 1 and 2, have been previously reported by Zubieta.13 Both organofunctionalized vanadium oxide assemblies exhibit solvent dependent shifts in their absorption profiles, assigned by Zubieta as charge transfer events (e.g. LMCT: O→V). Interested in comparing the electronic structure of these assemblies to the electrochemical profile of the POV-alkoxide clusters (vide infra), we collected the absorption spectra of complexes 1 and 2 in MeCN (note: MeCN is the solvent in which all solution-phase analysis is performed in this study).The absorption profiles of complexes 1 and 2 possess a broad absorption feature in the near ultraviolet region which begins at ∼500 nm. In the case of complex 1, a clear shoulder is observed at 358 nm (ε = 5470 M−1 cm−1). This peak shifts to 398 nm (ε = 4600 M−1 cm−1) in the spectrum of complex 2. Similar changes in the absorption profile of complexes 1 and 2 are reported in dimethylformamide: the absorption feature of complex 2 is red-shifted by ∼50 nm from that of 1 and exhibits a substantially decreased molar absorptivity in comparison to its methyl-congener.13 The shift in λmax is attributed to the differences in inductive properties of the peripheral ligands; the electron withdrawing nature of the nitro substituent in complex 2 stabilises the LMCT event (i.e. less energy is required to excite electron density from the surface oxide ligands to the d0 vanadium centres).
As noted in Fig. 5, the introduction of a single alkoxide functional group at the surface of the POV-alkoxide cluster results in substantial changes in the absorption profiles of the hexavanadate core. In contrast to their parent POV-assemblies, the broad absorption profile of complexes 1-OMe and 2-OMe have shifted bathochromically. The change in energy of absorption features again is consistent with the reduction of charge at the vanadium oxide core; this translates to a reduction in energy required for the LMCT event assigned to these absorption features. Complex 1-OMe now features a moderately sharp transition at 386 nm (ε = 6190 M−1 cm−1), with a shoulder at 520 nm (ε = 1300 M−1 cm−1). Complex 2-OMe possesses a spectrum with similar features; a transition at 381 nm (ε = 6360 M−1 cm−1) and 526 nm (ε = 965 M−1 cm−1). Notably, introduction of an alkyl substituent at the surface of the assembly results in absorption features for complexes 1-OMe and 2-OMe that are quite similar to one another. The similarities in energies of the absorption features of the alkylated derivatives of the cluster is striking, considering the substantial differences observed in the energies of electronic transitions of complexes 1 and 2. This result suggests that surface functionalisation decreases the electronic influence of the peripheral substituents bound to the cluster through the tridentate tris(hydroxymethyl)methane ligands.
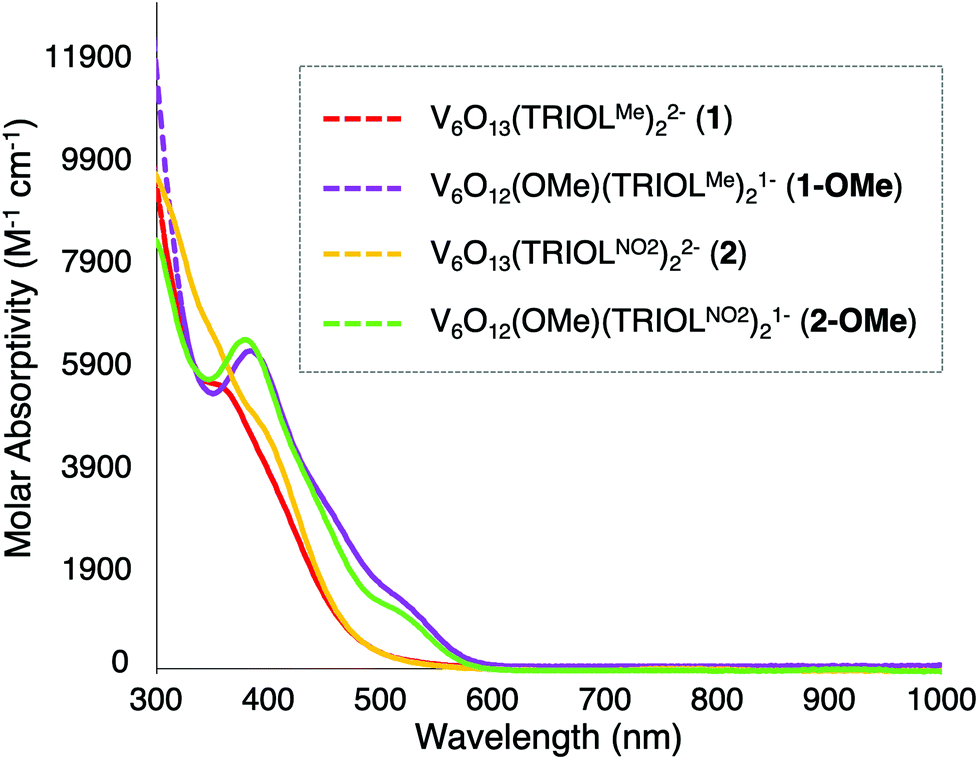 | ||
| Fig. 5 Electronic absorption spectra of complexes 1, 1-OMe, 2, and 2-OMe collected in MeCN at 21 °C. | ||
Results obtained via time-dependent density functional theory (TD DFT) calculations display similar absorbance features in the near ultraviolet region, corresponding to LMCT events from orbitals of bridging oxygen and TRIOL ligands to empty vanadium d-orbitals. Bathochromic shifts in the first absorption event are observed upon formation of the alkylated species (Fig. S13 and S14† for 1/1-OMe and 2/2-OMe respectively), in agreement with results obtained experimentally. Additionally, a difference in absorbance is observed between 1-OMe and 2-OMe, suggesting that, in contrast to the previous hypothesis, alkylating the surface of the clusters does not decrease the electronic influence peripheral ligands imposes upon the metal oxide core. Further studies into the electronic communication of ligands and metal oxide core are on-going in our laboratory in order explain this phenomenon.
To assess the electronic consequences of the introduction of a second methoxide group, we turned our attention to analysis of the electronic absorption spectra of [V6O11(OMe)2(TRIOLR)2]0 (R = Me, NO2). It is worth recalling that the neutral, difunctionalised POV-alkoxides are not sufficiently soluble in organic solvent for analysis. Thus, characterisation of these clusters was performed via diffuse reflectance spectroscopy. Utilization of the Kubelka–Munk transformation allows for the comparison of the reflectance data to the solution phase electron absorption spectra. As expected, bathochromic shifts in the absorption features relative to the monofunctionalised species are observed for difunctionalised assemblies (Fig. S15†), characteristic of further reduction of charge at the metal oxide core. Additionally, the similarities in reflectance spectra of complexes [V6O11(OMe)2(TRIOLMe)2]0 and [V6O11(OMe)2(TRIOLNO2)2]0 underscore a further decrease in the influence imparted on the metal oxide core by the tris(hydroxymethyl)methane ligand upon functionalisation at the surface of the metal oxide cluster.
Characterisation of the isolated, reduced difunctionalized clusters, complexes 1-(OMe)2 and 2-(OMe)2, via electronic absorption spectroscopy was also performed (Fig. S16†). These clusters exhibit broad, low energy and low intensity absorption features assigned to IVCT bands, characteristic of mixed valent POV-alkoxides.13,41
Electrochemical consequences of surface functionalisation
With the series of organofunctionalised Lindqvist-type POV clusters in hand, we next turned our attention to analysis of the electronic consequences of surface alkylation of the hexavanadate core via CV (Fig. 6, Table 2). The parent POV-alkoxide clusters, complexes 1 and 2 possess two (quasi)reversible, one-electron redox events (note that reversibility is judged based on the ratio of peak heights for the cathodic and anodic processes as indicated in parentheses in Table 2). This is in contrast to the original report published by Zubieta and coworkers, which describe the observation of only a single redox process for the hexavanadate assemblies.13 Indeed, under rigorously anhydrous conditions, the second reduction event can be observed. The disparate redox potentials of complexes 1 and 2 are attributed to the differences on the inductive properties of the methyl and nitro moieties appended to the surface of the cluster through the tris(hydroxymethyl)methane ligands;13 the electron donating character of the alkyl substituent increases the electron density at the cluster core, rendering reduction of 1 more challenging than that of its nitro-functionalised congener (E1/2 (1) = −0.923, −1.694 V vs. Fc+/0; E1/2 (2) = −0.617, −1.418 V vs. Fc+/0).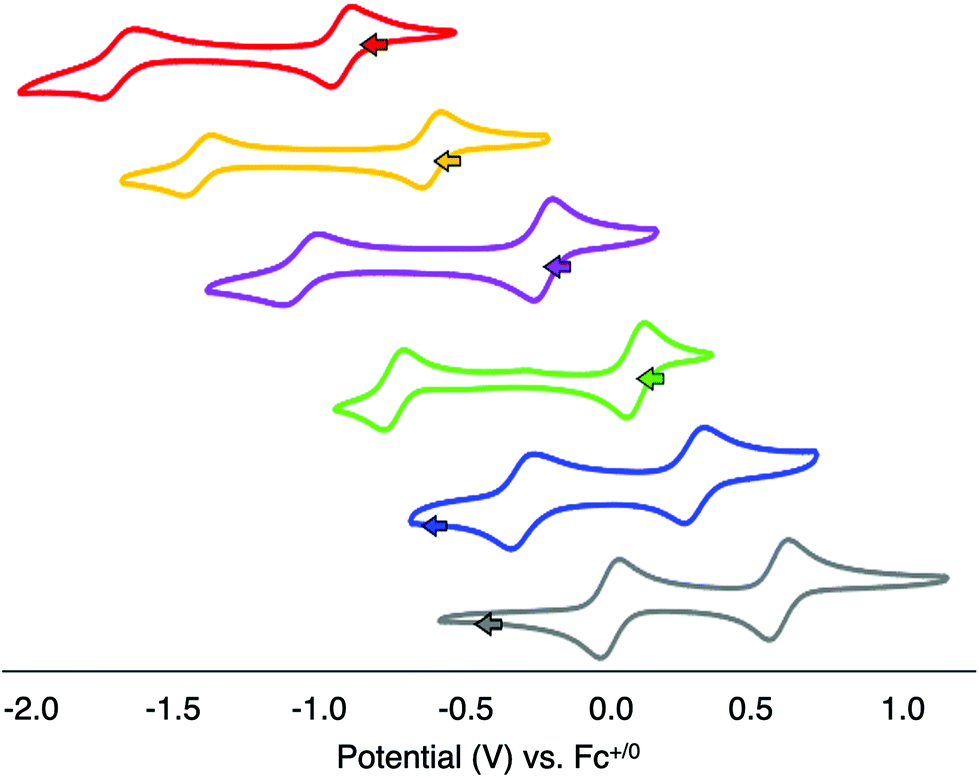 | ||
| Fig. 6 Cyclic voltammograms of complexes 1 (red, top), 2 (orange), 1-OMe (purple), 2-OMe (green), 1-(OMe)2 (blue), and 2-(OMe)2 (gray, bottom). All voltammograms were collected using 1 mM solutions of the complexes in acetonitrile with 0.1 M [nBu4N][PF6] as the supporting electrolyte at a scan rate of 200 mV s−1. | ||
| Complex | OCPa | 1st reduction VV6/VIVVV5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
2nd reduction VIVVV5/VIV2VV4b |
ΔE1/2 | K c |
|---|---|---|---|---|---|
| a Open circuit potential reported in V (vs. Fc+/0). b Redox potentials are reported in V (vs. Fc+/0). Values in parentheses correspond to ratios of the cathodic and anodic peak heights (ic/ia) for each redox event. | |||||
| [V6O13(TRIOLMe)22− (1) | −0.817 | −0.923 (1.03) | −1.694 (1.64) | 0.769 V | 1.5 × 1013 |
| [V6O13(TRIOLNO2)22− (2) | −0.567 | −0.617 (0.97) | −1.418 (0.99) | 0.801 V | 5.3 × 1013 |
| [V6O12(OMe)(TRIOLMe)21− (1-OMe) | −0.181 | −0.231 (0.88) | −1.066 (0.95) | 0.835 V | 2.1 × 1014 |
| [V6O12(OMe)(TRIOLNO2)21− (2-OMe) | 0.147 | +0.087 (1.06) | −0.746 (0.98) | 0.833 V | 1.9 × 1014 |
| [V6O11(OMe)2(TRIOLMe)22− (1-(OMe)2) | −0.587 | +0.289 (0.99) | −0.302 (0.97) | 0.591 V | 1.4 × 1010 |
| [V6O11(OMe)2(TRIOLNO2)22− (2-(OMe)2) | −0.493 | +0.581 (0.99) | −0.002 (0.98) | 0.583 V | 9.8 × 109 |
With open circuit potential (OCP) values for complexes 1 and 2 shifted anodically of the first reduction event, we can assign the redox processes observed for the parent POV-alkoxide clusters as follows: the first reduction event corresponds to the one electron reduction of the isovalent parent cluster (e.g. VV6/VIVVV5), while the second reduction event results from the injection of a second electron into the hexavanadate core (e.g. VIVVV5/VIV2VV4). The difference between E1/2 values of complexes 1 and 2 (ΔE1/2) are 0.769 V and 0.801 V, respectively (Table 2). The large separation between electrochemical processes observed in these clusters suggests that vanadium centres within the cluster core exhibit a pronounced electronic interaction. Reduction of one vanadyl moiety results in a change of electron density communicated across the cluster core as a result of the delocalised electronic structure. Indeed, in previous work, Zubieta has reported the synthesis and characterisation of the mixed valent cluster, [V6O13(TRIOLNO2)2]3− (formal e-distrib. = VIVVV5).13 The electronic absorption spectrum of this reduced POV-alkoxide cluster possesses an IVCT band, assigned to electron transfer between VIV (d1) and VV (d0) ions of the Lindqvist core. This data supports a delocalised electronic structure for the reduced hexavanadate assembly.
Indeed, modelling the first reduction event of complex 1 and 2 reveals the first electron is delocalised across the six vanadium centres of the Lindqvist core, in agreement with the previously reported electronic absorption spectra of reduced variants of these POV-alkoxide clusters (Fig. 7). This delocalization indicates that the notation VIVVV5, which suggests the localisation of the VIV centre, is not in agreement with the observed electron structure. Thus, an oxidation state distribution notation of V4.8+6 might better describe the electronic structure of the monoreduced cluster. The second reducing equivalent added to the Lindqvist core localised the two electrons in opposite vanadium centres in 1 (Fig. S17†) and in four vanadium centres in a ring for 2, with a weak spin coupling between the vanadium and the μ2-O2− moieties. It is, however, worth noting that the M06 exchange–correlation functional used in this study might lead to overlocalisation of the unpaired electrons, however M06-L single points only reveal a slightly more delocalised picture but still with the spin mainly localised in two vanadium centres.
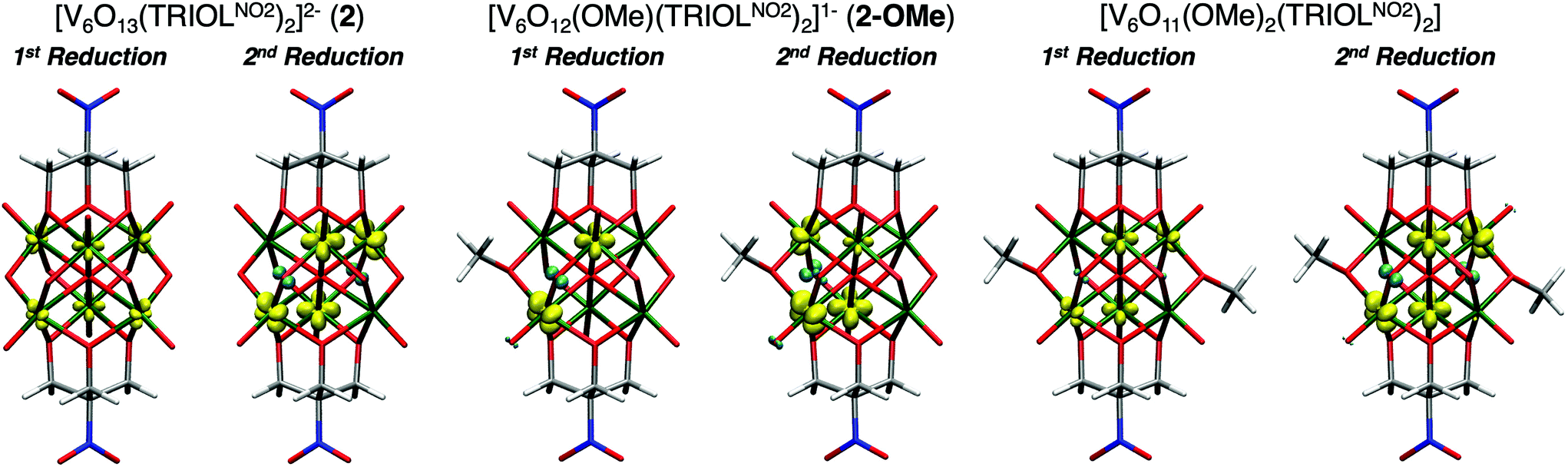 | ||
| Fig. 7 Spin density of reduced 2, 2-OMe, and para-[V6O11(OMe)2(TRIOLNO2)2] at M06/def2-TZVP//M06-L/def2-TZVP,def2-SVP level of theory. Vanadium in dark green, oxygen in red, nitrogen in blue, carbon in grey, and hydrogen in white. Alpha spin density in yellow and beta spin density in cyan. Isosurface = 0.4. Additional spin density plots for reduced variants of 1-OMe and meta-1-(OMe)2 can be found in Fig. S17.† | ||
Comproportionation equilibrium constants (Kc) are useful parameters for determining the extent of electron delocalisation in mixed-valent, multinuclear assemblies.43Kc can be calculated from the electrochemical data described above using the following equation (eqn (1)):
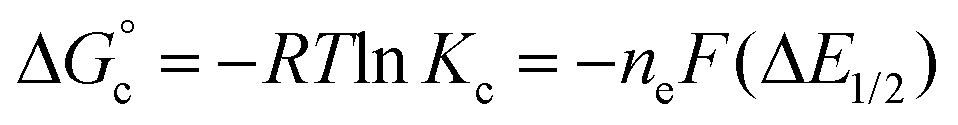 | (1) |
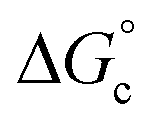 is the free energy of the comproportionation reaction, R corresponds to the universal gas constant, T is temperature (294 K), ne indicates number of electrons transferred (1 e−), F is Faraday's constant, and ΔE1/2 is the potential difference between adjacent redox couples (e.g. 0.769 V for complex 1, 0.801 V for complex 2).43 It is important to note that electrochemical profiles of multimetallic transition metal complexes (and thus, their Kc values) have been described by our group and others as highly sensitive to chemical environment of a sample during electrochemical analysis (e.g. solvent, supporting electrolyte).44–47 For the purpose of our discussion, we have limited analysis of the electrochemical properties of the vanadium oxide cluster core to CV measured in MeCN with [nBu4N][PF6] as the supporting electrolyte. This allows for direct comparison of differences in the electronic profiles of disparate clusters across identical reaction conditions. The calculated values of Kc for complex 1 and 2 are 1.5 × 1013 and 5.3 × 1013, respectively, indicating substantial delocalisation within the cluster core. This finding is consistent with the suggested formalism for oxidation state distribution of the cluster via Density Functional Theory of V4.8+6.
is the free energy of the comproportionation reaction, R corresponds to the universal gas constant, T is temperature (294 K), ne indicates number of electrons transferred (1 e−), F is Faraday's constant, and ΔE1/2 is the potential difference between adjacent redox couples (e.g. 0.769 V for complex 1, 0.801 V for complex 2).43 It is important to note that electrochemical profiles of multimetallic transition metal complexes (and thus, their Kc values) have been described by our group and others as highly sensitive to chemical environment of a sample during electrochemical analysis (e.g. solvent, supporting electrolyte).44–47 For the purpose of our discussion, we have limited analysis of the electrochemical properties of the vanadium oxide cluster core to CV measured in MeCN with [nBu4N][PF6] as the supporting electrolyte. This allows for direct comparison of differences in the electronic profiles of disparate clusters across identical reaction conditions. The calculated values of Kc for complex 1 and 2 are 1.5 × 1013 and 5.3 × 1013, respectively, indicating substantial delocalisation within the cluster core. This finding is consistent with the suggested formalism for oxidation state distribution of the cluster via Density Functional Theory of V4.8+6.
Notably, the calculated Kc values of complexes 1 and 2 are orders of magnitude larger than those reported POV-alkoxide clusters fully substituted with alkoxide ligands (e.g. [V6O7(OMe)12]n, avg. ΔE1/2 = 0.52 V, Kc = 3.7 × 108–1.8 × 109; electrochemistry measured in MeCN with [nBu4N][PF6] as the supporting electrolyte).48 This observation suggests that the extent of surface functionalisation of the cluster core influences electronic communication between vanadium centres. To probe this hypothesis, we examined the electrochemical profiles of the mono- and di-functionalised POV-alkoxide clusters, complexes X-OMe and X-(OMe)2 (X = 1, 2).
The CV of complex 1-OMe possesses two reversible redox events (E1/2 = −0.231, −1.066 V vs. Fc+/0). Both reduction events are shifted anodically from that of the parent cluster, complex 1, by approximately 0.7 V. This positive shift in reduction potential can be attributed to the overall decrease in charge of the cluster following the substitution of a methoxide for bridging oxide ligand; the lower charge of the POV cluster reduces the amount of energy required to add a single electron to the assembly. Support for the overall reduction in charge of the most oxidised form of the cluster as justification for the anodic shifts in the electrochemical profiles following surface functionalisation can be obtained from the CV of complex 2-OMe; the electrochemical profile of this cluster possesses two reversible reduction events (E1/2 = 0.087, −0.746 V vs. Fc+/0), likewise shifted from that of complex 2 by ∼+0.7 V.
The discrepancies in reduction potentials of complexes 1-OMe and 2-OMe are inconsistent with our analysis of the electronic absorption profile of the mono-alkylated POV-alkoxide clusters (vide supra). In previous work, Zubieta has qualified that the correlation of shifts in the electronic transitions observed in the absorption spectra of complexes 1 and 2 to their first reduction potentials indicates that reducing equivalents are placed into molecular orbitals that are influenced by the electronic properties of the tris(hydroxymethyl)methane ligands. The similarities in the absorption spectra prompted a hypothesis that the potentials of reduction for complexes 1-OMe and 2-OMe might be similar to one another. However, the reduction potentials of complex 2-OMe are shifted anodically by ∼0.3 V from that of 1-OMe. It is worth noting that this shift in reduction potential is of similar magnitude to that observed in between complexes 1 and 2: this result suggests that the inductive effect of the capping ligands remains a substantial contributor to the energetics of the cluster core, despite the minimal differences in the absorption profile of the assembly.
The separation of individual reduction events in complexes 1-OMe and 2-OMe (ΔE1/2 = 0.835 and 0.833 V, respectively) is quite similar to the ΔE1/2 values obtained for complexes 1 and 2 (Table 2). However, calculation of the Kc values for complexes 1-OMe and 2-OMe (2.1 × 1014 and 1.9 × 1014, respectively) reveal an increase of an order of magnitude over that of the parent cluster complexes. To our surprise, this result actually suggests that alkylation of a single bridging oxide unit at the surface of the cluster enhances electronic communication between triads of vanadyl ions in the cluster core.
The CVs of the bis-alkylated derivatives, complexes 1-(OMe)2 and 2-(OMe)2, again possess two reversible redox events. The first reduction process is shifted by ∼+0.5 V from that of their mono-alkylated congeners (Table 2), while the second redox event is shifted by ∼+0.7 V. The relative magnitude of both anodic shifts are again consistent with the decreased charge of the cluster upon introduction of a second methoxide moiety. However, the 200 mV discrepancy suggests that the introduction of the second alkoxide ligand has less influence over the energetics required for adding the first electron to the hexavanadate assembly. In any case, this difference in the change in redox potentials manifests in a significant decrease in ΔE1/2 of the two reduction events (ΔE1/2 (1-(OMe)2 = 0.591 V; ΔE1/2 (2-(OMe)2 = 0.583 V). These values translate to comproportionation constants for complexes 1-(OMe)2 and 2-(OMe)2 of 1.4 × 1010 and 9.8 × 109, respectively. The Kc are orders of magnitude lower than those reported for the unfunctionalised POV-alkoxide clusters and the mono-alkylated derivatives, resembling more closely those reported for [V6O7(OMe)12]n (vide supra).
We propose that this decrease in electronic communication between the two halves of the POV cluster observed upon alkylation results is due in part to the fact that bridging alkoxide ligands are less efficient at mediating electronic exchange in comparison to bridging oxide moieties. This hypothesis is consistent with a report by Hartl and coworkers, describing an increase in ΔE1/2 of POV-alkoxide clusters upon the substitution of a single bridging alkoxide ligand for a bridging oxide moiety (e.g. [V6O7(OMe)12]nvs. [V6O8(OMe)11]n).41 The authors attribute this change exclusively to strong antiferromagnetic exchange in low-valent POV clusters which possess bridging oxide moieties, a property which is diminished upon surface functionalisation of the vanadium oxide assembly. It is worth noting that the increased Kc values of complexes 1-OMe and 2-OMe offer evidence contradicting this hypothesis; at best these clusters exhibit comparable comproportionation constants to that of the parent POV-alkoxides, compounds 1 and 2. Ongoing investigations are focused on magnetic analysis of these molecular assemblies to ascertain the role exchange coupling plays in dictating electronic delocalisation across the cluster core.
It is worth noting that across the series of sequentially functionalised POV-alkoxide clusters, the change in potential of the first reduction event of similarly substituted clusters (e.g. complex 1-OMevs.2-OMe) differing only by the identity of the methyl or nitro functional group remains ∼0.3 V. Analysis of the frontier orbitals reveals that the LUMO of 2 lies 0.34 eV lower in energy than the LUMO of 1 (Fig. S18 and 19;† frontier orbitals for all reported complexes can be found in Fig. S18–S23†), justifying the anodic shift observed when comparing the electrochemical profile of the parent POV-alkoxide clusters. This is also true for the 1-OMe/2-OMe and 1-(OMe)2/2-(OMe)2 pairs which present a similar energy difference between the methyl and nitro TRIOL ligands. These results are in agreement with the experimental redox potentials where the more electron donating character of the methyl-backed triol derivatives render reduction of the cluster more challenging relative to the nitro-functionalised triol species. Additionally, we computed the redox potentials against Fc+/Fc in acetonitrile, however they are systematically overestimated by 390 mV on average towards lower potentials (Table S5†). Nevertheless, the calculated redox potentials reproduce the experimentally observed trend shifting the redox potential towards more positive values upon alkylation observed in Fig. 6 (e.g. LUMO energy of 1, 1-OMe, and 1-(OMe)2 are −2.95, −3.52, and −4.16 eV respectively, in agreement with the observed shift in the first reduction potential of each complex).
These observations are in accordance with the initial findings of Zubieta and coworkers, describing the fact that the electronic structure of [V6O13(TRIOLR)2]2− is highly sensitive to the character of the “R” substituent on the tris(hydroxymethyl)methane ligand.13 In stark contrast, our research group previously reported the synthesis and electrochemical characterisation of a family of tris(hydroxymethyl)methane substituted POV-alkoxide clusters [V6O7(OEt)9(TRIOLR)]; in examining the redox profiles of complexes [V6O7(OEt)9(TRIOLEt)]n and [V6O7(OEt)9(TRIOLNO2)]n, the peripheral functional group modulates the potential of redox events of the cluster core by only −0.10 V.37 Broadly speaking, the redox profile of the cluster core bearing twelve alkoxide functionalities appears relatively impervious to the electron donating or electron withdrawing properties of the “R” substituent of the tris(hydroxymethyl)methane ligand. We attribute this property to a relative decoupling of the electronic structure of the cluster core to its peripheral ligands. It remains unclear at what point the electronic structure of the cluster changes such that the redox properties become more dependent on molecular orbitals localised to the vanadium oxide core, however at the extent of surface functionalisation available through these studies, we can see the TRIOL substituent still exhibits a high degree of control over the resultant redox chemistry of the high-valent POV-alkoxide clusters. This translates to the isolation of a series of organo-soluble POV-alkoxide clusters with tuneable redox potentials that range a ∼3 V window.
Conclusions
Here, we describe the synthesis of a family of surface functionalised POV-alkoxide clusters. The development of post-synthetic modification strategies via the addition of MeOTf to the parent high-valent Lindqvist-type assemblies gives access to homogeneous samples of sequentially substituted clusters. Isolation of these complexes presents a platform to interrogate the physicochemical consequences of organofunctionalisation of the hexavanadate core. Structural analysis of all clusters via SCXRD reveals only minor perturbations to the Lindqvist assembly upon surface alkylation. These changes in bond metrics are localised to the V–Ob distances corresponding to μ2-O2− ligands that are transformed to μ2-OMe1− moieties. As expected, a significant elongation of the V–Ob bonds are noted, which slightly distorts the alkoxide ligand from the Lindqvist lattice (as evidence by a decrease in the magnitude of the V–Ob–V angles).Further characterisation of the series of POV-alkoxide clusters via electrochemical analysis indicate a handful of notable findings, that with appropriate caution, can be used to consider the consequence of surface functionalisation of metal oxide nanomaterials. First, general trends highlighted for the established family of POV-alkoxide clusters reveal that the extent of surface functionalisation appears to diminish electronic communication across the cluster core. It is important to note that in the case of the mono-functionalised POV-alkoxide clusters presented in this work, complexes 1-OMe and 2-OMe, comproportionation constants increase from the values determined for the parent clusters. This observation deviates from the general trend observed when comparing these values for the parent, difunctionalised (complexes 1-(OMe)2 and 2-(OMe)2), and fully-substituted (e.g. [V6O7(OR)12]n) vanadium oxide assemblies. While collectively, these serve as interesting observations, it is challenging to predict how this would translate to nanoscale assemblies of metal oxides. In the case of POV-alkoxide clusters, the low surface to volume ratio naturally results in a more substantial influence of the ligand shell on the electronic properties of the cluster core. The second key observation relates to the shift in redox potential (band energy) of the cluster as a function of surface ligation. It is likely that this trend holds in the case of nanocrystals, as substitution of a terminal oxido moiety (i.e. O2−) for a monoanionic alkoxide ligand requires charge balance the injection of electron density into the conduction band (i.e. reduction of a metal ion to support lower charge density of surface ligands). The result is a stabilised, reduced nanoassembly, a trend supported by the anodic shifts in redox potentials of the sequentially functionalised POV-alkoxide clusters. This result reveals that judicial selection of ligands offers additional opportunities to engineer defects in metal oxide nanomaterials.
Collectively, the synthetic, spectroscopic, and computational analyses summarised in this work present new insight into the physicochemical consequences of surface functionalisation of nanoscopic metal oxide assemblies. Coupled with electronic characterisation of the cluster core, these studies provide novel strategies for engineering redox potentials of vanadium oxide clusters. Ongoing efforts in our laboratory are focused on developing an understanding of how surface functionalisation translates to selective in hydrogen uptake mechanisms of these cluster complexes, translating these structure–function investigations to applications in small molecule activation and catalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. A. F, and E. M. M. acknowledge support from the Department of Energy Office of Basic Science through grant DE-SC0002106. M. D. K. acknowledges financial support from the NSF through grant CHE-1900125 and the University of Rochester. All computations supporting their project were performed on High Performance Computing systems at the University of South Dakota funded by NSF MRI award OAC-1626516. S. M. G. R. and P. M. are grateful to the Department of Chemistry of the University of South Dakota for start-up funds and graduate support. P. M. and S. M. G. R. acknowledge that the land the computational part of this research was performed on is the original homelands of the Dakota, Lakota, and Nakota tribal nations. P. M. and S. M. G. R. acknowledge the painful history of genocide and forced removals from this territory, and we honor and respect the many diverse Indigenous peoples still connected to this land on which we gather, giving thanks for our opportunity to gather on this land today.References
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed
.
- J. De Roo, I. Van Driessche, J. C. Martins and Z. Hens, Nat. Mater., 2016, 15, 517–521 CrossRef CAS PubMed
- J. Pal and T. Pal, Nanoscale, 2015, 7, 14159–14190 RSC
- S. Schauermann, N. Nilius, S. Shaikhutdinov and H.-J. Freund, Acc. Chem. Res., 2013, 46, 1673–1681 CrossRef CAS PubMed
- F. Zaera, Chem. Soc. Rev., 2013, 42, 2746–2762 RSC
- S. Wang, Z. Wang and Z. Zha, Dalton Trans., 2009, 9363–9373, 10.1039/B913539A
- L. Chen, Z. Liu, Z. Guo and X.-J. Huang, J. Mater. Chem. A, 2020, 8, 17326–17359 RSC
- Q. Zhang, E. Uchaker, S. L. Candelaria and G. Cao, Chem. Soc. Rev., 2013, 42, 3127–3171 RSC
- P. G. Bruce, B. Scrosati and J.-M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS PubMed
- X. Yu, T. J. Marks and A. Facchetti, Nat. Mater., 2016, 15, 383–396 CrossRef CAS PubMed
- X. Liang, S. Bai, X. Wang, X. Dai, F. Gao, B. Sun, Z. Ning, Z. Ye and Y. Jin, Chem. Soc. Rev., 2017, 46, 1730–1759 RSC
- A. Heuer-Jungemann, N. Feliu, I. Bakaimi, M. Hamaly, A. Alkilany, I. Chakraborty, A. Masood, M. F. Casula, A. Kostopoulou, E. Oh, K. Susumu, M. H. Stewart, I. L. Medintz, E. Stratakis, W. J. Parak and A. G. Kanaras, Chem. Rev., 2019, 119, 4819–4880 CrossRef CAS PubMed
- Q. Chen, D. P. Goshorn, C. P. Scholes, X. L. Tan and J. Zubieta, J. Am. Chem. Soc., 1992, 114, 4667–4681 CrossRef CAS
- F. Ahangaran and A. H. Navarchian, Adv. Colloid Interface Sci., 2020, 286, 102298 CrossRef CAS PubMed
- T. Rajh, L. X. Chen, K. Lukas, T. Liu, M. C. Thurnauer and D. M. Tiede, J. Phys. Chem. B, 2002, 106, 10543–10552 CrossRef CAS
- R. Deshmukh and M. Niederberger, Chem. – Eur. J., 2017, 23, 8542–8570 CrossRef CAS PubMed
- Z. B. Shifrina, V. G. Matveeva and L. M. Bronstein, Chem. Rev., 2020, 120, 1350–1396 CrossRef CAS PubMed
- E. Schechtel, R. Dören, H. Frerichs, M. Panthöfer, M. Mondeshki and W. Tremel, Langmuir, 2019, 35, 12518–12531 CrossRef CAS PubMed
- A. M. Nauth, E. Schechtel, R. Dören, W. Tremel and T. Opatz, J. Am. Chem. Soc., 2018, 140, 14169–14177 CrossRef CAS PubMed
- K. Davis, B. Cole, M. Ghelardini, B. A. Powell and O. T. Mefford, Langmuir, 2016, 32, 13716–13727 CrossRef CAS PubMed
- N. Guijarro, M. S. Prévot and K. Sivula, Phys. Chem. Chem. Phys., 2015, 17, 15655–15674 RSC
- N. I. Gumerova and A. Rompel, Nat. Rev. Chem., 2018, 2, 0112 CrossRef CAS
- T. M. Anderson and C. L. Hill, Inorg. Chem., 2002, 41, 4252–4258 CrossRef CAS PubMed
- M. Nyman and P. C. Burns, Chem. Soc. Rev., 2012, 41, 7354–7367 RSC
- B. E. Petel and E. M. Matson, Inorg. Chem., 2020 DOI:10.1021/acs.inorgchem.0c02052
- A. J. Kibler and G. N. Newton, Polyhedron, 2018, 154, 1–20 CrossRef CAS
- A. V. Anyushin, A. Kondinski and T. N. Parac-Vogt, Chem. Soc. Rev., 2020, 49, 382–432 RSC
- J. Zhang, Y. Huang, G. Li and Y. Wei, Coord. Chem. Rev., 2019, 378, 395–414 CrossRef CAS
- A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009–6048 CrossRef CAS PubMed
- P. Gouzerh and A. Proust, Chem. Rev., 1998, 98, 77–112 CrossRef CAS PubMed
- L. A. Combs-Walker and C. L. Hill, Inorg. Chem., 1991, 30, 4016–4026 CrossRef CAS
- C. Daniel and H. Hartl, J. Am. Chem. Soc., 2005, 127, 13978–13987 CrossRef CAS PubMed
- Y. Hayashi, Coord. Chem. Rev., 2011, 255, 2270–2280 CrossRef CAS
-
M. Pope, Heteropoly and Isopoly Oxometalates, 1983 Search PubMed
- K. Uehara, T. Miyachi, T. Nakajima and N. Mizuno, Inorg. Chem., 2014, 53, 3907–3918 CrossRef CAS PubMed
- A. M. Kosswattaarachchi, L. E. VanGelder, O. Nachtigall, J. P. Hazelnis, W. W. Brennessel, E. M. Matson and T. R. Cook, J. Electrochem. Soc., 2019, 166, A464–A472 CrossRef CAS
- B. E. Schurr, O. Nachtigall, L. E. VanGelder, J. Drappeau, W. W. Brennessel and E. M. Matson, J. Coord. Chem., 2019, 72, 1267–1286 CrossRef CAS
- M. Piepenbrink, M. U. Triller, N. H. J. Gorman and B. Krebs, Angew. Chem., Int. Ed., 2002, 41, 2523–2525 CrossRef CAS PubMed
- D. Hou, G.-S. Kim, K. S. Hagen and C. L. Hill, Inorg. Chim. Acta, 1993, 211, 127–130 CrossRef CAS
- A. Adach, M. Daszkiewicz and M. Cieślak-Golonka, Polyhedron, 2012, 47, 104–111 CrossRef CAS
- C. Daniel and H. Hartl, J. Am. Chem. Soc., 2009, 131, 5101–5114 CrossRef CAS PubMed
- J. Spandl, C. Daniel, I. Brüdgam and H. Hartl, Angew. Chem., Int. Ed., 2003, 42, 1163–1166 CrossRef CAS PubMed
- M. D. Ward, Chem. Soc. Rev., 1995, 24, 121–134 RSC
- R. Hernández Sánchez, S.-L. Zheng and T. A. Betley, J. Am. Chem. Soc., 2015, 137, 11126–11143 CrossRef PubMed
- F. Barrière, N. Camire, W. E. Geiger, U. T. Mueller-Westerhoff and R. Sanders, J. Am. Chem. Soc., 2002, 124, 7262–7263 CrossRef PubMed
- E. Schreiber, N. A. Hartley, W. W. Brennessel, T. R. Cook, J. R. McKone and E. M. Matson, ACS Appl. Energy Mater., 2019, 2, 8985–8993 CrossRef CAS
- D. M. D'Alessandro and F. R. Keene, Dalton Trans., 2004, 3950–3954, 10.1039/B413980A.
- E. Schreiber, B. E. Petel and E. M. Matson, J. Am. Chem. Soc., 2020, 142, 9915–9919 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details describing the synthesis and characterisation (1H NMR, ESI-MS, and IR data) of complexes 1-OMe, 1-(OMe)2, 2-OMe, 2-(OMe)2; electronic absorption spectra of complexes 1-(OMe)2 and 2-(OMe)2; crystallographic parameters of complexes 1-OMe, 1-(OMe)2, 2-OMe, and 2-(OMe)2; computation details including energies, electrostatic potentials, orbitals, spin density distributions, and optimized geometries. CCDC 2053154 (1-OMe); 2053156 (1-(OMe)2); 2053155 (2-OMe); 2053153 (2-(OMe)2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0nr09201k |
| This journal is © The Royal Society of Chemistry 2021 |