
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanism of inhibition of SARS-CoV-2 Mpro by N3 peptidyl Michael acceptor explained by QM/MM simulations and design of new derivatives with tunable chemical reactivity†
Kemel
Arafet
 a,
Natalia
Serrano-Aparicio
a,
Alessio
Lodola
b,
Adrian J.
Mulholland
c,
Florenci V.
González
d,
Katarzyna
Świderek
*a and
Vicent
Moliner
*a
a,
Natalia
Serrano-Aparicio
a,
Alessio
Lodola
b,
Adrian J.
Mulholland
c,
Florenci V.
González
d,
Katarzyna
Świderek
*a and
Vicent
Moliner
*a
aDepartament de Química Física i Analítica, Universitat Jaume I, 12071 Castelló, Spain. E-mail: swiderek@uji.es; moliner@uji.es
bDipartimento di Scienze degli Alimenti e del Farmaco, Università degli Studi di Parma, Italy
cCentre for Computational Chemistry, School of Chemistry, University of Bristol, UK
dDepartament de Química Inorgànica i Orgànica, Universitat Jaume I, 12071 Castelló, Spain
First published on 27th November 2020
Abstract
The SARS-CoV-2 main protease (Mpro) is essential for replication of the virus responsible for the COVID-19 pandemic, and one of the main targets for drug design. Here, we simulate the inhibition process of SARS-CoV-2 Mpro with a known Michael acceptor (peptidyl) inhibitor, N3. The free energy landscape for the mechanism of the formation of the covalent enzyme-inhibitor product is computed with QM/MM molecular dynamics methods. The simulations show a two-step mechanism, and give structures and calculated barriers in good agreement with experiment. Using these results and information from our previous investigation on the proteolysis reaction of SARS-CoV-2 Mpro, we design two new, synthetically accessible N3-analogues as potential inhibitors, in which the recognition and warhead motifs are modified. QM/MM modelling of the mechanism of inhibition of Mpro by these novel compounds indicates that both may be promising candidates as drug leads against COVID-19, one as an irreversible inhibitor and one as a potential reversible inhibitor.
Introduction
A novel coronavirus (severe acute respiratory syndrome coronavirus-2 – SARS-CoV-2) has been identified as responsible for the COVID-19 pandemic. There is a pressing need for effective antiviral treatments. Many researchers around the world are working to develop SARS-CoV-2 antiviral compounds, e.g. following previous SARS-CoV and MERS-CoV outbreaks and research. Remarkable progress has been achieved in just a few months with regard to the understanding of the phylogeny and genomic organization of SARS-CoV-2 as well as its molecular mechanisms of infection and replication.1 Knowledge of the life cycle of SARS-CoV-2 provides information about possible targets for drug development.2 These include inhibition of the viral–host interaction, endosome maturation, viral/endosome membrane fusion, and viral polypeptide maturation.3 Intense work has focused on identification and testing of compounds already approved for the treatment of other diseases such as remdesivir, a drug developed previously against Ebola virus (EBOV),4 dexamethasone and antimalarial drugs.5 While some studies show promise, there is a clear need for new compounds, specific to SARS-CoV-2, both as drug leads and as biochemical probes.6–9The present study focuses on the atomistic characterization of the inhibition mechanism of one of the proteins responsible for the virus replication and maturation, the main coronavirus protease (SARS-CoV-2 Mpro, also called 3CLpro). Mpro, together with the papain-like protease PLpro, are cysteine protease (CPs) that process the polyproteins that are translated from the viral RNA in the replication of the SARS-CoV-2 virus. Thus, inhibiting activity of these enzymes would block the viral life cycle. In addition, a distinguishing feature of SARS-CoV-2 Mpro with respect to human proteases is its ability to cleave peptides after a glutamine residue. This feature, which is shared also by SARS-CoV 3CL protease,10 has prompted a search for inhibitors incorporating a glutamine residue/mimic in their structure (see below) with the aim of obtaining selectivity in addition to potency.5 The current scenario explains recent efforts devoted to the identification of inhibitors of this enzyme, aided by X-ray crystallography,5,11,12 and computational modelling and simulation. An example is the Covid Moonshot initiative, which thanks to the contributions of scientists operating all over the world, has identified several potential Mpro inhibitors hits which are now under biochemical evaluation.13
The best characterized Mpro inhibitors so far act with a covalent mechanism. They share a similar recognition moiety, i.e. a peptidomimetic scaffold of moderate size with a glutamine or an isostere at the P1 position and a branched lipophilic group at P2,5,11,12 and are equipped with a reactive ‘warhead’, i.e. an electrophilic group responsible for the covalent modification of the Mpro (see Scheme 1). Warheads so far employed for the design of SARS-CoV-2 inhibitors ranged from classical Michael acceptors (MAs) to activated carbonyl derivatives, including alpha-ketoamides and aldehydes.
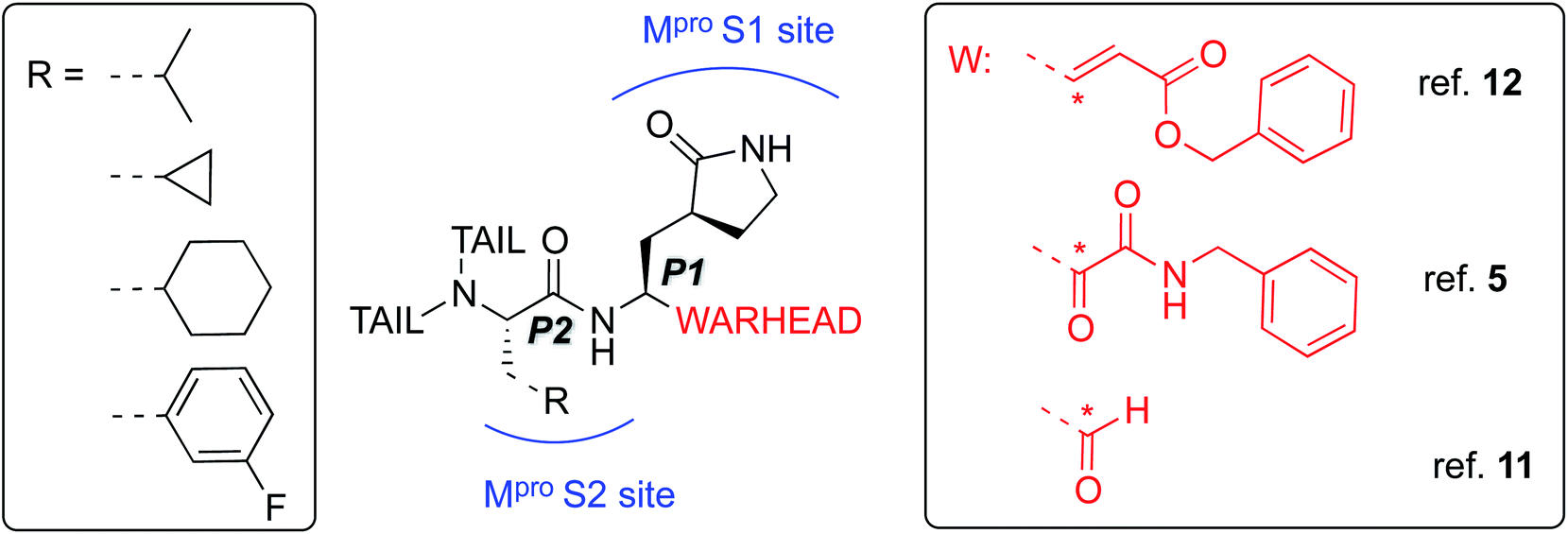 | ||
| Scheme 1 Schematic representation of the main classes of covalent inhibitors of CoVs Mpro so far reported. The reactive center on the warhead is marked with an asterisk. R emerging from P2 residue represents a lipophilic group of moderate size. The tail region is a highly variable portion in term of size and shape and involved other Mpro sub-pockets (i.e. S3 and S4) not represented in the scheme. | ||
For covalent modification of cysteine residues, the MA class is often the first choice with several examples of approved drugs containing this group.14–17 Despite the potential problem of off–target interactions due to the electrophilicity of α,β unsaturated systems, MAs are widely employed against cysteine proteases because they ensure the covalent inhibition of the enzyme.18 Compounds equipped with less reactive warheads (i.e. carbonyl-based compounds or nitriles) act as reversible inhibitors, as they form metastable adducts (such as hemithioketal or thioimidate species) with cysteine residues. In terms of target engagement, duration of inhibition and efficacy, MAs have important potential advantages over other warheads.16–18 Previous work on other viral Mpros exemplifies this: peptidyl MAs were the first class of mechanism-based covalent inhibitors of CoV Mpro enzymes described in 2005.19 This seminal work prompted Jin and colleagues12 to test this library of compounds on the SARS-CoV-2 Mpro. Among those compounds, they found one, N3 (see Scheme 2), which shows promising inhibitory activity against the Mpro of SARS-CoV-2.
 | ||
| Scheme 2 Chemical structures of known (N3) and proposed (B1 and B2) Michael acceptor inhibitors of SARS-CoV-2 Mpro. The modifications from N3 to B1 and B2 are highlighted in blue. | ||
Kinetic analysis of the inhibition of Mpro of SARS-CoV-2 by N3 suggests a mechanism of two steps leading to irreversible inactivation (see Scheme 3): protein-inhibitor association to form a noncovalent complex (E:I), followed by covalent bond formation (E-I).5 The X-ray diffraction structure indicates a covalent bond between the S atom of Cys145 of protomer A and the Cβ atom of the vinyl group (see Scheme 3), thus confirming that N3 is a MA inhibitor12 and confirming that SARS-CoV-2 Mpro is a CP with an active site catalytic dyad (C145/H41) similar to other CPs.
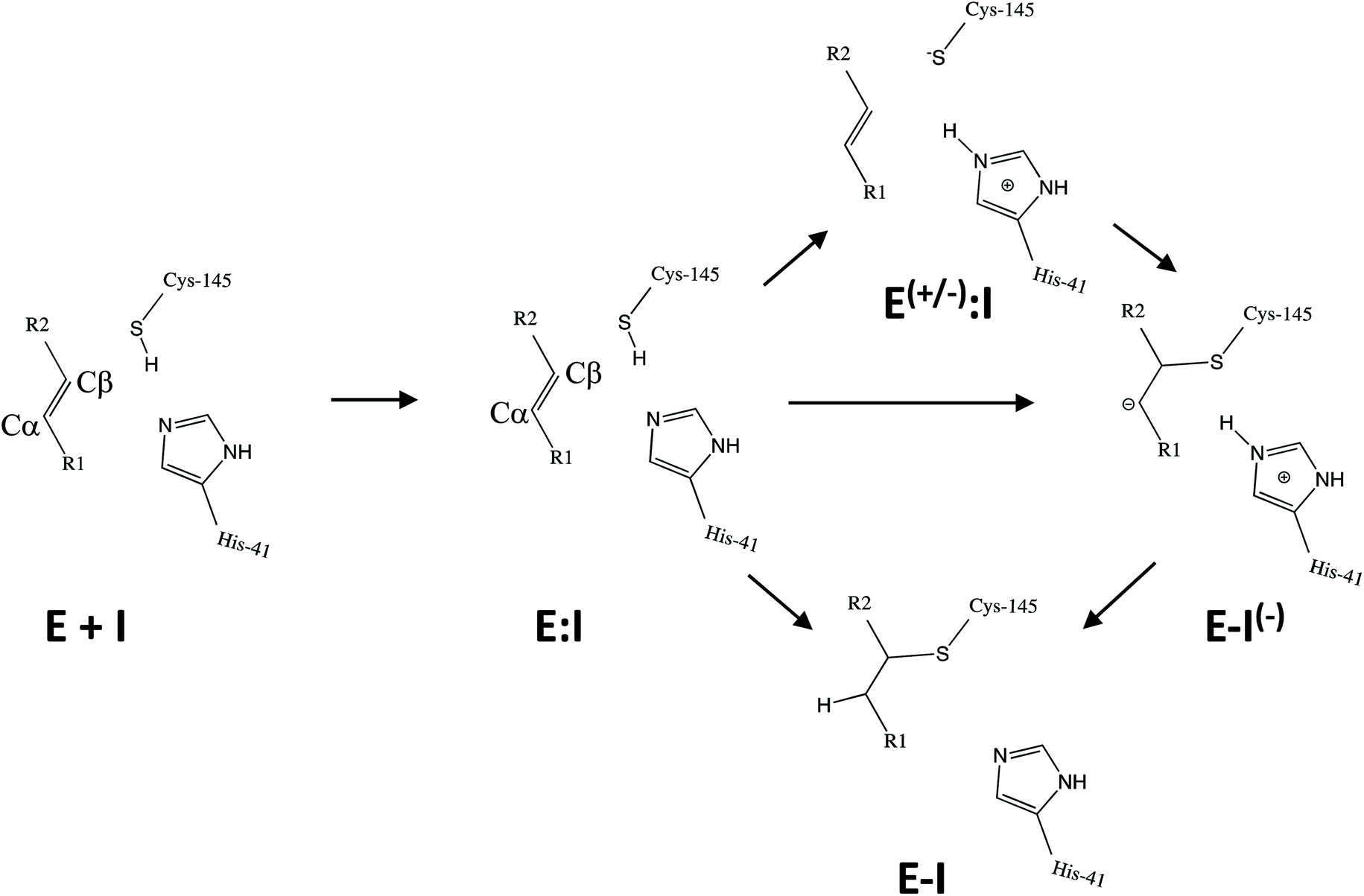 | ||
| Scheme 3 Possible general mechanisms of SARS-CoV-2 Mpro cysteine protease inhibition by Michael acceptor inhibitors: concerted or stepwise Michael addition. R1 and R2 represent different substituents, as shown in compounds depicted in Scheme 2. | ||
Crystallographic electron density maps of N3 (ref. 12) indicate hydrogen bond and van der Waals interactions between the inhibitor and residues in the substrate-binding pockets of Mpro. An exception is the solvent-exposed Val at P3, suggesting that this site can tolerate substituents of different shape and size.12
Mechanistically, it is proposed that the chemical reaction leading to Mpro inactivation requires the imidazole group of H41 to activate the SH group of C145 to form a highly nucleophilic CysS−/HisH+ ion pair that would readily react with the inhibitor.20 This equilibrium may be tipped in favour of the ion pair by ligand binding, and may depend on the features of the ligand itself. In this regard, the inactivation of SARS-CoV-2 Mpro by covalent (peptidyl) inhibitors, including N3, can be considered as equivalent to the acylation step of the proteolysis reaction in CPs. According to our previous QM/MM study on the proteolysis reaction catalyzed by cruzain CP, the proton from the cationic HisH+ is transferred to the N atom of the scissile peptide bond, followed by Cys attack on the carbonyl carbon atom of the peptide.21 However, our recent study on the proteolysis reaction of SARS-CoV-2 Mpro using as substrate the polypeptide Ac–Val–Lys–Leu–Gln–ACC (ACC being a fluorescent tag 7-amino-4-carbamoylmethylcoumarin) suggests that the mechanism of action of this enzyme differs from most other CPs.22 First of all, the enzyme:substrate initial complex would correspond to the neutral C145/H41 dyad (equivalent to E:I in Scheme 3) instead of the ionic pair dyad C145−/H41+ (E(+/−):I in Scheme 3). From this stable state, the acylation reaction consists of a proton transfer from C145 to the H41 concomitant with the nucleophilic attack on the carbonyl carbon atom of the peptide bond by the sulfur atom of C145, leading to a pseudo-stable intermediate. Then, the cleavage of the peptide bond by Mpro is assisted by proton transfer from the protonated H41+ to the nitrogen atom of the substrate, forming an acyl-enzyme covalent intermediate. This last step of the acylation was calculated to be almost barrierless with the substrate employed in our previous study.22 In the inhibition reaction by MA compounds shown in Scheme 2, the proton will be transferred from the protonated H41+ to the Cα of the inhibitor, thus leading to the covalent E-I adduct (Scheme 3). Consequently, in the design of covalent inhibitors, focus must be put on obtaining an exergonic process for the E-I formation, with low activation energy barriers. The energy barriers from E-I back to reactants E:I will determine the irreversible vs. reversible character of the inhibitors, with potentially paramount importance for finding the optimal balance between efficacy and safety.23 To reach this goal, in addition to the presence of a reactive warhead, the interactions between the recognition moiety of the inhibitor and the different sub-sites of the binding pocket of the protein must be taken into account.17 Design can be guided by the results derived from previous studies on this and related CPs. QM/MM simulations provide a good tool to investigate the reactivity of covalent inhibitors within their protein targets.24–30
Here, we focus firstly on the inhibition of the SARS-CoV-2 Mpro by the covalent (peptidyl) irreversible inhibitor N3, modelling the reaction with QM/MM techniques. Building upon these findings, on information derived from other CP inhibitors, and on our previous study on the proteolysis reaction of Mpro,22 we then designed, and tested computationally, two MA inhibitors to block the enzyme: compounds B1 and B2 in Scheme 2. B1 was designed according to some of the modifications made by Zhang et al.5 on their broad-spectrum peptidomimetic α-ketoamides inhibiting of the main proteases of betacoronaviruses, alphacoronaviruses and the 3C proteases of enteroviruses.31 In addition, although the inhibition of CPs has been proposed to depend on interactions between the peptidic framework (the P2) of the inhibitor and the S2 pocket of the enzyme,29,32–35 this is probably not the case in SARS-CoV-2 Mpro, according to the QM-MM protein-substrate interactions found in our previous study of the proteolysis reaction:22 S2 appears to be a small hydrophobic pocket without strong hydrogen bond interactions. Therefore, the isopropyl group of Leu at P2 site was replaced by a cyclopropyl group. This change is in accordance with the changes by Zhang et al. to the original peptidomimetic α-ketoamides to enhance anti-viral activity against beta coronaviruses (such as SARS-CoV and SARS-CoV-2).5 The tail of the N3 compound was replaced by an amino pyridone moiety carbamoylated by a tert-butyloxycarbonyl group, because this group is expected not to be a substrate of cellular proteases, and so offers potential advantages in term of pharmacokinetic properties.5 Our previous Mpro proteolysis reaction study indicated that the S3 subsite is completely exposed to the solvent; only three interactions between the peptide backbone atoms of Lys3 of the substrate and the protein were observed.22 In addition, the lack of strong hydrogen bond interactions in the S4 sub-site supports the strategy of reducing the size of the inhibitor.
In the case of compound B2, a more dramatic modification was introduced: we decided to change the warhead to a nitroalkene, based on the potent reversible inhibitory activity of a family of dipeptidyl nitroalkene derivatives against the CPs cruzain and rhodesain by one of us,14 together with our previous QM/MM study on the inhibition mechanism of three CPs belonging to the papain family (cruzain, rhodesain, and cathepsin L).29 Based upon the mechanism depicted in Scheme 3, the protonation of intermediate E-I(−) might be less favored in the case of the nitroalkane carbanion because the acidity of the corresponding acid, namely the nitroalkane, is higher, and thus, the basicity of the carbanion is lower; potentially being a reversible covalent inhibitor. The glutamine residue in P1 was introduced due to the strong favorable interactions that we observed in the study of the proteolysis of the Ac–Val–Lys–Leu–Gln–ACC substrate by Mpro,22 and based on the substrate specificity of SARS-CoV Mpro, i.e. requiring glutamine in the P1 position.36 P2 was kept the same as in N3, while the rest of the inhibitor in positions P3, P4 and P5 was replaced by a smaller moiety, with the aim of improving the physicochemical properties as well as synthetic accessibility. Importantly, both designed compounds B1 and B2 should be readily prepared through synthetic approaches inspired by published synthetic routes of similar compounds.13,37
Methods
The atomic coordinates of SARS-CoV-2 Mpro were taken from the X-ray structure of its complex with the N3 inhibitor, available in the Protein Data Bank (PDB ID 6LU7).12 The biological assembly (homodimer) was built using Discovery Studio Visualizer 19. Inhibitor N3 was replaced by two Michael acceptor inhibitors (compounds B1 and B2) to create two new covalent enzyme-inhibitor models (E-I in Scheme 2). Once the enzyme-inhibitor models were set up, solvated with a box of water molecules and equilibrated by means of preliminary MM molecular dynamics (MD) simulations (Fig. S1a, S2a and S3a†), QM/MM free energy surfaces (FESs) were calculated, in terms of Potentials of Mean Force (PMFs), for every step of the reaction, using umbrella sampling38 combined with the Weighted Histogram Analysis Method (WHAM)39 see ESI† for details. The QM region consisted of 75 atoms for the inhibitor N3 and compound B1, and 57 atoms for the compound B2 including P1′, P1 and P2 fragments of the inhibitors and the two catalytic residues C145 and H41. Four quantum ‘link’ atoms were inserted where the QM-MM frontier crosses a covalent bond (see Fig. S1b, S2b and S3b†), as in our study on proteolysis of SARS-CoV-2 Mpro enzyme.22 The Austin Model 1 (AM1)40 semiempirical method was used to treat the QM region in the initial exploration of the FESs. The Minnesota Density Functional M06-2X41 (with the standard 6-31+G(d,p) basis set42) as implemented in the Gaussian09 program,43 was used to treat the QM region, to calculate the final corrected high level FESs (see ESI† for details), as well as for the geometry optimizations of the different transition states. This is a good choice of functional and basis set, based on our previous tests and experience,44–46 including the study of the proteolysis reaction of SARS-CoV-2 Mpro.22 The protein and water molecules were treated with the AMBER ff03 (ref. 47) and TIP3P48 force fields, respectively. QM/MM MD simulations were performed using the fDynamo library,49 using procedures that we have previously extensively tested and validated. Structures of all the important states involved in the reaction (minima and transition state structures) were then optimized at the M06-2X:6-31+G(d,p)/MM level, starting from representative AM1/MM snapshots from the FESs, with Gaussian09 (ref. 43) coupled to the fDynamo library. The corrected free energy surfaces are designated M06-2X/6-31+G(d,p):AM1/MM. These structures are deposited in the ESI.†The inhibitor:Mpro binding energy was estimated by docking calculations for N3, B1 and B2, with the Glide program,50 starting from the QM/MM structures of the enzyme:inhibitor non-covalent reactant complex, E:I in Scheme 2 (see ESI† for details).
Results and discussion
Inhibition reaction of SARS-CoV-2 Mpro with N3
The first step of our program towards the design of new SARS-CoV Mpro inhibitors was the study of the reaction with the N3 inhibitor originally proposed by Yang et al.19 As described in the Methods section, the enzyme-inhibitor covalent E-I complex was equilibrated by MM and QM/MM MD simulations. A schematic representation of the equilibrated structure of the active site is shown in Fig. 1, where important interactions found in the MD simulations and the X-ray structure obtained by Jin et al.12 are indicated as blue and red dashed lines, respectively. The pattern of interactions between the enzyme and the inhibitor in our equilibrated structure is quite close to that observed crystallographically, thus supporting our starting structure for the exploration of the full mechanism. The MD results confirm the absence of hydrogen bond interactions with some of the side chains of the residues of N3 (P2–P5) which, considering the demonstrated efficiency of this inhibitor, can be used as a guide for the design of improved compounds not requiring hydrogen bond interactions with these sites, as mentioned in the Introduction. | ||
| Fig. 1 Schematic representation of N3 in the covalent E-I complex in the active site of SARS-CoV-2 Mpro. The dashed lines indicate hydrogen bond interactions between the inhibitor the protein found in MD simulations (blue lines) and the X-ray structure (red lines) obtained by Jin et al.12 Interatomic distances, computed as average values over the MD simulations (in blue) and from the X-ray structure (in red), are reported in Å. | ||
Once the covalent enzyme-inhibitor E-I complex was equilibrated, the Michael addition reaction and the proton transfer from the protonated H41 to the Cα atom of the inhibitor (see Scheme 3) was explored backwards from E-I to E:I by QM/MM MD simulations. Appropriate combinations of interatomic distances were employed to generate the potential energy surfaces (PESs) and the free energy surfaces (FESs) of every chemical step. These reaction coordinates were chosen based on previous experience and testing for this and similar reactions (see ESI† for details, including convergence tests). The FESs obtained (see Fig. S7 in ESI†) show that the most stable protonation state of the C145/H41 dyad corresponds to that in which both residues are neutral, designated E:I. This result is in contrast with previous computational studies of proteolysis21 and inhibition25,29,51 and earlier suggestions for other SARS-CoV main proteases that inhibitor binding may favor formation of the ion pair,52 but it is in agreement with our previous study of the proteolysis reaction of Mpro.22 From this initial state, the proton transfer from C145 to H41 to form the ionic dyad E(+/−):I precedes the Michael addition that forms the covalent bond between the sulphur atom of C145 and the Cβ of the inhibitor, to form E:I(−). Finally, the proton transfer from His41 to Cα of the substrate takes place as an almost barrierless process to produce the final, stable, E-I covalent complex. The resulting free energy profile is shown in Fig. 2, and details of the active site in the key states in the inhibition process are presented in Fig. 3. The reaction is a stepwise process, kinetically controlled by the carbon–sulphur bond formation, viaTS2, with a free energy barrier of 11.2 kcal·mol−1, and a reaction energy of −17.9 kcal mol−1. The low activation free energy of the inhibition reaction with N3 is in agreement with the experiments that revealed a process so fast that the enzyme inactivation-rate constant for covalent bond formation could not be measured.12 Regarding the thermodynamics, the resulting energy profile is in agreement with the irreversible character of the inhibition considering that activation barrier for the reverse retro-Michael reaction is nearly 30 kcal mol−1 (computed as the difference between E-I and TS2). On the other side, the free energy of activation for the initial proton transfer from C145 to H41 is very low (1.4 kcal mol−1) and the relative energy of the ion pair dyad, E(+/−):I, is only 1.3 kcal mol−1 higher than the starting E:I state (see red line in Fig. 2a). It is important to point out that while preparing the present manuscript, a QM/MM MD study on the mechanism of N3 appeared, where a different DFT Hamiltonian (B3LYP) was used to describe the QM region, as well as a different sampling method.53 Comparison of the two studies reveals some analogies but also some important differences. The stepwise and the exergonic character of the reaction, and the finding that the neutral E:I form is more stable than the ion pair E(+/−):I, are common to both studies. However, while our E(+/−):I is just 1.3 kcal mol−1 higher in energy than E:I, the difference obtained in the ref. 53 is much higher (10.3 kcal mol−1). Similarly, those workers found a much higher energy barrier for the C145–Cβ bond formation from E:I (20.9 kcal·mol−1). These differences may arise because of the different level of QM theory used. The mechanism is also slightly different because a water molecule is used in the transfer of the proton from His41 to Cα of the inhibitor while, as commented above, our simulations show that the direct transfer can take place, in an almost barrierless process, as previously for other CP inhibition reactions by Michael acceptors.29,51 We note that, all other things being equal, the lower barrier found here (not involving an intervening water molecule), indicates that the mechanism we find would dominate the experimentally observed kinetics.
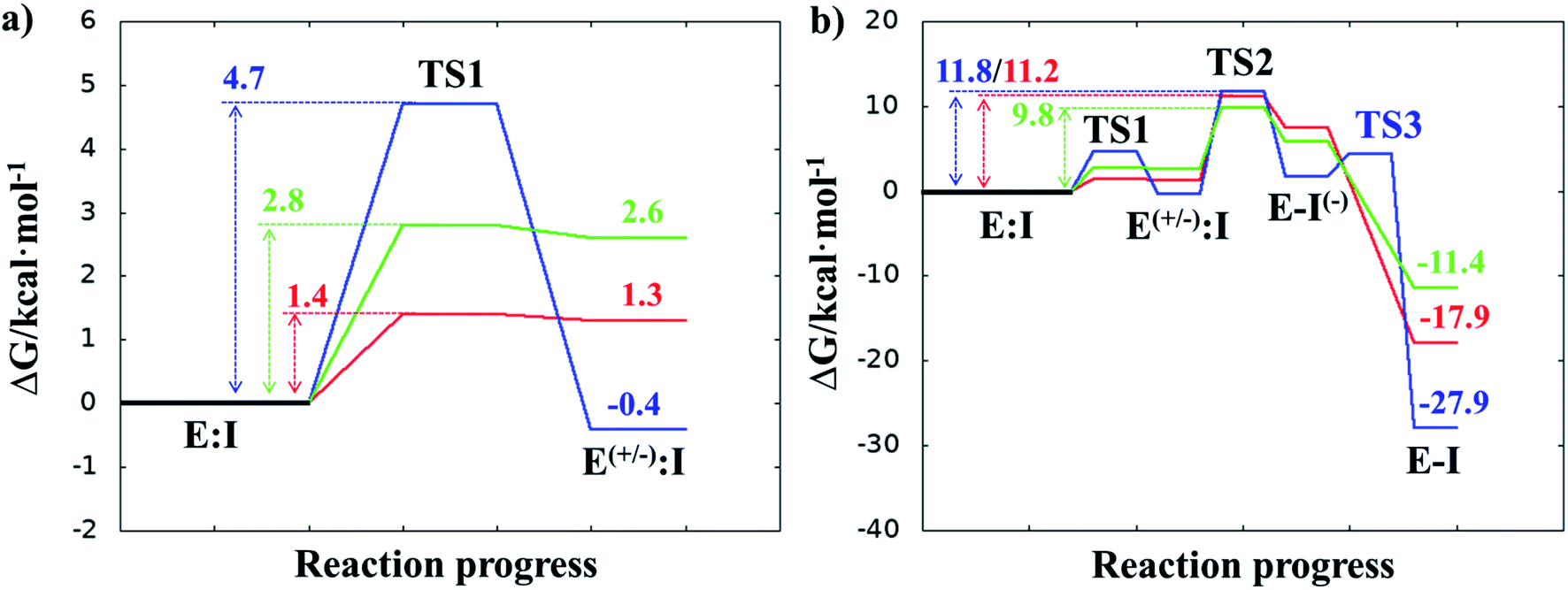 | ||
| Fig. 2 M06-2X/6-31+G(d,p):AM1/MM free energy profiles for covalent complex formation with SARS-CoV-2 Mpro and: N3 (red line); compound B1 (blue line); and compound B2 (green line). Panel (a) shows the formation of the ion pair E(+/−):I; and the full inhibition reaction is shown in panel (b). Energies are in kcal·mol−1. | ||
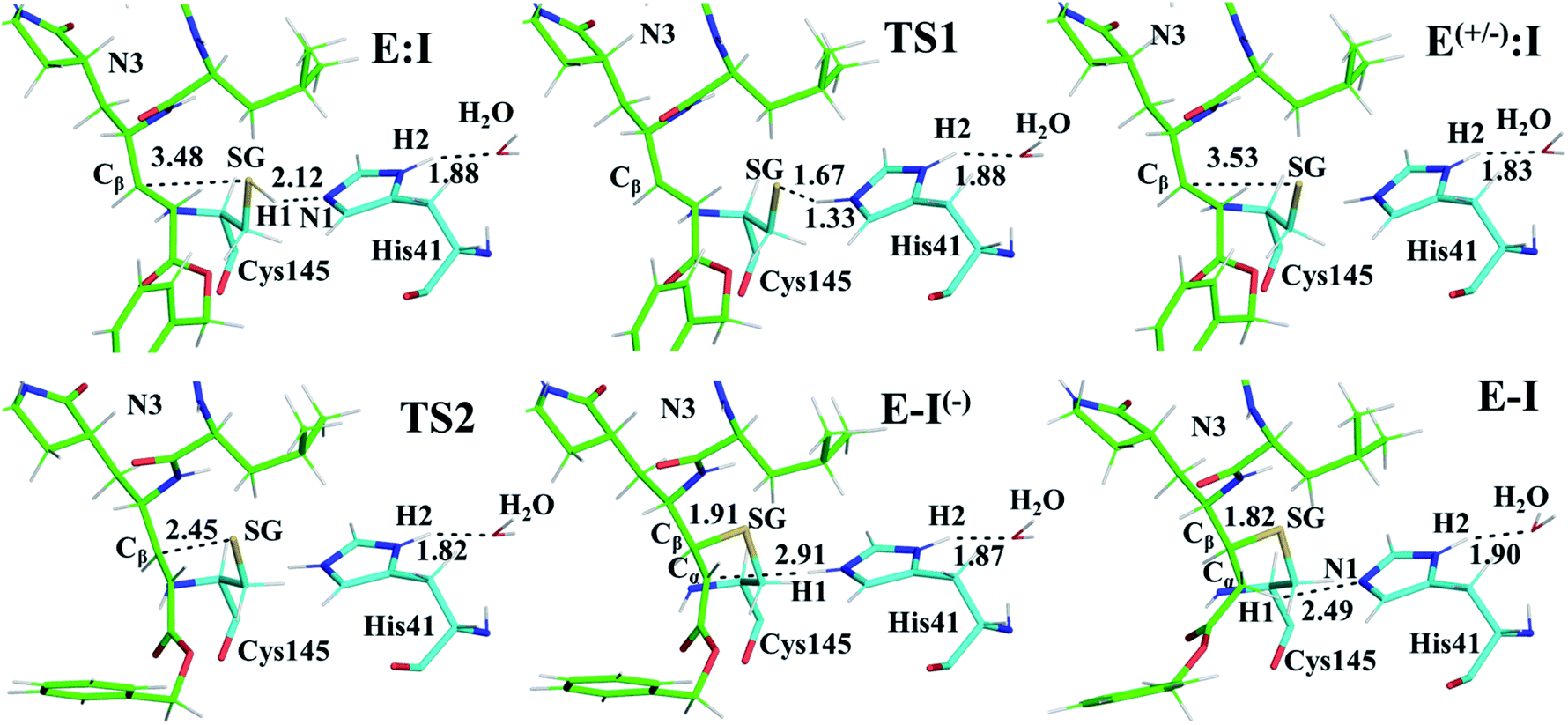 | ||
| Fig. 3 Detail of the M06-2X/6-31+G(d,p)/MM optimized structures of the important states in the inhibition process of Mpro by N3. Carbon atoms of the inhibitor are shown in green while those of the catalytic residues Cys145 and His41 are in blue. Key distances are in Å. | ||
Structural analysis of the structures of the states in the reaction (Fig. 3, Tables S5 and S6†) confirms the mechanism and suggests that the active site of the Mpro does not undergo dramatic changes during the chemical steps. The two catalytic residues are well oriented in the reactive non-covalent complex E:I in which the inhibitor is well anchored to the active site. Structures of TS1 and TS2 were optimized at the M06-2X/6-31+G(d,p)/MM level and the minimum energy path, computed as the IRC path, confirms the predictions derived from the M06-2X:AM1/MM FESs (see Tables S5–S7†).
In order to analyse the non-covalent enzyme:inhibitor reactant complexes, E:I, the interaction energies (electrostatic plus Lennard-Jones) between residues of Chain-A of Mpro and each fragment of the N3 were computed as an average over 1000 structures of the equilibration AM1/MM MD simulation (Fig. 4). The pattern of interactions is similar to that of E-I, shown in Fig. 1, confirming that the inhibitor and enzyme undergo no large structural changes during the chemical steps of the inhibition process. It is important to point out that, while there are protein residues that clearly bind the inhibitor in the active site, which could have been predicted by the X-ray geometrical analysis of the E–I complex (N142 and G143 in P1′  S1′, H163, E166, F140 and H164 in P1
S1′, H163, E166, F140 and H164 in P1  S1, Q189 in P2
S1, Q189 in P2  S2, E166 in P3
S2, E166 in P3  S3, T190 in P4
S3, T190 in P4  S4 or Q192 in P5
S4 or Q192 in P5  S5), some of these residues do not form binding interactions in E:I. For instance, interatomic distances (Table S5†), suggest that E166 forms a hydrogen bond with the backbone of P3, but no net stabilizing interaction was found (Fig. 4 and S8†) in E:I. Therefore, design of N3 analogues to generate a more stable enzyme-inhibitor initial complex should not be limited to geometrical analysis of X-ray structures or those derived from MD simulations of the reactant complex. Overall, our results suggest that P1 is the most important fragment to consider in the design of new efficient inhibitors. This accords with the conclusions of our QM/MM study of the proteolysis reaction of Mpro.22
S5), some of these residues do not form binding interactions in E:I. For instance, interatomic distances (Table S5†), suggest that E166 forms a hydrogen bond with the backbone of P3, but no net stabilizing interaction was found (Fig. 4 and S8†) in E:I. Therefore, design of N3 analogues to generate a more stable enzyme-inhibitor initial complex should not be limited to geometrical analysis of X-ray structures or those derived from MD simulations of the reactant complex. Overall, our results suggest that P1 is the most important fragment to consider in the design of new efficient inhibitors. This accords with the conclusions of our QM/MM study of the proteolysis reaction of Mpro.22
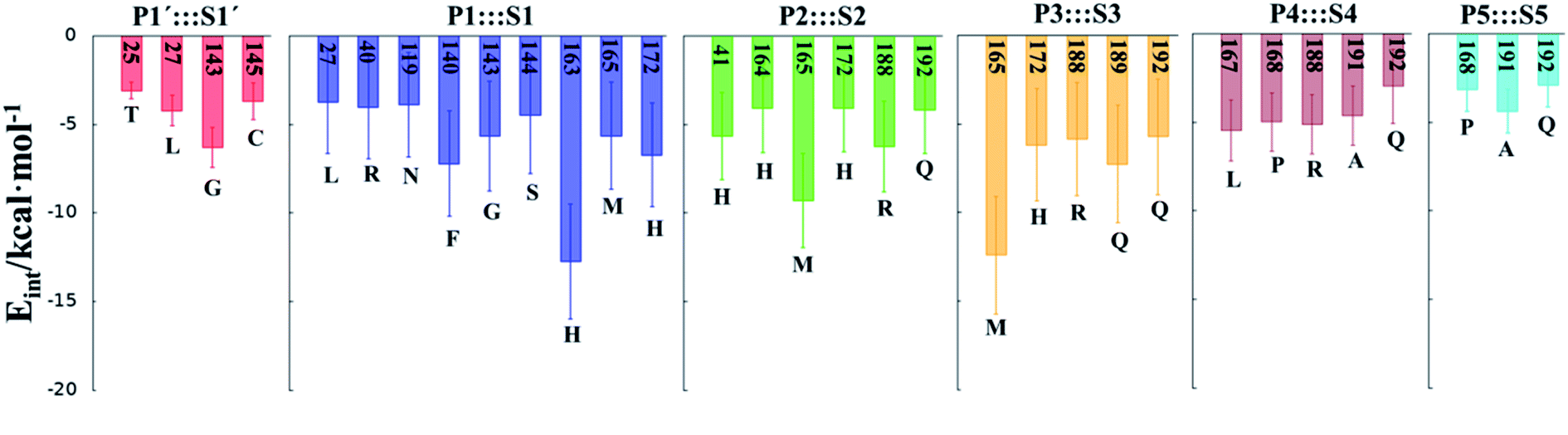 | ||
| Fig. 4 Main favorable average interaction energies (electrostatic plus Lennard-Jones) between residues of Chain-A and each fragment of the N3 computed in the E:I state. Results obtained as an average over 1000 structures of the AM1/MM MD simulations. | ||
Designed inhibitors of SARS-CoV-2 Mpro: compounds B1 and B2
After the study of the Michael addition with inhibitor N3, the inhibition reactions of Mpro with compounds B1 and B2 were simulated using the same methods. The calculations analyzed the stability of the E-I complexes, with special attention to the protein-inhibitor interactions. A schematic representation of the equilibrated E-I structures of the active site after the QM/MM MD simulation is shown in Fig. 5. The interactions between the enzyme and the two proposed inhibitors, indicated as dashed blue lines in Fig. 5, confirm the predictions of the design. In both cases, the interactions between the protein and the inhibitor are dominated by the P1′ S1′ and the P1
S1′ and the P1 S1. Apart from these interactions, in the case of compound B1, hydrogen bond interactions in P4 (with Gln192) are found, while in the case of compound B2 more strong interactions appear, especially in P3 (with Glu166). These interactions keep both compounds posed appropriately for covalent bond formation to take place.
S1. Apart from these interactions, in the case of compound B1, hydrogen bond interactions in P4 (with Gln192) are found, while in the case of compound B2 more strong interactions appear, especially in P3 (with Glu166). These interactions keep both compounds posed appropriately for covalent bond formation to take place.
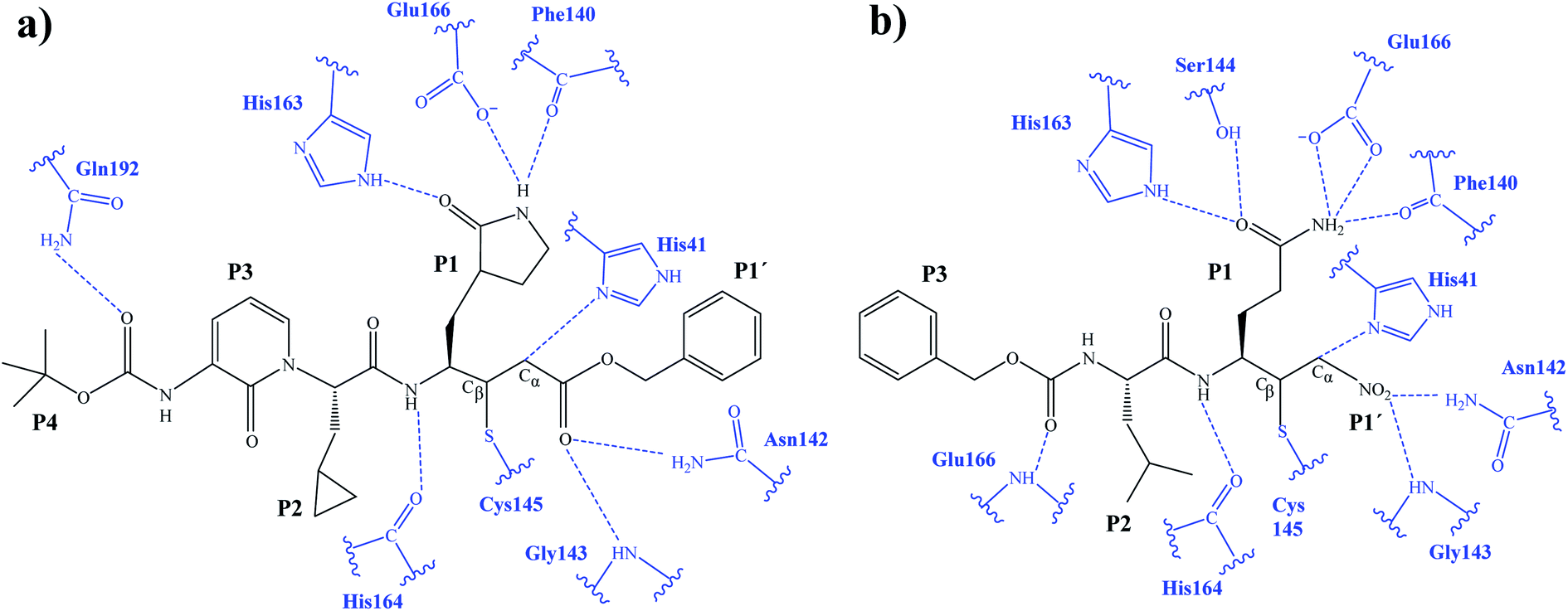 | ||
| Fig. 5 Schematic representation of the active site in the E-I complex and details of the H-bond interactions between the inhibitor and the active site of the SARS-CoV-2 Mpro from QM/MM MD simulations of compound B1 (a) and compound B2 (b). | ||
After equilibration of the two covalent E-I structures, the chemical reaction steps of the (reverse) inhibition process were studied by exploration of the PESs and FESs (Fig. S9 and S11†). The free energy profiles are shown in Fig. 2, together with that for N3. The reaction in both cases follows the same mechanism as for N3. The activation energy barriers obtained with compound B1 and B2, are both determined by the rate-limiting transition state of the C–S bond formation, TS2. Nevertheless, while the barrier (for covalent complex formation, i.e. for the forward reaction) of B1 (11.8 kcal mol−1) is very close to that obtained with N3 (11.2 kcal mol−1), that of B2 is slightly lower (9.8 kcal mol−1). In contrast, the reaction energies of B1 and B2 are very different: −27.9 and −11.4 kcal mol−1, respectively. These results suggest, first, that both designed inhibitors should present similar reactivity to N3 for covalent complex formation, with B2 being slightly more reactive. Second, while compound B1 would be an irreversible inhibitor, compound B2 is predicted to show more reversible inhibitor character than B1 and N3. As can be seen from Fig. 2a, there are also differences regarding the relative energy of the initial non-covalent states, E:Ivs.E(+/−):I. While in the case of compound B1 the neutral dyad is 0.4 kcal·mol−1 less stable than the ionic pair, the E(+/−):I of compound B2 is 2.6 kcal mol−1 higher in energy than the E:I. As observed in Fig. 2a, E(+/−):I of compound N3 is also higher in energy, by 1.3 kcal mol−1, than the E:I. These results, consistent with the results obtained with N3, with the previously studied proteolysis reaction with a reactive substrate,22 and with previous studied on other SARS-CoV Mpro enzymes52 indicate that the protonation state of C145 and H41 in the initial non-covalent binary complex depends on the substrate substituents in the P1′, P1 and P2 positions. It is remarkable how, according to the P1′ S1′ interatomic distances, the interaction between the nitro group of B2 and G143 appears to be stronger in the E:I than in E(+/−):I, in contrast to B1, which shows a only small decrease in the distances between the carbonyl group and residues G143; N3 that does not show any clear change in the interactions with residues G143, S144 and C145 (see Tables S5, S8 and S11†). The different electronic distributions in these molecules (a less basic carbanion in B2 as compared to N3 and B1) may slightly shift the pKa of the two catalytic residues. Nevertheless, as shown in Fig. 2, this influence is not significant for the inhibition process of Mpro. Finally, while the final proton transfer from H41 to the Cα of the inhibitor takes place as a barrierless process in N3 and B2, a transition state, TS3, is found for the reaction with B1, with a very small energy barrier (2.7 kcal mol−1).
S1′ interatomic distances, the interaction between the nitro group of B2 and G143 appears to be stronger in the E:I than in E(+/−):I, in contrast to B1, which shows a only small decrease in the distances between the carbonyl group and residues G143; N3 that does not show any clear change in the interactions with residues G143, S144 and C145 (see Tables S5, S8 and S11†). The different electronic distributions in these molecules (a less basic carbanion in B2 as compared to N3 and B1) may slightly shift the pKa of the two catalytic residues. Nevertheless, as shown in Fig. 2, this influence is not significant for the inhibition process of Mpro. Finally, while the final proton transfer from H41 to the Cα of the inhibitor takes place as a barrierless process in N3 and B2, a transition state, TS3, is found for the reaction with B1, with a very small energy barrier (2.7 kcal mol−1).
M06-2X/6-31+G(d,p)/MM optimized structures of the stable states in the reactions with both inhibitors are presented in Fig. 6 (structures of the TSs, E:I and E-I are given in the ESI,† together with a list of key interatomic distances of average structures obtained from the AM1/MM MD simulations and single optimized structures at the M06-2X/6-31+G(d,p)/MM level). As observed for N3, the enzyme-inhibitor interactions do not change significantly during the inhibition reaction with B1 and B2 (Fig. 6, Tables S8 and S11†). In both cases, designed inhibitors bind stably in the active site of the enzyme. Analysis of the favorable protein-inhibitor interactions in the E:I state, computed as the sum of QM/MM electrostatic and Lennard-Jones terms, shown in Fig. 7, confirms the predictions made in the design of B1 and B2 and the conclusions from the geometrical analysis of the optimized structures. In both cases the interactions between the protein and the inhibitors are dominated by those in the P1 S1 site.
S1 site.
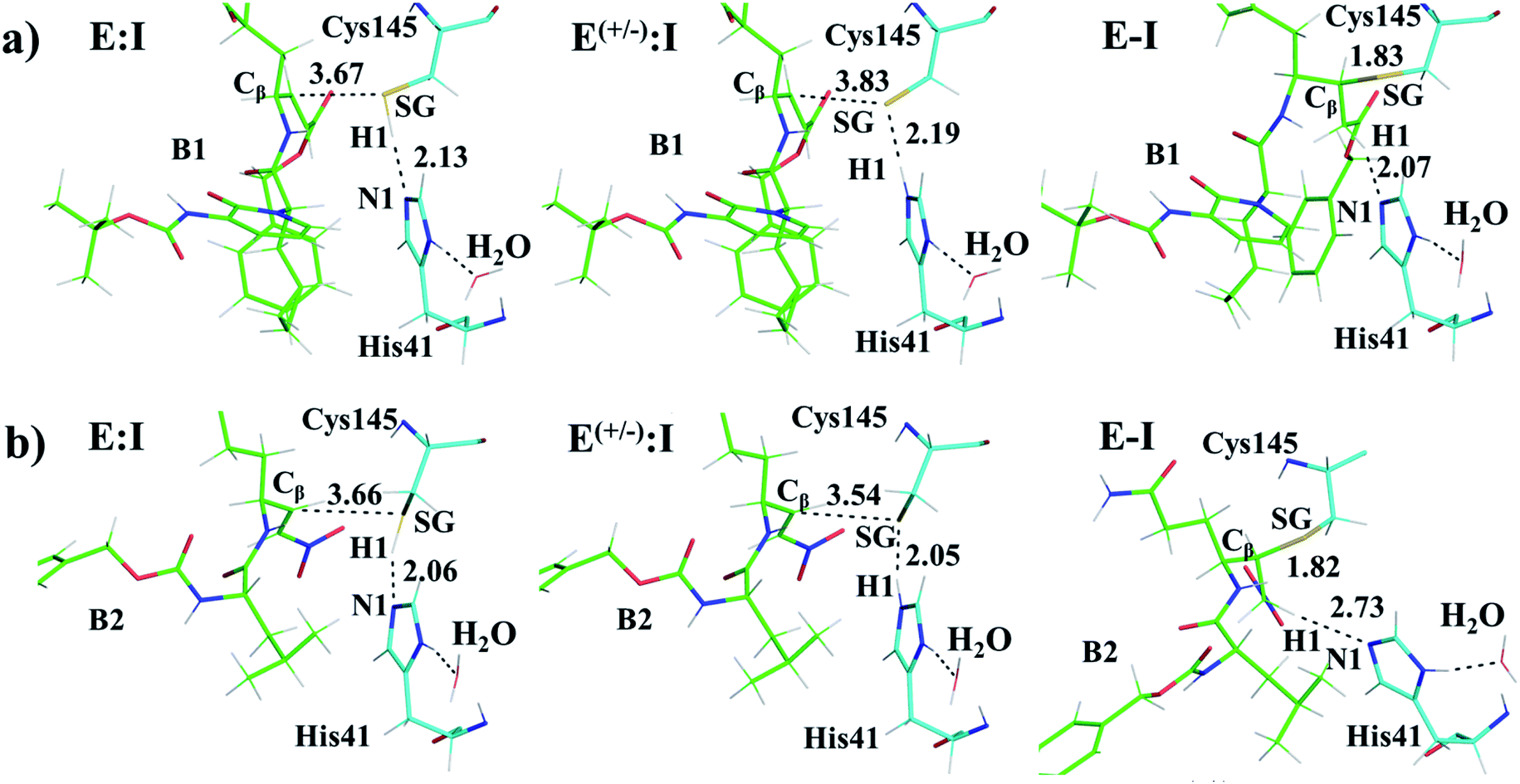 | ||
| Fig. 6 Detail of the M06-2X/6-31+G(d,p)/MM optimized structures of E:I, E(+/−):I and E-I appearing along the inhibition process of Mpro by compound B1 (a) and compound B2 (b). Carbon atoms of the inhibitors are shown in green while those of the catalytic residues Cys145 and His41 are in blue. Key distances are in Å. | ||
 | ||
| Fig. 7 Main favorable average interaction energies (electrostatic plus Lennard-Jones) between residues of Chain-A and each fragment of the compound B1 (a) and compound B2 (b) computed in the E:I state. Results obtained as an average over 1000 structures from the AM1/MM MD simulations. | ||
The averaged structures of E:I and TS2 in the three compounds (Tables S5, S8 and S11†), from the QM/MM MD simulations, show how the smaller the reduction in the distance between the SG atom of C145 and the Cβ atom of the inhibitors (the reaction coordinate that controls the rate-limiting step) from E:I complex to the TS2 (1.11, 1.09 and 0.74 Å for B1, N3, and B2, respectively), the lower the barrier (11.8, 11.2 and 9.8 kcal mol−1 for B1, N3 and B2, respectively). These results suggest that B2 adopts a more reactive conformation in the E:I state, closer to TS2 than those of N3 or B1, which may contribute to its lower overall activation energy barrier. These results explain why the inhibitor in which the ion pair is most disfavored, but structurally closer to the rate-limiting TS2, also has the lowest barrier.
Binding poses for designed inhibitors B1 and B2
Binding of inhibitors to the Mpro active site through noncovalent interactions (to form E:I, Scheme 3) is a key event that precedes the chemical step of the inhibitory process. The ability of B1 and B2 to fit the Mpro active site was therefore examined, starting from the structures of the reactants generated by QM/MM MD simulations. N3 was used as a control. The best docking pose for each ligand (see Methods) indicates that all the inhibitors assume a binding pose consistent with the X-ray structure of the Mpro-N3 adduct, i.e. with the warhead properly oriented to react with the catalytic cysteine, the polar sidechain at P1 site forming hydrogen bonds with S1 residues, and the lipophilic chain at P2 site undertaking several van der Waals contacts with S2 residues (Fig. S13†). Crucially, the binding poses of B1 and B2 resemble that of the reference inhibitor N3 (Fig. S13†), suggesting that modifications at level of the tail or of the warhead do not affect the accommodation of the critical P1 and P2 residues. Within the known limits of empirical scoring functions, the present docking analysis supports the proposal that B1 and B2 can bind to Mpro in a productive orientation in its active site.Conclusions
Here, we first explored the inhibition of SARS-CoV-2 Mpro with a known covalent (peptidyl) inhibitor, N3,19 by QM/MM MD simulations. The results are in good agreement with experimental crystal structures and kinetics, and reveal the chemical mechanism of covalent reaction. This provides an atomically detailed description of the process of formation of the covalent enzyme-inhibitor complex. We used these results, together with information from other CP protease inhibitors and from our recent study of the proteolysis reaction of SARS-CoV-2 Mpro,22 to design and computationally test two putative inhibitors of SARS-CoV-2 Mpro based on the scaffold of N3. In the first designed compound, B1, the recognition portion of N3 was modified while both the recognition part and the warhead of N3 (to a nitroalkene) were changed to generate a second compound, B2 (Scheme 2).The calculated free energy landscape for formation of the covalent enzyme-inhibitor intermediate indicate that the reaction, with all three compounds, proceeds in a stepwise manner: in the first step, Cys145 is activated by His41, forming the ion pair E(+/−):I, followed in the second step by attack of the sulfur atom of Cys145 on the Cβ atom of the inhibitor and proton transfer from His41 to the Cα atom of the inhibitor, leading a stable covalent E-I intermediate. The rate-limiting step of the process, in all three cases, corresponds to the enzyme-inhibitor covalent bond formation, with an activation free energy of 11.2, 11.8 and 9.8 kcal mol−1 for N3, B1 and B2, respectively. The low activation free energy of the inhibition reaction with N3 is consistent with kinetic experiments,12 while the values obtained with compound B1 and B2, indicate that both are also reactive. Further, the lower activation energy for B2 suggests that it would react faster with Mpro: this is a potential advantage in biological media in which the compounds have to compete with high concentration of the natural substrate. Within cells, covalent inhibitors must react with the target quickly, to avoid competing reactions with free bio-nucleophiles, such as glutathione, or with proteases (e.g. in the case of peptidyl compounds) that can reduce their active concentrations.54 From the thermodynamic point of view, the exergonic reaction with N3 (reaction energy −17.9 kcal mol−1) is consistent with its experimentally observed stability (e.g. revealed by X-ray crystallographic structures).12 The inhibition reactions of Mpro with B1 and B2 are also exergonic but are very different from each other (−27.9 and −11.4 kcal mol−1, respectively), suggesting that compound B1 would be an irreversible inhibitor, but compound B2 has a more reversible character. Analysis of the QM-MM interaction energies between the different residues of the inhibitor and the residues located in the substrate-binding pockets of Mpro confirms the predictions made in the design of B1 and B2, and the conclusions from geometrical analysis of the structures optimized at the DFT/MM level. In both cases, the interactions between the protein and the inhibitors are dominated by those in the P1 S1 site. Finally, docking carried out with the noncovalent enzyme:inhibitor reactant complex structures from our QM/MM structures support the proposal that B1 and B2 can bind Mpro in a reactive conformation in its active site.
S1 site. Finally, docking carried out with the noncovalent enzyme:inhibitor reactant complex structures from our QM/MM structures support the proposal that B1 and B2 can bind Mpro in a reactive conformation in its active site.
In summary, our QM/MM study of the inhibition of Mpro by N3 and two covalent (peptidyl) MA compounds, B1 and B2, which we designed based on these simulations and medicinal chemistry experience, indicates that a lower alkylation barrier than N3 can be obtained by modulating either the recognition portion or the warhead. Interactions between the recognition moiety and Mpro active site affect the chemical step because they dictate the pose of the inhibitor in the active site of the enzyme. Our results show that B1 has a more irreversible character than N3 while B2 is more reversible. This different behavior in silico suggest that both compounds should be tested and compared to N3 as promising candidates as drug leads against COVID-19. Both designed compounds can be easily prepared through synthetic approaches inspired by published synthetic routes of similar compounds.14,37
Author contributions
The study was designed by K. Ś., V. M., A. L. and A. J. M. The results were discussed and analyzed by all authors, and contributed to writing the manuscript. K. A., N. S. and K. Ś. carried out the QM/MM calculations. A. L. carried out the docking calculations. All authors contributed to discussion and design of inhibitors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Spanish Ministerio de Ciencia, Innovación y Universidades (Grant PGC2018-094852-B-C21 and PID2019-107098RJ-I00), Generalitat Valenciana (Grant AICO/2019/195 and SEJI/2020/007) Universitat Jaume I (UJI B2017-31, UJI B2018-41, UJI-A2019-04 and SomUJIContraCovid crowdfunding campaign), KŚ thanks the Spanish Ministerio de Ciencia, Innovación y Universidades for a Juan de la Cierva – Incorporación (ref. IJCI-2016-27503) contract. K. A. thanks Universitat Jaume I (POSDOC-A/2018/30) and Generalitat Valenciana (APOSTD/2020/015) for post-doctoral contracts. NS thanks the MINECO for doctoral FPI grant (BES-2016-078029). The authors thankfully acknowledge the computer resources at Pirineus and the technical support provided at Pirineus and by Barcelona Supercomputing Center (QSB-2020-2-0004), as well as the local computational resources of the Servei d’Informàtica of Universitat Jaume I. AJM thanks EPSRC for support (CCP-BioSim, grant number EP/M022609/1) and also the British Society for Antimicrobial Chemotherapy (grant number BSAC-COVID-30).References
- G. Zhu, C. Zhu, Y. Zhu and F. Sun, Current Research in Microbial Sciences, 2020, 1, 53–61 CrossRef.
- A. K. Ghosh, M. Brindisi, D. Shahabi, M. E. Chapman and A. D. Mesecar, ChemMedChem, 2020, 15, 907–932 CrossRef CAS.
- R. T. Eastman, J. S. Roth, K. R. Brimacombe, A. Simeonov, M. Shen, S. Patnaik and M. D. Hall, ACS Central Science, 2020, 6, 672–683 CrossRef CAS.
- T. Warren, R. Jordan, M. Lo, V. Soloveva, A. Ray, R. Bannister, R. Mackman, M. Perron, K. Stray, J. Feng, Y. Xu, J. Wells, K. Stuthman, L. Welch, E. Doerffler, L. Zhang, K. Chun, H. Hui, S. Neville, W. Lew, Y. Park, D. Babusis, R. Strickley, P. Wong, S. Swaminathan, W. Lee, D. Mayers, T. Cihlar and S. Bavari, Open Forum Infect. Dis., 2015, 2, LB-2 CrossRef.
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368, 409–412 CrossRef CAS.
- J. H. Beigel, K. M. Tomashek, L. E. Dodd, A. K. Mehta, B. S. Zingman, A. C. Kalil, E. Hohmann, H. Y. Chu, A. Luetkemeyer, S. Kline, D. Lopez de Castilla, R. W. Finberg, K. Dierberg, V. Tapson, L. Hsieh, T. F. Patterson, R. Paredes, D. A. Sweeney, W. R. Short, G. Touloumi, D. C. Lye, N. Ohmagari, M.-d. Oh, G. M. Ruiz-Palacios, T. Benfield, G. Fätkenheuer, M. G. Kortepeter, R. L. Atmar, C. B. Creech, J. Lundgren, A. G. Babiker, S. Pett, J. D. Neaton, T. H. Burgess, T. Bonnett, M. Green, M. Makowski, A. Osinusi, S. Nayak and H. C. Lane, N. Engl. J. Med., 2020, 383, 1813–1826 CrossRef CAS.
- L. Lisi, P. M. Lacal, M. L. Barbaccia and G. Graziani, Biochem. Pharmacol., 2020, 114169, DOI:10.1016/j.bcp.2020.114169.
- J. D. Norrie, Lancet, 2020, 395, 1525–1527 CrossRef CAS.
- L. Riva, S. Yuan, X. Yin, L. Martin-Sancho, N. Matsunaga, L. Pache, S. Burgstaller-Muehlbacher, P. D. De Jesus, P. Teriete, M. V. Hull, M. W. Chang, J. F.-W. Chan, J. Cao, V. K.-M. Poon, K. M. Herbert, K. Cheng, T.-T. H. Nguyen, A. Rubanov, Y. Pu, C. Nguyen, A. Choi, R. Rathnasinghe, M. Schotsaert, L. Miorin, M. Dejosez, T. P. Zwaka, K.-Y. Sit, L. Martinez-Sobrido, W.-C. Liu, K. M. White, M. E. Chapman, E. K. Lendy, R. J. Glynne, R. Albrecht, E. Ruppin, A. D. Mesecar, J. R. Johnson, C. Benner, R. Sun, P. G. Schultz, A. I. Su, A. García-Sastre, A. K. Chatterjee, K.-Y. Yuen and S. K. Chanda, Nature, 2020, 586, 113–119 CrossRef CAS.
- T. Muramatsu, C. Takemoto, Y.-T. Kim, H. Wang, W. Nishii, T. Terada, M. Shirouzu and S. Yokoyama, Proc. Natl. Acad. Sci. U.S.A., 2016, 113, 12997 CrossRef CAS.
- W. Dai, B. Zhang, X.-M. Jiang, H. Su, J. Li, Y. Zhao, X. Xie, Z. Jin, J. Peng, F. Liu, C. Li, Y. Li, F. Bai, H. Wang, X. Cheng, X. Cen, S. Hu, X. Yang, J. Wang, X. Liu, G. Xiao, H. Jiang, Z. Rao, L.-K. Zhang, Y. Xu, H. Yang and H. Liu, Science, 2020, 368, 1331 CrossRef CAS.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Nature, 2020, 582, 289–293 CrossRef CAS.
- J. Chodera, A. A. Lee, N. London and F. von Delft, Nat. Chem., 2020, 12, 581 CrossRef CAS.
- A. Latorre, T. Schirmeister, J. Kesselring, S. Jung, P. Johe, U. A. Hellmich, A. Heilos, B. Engels, R. L. Krauth-Siegel, N. Dirdjaja, L. Bou-Iserte, S. Rodriguez and F. V. Gonzalez, ACS Med. Chem. Lett., 2016, 7, 1073–1076 CrossRef CAS.
- P. A. Jackson, J. C. Widen, D. A. Harki and K. M. Brummond, J. Med. Chem., 2017, 60, 839–885 CrossRef CAS.
- M. Gehringer and S. A. Laufer, J. Med. Chem., 2019, 62, 5673–5724 CrossRef CAS.
- A. Voice, G. Tresadern, H. van Vlijmen and A. Mulholland, J. Chem. Inf. Model., 2019, 59, 4220–4227 CrossRef CAS.
- R. A. Bauer, Drug Discovery Today, 2015, 20, 1061–1073 CrossRef CAS.
- H. Yang, W. Xie, X. Xue, K. Yang, J. Ma, W. Liang, Q. Zhao, Z. Zhou, D. Pei, J. Ziebuhr, R. Hilgenfeld, K. Y. Yuen, L. Wong, G. Gao, S. Chen, Z. Chen, D. Ma, M. Bartlam and Z. Rao, PLoS Biol., 2005, 3, e324 CrossRef.
- J. W. Keillor and R. S. Brown, J. Am. Chem. Soc., 1992, 114, 7983–7989 CrossRef CAS.
- K. Arafet, S. Ferrer and V. Moliner, ACS Catal., 2017, 7, 1207–1215 CrossRef CAS.
- K. Świderek and V. Moliner, Chem. Sci., 2020, 11, 10626–10630 RSC.
- E. Awoonor-Williams, A. G. Walsh and C. N. Rowley, Biochim. Biophys. Acta, Proteins Proteomics, 2017, 1865, 1664–1675 CrossRef CAS.
- A. Lodola, M. Mor, J. Sirirak and A. J. Mulholland, Biochem. Soc. Trans., 2009, 37, 363–367 CrossRef CAS.
- K. Arafet, S. Ferrer and V. Moliner, Biochemistry, 2015, 54, 3381–3391 CrossRef CAS.
- R. E. Amaro and A. J. Mulholland, Nat. Rev. Chem., 2018, 2, 0148 CrossRef CAS.
- D. Callegari, K. E. Ranaghan, C. J. Woods, R. Minari, M. Tiseo, M. Mor, A. J. Mulholland and A. Lodola, Chem. Sci., 2018, 9, 2740–2749 RSC.
- N. Serrano-Aparicio, K. Świderek and V. Moliner, Eur. J. Med. Chem., 2019, 164, 399–407 CrossRef CAS.
- K. Arafet, F. V. González and V. Moliner, Chem.–Eur. J., 2020, 26, 2002–2012 CrossRef CAS.
- A. Lodola, D. Callegari, L. Scalvini, S. Rivara and M. Mor, Methods Mol. Biol., 2020, 2114, 307–337 CrossRef CAS.
- L. Zhang, D. Lin, Y. Kusov, Y. Nian, Q. Ma, J. Wang, A. von Brunn, P. Leyssen, K. Lanko, J. Neyts, A. de Wilde, E. J. Snijder, H. Liu and R. Hilgenfeld, J. Med. Chem., 2020, 63, 4562–4578 CrossRef CAS.
- S. A. Gillmor, C. S. Craik and R. J. Fletterick, Protein Sci., 1997, 6, 1603–1611 CrossRef CAS.
- E. Dunny, W. Doherty, P. Evans, J. P. G. Malthouse, D. Nolan and A. J. S. Knox, J. Med. Chem., 2013, 56, 6638–6650 CrossRef CAS.
- S. Royo, S. Rodriguez, T. Schirmeister, J. Kesselring, M. Kaiser and F. V. Gonzalez, ChemMedChem, 2015, 10, 1484–1487 CrossRef CAS.
- X. Zhai and T. D. Meek, Biochemistry, 2018, 57, 3176–3190 CrossRef CAS.
- L. Zhu, S. George, M. F. Schmidt, S. I. Al-Gharabli, J. Rademann and R. Hilgenfeld, Antivir. Res., 2011, 92, 204–212 CrossRef CAS.
- P. S. Dragovich, T. J. Prins, R. Zhou, S. E. Webber, J. T. Marakovits, S. A. Fuhrman, A. K. Patick, D. A. Matthews, C. A. Lee, C. E. Ford, B. J. Burke, P. A. Rejto, T. F. Hendrickson, T. Tuntland, E. L. Brown, J. W. Meador, R. A. Ferre, J. E. V. Harr, M. B. Kosa and S. T. Worland, J. Med. Chem., 1999, 42, 1213–1224 CrossRef CAS.
- G. M. Torrie and J. P. Valleau, J. Comput. Phys., 1977, 23, 187–199 CrossRef.
- S. Kumar, D. Bouzida, R. H. Swendsen, P. A. Kollman and J. M. Rosenberg, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healy and J. J. P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902–3909 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- W. J. Hehre, L. Radom, P. v. R. Schleyer and J. A. Pople, Ab Initio Molecular Orbital Theory, John Wiley, New York, 1986 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09 (Revision A.1), 2009 Search PubMed.
- K. Świderek, I. Tuñón, S. Martí and V. Moliner, ACS Catal., 2015, 5, 1172–1185 Search PubMed.
- K. Świderek, I. Tuñón, V. Moliner and J. Bertran, ACS Catal., 2015, 5, 2587–2595 Search PubMed.
- A. Krzemińska, V. Moliner and K. Świderek, J. Am. Chem. Soc., 2016, 138, 16283–16298 CrossRef.
- Y. Duan, C. Wu, S. Chowdhury, M. C. Lee, G. Xiong, W. Zhang, R. Yang, P. Cieplak, R. Luo, T. Lee, J. Caldwell, J. Wang and P. Kollman, J. Comput. Chem., 2003, 24, 1999–2012 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- M. J. Field, M. Albe, C. Bret, F. Proust-De Martin and A. Thomas, J. Comput. Chem., 2000, 21, 1088–1100 CrossRef CAS.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS.
- J. R. A. Silva, L. Cianni, D. Araujo, P. H. J. Batista, D. de Vita, F. Rosini, A. Leitao, J. Lameira and C. A. Montanari, J. Chem. Inf. Model., 2020, 60, 1666–1677 CrossRef CAS.
- A. Paasche, A. Zipper, S. Schäfer, J. Ziebuhr, T. Schirmeister and B. Engels, Biochemistry, 2014, 53, 5930–5946 CrossRef CAS.
- C. A. Ramos-Guzmán, J. J. Ruiz-Pernía and I. Tuñón, 2020, DOI:10.26434/chemrxiv.12895064.v1.
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery, 2011, 10, 307–317 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational methods, FF parameters for inhibitors, detail of active site and QM-MM partitioning, M06-2X:AM1/MM FESs, list of key interatomic distances in the states along the reaction path at AM1/MM and optimized at M06-2X/MM level, protein-substrate non-bonding interaction energies, per residue, Cartesian coordinates of the QM subset of atoms and full structures of the rate-limiting TSs, E:I and E-I optimized at M06-2X/6-31+G(d,p)/MM level, and figure of inhibitors docked into the active site of SARS-CoV-2 Mpro. See DOI: 10.1039/d0sc06195f |
| This journal is © The Royal Society of Chemistry 2021 |