
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence“Click-switch” – one-step conversion of organic azides into photochromic diarylethenes for the generation of light-controlled systems†
Steffy
Becht
 a,
Reena
Sen
b,
Simon M.
Büllmann
a,
Andreas
Dreuw
b and
Andres
Jäschke
*a
a,
Reena
Sen
b,
Simon M.
Büllmann
a,
Andreas
Dreuw
b and
Andres
Jäschke
*a
aInstitute of Pharmacy and Molecular Biotechnology (IPMB), Heidelberg University, Im Neuenheimer Feld 364, 69120 Heidelberg, Germany. E-mail: jaeschke@uni-hd.de
bTheoretical and Computational Chemistry, Interdisciplinary Center for Scientific Computing (IWR), Heidelberg University, Im Neuenheimer Feld 205A, 69120 Heidelberg, Germany
First published on 29th July 2021
Abstract
Diarylethenes (DAEs) are an established class of photochromic molecules, but their effective incorporation into pre-existing targets is synthetically difficult. Here we describe a new class of DAEs in which one of the aryl rings is a 1,2,3-triazole that is formed by “click” chemistry between an azide on the target and a matching alkyne–cyclopentene–thiophene component. This late-stage zero-length linking allows for tight integration of the DAE with the target, thereby increasing the chances for photomodulation of target functions. Nineteen different DAEs were synthesized and their properties investigated. All showed photochromism. Electron-withdrawing groups, and in particular −M-substituents at the triazole and/or thiophene moiety resulted in DAEs with high photo- and thermostability. Further, the chemical nature of the cyclopentene bridge had a strong influence on the behaviour upon UV light irradiation. Incorporation of perfluorinated cyclopentene led to compounds with high photo- and thermostability, but the reversible photochromic reaction was restricted to halogenated solvents. Compounds containing the perhydrogenated cyclopentene bridge, on the other hand, allowed the reversible photochromic reaction in a wide range of solvents, but had on average lower photo- and thermostabilities. The combination of the perhydrocyclopentene bridge and electron-withdrawing groups resulted in a DAE with improved photostability and no solvent restriction. Quantum chemical calculations helped to identify the photoproducts formed in halogenated as well as non-halogenated solvents. For two optimized DAE photoswitches, photostationary state composition and reaction quantum yields were determined. These data revealed efficient photochemical ring closure and opening. We envision applications of these new photochromic diarylethenes in photonics, nanotechnology, photobiology, photopharmacology and materials science.
Introduction
Diarylethenes (DAEs) are among the most commonly used classes of photochromic compounds. These molecules undergo a light-induced reversible electrocyclic ring closure reaction (Fig. 1(a)) which has little effect on end-to-end distance and dipole moment, but dramatically changes electronic properties, rigidity and rotational degrees of freedom. Since their first description in the late 1960s, literally thousands of DAE derivatives have been synthesized, characterized, and applied in many fields.1 In addition to their originally intended application in optical information storage materials, DAEs have been incorporated into biomolecules,2 molecular machines,3–5 crystals,6 surfaces7 and nanoparticles,8–10 as the reversible induction of structural changes by light promises attractive options for the modulation of chemical, physical, or biological properties. The incorporation of DAEs into target molecules typically requires multi-step organic synthesis, thereby limiting the application of these compounds. Often it would be desirable to attach a DAE to a pre-existing target structure – an organic molecule, a biopolymer, a 3D-nanotechnology object, a functional surface, or a macroscopic object. Ideally this would be possible in a fully site-specific manner, and orthogonal to the functional groups present in the target.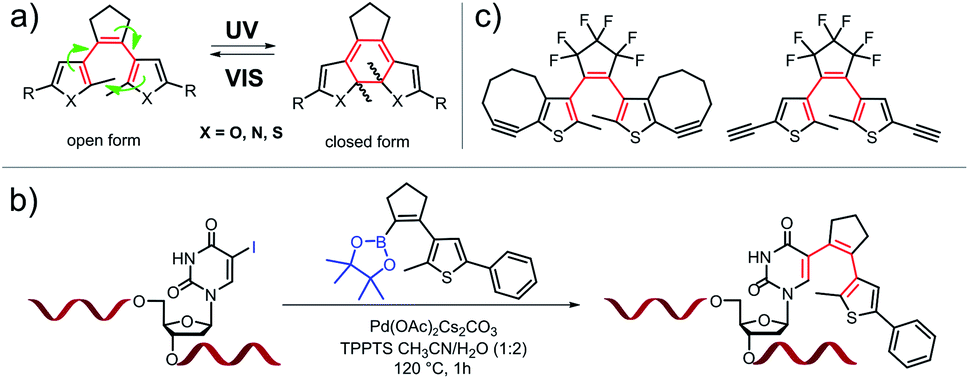 | ||
| Fig. 1 Examples of known DAEs: (a) classical symmetric DAE structure and its reversible cyclization upon UV light irradiation (R: e.g. phenyl, OF: open form, CF: closed form), (b) “zero-length” linker synthesis of oligonucleotide-DAE photoswitches,11 (c) examples of alkyne-substituted dithienyl-ethenes.12,13 | ||
Examples for this approach are based on activated esters,14,15 acetylenes,12,13 carboxylic acids, and hydrazides,16,17 but in these cases the DAE is typically appended to the target via linkers on the periphery of the photochromic unit. This connectivity provides flexibility and distance to the chromophore, which in turn attenuates or even eliminates the potential effects of DAE switching on the target's function.8–10,15 A “zero-length”-linker would be more suitable for the tight integration of the DAE into the target's overall architecture. In a previous approach from our laboratory,11 a precursor was synthesized that allowed the formation of a DAE in pre-assembled DNA strands by late-stage Suzuki–Miyaura cross coupling. A site-specifically placed iodo-substituent on a nucleobase was used to create a photoswitch where this nucleobase acted as one of the two aryl residues of the DAE (Fig. 1(b)). The approach of tight integration by late-stage zero-length linking should be amenable to considerable expansion. Theoretically, the various types of “click” chemistry18 developed over the past two decades appear perfectly suited for such tight integration, and in particular those that involve azides, as various approaches for their site-specific incorporation have been developed (and in part commercialized) for biopolymers,19 cyclodextrins,20 nanoparticles,21,22 surfaces,23,24 and even some complex natural products.25,26 While there are examples of attaching DAEs to targets by azide–alkyne based click chemistry, these again attach them to the periphery of the chromophore or via flexible linkers12,13 (Fig. 1(c)). We are not aware of a single example where one of the DAE aryl rings is actually formed in a click reaction with a functional group of the target.
Here we report a new class of DAEs, in which one of the aryl rings is a 1,2,3-triazole. Importantly, this ring is formed in one step by copper-catalyzed alkyne–azide cycloaddition (CuAAC) using an azide group on the target and an alkyne that contains the DAE's bridge and another aryl ring (Fig. 2).
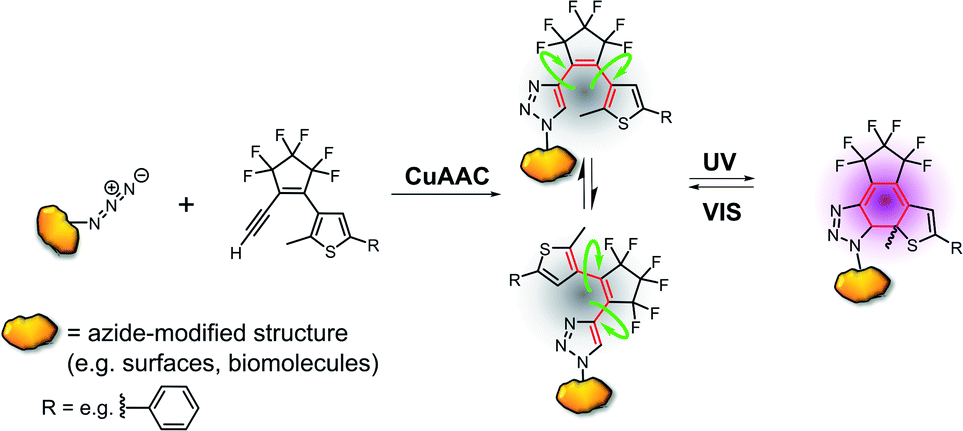 | ||
| Fig. 2 Novel triazole DAE system: click reaction between an azide-modified target and a photoswitch precursor (cyclopentene-acetylene) forms a triazole diarylethene that is switchable between the flexible open and the rigid closed isomer, R: e.g. –phenyl. | ||
We report the synthesis of 19 new DAEs, their analysis, and the optimization of their properties for applications as photoswitches. Our approach paves the way for the easy introduction of DAE photochromic units at strategically chosen positions of the target with zero-length connectivity and may facilitate applications in nanotechnology, photobiology, photopharmacology and materials science. By incorporating additional functional groups into the alkyne component, the approach can be easily expanded to allow the attachment of further building blocks (Fig. S1†).
Results and discussion
Design and synthesis of the alkyne component
At the outset of this study, it was unknown which structural features would render triazolyl-DAEs photochromic, reversible, fatigue-resistant, photo- and thermostable. To limit the combinatorial explosion of DAE diversity, we restricted ourselves to the following design features (Fig. 2): while one aryl of the DAE is represented by the triazole formed in the CuAAC, the second aryl should be a member of a small library of substituted 2-methylthiophenes, by far the most common aryl moiety in DAEs. With respect to the bridge moieties, we decided to stick with the two classical and most common ones, namely perfluorocyclopentene and cyclopentene (or perhydrocyclopentene). Thus, the first task at hand was to develop a synthetic route towards the alkyne component which consists of three elements: (i) the acetylene for the CuAAC reaction, (ii) the cyclopentene bridge, and (iii) the thiophene ring where limited diversity is introduced by further substitution (Fig. 2).Coupling of acetylene groups to a perhydrocyclopentene ring has been reported, but the symmetrical 1,2-diacetylene-cyclopentene was unstable and turned brownish-black within minutes at room temperature.27 For the synthesis of asymmetric cyclopentene acetylenes we adapted common synthesis routes for asymmetric diarylethenes by the introduction of an acetylene instead of the second aryl.
The synthesis of perfluorocyclopentene-bridged alkynes 6a–d proceeded via compounds 5a–d which are known intermediates in the synthesis of asymmetric DAEs28–30 (Scheme 1(a)).
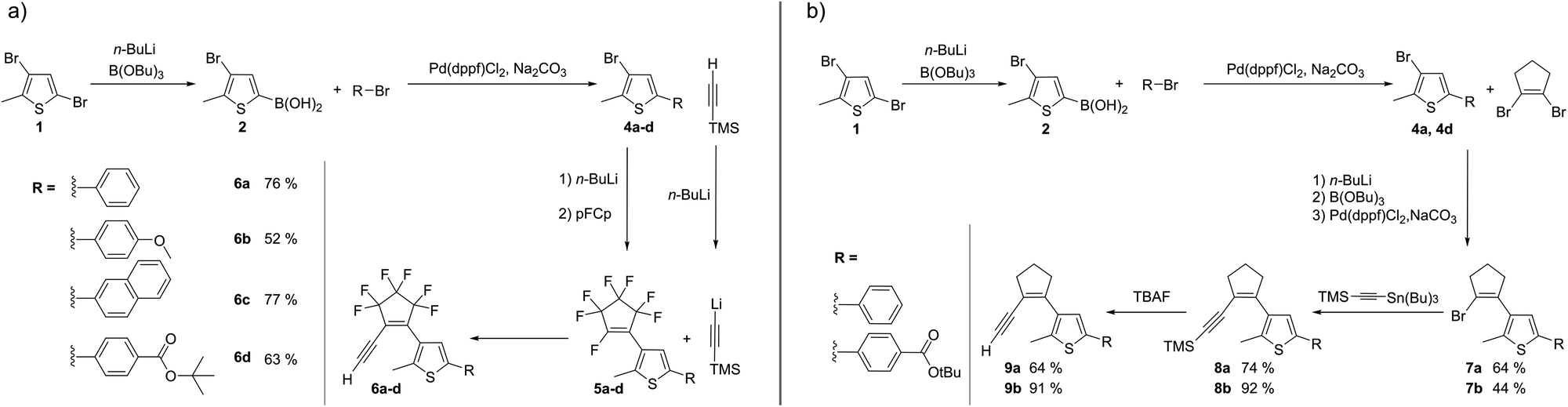 | ||
| Scheme 1 (a) Synthesis of perfluorocyclopentene acetylenes 6a–d. (b) Synthesis of perhydrocyclopentene acetylenes 9a and 9b. | ||
Trimethylsilyl-acetylene (TMS-acetylene) was introduced by lithiation analogous to the common introduction of the aryl moieties in high yields. The TMS protecting group was cleaved in situ, due to the release of fluorine ions in the coupling step, without compromising product formation. Compound 6d was synthetized at lower temperatures (−90 °C (ref. 31)) to introduce the tert-butyl ester group.
Synthesis of perhydrocyclopentene acetylenes 9a and 9b was more challenging since acetylene coupling via Li–F-exchange reaction was not applicable. In the synthesis of DAEs of this type, the second aryl moiety is usually attached to the bridge by cross-coupling.32,33 Sonogashira, Negishi and Stille conditions all gave the desired products, with the Stille coupling of (trimethylsilylethynyl)-tributylstannane (Scheme 1(b)) giving the highest yields and purities.
In contrast to the reported diacetylene cyclopentenes,27 the acetylene-thienyl-cyclopentenes 6a–d and 9a–b showed sufficient stability for application in click reactions (see below). Perhydrocyclopentenes 9a–b should be stored at −20 °C, as they turned brownish after a few days at room temperature. The perfluorinated derivatives 6a–d were stable for months at room temperature, if undissolved and protected from light.
CuAAC synthesis of perfluorocyclopentene triazole photoswitches
The click reaction of the cyclopentene acetylene precursors was first investigated with perfluorocyclopentene acetylenes (6a–d) and small organic azides (10a–l). The lipophilic nature of the acetylenes required unpolar conditions, and the reaction was catalyzed by the [(1,10-phenanthroline)bis(triphenyl-phosphine)] copper(I)-nitrate dichloromethane adduct and additional copper(I) iodide (Scheme 2) at room temperature. This catalyst had been used even in solvent-free reactions34 and turned out to be – in combination with copper(I) iodide – very efficient in the conversion of our new acetylenes in THF. 15 different triazole photoswitch derivatives (pFPS 1–15, abbreviation for perFluorocyclopentene-PhotoSwitch) were synthetized and characterized in order to derive first structure–property relationships for this novel DAE class, and to understand the effects of the reactant structure on the CuAAC reaction. In the first series, we kept the thiophene substituent R2 constant and varied the azide substituent R1 (for R2 = phenyl: pFPS 1–10), while in a second series R1 was kept constant (benzyl) and R2 was varied (pFPS 11–13). A third set focused on the influence of R1 and R2 substituents with mesomeric effects (pFPS 13–15).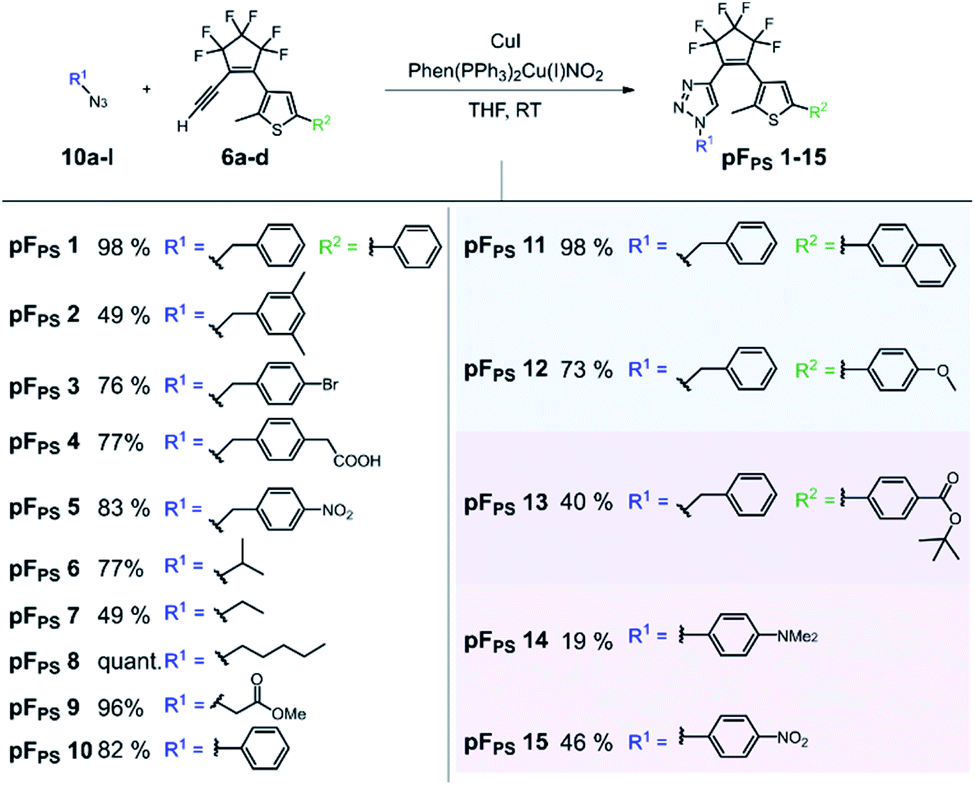 | ||
| Scheme 2 CuAAC reaction yields of perfluorocyclopentene acetylene precursors (6a–d) with small organic azides (10a–l). 15 different triazole photoswitches were synthesized (pFPS 1–15). | ||
CuAAC allowed the synthesis of photoswitches from aromatic and aliphatic (including benzylic) azides, whereby the latter ones were obtained in higher yields. In most cases the conversion was complete after 1–3 hours, and stirring overnight did not induce significant side reactions. The phenyl thiophene-derivatized alkyne generally gave higher yields than the p-benzoic acid tert-butyl ester thiophene building block, and the combination of the latter with an electron-donating N,N-dimethylaminophenyl substituent was particularly low-yielding. Nevertheless, all target compounds could be synthesized by CuAAC.
Photochromism of pFPS 1
Upon irradiation with UV light at 254 nm or 310 nm, at different concentrations, light intensities and irradiation times, solutions of the first triazole photoswitch pFPS 1 showed no significant photochromism in a range of commonly used non-halogenated solvents (methanol, ethyl acetate, dioxane or DMSO); they either retained their slightly brownish color, or their color changed irreversibly to faint yellow. However, solutions in halogenated solvents (CHCl3, CH2Cl2, CH2Br2, and CCl4) turned deeply red immediately, within seconds of UV light exposure.Absorption spectroscopy revealed the formation of a new absorption band in the visible range upon UV irradiation, as is typical for DAEs (Fig. 3). Upon white-light illumination, the red color disappeared, indicative of the electrocyclic ring opening.
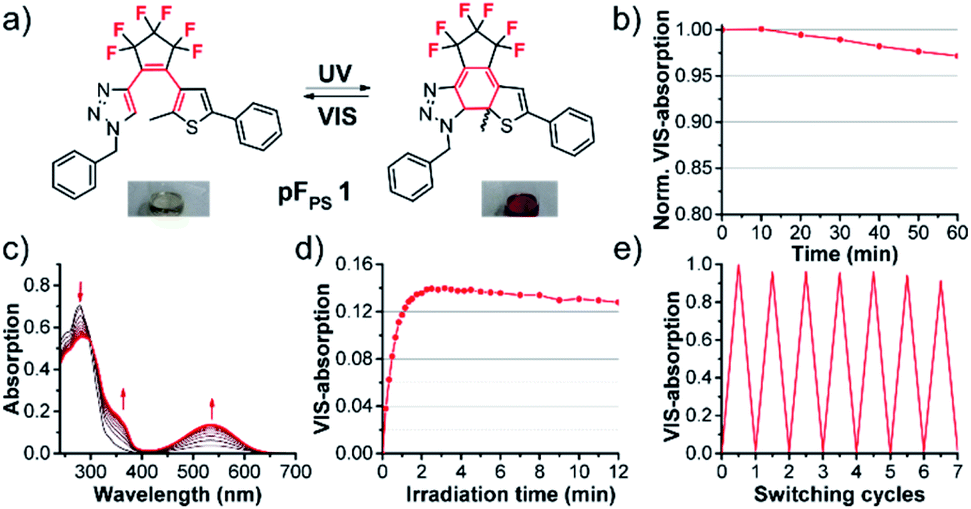 | ||
| Fig. 3 Photochromism of pFPS 1 (60 μM solution in CHCl3, irradiation at 310 nm, LED): (a) reversible electrocyclic ring closure, insets: photo of vial with colored solution, (b) thermostability of the closed form, monitored for 1 h at 20 °C at the VIS absorption maximum (534 nm), (c) evolution of the absorption spectra upon UV irradiation from the open form (black) towards the (pseudo-)photostationary state (p)PSS (red), (d) kinetic trace of the absorbance at 534 nm (VIS absorption maximum of the closed isomer) during prolonged UV irradiation, (e) reversibility measurement by alternating 310 nm and white-light illumination. | ||
The process was found to be highly reversible, and the colored closed-ring form showed high photo- and thermostability (τ20 °C = 16.0 h in CHCl3), indicating that this first triazolyl DAE performed very well, at least in halogenated solvents. We therefore characterized all 15 pF switches in CHCl3, in order to obtain a basic understanding of the relationships between structure and switching properties. Concurrently, we attempted to design a structure that may allow a photochromic reaction in a wider range of solvents.
Understanding the role of the triazole substituents
Ten derivatives were analyzed that differed only in the triazole substituent (pFPS 1–10, Fig. 4). All compounds exhibited photochromism in chloroform, and among the alkyl-substituted compounds there was only little variation in the wavelength of the VIS absorption maximum (between 528 nm, pFPS 9, and 535 nm, pFPS 2, Fig. S2†), and the apparent extinction coefficient in the (pseudo)-photostationary state ((p)PSS, εVISmax, app, between 0.18 × 104 L (mol cm)−1 (pFPS 6) and 0.40 × 104 L (mol cm)−1 (pFPS 9), Fig. S2†). The bleaching behavior (e.g., the absorbance vs. time trace after reaching the (p)PSS) showed slight differences, with compound pFPS 2 being the most and compound pFPS 4 the least photostable one. The largest differences were measured for the thermal stability of the closed-ring form (CF). Here, pFPS 9 was the most and pFPS 4, again, the least stable one (Table S1†). This low performance of pFPS 4 may be a consequence of the interaction between the acidic proton and the triazole ring. Attachment of an aromatic residue (pFPS 10) caused a large bathochromic shift of the VIS absorption band to 558 nm, an εVISmax, app of 0.32 × 104 L (mol cm)−1, and good (but not excellent) bleaching resistance and thermal stability. These observations correlate well with the structural diversity of the DAEs. Most obvious is the similarity of absorption vs. time traces within the group of benzyl derivatives, and the higher diversity of the traces among the derivatives with larger structural differences. Among the non-benzylic substituents, pFPS 9 shows an exceptionally high thermostability, which may be caused by the electron withdrawing ester group in proximity to the triazole.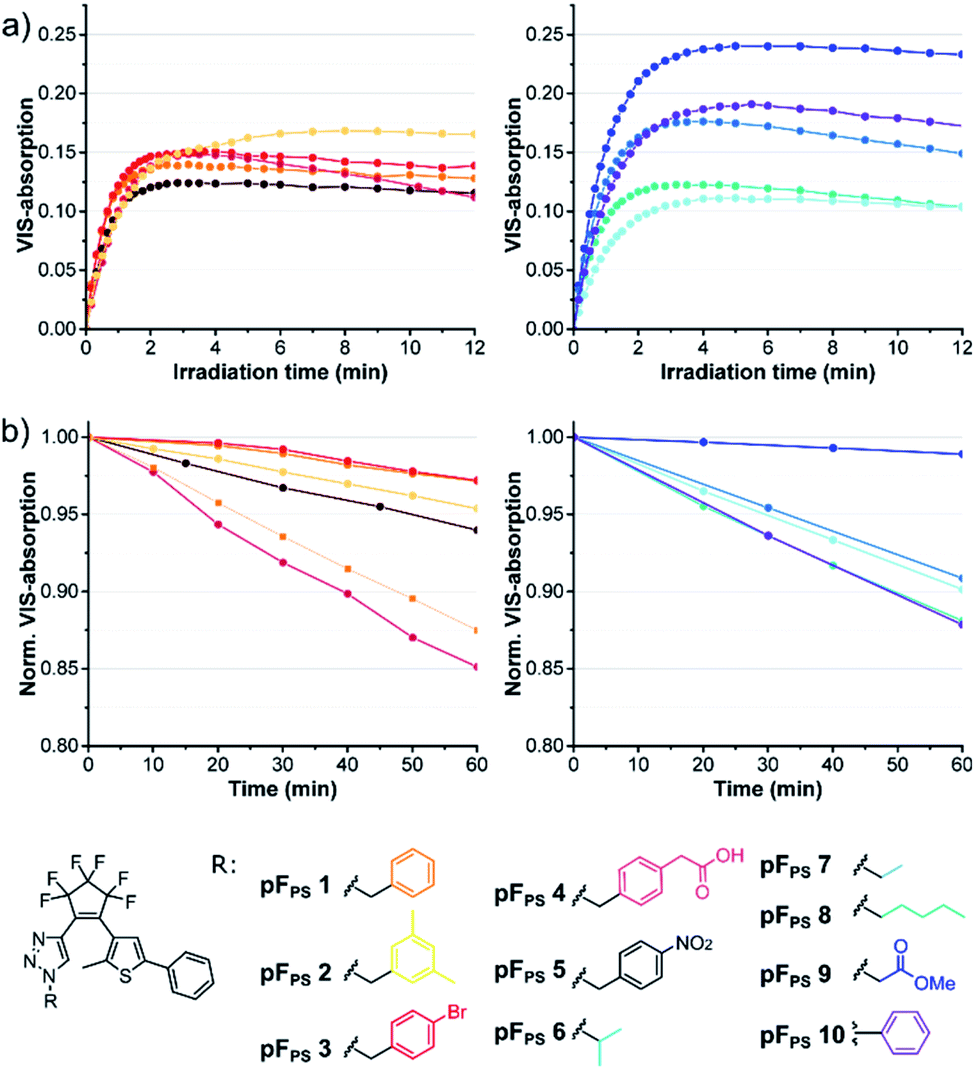 | ||
| Fig. 4 Comparison of the photochromic properties of various alkyl triazole photoswitches pFPS 1–10 (60 μM solutions in chloroform): (a) kinetic trace of the absorbance at the VIS maximum, (b) thermostability (20 °C) of the closed-ring forms. | ||
Role of the thiophene substitution
Comparing pFPS 1 and pFPS 13 (Fig. 5) with each other revealed the effects of adding an ester group to the p-position of the phenyl ring: increased absorption of the open form (OF), small bathochromic shift of the (p)PSS maximum (2 nm), increased (p)PSS absorption (from 0.14 to 0.23), similar photostability, and only slightly reduced thermostability. If an electron-donating methoxy group was added instead of the electron-withdrawing ester (pFPS 12), λmax was bathochromically shifted by 11 nm, εVISmax, app was increased to 0.47 × 104 L (mol cm)−1), and a very high thermal stability (τ20 °C = 21.2 h) was observed (Fig. 5). In addition, the spectral differences between open and closed form of this switch were particularly large. On the other hand, photostability was significantly reduced for this electron-rich photochrome. A similar trend was seen when the π-system of pFPS 1 was extended from phenyl to naphthyl (pFPS 11).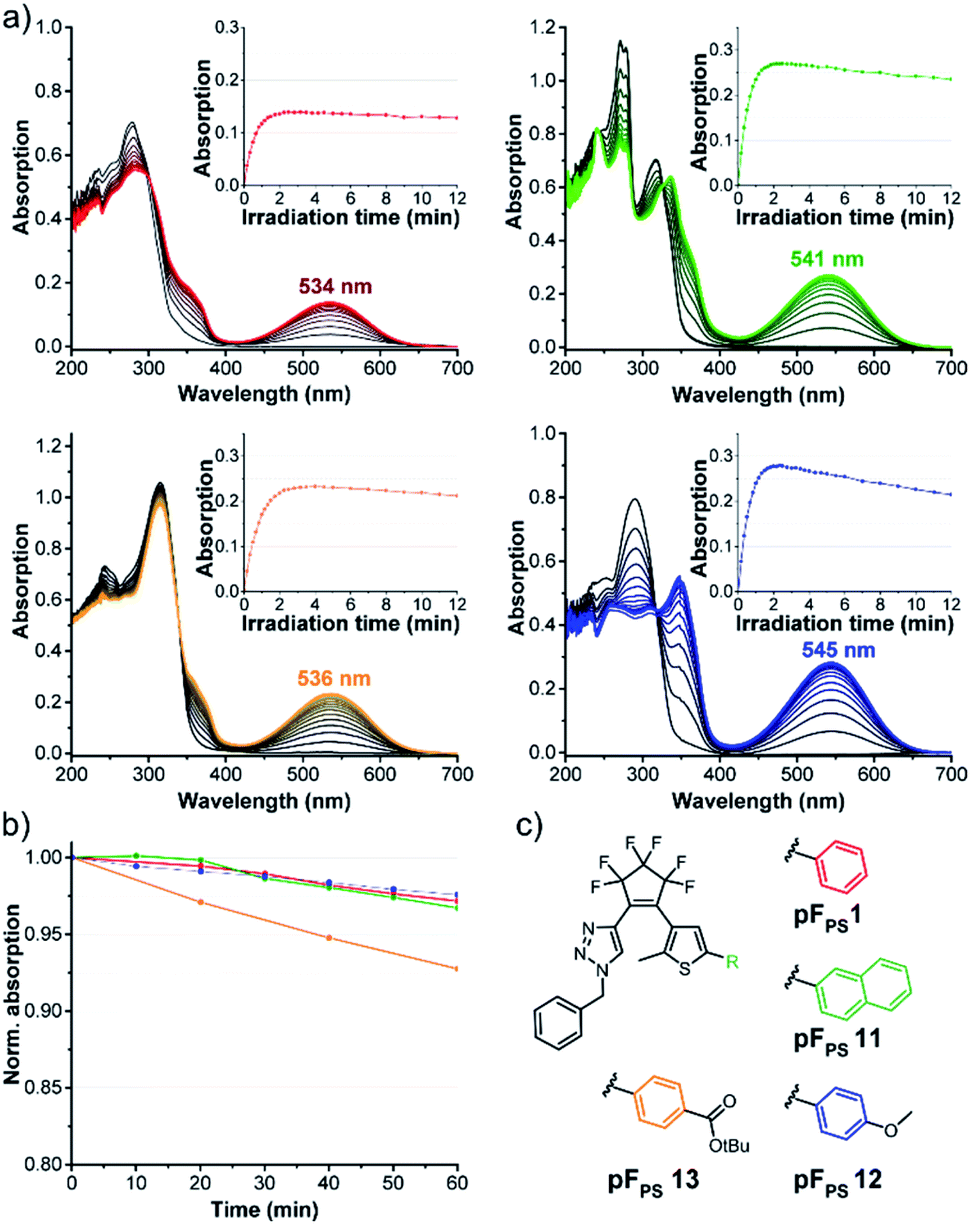 | ||
| Fig. 5 Photochromic properties of various thienyl substitutions: (a) time-dependent changes of the absorption spectra of 60 μM solutions of pFPS 1, 11–13 upon UV light irradiation (310 nm, LED) in chloroform towards the (pseudo)-photostationary state; insets show the absorption vs. time trace measured at the VIS absorption maximum during extended UV irradiation to analyse the photostability, (b) thermostability (20 °C) of the CF isomers, (c) structures. | ||
Thus, the substituents at the thiophene ring exert a much stronger influence on the photochromic properties than those on the triazole ring described above (Fig. 4).
Structure optimization using mesomeric effects
During the systematic variation of the triazole substituents (Fig. 4) we observed that the change from alkyl to aryl substituents has a strikingly large influence on the VIS absorption wavelength of the closed isomers (see also Fig. S2†). This behavior indicates that the conjugation of the phenyl ring extends into the chromophore of the closed ring form, and implies that +M or −M substituents attached to the phenyl ring might efficiently modulate the properties of these DAEs. We tested this hypothesis using compounds with benzoic acid tert-butylester as the thiophene substituent. The rationale behind this choice was the option to later use the ester function for further late-stage functionalization or cross-linking, e.g., by amide coupling. We furthermore expected the ester to lead to higher photostabilities, compared to an +M substituent (compare pFPS 13 and pFPS 12, Fig. 5). We directly compared an electron-withdrawing aromatic para-nitrophenyl substituent (pFPS 15) with an electron-donating aromatic para-N,N-dimethylaminophenyl (pFPS 14) and a benzyl substituent (pFPS 13) at the triazole (Fig. 6). Again, all compounds were photochromic. pFPS 14 showed broad absorption features (with two maxima), low εVISmax, app (0.18 × 104 L (mol cm)−1), strong bleaching and low thermal stability. Overall, these results indicated that increased electron density at the triazole ring (+M effect) results in decrease photo- and thermostability. For pFPS 15, a very strong band (εVISmax, app = 0.53 × 104 L (mol cm)−1) with a strongly bathochromically shifted maximum at 565 nm, high thermo- and photostability was observed. Thus, the combination of two aromatic −M substituents R1 and R2, as in pFPS 15, creates particularly performant photoswitches, at least in halogenated solvents.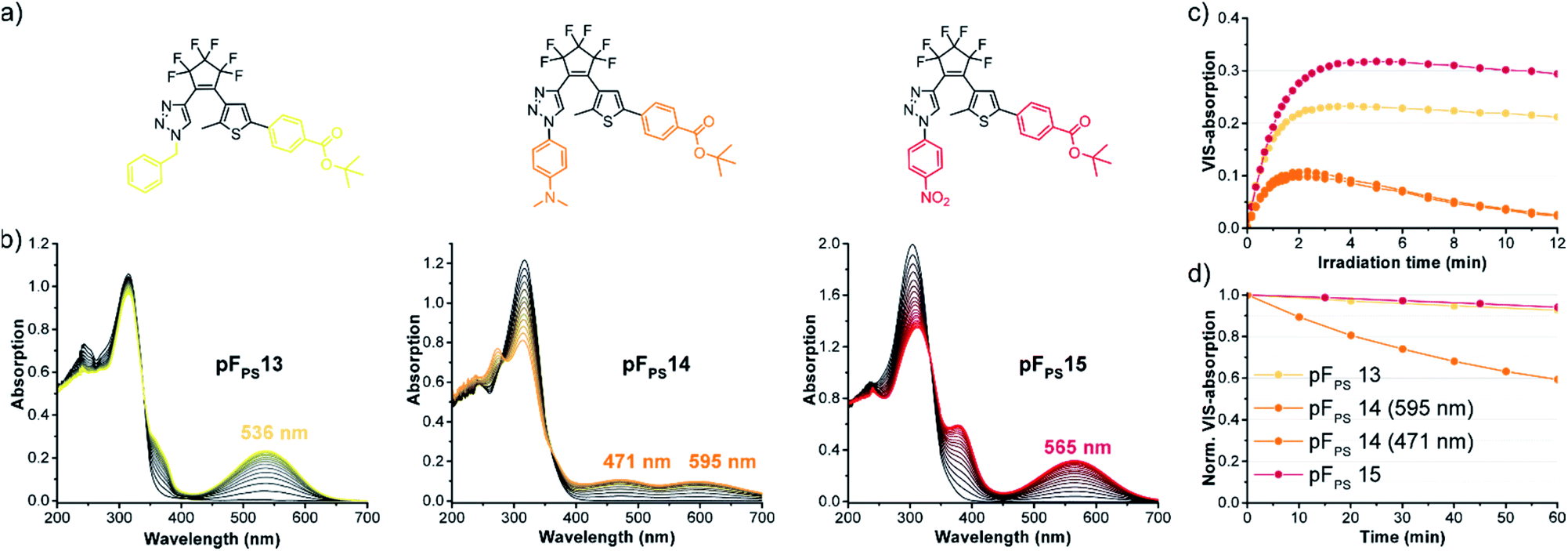 | ||
| Fig. 6 Effect of electron-donating and electron-withdrawing substituents at the triazole ring on the photochromic properties: (a) structures of pFPS 13–15, (b) time-dependent changes of the absorption spectra of solutions (60 μM in CHCl3) during UV light irradiation (310 nm UV light) to the (pseudo)-photostationary state, (c) kinetic trace of the VIS absorption bands during extended UV irradiation, (d) thermostability (20 °C) of the CF isomers. | ||
However, none of the 15 pF compounds showed reversible photochromism in non-halogenated solvents. Overall, the addition of functional groups to a phenyl substituent has much stronger overall influence on the photochromic properties on the triazole side, compared to the thiophenyl side (Table S1†). In summary, pFPS 15 can be considered an optimized structure among the pF-photoswitches.
Synthesis and photochromism of perhydrocyclopentene photoswitch pHPS 1
To investigate the influence of the bridge moiety on the properties of triazolyl-DAEs, we converted perhydrocyclopentene acetylenes 9aand 9b under the same CuAAC reaction conditions in yields similar to those of the pF-switches (see details in Scheme S1†). Next, we characterized benzyl-phenyl compound pHPS 1 and compared its properties to its sister compound pFPS 1 (Fig. 7). | ||
| Fig. 7 Comparison of triazole photoswitches with perfluorinated (pFPS 1) and perhydrogenated (pHPS 1) cyclopentene bridge: (a) structures of the respective OF and CF isomers, (b) time-dependent changes in the absorption spectra upon UV light irradiation in different solvents (310 nm, 60 μM conc.) to the (p)PSS, (c) reversibility of pHPS 1 in methanol, (d) kinetic trace of the VIS absorption bands (CF isomers) during extended UV light irradiation, (e) thermostability (20 °C), see also Table S4.† | ||
For pHPS 1, the VIS absorption band formed upon UV irradiation in chloroform was hypsochromically shifted by 47 nm, compared to the band of the perfluorinated analog pFPS 1 (Fig. 7(b)). The switching process was found to be reversible, although a high photobleaching rate led to a fast decrease of the VIS absorption over 10 reaction cycles (Fig. 7(c)). The low photostability was also detected in the kinetic trace of the VIS absorption during extended UV irradiation (Fig. 7(d)). Advantageously, however, pHPS 1 showed photochromism not only in halogenated solvents, but also in methanol and DMSO. The VIS absorption bands formed in these solvents appeared at exactly the same wavelengths as in chloroform. The highest εVISmax, app was measured in DMSO (0.60 × 104 L (mol cm)−1), however the bleaching rate was high in all three tested solvents. For pHPS 1, chloroform was inferior to the other solvents with respect to εVISmax, app and bleaching. The highest thermal stability of the closed-ring form was measured in DMSO, too (80% remaining after 1 h, τ = 2.6 h, 20 °C). Overall, with the exception of εVISmax, app, perhydrocyclopentene switch pHPS 1 was inferior to its perfluorinated sister compound (Fig. 7(c–e)). The electronegative fluorine atoms had a positive effect on photo- and thermostability. Nevertheless, the perfluorocyclopentene bridge caused an incompatibility with most solvents, while perhydrocyclopentene was found to overcome this solvent restriction.
Investigation of the irreversible reaction of pFPS 1 in non-halogenated solvents
While solvent-dependent photochromism of DAEs is known,35–37 the selective photochromism in halogenated solvents only, as observed for all our perfluorocyclopentene diarylethenes, had not been described before (to our knowledge). We therefore investigated the photochromism of compound pFPS 1 in a wider panel of solvents.When irradiated in methanol, DMSO, ethyl acetate, or dioxane, pFPS 1 formed a new intense absorption band outside the visible spectrum (Fig. S3,† blue spectra). The formation of this absorption band (λmax = 420 nm in DMSO, ∼372 ± 1 nm in all other solvents) was irreversible: neither irradiation with a white-light lamp nor with LEDs emitting near this maximum had any effect on the absorption spectrum of the solution. Furthermore, no bleaching was observed during extended 310 nm illumination. Analysis of the absorption vs. time traces (at 373 nm, the newly formed maximum during the irradiation) in DMSO revealed a sigmoidal course in the early phase of the reaction (Fig. 8). This observation may indicate a fast consecutive reaction that consumes the initially formed CF isomer.1,38 Indeed, in DMSO the intermittent formation of CF was revealed by the A510nmvs. time trace that quickly (t < 1 min) reached a maximal value and then declined, while the inflection point of the curve at A373nm correlated exactly with the maximum of A510nm (Fig. S4a†). In methanol, the rate of the consecutive reaction was much higher than in DMSO, the concentration of CF remained very low, and the initial slope of the curve was much steeper (Fig. 8).
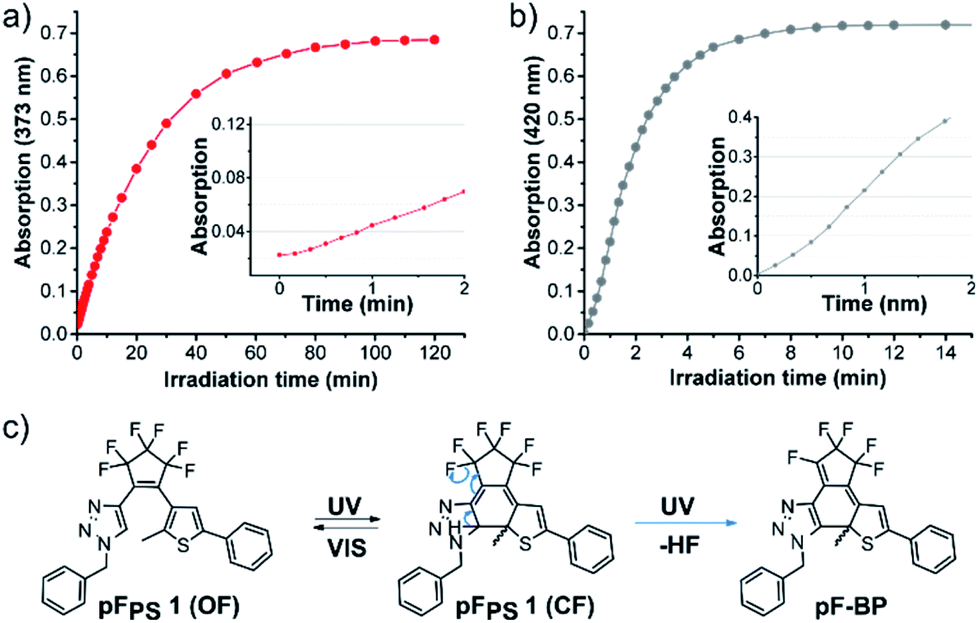 | ||
| Fig. 8 Kinetic trace of absorption bands formed in pFPS 1 solutions during UV light irradiation (310 nm, 60 μM in (a) methanol (absorption band at 373 nm) and (b) DMSO (absorption band at 420 nm), (c) hypothetic reactions of pFPS 1 in these solvents and the assumed structure of the specific side product pF-BP. | ||
On the other hand, in dibromomethane the side reaction was slowed down considerably, so that both processes (the formation of the VIS absorption bands of the closed form and the UV-band of the byproduct) could be well observed on the same time scale (Fig. S4c†). Samples taken during irradiation of a methanolic solution of pFPS 1 were analyzed by HPLC (Fig. S5†). Concurrent to the decrease of the peak of the OF isomer (12.9 min retention time), a new peak appears at 14.3 min that absorbs strongly at 373 nm, suggesting that only one product is formed in significant amounts. This product was isolated by column chromatography (ESI, chapter 2.2.5†) to obtain a yellow solid with a mass (508.0879 m/z (M + Na)) 20 m/z units smaller than the mass of pFPS 1(528.0932 m/z (M + Na)). 1H-NMR spectra indicated diastereotopic CH2-protons and the 19F-spectrum revealed a pattern of nine distinct fluorine signals (Fig. S6†), both findings suggesting the elimination of HF, as observed for the CF of DAEs by others.39–41 The electronic structure of the closed triazole photoswitches could favor HF-elimination, as the high electronegativity of the six fluorine and three nitrogen atoms should acidify the triazole proton, leading to the reaction product proposed in Fig. 8(c) and S7.† Products of the often-described annulation (formally by a 1,2-dyotropic rearrangement)1,38,42,43 and the HF elimination would still have an extended π-system with a fulvene-like structure which can be expected to be strongly colored, too,44 which is not the case for our isolated side product. Considering additionally the energetically favorable restoration of the aromatic triazole, and the small difference in the chemical shifts of the CH2-protons between pFPS 1 and the bleaching product, the elimination from the closed isomer is – in our opinion – the more likely process.
HPLC analysis of the UV-induced photocyclization of perhydro sister compound pHPS 1 revealed the formation of a peak eluting at 15.1 min with strong absorption at 486 nm, indicative of the closed isomer (Fig. S5†), while no signal of a specific rearrangement product was observed. After reaching the (p)PSS, the absorption decreases over a wide wavelength range (>250 nm), suggestive of unspecific bleaching.
Thus, the cyclopentene bridge has a strong influence on the photochromic properties of the triazole photoswitches, and it also determines the scope of solvents that can be used in the desired application. Why the HF elimination is prevented in halogenated solvents is not clear at this point. A coordination effect between the solvent and the triazole ring is conceivable as well as energetic effects by solvation, raising the possibility to modulate this aspect by structural modifications at the triazole ring. In conclusion, an important design consideration for photochromism that is not limited to chlorinated solvents is the use of cyclopentene bridges that do not allow 1,4-elimination reactions.
Computational investigation of the photoswitching mechanism of pFps 1
The photochemical switching mechanism underlying the photochromism of DAEs has been investigated previously, in particular for derivatives with two thiophene rings as aryl moieties.1,45–47 The switching mechanism has been shown to proceed efficiently via a conical intersection.48–50 In the DAEs synthesized here, one of the thiophene rings is replaced by triazole with substantially different molecular properties, and the first question to address is thus whether this has an influence on the switching mechanism.Among our different perfluorocyclopentenyl photoswitches, pFPS 1 was taken as representative for further quantum chemical investigation at standard time-dependent density functional theory (DFT)51,52 level using ωB97X-D53 as the exchange–correlation functional and cc-pVDZ basis set. The conductor-like polarizable continuum model (CPCM)54 was used to incorporate bulk solvent effects. Since the open-form of pFPS 1 is highly flexible, among other stable isomers (Table S2†), most possess an outward skewed geometry with the –CH3 group on the thiophene sticking out of the molecular plane (see Fig. 9, pFPS 1(OF)). The terminal C–C separation along the reaction coordinate in this geometry is 3.4 Å, and this geometry has been chosen for our calculation of the vertical excited states of pFPS 1.
 | ||
| Fig. 9 Frontier MOs: HOMO and LUMO from wB97X-D/cc-pvdz/PCM full TDDFT calculations for the two different solvents. (a) OF and CF in CHCl3 (b) OF and BP in MeOH. | ||
To investigate the light-induced ring closure mechanism upon vertical excitation of the OF ground state to the first excited S1 state, a relaxed scan of the S1 potential energy surface has been performed along the C–C distance formed during ring closure (Fig. 10). According to our calculations, the reactive first excited state of both conformers corresponds essentially entirely to the HOMO–LUMO transition. The frontier orbitals are shown in Fig. 9. Since a conical intersection is expected to be involved in the reaction, spin-flip-TDDFT55 has been used for this calculation. As can be seen in Fig. 10, a conical intersection is present at a C–C distance of 2.1 Å. Hence, upon vertical excitation of the OF, the molecules undergo a barrierless ring-closure reaction towards the conical intersection, at which they can switch efficiently into the closed-form electronic ground state. The C–C bond length of the newly formed bond is found to be 1.53 Å. Thus, our analysis revealed that pFPS 1 indeed undergoes an electrocyclic ring closure through a conical intersection in analogy to other DAEs,48–50 indicating that the introduction of a triazole ring in the DAE does not disturb their common photoswitching mechanism.
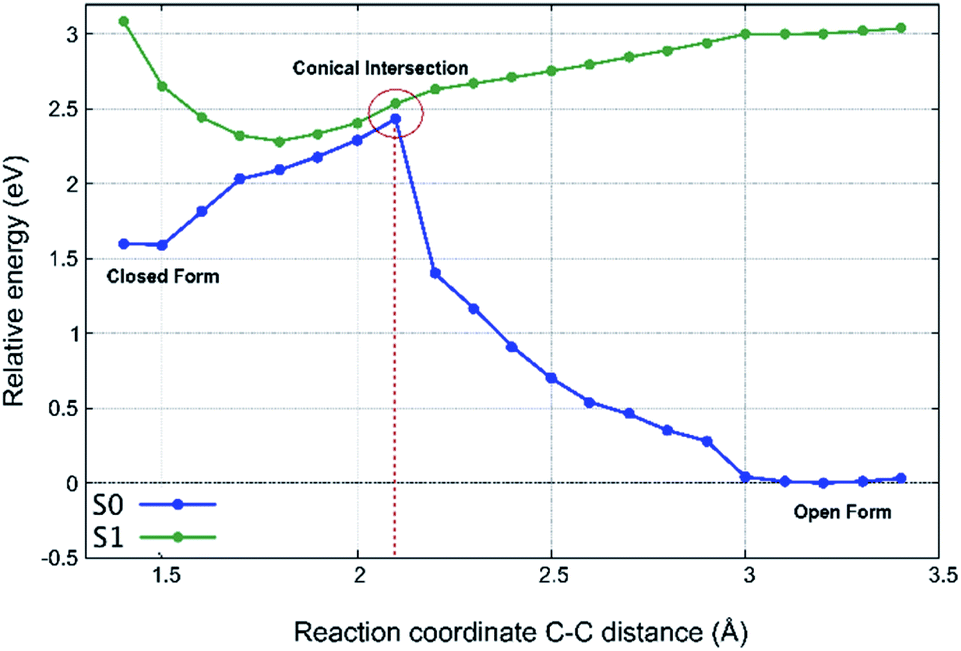 | ||
| Fig. 10 Potential energy scan for the first excited state (S1) and the ground state (S0) along relaxed S1 geometries along the reaction coordinate. The intersection denotes the approximate crossing point of the surfaces. | ||
The –CH2 group in the benzyl substituent on the triazole ring breaks the extended π-conjugation which exists throughout the structure. Furthermore, it can be seen that there is no contribution from the benzyl fragment to the frontier orbitals (Fig. 9). As a result, the first singlet electronic transition remains unaffected from the presence of the benzyl substituent. This phenomenon can also be observed in the other derivatives where the π-conjugation is hindered due to a –CH2 bridge (pFPS 1–5, Fig. 4). Derivatives with an extended conjugation, on the other hand, show markedly different spectra of the CFs in halogenated solvents (pFPS 14 and pFPS 15 with phenyl substituents).
Identification of the irreversible intermediate in non-halogenated solvents
pFPS 1 and the other perfluorinated derivatives exhibit an entirely different behavior in halogenated vs. non-halogenated solvents (see Fig. S3†). To model the effect of the CHCl3 solvent, the ground state geometries of the open and closed forms were coordinated with one CHCl3 molecule (Fig. 9(a)), and their geometry re-optimized, followed by a full TD-DFT/ωB97x-D/cc-pvdz/PCM calculation for their corresponding absorption spectra (Fig. 11).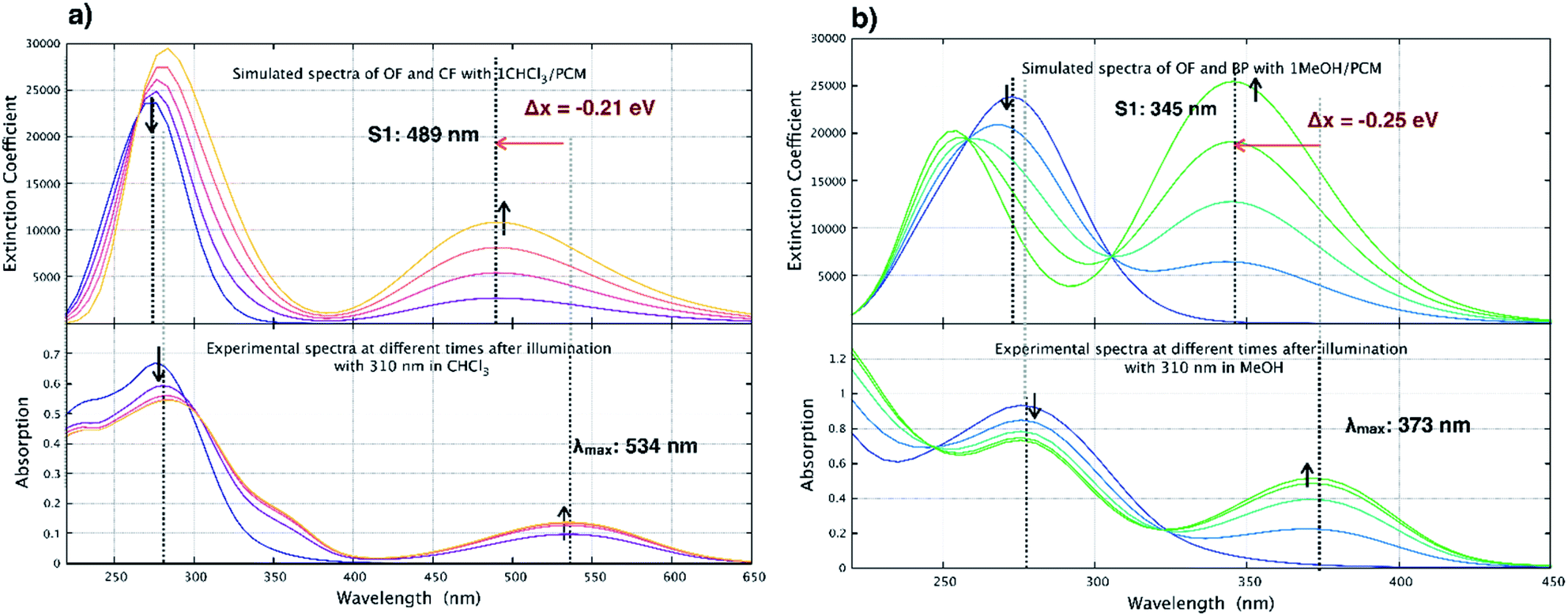 | ||
| Fig. 11 Comparison between experimental spectra and simulated spectra in (a) CHCl3 and (b) MeOH. The computational spectra are plotted for mixtures of OF and corresponding end product in varying ratios in the two solvents using wB97X-D/cc-pvdz/PCM for geometries in Fig. 10. | ||
The absorption maximum for the OF of pFps 1 is calculated to be at 276 nm, which is in excellent agreement with the experimental value of 280 nm. The absorption maximum of the CF is calculated to be at 489 nm, however, this peak is blue shifted by 28 nm (0.21 eV) relative to the measured value, which is the typical accuracy of the chosen methodology.56,57 The same trends are found for the OF and CF of the parent pHps 1, which is shown in Table S3.†
In Fig. 11(a), the time-dependent changes in the absorption spectrum of the open form of pFPS 1 are modeled in chloroform assuming a quantitative conversion to the CF. Therefore, spectra of OF and CF are plotted together with 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:1, and 1:3 superpositions thereof. Comparison with the corresponding experimental spectra demonstrates an excellent agreement, supporting the above-described photoswitching mechanism in halogenated solvents.
:1, 1:1, and 1:3 superpositions thereof. Comparison with the corresponding experimental spectra demonstrates an excellent agreement, supporting the above-described photoswitching mechanism in halogenated solvents.
In non-halogenated solvents, for example MeOH, experimental evidence is given that irreversible elimination of HF takes place, thereby quenching the photoswitching reaction. To support this assumption, the time-dependent changes in the absorption spectra of pFPS 1 have been modeled following the same procedure as above, now with the elimination byproduct pF-BP (Fig. 9(b), for further structural details see Fig. S7†) as final state of the time evolution. The elimination extends the aromaticity to the perfluorocyclopentene ring due to increased π-conjugation. The simulated absorption maximum for the elimination product is at 345 nm (Fig. 11), which is slightly blue-shifted relative to the experimental value of 373 nm, well within the typical range of error of the method.56,57
Again, the agreement between the simulated and measured absorption spectra in CH3OH is good, corroborating the elimination reaction to be the main reaction path in non-halogenated solvents.
The origin of this very unusual solvent dependency needs further theoretical modeling of the solute–solvent interactions that go beyond the current work. This will likely require extensive sampling of the solvent environment, the weak but explicit solute–solvent interactions, and possibly even the interaction with several solvent molecules, which altogether make such simulations extremely demanding, if they are currently possible at all.
Optimization of the triazole photoswitch structure
Further investigations with different derivatives of pHPS 1 revealed that among the perhydrocyclopentene triazole DAEs, the substituents have an even stronger influence on the photochromic properties than in the perfluorocyclopentene series (Fig. S8 and Table S4†). Thus, the structure of the pH photoswitches could be further optimized, applying the knowledge about the structure–property relationships obtained in the pF series. As −M substituents on both rings were found to be beneficial, we synthesized and characterized the perhydrocyclopentene analog of our optimized compound pFPS 15, combining a p-nitrophenyl substituent on the triazole and p-benzoic acid tert-butyl ester on the thiophene ring. The resulting compound pHPS 2 (Fig. 12(a)) shows actually much higher photostability than pHPS 1, retaining 96% of its maximal VIS absorption after 12 minutes of UV light irradiation in DMSO (Fig. 12(c) and Table S4†).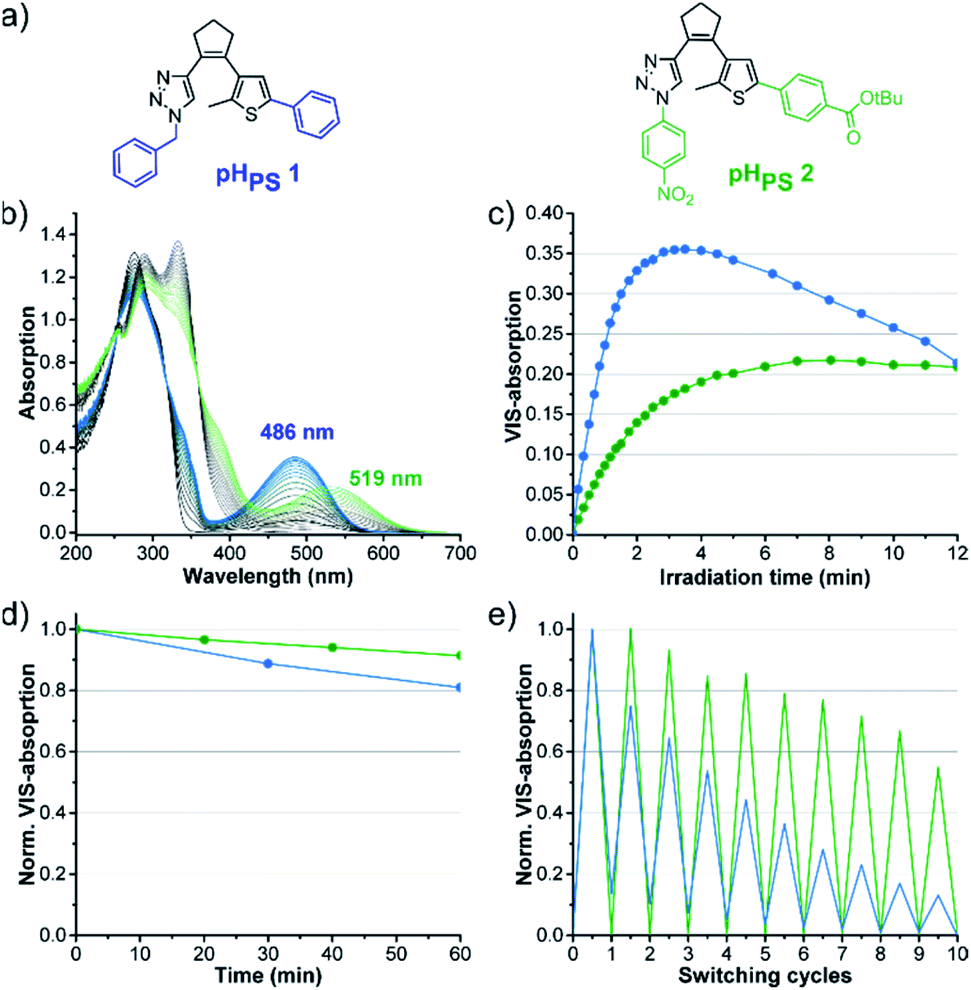 | ||
| Fig. 12 Effect of structure optimization on the photochromic properties: (a) structures of pHPS 1 and pHPS 2, (b) absorption spectra change of 60 μM solutions of pHPS 1 and pHPS 2 during UV light irradiation (310 nm, LED) in DMSO to the (p)PSS, (c) kinetic trace of the VIS absorption bands during prolonged UV illumination, (d) thermostability (20 °C), (e) reversibility measurement (normalized VIS absorption). | ||
The thermostability was improved, too (τ20 °C: 5.9 h vs. 2.6 h for pHPS 1, Fig. 12(d), Table S4†), and a higher degree of reversibility could be reached (Fig. 12(e)). While the properties are still far from perfect, these data indicate the directions for further optimization, e.g., by the addition of further electron-withdrawing groups on the triazole-phenyl ring.
Photostationary state composition and quantum yields
For most of the novel DAEs described here, the solvent dependence or restriction of photochromism in combination with the imperfect thermo- and photostability prevented an exact determination of (p)PSS composition and quantum yields. For our two optimized compounds, pFPS 15 and pHPS 2, however, we could estimate these parameters. For the perhydrocyclopentene switch pHPS 2, OF and CF of (p)PSS mixtures in DMSO were separated by reversed-phase HPLC and the composition determined from the integrated peak areas, taking into account decomposition during the analysis (Table 1 and Fig. S9†). The (p)PSSUV was found to contain 58–60% CF, while the PSSVis contained ∼0% CF, as indicated by the reduction to zero of the 519 nm absorbance band upon vis light illumination (Fig. 12(e)). Ring closing and ring opening quantum yields were determined as reported previously58,59 and indicate a rather efficient photochemistry. A most striking feature is the high quantum yield of ring opening upon irradiation with 505 nm visible light (0.56), which compares favorably with classical dithienylethenes where this value is often smaller than 0.01.| PSSUVa [%] | PSSVisb [%] | ϕ OC UV [%] | ϕ OC UV [%] | ϕ CO Vis [%] | ϕ OC/ϕCOUVf | |
|---|---|---|---|---|---|---|
| a Photostationary state after irradiation with UV light, in % CF. b Photostationary state after irradiation with Vis light, in % CF. c Ring cyclization quantum yields at 325 nm and 300 nm, respectively. d Ring opening quantum yields at 325 nm and 300 nm, respectively. e Ring cyclization quantum yields at 505 nm and 567 nm, respectively. f Ratio of ring closing to ring opening quantum yields upon UV irradiation. | ||||||
| pHPS 2 | 58–60 | 0 | 31 | 29 | 56 | 1.07 |
| pFPS 15 | 55–58 | 0 | 39 | 40 | 45 | 0.98 |
As perfluorocyclopentene switch pFPS 15 could not be analyzed by HPLC, we prepared 15 ml of a 100 μM solution in CDCl3, irradiated to the PSSUV, then quickly concentrated the reaction mixture in vacuo, and analyzed it by 1H-NMR spectroscopy (Table 1 and Fig. S10†). This analysis revealed a PSSUV composition of 55–58% CF, while the reversibility measurements (Fig. 3e) indicate a PSSVis content of ∼0% CF. The ring opening quantum yield (in CHCl3) was slightly lower than for pHPS 2.
Conclusions
A new class of DAEs was designed which allows the generation of the photochromic moiety by CuAAC in direct proximity to the attachment point, using azide groups present in the target structures. Several systematically varied DAEs were synthesized from small organic azides, and the structure–property relationships of the compounds were determined by absorption spectroscopy and DFT calculations. Based on this knowledge, a first structural optimization was carried out, and the optimized DAEs already had properties which may permit a wide range of applications. The high conversion in the photostationary states and the high reaction quantum yields make these novel DAEs particularly attractive for applications in photonics, where fast and efficient back-and-forth switching is desired.60 However, there is still plenty of room for further structural modifications. Our next step will be the application of the acetylene precursors developed here for the modification of azido-derivatized amino acids and peptides.Data availability
The data supporting the findings of this study are available within the paper and its ESI† files.Author contributions
S. B. and A. J. conceived the project. S. B. designed, synthesized and characterized the compounds and analyzed the data. S. M. B. determined quantum yields and photostationary state compositions. R. S. and A. D. conducted the theoretical investigations and supported the elucidation of the structure of pF-BP. S. B. and A. J. wrote the initial draft of the manuscript. All authors contributed to the writing, editing and proof-reading process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Federal Ministry of Education and Research (BMBF), grant # 031A170B. The authors gratefully acknowledge technical support by S. Suhm, T. Timmermann and H. Rudy. We furthermore thank A. H. Becker, S. Braun, B. Bühler, S. Feth, N. Kipper, F. S. Menke and L. Petersen for assistance in the synthesis. The computational work was conducted within the Collaborative Research Center “N-heteropolycycles as functional materials” (SFB1249) of the German Science Foundation. Reena Sen received financial support from the Heidelberg graduate school, “Mathematical and Computational Methods in the Sciences” (GSC 220).References
- M. Irie, T. Fulcaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS.
- Z. L. Pianowski, Chem.–Eur. J., 2019, 25, 5128–5144 CrossRef CAS PubMed.
- S. Corra, M. Curcio, M. Baroncini, S. Silvi and A. Credi, Adv. Mater., 2020, 32, 1906064 CrossRef CAS PubMed.
- M. Q. Li, L. J. Chen, Y. S. Cai, Q. F. Luo, W. L. Li, H. B. Yang, H. Tian and W. H. Zhu, Chem, 2019, 5, 634–648 CAS.
- H. Zhang, X. X. Kou, Q. Zhang, D. H. Qu and H. Tian, Org. Biomol. Chem., 2011, 9, 4051–4056 RSC.
- T. I. Lansakara, F. Tong, C. J. Bardeen and A. V. Tivanski, Nano Lett., 2020, 20, 6744–6749 CrossRef CAS PubMed.
- I. Hnid, D. Frath, F. Lafolet, X. N. Sun and J. C. Lacroix, J. Am. Chem. Soc., 2020, 142, 7732–7736 CrossRef CAS PubMed.
- H. Yamaguchi, M. Ikeda, K. Matsuda and A. Irie, Bull. Chem. Soc. Jpn., 2006, 79, 1413–1419 CrossRef CAS.
- T. Kudernac, S. J. van der Molen, B. J. van Wees and B. L. Feringa, Chem. Commun., 2006, 3597–3599 RSC.
- A. Ghavidast and N. O. Mahmoodi, J. Mol. Liq., 2016, 216, 552–564 CrossRef CAS.
- H. Cahova and A. Jäschke, Angew. Chem., Int. Ed., 2013, 52, 3186–3190 CrossRef CAS.
- A. V. Strizhak, K. Sharma, O. Babii, S. Afonin, A. S. Ulrich, I. V. Komarov and D. R. Spring, Org. Biomol. Chem., 2018, 16, 8559–8564 RSC.
- F. Hu, M. Hu, W. J. Liu, J. Yin, G. A. Yu and S. H. Liu, Tetrahedron Lett., 2015, 56, 452–457 CrossRef CAS.
- K. Fujimoto, M. Kajino, I. Sakaguchi and M. Inouye, Chem.–Eur. J., 2012, 18, 9834–9840 CrossRef CAS.
- N. Soh, K. Yoshida, H. Nakajima, K. Nakano, T. Imato, T. Fukaminato and M. Irie, Chem. Commun., 2007, 5206–5208 RSC.
- O. Babii, S. Afonin, M. Berditsch, S. Reisser, P. K. Mykhailiuk, V. S. Kubyshkin, T. Steinbrecher, A. S. Ulrich and I. V. Komarov, Angew. Chem., Int. Ed., 2014, 53, 3392–3395 CrossRef CAS PubMed.
- K. Fujimoto, T. Maruyama, Y. Okada, T. Itou and M. Inouye, Tetrahedron, 2013, 69, 6170–6175 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- M. Aigner, M. Hartl, K. Fauster, J. Steger, K. Bister and R. Micura, Chembiochem, 2011, 12, 47–51 CrossRef CAS PubMed.
- M. Gerowska, L. Hall, J. Richardson, M. Shelbourne and T. Brown, Tetrahedron, 2012, 68, 857–864 CrossRef CAS.
- L. H. Liu and M. D. Yan, Acc. Chem. Res., 2010, 43, 1434–1443 CrossRef CAS.
- J. Park, H. S. N. Jayawardena, X. Chen, K. W. Jayawardana, M. Sundhoro, E. Ada and M. D. Yan, Chem. Commun., 2015, 51, 2882–2885 RSC.
- J. P. Collman, N. K. Devaraj and C. E. D. Chidsey, Langmuir, 2004, 20, 1051–1053 CrossRef CAS.
- A. Devadoss and C. E. D. Chidsey, J. Am. Chem. Soc., 2007, 129, 5370–5371 CrossRef CAS.
- S. J. Luchansky, H. C. Hang, E. Saxon, J. R. Grunwell, C. Y. Danielle, D. H. Dube and C. R. Bertozzi, Methods Enzymol., 2003, 362, 249–272 CAS.
- J. W. Chin, S. W. Santoro, A. B. Martin, D. S. King, L. Wang and P. G. Schultz, J. Am. Chem. Soc., 2002, 124, 9026–9027 CrossRef CAS.
- A. J. Ashe and F. J. Drone, J. Am. Chem. Soc., 1987, 109, 1879–1880 CrossRef CAS.
- A. Peters, C. Vitols, R. McDonald and N. R. Branda, Org. Lett., 2003, 5, 1183–1186 CrossRef CAS.
- S. Kobatake and M. Irie, Tetrahedron, 2003, 59, 8359–8364 CrossRef CAS.
- F. Sun, F. H. Zhang, H. B. Guo, X. H. Zhou, R. J. Wang and F. Q. Zhao, Tetrahedron, 2003, 59, 7615–7621 CrossRef CAS.
- W. E. Parham and L. D. Jones, J. Org. Chem., 1976, 41, 2704–2706 CrossRef CAS.
- M. Singer and A. Jäschke, J. Am. Chem. Soc., 2010, 132, 8372–8377 CrossRef CAS PubMed.
- T. C. Pijper, T. Kudernac, W. R. Browne and B. L. Feringa, J. Phys. Chem. C, 2013, 117, 17623–17632 CrossRef CAS.
- D. Wang, M. M. Zhao, X. Liu, Y. X. Chen, N. Li and B. H. Chen, Org. Biomol. Chem., 2012, 10, 229–231 RSC.
- V. Aubert, E. Ishow, F. Ibersiene, A. Boucekkine, J. A. G. Williams, L. Toupet, R. Metivier, K. Nakatani, V. Guerchais and H. Le Bozec, New J. Chem., 2009, 33, 1320–1323 RSC.
- L. Ordronneau, J. Boixel, V. Aubert, M. S. Vidal, S. Moya, P. Aguirre, L. Toupet, J. A. Williams, H. Le Bozec and V. Guerchais, Org. Biomol. Chem., 2014, 12, 979–992 RSC.
- H. Xi, Z. Zhang, W. Zhang, M. Li, C. Lian, Q. Luo, H. Tian and W. H. Zhu, J. Am. Chem. Soc., 2019, 141, 18467–18474 CrossRef CAS.
- K. Higashiguchi, K. Matsuda, S. Kobatake, T. Yamada, T. Kawai and M. Irie, Bull. Chem. Soc. Jpn., 2000, 73, 2389–2394 CrossRef CAS.
- M. Kathan, F. Eisenreich, C. Jurissek, A. Dallmann, J. Gurke and S. Hecht, Nat. Chem., 2018, 10, 1031–1036 CrossRef CAS.
- S. Fredrich, A. Bonasera, V. Valderrey and S. Hecht, J. Am. Chem. Soc., 2018, 140, 6432–6440 CrossRef CAS PubMed.
- S. Kobatake, H. Imagawa, H. Nakatani and S. Nakashima, New J. Chem., 2009, 33, 1362–1367 RSC.
- M. Herder, B. M. Schmidt, L. Grubert, M. Patzel, J. Schwarz and S. Hecht, J. Am. Chem. Soc., 2015, 137, 2738–2747 CrossRef CAS PubMed.
- M. Irie, T. Lifka, K. Uchida, S. Kobatake and Y. Shindo, Chem. Commun., 1999, 747–748 RSC.
- J. H. Day, Chem. Rev., 1953, 53, 167–189 CrossRef CAS.
- M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed.
- H. Tian and S. J. Yang, Chem. Soc. Rev., 2004, 33, 85–97 RSC.
- P. R. Hania, A. Pugzlys, L. N. Lucas, J. J. D. de Jong, B. L. Feringa, J. H. van Esch, H. T. Jonkman and K. Duppen, J. Phys. Chem. A, 2005, 109, 9437–9442 CrossRef CAS PubMed.
- A. Jarota, E. Pastorczak and H. Abramczyk, Phys. Chem. Chem. Phys., 2020, 22, 5408–5412 RSC.
- M. Isegawa and K. Morokuma, J. Phys. Chem. A, 2015, 119, 4191–4199 CrossRef CAS.
- M. Boggio-Pasqua, M. Ravaglia, M. J. Bearpark, M. Garavelli and M. A. Robb, J. Phys. Chem. A, 2003, 107, 11139–11152 CrossRef CAS.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- A. Dreuw and M. Head-Gordon, Chem. Rev., 2005, 105, 4009–4037 CrossRef CAS.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef CAS.
- Y. H. Shao, M. Head-Gordon and A. I. Krylov, J. Chem. Phys., 2003, 118, 4807–4818 CrossRef CAS.
- M. Isegawa, R. Peverati and D. G. Truhlar, J. Chem. Phys., 2012, 137, 244104 CrossRef.
- Y. H. Shao, Y. Mei, D. Sundholm and V. R. I. Kaila, J. Chem. Theory Comput., 2020, 16, 587–600 CrossRef.
- S. M. Büllmann, T. Kolmar, P. Slawetzky, S. Wald and A. Jäschke, Chem. Commun., 2021, 57, 6596–6599 RSC.
- T. Kolmar, S. M. Büllmann, C. Sarter, K. Höfer and A. Jäschke, Angew. Chem., Int. Ed., 2021, 60, 8164–8173 CrossRef CAS.
- D. L. Kellis, C. Sarter, B. L. Cannon, P. H. Davis, E. Graugnard, J. Lee, R. D. Pensack, T. Kolmar, A. Jäschke, B. Yurke and W. B. Knowlton, ACS Nano, 2019, 13, 2986–2994 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures, schemes and tables, experimental procedures, NMR spectra, computational details. See DOI: 10.1039/d1sc02526k |
| This journal is © The Royal Society of Chemistry 2021 |