
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA critical review on emerging photocatalysts for syngas generation via CO2 reduction under aqueous media: a sustainable paradigm
Deepak Kumar
Chauhan
,
Neha
Sharma
and
Kamalakannan
Kailasam
 *
*
Advanced Functional Nanomaterials, Institute of Nano Science and Technology (INST), Knowledge City, Sector-81, Manauli, SAS Nagar, 140306 Mohali, Punjab, India. E-mail: kamal@inst.ac.in; kkamal17@gmail.com
First published on 5th May 2022
Abstract
In a holistic view, global energy generally depends on burning fossil fuels that intensifies the worldwide energy crisis and the levels of CO2 in the atmosphere. Undesirable CO2 levels in the atmosphere are a major concern for alleviating global warming in particular. Impeding CO2 emission in the atmosphere is quite important for sustainable development. Utilizing solar energy for photocatalytically driven CO2 reduction to value-added products or chemical feedstocks can lead to CO2 consumption in a more renewable way and reduce pollution levels. Photocatalytic CO2 reduction in water (H2O) to produce synthesis gas (syngas, CO + H2) is considered a highly advantageous and pivotal intermediate for the upgradation of valuable hydrocarbon fuels via the Fischer–Tropsch reaction. This timely mini-review aims to expatiate on the recent advances in syngas production via photocatalytic CO2 reduction under aqueous media following up on the compendious background of syngas production. Furthermore, we make firm efforts to spotlight various photocatalytic systems, and their structure–activity relationships for syngas production. However, in addition, emphasis has been given to rationalize the stream proportion of the syngas mixture i.e. CO/H2 or H2/CO. This could promptly be assessed via various requisite parameters such as initial feed concentration (CO2/H2O) and the cooperative effect of active metallic sites, liners and sensitizers. Lastly, future aspects summarizing the conceptual idea/concern for tuneable syngas production via the photoreduction of CO2 are presented.
 Deepak Kumar Chauhan | Deepak Kumar Chauhan received his BSc (2011) and MSc (2013) degrees from the University of Lucknow. Currently, he is registered as a PhD student at the Indian Institute of Science Education and Research (IISER), Mohali, Punjab, India, and works at the Institute of Nano Science and Technology (INST) Mohali, Punjab, India, under the supervision of Dr Kamalakannan Kailasam. His research interest is photocatalytically driven water splitting, simultaneous biomass conversion into value-added products/fine chemicals and CO2 sorption and reduction. |
 Neha Sharma | Neha Sharma is a PhD student at the Institute of Nano Science and Technology under the direction of Dr Kamalakannan Kailasam. She received MSc from S. G. G. S. Khalsa College, Mahilpur (affiliated to Punjab University). Her research focuses on the development and characterization of novel organic porous materials for photocatalysis, organocatalysis, CO2 sorption and conversion. |
 Kamalakannan Kailasam | Kamalakannan Kailasam is working as a Scientist-F/Professor at the Institute of Nano Science and Technology (INST), Mohali, Punjab, India. He grew up in Andagalore Gate, Namakkal District in Tamil Nadu, India. He completed his BSc (1999) and MSc (2002) degrees at Bishop Heber College affiliated to Bharathidasan University, Tiruchirappalli. He went on to earn a PhD with Late Prof. Klaus Müller in Universität Stuttgart, Germany, in 2008. He was a postdoctoral fellow at the Max Planck Institute for Colloids and Interfaces, Potsdam, Germany, in the Colloid Chemistry department from 2009 to 2010. Then he moved with Professor Arne Thomas in 2010 to Technische Universität Berlin, Germany, and joined INST Mohali as a scientist in April 2015. He is a materials chemist and leads the “Advanced Functional Nanomaterials” group working on material development for various energy and environmental applications. In particular, his interest lies in photocatalytic conversions, which includes water splitting, biomass to fine chemicals and CO2 reduction. In addition, his group has broad interests in organic photovoltaics, fuel cells, humidity and VOC sensing and biotechnology applications. |
1. Introduction
Currently, rising population and worldwide energy demand are two major issues. Such a tremendous energy supply chain generally relies on the large-scale depletion of non-renewable fossil fuels. Profound mining of non-renewable sources (fossil fuels) causes the piling up of carbon dioxide (CO2) levels in the atmosphere, which is an alarm call for global warming and has an adverse impact on the environment.1–4A special report by the Intergovernmental Panel on Climate Change (IPCC) predicted that CO2 levels in the atmosphere could cause temperatures to increase by up to 1.5 °C by 2050, which may have devastating consequences for humanity and the natural environment across the globe. Therefore, it is urgent to mitigate the anthropogenic emission of CO2 in the atmosphere for the sustainable development of mankind.5
As a way to subdue this problem, CO2 reduction to value-added products is a key consideration for sustainable feed-stocks and energy sources. In the last three decades, CO2 reduction has drawn significant attention for converting CO2 into promising products or fuels such as CO, CH4, HCHO, HCOOH, CH3OH, C2H4, and C2H6.6–9
The CO2 reduction process for CO production is one such interesting pathway. The CO2 redox reaction with H2O has shown the potential to produce syngas (i.e. CO and H2), which benefits the reaction pathway since the obtained syngas is a crucial intermediate of the production of valuable hydrocarbons, methanol, alcohols and fuel additives via the Fischer–Tropsch process.10,11
Currently, syngas production mainly depends on a variety of sources, including fossil fuels such as coal, natural gas and oil, via thermo-catalytic systems operating at relatively high temperatures and pressures.12,13 For that reason, it is a challenging task to hunt for more viable approaches that use renewable energy sources (solar, wind and biomass) for the conversion of CO2 to syngas.
The utilization of solar energy for the reduction of CO2 to syngas is the most propitious and highly sustainable strategy.14–17 The strategies reported include photocatalytic or photo-electrochemical (PEC) CO2 reduction by H2O and solar light-driven CO2 reduction via CH4i.e. dry methane reforming.18–21 It is interesting to point out that dry methane reforming is a very promising strategy to realize the effective reduction of CO2 to syngas for practical application in chemical feedstocks for sustainable energy production using natural gas.19,22,23 In spite of its promising feature of driving the photo-reduction of CO2, the practical application of the dry methane reforming process still requires external thermal energy input. This makes the process highly vulnerable and thermodynamically unfavourable as it requires high endothermic enthalpy for the reaction.24,25 Thus, herein, the discussion of the photocatalytic dry methane reforming process is completely out of our focus due to the above concerns.
Solar-driven CO2 reduction under aqueous (H2O) solution represents an ideal strategy for syngas production.21,26,27 This manifests many advantages such as: (1) this reaction may require a boundless source of energy i.e. solar light, which is abundant, and (2) this reaction can be initiated by H2O and CO2 and (3) requires ambient conditions such as low temperature and pressure.
In brief, the solar-driven production of syngas is a renewable and promising strategy to respond to the energy and environmental crises.21,28 As we have mentioned earlier, different approaches have been adopted from time to time for syngas production, but the solar-driven approach is one of the pioneering ones.16,26,29 Over a decade, numerous studies have been conducted on photocatalytic CO2 reduction and significant efforts have been made to develop new photocatalytic systems to increase photocatalytic performance.30–34
In addition, there are plenty of reviews and perspectives on photocatalytic CO2 reduction that mainly discuss the challenges in CO2 photoreduction for solar fuel production, the different types of photocatalytic systems for CO2 reduction, improvement in the photo selectivity of solar fuels, and advancements in the structural engineering of photocatalysts for solar-driven CO2 reduction into fuels.4,31,35–38 However, an exclusive review on photocatalytic syngas generation via CO2 reduction under aqueous media is not available. In view of the significance of photo-driven syngas production, this review concisely underlines the recent advances of photocatalytic syngas generation over various photocatalysts.
Herein, we have discussed and emphasized different photoactive materials including MOFs,28,39 organic polymers,21,40–42 POMs,43,44 LDHs,45,46 metal oxides,47,48 metal complexes,49,50 and single atom metals51,52 for CO2 reduction into syngas and have thus briefly described their photocatalytic activities to rationalize the stream proportion of the syngas mixture i.e. CO/H2 or H2/CO. In the end, conclusive future perspective and challenges are briefly highlighted, which defines new avenues and provides excellent opportunities for materials scientists to get deep insight into the development of sustainable photocatalytic systems for solar-driven syngas production.
2. Background and recent advancements in syngas production
Syngas is a gaseous mixture of hydrogen (H2) and carbon monoxide (CO), and is quite an indispensable component used as an intermediate for the industrial production of ammonia, methanol, synthetic petroleum products, and other chemical commodities via the Fischer–Tropsch reaction.53–57 Being an attractive feedstock for bulk chemical production, syngas has drawn the attention of research communities since 1900.13 Currently, the production of syngas predominantly relies on the conventional reforming of non-renewable sources, including fossil fuels such as natural gas, oil, and coal, generally at high temperatures and pressures.13,58–60 Typical methods involved in syngas production include steam reforming,61 partial oxidation,62 and autothermal reforming or oxidative steam reforming.63 For the first time ever, the syngas mixture (CO + H2) was manufactured via the reaction between steam and incandescent coke at 1000 °C (eqn (1)).64| C + H2O → CO + H2 | (1) |
Later, the syngas mixture was considered as a feedstock for the catalytic synthesis of methanol via a mixture of zinc oxide (ZnO) and chromia as a catalyst (eqn (2)).65
| 2H2 + CO → CH3OH | (2) |
During the 1920s to 1960s, the focus of the research communities was shifted towards the use of natural gases including methane and lighter naphtha instead of incandescent coke for syngas production via steam reforming (eqn (3)).66,67 The process was typically operated with an excess molar ratio of steam to hydrocarbons (H2O/HCs) at elevated temperatures (400–800 °C) and pressures.
| CH4 + H2O → CO + 3H2 | (3) |
Although steam reforming has had a huge importance and impact in the industrial process for syngas production,67 continuous progressive efforts in this process have featured various advancements. Advent of autothermal reforming and partial oxidation showed extreme advancements over ordinary steam reforming. Autothermal reforming was established in the 1950s by Haldor Topsoe where the combination of the partial oxidation process with steam reforming showed an advantageous performance for syngas production.68,69 In this process no external heat was supplied to the reactors for syngas production. However, the heat generated in an inlet zone of the reactor via the partial oxidation process was supplied to the second reactor for the steam reforming process. For example, the partial oxidation process of n-hexane (C6H14) as a feedstock is illustrated by the following reaction (eqn (4)):70
| C6H14 + 3O2 → 6CO + 7H2 | (4) |
Apparently, the development and optimization of this technology has led to the most efficient and cost-effective operation generally at low molar ratio of steam/HC feed to produce CO-rich syngas. This demonstrated advancements in steam reforming technology that improved the overall reactor efficiency for syngas production.
Nevertheless, the different methods that are discussed here have shown progressive advancements in syngas production, although derived from non-renewable carbon sources such as fossil fuels or natural gases, which have limited reservoirs on Earth.60,68 Moreover, excessive utilization of fossil fuels has already led to global warming across the world. Therefore, over the last few years, significant efforts have been made towards the development of clean and alternative routes for sustainable syngas or energy production and purification technology. In such scenarios, exploitation of renewable sources (biomass, CO2 and H2O) could make an apparent advancement in syngas production.71–73
Biomass or its derivatives, as a renewable source of carbon content, could also feature in the production of syngas via high temperature gasification processes.71 This is mainly achieved by reacting the biomass material at relatively high temperatures (>700 °C) without combustion, with a controlled amount of oxygen and/or steam. However, during the combustion of biomass, the presence of volatile contaminants such as NOx, SOx, NH3, H2S and other particulates in the generated syngas poses various issues including equipment corrosion, catalyst deactivation and most importantly environmental pollution.74 Therefore, further applications in downstream processes require clean and contaminant-free syngas production.
Moreover, the high abundance of CO2, a renewable carbon source, in the atmosphere is a major concern for global warming.75,76 Researchers have made plenty of efforts to mitigate the existence of CO2 in the atmosphere by means of various catalytic approaches for the production of fuels and chemical commodities.77–80 Several reports have demonstrated that CO2 is the vital C1 reactant for the production of the syngas mixture via the dry reforming of methane.81 However, the process is generally operated at higher temperatures in industries.
The recycling of CO2 with H2O into syngas is a very advantageous process. Syngas production from CO2 and H2O could manifest great potential, as mentioned below:
(i) to operate the process generally at lower temperatures,
(ii) to supply clean liquid and gaseous fuels, and
(iii) to maximize the efficiency of energy utilization for fuel production via the Fischer–Tropsch reaction.
The driving potential for the production of syngas from the CO2–H2O mixture can feasibly be attained via renewable sources of energy such as solar, hydro and nuclear.
The electrochemical fission of CO2 and H2O to syngas feedstocks has witnessed phenomenal advancements that have been acclaimed as an efficient way to recycle waste carbon into valuable products.82 Moreover, focusing on the production of syngas widens the opportunities for the development of electrocatalysts. So far, various electrocatalysts have been employed for syngas production from the CO2–H2O mixture.83–85 Unlike the thermochemical process, the electrochemical process, has in recent years advanced the production of syngas in terms of its selectivity, efficiency, and low operational cost towards practical implementation under relatively ambient conditions using electricity. The electrochemical process for syngas production could be further improved by preparing more sophisticated electrocatalysts, electrolytes, and cell designs.
Moving a single step ahead, integrating the electrochemical process with solar light irradiation could rationally modify the entire electrochemical process, propelling to fabricate a PEC device for syngas generation, which has received considerable accolades in recent years.20,86 The concept of a PEC cell is inspired by natural photosynthesis. In PEC cells, the essential energy to commence the redox reactions at electrodes for chemical production may generally be supplied via light irradiation. The progressive advantage of PEC over conventional electrochemical processes reinforced the PEC technology to be highly feasible and sustainable as it provides huge variations in catalytic and semiconductor/liquid interface systems for syngas generation.87 Despite remarkable advancements in the PEC strategy for syngas production, it remains challenging to develop more efficient and robust PEC catalytic systems that can surpass the overpotential of CO2 (inert molecule, requires high activation energy) or can activate CO2 at lower overpotentials feasibly and selectively to produce syngas with a tuneable ratio of CO/H2, yielding further downstream products.
The solar-driven production of the syngas mixture (CO + H2) via CO2 reduction in aqueous media manifests the most efficient and widely sustainable route. Therefore, taking this aspect into consideration we have highlighted the concept of photocatalytic syngas production. Exploitation of renewable sources of energy i.e. solar light for the redox reactions is the most fascinating and sustainable feature in photocatalysis.88 It has emerged as a highly advanced and promising method for artificial photosynthesis and selective chemical synthesis under mild conditions.89
To date, numerous photocatalytic systems have been explored under which the production of the syngas mixture has commenced from the photoreduction of CO2 under aqueous media. A photocatalyst (semiconductor) is the staple integral part of the photocatalytic system that provides the catalytic unit sites for CO2 and H2O reduction and acts as a sunlight harvester. The detailed process of CO2 reduction to syngas and its various photocatalytic systems are discussed in the next section.
In spite of the ingenious beauty of the state of the art of the photocatalytic CO2 reduction to syngas, its large-scale production to realize solar fuels via CO2 reduction is still in the infancy stage. Over the past few years photocatalytic CO2 reduction to syngas has been considered as the most viable pathway describing the pioneering scientific endeavours, which is still being expanded by researchers for developing photocatalytic systems including photoreactor design and most efficient photocatalysts that can withstand for a longer time into the reactor system without being poisoned for sustainable production of syngas.
3. Photocatalysts for syngas production
Sunlight provides a huge single source of energy, and researchers have devoted plenty of time, resources and intellectual input towards best exploiting this resource.88 In addition, renewable and sustainable technology has attracted much consideration because photocatalytic reactions are generally carried out at low temperatures, normal pressure and without the requirement of high input energy.90 Therefore, leading research in the field of photocatalysis is accompanied by an impressive and mammoth number of publications.3.1. Fundamental concept of CO2 photoreduction
Before moving ahead, it is quite important to discuss the phenomenon of photocatalytic CO2 reduction in an aqueous environment, as well as the band structure and reduction potential for various reduced products in brief. This may introduce a general idea in the reader's mind for better understanding. In the field of photocatalysis, the band structure of the photocatalyst has a unique importance as it defines the absorbance of the incident light according to the band gap (Eg) of the photocatalyst. In sunlight, different wavelengths of light are present; therefore, when the condition hν > Eg is satisfied, the light-absorption phenomenon takes place.91,92The practical efficiency of the photocatalyst can be estimated by the effective charge separation followed by charge transfer. When the photon energy is shed over the semiconductor, excitons i.e. electrons and holes, are generated. Further, electrons from the valence band (VB) get excited to the conduction band (CB), leaving behind holes in the VB. These photogenerated electrons and holes then migrate to the surface of the semiconductor (Fig. 1a). After that, the reaction is initiated by the accepting an electron by a CO2 molecule from the CB of the semiconductor.93 However, the phenomenon of charge recombination also takes place when an electron and hole combine over the surface. This may lower the efficacy of the photocatalyst for the reaction. Note that the reduction of CO2 is an uphill reaction; therefore, the CB and VB position of the photocatalyst must bestride the reduction potential of CO2 and the oxidation potential of H2O.31,94
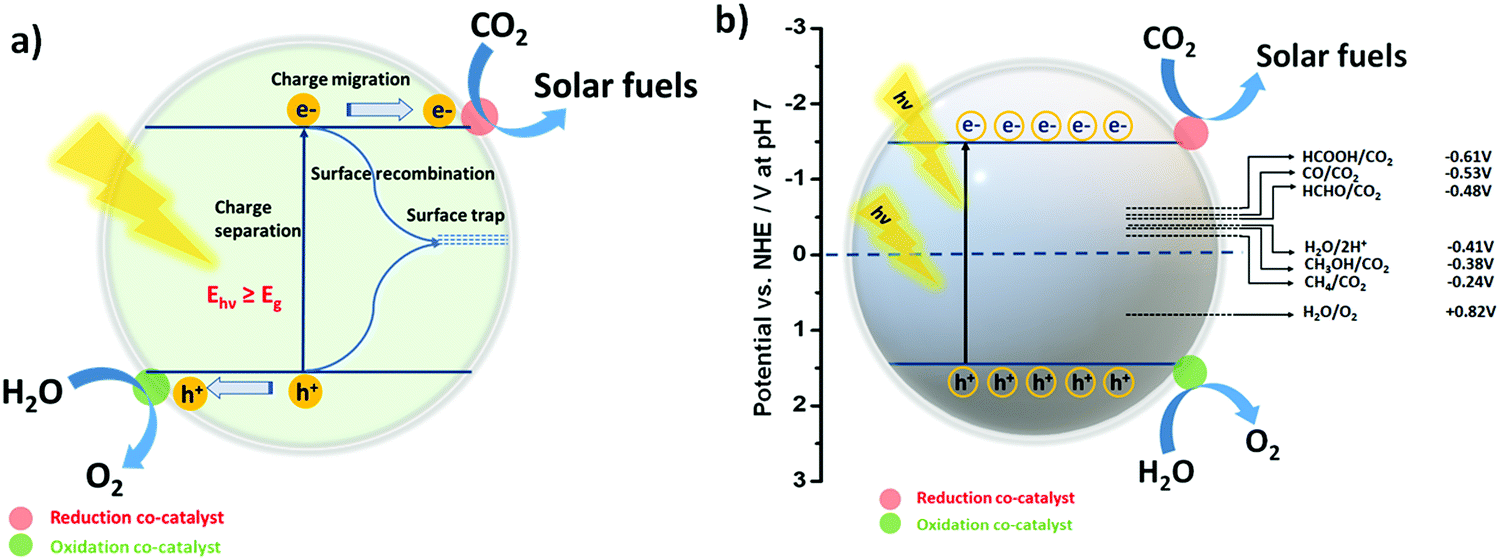 | ||
| Fig. 1 (a) Schematic illustration of the elementary processes of photogenerated charges over the photocatalytic surface. (b) Schematic illustration of photocatalytic CO2 and H2O conversion over a semiconducting photocatalyst for solar fuel production mediated by suitable redox co-catalysts. | ||
The symmetrical structure and strong bond energy ascertain the thermodynamically unfavourable photoreduction of CO2 due to the high negative reduction potential of CO2/˙−CO2 (−1.90 V vs. NHE, at pH 7.00). On the other hand, it must be realized that the proton-driven photoreduction of CO2 (CO2 reduction under H2O) could facilitate more favourable conditions for CO2 reduction at lower negative reduction potentials (vs. NHE, at pH 7.00) as the required reduction potential of the protons is less negative than the reduction potential of CO2.
Thus, these two processes such as the hydrogen evolution reaction (HER)95–97 and CO2 reduction reaction49,98 may always compete with each other. The formal electrochemical potentials of the reactions associated with the photoreduction of CO2 and H2O redox reactions are summarized in Table 1 and also shown in Fig. 1b.35
| Reaction | E 0 vs. NHE at pH = 7 (V) |
|---|---|
| CO2 + e− → CO2˙− | −1.90 |
| CO2 + 2e− + 2H+ → HCOOH | −0.61 |
| CO2 + 2e− + 2H+ → CO + H2O | −0.53 |
| CO2 + 4e− + 4H+ → HCHO + H2O | −0.48 |
| CO2 + 6e− + 6H+ → CH3OH + H2O | −0.38 |
| CO2 + 8e− + 8H+ → CH4 + H2O | −0.24 |
| 2H+ + 2e− → H2 | −0.41 |
| 2H2O + 4h+ → O2 + 4H+ | +0.82 |
3.2. Oxidation half-reaction: complementing CO2 reduction
Photocatalytic CO2 conversion is a redox process. In accordance with the CO2 photoreduction reaction via photo-excited electrons, the oxidation of water to produce O2 may also take place as the oxidation half reaction via photo-excited holes. Ideally, H2O acts as an electron donor and a hydrogen source for the photocatalytic CO2 reduction. The addition of sacrificial agents (alcohols, amines, and acids) generally boosts the electron donation ability, thus realizing the competing production of H2 coupled with CO2 reduction. Remarkably, the oxidation potential of water (H2O/O2) to O2 is +0.82 V (vs. NHE, at pH 7.00), which is a lot less positive than the VB potential of most semiconductors, making the oxidation process thermodynamically feasible (Fig. 1a).35 In order to ensure the continuation of the CO2 reduction process, the generated oxidizing agents such as OH• or O2 and the oxidized products must be desorbed immediately from the surface of the photocatalyst.Of note, so far, numerous photocatalysts including metal oxides, metal–organic complexes, metal–organic frameworks (MOFs), covalent organic frameworks (COFs), single metals and layered semiconducting materials have been employed for CO2 reduction into CO, HCOOH, CH3OH, CH4, and HCOH products.99,100 In the following section, we discuss the photocatalysts associated with photocatalytic CO2 reduction in H2O for the production of syngas (CO + H2) only and their corresponding activities along with the CO/H2 ratios, which are listed in Table 2.
| Entry | Photocatalyst | Photosensitizer | CO and H2 yield/conversion rate | CO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 (tuneable) :H2 (tuneable) |
Year | Ref. |
|---|---|---|---|---|---|---|
| Note: N.A.; data not available/provided. | ||||||
| 1 | CdSNRs/Fe(III)Salen | N.A. | CO: 143 μmol h−1 | 1:2.09 |
2020 | 101 |
| H2: 300 μmol h−1 | ||||||
| 2 | Fe(CO)3bpy | [Ru(bpy)3]Cl2 [Ir(ppy)2(bpy)]PF6 | CO: 35 μmol | 1:1.5 |
2015 | 102 |
| H2: 42 μmol | ||||||
| 3 | CoII(Ch) | [RuII(Me2phen)3]2+ | NA | 2.4:1 |
2016 | 103 |
| 4 | [Co5(btz)6(NO3)4(H2O)4] | [Ru(bpy)3]Cl2 | CO: 79.2 μmol | 1:16 to 1:2 |
2020 | 50 |
| H2: 140.6 μmol | ||||||
| 5 | Co(bpy)2Cl2 | [Ru(bpy)3]Cl2 | CO:62.3 μmol and H2: 69.9 μmol | 1:1.2 |
2017 | 49 |
| 6 | CoAl-LDH/MoS2 | [Ru(bpy)3]Cl2·6H2O | CO: 8070 μmol g−1 h−1 | 1:1.3 to 1:15 |
2020 | 45 |
| H2: 8415 μmol g−1 h−1 | ||||||
| 7 | (Pd/CoAl-7.57) | [Ru(bpy)3]Cl2·6H2O | When λ > 400 nm CO: 1300 μmol g−1 h−1 | 1:0.74 to 1:3 |
2019 | 46 |
| H2: 600 μmol g−1 h−1 | ||||||
| When λ > 600 nm CO: 4.1 μmol g−1 h−1 | ||||||
| H2: 5.4 μmol g−1 h−1 | ||||||
| 8 | Ce-0.15 | [Ru(bpy)3]Cl2·6H2O | CO: 5 μmol g−1 h−1 | 1:7.7 to 1:1.3 |
2021 | 104 |
| H2: 52 μmol g−1 h−1 | ||||||
| 9 | (Co6–MOF) | [Ru(bpy)3]Cl2·6H2O | CO: 39.6 μmol | 1.4:1 |
2017 | 105 |
| H2: 28.13 μmol | ||||||
| 10 | (Co/Ru)2.4-UiO-67(bpydc) | [Ru(bpy)3]Cl2·6H2O | CO: 4520.5 μmol g−1 | 1:2 |
2019 | 39 |
| H2: 9121.5 μmol g−1 | ||||||
| 11 | C-BMZIFs | [Ru(bpy)3]Cl2·6H2O | CO: 6883 μmol g−1 h−1 | 1.9:0.7 |
2018 | 106 |
| H2: 3600 μmol g−1 h−1 | ||||||
| 12 | Co-ZIF-9 | [Ru(bpy)3]Cl2·6H2O | CO: 41.8 μmol | 1.39:1 |
2014 | 107 |
| H2: 29.9 μmol | ||||||
| 13 | Fe0.5Ni0.5 MOFs | [Ru(bpy)3]Cl2·6H2O | Low conc. CO2 reduction | 1:1.1 |
2021 | 28 |
| CO: 5 μmol h−1 | ||||||
| H2: 5.5 μmol h−1 | ||||||
| 14 | Re-Bpy-sp2c-COF | N.A. | CO: 12.48 μmol H2:2.99 μmol |
4:1 to 1:0 |
2020 | 108 |
| 15 | CTF-TDPN | [Co(bpy)3]2+ | CO: 200 μmol g−1 h−1 | 1.4:1 |
2021 | 40 |
| H2: 140 μmol g−1 h−1 | ||||||
| 16 | Ni-COFs | [Ru(bpy)3]2+ | CO: 9.06140 μmol g−1 | 1:19 to 9:1 |
2020 | 41 |
| H2: 1.16140 μmol g−1 | ||||||
| 17 | CoO-Mo8 UNWs | [Ru(bpy)3]Cl2·6H2O | CO: 4165 μmol g−1 h−1 | 1:2.27 |
2020 | 44 |
| H2: 11555 μmol g−1 h−1 |
||||||
| 18 | Co2[Co20Mo16P24] | [Ru(bpy)3]Cl2 | CO: 31.42 μmol | 1:1.5 |
2020 | 43 |
| H2: 20.94 μmol | ||||||
| 19 | [Co(H2O)6][Co-POM] | [Ru(bpy)3]Cl2 | Pure CO2 | 1.8:1 and 1:5 |
2020 | 109 |
| CO: 24 mmol g−1 h−1 | ||||||
| H2: 13.3 mmol g−1 h−1 20% dil. CO2 | ||||||
| CO: 9.4 mmol g−1 h−1 | ||||||
| H2: 47.4 mmol g−1 h−1 | ||||||
| 20 | [Co2.67(SiW12O40)(H2O)4(Htrz)4]·Cl1.33 | [Ru(bpy)3]Cl2·6H2O | CO: 15705 μmol g−1 |
1:0.92 1:0.98 |
2019 | 110 |
| H2: 14523 μmol g−1 |
||||||
| [Co3(SiW12O40)(H2O)3(Htrz)6Cl]·Cl·6H2O | CO: 18501 μmol g−1 |
|||||
| H2: 18199 μmol g−1 |
||||||
| 21 | Mn SAs | [Ru(bpy)3]Cl2 | CO: 1470 μmol g−1 h−1 | 1.12 to 0.43 | 2020 | 111 |
| H2: 1310 μmol g−1 h−1 | ||||||
| 22 | CoN4-SiO2 | g-C3N4 | CO: 398 μmol g−1 | 1:2 |
2019 | 112 |
| H2: 804 μmol g−1 | ||||||
| 23 | Fe-SAs/N−C | [Ru(bpy)3]Cl2 | CO: 4500 μmol g−1 h−1 | 0.3 to 8.8 | 2020 | 51 |
| H2: 4950 μmol g−1 h−1 | ||||||
| 24 | TiO2 fiber (B) | N.A. | CO: 203.91 μmol g−1 | 1:1.9 |
2016 | 48 |
| H2: 398.84 μmol g−1 | ||||||
| 25 | 1.0Ag1.0Au/TiO2 | N.A. | CO: 2.3 μmol g−1 | 1:2 |
2020 | 113 |
| H2: 4.3 μmol g−1 | ||||||
| 26 | Cu2O/MnOx | N.A. | CO: 5.71 μmol h−1 | 1.38:1 |
2020 | 114 |
| H2: 4.11 μmol h−1 | ||||||
| 27 | Dye/TiO2/ReP:CoP | (E)-2-Cyano-3-(5′-(5′′-(p-(diphenylamino)phenyl)thiophen-2′′-yl)thiophen-2′-yl)-acrylic acid (Dye) | CO: 773 μmol g−1 | 3.5:1 |
2016 | 115 |
| H2: 221 μmol h−1 | ||||||
| 28 | Pt-Modified SCS-AgBiW2O8 | N.A. | CO: 3 × 10−3 mol g−1 L | 0.3:1 |
2012 | 116 |
| H2: 1 × 10−2 mol g−1 L | ||||||
| 29 | MTC3.17P-MS | N.A. | CO: 80 μmol g−1 h− | 1:2 |
2018 | 117 |
| H2: 160 μmol g−1 h−1 | ||||||
| 30 | Rh-Au@SrTiO3 | N.A. | CO:66.8 μmol g−1 h−1 | 1.3:1 |
2016 | 118 |
| H2: 50.5 μmol g−1 h−1 | ||||||
| 31 | 2% Ag/TiO2-SP | CO:103 μmol g−1 h− | 1:2.1 |
2012 | 119 | |
| H2: 220 μmol g−1 h−1 | ||||||
| 32 | Meso. TiO2 (by KIT-6 replication) | N.A. | CO:26.3 μmol g−1 H2:83.5 μmol g−1 |
1:3.1 |
2015 | 47 |
| 33 | NVs-PCN (PCN-23) | N.A. | CO: 8 μmol g−1 H2:2 μmol g−1 |
4:1 |
2021 | 42 |
| 34 | POP2-Fe | [Ru(bpy)3]Cl2 | CO: 3043 μmol g−1 | 1:1.23 |
2021 | 120 |
| H2: 3753 μmol g−1 | ||||||
| 35 | Pt/BP-OvMBWO | N.A. | CO: 20.5 μmol g−1 h−1 | 1:1–2:1 |
2021 | 121 |
| H2: 16.8 μmol g−1 h−1 | ||||||
| 36 | Ag/LaFeO3 (ALFO-600) | N.A. | CO: 2.41 μmol g−1 h−1 | 1:3 |
2022 | 122 |
| H2: 7.3 μmol g−1 h−1 | ||||||
3.3. Metal oxide photocatalysts for syngas production
3.3.1.1. Nanostructured TiO2 photocatalyst. Nano-engineering, in particular, helps to improve the light absorption capacity, and accentuate the photogenerated charge separation and transport properties in addition to increasing the available surface area of the TiO2 photocatalysts. Reports have suggested that the nanostructured TiO2 photocatalysts boosted the photo performance for syngas production via CO2 reduction, as shown in Fig. 2.
 | ||
| Fig. 2 Schematic illustration of photocatalytic syngas production over TiO2-based nanocomposite photocatalysts. Reproduced and modified with permission.47 Copyright 2015, Elsevier. Reproduced and modified with permission.48 Copyright 2016, Elsevier. Reproduced and modified with permission.113 Copyright 2020, American Chemical Society. Reproduced and modified with permission.115 Copyright 2017, Wiley-VCH. Reproduced and modified with permission.117 Copyright 2018, Royal Society of Chemistry. | ||
3.3.1.2. Metal-deposited TiO2 photocatalyst. It is evident from the previous literature that metal loading on TiO2 semiconductor has significantly enhanced the photo-generated charge separation and also improved the light absorption ability towards the visible light spectrum.130–132 Zhao et al.119 reported an efficient approach of ultrasonic spray pyrolysis (SP) method to prepare a mesoporous silver nanoparticle deposited TiO2 (Ag/TiO2) composite. The synthesized Ag/TiO2 material was further examined for the concurrent photocatalytic hydrogen production and CO2 reduction to CO i.e. syngas (CO + H2) from water using methanol as a hole scavenger and a solar simulator as a light source. Varying the reaction gas composition affected the molar ratio of H2/CO production rates during syngas synthesis and it was effectively tuned in the range from 2 to 10.
Syngas production could generally be improved by engineering TiO2 catalyst with bi-metallic nanoparticle (NP) deposition over it as compared to bare TiO2. In one of the studies, Renones et al.113 demonstrated that deposition of both Ag130 and Au133 metals over TiO2 semiconductor (Ag–Au/TiO2) imparts a significant activity for the production of syngas from CO2–H2O mixtures under visible light. Au and Ag both metals are known for localized surface plasmon resonance (LSPR) which tends to improve the light absorption efficiency by folds and also improve the photogenerated charge separation ability by means of capturing electrons, thus facilitating the multi-electron reduction–oxidation (red–ox) reactions involved in photocatalytic CO2 reduction. Of note, the Ag–Au/TiO2 (1 wt%, optimal metal loading) catalyst demonstrated a dual behaviour in terms of selectivity for CO2 reduced products under different wavelengths of light. Under UV light, the main CO2 reduction product was CH4 and the minor product was CO, accompanied by the generation of H2 from water reduction. On the contrary, under the visible light, the main CO2 reduction product was CO in all cases coupled with H2 production, resembling the critically indispensable syngas (CO + H2) product with a CO production rate of 2.3 μmol g−1 and H2 production rate of 4.3 μmol g−1.
3.3.1.3. Mesoporous and morphologically tuned TiO2 photocatalyst. Introduction of porosity and high surface area can amplify the photocatalytic performance in nanomaterials.134 Mesoporous TiO2 has gained increasing interest in the field of photocatalysis by improving the conversion efficiencies of solar energy, minimizing the recombination of photogenerated electron–hole pairs, and optimizing the mass and fast charge transport.135,136 Sol gel, a bottom-up approach, is one of the desirable protocols to develop mesoporosity into the TiO2 semiconductor for the efficient CO2 adsorption and photocatalytic reduction of CO2 to fractional energy products including syngas (CO + H2). Owing to this, Akhter et al.47 synthesized nanostructured or mesoporous TiO2 with an enhanced surface area (190 m2 g−1) and high adsorption capacity using KIT-6 silica template for the photocatalytic reduction of CO2 in the presence of H2O vapor to produce syngas (CO + H2) along with hydrocarbons. Mesoporous TiO2 showed high adsorption ability of reacting gases (CO2 and H2O) on the surface of the catalyst as compared to Aeroxide P25 TiO2. The study demonstrated that the key parameters including the UV light source, intensity, and initial feed ratios i.e. H2O
:CO2 directly influence the photocatalytic activity of the catalyst for fuel production.
However morphological tuning of TiO2 in 1D (i.e. rods, fibres and tubes) is another extension towards nano engineering, which features unique properties, diverse functions, advocating easy electron–hole separation and high rate of electron diffusion coefficient. Accompanying this, Renones et al.48 synthesized a hierarchical assembly of mesoporous TiO2 1-D nanofibers via combination of electrospinning and sol gel methods for the photocatalytic reduction of gas phase CO2 using H2O as a sacrificial electron donor under UV light irradiation. Among all the catalysts, profound CO2 reduction activity was achieved by the TiO2 Fibres B catalyst, which was 398.84 μmol g−1 (H2) and 203.91 μmol g−1 (CO), respectively, compared to the TiO2 Fibres-A catalyst (H2; 42.78 μmol g−1 and CO; 55.06 μmol g−1).
3.3.1.4. Binary-ternary composite based TiO2 photocatalyst. Varying wide range of photocatalysts (metal oxides, dyes, and metal complexes) with TiO2 as the semiconductor have shown huge potential for tailoring and design of highly desirable TiO2-based nanocomposites in order to enhance the light absorption ability, fast charge separation and transportation and further improvement of the activity for CO2 photoreduction.123,137
Hybridizing photosensitizers (dyes and metal complexes) with TiO2 semiconductor has been proven to be a highly novel strategy to nano engineer TiO2 based semiconductors for the photoreduction of CO2 under visible light. Molecular transition metal complexes-TiO2 has received much attention as a potential photosensitized hybrid nanomaterial for CO2 photoreduction.138,139 The advantages of using metal complexes for the CO2 reduction along with TiO2 based nanomaterials are follows: (1) improvement in light harvesting ability towards visible light region, (2) tuneable redox potentials via ligand modifications, (3) facile coordination of CO2 molecules to the metal, and (4) multi-electron reduction process by pumping an electron from the excited state of the metal-complex to the conduction band of the TiO2. Various transition metal complexes with TiO2 semiconductor, including Re,140 Ru,141 Mn(I),142 Cu(II),143etc., have been proposed as potential candidates for the photo-reduction of CO2. However, the practical applicability of the above-mentioned metal complexes with TiO2 for the tuneable syngas (CO + H2) production has not been given much attention. Therefore, in a very interesting study, Lee et al.115 demonstrated a controllable syngas production under visible-light irradiation by synthesizing a dye-sensitized TiO2 photocatalyst containing Re(I) and Co(III) metal complexes. In the ternary hybrid catalyst (dye/TiO2/ReP:CoP), the Re(I) metal complex (ReP) was shown as a CO-producing site and a Co(III) molecular catalyst (CoP) was referred to act as a H2-producing active site (Fig. 3a). Furthermore, dynamic electron transfer from the dye to TiO2 initiated the photoreduction process of CO2 in the N,N-dimethyl formamide (DMF)–water system to co-produce H2 and CO. However, after CO2 reduction the H2/CO ratio in the generated syngas was effectively controlled from 1:2 to 15:1 by the water content of the solvent or the molar Re(I)/Co(III) ratio of the metal complexes in to the dye/TiO2/ReP:CoP catalyst.
 | ||
| Fig. 3 (a) Schematic representation of the heterogeneous ternary photocatalytic system for syngas production. Reproduced and modified with permission.115 Copyright 2017, Wiley-VCH. (b) Mechanism of the photocatalytic CRR driven by MTCP-MSs. Reproduced and modified with permission.117 Copyright 2018, Royal Society of Chemistry. (c) The photoreaction processes of D-CMH. Reproduced and modified with permission.114 Copyright 2020, Royal Society of Chemistry. | ||
Furthermore, for tuneable syngas production, tailoring TiO2 semiconductors with metal oxides and metal alloys is in high demand. TiO2 nanocomposite arrays composed of TiO2 hollow spheres and MnOx and CuPt alloys (denoted as MTCP-MS) have been fabricated, with the hollow structure of the TiO2 catalyst with spatially separated oxidative inner surfaces containing the oxidation co-catalyst MnOx and the reductive outer surfaces containing the reduction co-catalyst CuPt, which was reported to be efficient in the production of syngas from photocatalytic CO2 reduction with a tuneable CO/H2 ratio (Fig. 3b). It was shown that CO/H2 ratio was perfectly tuned in a desirable 1:2 ratio with the MTC3.17P-MS catalyst, offering a CO evolution rate of 80 μmol g−1 h−1 and H2 evolution rate of 160 μmol g−1 h−1 by altering the components present at the outer reductive surfaces (co-catalysts, CuPt). Furthermore, a prominent CO evolution rate of 84.2 μmol h−1 g−1 was achieved with 0.108% CO energy conversion yield.117
Perovskites are another class of oxides of interest. SrTiO3 is a perovskite semiconductor, and offers many useful characteristics for CO2 photoreduction reactions. Therefore, Li. et al.118 designed a strategy to combine SrTiO3 with Au and Rh co-catalyst to construct a new photocatalyst system. Au, as a plasmonic nanostructured metal, exhibits strong light absorption via excitation of localized surface plasmon resonances (LSPR) and acts as a visible-light sensitizer. Rh acts as a photoelectron receiver and usually applied in dry methane reforming reactions.145 However, their mutual interactive effect over SrTiO3 exerted noticeable conversion efficiency and high selectivity for syngas production from reduction of the CO2–H2O mixture under visible-light irradiation. The production rate of CO and H2 was estimated to be 66.8 μmol g−1 h−1 and 50.5 μmol g−1 h−1 with a CO:H2 ratio of 1.3:1. As compared to Au@SrTiO3 and Rh@SrTiO3 catalysts, the synergistic effect of Rh and Au over the SrTiO3 surface showed 22- and 153-fold enhancement in the photocatalytic activity for syngas production, respectively.
Moreover, silver (Ag), bismuth (Bi), and tungsten (W)-containing complex oxides have shown huge importance in photocatalysis. For instance, silver bismuth tungstate (AgBiW2O8) nanoparticles with moderate band gap (indirect), excellent stability in aqueous media and suitable band edge positions feature in solar-driven HER and CO2RR. Tacconi et al.116 demonstrated mild syngas photo-production using AgBiW2O8 nanoparticles that were synthesized via the solution combustion procedure from their corresponding metal salts. It is essential to point out that the CO2 was generated in situ from formic acid solution, which was majorly responsible for the production of syngas (CO + H2).
3.4. Layered double hydroxide photocatalyst for syngas production
Layered double hydroxides (LDHs) are two dimensional inorganic crystalline nanostructured materials with a general formula of [M1−x2+M3+x(OH)2]x+ [Ax/pn−]x+·mH2O, where M2+ and M3+ are a metallic bivalent cation and a metallic trivalent cation, respectively, An− is an interlayer anion typically carbonate, nitrates and other charge balancing anions and X = M3+/(M2+ + M3+) is the surface charge.146,147 Their layered architecture, earth-abundant components, easy synthetic procedure, and light harvesting capability make LDHs attractive photocatalysts. However, poor quantum efficiency due to sluggish charge mobility and facile electron–hole recombination in pristine LDHs generally limit their practical application in photocatalysis. Several attempts have been made to construct a heterojunction at the interface of LDHs by combining them with different metals or semiconductor materials such as Pd, Ag, MoS2, and g-C3N4. Heterostructures of LDHs could facilitate the ease of charge transfer, thus advancing their broad applications in photocatalysis.148–152In a broader view, LDHs have been used in various photocatalytic reactions including water splitting, environmental remediation, CO2 reduction, and organic transformations.147 However, their application in CO2 photoreduction reaction is highly demanding. Generally, CH3OH, HCOOH, CH4 and CO are the major reduced products arising from the photoreduction of CO2.153 CO2 reduction into CO and H2 is a requisite task to produce syngas for upgradation of fuel via Fischer–Tropsch synthesis. In the wake of this necessity, Wang et al.46 reported Pd nanoparticle loaded CoAl-LDH (Pd/CoAl-LDH) in conjunction with ruthenium complex (a photosensitizer; [Ru(bpy)3]Cl2·6H2O) as a heterostructure photocatalyst for CO2 reduction to syngas under visible light irradiation. Pd is a good electron absorber, thus facilitating excellent charge separation and migration and known for producing H2 in the HER. Pd/CoAl-LDH and ruthenium complex ensured tuneable syngas production with a CO:H2 ratio ranging from 1:0.74 to 1:3 under visible light irradiation (λ > 400 nm) as shown in Fig. 4(a and b). Interestingly, it was shown that syngas production under visible light irradiation could further be expanded up to λ > 600 nm in the presence of the Pd/CoAl-7.57 catalyst (Fig. 4c). DFT and structure characterization techniques demonstrated the superficial role of Pd nanoparticles over CoAl-LDH for tuneable syngas production.
 | ||
| Fig. 4 (a) The selectivity of CoAl-LDH and Pd/CoAl-x for CO2 reduction under visible light irradiation in the presence of a [Ru(bpy)3]2+ sensitizer and triethanolamine (TEOA) as a sacrificial electron donor. (b) Selectivity of CO and H2 on CoAl-LDH, Pd/CoAl-0.55, Pd/CoAl-2.46 and Pd/CoAl-7.57. (c) Pd/CoAl-7.57 under irradiation with different wavelengths. Reproduced and modified with permission.46 Copyright 2020, Elsevier. (d) Scheme of the tuneable selectivity of syngas from photocatalytic CO2 reduction by LDH, Ce-x (x = 0.05, 0.10, 0.15, 0.20, 0.30 and 0.40) and CeO2 in conjunction with a Ru-complex photosensitizer. Ce-0.15 in CO2PR under different cut-off filter light irradiation. (e) Selectivity of CO and H2. Reproduced and modified with permission.155 Copyright 2020, Springer Nature. (f) Schematic illustration of photocatalytic CO2 reduction to tuneable syngas on CoAl-LDH/MoS2 heterostructures. Reproduced and modified with permission.45 Copyright 2020, Royal Society of Chemistry. | ||
Moreover, in this direction, extending the light absorption capability of LDHs beyond 600 nm is still a desirable task. Like metallic nanoparticles, the LDH heterostructure with other materials shows extended light capturing efficiency, thus improving the photocatalytic performance. Ceria (CeO2), as an n-type semiconductor, has received wide attention in photocatalysis for the CO2 reduction reaction.154 Owing to this, Tan et al.155 recently reported photocatalytic syngas production under visible-light irradiation up to 600 nm from the CO2–H2O mixture by constructing the CeO2–MgAl–LDH heterostructure (denoted as Ce-x, x = different molar ratio). Varying content of CeO2 on MgAl–LDH has shown significant yield of syngas mixture (CO + H2) with different molar ratios of CO/H2. The highest yield and selectivity of syngas was achieved from Ce-0.15 and the ratio of the syngas products i.e. CO/H2 was tuned ranging from 1/7.7 (LDH) to 1/1.30 (Ce-0.15). Of note, Ce-0.15 exerted excellent CO2 reduction to syngas under visible light irradiation (λ > 600 nm) as shown in Fig. 4(d and e) which can further be confirmed by various characterization techniques.
Molybdenum disulphide (MoS2), a metal dichalcogenide, is a 2D graphene-like layer-structured semiconductor material. Its intrinsic electronic structure provides high chemical stability and superior electronic mobility, which has received considerable attention in the photocatalytic H2 evolution reaction and CO2 reduction.156,157 Moreover, the photophysical properties and quantum efficiency of MoS2 could further be improved by constructing a heterostructure with other 2D layered semiconducting materials.158 Constructing a heterojunction between LDHs and MoS2 could be an obvious choice. Owing to this, Qui et al.45 fabricated a heterostructure by integrating CoAl-LDH and 2D MoS2via electrostatic interaction for efficient CO2 reduction into syngas under visible light irradiation. The photoactivity of the CoAl-LDH/MoS2 material manifested excellent CO2 reduction to syngas and the ratio of syngas products H2:CO was tuned ranging from 1.3:1 to 15:1 which was rationalized via controlling the concentration of the CoAl-LDH/MoS2 catalyst (Fig. 4f). In addition, photocatalytic activity of the material was also tested under visible light irritation up to 500 nm, which resulted in a high evolution rate of CO (4575 μmol g−1 h−1) from CO2 photoreduction.
Obvious efforts for solar-driven syngas production generally based on LDH photocatalysts have shown judicious potential for CO2 reduction to syngas. However, still extensive scientific endeavours are required to design LDH based photocatalysts, emphasizing the concept of constructing desirable heterojunctions with LDHs for feasible charge transfer, CO2 adsorption–activation and efficient syngas production.
3.5. Polyoxometalate photocatalyst for syngas production
Polyoxometalates (POMs) are a large class of inorganic molecular metal clusters with definite particle sizes and structural dimensions furnishing unique physical and chemical properties including physical solubility, acidity, redox ability, and high thermal and chemical stability. Plenty of POM structures and their constituent hybrids have been proposed to show exciting potential in electro-/photocatalytic CO2 conversion reactions.159–161 In addition, their excellent redox properties and phenomenal solution stability renders them suitable to carry out photocatalytic CO2 reduction in H2O as a solvent. Yang et al.44 reported (n-C4H9)N]4Mo8O26 POM denoted as Mo8 which was further incorporated with CoO nanowires for the facile production of syngas from photoreduction of CO2 and H2O under visible light irradiation, which manifested excellent H2 and CO evolution rates of 11555 and 4165 μmol g−1 h−1, respectively (Fig. 5a–c). Additionally, they also exhibited a rationally higher CO/H2 ratio than without CoO nanowires. Furthermore, this study demonstrated an ultimate synergistic role of CoO nanowires as Co active sites for tuneable syngas production and Co-based POM engineering for advanced CO2 photocatalysis.
 | ||
| Fig. 5 (a) A schematic picture of synthetic procedures towards CoOUNWs and CoO-Mo8 UNWs. (b) Proposed plausible mechanism. (c) Photochemical syngas production performance of different materials. Reproduced and modified with permission.44 Copyright 2020, Wiley-VCH. (d) The proposed CO2 photoreduction mechanism of the [Ru(bpy)3]/[Co20Mo16P] composite. (e) The influence of water content, from 0 vol% to 20 vol%. Total volume of solvent is unchanged. (f) The reduction performed with different CO2 content, ranging from 3% to 20%. In these systems, CO2 is diluted by Ar, for example, in 20% CO2, the CO2/Ar volume is 4:1. Reproduced and modified with permission.43 Copyright 2020, Wiley-VCH. | ||
Furthermore, Co metal has got extended attention as a linker between Keggin-type POMs and organic ligands for the synthesis of new POM-based hybrid materials for CO2 photoreduction. In a very impressive study, Yao et al.110 prepared two different new POM-based organic–inorganic hybrids with H4SiW12O40·2H2O as the molecular building block, Co cluster (bi and tri-nuclear) as the linker and 1,2,4-triazole (Htrz) as the organic ligand under hydrothermal conditions. Compound [Co2.67(SiW12O40) (H2O)4(Htrz)4]·Cl1.33 (1) and compound [Co3[SiW12O40(H2O)3(Htrz)6Cl]·Cl·6H2O (2) were utilized as heterogeneous photocatalysts in the photoreduction of CO2 to CO and H2 under visible light. However, the production of CO and H2 for (1) was 15705 and 14523 μmol g−1 and 18501 and 18199 μmol g−1 for (2), respectively. It was shown that different Co clusters responsible for innate properties result in excellent photocatalytic activity of (1) and (2).
The negative charges over the surface of polyoxometalates (POMs) might be helpful to couple the [Ru(bpy)3]2+ complex via electrostatic interactions. Taking advantage of it, Xu et al.43 synthesized a [Ru(bpy)3]/[Co20Mo16P24] composite via ionic POM based on Co(II) and P4Mo6 units (Co2[Co20Mo16P24]) and [Ru(bpy)3]2+ for efficient photoreduction of dilute CO2 to syngas under visible light irradiation. The as prepared [Ru(bpy)3]/[Co20Mo16P24] composite exhibited high efficiency for syngas generation with 523.6 TONs (74.9 mmol g−1 h−1) in pure CO2 and 964.9 TONs (137.9 mmol g−1 h−1) in diluted CO2 (Fig. 5d–f). However, the H2/CO ratio was widely tuned from 5.9:1 to 1:2. Likewise, Zhao et al.109 also demonstrated the photoreduction process of diluted CO2 to syngas. Polyoxometalate [Co-POM]2− and [Ru(bpy)3]2+ complex were integrated to synthesize a hybrid photocatalyst for dilute CO2 conversion to syngas under the visible light region. It was shown that in diluted CO2, a conversion efficiency of 56.8 mmol g−1 h−1 was achieved in syngas production.
It has been shown that POMs are efficient and promising photocatalysts for CO2 reduction to syngas. Incorporation of metal ions or clusters, organic linkers and metal complexes as sensitizers could impart enhanced photocatalytic activity and efficiency. Additionally, it provided insights into rational modification into the POM based heterogeneous photocatalysts for highly efficient CO2 reduction even in low concentration to generate syngas, which will pave a sustainable route for renewable energy production in the near future.
3.6. Single-atom photocatalyst for syngas production
Single atom catalysts (SACs) have feasibly become the most fascinating choice, advancing the atomic efficiency and catalytic performance by magnitude in various catalytic reactions when dispersed over the matrix support.162,163 Unusual ultrahigh ratio of low coordination-number metal atoms and unique electronic properties of SACs at the atomic level propel an excellent pathway for the fascinating catalytic property as compared to conventional catalysts.164 In addition, they address the concern over economic issues by minimizing the catalyst consumption, especially for noble metals such as Pt, Pd, Au, Rh, and Ir.Importantly, the applicability of SACs can promptly be facilitated by the matrix supports, providing a strong anchoring site, high surface area and feasible charge mobility, which can arguably bestow cogent catalysis and stability.164 Recently, transition metal atom-based SACs have shown great potential in wide applications. In consequence, they are extensively employed as heterogeneous catalysts in photocatalytic CO2 reduction reactions.165,166 Herein, we distinctively feature transition metal SACs promoting CO2 photo-reduction to syngas. Wang et al.51 demonstrated room-temperature synthesis of Fe single atoms anchored over nitrogen doped porous carbon support (denoted as Fe–SAs/N–C) via an electro-chemical filtration method for CO2 photoreduction into tuneable syngas. Coordination of Fe single atoms with N in the carbon matrix intensifies the CO2 reduction performance. By an instance, the as prepared Fe–SAs/N–C exhibited an outstanding photocatalytic performance of CO2 in assistance with [Ru(bpy)3]2+ for the production of tuneable syngas, resulting in estimated production rates of CO and H2 of 4500 and 4950 μmol g−1 h−1, respectively (Fig. 6a–c). Interestingly, the CO/H2 ratio was tuned ranging from 0.3 to 8.8.
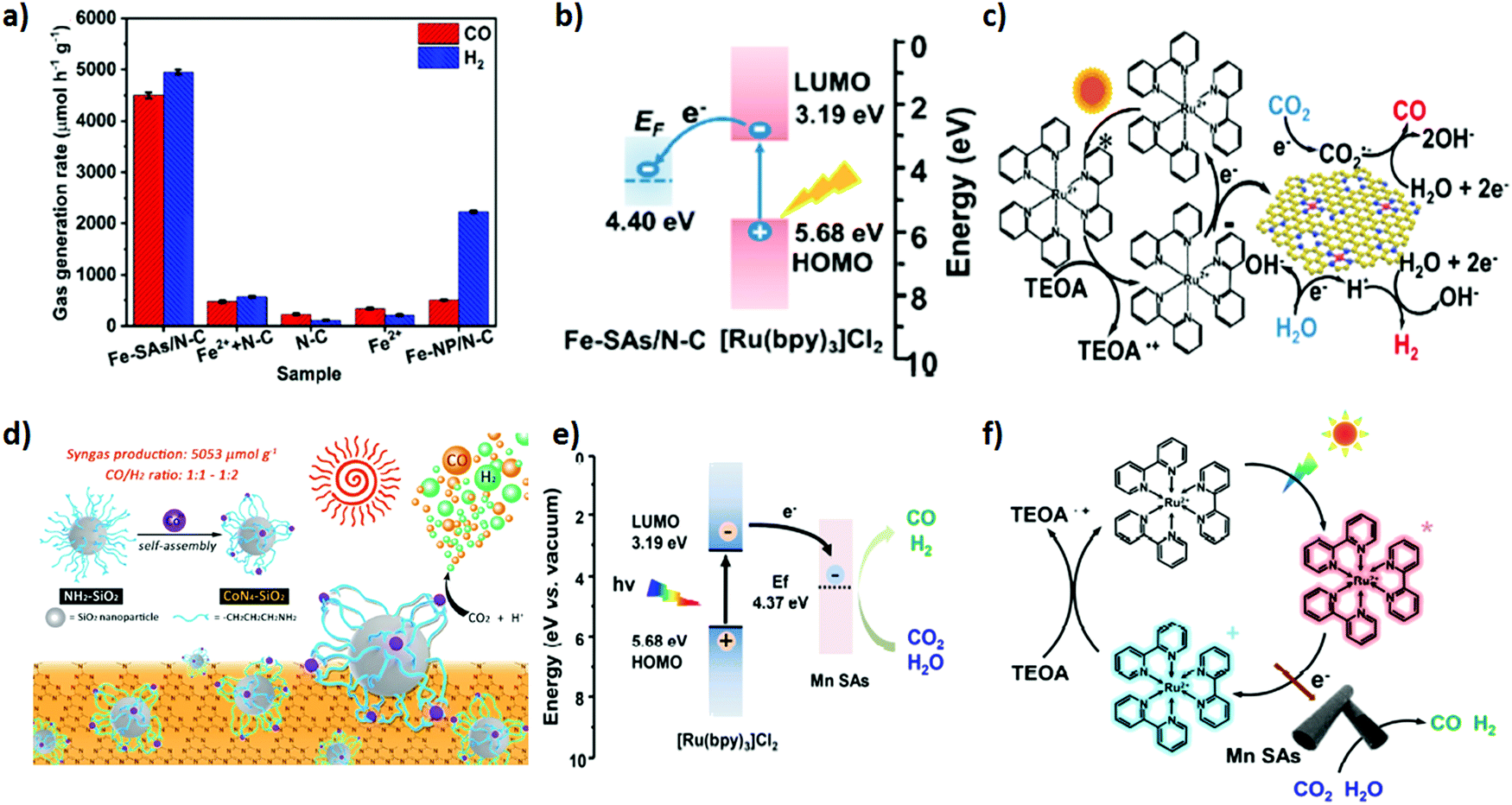 | ||
| Fig. 6 (a) CO2 photoreduction activities of Fe–SAs/N–C, Fe2+ + N–C, N–C, Fe2+ and FeNP/N–C under the same conditions with 5 mg catalyst, 5 mg [Ru(bpy)3]Cl2·6H2O in 6 mL mixed solution (acetonitrile/H2O/triethanolamine = 4/1/1 in volume) at 25 °C under visible light irradiation (≥420 nm). (b) Schematic energy-level diagram showing the electron transfer from [Ru(bpy)3]Cl2 to the Fe-SAs/N-C catalyst. EF: Fermi level; LUMO: lowest unoccupied molecular orbital; HOMO: highest occupied molecular orbital. (c) Schematic process for the photocatalytic reaction using [Ru(bpy)3]Cl2 as a light absorber and Fe–SAs/N–C as a catalyst. Reproduced and modified with permission.51 Copyright 2020, American Chemical Society. (d) The preparation of CoN4–SiO2 nanoparticles and the photocatalytic system. Reproduced and modified with permission.112 Copyright 2019, Royal Society of Chemistry. (e) Energy-level diagram of [Ru(bpy)3]Cl2 + MnSAs. (f) Schematic showing the photocatalytic process of [Ru(bpy)3]Cl2 + MnSAs. Reproduced and modified with permission.111 Copyright 2020, Elsevier. | ||
Previous reports suggested that (cobalt) Co is the promising active centre for CO2 reduction under light irradiation.167 In addition, the unique electronic structure and appealing properties of Co-SAC manifested an outstanding performance toward photocatalytic CO2 reduction. Hu et al.112 demonstrated photocatalytic CO2 reduction to syngas based on Co-SAC featuring a Co-N4 active core centre onto an aminated SiO2 support in association with g-C3N4 as a light harvester (Fig. 6d). 3-(Trimethoxysilyl) propan-1-amine (APTMS) on the SiO2 provided a very strong coordination environment for anchoring Co atoms into the core. The X-ray absorption spectroscopy (XAS) technique was applied to further study the coordination core structure of CoN4. The as prepared CoN4-SiO2 catalyst revealed outstanding stability and excellent photocatalytic activity for CO2 photo-reduction to syngas with production rates of CO 398 μmol g−1 and H2 804 μmol g−1. Of note, the CO/H2 ratio in the syngas mixture was maintained from 1:1 to 1:2.
Heteroatom doped (N, S) organic semiconducting polymers including polypyrrole, polyaniline and polythiophene are fascinating choices to fabricate the porous heteroatom doped carbon matrix by impregnating SACs. Doped heteroatoms into the carbon matrix provide an extra stabilization of SACs via simple Lewis acid–base interactions compared to the simple carbon matrix. Owing to this, recently, Yang et al.111 successfully devised an in situ polymerization approach to disperse Mn SACs over the polymeric N-doped carbon matrix. In general, anchored Mn atoms over the N-doped carbon matrix were extracted from MnO2 during in situ polymerization of pyrrole monomers. The resultant uniformly dispersed Mn single atom over the N-doped carbon matrix was exploited as a co-catalyst to realize the photocatalytic reduction of CO2 to syngas under visible light irradiation using [Ru(bpy)3]Cl2 as a light harvester (Fig. 6e and f). Apparently, the gas evolution rates of CO and H2 were estimated to be 1470 μmol h−1 g−1 and 1310 μmol h−1 g−1, respectively and the CO/H2 ratio in the syngas mixture was tuned from 1.12 to 0.43.
Of note, the unique electronic structure, appealing catalytic properties and strong affinity for CO2 make SACs a potent candidate for photocatalytic syngas production via CO2 reduction. Atomically dispersed metal active sites over the supportive surface can maximize the catalytic efficiency. It remains necessary to fabricate a variety of morphologically tuned SAC photocatalytic systems for the excessive high production of syngas with tuneable CO/H2 ratio for further implication in industrial applications.
3.7. Metal-complex photocatalyst for syngas production
Pioneering research by Lehn and co-workers embarked on the photocatalytic CO2 reduction by employing transition metal complexes as homogeneous catalysts.168,169 In recent years, various metal complex catalysts featuring noble metals including rhenium (Re),170 ruthenium (Ru)171 and iridium (Ir)172 have been well established for photocatalytic CO2 reduction, whereas inexpensive and earth-abundant first row transition metals such as iron (Fe),102 cobalt (Co),173 manganese (Mn)174 and nickel (Ni)175 have gained tremendous attention for photocatalytic CO2 reduction and its conversion. These metal complexes and their hybrid functionalities were applied in syngas production.Yao et al.49 remarkably defined that a mono-nuclear Co complex (Co(bpy)2Cl2 as catalyst) together with Ru(bpy)3Cl2 as a photosensitizer could efficiently be eligible for CO2 reduction under aqueous media to realize co-production of CO and H2i.e. syngas mixture. The facile charge transfer over the integrated homogeneous photocatalytic system resulted in excellent yields of CO (62.3 μmol) and H2 (69.9 μmol), and the corresponding turnover numbers (TONs) reached 6230 and 6990, respectively, under visible-light irradiation (Fig. 7(a and b)).
 | ||
| Fig. 7 (a) Diagram of the photocatalytic reduction of CO2 and H2O into CO and H2 by the catalyst Co (bpy)2Cl2. (b) Simplified energy levels and proposed electron transfer processes in the photocatalytic systems. Reproduced and modified with permission.49 Copyright 2017, Elsevier. (c) H2/CO ratio under the CO2-saturated CH3CN solution (5 mL) containing [Co5(btz)6(NO3)4(H2O)4] (0.08 μmol), [Ru(bpy)3]Cl2 (0.01 mmol) and TEOA (1 mL) at 20 °C and irradiated by λ > 420 nm. (d) Generation of CO and H2 for [Co5(btz)6(NO3)4(H2O)4] with 4 cycles under the CO2-saturated CH3CN solution (5 mL) containing [Co5(btz)6(NO3)4(H2O)4] (0.08 μmol), [Ru(bpy)3]Cl2 (0.01 mmol) and TEOA (1 mL) at 20 °C and irradiated by λ > 420 nm. Reproduced and modified with permission.50 Copyright 2020, Elsevier. | ||
Alsabeh et al.102 reported a non-precious iron metal complex catalysed syngas production via photoreduction of CO2. In particular, the photocatalytic reaction was triggered using tri-nuclear [Fe3(CO)12] complexes together with either [Ru(bpy)3]Cl2 (PS1) or [Ir(ppy)2(bpy)]PF6 (PS2) photosensitizer in the presence of visible light irradiation. In most cases, either a 1:1 CO:H2 ratio was observed or the selectivity was inclined slightly towards CO with combined TONs reaching nearly 100.
Poor stability of the mononuclear metal-complexes limits their practical use in industry applications. Stretching the research in the development of stable multinuclear metal complexes as a homogeneous catalyst is one of the advantageous protocols, accounting for an enhanced stability of the catalysts compared to mononuclear metal complexes. Recently, Sun et al.50 explicitly demonstrated an appealing activity of a cobalt-based complex (multi-nuclear) in CO2 reduction to syngas. For instance, they employed a pentanuclear complex [Co5(btz)6(NO3)4(H2O)4] (1, btz = benzotriazolate) as the homogeneous catalyst coupled with [Ru(bpy)3]Cl2 as a photosensitizer for visible-light-mediated CO2 reduction to syngas, which further resulted in a high stability and reactivity in both pure and diluted CO2. Compared to the mono nuclear cobalt complex, pentanuclear [Co5(btz)6(NO3)4(H2O)4] ascertained 219.8 μmol yield (2748 TONs) of syngas in pure CO2 under visible light irradiation, which was 212-fold that of a mononuclear cobalt complex. The catalytic activity was maintained up to 200 h, which was manifold that of most reported homogeneous molecular catalysts (Fig. 7(c and d)). In addition, high reactivity was also achieved in diluted CO2 content (5%). The ratio of H2/CO in syngas was considerably varied from 16:1 to 2:1.
Immobilizing the homogeneous catalysts onto the solid support is an alternative approach for stabilizing the catalysts. To consolidate this, Aoi et al.103 defined a photocatalytic system for photo-reduction of CO2 into CO and H2 by using a cobalt(II)chlorin complex adsorbed on multi-walled carbon nanotubes as a CO2 reduction catalyst and [Ru(II)Me2phen)3]2+ (Me2phen = 4,7-dimethyl-1,10-phenanthroline) as a photocatalyst to achieve the generation of CO and H2 with a ratio of 2.4:1 with a high turnover number of 710. This study presented a phenomenal route for CO2 reduction to syngas using an earth-abundant metal complex catalyst under visible light irradiation.
3.8. Metal–organic framework photocatalyst for syngas production
Metal–organic frameworks (MOFs) and their derivatives have been extensively studied as excellent catalysts for efficient CO2 adsorption and conversion due to their tuneable properties and promising catalytic performance.176,177 They are micro-mesoporous hybrid materials composed of metal ions or clusters and organic frameworks with controllable pore size distribution and high specific surface area. Both physical and chemical properties of MOFs can be easily tuned by changing the metal ions or organic linkers in the matrix. The high surface area of MOFs has given an ultimate solution for typical gas absorption and gas separation, besides heterogeneous catalysis.178 Until now, plenty of MOFs have been reported, which include different active metal-sites (such as Ti, Fe, Co, Ni, Mn, Zn or Cu) and varied organic linkers.179–182 Of note, excellent photocatalytic performance and light harvesting phenomenon of MOFs can further be improved by integrating MOFs with other functional materials to create new photoactive materials or composites.A number of studies have manifested that CO2 can be efficiently reduced into CH4 and CO via MOF assisted photocatalysis.183,184 However, in this section particularly we will discuss only photocatalytic CO production synergistically with H2 production for syngas generation from the CO2–H2O mixture.
Ru-based MOFs have shown splendid importance towards CO2 to CO production.185 However, incorporation of photosensitizers or single metallic sites can lead to an effortless photoreduction process for CO2 reduction into syngas. Cobalt (Co) has been examined to alter MOFs to boost CO2 reduction. Liu et al.39 synthesized (Co/Ru)n-UiO-67(bpydc) by a simple two-step self-assembly process to incorporate (Ru)n-UiO-67(bpydc) with Co metallic sites. Facile pumping of electrons from the ligand to metal (Co) accelerates the activity of the (Co/Ru)n-UiO-67(bpydc) photocatalyst towards efficient syngas production via reduction of CO2 and H2O with a yield of 13600 μmol g−1 in 16 h, which was much higher i.e. 29.2-fold as compared to its homogeneous analogues (Fig. 8a). However, the H2:CO ratio (2:1) was maintained by adjusting the Co/Ru ratio of 2.4 with 10% water content in the photocatalytic system (Fig. 8b). This work highlighted the importance of MOF functionalization via simple metal incorporation and provided a new perspective for the tuneable syngas production.
 | ||
| Fig. 8 (a) Proposed mechanism for photocatalytic syngas production with (Co/Ru)n-UiO-67(bpydc) as the catalyst under the visible light irradiation. (b) Time profile of H2 and CO evolution rate. Reproduced and modified with permission.39 Copyright 2019, Elsevier. | ||
Zeolitic imidazolate frameworks (ZIFs) are typical MOFs that are composed of tetrahedrally coordinated transition metal ions (e.g. Zn, Co, Fe, and Cu) linked via imidazolate linkers. ZIF-9 and ZIF-67 have shown critical CO2 photoreduction to CO.186–188 However, different studies suggested that incorporation of active metallic sites including Zn, Ni, and Co could play a promotional role in H2 evolution and CO2 reduction to CO. For example, for the first time Wang et al.107 reported a phenomenal photocatalyst for CO2 photoreduction by incorporation of Co-ZIF-9, a novel hexa-nuclear Co active site containing metal–organic framework (Co6-MOF) as a co-catalyst with [Ru(bpy)3]Cl2·6H2O as a photosensitizer. The photolysis of CO2via the photosensitized-MOF bestowed an excellent yield of 41.8 μmol CO and 29.9 μmol H2. In addition, Mu et al.106 prepared carbonized bimetallic ZIFs (C-BMZIFs) with different Zn/Co ratios through the pyrolysis of bimetallic Zn–Co ZIFs at 700 °C under an inert atmosphere as shown in Fig. 9 a. Herein, C-BMZIFs were utilized as a co-catalyst with [Ru(bpy)3]2+ as the photosensitizer for the photoreduction of CO2 to syngas (CO + H2). Among all the C-BMZIFs, the carbonized ZIF composite having a stoichiometry Zn/Co ratio of 5:1 demonstrated the highest TOF of CO2 conversion i.e. 9.9 × 10−3, 23-times larger than that of simple C-ZIF-67; on the other hand, the C-BMZIF with a Zn/Co ratio of 3:1 brought out a highest CO production rate of 1.1 × 104 μmol g−1 h−1 (Fig. 9b and c). Of note, it is curious to know that catalytic structure–selectivity relationship for tuneable syngas production with desirable CO/H2 ratio could be ascertained by the composition and the size of the active metallic site. Therefore, in C-BMZIFs, smaller Co active moieties favoured higher CO production, and H2 evolution was preferentially much higher on larger Co active moieties. Lastly, the CO/H2 ratio in the generated syngas was rationally tuned between 1.9 and 0.7.
 | ||
| Fig. 9 (a) Schematic illustration of the synthesis of C-ZIFs and the role of Co active sites in photocatalysis. (b) The corresponding TOFs based on the mass percentage of Co quantified by EDX. (c) The total evolution of CO and H2 within the 3 h photocatalytic reaction for C-BMZIFs with various Zn/Co ratios. Reproduced and modified with permission.106 Copyright 2018, Royal Society of Chemistry. | ||
3.9. Organic polymer photocatalyst for syngas production
Organic polymeric photocatalysts offer a unique and molecular-level structural layout of their optoelectronic and surface photocatalytic properties.189–191 Over the period of time, organic polymers including carbon nitrides,192 porous organic polymers,193 covalent triazine frameworks194 and covalent organic frameworks195 have undergone an impressive development in their potential to propel photo catalytic CO2 reduction to syngas. In comparison to inorganic photocatalysts, CO2 reduction to syngas over organic photocatalysts is still in the infancy stage.Carbon nitrides (PCN) are exceptional organic conjugated polymeric photocatalysts that have gained tremendous laurels primarily owing to their high chemical stability, easy synthesis from inexpensive precursors and suitable CB and VB positions, straddling the reduction potential of protons, CO2 and water oxidation.97,196 Despite the huge acclamation of PCN as a photocatalyst in the field of CO2 reduction, the photocatalytic efficiency of CN-based photosystems for CO2 reduction to syngas remains moderate. Mainly, the H2/CO ratio of reported PCN systems is uncontrollable. In this sense, development of CN-based photosystems has to be revamped to improve the efficiency of CO2 reduction to syngas and control the H2/CO ratio in the generated syngas.
In a latest report, Yang et al.42 demonstrated that defects such as nitrogen vacancies (NVs) intensify the structure–activity relationship between the PCN and CO2, manifesting exceptional photocatalytic activity for syngas production under visible light. In this work, numerous characterization techniques (XPS, EPR and in situ DRIFTS) with the combination of DFT calculations have been adopted, confirming that the NVs could possibly originate from surface N–(C)3 sites of PCN, which can promptly amplify the activation and reduction of CO2, while lowering the formation energy barrier for COOH* intermediates. Incredibly, it was seen that the syngas production activity accelerated nearly 10 times higher in the case of defect rich PCN (NVs-PCN) than that of pristine PCN under identical reaction conditions. In addition, the H2/CO ratio in syngas can be tuned from 0.24:1 to 6.8:1 by controlling the concentration of NVs. For that reason, it could explicitly be concluded that defects (NVs) over the PCN surface provide a modulation strategy to develop defect rich PCN based photocatalysts, showing huge advancement in structure–activity relationship for highly efficient CO2 reduction to syngas with tuneable H2/CO ratio.
COFs are highly porous, crystalline, and extended two- or three-dimensional (2D or 3D) ordered structures, constructed from organic building blocks and connected via covalent bonds. Excellent structural tenacity and diverse functionalities provide phenomenal physico-chemical stabilities with intriguing semiconducting properties in various photochemical reactions.197,198 Photocatalytic reduction of CO2 is one the challenging photochemical reactions. In recent years, COFs have emerged as a new class of organic porous semiconducting materials for CO2 photoreduction into various hydrocarbons and fuel fractions.195 However, photocatalytic CO2 reduction to syngas over COFs is still in infancy. Only few reports are available regarding the syngas production over COFs.
Very recently Fu et al.108 showed that a rhenium complex [Re(bpy)(CO)3Cl] coupled with a crystalline covalent organic framework (COF) i.e. Bpy-sp2c-COF afforded a much stronger CO2 absorption affinity and improved CO2 reduction over a bare [Re(bpy)(CO)3Cl] complex under visible light irradiation. However, [Re(bpy)(CO)3Cl incorporated Bpy-sp2c-COF resulted in a maximum rate of 1040 mmol g−1 h−1 for CO production with 81% selectivity, which was further increased to 86% with the increased production rate of CO up to 1400 mmol g−1 h−1, when a dye was added as a photosensitizer. Apparently, it was shown that addition of platinum (Pt) favoured the co-production of CO and H2, i.e. syngas. Moreover CO:H2 ratio in the syngas was adjusted in the range from 4:1 to 1:10 by adding different amounts of Pt over COF.
Importance of Co–metal complexes has already been discussed for CO2 photoreduction to syngas. However, the co-operative effect of these Co-based metal complexes with COFs could be rationalized for the syngas production. Triazine based COFs have unique features for CO2 reduction. He et al.199 synthesized an efficient photocatalytic system by integrating [Co(bpy)3]2+ as an active Co single site in covalent triazine frameworks (CTFs) for the photocatalytic production of syngas from CO2 reduction in aqueous media. Incorporation of Co single sites with CTFs synergistically enhanced the light absorption of the CTFs and enabled the excellent syngas (CO/H2 = 1.4:1) production with a corresponding yield of 3303 μmol g−1 in 10 h, which was almost 3-fold higher than that of bare CTF without Co single sites.
Moreover, it has been found that the co-operative effect between the d-block elements such as Fe51 and Ni200 could feature in bimetallic active centres, which resulted in pronounced/improved photoreduction of CO2. Aiming towards it, very recently, Han et al.41 rationally customized a COF by integrating Fe/Ni metal sites over the surface, which was successfully utilized for photoreduction of less concentrated CO2 into tuneable syngas (Fig. 10a–c). However, facile metal site (Fe/Ni) composition alteration and its adsorption affinities for CO2 and H2O manifested an encouraging parameter to tune the CO/H2 ratio, which was effectively tuned ranging from 1:19 to 9:1. This work demonstrated an ample choice for modulation in COF surface via bi-metallic active sites (Fe/Ni) for syngas production, which was further underpinned by experimental and theoretical studies.
 | ||
| Fig. 10 (a) Synthesis of M-COFs with 2,4,6-triformylphloroglucinol (Tpg) and 2,5-diaminobenzenesulfonic acid (Dbsa) as the precursors. (b) The dependence of CO/H2 ratios of metals in a series of Fe/Ni-COF samples in low-concentration CO2. (c) Schematic scheme for CO2 reduction into CO and H2. Reproduced and modified with permission.41 Copyright 2020, Wiley-VCH. | ||
Porous Organic Polymers (POPs) are being investigated extensively as a consequence of high/excellent porosity, thermal and chemical stability, adjustable composition, and diverse functionalization. POPs are highlighted as competitive candidates in various applications.190,201 Rationalizing POPs for photocatalytic CO2 reduction to fuels/chemicals enlightens the development route for POPs as well as CO2 reduced products.193 This could be justified as structural modulation of POPs alters the CB-VB positions, and thus accordingly changes the generation of CO2 reduced products.
Keeping in view of the above observation, Yao et al.120 demonstrated that Fe metal incorporated POPs i.e., ferric porphyrin-based POPs (i.e., POP1-Fe and POP2-Fe) could rationally be exploited for photocatalytic CO2 reduction to syngas. The POP1-Fe and POP2-Fe were fabricated using porphyrins and adjustable benzene/biphenyl as linker units. Importantly it was found that the inclusion of biphenyl linker in the ferric porphyrin system results in extended π-conjugation, enabling a lower CB potential suitable for CO generation in POP2-Fe. Moreover, experimental results confirmed that ferric porphyrin sites were responsible for CO generation, while the uncoordinated porphyrin units promoted the H2 generation. By changing the phenyl linker to the biphenyl linker, the ratio of CO/H2 was adjusted from 1:1 to 1:1.5 in POP2-Fe and at 450 nm wavelength, the ratio of CO/H2 was found to be 1:2. Such studies provide insights into the synthetic strategy for POP structure–activity performance for CO2 reduction towards the selective formation of syngas with tuneable CO/H2 ratios through facile regulatory linkers.
These works explicitly demonstrated a tremendous potential of organic polymers for the photocatalytic production of syngas via CO2 photoreduction. In addition, these studies also manifest a profound knowledge of cooperative effects of active metal sites, linkers and metal complexes with high accessibility due to their high surface area, resulting in high light absorption, facile charge transfer and efficient CO2 photo-reduction to syngas.
4. Photocatalytic syngas production: a mechanistic insight
A mechanistic insight towards syngas production via CO2 photoreduction pathways corroborates the reaction kinetics/dynamics and also underpins the rational modifications of photocatalysts for efficient production of syngas with a wide range of tunability in the CO:H2 ratio. To comprehend this, various advanced characterization techniques are quite beneficial to uncover the photocatalytic reaction process and also provide a structure–activity relationship between photocatalysts and molecular CO2. In addition, isotope labelling experiments and theoretical studies (DFT calculations) also provide a deep insight towards reaction pathways, which further solidifies the experimental conditions and outcomes.
4.1. In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS)
Recently, the in situ DRIFTS technique has been widely adapted to provide a deep insight into the reaction intermediate product generation and reaction pathways during the CO2 photoreduction, helping to investigate the plausible reaction mechanisms.202 Fundamentally, CO2 half reaction includes the following steps: (i) first the adsorption of CO2 molecules over the surface of photocatalysts; (ii) followed by activation to give a carboxyl intermediate COOH* (CO2* + H+ + e− → COOH*); (iii) the reduction and dissociation of carboxyl intermediate COOH* to CO* via a proton-electron transfer reduction process (COOH* + H+ + e− → CO* + H2O); and (iv) the desorption of CO (CO* → CO).NVs have shown a preferential active role in CO2 adsorption–activation over the PCN. As shown in Fig. 11 a, for CO2 reduction to CO over the defect rich PCN (NVs-PCN), the FTIR band at 2350 cm−1 can be attributed to the symmetric stretching vibration of CO2. The intensity of the observed bands in the range from 1300 to 1800 cm−1 of PCN-23 (defect rich PCN) is quite prominent compared to pristine PCN, manifesting that existence of NVs over the PCN promotes the adsorption–activation of CO2. Moreover, under the prolonged visible light irradiation the peaks from 1300 to 1800 cm−1 of PCN-23 are more in intensity, featuring an improved CO2 activation from photogenerated electrons. In particular, a new peak around 1559 cm−1 upon visible-light irradiation corresponds to COOH*, generally originating from co-adsorbed molecules of CO2 and H2O, ensuring that activated COOH* is one of the key intermediates during CO2 reduction to CO occurring via a 2 electron reduction process.42
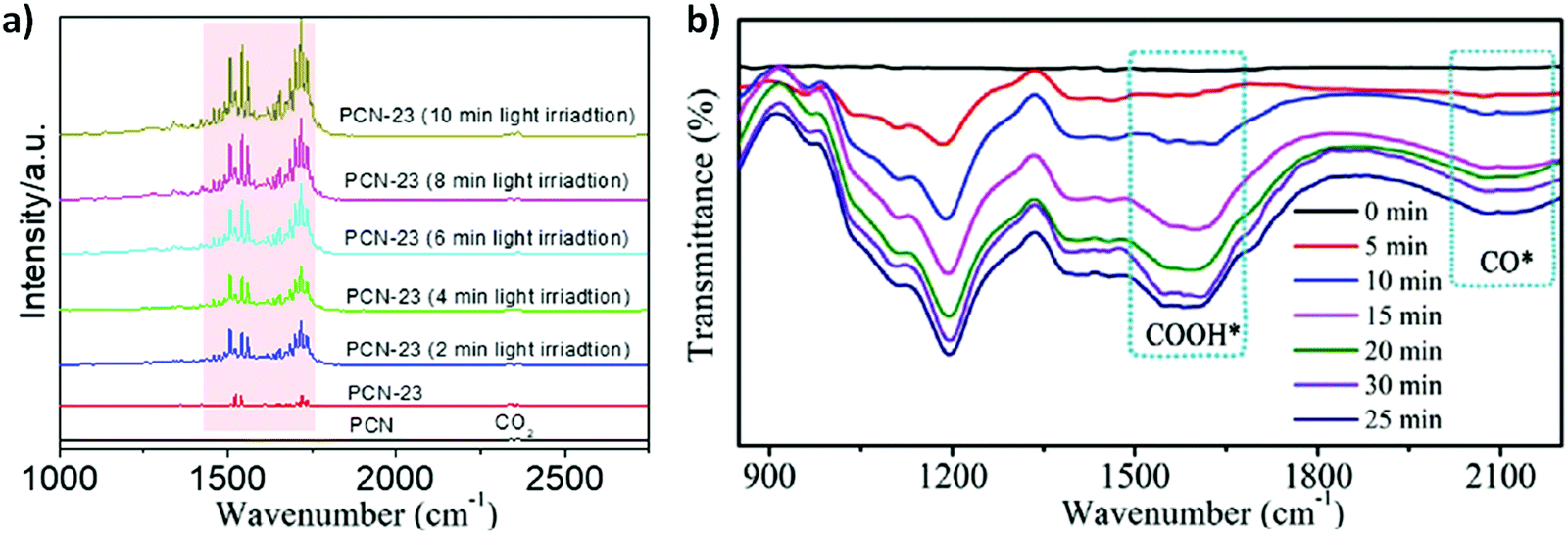 | ||
| Fig. 11 In situ DRIFTS spectra illustrating the photocatalyzed CO2 adsorption–activation over the surface of (a) PCN and defect rich PCN-23 (reproduced and modified with permission.42 Copyright 2021, Elsevier) and (b) 3DOM CdSQD/NC (reproduced and modified with permission.20 Copyright 2021, Wiley-VCH). | ||
Mechanistic investigation for photocatalytic CO2 to CO reduction over the surface of 3D ordered macroporous N-doped carbon (NC) supported CdS quantum dots (3DOM CdSQD/NC) was also performed by in situ DRIFTS analysis.203 It is shown in Fig. 11b that with an extension of visible light irradiation, new peaks at 1200 cm−1 and 1556 cm−1 appeared, in which the intensity was gradually increased, signifying the generation of the COOH* intermediate over the photocatalytic surface. For the time being, a new peak at 2091 cm−1 appeared and an increased intensity in the peak was observed with visible light irradiation time, conferring the production of CO as the final product.
4.2. In situ electron paramagnetic resonance (EPR)
The efficacy of the photocatalysts for CO2 photoreduction to CO could be facilitated by an easy electron transfer process. Moreover, the electron transfer process from the surface of the photocatalysts to CO2 molecules pronounced the excellent structure–activity relationship, which is beneficial and provides an information for the generation of intermediates during the photoreduction reaction under the light irradiation. The electron transfer process and active intermediate formation during CO2 to CO reduction under visible light irradiation over the NH2-Uio-66(Zr) photocatalyst was investigated by in situ EPR.204 As shown in Fig. 12, when visible light was shed over H2ATA, an ESR signal with a g value of 2.004 was observed, which can emerge due to the spatially confined –NH2 groups. In NH2-Uio-66(Zr), a new ESR signal appeared at g = 2.002 with increased intensity compared to H2ATA under light irradiation, signifying that the new signal can be ascribed to ZrIII. On the other hand, in the case of Uio-66(Zr) no ESR signal was observed on visible-light irradiation. These findings explicitly suggested that the ZrIII can only be generated over the visible light irradiated NH2-Uio-66(Zr) photocatalyst (Fig. 12a). Of note, it was observed that the ESR signal corresponding to ZrIII was quenched as the NH2-Uio-66(Zr) photocatalyst was irradiated under visible light in the presence of CO2 (Fig. 12b). This remarkably proved that the photocatalytic reduction of CO2 over NH2-Uio-66(Zr) involves the photogenerated ZrIII species, synergizing the CO2 activation and reduction by ZrIII species. | ||
| Fig. 12 (a) EPR spectra of NH2-Uio-66(Zr), Uio-66(Zr), and H2ATA under visible light irradiation. (b) EPR spectra of NH2-Uio-66(Zr) in different atmospheres under visible light irradiation. Reproduced and modified with permission.204 Copyright 2013, Wiley-VCH. | ||
4.3. Isotopic (13C) labelling
Furthermore, the mechanistic pathway of the reaction could further be well examined by isotope labelling experiment, which can be helpful in defining the origin of the products. As in the case of CO2 photoreduction to syngas, isotopic labelling experiments using 13CO2, D2O or H218O and identification of their reduced products by gas chromatography/mass spectrometry (GC-MS) provide concrete evidence of the reactant sources.7Feasible CO2 adsorption and facile charge transfer over the ultrathin ZnAl-LDH nanosheets with Zn ion vacancies remarkably showed the excellent activity for the photoredution of CO2 to CO in the presence of H2O vapor. An isotopic labelling technique was further adapted in order to identify the source of the originated CO. Photocatalytic reduction of isotopically labelled 13CO2 in the presence of H2O was performed over the ZnAl-2 photocatalyst.205 From Fig. 13 a, it is evident that using 13CO2 as a gas feed, a peak at m/z 29 corresponding to 13CO was observed after 10 h of irradiation, which was subsequently repeated for 4 cycles. On comparison with the results of 10 h light irradiation, the amount of 13CO increased in the case of 20 h light irradiation. This result confirmed that the production of CO was mainly resulted from the photocatalytic reduction of CO2 over ultrathin ZnAl-2 nanosheets. Similarly, isotopic labelling experiments were also performed over Ni single atoms on defect rich zirconia (Ni-SA-x/ZrO2) for photocatalytic CO2 reduction to CO under Xe lamp irradiation.202 Isotopic labelling experiment substantiated that the generation of CO was derived from CO2 reduction and with increasing time the amount of labelled 13CO (m/z =29) was also continuously increasing with irradiation time (Fig. 13b).
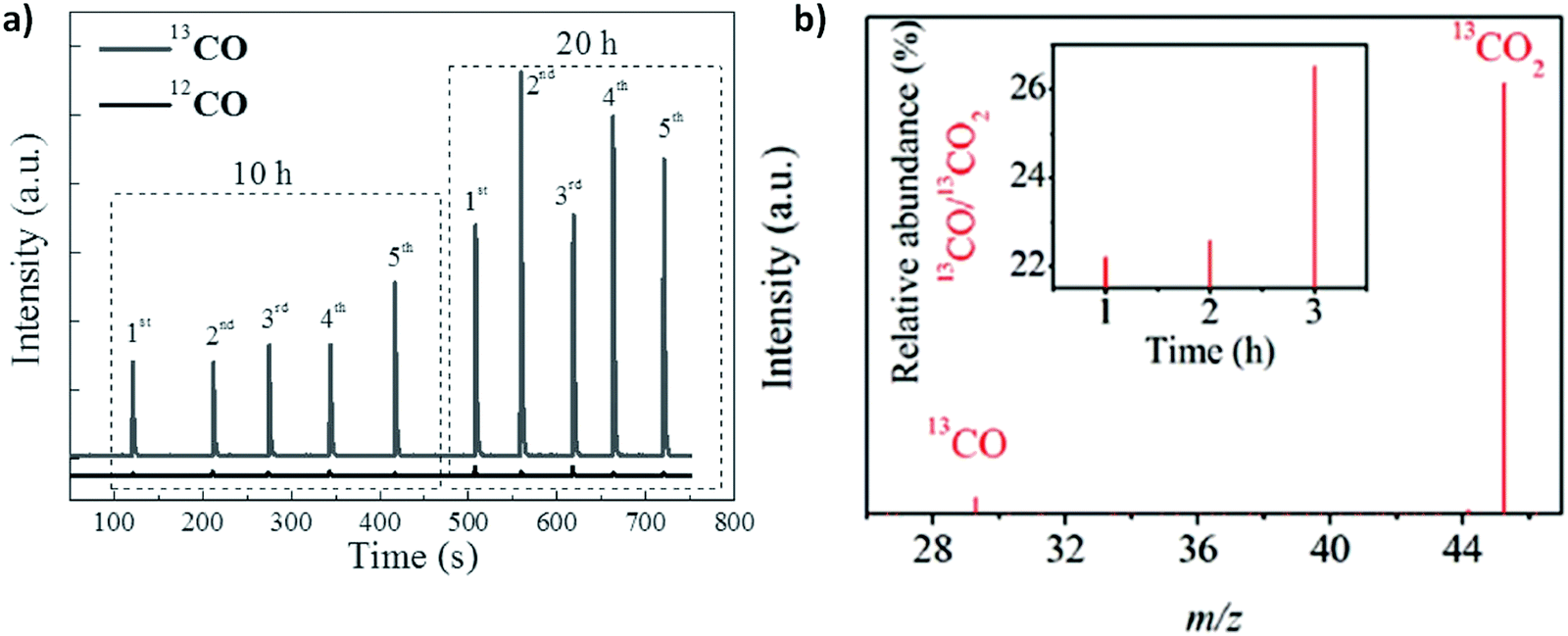 | ||
| Fig. 13 (a) Mass spectra of repeated analysis (m/z 29, 28) after 10 and 20 h of irradiation for the reduction of 13CO2 in the presence of H2O vapor over ZnAl-2. The weak peak at m/z 28 assigned to 12CO was also detected due to the impurity of the 13CO2 raw gas (99%). Reproduced and modified with permission.205 Copyright 2015, Wiley-VCH. (b) Mass spectrum of 13CO (m/z = 29) generated from the photoreduction of 13CO2 over Ni-SA-5/ZrO2 (inset: time profile of the relative abundance of 13CO/12CO). Reproduced and modified with permission.202 Copyright 2020, Wiley-VCH. | ||
4.4. Theoretical or DFT calculations
In addition to isotopic labelling and characterization techniques (e.g. in situ EPR, and in situ DRIFTS), DFT calculations may also bestow a significant improvement to obtain insight into the possible reaction pathways for CO2 photoreduction. Furthermore, the band structure of a photocatalyst and the interactive mechanism of CO2 molecules over the surface of the photocatalyst could possibly be examined with the DFT calculations prior to the practical experiments. Atomic level mechanistic interpretation of the CO2 photoreduction to CO over Ni-SA-5/ZrO2 was scrutinized by DFT calculations.202 Theoretical calculations and modeling were based on ZrO2(010) facets as shown in Fig. 14a. The lowest energy barrier pathway (<1 eV overall) for CO2 conversion to CO was estimated over Ni-SA/O. This model described that the adsorption of *COOH onto the photocatalyst was the rate-limiting step (highest energy barrier), while the subsequent transformation of *COOH to *CO likely to occur over a low energy barrier. Moreover, a deep insight towards the electron migration pathways involved in the photoreduction of adsorbed CO2 on Ni–SA/O was provided by the differential charge density diagrams shown in Fig. 14b. Firstly, the activation of CO2 molecules (steps 1 and 2) took place via electron transfer from isolated Ni sites to the π* orbital of CO2. After that, the generated H+ (via photocatalytic water splitting) reacted with the *CO2 intermediate, manifesting the production of the *COOH intermediate (steps 3 and 4). Further subsequent electron transfer promoted the disintegration of *COOH to *CO, followed by CO desorption from the photocatalytic surface (steps 5 and 6).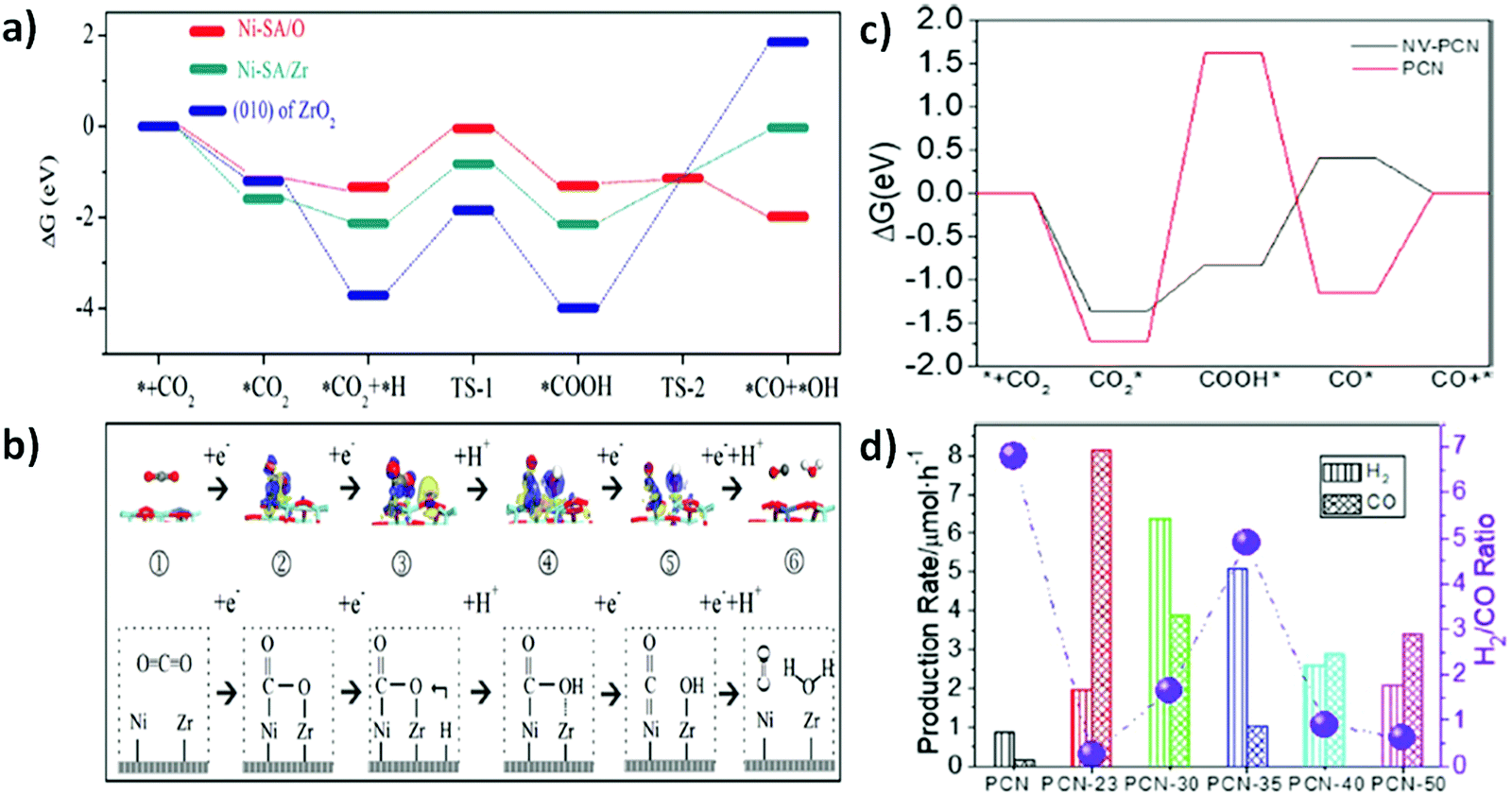 | ||
| Fig. 14 (a) Energy profile for CO2 reduction to CO over the (010) facets of ZrO2, Ni-SA/Zr, and Ni-SA/O. (b) Differential charge density diagrams and intermediates during CO2 reduction to CO over the Ni-SA/O model. Reproduced and modified with permission.202 Copyright 2020, Wiley-VCH. (c) Gibbs free energy diagram for PCN and NV-PCN for CO2 reduction to CO, and (d) photocatalytic activity for CO2 reduction over PCN and various defect-rich NV-PCN photocatalysts. Reproduced and modified with permission.42 Copyright 2021, Elsevier. | ||
In addition, as earlier it was mentioned that NVs in the PCN promote the CO2 adsorption–activation during the photoreduction steps involved in CO2 to CO production. The Gibbs free energy (ΔG) diagram of CO2-to- CO conversion over pristine PCN and NV-PCN is shown in Fig. 14c. ΔG values of pristine PCN were found to be much higher than that of NV-PCN, conferring that the CO2 activation to produce COOH* intermediates over NV-PCN is more favorable due to the low energy barrier. These theoretical studies confirmed that the NVs can significantly promote the activation–reduction of CO2 which is well matched with the experimental studies (Fig. 14d).42
5. Conclusion and future perspective
In recent decades, rising CO2 levels in the atmosphere have accelerated global warming and the energy crisis across the world. Deterring the CO2 levels in the atmosphere requires an urgent solution. Among the various techniques used, the photocatalytic conversion of CO2 to fuels or value-added chemicals may be a promising solution. Aiming at this, photocatalytic CO2 reduction to chemicals or fuels such as CO, CH4, HCHO, and CH3OH is an exceptionally sustainable process.The co-production of CO and H2 (syngas) via the solar-driven reductive transformation of CO2 under aqueous (H2O) media has been considered one of the most beneficial solutions. Syngas is a crucial intermediate for the production of synthetic fuels such as hydrocarbons, methanol, alcohols and fuel additives via the Fischer–Tropsch reaction.
Generally, the production of syngas predominantly depends on the electrochemical and thermochemical reduction of CO2 or/and H2O, operating at relatively high temperatures and pressures. On the contrary, photochemistry, a renewable strategy, advances the pathway for the production of syngas via the reduction of CO2 in aqueous media. In this review, therefore, we have attempted to introduce an ideal renewable pathway for the production of syngas via the photoreduction of CO2 under aqueous media. In addition, various photocatalysts such as metal oxides, LDHs, metal complexes, SACs, POMs, MOFs and COFs as well as structural engineering of the photocatalysts and their relative activity for syngas production with tuneable ratios of CO/H2 have been deliberately discussed.
Of note, solar-driven CO2 reduction under aqueous solution results in recent advancements towards syngas production. This could be summarized by the following points: (1) this reaction may require a boundless source of energy i.e. solar light; (2) this reaction can be initiated by only H2O and CO2 molecules and (3) requires comparatively ambient conditions such as low temperature and pressure. These are the superlative advantages laying an ideal and pioneering road map for the development of the sustainable production of syngas via CO2 photoreduction.
Moreover, in this review, the discussed photocatalysts have shown prodigious potential for syngas production via CO2 reduction. Utilization of such a photocatalytic system has witnessed great scientific advancements so far. TiO2-Based photocatalysts have been scrutinized the most for CO2 reduction into syngas. Nano-engineering of TiO2 photocatalysts by metal doping, heterostructure construction and morphological tuning affords excellent chemical and physical properties for the efficient production of syngas via CO2 reduction. Furthermore, the efficacy of the other photocatalysts including mixed metal oxides, LDHs, SACs, metal complexes, POPs, MOFs and organic polymers has also shown encouraging progress in recent years for the sustainable production of syngas via CO2 reduction (Scheme 1a).
 | ||
| Scheme 1 (a) Schematic illustration showing the various catalysts involved in the generation of syngas under light illumination. (b) Schematic diagram showing the overall essential considerations required for syngas production and the challenges that need to be overcome. | ||
In the latest inception of CO2 sorption and capture (CSC) technology, POMs, MOFs and COFs have set a promising paradigm for a highly sustainable process. Their high surface area, enhanced light harvesting ability, rigid 3D structure, and unique electronic features with tuneable band positions offer an excellent photocatalytic route for syngas generation via CO2 photoreduction.
Herein, it is indispensable to point out that despite several recent advances, the related development in photocatalytic syngas production via CO2 reduction is still in its infancy. Plenty of opportunities are available and many challenges need to be overcome and addressed. In this regard, the engineering of efficient, robust and low-cost photocatalysts with tuneable band structure is required to maximize the light absorption, improve charge separation, and achieve high efficiency for both fundamental research and the large-scale production of syngas via CO2 photoreduction.
Currently, in industries, syngas is produced on thermo-catalytic operational giant plants or reactors operating at relatively high temperatures and pressures. Replicating similar yields with photocatalytic plants or glass panel reactors for syngas production is highly challenging. In addition, compromised or low yields of the products and the relatively high cost of production may impede the practical application of bulk syngas production. Therefore, initial additional efforts such as glass panel reactors must be built with a focus on sufficient robustness to ensure long-term outdoor operation (under natural sunlight) to prepare the process for industrial use. Moreover, importantly, the ratio of syngas products i.e. CO/H2 plays a pivotal role in the production of upgraded fuels via the Fischer–Tropsch reaction. Therefore, the requisite of tuneable CO/H2 molar ratio in syngas is significantly crucial and should be addressed.
As mentioned above, the generation of syngas via CO2 photoreduction requires quite efficient, stable and visible light-absorbing photocatalysts. To view this mechanistically, the photocatalysts must first effectively promote CO2 adsorption–activation over the surface of photocatalysts. Structural engineering in photocatalysts, such as through surface vacancies, 2D-2D heterojunctions, porosity, and co-catalyst loading, facilitates syngas generation. In particular, it is of great importance to employ working techniques (e.g., in situ XPS, in situ EXAFS, in situ XANES, in situ DRIFT, and in situ PL) to examine any change in the physicochemical properties of photocatalysts to understand the mechanisms of interfacial charge separation and transfer as well as photocatalytic syngas generation over the surface via CO2 reduction. In addition, theoretical studies could further be employed to obtain deep insight into the structure–activity relationship between the photocatalysts and CO2 molecules, thus providing theoretical guidance for the design of high performance CO2 reduction photocatalysts for syngas generation (Scheme 1b).
In closing, the concept of renewable syngas production via photocatalytic CO2 reduction provides a new pathway towards sustainable fuel production that is still in its infancy. With the established advancement in the Fischer–Tropsch process for the synthesis of fuels, it remains to be seen where the photocatalytic process will lead to in the future. This may turn out to be a milestone in the field of sustainable energy production for the betterment of humankind.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
D. K. Chauhan and N. Sharma thank INST Mohali for financial support. Dr K. Kailasam thanks the Department of Science and Technology, India (DST) for the DST Nano Mission NATDP funded Technology Project, File No. SR/NM/NT-06/2016 for the financial support.References
- R. Pang, K. Teramura, M. Morishita, H. Asakura, S. Hosokawa and T. Tanaka, Commun. Chem., 2020, 3, 137 CrossRef CAS.
- T. Banerjee, F. Podjaski, J. Kröger, B. P. Biswal and B. V. Lotsch, Nat. Rev. Mater., 2021, 6, 168–190 CrossRef CAS.
- M. A. Raza, F. Li, M. Que, L. Zhu and X. Chen, Mater. Adv., 2021, 2, 7187–7209 RSC.
- J. Ran, M. Jaroniec and S.-Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed.
- M. R. Allen, O. P. Dube, W. Solecki, F. Aragón-Durand, W. Cramer, S. Humphreys, M. Kainuma, J. Kala, N. Mahowald, Y. Mulugetta, R. Perez, M. Wairiu and K. Zickfeld, Framing and Context, in Global Warming of 1.5 °C. An IPCC Special Report on the impacts of global warming of 1.5 °C above pre-industrial levels and related global greenhouse gas emission pathways, in the context of strengthening the global response to the threat of climate change, sustainable development, and efforts to eradicate poverty, ed. V. Masson-Delmotte, P. Zhai, H.-O. Pörtner, D. Roberts, J. Skea, P. R. Shukla, A. Pirani, W. Moufouma-Okia, C. Péan, R. Pidcock, S. Connors, J. B. R. Matthews, Y. Chen, X. Zhou, M. I. Gomis, E. Lonnoy, T. Maycock, M. Tignor, T. Waterfield, 2018, in press Search PubMed.
- Z. Sun, N. Talreja, H. Tao, J. Texter, M. Muhler, J. Strunk and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 7610–7627 CrossRef CAS PubMed.
- H. Shen, T. Peppel, J. Strunk and Z. Sun, Sol. RRL, 2020, 4, 1900546 CrossRef CAS.
- L. Děkanovský, J. Plutnar, J. Šturala, J. Brus, J. Kosina, J. Azadmanjiri, D. Sedmidubský, Z. Sofer and B. Khezri, ACS Catal., 2022, 12, 1558–1571 CrossRef.
- A. Kumar, V. Hasija, A. Sudhaik, P. Raizada, Q. Van Le, P. Singh, T.-H. Pham, T. Kim, S. Ghotekar and V.-H. Nguyen, Chem. Eng. J., 2022, 430, 133031 CrossRef CAS.
- S. Wang, P. Wang, D. Shi, S. He, L. Zhang, W. Yan, Z. Qin, J. Li, M. Dong, J. Wang, U. Olsbye and W. Fan, ACS Catal., 2020, 10, 2046–2059 CrossRef CAS.
- S. Lu, Y. Shi, N. Meng, S. Lu, Y. Yu and B. Zhang, Cell Rep. Phys. Sci., 2020, 1, 100237 CrossRef.
- C. Graves, S. D. Ebbesen, M. Mogensen and K. S. Lackner, Renewable Sustainable Energy Rev., 2011, 15, 1–23 CrossRef CAS.
- S. C. Reyes, J. H. Sinfelt and J. S. Feeley, Ind. Eng. Chem. Res., 2003, 42, 1588–1597 CrossRef CAS.
- F. Urbain, P. Tang, N. M. Carretero, T. Andreu, L. G. Gerling, C. Voz, J. Arbiol and J. R. Morante, Energy Environ. Sci., 2017, 10, 2256–2266 RSC.
- D. Esposito, Nat. Catal., 2018, 1, 738 CrossRef.
- H.-Y. Wang, W.-H. Xie, D.-D. Wei, R. Hu, N. Wang, K. Chang, S.-L. Lei, B. Wang and R. Cao, ChemSusChem DOI:10.1002/cssc.202200200.
- Z. Zhao, Z. Liu, T. Wang, F. Teng, W. Jiang, J. Li, Z. Zhang and Y. Yang, J. Mater. Chem. A, 2022, 10, 2924–2931 RSC.
- S. Shoji, A. S. Bin Mohd Najib, M.-W. Yu, T. Yamamoto, S. Yasuhara, A. Yamaguchi, X. Peng, S. Matsumura, S. Ishii, Y. Cho, T. Fujita, S. Ueda, K.-P. Chen, H. Abe and M. Miyauchi, Chem. Catal., 2022, 2, 321–329 CrossRef.
- A. V. Tavasoli, M. Preston and G. Ozin, Energy Environ. Sci., 2021, 14, 3098–3109 RSC.
- V. Andrei, B. Reuillard and E. Reisner, Nat. Mater., 2020, 19, 189–194 CrossRef CAS PubMed.
- Z. Fu, A. Vogel, M. A. Zwijnenburg, A. I. Cooper and R. S. Sprick, J. Mater. Chem. A, 2021, 9, 4291–4296 RSC.
- D. Pakhare and J. Spivey, Chem. Soc. Rev., 2014, 43, 7813–7837 RSC.
- N. A.-K. Aramouni, J. G. Touma, B. A. Tarboush, J. Zeaiter and M. N. Ahmad, Renewable Sustainable Energy Rev., 2018, 82, 2570–2585 CrossRef CAS.
- Z. Li, Q. Lin, M. Li, J. Cao, F. Liu, H. Pan, Z. Wang and S. Kawi, Renewable Sustainable Energy Rev., 2020, 134, 110312 CrossRef CAS.
- L. Yuliati and H. Yoshida, Chem. Soc. Rev., 2008, 37, 1592–1602 RSC.
- T. Takayama, K. Sato, T. Fujimura, Y. Kojima, A. Iwase and A. Kudo, Faraday Discuss., 2017, 198, 397–407 RSC.
- H. Zhang, J. Ming, J. Zhao, Q. Gu, C. Xu, Z. Ding, R. Yuan, Z. Zhang, H. Lin, X. Wang and J. Long, Angew. Chem., Int. Ed., 2019, 58, 7718–7722 CrossRef CAS PubMed.
- B. Han, X. Ou, Z. Zhong, S. Liang, X. Yan, H. Deng and Z. Lin, Appl. Catal., B, 2021, 283, 119594 CrossRef CAS.
- W. Liao, W. Chen, S. Lu, S. Zhu, Y. Xia, L. Qi, M.-Q. Yang and S. Liang, ACS Appl. Mater. Interfaces, 2021, 13, 38239–38247 CrossRef CAS PubMed.
- O. Ola and M. M. Maroto-Valer, J. Photochem. Photobiol., C, 2015, 24, 16–42 CrossRef CAS.
- N.-N. Vu, S. Kaliaguine and T.-O. Do, Adv. Funct. Mater., 2019, 29, 1901825 CrossRef.
- J. Albero, Y. Peng and H. García, ACS Catal., 2020, 10, 5734–5749 CrossRef CAS.
- S. Samanta and R. Srivastava, Mater. Adv., 2020, 1, 1506–1545 RSC.
- K. Bramhaiah and S. Bhattacharyya, Mater. Adv., 2022, 3, 142–172 RSC.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- A. Behera, A. K. Kar and R. Srivastava, Mater. Horiz., 2022, 9, 607–639 RSC.
- E. Gong, S. Ali, C. B. Hiragond, H. S. Kim, N. S. Powar, D. Kim, H. Kim and S.-I. In, Energy Environ. Sci., 2022, 15, 880–937 RSC.
- M. Muringa Kandy, A. Rajeev K and M. Sankaralingam, Sustainable Energy Fuels, 2021, 5, 12–33 RSC.
- M. Liu, Y.-F. Mu, S. Yao, S. Guo, X.-W. Guo, Z.-M. Zhang and T.-B. Lu, Appl. Catal., B, 2019, 245, 496–501 CrossRef CAS.
- Y. He, X. Chen, C. Huang, L. Li, C. Yang and Y. Yu, Chin. J. Catal., 2021, 42, 123–130 CrossRef CAS.
- B. Han, X. Ou, Z. Zhong, S. Liang, H. Deng and Z. Lin, Small, 2020, 16, 2002985 CrossRef CAS PubMed.
- P. Yang, L. Shang, J. Zhao, M. Zhang, H. Shi, H. Zhang and H. Yang, Appl. Catal., B, 2021, 297, 120496 CrossRef CAS.
- H. Xu, S. You, Z. Lang, Y. Sun, C. Sun, J. Zhou, X. Wang, Z. Kang and Z. Su, Chem. – Eur. J., 2020, 26, 2735–2740 CrossRef CAS PubMed.
- H. Yang, D. Yang and X. Wang, Angew. Chem., Int. Ed., 2020, 59, 15527–15531 CrossRef CAS PubMed.
- C. Qiu, X. Hao, L. Tan, X. Wang, W. Cao, J. Liu, Y. Zhao and Y.-F. Song, Chem. Commun., 2020, 56, 5354–5357 RSC.
- X. Wang, Z. Wang, Y. Bai, L. Tan, Y. Xu, X. Hao, J. Wang, A. H. Mahadi, Y. Zhao, L. Zheng and Y.-F. Song, J. Energy Chem., 2020, 46, 1–7 CrossRef.
- P. Akhter, M. Hussain, G. Saracco and N. Russo, Fuel, 2015, 149, 55–65 CrossRef CAS.
- P. Reñones, A. Moya, F. Fresno, L. Collado, J. J. Vilatela and V. A. de la Peña O’Shea, J. CO2 Util., 2016, 15, 24–31 CrossRef.
- Y. Yao, Y. Gao, L. Ye, H. Chen and L. Sun, J. Energy Chem., 2018, 27, 502–506 CrossRef.
- M. Sun, C. Wang, C.-Y. Sun, M. Zhang, X.-L. Wang and Z.-M. Su, J. Catal., 2020, 385, 70–75 CrossRef CAS.
- Z. Wang, J. Yang, J. Cao, W. Chen, G. Wang, F. Liao, X. Zhou, F. Zhou, R. Li, Z.-Q. Yu, G. Zhang, X. Duan and Y. Wu, ACS Nano, 2020, 14, 6164–6172 CrossRef CAS PubMed.
- J. Jiang, D. Duan, J. Ma, Y. Jiang, R. Long, C. Gao and Y. Xiong, Appl. Catal., B, 2021, 295, 120261 CrossRef CAS.
- M. T. Arslan, B. A. Qureshi, S. Z.-A. Gilani, D. Cai, Y. Ma, M. Usman, X. Chen, Y. Wang and F. Wei, ACS Catal., 2019, 9, 2203–2212 CrossRef CAS.
- S. R. Foit, I. C. Vinke, L. G.-J. de Haart and R.-A. Eichel, Angew. Chem., Int. Ed., 2017, 56, 5402–5411 CrossRef CAS PubMed.
- A. Paredes-Nunez, D. Lorito, N. Guilhaume, Y. Schuurman and F. C. Meunier, Catal. Today, 2019, 336, 84–89 CrossRef CAS.
- M. Ziaei, M. Panahi, M. A. Fanaei, A. Rafiee and K. R. Khalilpour, J. CO2 Util., 2020, 35, 14–27 CrossRef CAS.
- M. Ao, G. H. Pham, J. Sunarso, M. O. Tade and S. Liu, ACS Catal., 2018, 8, 7025–7050 CrossRef CAS.
- S. You, Y. S. Ok, D. C.-W. Tsang, E. E. Kwon and C.-H. Wang, Crit. Rev. Environ. Sci. Technol., 2018, 48, 1165–1213 CrossRef CAS.
- F. Guerrero, L. Espinoza, N. Ripoll, P. Lisbona, I. Arauzo and M. Toledo, Front. Chem., 2020, 8, 145 CrossRef CAS PubMed.
- B. Ayodele, S. I. Mustapa, T. A.-R. Tuan Abdullah and S. F. Salleh, Front. Energy Res., 2019, 7, 118 CrossRef.
- V. R. Choudhary and A. M. Rajput, Ind. Eng. Chem. Res., 1996, 35, 3934–3939 CrossRef CAS.
- S. S. Bharadwaj and L. D. Schmidt, Fuel Process. Technol., 1995, 42, 109–127 CrossRef CAS.
- I. Dybkjaer, Fuel Process. Technol., 1995, 42, 85–107 CrossRef.
- H. T. Briscoe, The production of synthesis gas by steam reforming of methane general chemistry for colleges, The Riverside Press, Cambridge, MA, 1943, pp. 138–139 Search PubMed.
- R. G. Herman, K. Klier, G. W. Simmons, B. P. Finn, J. B. Bulko and T. P. Kobylinski, J. Catal., 1979, 56, 407–429 CrossRef CAS.
- P. J. Byrne Jr., E. J. Gohr, N. J. Elizabeth and R. T. Haslam, Ind. Eng. Chem., 1932, 24, 1129–1135 CrossRef.
- J. R. Rostrup-Nielsen, J. Catal., 1973, 31, 173–199 CrossRef CAS.
- J. R. Rostrup-Nielsen, Catal. Today, 1993, 18, 305–324 CrossRef.
- T. S. Christensen and I. I. Primdahl, Hydrocarb. Process. States.
- R. P. O’Connor, E. J. Klein and L. D. Schmidt, Catal. Lett., 2000, 70, 99–107 CrossRef.
- J. Ren, J.-P. Cao, X.-Y. Zhao, F.-L. Yang and X.-Y. Wei, Renewable Sustainable Energy Rev., 2019, 116, 109426 CrossRef CAS.
- K. Göransson, U. Söderlind, J. He and W. Zhang, Renewable Sustainable Energy Rev., 2011, 15, 482–492 CrossRef.
- V. N. Nguyen and L. Blum, Chem. Ing. Tech., 2015, 87, 354–375 CrossRef CAS.
- P. Mondal, G. S. Dang and M. O. Garg, Fuel Process. Technol., 2011, 92, 1395–1410 CrossRef CAS.
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 RSC.
- F. A. Rahman, M. M.-A. Aziz, R. Saidur, W. A.-W. A. Bakar, M. R. Hainin, R. Putrajaya and N. A. Hassan, Renewable Sustainable Energy Rev., 2017, 71, 112–126 CrossRef.
- D. Voiry, H. S. Shin, K. P. Loh and M. Chhowalla, Nat. Rev. Chem., 2018, 2, 105 CrossRef CAS.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- N. Sharma, B. Ugale, S. Kumar and K. Kailasam, Front. Chem., 2021, 9, 737511 CrossRef CAS PubMed.
- J. Roeser, K. Kailasam and A. Thomas, ChemSusChem, 2012, 5, 1793–1799 CrossRef CAS PubMed.
- J. Baltrusaitis and W. L. Luyben, ACS Sustainable Chem. Eng., 2015, 3, 2100–2111 CrossRef CAS.
- P. Kang, Z. Chen, A. Nayak, S. Zhang and T. J. Meyer, Energy Environ. Sci., 2014, 7, 4007–4012 RSC.
- W. Sheng, S. Kattel, S. Yao, B. Yan, Z. Liang, C. J. Hawxhurst, Q. Wu and J. G. Chen, Energy Environ. Sci., 2017, 10, 1180–1185 RSC.
- S. Hernández, M. Amin Farkhondehfal, F. Sastre, M. Makkee, G. Saracco and N. Russo, Green Chem., 2017, 19, 2326–2346 RSC.
- W. Yang, J.-H. Zhang, R. Si, L.-M. Cao, D.-C. Zhong and T.-B. Lu, Inorg. Chem. Front., 2021(8), 1695–1701 Search PubMed.
- X. Gu, L. Qian and G. Zheng, Mol. Catal., 2020, 492, 110953 CrossRef CAS.
- C. Li, T. Wang, B. Liu, M. Chen, A. Li, G. Zhang, M. Du, H. Wang, S. F. Liu and J. Gong, Energy Environ. Sci., 2019, 12, 923–928 RSC.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343(6174), 1239176 CrossRef PubMed.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- H. Wang, W. Liu, S. Jin, X. Zhang and Y. Xie, ACS Cent. Sci., 2020, 6, 1058–1069 CrossRef CAS PubMed.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- B. Liu, X. Zhao, C. Terashima, A. Fujishima and K. Nakata, Phys. Chem. Chem. Phys., 2014, 16, 8751–8760 RSC.
- X. Wang, F. Wang, Y. Sang and H. Liu, Adv. Energy Mater., 2017, 7, 1700473 CrossRef.
- S. Xu and E. A. Carter, Chem. Rev., 2019, 119, 6631–6669 CrossRef CAS PubMed.
- V. R. Battula, S. Kumar, D. K. Chauhan, S. Samanta and K. Kailasam, Appl. Catal., B, 2019, 244, 313–319 CrossRef CAS.
- D. K. Chauhan, V. R. Battula, S. Jain and K. Kailasam, J. Cleaner Prod., 2021, 307, 127162 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- S. Boddu, S. T. Nishanthi and K. Kailasam, Visible Light Photocatal, 2018, 421–446 CAS.
- S. Nahar, M. F.-M. Zain, A. A.-H. Kadhum, H. A. Hasan and M. R. Hasan, Mater., 2017, 10 Search PubMed.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- R. M. Irfan, T. Wang, D. Jiang, Q. Yue, L. Zhang, H. Cao, Y. Pan and P. Du, Angew. Chem., Int. Ed., 2020, 59, 14818–14824 CrossRef CAS PubMed.
- P. G. Alsabeh, A. Rosas-Hernández, E. Barsch, H. Junge, R. Ludwig and M. Beller, Catal. Sci. Technol., 2016, 6, 3623–3630 RSC.
- S. Aoi, K. Mase, K. Ohkubo and S. Fukuzumi, Catal. Sci. Technol., 2016, 6, 4077–4080 RSC.
- L. Tan, K. Peter, J. Ren, B. Du, X. Hao, Y. Zhao and Y.-F. Song, Front. Chem. Sci. Eng., 2021, 15, 99–108 CrossRef CAS.
- J. Zhao, Q. Wang, C. Sun, T. Zheng, L. Yan, M. Li, K. Shao, X. Wang and Z. Su, J. Mater. Chem. A, 2017, 5, 12498–12505 RSC.
- Q. Mu, W. Zhu, G. Yan, Y. Lian, Y. Yao, Q. Li, Y. Tian, P. Zhang, Z. Deng and Y. Peng, J. Mater. Chem. A, 2018, 6, 21110–21119 Search PubMed.
- S. Wang, W. Yao, J. Lin, Z. Ding and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 1034–1038 CrossRef CAS PubMed.
- Z. Fu, X. Wang, A. M. Gardner, X. Wang, S. Y. Chong, G. Neri, A. J. Cowan, L. Liu, X. Li, A. Vogel, R. Clowes, M. Bilton, L. Chen, R. S. Sprick and A. I. Cooper, Chem. Sci., 2020, 11, 543–550 RSC.
- X. Zhao, J. Zhou, C.-Y. Sun, S.-Q. You, X.-L. Wang and Z.-M. Su, Nanotechnology, 2020, 31, 255402 CrossRef CAS PubMed.
- W. Yao, C. Qin, N. Xu, J. Zhou, C. Sun, L. Liu and Z. Su, CrystEngComm, 2019, 21, 6423–6431 RSC.
- J. Yang, Z. Wang, J. Jiang, W. Chen, F. Liao, X. Ge, X. Zhou, M. Chen, R. Li, Z. Xue, G. Wang, X. Duan, G. Zhang, Y.-G. Wang and Y. Wu, Nano Energy, 2020, 76, 105059 CrossRef CAS.
- J.-C. Hu, M.-X. Gui, W. Xia, J. Wu, Y.-N. Zhou, N. Feng, J. Xiao, H. Liu, C.-H. Tung, L.-Z. Wu and F. Wang, J. Mater. Chem. A, 2019, 7, 10475–10482 RSC.
- P. Reñones, L. Collado, A. Iglesias-Juez, F. E. Oropeza, F. Fresno and V. A. de la Peña O’Shea, Ind. Eng. Chem. Res., 2020, 59, 9440–9450 CrossRef.
- H. Huo, D. Liu, H. Feng, Z. Tian, X. Liu and A. Li, Nanoscale, 2020, 12, 13912–13917 RSC.
- J.-S. Lee, D.-I. Won, W.-J. Jung, H.-J. Son, C. Pac and S. O. Kang, Angew. Chem., Int. Ed., 2017, 56, 976–980 CrossRef CAS PubMed.
- N. R. de Tacconi, H. K. Timmaji, W. Chanmanee, M. N. Huda, P. Sarker, C. Janáky and K. Rajeshwar, Chem. Phys. Chem., 2012, 13, 2945–2955 CrossRef CAS PubMed.
- A. Li, T. Wang, X. Chang, Z.-J. Zhao, C. Li, Z. Huang, P. Yang, G. Zhou and J. Gong, Chem. Sci., 2018, 9, 5334–5340 RSC.
- D. Li, S. Ouyang, H. Xu, D. Lu, M. Zhao, X. Zhang and J. Ye, Chem. Commun., 2016, 52, 5989–5992 RSC.
- C. Zhao, A. Krall, H. Zhao, Q. Zhang and Y. Li, Int. J. Hydrogen Energy, 2012, 37, 9967–9976 CrossRef CAS.
- X. Yao, K. Chen, L.-Q. Qiu, Z.-W. Yang and L.-N. He, Chem. Mater., 2021, 33, 8863–8872 CrossRef CAS.
- C. Chen, J. Hu, X. Yang, T. Yang, J. Qu, C. Guo and C. M. Li, ACS Appl. Mater. Interfaces, 2021, 13, 20162–20173 CrossRef CAS PubMed.
- Z. Li, Y. Yang, J. Tian, J. Li, G. Chen, L. Zhou, Y. Sun and Y. Qiu, ChemSusChem, 2022, n/a, e202102729 Search PubMed.
- N. Shehzad, M. Tahir, K. Johari, T. Murugesan and M. Hussain, J. CO2 Util., 2018, 26, 98–122 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- R. Daghrir, P. Drogui and D. Robert, Ind. Eng. Chem. Res., 2013, 52, 3581–3599 CrossRef CAS.
- M. Pelaez, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos, P. S.-M. Dunlop, J. W.-J. Hamilton, J. A. Byrne, K. O’Shea, M. H. Entezari and D. D. Dionysiou, Appl. Catal., B, 2012, 125, 331–349 CrossRef CAS.
- S. A. Rawool, K. K. Yadav and V. Polshettiwar, Chem. Sci., 2021, 12, 4267–4299 RSC.
- K. Li, C. Teng, S. Wang and Q. Min, Front. Chem., 2021, 9, 637501 CrossRef CAS PubMed.
- K. H. Do, D. Praveen Kumar, A. Putta Rangappa, J. Wang, Y. Hong, E. Kim, D. Amaranatha Reddy and T. Kyu Kim, Mater. Today Chem., 2021, 22, 100589 CrossRef CAS.
- J. Low, S. Qiu, D. Xu, C. Jiang and B. Cheng, Appl. Surf. Sci., 2018, 434, 423–432 CrossRef CAS.
- R. Nematollahi, C. Ghotbi, F. Khorasheh and A. Larimi, J. CO2 Util., 2020, 41, 101289 CrossRef CAS.
- S. B. Patil, P. S. Basavarajappa, N. Ganganagappa, M. S. Jyothi, A. V. Raghu and K. R. Reddy, Int. J. Hydrogen Energy, 2019, 44, 13022–13039 CrossRef CAS.
- H. Zhao, X. Zheng, X. Feng and Y. Li, J. Phys. Chem. C, 2018, 122, 18949–18956 CrossRef CAS.
- K. Kailasam, J. D. Epping, A. Thomas, S. Losse and H. Junge, Energy Environ. Sci., 2011, 4, 4668–4674 RSC.
- L. Li, X. Chen, L. Wang, C. Tao, X. Wu, J. Du and Z. Liu, J. Alloys Compd., 2020, 845, 156138 CrossRef CAS.
- T. Zhang, J. Low, K. Koh, J. Yu and T. Asefa, ACS Sustainable Chem. Eng., 2018, 6, 531–540 CrossRef CAS.
- L. Wei, C. Yu, Q. Zhang, H. Liu and Y. Wang, J. Mater. Chem. A, 2018, 6, 22411–22436 RSC.
- D.-I. Won, J.-S. Lee, Q. Ba, Y.-J. Cho, H.-Y. Cheong, S. Choi, C. H. Kim, H.-J. Son, C. Pac and S. O. Kang, ACS Catal., 2018, 8, 1018–1030 CrossRef CAS.
- E. Lam and E. Reisner, Angew. Chem., Int. Ed., 2021, 60, 23306–23312 CrossRef CAS PubMed.
- M. Jo, S. Choi, J. H. Jo, S.-Y. Kim, P. S. Kim, C. H. Kim, H.-J. Son, C. Pac and S. O. Kang, ACS Omega, 2019, 4, 14272–14283 CrossRef CAS PubMed.
- H. Huang, J. Lin, G. Zhu, Y. Weng, X. Wang, X. Fu and J. Long, Angew. Chem., Int. Ed., 2016, 55, 8314–8318 CrossRef CAS PubMed.
- S.-J. Woo, S. Choi, S.-Y. Kim, P. S. Kim, J. H. Jo, C. H. Kim, H.-J. Son, C. Pac and S. O. Kang, ACS Catal., 2019, 9, 2580–2593 CrossRef CAS.
- L. Wang, S. Duan, P. Jin, H. She, J. Huang, Z. Lei, T. Zhang and Q. Wang, Appl. Catal., B, 2018, 239, 599–608 CrossRef CAS.
- M. E. Aguirre, R. Zhou, A. J. Eugene, M. I. Guzman and M. A. Grela, Appl. Catal., B, 2017, 217, 485–493 CrossRef CAS.
- C. Wang, Y. Su, A. Tavasoli, W. Sun, L. Wang, G. A. Ozin and D. Yang, Mater. Today Nano, 2021, 14, 100113 CrossRef CAS.
- L.-X. Zhang, J. Hu, Y.-B. Jia, R.-T. Liu, T. Cai and Z. P. Xu, Nanoscale, 2021, 13, 7533–7549 RSC.
- L. Mohapatra and K. Parida, J. Mater. Chem. A, 2016, 4, 10744–10766 RSC.
- G. Zhang, X. Zhang, Y. Meng, G. Pan, Z. Ni and S. Xia, Chem. Eng. J., 2020, 392, 123684 CrossRef CAS.
- M. J. Wu, J. Z. Wu, J. Zhang, H. Chen, J. Z. Zhou, G. R. Qian, Z. P. Xu, Z. Du and Q. L. Rao, Catal. Sci. Technol., 2018, 8, 1207–1228 RSC.
- J. Li, Y. Xu, S. Wang and H. Zhang, J. Phys. Chem. C, 2019, 123, 15483–15494 CrossRef CAS.
- J. Tao, X. Yu, Q. Liu, G. Liu and H. Tang, J. Colloid Interface Sci., 2021, 585, 470–479 CrossRef CAS PubMed.
- S. Tonda, S. Kumar, M. Bhardwaj, P. Yadav and S. Ogale, ACS Appl. Mater. Interfaces, 2018, 10, 2667–2678 CrossRef CAS PubMed.
- Z. Bi, R. Guo, X. Hu, J. Wang, X. Chen and W. Pan, Nanoscale, 2022, 14, 3367–3386 RSC.
- A. Hezam, K. Namratha, Q. A. Drmosh, D. Ponnamma, J. Wang, S. Prasad, M. Ahamed, C. Cheng and K. Byrappa, ACS Appl. Nano Mater., 2020, 3, 138–148 CrossRef CAS.
- L. Tan, K. Peter, J. Ren, B. Du, X. Hao, Y. Zhao and Y.-F. Song, Front. Chem. Sci. Eng., 2020 DOI:10.1007/s11705-020-1947-4.
- S. Guo, Y. Zhang, S. Tang, B. Wang, Y. Wang, Y. Song, X. Xin, Y. Zhang and X. Li, J. Alloys Compd., 2021, 864, 158581 CrossRef CAS.
- S. Singh, A. Modak, K. K. Pant, A. Sinhamahapatra and P. Biswas, ACS Appl. Nano Mater., 2021, 4, 8644–8667 CrossRef CAS.
- S. Subramanian, Q. T. Campbell, S. K. Moser, J. Kiemle, P. Zimmermann, P. Seifert, F. Sigger, D. Sharma, H. Al-Sadeg, M. Labella, D. Waters, R. M. Feenstra, R. J. Koch, C. Jozwiak, A. Bostwick, E. Rotenberg, I. Dabo, A. W. Holleitner, T. E. Beechem, U. Wurstbauer and J. A. Robinson, ACS Nano, 2020, 14, 16663–16671 CrossRef CAS PubMed.
- G. Paille, M. Gomez-Mingot, C. Roch-Marchal, B. Lassalle-Kaiser, P. Mialane, M. Fontecave, C. Mellot-Draznieks and A. Dolbecq, J. Am. Chem. Soc., 2018, 140, 3613–3618 CrossRef CAS PubMed.
- Y. Liu, C. Tang, M. Cheng, M. Chen, S. Chen, L. Lei, Y. Chen, H. Yi, Y. Fu and L. Li, ACS Catal., 2021, 11, 13374–13396 CrossRef CAS.
- R. Sivakumar, J. Thomas and M. Yoon, J. Photochem. Photobiol., C, 2012, 13, 277–298 CrossRef CAS.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- W. Guo, Z. Wang, X. Wang and Y. Wu, Adv. Mater., 2021, 33, 2004287 CrossRef CAS PubMed.
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef.
- Z. Lin, M. Escudero-Escribano and J. Li, J. Mater. Chem. A, 2022, 10, 5670–5672 RSC.
- P. Sharma, S. Kumar, O. Tomanec, M. Petr, J. Zhu Chen, J. T. Miller, R. S. Varma, M. B. Gawande and R. Zbořil, Small, 2021, 17, 2006478 CrossRef CAS PubMed.
- P. Huang, J. Huang, S. A. Pantovich, A. D. Carl, T. G. Fenton, C. A. Caputo, R. L. Grimm, A. I. Frenkel and G. Li, J. Am. Chem. Soc., 2018, 140, 16042–16047 CrossRef CAS PubMed.
- L. Jean-Marie and Z. Raymond, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 701–704 CrossRef PubMed.
- J. Hawecker, J.-M. Lehn and R. Ziessel, Helv. Chim. Acta, 1986, 69, 1990–2012 CrossRef CAS.
- H. Takeda, K. Koike, H. Inoue and O. Ishitani, J. Am. Chem. Soc., 2008, 130, 2023–2031 CrossRef CAS PubMed.
- W.-M. Liao, J.-H. Zhang, Y.-J. Hou, H.-P. Wang and M. Pan, Inorg. Chem. Commun., 2016, 73, 80–89 CrossRef CAS.
- K. Kamada, J. Jung, T. Wakabayashi, K. Sekizawa, S. Sato, T. Morikawa, S. Fukuzumi and S. Saito, J. Am. Chem. Soc., 2020, 142, 10261–10266 CrossRef CAS PubMed.
- T. Ouyang, C. Hou, J.-W. Wang, W.-J. Liu, D.-C. Zhong, Z.-F. Ke and T.-B. Lu, Inorg. Chem., 2017, 56, 7307–7311 CrossRef CAS PubMed.
- A. Sinopoli, N. T. La Porte, J. F. Martinez, M. R. Wasielewski and M. Sohail, Coord. Chem. Rev., 2018, 365, 60–74 CrossRef CAS.
- L.-L. Gracia, L. Luci, C. Bruschi, L. Sambri, P. Weis, O. Fuhr and C. Bizzarri, Chem. – Eur. J., 2020, 26, 9929–9937 CrossRef CAS PubMed.
- L. Zhang and J. Zhang, Front. Energy, 2019, 13, 221–250 CrossRef.
- L. Zhu, X.-Q. Liu, H.-L. Jiang and L.-B. Sun, Chem. Rev., 2017, 117, 8129–8176 CrossRef CAS PubMed.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- D. A. Reddy, Y. Kim, M. Gopannagari, D. P. Kumar and T. K. Kim, Sustainable Energy Fuels, 2021, 5, 1597–1618 RSC.
- A. V. Desai, S. Sharma, S. Let and S. K. Ghosh, Coord. Chem. Rev., 2019, 395, 146–192 CrossRef CAS.
- B. Lee, D. Moon and J. Park, Angew. Chem., Int. Ed., 2020, 59, 13793–13799 CrossRef CAS PubMed.
- Z. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315–1329 RSC.
- H.-B. Huang, Z.-B. Fang, R. Wang, L. Li, M. Khanpour, T.-F. Liu and R. Cao, Small, 2022, n/a, 2200407 CrossRef PubMed.
- J.-R. An, Y. Wang, W.-W. Dong, X.-J. Gao, O.-Y. Yang, Y.-L. Liu, J. Zhao and D.-S. Li, ACS Appl. Energy Mater., 2022, 5, 2384–2390 CrossRef CAS.
- X. Deng, Y. Qin, M. Hao and Z. Li, Inorg. Chem., 2019, 58, 16574–16580 CrossRef CAS PubMed.
- S. Wang and X. Wang, Appl. Catal., B, 2015, 162, 494–500 CrossRef CAS.
- B. Chen, Z. Yang, Y. Zhu and Y. Xia, J. Mater. Chem. A, 2014, 2, 16811–16831 RSC.
- J. Qin, S. Wang and X. Wang, Appl. Catal., B, 2017, 209, 476–482 CrossRef CAS.
- V. S. Vyas and B. V. Lotsch, Nature, 2015, 521, 41–42 CrossRef CAS PubMed.
- Z. Zhang, J. Jia, Y. Zhi, S. Ma and X. Liu, Chem. Soc. Rev., 2022, 51, 2444–2490 RSC.
- J. Byun, Y. Hong and K. A.-I. Zhang, Chem. Catal., 2021, 1, 771–781 CrossRef.
- A. Kumar, P. Raizada, V. Kumar Thakur, V. Saini, A. Aslam Parwaz Khan, N. Singh and P. Singh, Chem. Eng. Sci., 2021, 230, 116219 CrossRef CAS.
- Y.-Z. Cheng, X. Ding and B.-H. Han, ChemPhotoChem, 2021, 5, 406–417 CrossRef CAS.
- S. Zhang, S. Wang, L. Guo, H. Chen, B. Tan and S. Jin, J. Mater. Chem. C, 2020, 8, 192–200 RSC.
- H. L. Nguyen and A. Alzamly, ACS Catal., 2021, 11, 9809–9824 CrossRef CAS.
- D. K. Chauhan, S. Jain, V. R. Battula and K. Kailasam, Carbon N. Y., 2019, 152, 40–58 CrossRef CAS.
- X. Li, C. Yang, B. Sun, S. Cai, Z. Chen, Y. Lv, J. Zhang and Y. Liu, J. Mater. Chem. A, 2020, 8, 16045–16060 RSC.
- S. Kandambeth, K. Dey and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 1807–1822 CrossRef CAS PubMed.
- X. C. Yajun He Chi Huang, L. Li, C. Yang and Yan Yu, Chin. J. Catal., 42, 123–130 Search PubMed.
- W. Zhong, R. Sa, L. Li, Y. He, L. Li, J. Bi, Z. Zhuang, Y. Yu and Z. Zou, J. Am. Chem. Soc., 2019, 141, 7615–7621 CrossRef CAS PubMed.
- Y. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083–2094 RSC.
- X. Xiong, C. Mao, Z. Yang, Q. Zhang, G. I.-N. Waterhouse, L. Gu and T. Zhang, Adv. Energy Mater., 2020, 10, 2002928 CrossRef CAS.
- F. Wang, T. Hou, X. Zhao, W. Yao, R. Fang, K. Shen and Y. Li, Adv. Mater., 2021, 33, 2102690 CrossRef CAS PubMed.
- D. Sun, Y. Fu, W. Liu, L. Ye, D. Wang, L. Yang, X. Fu and Z. Li, Chem. – Eur. J., 2013, 19, 14279–14285 CrossRef CAS PubMed.
- Y. Zhao, G. Chen, T. Bian, C. Zhou, G. I.-N. Waterhouse, L.-Z. Wu, C.-H. Tung, L. J. Smith, D. O’Hare and T. Zhang, Adv. Mater., 2015, 27, 7824–7831 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |