
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural variation of self-assembled lanthanide supramolecular complexes induced by reaction conditions
King-Him
Yim
a,
Chi-Tung
Yeung
 a,
Ho-Yin
Wong
a and
Ga-Lai
Law
*ab
a,
Ho-Yin
Wong
a and
Ga-Lai
Law
*ab
aState Key Laboratory of Chemical Biology and Drug Discovery, Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong. E-mail: ga-lai.law@polyu.edu.hk
bThe Hong Kong Polytechnic University Shenzhen Research Institute, Shenzhen 518000, PR China
First published on 6th May 2021
Abstract
The chemistry of lanthanide supramolecular self-assembly has received much attention with many extraordinary structures discovered due to the unique photophysical properties such as the Laporte forbidden f–f transition, large Stokes shift and long luminescence lifetime of lanthanides. Recent investigations have demonstrated the formation of architectures that are highly sensitive towards different stimuli, such as concentration, light, solvent and counter-anions. Various stimuli have been employed extensively for the preparation of desired supramolecular topologies with specific properties. Moreover, transformation from lower order- to higher order-supramolecular systems has also been observed by various stimuli due to the labile nature of lanthanide ions. This review summarizes recent research on the factors that govern the formation of self-assembled lanthanide supramolecules and aims to provide readers with the information required for designing functional lanthanide supramolecular systems.
 King-Him Yim (middle), Chi-Tung Yeung (left) and Ho-Yin Wong (right) | Mr King-Him Yim obtained his BSc in chemical technology from The Hong Kong Polytechnic University in 2017. He is currently a PhD student under the supervision of Dr Ga-Lai Law. His research focuses on the design of self-assembled lanthanide supramolecular architectures. Dr Chi-Tung Yeung received his PhD in applied chemistry from the City University of Hong Kong under the supervision of Prof. Hoi-Lun Kwong. Currently, he works at the Hong Kong Polytechnic University as a research fellow. His main research interests focus on rare earth chemistry, supramolecular chemistry, organocatalysis and asymmetric catalysis. Dr Ho-Yin Wong gained his BSc in Chemical Technology from the Hong Kong Polytechnic University in 2015. After that, he finished his PhD degree in chemistry at the same institute under the supervision of Dr Ga-Lai Law. His research interest focuses on the development of lanthanide compounds with luminescence and magnetic properties. |
 Ga-Lai Law | Dr Ga-Lai Law received her undergraduate degree and MChem from The University of Manchester, and later a Ph.D. from The University of Hong Kong with Prof. Wing-tak Wong, researching on “Synthesis and photo-physical studies of organic lanthanide complexes” Following this, she carried out two postdoctoral studies at Durham University with Prof. David Parker and at The University of California, Berkeley, with Prof. Kenneth N Raymond. Currently, she is an associate professor at the Hong Kong Polytechnic University continuing her passion in the area of lanthanide chemistry. Her current research now focuses on utilising lanthanide properties as toolboxes for chemical imaging, bio-applications and the development of new luminescent materials. |
1. Introduction
Self-assembly of supramolecular edifices is an important process in biological systems because of its ability to construct a more stable, functional, and non-covalently linked three-dimensional architecture starting from simple one-dimensional molecules.1 For example, the cell membrane is formed by the self-assembly of a phospholipid bilayer. Inspired by these smart natural systems, artificial supramolecular self-assembled structures have received intense attention in the past few decades.2 Similar to those natural systems, artificial supramolecular systems are constructed by self-assembly processes, in which the most thermodynamically favored products are formed by spontaneous assembly between organic ligands and lanthanide ions. These artificial systems are often formed by noncovalent interactions including hydrogen bonding, van der Waals forces and metal–ligand coordination.3–5 Among these different types of noncovalent interactions, metal–ligand coordination has been extensively applied as the main driving force for constructing artificial supramolecular systems. A large number of supramolecular structures based on transition metals have been reported.6–12 These structures have been studied well and have shown many promising applications as catalysts,13,14 magnetic materials,15 and molecular sensors and in bioimaging.16,17Apart from transition metals, lanthanide ions are also applied in preparing artificial supramolecular systems. Lanthanides exhibit unique photophysical and magnetic properties that can be attributed to their electronic configuration, in which the 4f orbitals are gradually filled across the series.18,19 Unlike transition metals, lanthanide ions exhibit characteristic excitation and emission spectra, which are less likely to be perturbed by the coordination environment.20 The use of lanthanide ions can lead to the formation of unique and stable species that show promising functional and responsive applications.
Many elegant and versatile lanthanide coordination assemblies have been reported, including helicates,21,22 cages,23,24 cubes,25 metal–organic frameworks26,27 and interlocked structures.28 However, the driving force and directing factor of lanthanide supramolecular chemistry are less understood. Construction of lanthanide self-assembled supramolecular architectures are more challenging compared to that of transition metals due to coordination diversity and the lack of stereochemical preferences.29,30 Lanthanide ions are considered as hard Lewis acids because of their large ionic radii and high charge density, causing them to accommodate large coordination numbers from 6 to 12, however by far, the most common coordination numbers are 8 to 9.30 Lanthanide ions highly prefer to saturate the inner coordination sphere. In the presence of methanol or water, lanthanide ions are labile, and ligands with weak binding power can be easily displaced by solvents.31,32 This causes the prediction of lanthanide self-assembled supramolecules to be a challenging prospect as it is difficult to successfully control the formed supramolecular topology.
To date, a number of excellent reviews that discuss lanthanide self-assembled supramolecular architects have been published.20,30,33,34 They focus on the coordination chemistry and photophysical properties of lanthanide ions and many latest examples of lanthanide supramolecular systems are well documented. However, the applications of these lanthanide self-assembled systems, such as in catalysis and molecular recognition, are all structure-dependent; therefore, beyond the chemical and physical properties of lanthanide ions, understanding the factors that govern the structural formation is also essential for the construction of functional supramolecular complexes. By fine-tuning the self-assembly process of the same building blocks, diverse supramolecular materials can be generated with variable structural properties and functions.
In order to provide new insight into the formation of lanthanide self-assembly supramolecular complexes, this review will focus on the factors that affect the structural formation of non-polymeric self-assembled complexes at the higher supramolecular level (number of metal ions per complex ≥2). Recent examples of supramolecular transformation will also be discussed.
2. Concentration-dependent supramolecular formation
The phenomenon of concentration-dependent supramolecular formation can be attributed to the labile feature of non-covalent coordination between lanthanide ions and organic ligands with weak coordinating power, in which the ligand and lanthanide ions will self-assemble randomly, giving rise to several constitutional isomers which can coexist in the solution state if there is no clear thermodynamic preference for one product over the other. According to Le Chatelier's principle, changes in concentration can trigger a supramolecular transformation even if the metal-to-ligand ratio remains unchanged. In most cases, an increase in concentration tends to increase the chance of aggregation of building blocks which promotes the formation of higher order supramolecular topology. To date, numerous examples based on transition metals have been well documented.2 However, such transformation strongly depends on the ligand design and it only occurs if the ligand design favors the formation of a higher order supramolecular product.An early example of lanthanide concentration-dependent supramolecular transformation was studied by Mazzanti's group in 2010.35 An enantiomerically pure ligand L1 was prepared in seven steps. Upon coordination with Eu3+ at a low concentration (7 mM), diastereomeric complexes (Δ)-[EuL12]+ and (Λ)-[EuL12]+ were formed and partial stereoselectivity at a ratio of 1.8 (Λ/Δ) was observed by NMR spectroscopy. At a higher concentration (14 mM), the NMR spectra showed an additional set of signals, suggesting the formation of a new species. These signals were assigned to the trinuclear complex (ΔΔΔ)-[EuL12]33+ and no other possible chiral trinuclear complexes, such as ΔΔΛ, ΔΛΛ and ΛΛΛ, could be observed (Fig. 1a). A molecular model suggested that the diastereoselectivity of the self-assembly process could be attributed to the phenyl substituents on the oxazoline ring, in which steric constraints were observed for ΔΔΛ, ΔΛΛ and ΛΛΛ isomers when mononuclear Λ complexes were self-assembled in proximity.
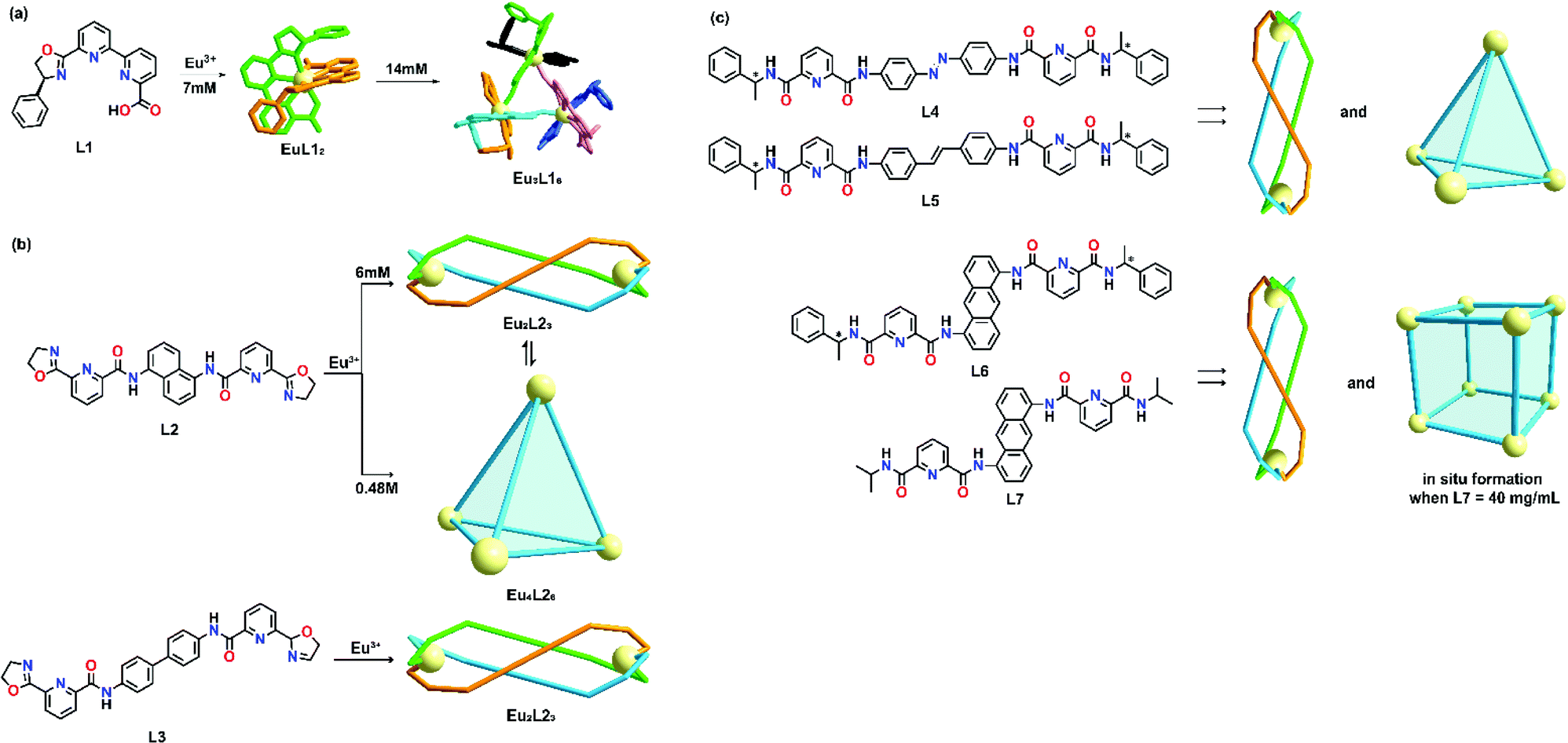 | ||
| Fig. 1 Concentration-dependent supramolecular formation of (a) L2, reproduced with permission from ref. 35. Copyright 2010 Wiley, (b) L2 and L3, reproduced with permission from ref. 36. Copyright 2017 The Royal Society of Chemistry, (c) L4, L5, L6 and L7, reproduced with permission from ref. 25. Copyright 2017 American Chemical Society. | ||
More recently, Sun's group demonstrated another example of concentration-dependent supramolecular formation in which the number of metal ions per complex could be increased from 2 to 4 with the use of two bis-tridentate oxazoline-based ligands, L2 and L3, incorporated with either a 1,5-diaminonaphthalene or benzidine as the central spacer.36 In this study, it was observed that coordination of L2 with Eu(OTf)3 at a low concentration (6 mM) in CD3CN would give rise to single helicate species Eu2L23. However, a mixture of both the Eu2L23 helicate and Eu4L26 cage was observed by proton NMR when the ligand concentration was increased 10-fold (Fig. 1b). The single species Eu4L26 was obtained when the concentration of L2 was increased to 0.48 M. Eu4L26 was found to be kinetically stable and would not rearrange back to Eu2L23 helicate upon dilution. When the spacer was changed to a more linear and flexible benzidine, only Eu2L33 helicate was observed under both diluted and concentrated conditions. This study implied that the ligand design also contributed significantly to concentration-dependent supramolecular formation. Ligands with shorter and more rigid spacers favor the formation of both a 1D helicate and 3D tetrahedral cage which have a higher chance to undergo concentration-dependent transformation compared with ligands with longer and more flexible spacers.
Based on the previous result, the relationship between ligand design and concentration effect was further studied by the same group. Four different pairs of ligands were prepared and the relationship between the supramolecular formation and the offset distance between two metal chelating groups was studied.25 It was found that increase in offset distance promoted the formation of a higher order architecture and Sun's group demonstrated a concentration-dependent supramolecular transformation from a bimetallic helicate to a tetranuclear cage by employing L4 and L5 (Fig. 1c). For L6 and L7, a bimetallic helicate and an octanuclear cube could be formed by varying the ligand concentration. Crystallization was found to be a useful technique for mimicking high ligand concentration. Pure Ln8L612 and Ln8L712 could be prepared by crystallization without performing the in situ synthesis at high ligand concentrations in CH3NO2.
3. Anion- and solvent-directed supramolecular formation
Counter-anions and solvents have been proven to affect the self-assembly process in transition metal-based supramolecular architectures. Anionic species with different shapes and sizes can act as templates to fill the inner cavity of the supramolecular structures. By incorporating different types of anions, various supramolecular structures with different properties can easily be prepared. In terms of the solvent effect, the solvent polarity may contribute to the result of supramolecular formation. Solvent molecules may also act as a template in the self-assembly process by interacting with organic building blocks, coordinating with the metal center and being encapsulated by the supramolecular cage.One of the successful examples of anion-directed supramolecular formation was reported by Liu's group in 2013.37 They designed and synthesized a flexible dentate-bridging ligand L8. Upon self-assembly of L8 with La3+, two different supramolecular clusters could be prepared depending on the anion template. Larger La6L86 hexanuclear circular helicates could be prepared when tetrahedral ClO4− was used, while a more compact La4L84 quadruple-stranded helicate was formed with the use of trigonal-planar NO3− (Fig. 2a). X-ray crystal structures showed that ClO4− and NO3− exhibited two different types of intermolecular interaction with the building blocks, which contributed to the formation of two different supramolecular topologies. For La6L86 hexanuclear circular helicates, two ClO4− anions interact with three coordinated methanol and the ligand through a hydrogen bond, while for La4L84 quadruple-stranded helicate, a NO3− anion coordinated to the La ions in a bridging mode to serve as a nucleation site. A supramolecular transformation from circular helicate to quadrupole-stranded helicate can also be achieved by adding NO3− anions as the templating anion.
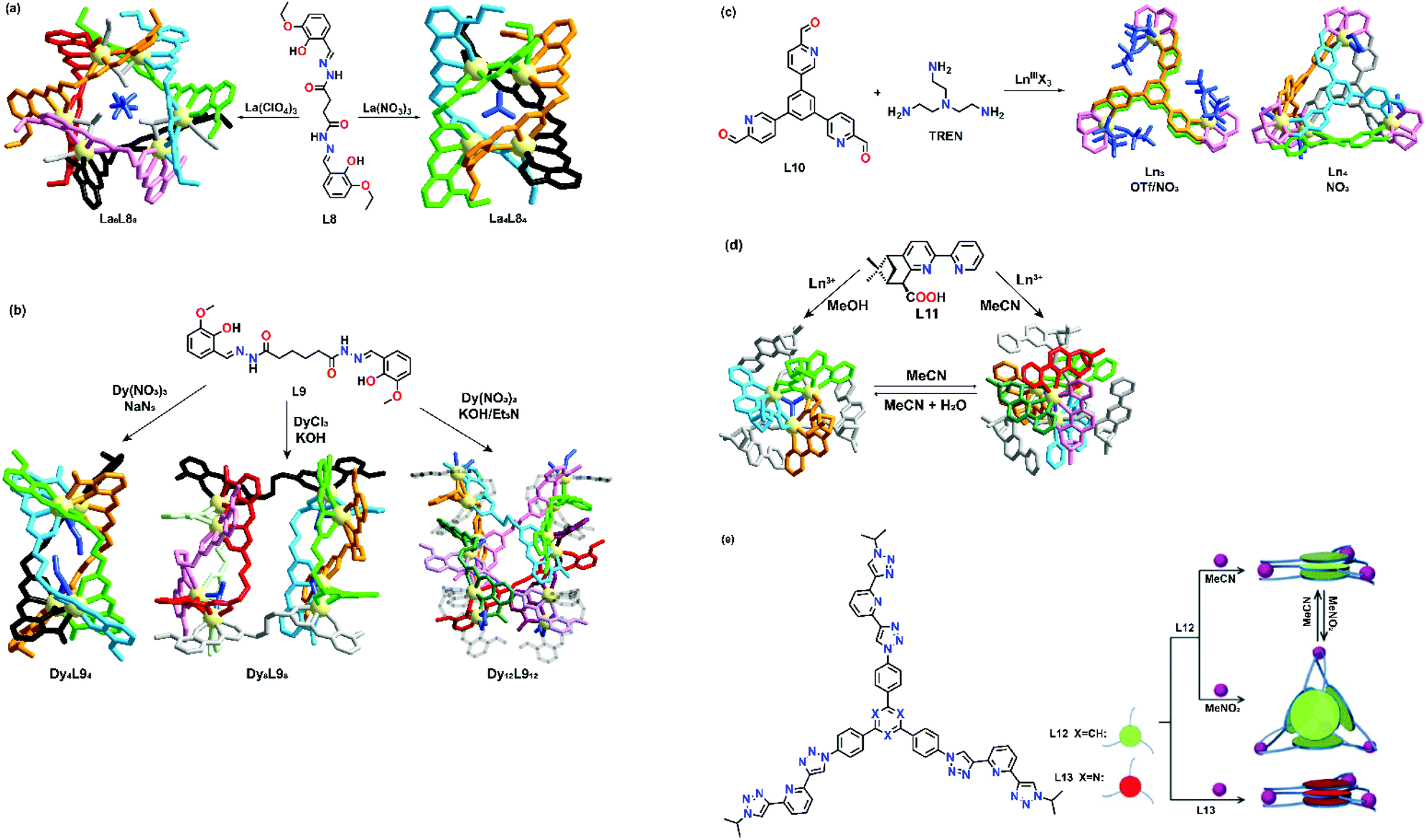 | ||
| Fig. 2 Anion- and solvent-directed supramolecular formation of (a) L8, reproduced with permission from ref. 37. Copyright 2019 Wiley, (b) L9, reproduced with permission from ref. 38. Copyright 2019 American Chemical Society, (c) L10, reproduced with permission from ref. 39. Copyright 2020 American Chemical Society, (d) L11, reproduced with permission from ref. 40. Copyright 2008 American Chemical Society and (e) L12 and L13, reproduced with permission from ref. 41. Copyright 2019 Wiley. | ||
Another example was shown by Zhang et al. who reported an example of supramolecular construction by varying the reaction conditions to give supramolecular products from helicates to octahedral cages. A flexible dihydrazone-based ligand L9 was employed and the ligand was allowed to coordinate with Dy3+ ions.38 Upon coordination with Dy(NO3)3·6H2O in NaN3, a quadruple-stranded helicate structure was obtained. When the anion and solvent were changed into Cl− and KOH, respectively, a dual triple-stranded helicate structure was formed (Fig. 2b). Moreover, two octahedral cage structures were formed when L9 was allowed to coordinate with Dy(NO3)3·6H2O in either KOH or Et3N. Solvent molecules and counter-anions were found in all crystal structures to stabilize the supramolecular products. Zhang et al. successfully demonstrated a new strategy for constructing interesting topologies. Under the same metal and ligand combination, a slight variation of the reaction conditions and anion could result in a drastic change in the final supramolecular structures.
A recent research study reported by Sun's group also showed anion-directed supramolecular formation. With the use of tris(2-aminoethyl)amine (TREN), a series of multinuclear lanthanide complexes were formed based on L10. Upon coordination of L10 with Ln(OTf)3·6H2O (Ln = La3+, Sm3+, Eu3+), pillar-typed triangular prisms were formed.39 However, when OTf− was replaced with NO3−, a mixture of Ln3L102(TREN)3 and Ln4L104(TREN)4 was observed in ESI-HRMS, which corresponds to the structure of a triangular prism and tetrahedral cage as evidenced by the X-ray crystal structures (Fig. 2c). Although the Eu center was nine-coordinated in both topologies, X-ray crystal structures showed that the Eu3+ coordination environment of the triangular prisms was different from that of the tetrahedral cage. In the triangular prisms, the Eu ion coordinated with two pyridyl N atoms, four N atoms of the TREN unit and three O atoms of three triflates, while the Eu3+ ion coordinated with three pyridyl N atoms, four N atoms of the TREN unit and two O atoms of one NO3− in the tetrahedral cage. Coordination of NO3− to the Eu3+ ions reduced the T symmetry of the cage, causing the six edges to become unequal.
A solvent-triggered supramolecular formation was observed by Mamula's group in 2007.40 In the study, a carboxylic derivative L11 was synthesized and used as a building block to coordinate with lanthanide ions. Upon coordination, two distinct complexes could be formed depending on the reaction solvent. Trinuclear structures [Ln3L116(OH)(H2O)3](ClO4)2 (Ln = La3+, Pr3+, Nd3+, Sm3+, Eu3+, Gd3+, Tb3+, Dy3+, Ho3+ and Er3+) were obtained when methanol was used, while tetranuclear complexes [Ln4L119(OH)](ClO4)2 (Ln = La3+, Pr3+, Nd3+, Sm3+, Eu3+, Gd3+ and Tb3+) were obtained when acetonitrile was used (Fig. 2d). Two structures were found to have undergone interconversion by either dissolving the trinuclear complexes in acetonitrile or adding a small amount of water into the tetranuclear complexes. Various factors that affect the interconversion were also studied in detail.
Another example of solvent-controlled supramolecular formation was also observed by Sun's group.41L12 and L13, two tri(tridentate) ligands with a small variation in the position of the central tridentate spacer, were designed and prepared. When an equimolar amount of Eu(OTf)3 was added to either L12 or L13 in a mixed solvent of acetonitrile and methanol (v/v, 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), Eu3(L12)3 and Eu3(L13)3 sandwich type structures were formed and the stoichiometric ratio and structures were confirmed by HRMS and molecular modelling, respectively. Interestingly, when L12 was allowed to coordinate with Eu(OTf)3 in nitromethane (with a lower polarity), an Eu4(L12)4 tetrahedral cage was formed. When the nitromethane solution of Eu4(L12)4 was evaporated and re-dissolved in acetonitrile by heating at 60 °C for 1.5 h, Eu4(L12)4 tetrahedral species could be converted into an Eu3(L12)3 sandwich structure (Fig. 2e). This system successfully demonstrated that supramolecular transformation is affected by solvent polarity.
:1), Eu3(L12)3 and Eu3(L13)3 sandwich type structures were formed and the stoichiometric ratio and structures were confirmed by HRMS and molecular modelling, respectively. Interestingly, when L12 was allowed to coordinate with Eu(OTf)3 in nitromethane (with a lower polarity), an Eu4(L12)4 tetrahedral cage was formed. When the nitromethane solution of Eu4(L12)4 was evaporated and re-dissolved in acetonitrile by heating at 60 °C for 1.5 h, Eu4(L12)4 tetrahedral species could be converted into an Eu3(L12)3 sandwich structure (Fig. 2e). This system successfully demonstrated that supramolecular transformation is affected by solvent polarity.
4. Cationic radii-dependent supramolecular formation
As stated in the Introduction, lanthanides are a series of metals with an incremental filling of the 4f orbitals across the row. This governs their unique optical and magnetic properties. Due to the variable coordination number and the lack of stereochemical preference of the lanthanide ion, minor structural variations in ligand design and subtle differences in ionic radii can render a high effect on the self-assembled supramolecular structures. In general, it is considered that the phenomenon of lanthanide contraction, i.e., a greater nuclear effect is experienced by the outer 5s25p6 orbitals as the nuclear charge increases, causes lanthanide ions to have similar sizes and reactivity profiles, which in turn simultaneously increases the difficulty in controlling the selectivity of lanthanide complexes. However, in the past years, more examples that show a deviation from this have been reported, demonstrating that lanthanides with different ionic radii alter the impact on supramolecule formations.An early study on radii-dependent lanthanide self-assembly coordination behavior was reported by Liu and co-workers in 2003.42 Upon coordination of lanthanide nitrate (Ln = La3+, Nd3+, Sm3+, Eu3+ and Ho3+) with a heptadentate ligand L14, a dinuclear lanthanide metal complex [Ln2L14(NO3)6] was formed, evidenced by IR and proton NMR. Upon crystallization, La and Nd complexes showed two different X-ray structures in the solid state. For [La2L14(NO3)6(H2O)]·H2O, two [LaL14(H2O)]3+ were connected by a hexanitrato anion [La(NO3)6]3− to form a trinuclear complex, while for the Nd complex, only a single [NdL14(H2O)]3+ complex and a hexanitrato complex anion [Nd(NO3)6]3− were connected by a bridging molecule NO3−. Detailed NMR studies showed that the ligand coordinated with La3+ more strongly than Nd3+, demonstrating the selectivity of the ligand towards different lanthanide ions. Both La and Nd complexes were found to exhibit different structural properties. For example, La complexes were in a parallel chain-like shape with no hydrogen bonds found in the structure, while for Nd complexes, a chiral cavity is formed by hydrogen bonds between two adjacent supramolecular chains (Fig. 3a).
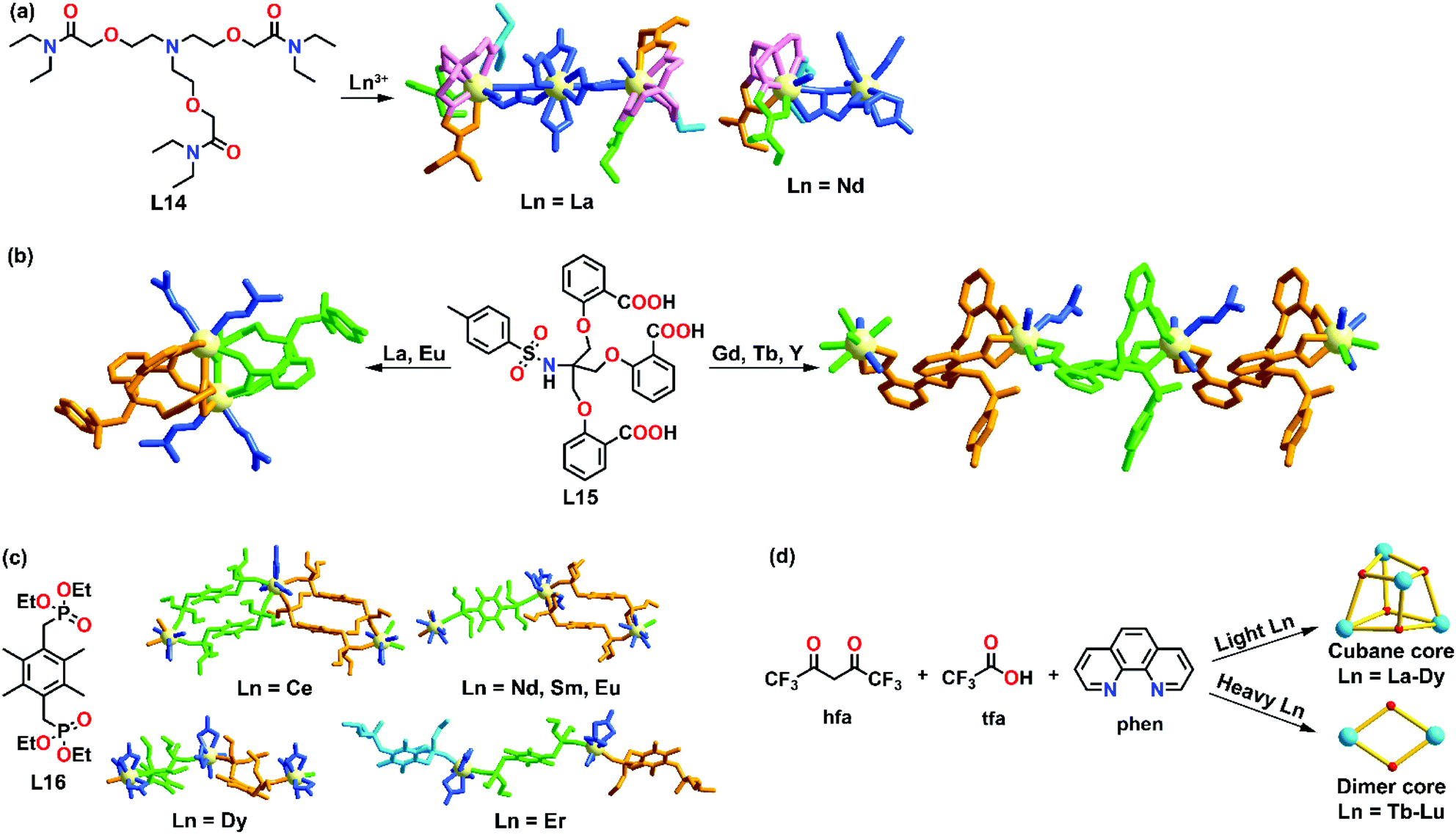 | ||
| Fig. 3 Cationic radii-dependent supramolecular formation of (a) L14, reproduced with permission from ref. 42. Copyright 2003 American Chemical Society, (b) L15, reproduced with permission from ref. 43. Copyright 2010 The Royal Society of Chemistry, (c) L16, reproduced with permission from ref. 44. Copyright 2011 The Royal Society of Chemistry and (d) a series of cubanes and cluster reported by our group, reproduced with permission from ref. 45. Copyright 2018 American Chemical Society. | ||
Based on these findings, Liu and co-workers further investigated the effect of ionic radii on supramolecular formation by utilizing a flexible tripodal salicylic acid L15 as a ligand.43 Five different lanthanide ions (La3+, Eu3+, Gd3+, Tb3+ and Y3+) were coordinated with L15 to form complexes with the formulae [La2L152(DMF)4]·4DMF·4EtOH·2H2O, [Eu2L152(DMF)4]·2DMF, {[GdL15(DMF)(H2O)2]·DMF}∞, {[TbL15(DMF)(H2O)2]·DMF}∞ and {[YL15(DMF)(H2O)2]·DMF}∞ (Fig. 3b). Single X-ray diffraction studies revealed that the La and Eu complexes were cage-like homodinuclear products, while the Gd, Tb and Y complexes were helical 1D coordination polymers. These results demonstrated that the early lanthanides (La3+ and Eu3+) and late lanthanides (Gd3+, Tb3+, Y3+) showed a difference in their supramolecular formation which was influenced by the differences in the ionic radii.
A similar lanthanide radii-controlled supramolecular formation of polymers was also reported by Liu's group. L16 was synthesized where a bi-phosphonate unit was employed as a bridging ligand.44 Upon coordination with lanthanide nitrate (Ln = Ce3+, Nd3+, Sm3+, Eu3+, Dy3+ and Er3+), four different supramolecular structures were observed depending on the ionic radii: a ribbon like polymer was formed for Ce3+; a semi-ribbon polymer for Nd3+, Sm3+ and Eu3+, a zigzag like polymer for Dy3+ and a dinuclear-tri-ligand short chain was observed for Er3+ (Fig. 3c). All the structures were characterized by IR spectroscopy, elemental analysis and X-ray crystallography. The Eu complex also exhibited luminescence properties and the quantum yield was found to be 28%.
Our group also reported an example of a metal-dependent supramolecular formation system.45 Two different clusters were formed based on different lanthanide ions. Under the same reaction conditions, a series of cubanes, [Ln4(μ3-OH)4(μ-tfa)4(hfa)4(phen)4] (tfa = trifluoroacetate, hfa = hexafluoroacetylacetonate, phen = 1,10-phenanthroline), were formed when Ln = La3+–Dy3+, while dinuclear clusters [Ln2(μ OH)2(hfa)4(phen)2] were self-assembled when Ln = Tb3+–Lu3+ (Fig. 3d). The difference in supramolecular formation could be explained by a change in the preferred coordination geometry. In this supramolecular system, lighter lanthanides favored the formation of nine-coordinated cubanes, while the heavier lanthanide ions preferred to form eight-coordinated dimers.
Apart from simple lanthanide clusters and 2D polymers, the formation of higher order 3D structures is also affected by lanthanide contraction. Hamacek's group was one of the pioneers to investigate the formation of lanthanide supramolecular 3D tetrahedral cages with extensive experience in designing tetrahedral cages. In 2008, Hamacek and co-workers reported an example of lanthanide supramolecular tetrahedral cages.46 With the use of a tripodal ligand L17, the tetrahedral cage Ln4L174 (Ln = Eu3+, Tb3+ and Lu3+) could be self-assembled, which was evidenced by proton NMR and ESI-HRMS. However, for a La complex, only large unresolved peaks in proton NMR could be observed and thus they proposed that the formation of La4L174 was not possible.
Hamacek's group further investigated the formation of a tetrahedral cage based on the design of L17.47 They synthesized L18 and L19, which were also characterized by NMR spectroscopy, mass spectroscopy and spectrophotometric titrations. This work showed their attempt to study the effect of lighter and heavier lanthanide ions on the self-assembly process. For lighter lanthanide ions (Ln = La3+, Nd3+ and Eu3+), the major product was found to be mono- or dinuclear species upon coordination with L19. The [Eu4L194]12+ tetrahedral cage could only be observed at high concentrations, and was found to dissociate upon dilution. In contrast, the coordination of L19 with heavier lanthanide ions (Ln = Tb3+, Er3+ and Lu3+) provided a well-defined and thermodynamically stable tetranuclear cage. The result showed that the self-assembly process of L19 with lanthanide ions differentiated the lanthanides along the series and it exhibited highly selective tetrahedral cage formation towards heavier lanthanide ions.
A more recent example of radii-dependent self-assembly of chiral lanthanide complexes was well demonstrated by Li et al. They designed and synthesized a pair of 3-methoxysalicylhydrazone-based homochiral ligands L20.48 Under the same reaction conditions, coordination of L20 with La3+ gave rise to a trinuclear complex, while a pentanuclear cluster was formed when Dy3+ was used (Fig. 4b). The result showed that the resulting supramolecular product was governed by the size of the lanthanide ion. The magnetic properties of the Dy complex were also examined and it exhibited typical single-molecular magnet behavior.
 | ||
| Fig. 4 Cationic radii-dependent supramolecular formation of 2D and 3D structures. (a) Structures of L17, L18 and L19. (b) Two different frameworks formed based on the size of lanthanide ions by L20, reproduced with permission from ref. 48. Copyright 2020 The Royal Society of Chemistry, (c) cationic radii-dependent supramolecular formation of L21, reproduced with permission from ref. 49. Copyright 2020 The Royal Society of Chemistry. | ||
Lanthanide contraction affects not only supramolecular formation, but also supramolecular transformation from one structure to another. Recently, Sun and co-workers reported an example of metal-dependent supramolecular transformation.49 A porphyrin-cored tetrakis-tridentate ligand L21 containing chiral pyridine-2,6-dicarboxamide-coordinating units was prepared and the supramolecular formation was highly sensitive to lanthanide ions. When L21 was coordinated with La3+, a La6(Zn-L21R)3 triangular prism was formed, while an Eu8(Zn-L21R)6 cube was formed when Eu3+ was used. A structural transformation from the La6(Zn-L21R)3 triangular prism to the Eu8(Zn-L21R)6 cube could be achieved by the addition of Eu (Fig. 4c). However, they reported that direct synthesis of Yb8(Zn-L21R)6 was not possible as the reaction mixture remained opaque after prolonged heating. Post-synthetic metal exchange occurred when Yb(OTf)3 was added to the Eu8(Zn-L21R)6 complex and stirred for 4 h. Such metal directed supramolecular transformation allowed the preparation of NIR-emitting Yb8L6 complexes.
5. Stoichiometric ratio-dependent supramolecular formation
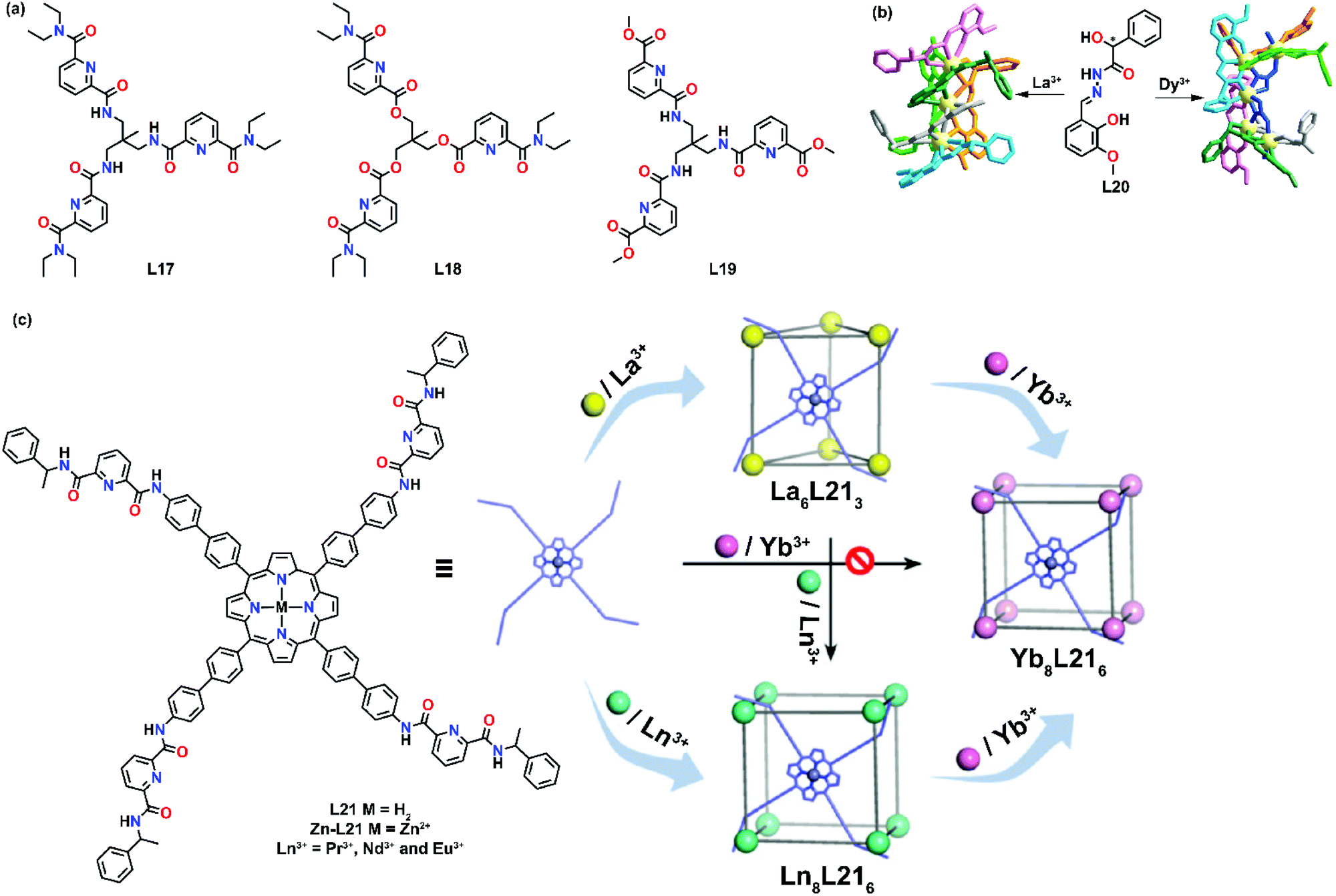
Unlike those preorganized ligands such as macrocycles and cyclen,50–52 the coordinating environment in dipodal ligands, tripodal ligands or even simple organic clusters is not well defined. For example, a lanthanide ion could have a coordination number of 9 when coordinated with one tridentate ligand and six solvent molecules when fewer ligands are employed, while the same coordination number also applies when three tridentate ligands are used to fulfill the coordination. Based on the stoichiometric ratio of the reaction conditions, ligands and metals will self-assemble to form the most thermodynamically stable product. With the use of simple organic building blocks, diverse supramolecular products can be formed by varying the metal-to-ligand ratio.Mazzanti's group is one of the frontiers in this aspect and they have reported several examples of stoichiometric cation-dependent supramolecular formation. In 2002, they demonstrated one of the examples of cation-controlled self-assembly of a hexameric europium wheel.53 Upon coordination of the simple asymmetric tetradentate ligand L22 with Eu(OTf)3 in a ratio of 2:1, a mononuclear eight-coordinated complex [Eu(L22)2](OTf) was formed (Fig. 5a). Interestingly, upon addition of a small amount of Eu(OTf)3 to [Eu(L22)2](OTf), another rigid species was formed. At a [Eu(L22)2](OTf):Eu(OTf)3 ratio of 0.16, the proton NMR spectrum showed only the presence of the rigid solution species which corresponded to the hexameric europium wheel [Eu⊂(EuL222)6](OTf)9 as evidenced by the X-ray crystal structure. In this system, the stochiometric ratio-dependent supramolecular formation is strongly influenced by the ligand design. In each [Eu(L22)2], one carboxylic group was responsible for bridging with the adjacent [Eu(L22)2] unit to form a ring, while the ring-encapsulated Eu3+ ion was coordinated by another carboxylic unit. Such a coordination environment caused the instability of [Eu⊂(EuL222)6](OTf)9 in methanol, in which disruption of the cyclic structure was observed due to the displacement of bridging carboxylates by methanol molecules.
 | ||
| Fig. 5 Structural properties of stoichiometric ratio-dependent supramolecular complexes: (a) Formation of hexameric lanthanide wheel by L22, reproduced with permission from ref. 54. Copyright 2007 American Chemical Society, (b) structure of L23–L26 and view of circular single-stranded helicate [Ln3(L23)3]9+, reproduced with permission from ref. 56. Copyright 2005 The Royal Society of Chemistry, (c) schematic representation of stoichiometric ratio-dependent supramolecular formation by L8, reproduced with permission from ref. 57. Copyright 2015 Wiley. (d) Structure of L27. | ||
A further study on this interesting hexameric lanthanide wheel was performed by the same group. With the use of the same ligand L22, they reported the evolution of supramolecular complexes from a homometallic wheel to a heterometallic wheel.54 A detailed study of the formation mechanism was also reported.
The formation of a heterometallic wheel was found to be highly sensitive to the ionic radii. Upon reaction of Ln(OTf)3 (Ln = Tb3+, Er3+, Yb3+, and Lu3+) with the monometallic [EuL222], the components self-assembled to form a highly selective heterobimetallic species [Ln⊂(EuL22)6](OTf)9 (Ln = Tb3+, Er3+, Yb3+ and Lu3+). When lanthanides with ionic radii either larger than or very similar to Eu3+, such as La3+, Sm3+ and Tb3+, were used, a very complicated proton NMR signal was observed and thus the formation of a heterometallic wheel was ruled out. The result showed that the formation of a heterometallic wheel was controlled by the size of the lanthanide ion. Smaller ions preferred to occupy the central site, while larger ions preferred the peripheral site.
Mazzanti's group extended the study of L1 in 2012.55 Concentration-dependent supramolecular formation was found in L1 and Mazzanti and co-workers further investigated the effect of the metal to ligand ratio on L1. Addition of Eu(OTf)3 to L1R in a 12:7 ratio gave rise to a heptameric enantiopure Eu wheel [Eu⊂(Λ-Eu(L1R)2Δ-Eu(L1R)2)3](OTf)9 and the structure was confirmed by X-ray crystallography. Moreover, excess addition of Eu(OTf)3 to a (ΔΔΔ/ΛΛΛ)-[EuL12]33+ trinuclear complex promoted the transformation into a heptanuclear complex. Similar to the case of L22 reported by the same group, the ligand design also played an important role in the supramolecular transformation. Six Eu(L1R)2 were connected by a bridging carboxylic oxygen to form a ring structure. One Eu3+ ion was encapsulated by six carboxylate oxygens to form a heptameric enantiopure Eu wheel.
Piguet and co-workers reported another example of stoichiometric ratio-dependent supramolecular formation.56 Four ligands (L23–L26) with semi-flexible secondary amide linkers were synthesized (Fig. 5b). For L25, [Ln2(L25)3]6+ was found upon coordination in a ratio of Ln:L25 0.67. While for L23, L24 and L26, a mixture of three complexes [Ln2(L)3]6+, [Ln2(L)2]6+ and [Ln3(L)3]9+ (L = L23, L24 and L26) was observed under the same reaction conditions. When the metal-to-ligand ratio was increased from 0.67 to 1, pure [Ln3(L23)3]9+ was observed as the only single product. X-ray crystal studies showed that [Ln3(L23)3]9+ existed as a circular trimetallic single-stranded helicate. No templating effects or intramolecular interactions were observed in [Ln3(L26)3]9+ but they suggested that [Ln3(L)3]9+ might be stabilized by three weak intermolecular (pyridine)CH–O(triflate) interactions.
L8 was found to exhibit anion-dependent self-assembly of a hexanuclear circular helicate and a quadruple-stranded helicate. In 2015, Konar's group further demonstrated the effect of the stoichiometric ratio on the self-assembly of L8 with lanthanide ions.57 Upon coordination with Eu(NO3)3·6H2O in a 1:1 metal-to-ligand ratio, a dinuclear triple-stranded helicate [Eu2L83] was isolated. When the ratio was varied to 2:1, a tetranuclear quadrupole-stranded helicate [Eu4L84(NO3)]3+ was observed (Fig. 5c). The X-ray crystal structures of the two supramolecular complexes displayed contrasting Eu-coordinating environments. Two Eu3+ ions were located along the three-fold axis in [Eu2L83], while four Eu3+ ions were arranged in a triangular fashion along the four-fold axis in [Eu4L84(NO3)]3+. The differences in the coordinating environment could be attributed to the ligand design. For the triple-stranded helicate [Eu2L83], ligands were homotopic and each Eu3+ center was filled by three phenol oxygens, three carbonyl oxygens and three imine nitrogens. While in the tetranuclear quadrupole-stranded helicate [Eu4L84(NO3)]3+, the ligands were asymmetrical and the Eu3+ center was coordinated in a tridentate and tetradentate mode that could be attributed to the involvement of the ethoxy-O and the η2 bridging mode of the phenoxido-O group to the Eu3+ center.
In continuation of their tripodal ligand studies, Hamacek and co-workers demonstrated the effect of the stoichiometric ratio on the formation of complexes by designing an asymmetrical tripodal ligand L27.58 Detailed NMR titration was done with Lu3+ as the metal source to observe the supramolecular formation. Upon addition of Lu3+ to L27 at [Lu]/[L27] ratios of 0.3, 0.65 and 1, [LuL273]3+, [Lu2L273]6+, and [Lu2L272]6+ were formed, respectively. [Lu2L27]6+ or [Lu3L27]9+ was formed when Lu3+ was in excess. A similar result was also observed for the symmetrical tripodal ligand L17.59 For example, reaction of L27 with Eu3+ at a ratio of 1:1 resulted in the formation of tetrahedral [Eu4L274].
6. UV-induced supramolecular transformation
Light has been utilized extensively to trigger the supramolecular transformation of transition metal complexes. One of the common strategies is to introduce a photochromic moiety into the ligand. Upon UV irradiation, the photochromic moiety undergoes a structural change, resulting in the transformation of metallosupramolecules. UV-induced supramolecular transformation has been shown to be a useful tool to prepare smart materials as their size and shape can be changed easily upon irradiation, and it is considered as a promising candidate in advanced information technology applications such as optical storage and sensors.In 2018, Sun's group synthesized dithienylethene-based L28 and a dinuclear triple-stranded europium helicate was prepared based on a chiral-induction strategy.60 The chiral information was successfully transmitted to the metal center to give either Δ or Λ metal, leading to an overall P or M helicate (Fig. 6a). Similar to the result of Li's group, the helicate also underwent a reversible photochromic transformation in the solution state. Upon irradiation at 334 nm, a distinct color change from colorless to dark blue took place and the photochromic transformation was evidenced by a characteristic shift of the signals in the proton NMR spectra. The helicate could be converted back to the original state by irradiating the solution at 630 nm for 5 min.
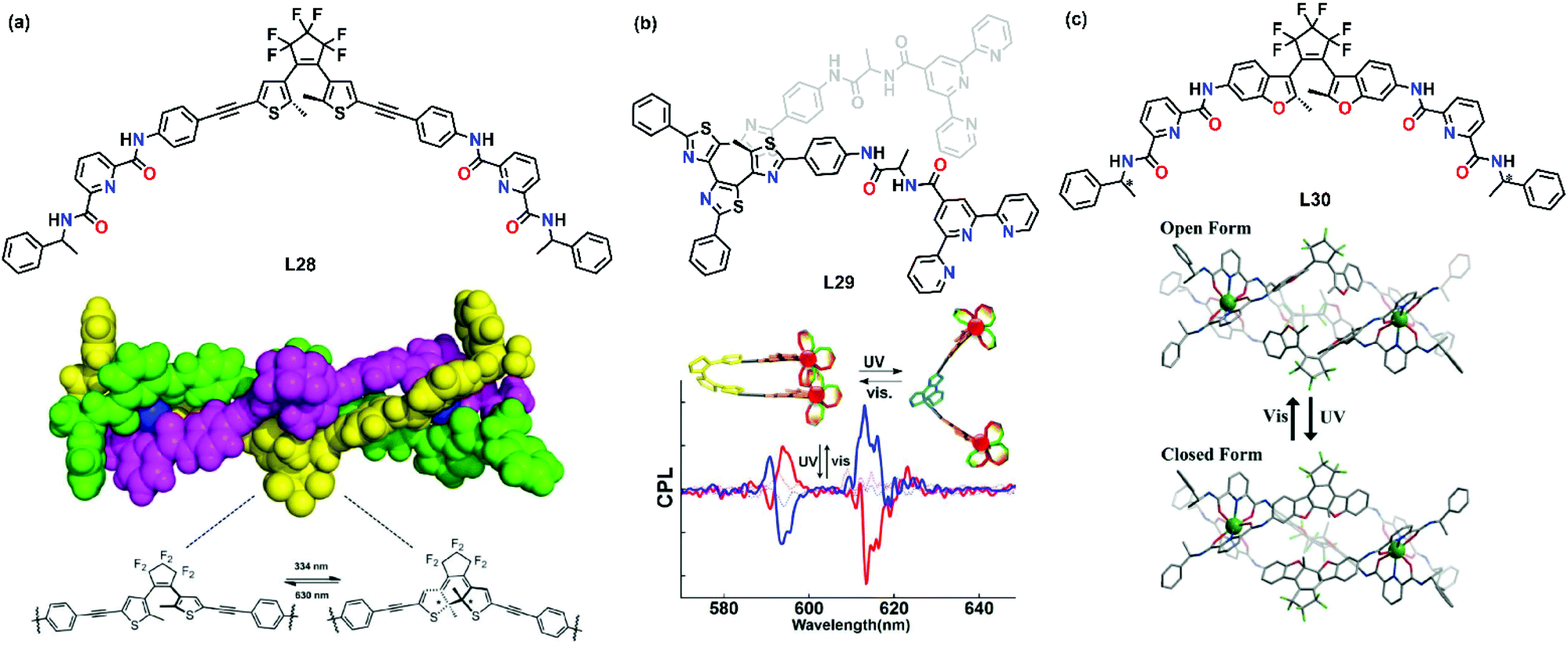 | ||
| Fig. 6 UV-induced supramolecular transformation of (a) L28, reproduced with permission from ref. 60. Copyright 2018 The Royal Society of Chemistry, (b) L29, reproduced with permission from ref. 61. Copyright 2018 American Chemical Society and (c) L30, reproduced with permission from ref. 62. Copyright 2018 The Royal Society of Chemistry. | ||
Hashimoto et al. demonstrated the on–off photoswitching of circularly polarized luminescence in a binuclear supramolecular system. They introduced terpyridine units at both ends of a helical tetrathiazole scaffold to form L29.61 Upon coordination, L29 and Eu3+ self-assembled to form a helical photochromic dinuclear complex with thenoyltrifluoroacetonate (tta) as the auxiliary ligand. It was found that the intramolecular hydrogen bond was essential for bringing two metal chelating sites close together and helped the transfer of chiral information from the point chirality of the amino acid spacer to the metal center. Thus, the asymmetric tta auxiliary ligand coordinated with Eu3+ in a chiral manner. Interestingly, a photoinduced helical to nonhelical transformation was observed and the chiral transfer was switched off upon irradiation of UV (Fig. 6b). This work demonstrated an example of photoswitching of the emission intensity owing to changes in the electronic structure of ligands.
In a more recent example, Li's group reported and developed lanthanide-based chiroptical photoswitches.62 Ligand L30 was designed based on the photochromic diarylethene moieties as central bridging linkers and two chiral pyridine-2,6-dicarboxamide (pcam) as chelating units. Coordination with Eu(OTf)3 resulted in the self-assembly of a pair of chiral triple-stranded helicates [Eu2(o-L30RR)3](OTf)6 and [Eu2(o-L30SS)3](OTf)6. Upon irradiation with UV and visible light, a reversible change of the diarylethene between a colorless open-ring state and a pink closed-ring state was observed (Fig. 6c). This photochromic process resulted in a variation of the electronic structures and it showed promising application in light-responsive optical and chiroptical smart photoswitches.
7. Conclusions and perspectives
Over the last decade, there has been a rapid development in the formation of self-assembled lanthanide supramolecules. A large number of impressive coordination-driven lanthanide supramolecular structures have been reported. Recent research has shown that the structural formation of supramolecular assemblies can be changed using various reaction conditions. Using recent examples, this review discussed the factors that affect the structural formation, such as concentration, solvent, metal cation, anion and light.By varying the reaction conditions, the structural properties have been shown to change dramatically even if the same building blocks are used. In terms of transition metal complexes, these structural conversions induced by various reaction stimuli have been shown to be a useful tool for preparing molecular probes and catalysts. For example, the size of the cavity can be changed by supramolecular transformation for specific host–guest encapsulation, or switchable catalysis systems can be prepared by changing the arrangement of functional catalytic sites of the supramolecule. This method has been proven to be an excellent approach for preparing a wide variety of supramolecular materials, and it can also be applied for lanthanides. For instance, the coordination environment of the lanthanide ion can be changed by varying the counter-anion; it can even change the structural hierarchy (from lower-order to higher-order) by increasing the ligand concentration.
There are still some difficulties in the development of this fast-growing research field. One of the challenges concerns the accurate prediction of the resulting supramolecular complexes. Since the application of supramolecular complexes, such as molecular recognition and catalysis, is structure dependent, a slight variation of complexes will result in different host–guest interactions. However, unlike transition metals, which exhibit strong directional coordination ability, lanthanide ions have variable coordination numbers, making the formation of the desired supramolecular complexes more difficult. Another challenge involves the stability of lanthanide supramolecular complexes in water. Most of the reported examples are not water-soluble. To date, only a limited number of water-soluble lanthanide supramolecular complexes (with a number of metal ions per complex ≥2) have been reported.63 As stated in the Introduction, ligands will easily exchange with water molecules if ligands with weak binding ability are used; thus, the application in biological aspects is limited. For further investigation of structural variations, more efforts should be devoted to investigating the ligand design principle and improving the stability of lanthanide complexes in solution state, especially in aqueous media.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
G.-L. L. acknowledges the financial support from the National Natural Science Foundation of China (NSFC, 21875201). We also acknowledge support from the Hong Kong Polytechnic University (University Research Facility in Chemical and Environmental Analysis [UCEA] and University Research Facility in Life Sciences [ULS]).References
- Y.-C. Lee, Introduction to bionanotechnology, Springer, Singapore, Singapore, 2020 Search PubMed.
- W. Wang, Y.-X. Wang and H.-B. Yang, Supramolecular transformations within discrete coordination-driven supramolecular architectures, Chem. Soc. Rev., 2016, 45, 2656–2693 RSC.
- J. M. Lehn, Toward self-organization and complex matter, Science, 2002, 295, 2400–2403 CrossRef CAS PubMed.
- A. Aliprandi, M. Mauro and L. De Cola, Controlling and imaging biomimetic self-assembly, Nat. Chem., 2015, 8, 10–15 CrossRef PubMed.
- L.-J. Chen, H.-B. Yang and M. Shionoya, Chiral metallosupramolecular architectures, Chem. Soc. Rev., 2017, 46, 2555–2576 RSC.
- R. M. Yeh and K. N. Raymond, Supramolecular asymmetric induction in dinuclear triple-stranded helicates1, Inorg. Chem., 2006, 45, 1130–1139 CrossRef CAS PubMed.
- M. C. Young, A. M. Johnson and R. J. Hooley, Self-promoted post-synthetic modification of metal–ligand M2L3 mesocates, Chem. Commun., 2014, 50, 1378–1380 RSC.
- A. P. Paneerselvam, S. S. Mishra and D. K. Chand, Linear and circular helicates: A brief review, J. Chem. Sci., 2018, 130, 1–18 CrossRef CAS.
- D. Zhang, M. J. Ronson, A. Martinez, L. Guy and J. R. Nitschke, Anion binding in water drives structural adaptation in an azaphosphatrane-functionalized FeII4L4 tetrahedron, J. Am. Chem. Soc., 2017, 139, 6574–6577 CrossRef CAS PubMed.
- J. S. Train, A. B. Wragg, A. J. Auty, A. J. Metherell, D. Chekulaev, C. G. P. Taylor, S. P. Argent, J. A. Weinstein and M. D. Ward, Photophysics of cage/guest assemblies: photoinduced electron transfer between a coordination cage containing osmium(II) luminophores, and electron-deficient bound guests in the central cavity, Inorg. Chem., 2019, 58, 2386–2396 CrossRef CAS PubMed.
- L. L. K. Taylor, I. A. Riddell and M. M. J. Smulders, Self–assembly of functional discrete three–dimensional architectures in water, Angew. Chem., Int. Ed., 2019, 58, 1280–1307 CrossRef CAS PubMed.
- M. Pan, K. Wu, J.-H. Zhang and C.-Y. Su, Chiral metal–organic cages/containers (MOCs): From structural and stereochemical design to applications, Coord. Chem. Rev., 2019, 378, 333–349 CrossRef CAS.
- X. Jing, C. He, Y. Yang and C. Duan, A metal–organic tetrahedron as a redox vehicle to encapsulate organic dyes for photocatalytic proton reduction, J. Am. Chem. Soc., 2015, 137, 3967–3974 CrossRef CAS PubMed.
- C. Zhao, Q.-F. Sun, W. M. Hart-Cooper, A. G. DiPasquale, F. D. Toste, R. G. Bergman and K. N. Raymond, Chiral amide directed assembly of a diastereo-and enantiopure supramolecular host and its application to enantioselective catalysis of neutral substrates, J. Am. Chem. Soc., 2013, 135, 18802–18805 CrossRef CAS PubMed.
- F. Habib and M. Murugesu, Lessons learned from dinuclear lanthanide nano-magnets, Chem. Soc. Rev., 2013, 42, 3278–3288 RSC.
- C. P. Montgomery, B. S. Murray, E. J. New, R. Pal and D. Parker, Cell-penetrating metal complex optical probes: targeted and responsive systems based on lanthanide luminescence, Acc. Chem. Res., 2009, 42, 925–937 CrossRef CAS.
- Y. Liu, X. Wu, C. He, R. Zhang and C. Duan, A truncated octahedral nanocage for fluorescent detection of nucleoside, Dalton Trans., 2008, 43, 5866–5868 RSC.
- J.-C. G. Bünzli, Benefiting from the unique properties of lanthanide ions, Acc. Chem. Res., 2006, 39, 53–61 CrossRef PubMed.
- Y. Zhou, H. Li, T. Zhu, T. Gao and P. Yan, A highly luminescent chiral tetrahedral Eu4L4(L′)4 cage: chirality induction, chirality memory, and circularly polarized luminescence, J. Am. Chem. Soc., 2019, 141, 19634–19643 CrossRef CAS PubMed.
- H.-Y. Wong, W.-S. Lo, K.-H. Yim and G.-L. Law, Chirality and chiroptics of lanthanide molecular and supramolecular assemblies, Chem, 2019, 5, 3058–3095 CAS.
- C.-T. Yeung, W. T. K. Chan, S.-C. Yan, K.-L. Yu, K.-H. Yim, W.-T. Wong and G.-L. Law, Lanthanide supramolecular helical diastereoselective breaking induced by point chirality: mixture or P-helix, M-helix, Chem. Commun., 2015, 51, 592–595 RSC.
- O. Kotova, S. Comby, K. Pandurangan, F. Stomeo, J. E. O'Brien, M. Feeney, R. D. Peacock, C. P. McCoy and T. Gunnlaugsson, The effect of the linker size in C2-symmetrical chiral ligands on the self-assembly formation of luminescent triple-stranded di-metallic Eu(III) helicates in solution, Dalton Trans., 2018, 47, 12308–12317 RSC.
- C.-T. Yeung, K.-H. Yim, H.-Y. Wong, R. Pal, W.-S. Lo, S.-C. Yan, M. Y.-M. Wong, D. Yufit, D. E. Smiles, L. J. McCormick, S. J. Teat, D. K. Shuh, W.-T. Wong and G.-L. Law, Chiral transcription in self-assembled tetrahedral Eu4L6 chiral cages displaying sizable circularly polarized luminescence, Nat. Commun., 2017, 8, 1128 CrossRef PubMed.
- L.-L. Yan, C.-H. Tan, G.-L. Zhang, L.-P. Zhou, J.-C. Bünzli and Q.-F. Sun, Stereocontrolled self-assembly and self-sorting of luminescent europium tetrahedral cages, J. Am. Chem. Soc., 2015, 137, 8550–8555 CrossRef CAS.
- X.-Z. Li, L.-P. Zhou, L.-L. Yan, D.-Q. Yuan, C.-S. Lin and Q.-F. Sun, Evolution of luminescent supramolecular lanthanide M2nL3n complexes from helicates and tetrahedra to cubes, J. Am. Chem. Soc., 2017, 139, 8237–8244 CrossRef CAS PubMed.
- J.-M. Zhou, W. Shi, N. Xu and P. Cheng, Highly selective luminescent sensing of fluoride and organic small-molecule pollutants based on novel lanthanide metal–organic frameworks, Inorg. Chem., 2013, 52, 8082–8090 CrossRef CAS PubMed.
- Z. Min, M. A. Singh-Wilmot, C. L. Cahill, M. Andrews and R. Taylor, Isoreticular lanthanide metal–organic frameworks: syntheses, structures and photoluminescence of a family of 3D phenylcarboxylates, Eur. J. Inorg. Chem., 2012, 2012, 4419–4426 CrossRef CAS.
- J.-F. Ayme, G. Gil-Ramírez, D. A. Leigh, J.-F. Lemonnier, A. Markevicius, C. A. Muryn and G. Zhang, Lanthanide template synthesis of a molecular trefoil knot, J. Am. Chem. Soc., 2014, 136, 13142–13145 CrossRef CAS PubMed.
- L. G. Nielsen, A. K. R. Junker and T. J. Sørensen, Composed in the f-block: solution structure and function of kinetically inert lanthanide (III) complexes, Dalton Trans., 2018, 47, 10360–10376 RSC.
- D. E. Barry, D. F. Caffrey and T. Gunnlaugsson, Lanthanide-directed synthesis of luminescent self-assembly supramolecular structures and mechanically bonded systems from acyclic coordinating organic ligands, Chem. Soc. Rev., 2016, 45, 3244–3274 RSC.
- A. Thibon and V. C. Pierre, Principles of responsive lanthanide-based luminescent probes for cellular imaging, Anal. Bioanal. Chem., 2009, 394, 107–120 CrossRef CAS.
- G. Liu and B. Jacquier, Spectroscopic Properties of Rare Earths in Optical Materials, Berlin, Heidelberg, Springer Berlin/Heidelberg, Berlin, Heidelberg, 2005 Search PubMed.
- J.-C. G. Bünzli and C. Piguet, Taking advantage of luminescent lanthanide ions, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- F. Zinna and L. Di Bari, Lanthanide circularly polarized luminescence: bases and applications, Chirality, 2015, 27, 1–13 CrossRef CAS PubMed.
- G. Bozoklu, C. Marchal, C. Gateau, J. Pécaut, D. Imbert and M. Mazzanti, Diastereoselective self-assembly of a homochiral europium triangle from a bipyoxazoline-carboxylate ligand, Chem. – Eur. J., 2010, 16, 6159–6163 CrossRef CAS PubMed.
- C.-L. Liu, L.-P. Zhou, D. Tripathy and Q.-F. Sun, Self-assembly of stable luminescent lanthanide supramolecular M4L6 cages with sensing properties toward nitroaromatics, Chem. Commun., 2017, 53, 2459–2462 RSC.
- B. Wang, Z. Zang, H. Wang, W. Dou, X. Tang, W. Liu, Y. Shao, J. Ma, Y. Li and J. Zhou, Multiple lanthanide helicate clusters and the effects of anions on their configuration, Angew. Chem., Int. Ed., 2013, 52, 3756–3759 CrossRef CAS PubMed.
- Y. Zhang, B. Ali, J. Wu, M. Guo, Y. Yu, Z. Liu and J. Tang, Construction of metallosupramolecular coordination complexes: from lanthanide helicates to octahedral cages showing single-molecule magnet behavior, Inorg. Chem., 2019, 58, 3167–3174 CrossRef CAS PubMed.
- S.-L. Han, J. Yang, D. Tripathy, X.-Q. Guo, S.-J. Hu, X.-Z. Li, L.-X. Cai, L.-P. Zhou and Q.-F. Sun, Self-assembly of lanthanide-covalent organic polyhedra: chameleonic luminescence and efficient catalysis, Inorg. Chem., 2020, 59, 14023–14030 CrossRef CAS PubMed.
- M. Lama, O. Mamula, G. S. Kottas, L. De Cola, H. Stoeckli-Evans and S. Shova, Enantiopure, supramolecular helices containing three-dimensional tetranuclear lanthanide(III) arrays: synthesis, structure, properties, and solvent-driven trinuclear/tetranuclear interconversion, Inorg. Chem., 2008, 47, 8000–8015 CrossRef CAS PubMed.
- S. J. Hu, X. Q. Guo, L. P. Zhou, L. X. Cai and Q. F. Sun, Coordination–assembled lanthanide–organic Ln3L3 sandwiches or Ln4L4 tetrahedron: structural transformation and luminescence modulation, Chin. J. Chem., 2019, 37, 657–662 CrossRef CAS.
- X. Li, W. Liu, Z. Guo and M. Tan, Trinuclear to dinuclear: a radii dependence lanthanide (III) self-assembly coordination behavior of an amide-type tripodal ligand, Inorg. Chem., 2003, 42, 8735–8738 CrossRef CAS PubMed.
- Y.-W. Wang, Y.-L. Zhang, W. Dou, A.-J. Zhang, W.-W. Qin and W.-S. Liu, Synthesis, radii dependent self-assembly crystal structures and luminescent properties of rare earth (III) complexes with a tripodal salicylic derivative, Dalton Trans., 2010, 39, 9013–9021 RSC.
- X. Hu, W. Dou, C. Xu, X. Tang, J. Zheng and W. Liu, Lanthanide radii controlled one-dimensional polymer and dinuclear complexes and their fluorescent properties, Dalton Trans., 2011, 40, 3412–3418 RSC.
- H.-Y. Wong, W. T. K. Chan and G.-L. Law, Assembly of lanthanide (III) cubanes and dimers with single-molecule magnetism and photoluminescence, Inorg. Chem., 2018, 57, 6893–6902 CrossRef CAS.
- J. Hamacek, G. Bernardinelli and Y. Filinchuk, Tetrahedral assembly with lanthanides: toward discrete polynuclear complexes, Eur. J. Inorg. Chem., 2008, 2008, 3419–3422 Search PubMed.
- S. Zebret, C. Besnard, G. Bernardinelli and J. Hamacek, Thermodynamic discrimination in the formation of tetranuclear lanthanide helicates, Eur. J. Inorg. Chem., 2012, 2012, 2409–2417 CrossRef CAS.
- G. Li, X. Zhao, Q. Han, L. Wang and W. Liu, Radii-dependent self-assembly of chiral lanthanide complexes: synthesis, chirality, and single-molecule magnet behavior, Dalton Trans., 2020, 49, 10120–10126 RSC.
- X.-Z. Li, L.-P. Zhou, S.-J. Hu, L.-X. Cai, X.-Q. Guo, Z. Wang and Q.-F. Sun, Metal ion adaptive self-assembly of photoactive lanthanide-based supramolecular hosts, Chem. Commun., 2020, 56, 4416–4419 RSC.
- S. Faulkner and B. P. Burton-Pye, pH Dependent self-assembly of dimetallic lanthanide complexes, Chem. Commun., 2005, 259–261 RSC.
- I. Lukeš, J. Kotek, P. Vojtíšek and P. Hermann, Complexes of tetraazacycles bearing methylphosphinic/phosphonic acid pendant arms with copper(II), zinc(II) and lanthanides(III). A comparison with their acetic acid analogues, Coord. Chem. Rev., 2001, 216–217, 287–312 CrossRef.
- S. E. Plush, N. A. Clear, J. P. Leonard, A.-M. Fanning and T. Gunnlaugsson, The effect on the lanthanide luminescence of structurally simple Eu(iii) cyclen complexes upon deprotonation of metal bound water molecules and amide based pendant arms, Dalton Trans., 2010, 39, 3644–3652 RSC.
- Y. Bretonnière, M. Mazzanti, J. Pécaut and M. M. Olmstead, Cation-controlled self-assembly of a hexameric europium wheel, J. Am. Chem. Soc., 2002, 124, 9012–9013 CrossRef PubMed.
- X.-Y. Chen, Y. Bretonnière, J. Pécaut, D. Imbert, J.-C. Bünzli and M. Mazzanti, Selective self-assembly of hexameric homo-and heteropolymetallic lanthanide wheels: Synthesis, structure, and photophysical studies, Inorg. Chem., 2007, 46, 625–637 CrossRef CAS PubMed.
- G. l. Bozoklu, C. Gateau, D. Imbert, J. Pécaut, K. Robeyns, Y. Filinchuk, F. Memon, G. Muller and M. Mazzanti, Metal-controlled diastereoselective self-assembly and circularly polarized luminescence of a chiral heptanuclear europium wheel, J. Am. Chem. Soc., 2012, 134, 8372–8375 CrossRef CAS PubMed.
- J.-M. Senegas, S. Koeller, G. Bernardinelli and C. Piguet, Isolation and characterization of the first circular single-stranded polymetallic lanthanide-containing helicate, Chem. Commun., 2005, 17, 2235–2237 RSC.
- A. Malviya, H. S. Jena, A. K. Mondal and S. Konar, Europium–based dinuclear triple–stranded helicate vs. tetranuclear quadruple–stranded helicate: effect of stoichiometric ratio on the supramolecular self–assembly, Eur. J. Inorg. Chem., 2015, 2015, 2901–2907 CrossRef CAS.
- B. El Aroussi, N. Dupont, G. r. Bernardinelli and J. Hamacek, Unsymmetrical tripodal ligand for lanthanide complexation: structural, thermodynamic, and photophysical studies, Inorg. Chem., 2010, 49, 606–615 CrossRef CAS.
- J. Hamacek, C. Besnard, T. Penhouet and P.-Y. Morgantini, Lanthanide-mediated supramolecular cages and host–guest interactions, Chem. – Eur. J., 2011, 17, 6753–6764 CrossRef CAS.
- L.-X. Cai, L.-L. Yan, S.-C. Li, L.-P. Zhou and Q.-F. Sun, Stereocontrolled self-assembly and photochromic transformation of lanthanide supramolecular helicates, Dalton Trans., 2018, 47, 14204–14210 RSC.
- Y. Hashimoto, T. Nakashima, M. Yamada, J. Yuasa, G. n. l. Rapenne and T. Kawai, Hierarchical emergence and dynamic control of chirality in a photoresponsive dinuclear complex, J. Phys. Chem. Lett., 2018, 9, 2151–2157 CrossRef CAS PubMed.
- J. Zhang, Y. Zhou, Y. Yao, Z. Cheng, T. Gao, H. Li and P. Yan, A light triggered optical and chiroptical switch based on a homochiral Eu2L3 helicate, J. Mater. Chem. C, 2020, 8, 6788–6796 RSC.
- Z. Wang, L. He, B. Liu, L.-P. Zhou, L.-X. Cai, S.-J. Hu, X.-Z. Li, Z. Li, T. Chen, X. Li and Q.-F. Sun, Coordination-assembled water-soluble anionic lanthanide organic polyhedra for luminescent labeling and magnetic resonance imaging, J. Am. Chem. Soc., 2020, 142, 16409–16419 CrossRef CAS.
| This journal is © the Partner Organisations 2021 |