
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-assembled silk fibroin hydrogels: from preparation to biomedical applications
Ozgun Can
Onder
 *a,
Syeda Rubab
Batool
b and
Muhammad Anwaar
Nazeer
b
*a,
Syeda Rubab
Batool
b and
Muhammad Anwaar
Nazeer
b
aPolymer Performance Materials Group, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, 5600 MB Eindhoven, The Netherlands. E-mail: ozgnondr@gmail.com
bSchool of Engineering and Technology, National Textile University, 37610 Faisalabad, Pakistan
First published on 3rd August 2022
Abstract
Silk fibroin (SF) from Bombyx mori silkworm has been used as a textile fiber for centuries. In addition, for decades, SF has been used as a suture material. SF's unique properties, such as exceptional host tissue response, appropriate mechanical properties, tunable degradation, simple processing method, and low cost, make it an attractive biomaterial. Accordingly, SF-based hydrogels have attracted much interest in the past years for biomedical applications. The tendency of SF chains to associate and form physical networks has been exploited very frequently in the last decade to prepare self-assembled SF hydrogels. This review describes the methods to prepare self-assembled SF hydrogels focusing on gelation mechanisms and biomedical applications thereof. The advantages and limitations of the methods used to induce SF assembly were discussed. In addition, the use of self-assembled SF hydrogels in interpenetrating network systems and the 3D printing applications were reviewed.
 Ozgun Can Onder | Ozgun Can Onder is a senior researcher at the Eindhoven University of Technology. Ozgun studied Chemistry and Molecular Biology at Bogazici University and trained as a synthetic chemist. Ozgun obtained his PhD in 2018 from Koc University, specializing in the preparation of polyester-based foams. He also participated in industrial projects related to the synthesis and characterization of polyurethanes and gained an understanding of the structure–morphology–properties relationships. Between 2018 and 2022, he worked at the Slovenian National Institute of Chemistry as a researcher, where he developed macroporous polypeptide-based biomaterials, mentored graduate students, and collaborated with industrial partners for EU-funded projects. |
 Syeda Rubab Batool | Syeda Rubab Batool completed her Doctor of Pharmacy from Baqai Medical University, Pakistan, in 2013. After that, she moved to Koc University, Turkey, where she completed her PhD in Biomedical Sciences and Engineering in 2021. Her current research focuses on the preparation of innovative biomaterials and stimuli-responsive hydrogels for various biomedical, tissue engineering, and regenerative medicine applications. |
 Muhammad Anwaar Nazeer | Muhammad Anwaar Nazeer completed his PhD in Biomedical Sciences and Engineering from Koc University. He continued working as a postdoc at Koc University for 2 more years. He is currently working as an assistant professor at National Textile University, Pakistan. His research interests include the synthesis of biocompatible polymers, the development of scaffolds for medical textile, tissue engineering, and regenerative medicine applications. |
1. Introduction
The cocoons of silkworms have evolved over millions of years to protect the larvae from predators as they metamorphose into moths. The cocoon silks spun by the domestic silkworm Bombyx mori (B. mori) have been used in textiles for thousands of years.1 Silk has long been a vital textile stock due to its unique feel, luster, dyeability, tensile strength, and elasticity. In recent years its applications substantially increased, especially in the biomedical and cosmetics sectors. Nearly 1000 metric tons of B. mori silk are produced and processed annually.2The silk fibroin (SF) isolated from B. mori cocoons is a widely investigated, natural silk protein used for numerous biomedical applications due to its biocompatibility, biodegradability, and mechanical stability. In addition, it is also advantageous to use silk for biomedical applications because of the available large-scale processing infrastructure of traditional silk textile industries. Regenerated SF has been approved by the U.S. Food and Drug Administration (FDA) for clinical use in biomedical applications.3,4
Hydrogels are water-swollen three-dimensional viscoelastic macromolecular networks, crosslinked through either covalent bonds or non-covalent interactions such as electrostatic interactions, hydrogen bonding, hydrophobic association, and multivalent coordination.5,6 Hydrogels have attracted extensive attention due to their versatility in composition, preparation, and resulting tunable properties such as swelling capacity, an interfacial affinity for target compounds, stimuli response, degradability, viscoelasticity, and network architecture.7,8 Polymeric hydrogels have been used as functional materials in a variety of applications. Biomedical applications can benefit from the properties above, as hydrogels resemble biological tissues because of their porosity, high water content, and tissue-like elasticity.9,10 Therefore, biocompatible hydrogels have emerged as promising biomaterials in a variety of biomedical applications such as tissue engineering,11 and biosensing.12 They are also used as biointerface materials13 and drug delivery systems.14
SF hydrogels have recently received significant attention in the biomedical field. They have been used in various applications, including bone-filling materials, sustained delivery systems, and three-dimensional cell culture carriers as shown in Fig. 1.15–17 For the preparation of SF hydrogels, several chemical and physical crosslinking methodologies have been developed. SF chains in aqueous solutions tend to associate and form physical 3D networks, leading to the formation of self-assembled hydrogels. The physical crosslinking approach is quite useful for biomedical applications because it is cheaper, safer, and the use of toxic chemical agents is not required. In addition, the complete removal of residual chemical crosslinkers after purification is not a concern. Furthermore, shear-thinning platforms can be developed for minimally invasive operations by utilizing the tendency of SF chains to associate in aqueous solutions. Several excellent reviews regarding the preparation of SF-based hydrogels for biomedical applications have been published in recent years.18–20 However, there is no comprehensive review article on self-assembled SF hydrogels to the best of our knowledge. This report aims (i) to overview the strategies to prepare self-assembled SF hydrogel-based biomaterials, (ii) to investigate the principles of SF gelation in aqueous solutions, and (iii) to describe the applications of self-assembled SF hydrogels in the biomedical sector. For this purpose, we will start with a quick explanation of the cocoon formation process and chemical composition of cocoons. Then we will overview the regeneration process of SF. The methods used to induce the association of SF chains will be reviewed. The use of self-assembled SF hydrogels in interpenetrating network systems and bioink formulations for 3D printing applications will be discussed.
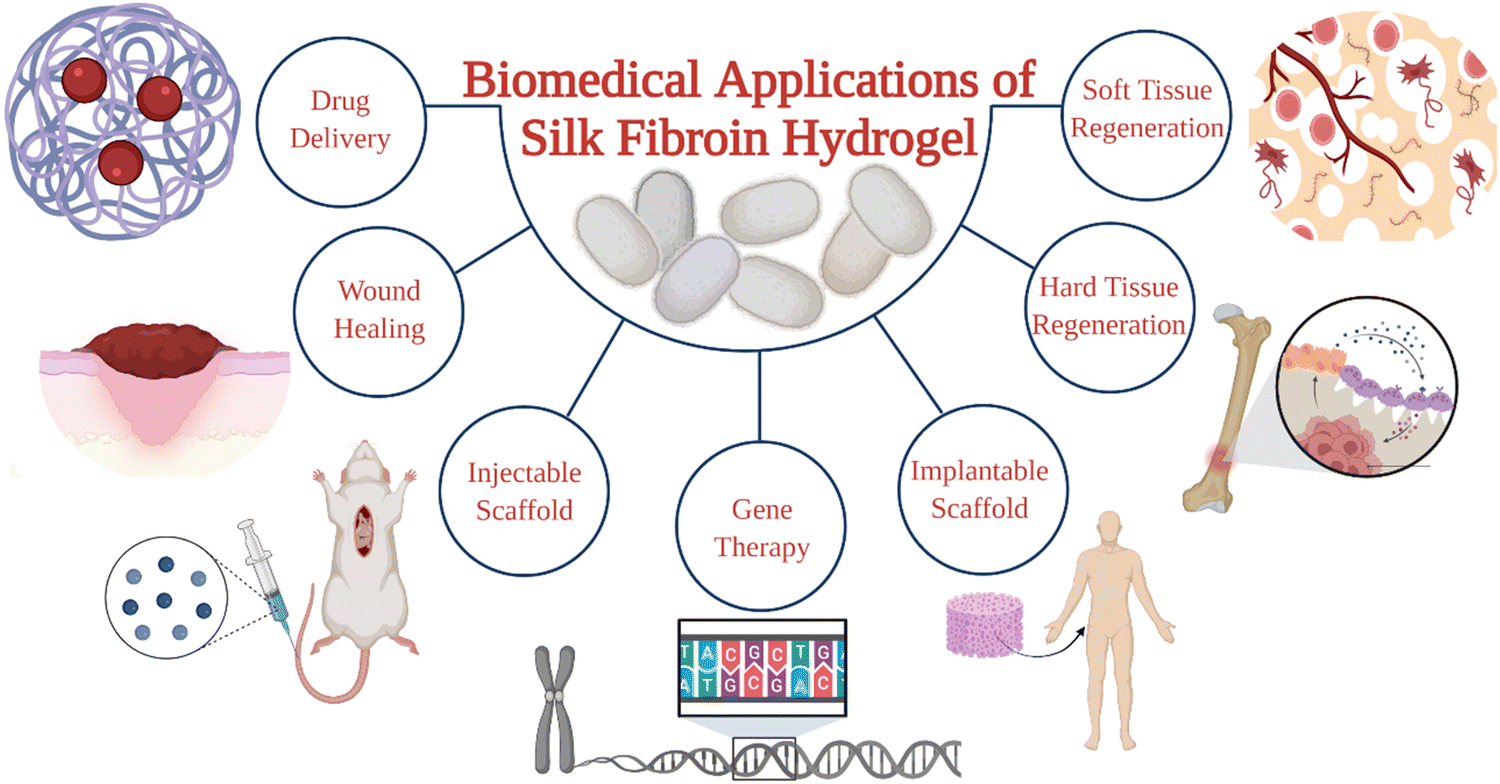 | ||
| Fig. 1 Biomedical applications of silk fibroin hydrogels. | ||
1.1 Overview of the cocoon formation process
B. mori is the domestic silk moth that feeds on the mulberry plants. These moths live for only several weeks. During that span, moths pass through four development stages: egg, larva, pupa, and adult, as presented in Fig. 2A. The life cycle begins when the female silk moth lays eggs. The eggs hatch into larvae that feed on the mulberry leaves. At this stage, the larva spins silk threads around itself and forms a cocoon to protect itself from predators while continuing its development into a pupa. In the pupa stage, the silk cocoon can be collected by placing the pupa in boiling water, so that the silk thread can be used for industrial applications.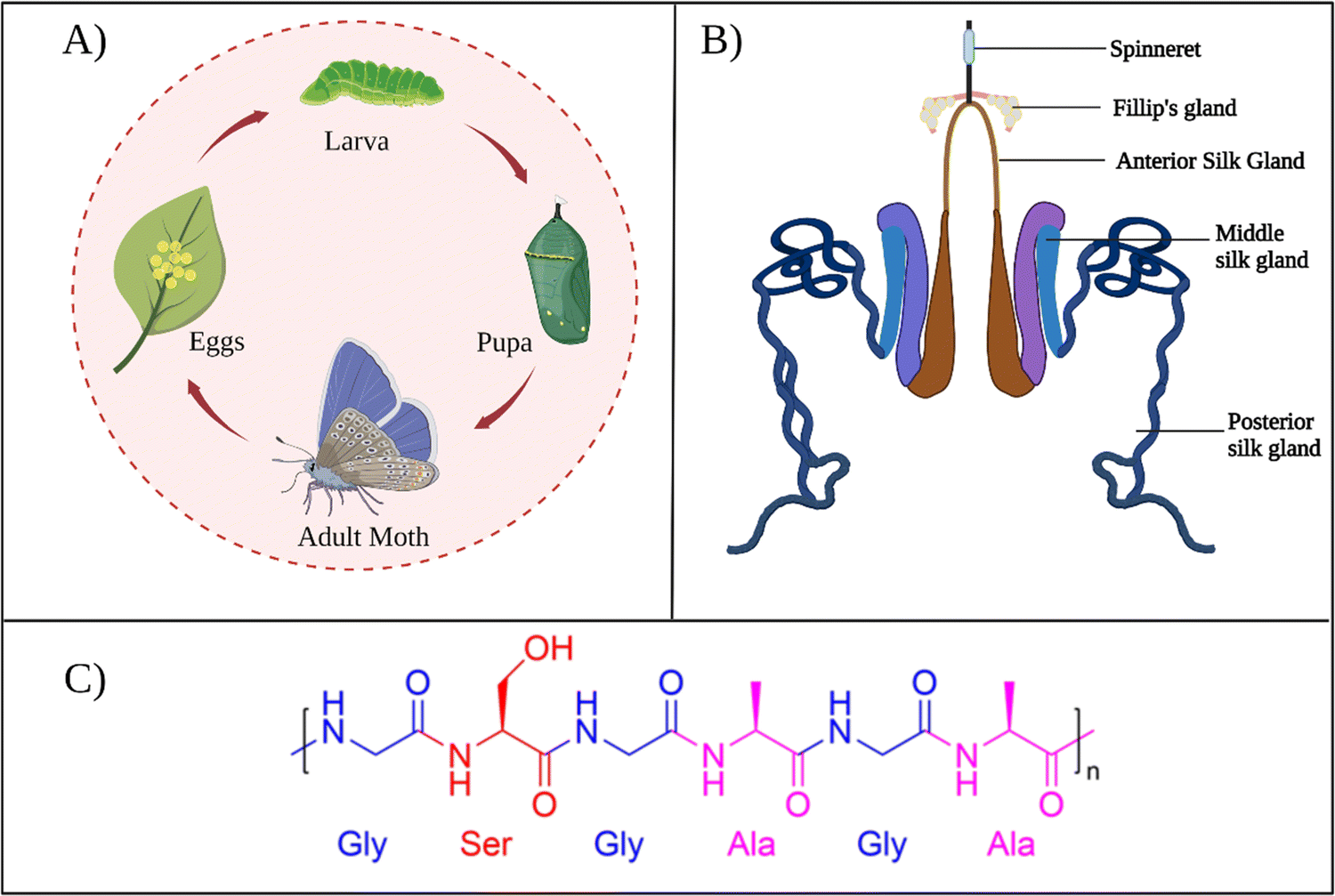 | ||
| Fig. 2 (A) The life cycle of B. mori. (B) B. mori silk gland organization. Reproduced with permission from Elsevier.27 (C) Chemical structure of common repeat unit of SF. | ||
During the development of larvae, the silk gland grows to be an organ over 15 cm long. Silk fibers are produced through unique biomolecular machinery contained in these glands. The silk glands of B. mori exist as two tubular structures (Fig. 2B). According to its morphology and function, the silk gland contains three compartments: the anterior silk gland, middle silk gland, and posterior silk gland.21 First, SF components are secreted into the posterior gland as an aqueous solution during cocoon production. These components are fibroin heavy chain (350 kDa), fibroin light chain (25 kDa), and P25 glycoprotein (30 kDa). A disulfide bond links fibroin heavy and light chains in a 1–1 ratio. The formed heavy-light chain complex also binds glycoprotein P25 in a ratio of 6–1 via hydrophobic interactions to form a high molecular mass elementary unit.22 This fibroin unit is then transferred from the posterior to the middle gland. In the middle gland, glue-like sericin proteins are produced. As fibroin passes through the middle gland, it is coated via sericins.23 Sericin-coated fibroin then flows through the narrow anterior silk gland duct. The viscosity of the fibroin solution increases as it passes through the anterior gland. At the end of the anterior gland, the tubules come together to form a spinneret, from which silk fibers are pulled via larval head movement. The spinneret consists of a silk press and a duct where the fiber exits through a pore. The combined actions of strain and shear, together with the dehydration and induction of the ions, induce the conversion of viscous fibroin solution into a solid filament at the spinneret.24 A fully grown cocoon comprises a single fiber 1300–1500 m long and 10–20 μm in diameter.25 The soluble silk proteins exist in a metastable state within silk glands, and chains adopt disordered conformations. During fiber formation, the protein chains undergo major structural rearrangements, and chains are transformed from largely disordered states to β-sheet-rich conformations, serving as the underlying basis for the high strength of silk fibers.26
1.2 Chemical composition of the cocoons
In general, as produced cocoons are composed of fibroin (>70 wt%) and sericin (25–30 wt%) proteins.28 The sericin proteins located outside the fibroin proteins act as the glue that brings the two fibroin units together. The chemical composition of these proteins was characterized extensively. The primary structure of a heavy chain consists of two flanking hydrophilic blocks at C- and N-terminals.29 In between are hydrophobic blocks intertwined by small hydrophilic domains. The hydrophobic domains are mainly composed of dipeptide units in the form of GX where X can be alanine, serine, tyrosine, valine, or threonine. The most common repeat units include GAGAGS, GAGAGY, and GAGAGVGY. The presence of these repeat units is very critical for the self-organization of SF chains into the well-defined β-sheet crystalline structures.30 Overall, the heavy chain comprises 45.9% glycine, 30.3% alanine, 12.1% serine, 5.3% tyrosine, 1.8% valine, and only 4.7% of the other 15 amino acids.31 The proportion of hydrophobic residues in the heavy chain is approximately 79%. The heavy chains of SF materials are generally responsible for the extraordinary mechanical performance, as they can self-assemble into discrete β-sheet crystallites.The light chain has a standard amino acid composition and a non-repetitive sequence. It contains a very different amino acid composition of 15% aspartate, 14% alanine, 11% glycine, and 11% serine.32 Light chains do not play a significant role during the self-assembly of SF chains because of their small size and the lack of repetitive sequences. So far, light chain amino acid sequences are not observed in the β-sheet domains of SF. The sericin proteins have a wide range of molecular weights between 40–400 kDa. Sericin chains are mainly composed of serine (25%), glycine (18%), aspartic acid (18%), and threonine (7%) amino acids.33 Sericins are soluble in water due to a high content of hydrophilic amino acids.
1.3 Regeneration of fibroin from the cocoons
For a long time, sericin has been considered to induce allergic and immunological reactions in humans.34 However, it was demonstrated that, sericin does not cause these reactions itself.35 Instead, the immune response is induced only when sericin and fibroin molecules are used together.36,37 Therefore, all sericin should be removed from fibroin intended for biological applications. This removal step is called degumming. Since the hydrophilicity of sericin is much more excellent than fibroin, it can be selectively removed from the cocoons. Various degumming conditions are reported in the literature. For example, selective dissolution of sericin can be conducted in alkali (e.g., sodium carbonate), acids (e.g., citric acid), or high concentration denaturant solutions (e.g., urea), or proteases.38 The most commonly used degumming method is alkali treatment. This process most commonly involves boiling cocoons in an aqueous sodium carbonate for 10–60 min.39 Silk fibroin regeneration process was presented schematically in Fig. 3A.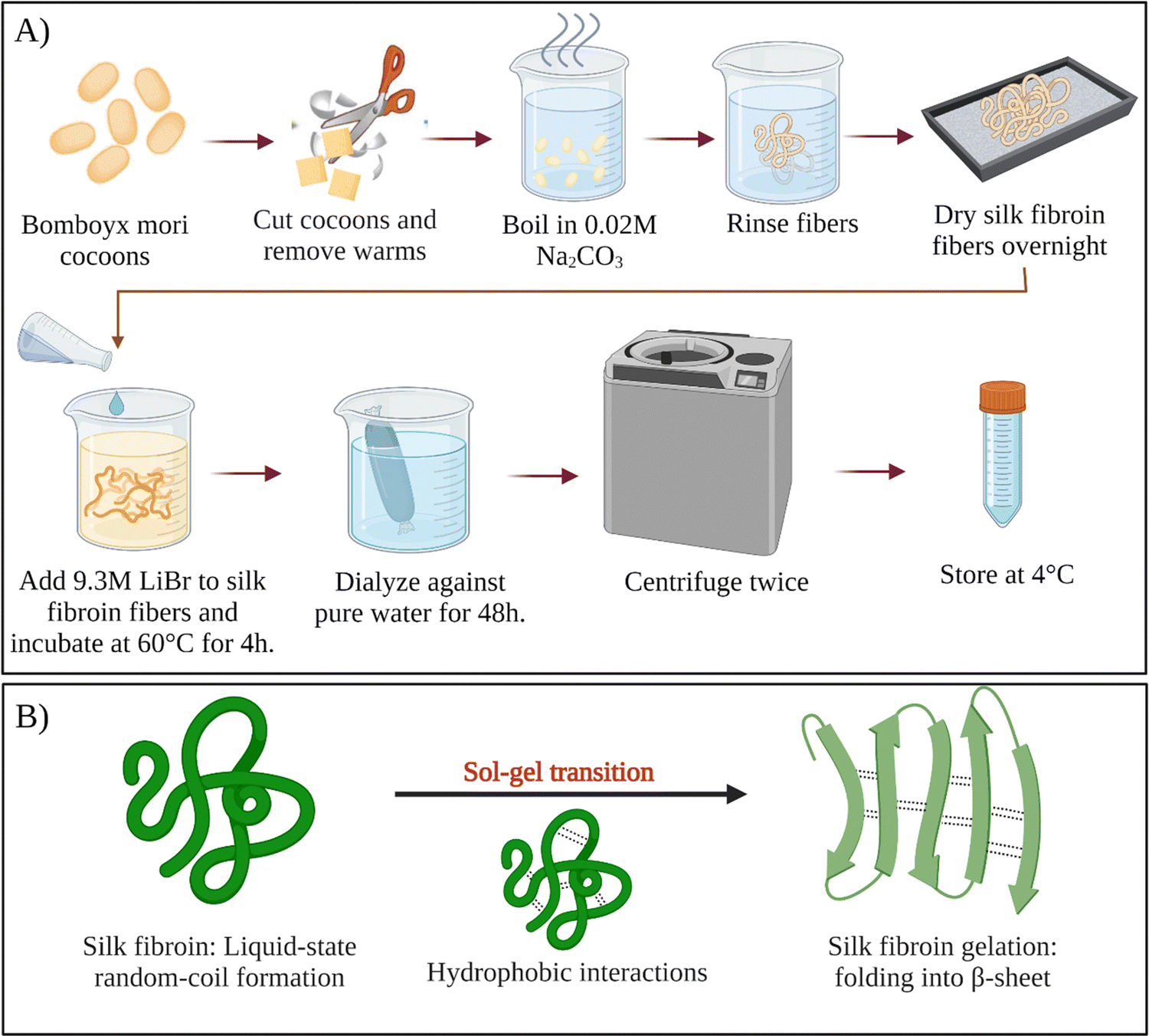 | ||
| Fig. 3 (A) Schematic of silk fibroin regeneration process. (B) Sol–gel transition of silk fibroin solution. | ||
After degumming, the cocoons are rinsed with water several times and dried. Fibroin in the cocoon is found in the β-sheet structure, which is stabilized by the intermolecular hydrogen bonds and hydrophobic interactions. Because of the strong intermolecular interactions, fibroin cannot be solubilized in the water right away. To solubilize fibroin in aqueous solutions, chaotropic reagents are used to disrupt the hydrogen bonds between fibroin chains (e.g., LiBr, LiSCN, CaCl2–ethanol).40 Most commonly, extracted dry cocoons are dissolved in 9.3 M LiBr aqueous solution at 60 °C. After that, the solution is filtered or centrifuged. Filtered solution or the supernatant are dialyzed continuously for several days in distilled water to remove the salts and low molecular weight impurities. Depending on the conditions, the resulting fibroin solution (usually a few weight percent) is stable at 4 °C for several weeks. To obtain higher concentrations of fibroin, reverse dialysis can be performed. For example, an aqueous solution of polyethylene glycol (PEG) could be used instead of pure water during dialysis.41 As-obtained regenerated aqueous fibroin solutions can be processed into several forms, such as films,42 meshes,43 sponges,44 and hydrogels.45–47
1.4 Characteristics of SF as biomaterials
SF derived from the B. mori, as a natural protein biopolymer, has been widely studied in biomedical applications, mainly due to its mechanical performance, tunable degradation ability, and its biocompatibility with various biological tissues such as skin and cartilage.48,49The proper balance of breaking strength, modulus, and elongation makes SF ductile and tough.50 Naturally produced SF fiber has a high ultimate tensile strength of 300–740 MPa. It also has a large breaking strain and high toughness exceeding those of synthetic fibers such as Kevlar. The strength-to-density ratio of silk is up to ten times higher than steel.51 Considering these outstanding mechanical features, the high tensile strength of silk fiber is suitable for suture, while its flexibility is appropriate for preparing load-bearing applications. The strength and stiffness of silk are dictated mainly by β-sheet crystallites due to intermolecular hydrogen bonds, van der Waals forces, and hydrophobic interactions. These contribute significantly to the stability of the structure. Whereas the toughness of the material is attributed to the presence of a semi-amorphous matrix. Therefore, collectively, the semi-amorphous matrix and β-sheet crystallites govern the mechanical behavior of SF-based materials.52
As a protein-based biomaterial, silk fibroin degrades in vitro and in vivo by proteolytic enzymes such as protease XIV, α-chymotrypsin, proteinase K, papain, and collagenase.53 During the enzymatic degradation, the hydrophilic amorphous domains degrade firstly. These domains include the C-termini, N-termini, linker segments in the heavy chain, and the light chain. Subsequently, the more crystalline domains with packed structures start to degrade.54 SF-based materials with higher β-sheet contents degrade slower. In addition, the degradation time of SF depends on some other factors such as external pressure, concentration, treatment conditions, and characteristics of the implanted site.55 The degradation behavior of natural fibers is also different from regenerated silk materials. The degradation rate of the natural fibers is much slower due to their compact semicrystalline structure with a highly oriented fibrous network.56 The biodegradation of SF can be investigated both in vitro and vivo. In vitro, it can be studied based on mass loss, morphological changes, and analysis of degraded by-products.57In vivo, (i) mechanical characteristics tests following implantation in animal models for a specific time, (ii) structural integrity analysis via histological examinations, (iii) fluorescent staining and various biochemical assays can be used to study silk biodegradability.58
In terms of biodegradability, the use of SF in biomedical applications has several advantages compared to other synthetic and natural polymers. Even though the use of biodegradable polyesters such as PLA, PCL, PGA is approved by FDA for biomedical applications,59–62 the release of acidic by-products during their degradation is an issue of concern. However, there are no such issues associated with silk degradation. Because the enzymatic degradation of SF leads to the formation of smaller polypeptides and eventually amino acids, which are easily absorbed or metabolized in vivo. Unlike other natural polymers such as gelatin and collagen, SF-based materials can retain their mechanical strength over a long time in vivo, which is advantageous in tissue engineering applications where slow degradation and load-bearing capacity are required.63 The degradation of SF occurs via surface erosion, unlike that of biodegradable polyesters, which undergo bulk hydrolysis degradation.64 Owing to its slow loss of mechanical strength, non-toxic by-products, and controllable degradation rate, SF presents distinct benefits for biomedical applications compared with other synthetic or natural polymers.
Apart from its robust mechanical properties and biodegradability, degummed SF has excellent biocompatibility. In some studies, the presence of outer sericin coating was found to cause immunogenic reactions. Therefore, removing sericin via a degumming process is an essential step for SF processing. SF is an FDA-approved biomaterial and is widely used as a suture material. There are various SF-based commercial products in the market such as Seri® and SeriACLTM®.65
In previous years, many in vitro and in vivo studies confirmed the biocompatibility of SF-based materials. For example biocompatibility of SF-based materials was confirmed by different research groups in vitro by using various cell cultures such as osteoblasts, epithelial cells, fibroblasts, keratinocytes, mesenchymal stem cells (MSCs), and hepatocytes.66
Long-term in vivo biocompatibility of biomaterials based on SF is also assessed by various research groups. For example, after implantation of the silk fibroin films seeded with autologous rat (MSCs) in Lewis rats, a very low inflammatory potential was observed. Histological and immunohistochemical evaluation after 6 weeks revealed the presence of circumferentially oriented fibroblasts, few blood vessels, macrophages at the implant-host interface, and the absence of giant cells. In addition, the inflammatory tissue reaction was less pronounced around the fibroin films compared to collagen or PLA films.67
In another study, various SF scaffolds were implanted subcutaneously or intramuscularly in nude or Lewis rats. In order to investigate the acute and long-term immunogenic potential, gross observations, histological and immunohistochemical evaluations, and real-time PCR analyses were conducted throughout 52 weeks. And, it was found that all the scaffolds were well tolerated throughout the study without any abnormalities.68
In addition to implantable systems, the biocompatibility of injectable systems was also investigated. As an example, the biocompatibility of SF hydrogels was studied in a rabbit critical-sized distal femur defect model over 12 weeks. It was revealed that the injected SF hydrogels did not elicit an immunological reaction and promoted bone remodeling and maturation. Surprisingly, the defects treated by SF hydrogel were more similar to the native bone than that for PLGA-treated defects.69
2. Self-assembled SF hydrogels
Fibroin molecules in the regenerated aqueous solution are primarily found in helical and random-coil conformations. However, this conformation is metastable because of the tendency of fibroin chains to associate and form β-sheet structures in aqueous solutions.70 The less stable helical and random-coil conformations of fibroin are also called Silk I. The more stable β-sheet-rich conformation of fibroin is called Silk II.71 With time, the conformation of fibroin molecules transforms from random-coil to β-sheet, then the β-sheet structures aggregate to form physical crosslinks that organize into hydrogels (Fig. 3B). Gelation kinetics is a function of several parameters, including temperature, pH, concentration, and the presence of residual chaotropic reagents (due to incomplete dialysis). The regenerated solution cannot be stored indefinitely, after some point it can inevitably form a gel due to self-assembly and organization of SF chains. However, it is known that at 4 °C, the regenerated solution can be stored for several weeks depending on the fibroin concentration.39Nogueira et al. have studied the effect of dialysis time and temperature on the gelation kinetics of the regenerated fibroin solution.70 They used a CaCl2–ethanol system to solubilize fibroin, and they conducted dialysis at different temperatures to remove CaCl2–ethanol. They monitored the residual Ca2+ concentration and recorded the sol–gel transition time. The presence of Ca ions in the fibroin solution decreased the tendency of association. As the Ca2+ concentration decreased during dialysis, intermolecular interactions between chains became more and more favorable, and at some point, the gelation took place. It was found that the gelation time gradually decreased with increasing temperature. When the temperature was increased from 10 to 25 °C, the gelation time was decreased from 22 to 3 days. This decrease in gelation time is due to the enhanced fibroin association at higher temperatures due to increased mobility of SF chains. It was also found that when the temperature increased from 10 to 25 °C, the residual concentration of Ca ions increased from 5.70 to 11.09 mg mL−1 at the time of gelation. Despite a significantly higher Ca2+ concentration at 25 °C, the gelation time was only three days which is significantly lower than the gelation time at 10 °C (22 days). Therefore, it was evident that the effect of temperature on gelation kinetics is higher than that of residual Ca2+ concentration. Therefore, it can be concluded that the duration and temperature of the dialysis process are critical parameters during the regeneration process. Moreover, dialysis should be stopped before the solution gels if further processing of the fibroin solution is desired, which is generally the case.
Ma et al. studied the biomineralization of Calcite crystals within SF hydrogels.72 For this purpose, 5, 10, 15, and 25 wt% regenerated fibroin solutions were prepared. Then, CaCl2 was dissolved in each solution at a concentration of 20 mM. The obtained solutions were incubated at 60 °C. Even though the exact gelation time of each solution was not explicitly mentioned, it was said that after 20 h, hydrogels were obtained in all solutions. Obtained hydrogels were immersed in Na2CO3 solutions for 12 h to induce the mineralization of CaCO3 crystals. Furthermore, the effect of fibroin concentration on the morphology of the crystals was analyzed.
Jin et al. studied the biomineralization of hydroxyapatite in fibroin hydrogels for bone tissue engineering applications.73 They prepared 5 wt% fibroin solutions with varying Ca2+ concentrations of 0, 10, 30, and 80 mM. Then, the solutions were incubated at 60 °C for 24 h to induce gelation. The obtained hydrogels were immersed in Na2HPO4 solution at pH 8.5 to promote crystallization and growth of hydroxyapatite crystals. They investigated the effect of phosphate concentration and mineralization time on osteoblast proliferation and physicochemical properties of hydrogels.
Li et al. utilized a freeze-thawing approach to speed up the thermal gelation of fibroin solutions.74 They prepared 1, 2, and 4 wt% fibroin solutions. Then, the solutions were frozen at −7 °C to induce the association of fibroin chains. The solutions were frozen for 6, 24, or 48 h. After thawing the solutions at room temperature, the sol–gel transition of the solutions was optically monitored. It was found that the gelation of 24 h frozen 1 and 2 wt% solutions was observed after 12 and 6 h, respectively. They analyzed the effect of freezing time on gelation rate for 2 wt% fibroin solutions. When the freezing time was decreased to 6 h, the gelation was significantly slowed down, and it took more than two days for the system to gel. When the solution was frozen for 48 h, the frozen solution became insoluble after thawing due to a more significant association of the chains. For the 4 wt% solution, the frozen gel also became water-insoluble. It was suggested that slightly associated chains form during freezing, which can act as potential nucleation sites and facilitate the nucleation and growth of β-sheet crystals, resulting in a decrease in gelation time of the solutions. Due to extensive association, the frozen solutions became water-insoluble and failed to form a gel when the concentration was greater than 2 wt%, or the freezing time was greater than 24 h. Therefore, this method is limited to fibroin solutions with concentrations less than 2 wt%.
In all the examples provided above, the thermal gelation of SF via self-assembly was observed. Regenerated fibroin molecules exhibit strong intra- and intermolecular interactions, facilitating their self-assembly into antiparallel β-sheets and ultimately leading to physical gelation of the solution. However, as seen from the examples above, the thermal gelation process can take several hours to days, depending on SF concentration, solution compositions, and storage conditions. This long duration can limit its applications. Therefore, several strategies were developed to speed up the physical gelation of fibroin via self-assembly of SF chains as summarized in Fig. 4B.
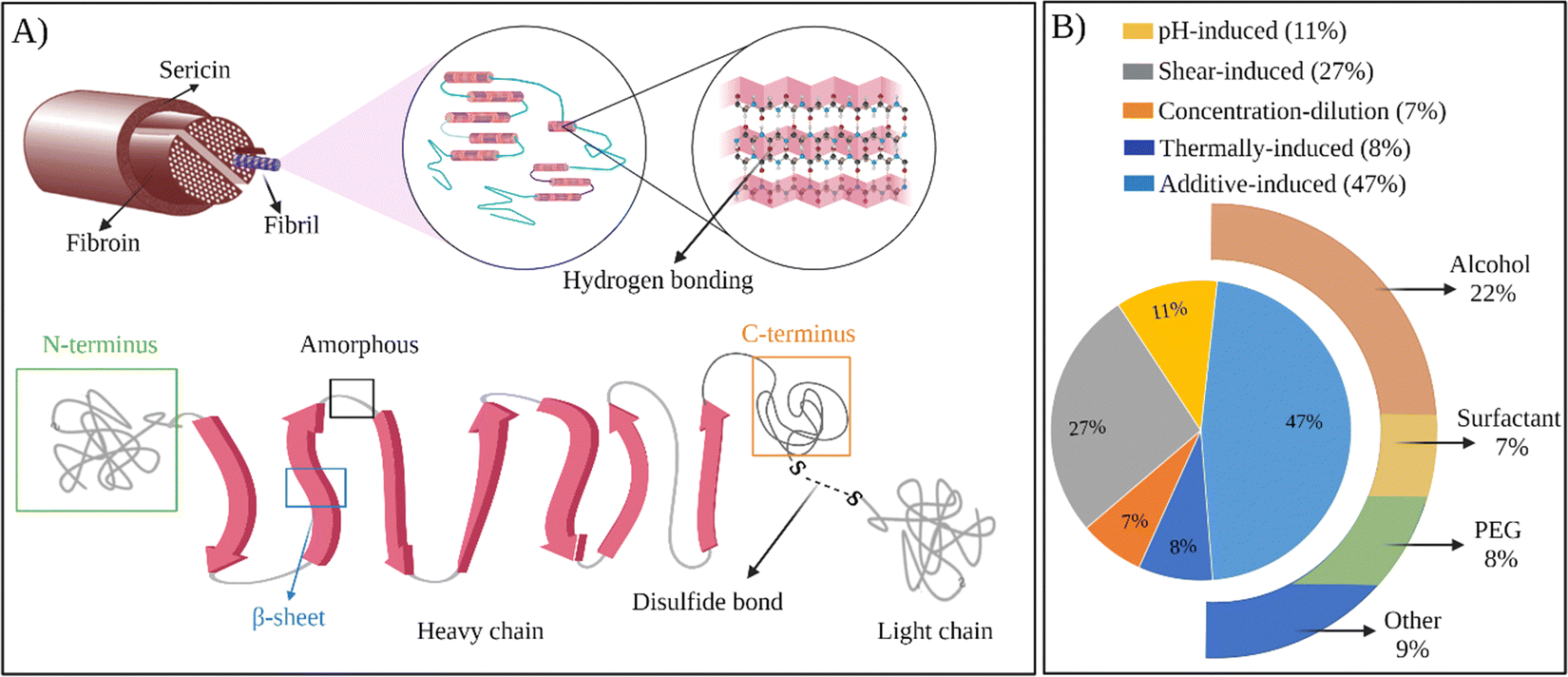 | ||
| Fig. 4 (A) The structure of SF at different scales. (B) Chart representing the usage of methods to prepare self-assembled SF hydrogels. | ||
2.1 pH-Induced self-assembly
The isoelectric point (pI) is the pH of a solution at which the net primary charge of a protein becomes zero.75 Above pI, protein molecules attain a net negative charge, and electrostatic repulsion of the chains can occur, which may decrease the tendency of intermolecular association.76 It is known that lowering the pH from neutral toward the pI reduces the electrostatic repulsion and may expose hydrophobic groups of the proteins. These changes can lead to enhanced protein-protein interactions and induce self-assembly.In their studies, Hirabayashi et al. studied the effect of pH on the gelation rate of 3 wt% fibroin solutions at 20 °C.77 It was found that, when the pH was decreased from 6 to 4, the number of days required for the gelation was decreased from 8 to 1. Ayub et al. also studied the effect of pH on the gelation rate of 3 wt% fibroin solutions at 20 °C. It was found that when the pH was between 6 and 13, the gelation time was around 7–8 days. However, when the pH was between 2 and 4, the gelation took place within one day. It was also reported that when the pH was above 13 or below 1.5, gelation did not take place.78 The reason for failing to form a gel was explained by the extensive repulsion between fibroin chains. However, the hydrolysis of peptide bonds might also contribute to the failure of gelation in these very acidic or basic aqueous solutions. pI of silk fibroin is around 3.8; therefore, when the pH is decreased near pI, carboxylate anions become protonated, decreasing the fibroin chains' net charge. The resulting increased tendency for association leads to the faster gelation of fibroin solutions in these studies.
Nagarkar et al. investigated the mechanism of sol–gel transition of fibroin solutions upon lowering the pH below its pI. They performed rheology and light scattering experiments. Upon lowering the pH from 8.2 to 2.0, 20 nm-sized aggregates consisting of an association of 3–4 fibroin chains formed immediately. With time, these aggregates increased both in size and mass. Moreover, at the time of gelation, the aggregates rapidly agglomerated to form a heterogeneous structure. It was found that the gelation proceeds in two steps. Initially, there is a gradual increase in light scattering and elasticity until the gelation point is reached. Then, these parameters start to increase rapidly, suggesting a nucleation and growth mechanism.79
Fini et al. used fibroin hydrogels as scaffolds to promote the healing of critically sized cancellous defects.69 Fibroin hydrogels were obtained by decreasing the pH of the regenerated fibroin solution to 3.8 and incubating the solution overnight at 50 °C. It was found that the hydrogels did not cause any inflammation, and stimulation of β1 growth factors was achieved. After 12 weeks, complete healing of defects in the trabecular bone of rabbits was achieved. It was also found that fibroin hydrogels significantly improved bone remodeling and maturation compared to the synthetic poly(lactic acid)-PEG hydrogels.
Kambe et al. assessed the use of SF hydrogels as implants to treat ischemic tissue disorders.80 They produced fibroin hydrogels with or without vascular inducing peptide (VIP). During preparation, they mixed aqueous solutions of SF and VIP with citrate buffer at pH 3.0. After incubation at 37 °C overnight, hydrogels were obtained. After the subcutaneous implantation of SF hydrogels in rats for several weeks, it was revealed that endothelial cells successfully migrated through the hydrogels, leading to the infiltration of macrophage and fibroblast cells. With time, macrophage and fibroblasts degraded the fibroin network and produced collagen. Thus, the fibroin hydrogel was gradually replaced by the regenerated tissue. For the hydrogels containing VIP, it was observed that the cell infiltration was accelerated, and the number of blood vessels in the regenerated tissue was doubled compared to the native fibroin gels.
2.2 Additive-induced self-assembly
Gelation time of the aqueous fibroin solutions was decreased from several days to less than a day by reducing the pH below its pI. However, it is still quite long; therefore, various additive-induced self-assembly approaches were undertaken to improve the gelation kinetics of SF solutions.Numata et al. studied the effect of fibroin concentration and ethanol content of the solution on the gelation behavior by rheometry.83 They prepared a stock solution of fibroin at 6.3 wt% concentration. Then, they mixed this solution with different volumes of ethanol. Furthermore, they obtained a series of solutions with decreasing fibroin concentration and increasing ethanol content. To determine the gelation point, they analyzed the time evolution of storage modulus G′ and the loss modulus G′′ of the obtained solutions. In Table 1, the gelation time of each solution is presented. It was observed that, as the ethanol content of the solution increases, gelation time decreases significantly. For example, gelation time was decreased from around 24 h to 5 s when the ethanol content was increased from 10 to 50% (v/v). However, when the ethanol content was increased further, the gelation time started to level off and increase, presumably due to decreased fibroin concentration.
| SF concentration (w/v%) | Ethanol content (v/v%) | Gelation time (s) |
|---|---|---|
| 5.7 | 10 | 8.6 × 104 |
| 5.0 | 20 | 2.2 × 104 |
| 4.4 | 30 | 1.2 × 103 |
| 3.8 | 40 | 5.6 × 102 |
| 3.2 | 50 | 5 |
| 2.5 | 60 | 4 |
| 1.9 | 70 | 15 |
| 1.3 | 80 | 6.6 × 103 |
| 0.7 | 90 | No gelation |
Kaewprasit et al. systematically investigated the effect of different alcohol types such as monohydric (methanol, ethanol, propanol, and butanol) and polyhydric alcohols (1,3-propanediol, and glycerol) on the SF gelation at 25 °C (Fig. 5A).84 They used 5 wt% fibroin solutions, and the alcohol content of the solutions was fixed at 10 wt%. The gelation of the system was monitored by optical density analysis. The gelation of fibroin in the absence of alcohol occurred around 7–8 days. This time was remarkably shortened by alcohol addition. For monohydric alcohols, the gelation time of solutions containing methanol, ethanol, and propanol was around 9.1, 8.7, and 8.2 h, respectively. Interestingly, the addition of butanol resulted in significantly faster gelation (0.7 h). As the polarity of alcohol decreases, the extent of hydrophobic interactions between alcohol and fibroin chains increases, leading to faster chain association. In the case of polyhydric alcohols, presumably due to their higher polarities, the gelation time increased significantly compared to their monohydric counterparts. In their another study, Kaewprasit et al. prepared a series of SF hydrogels via alcohol-induced self-assembly.85 Methanol, butanol, and glycerol were used as triggers to induce self-assembly. They conducted cell attachment and proliferation tests with L929 cell lines on hydrogels produced with different alcohol types. Firstly, all the hydrogels were found to be non-toxic. L929 cells were found to attach to the hydrogel surface one day after the seeding and start spreading. The proliferation and differentiation of the fibroblasts were also achieved within five days throughout the hydrogel scaffolds.
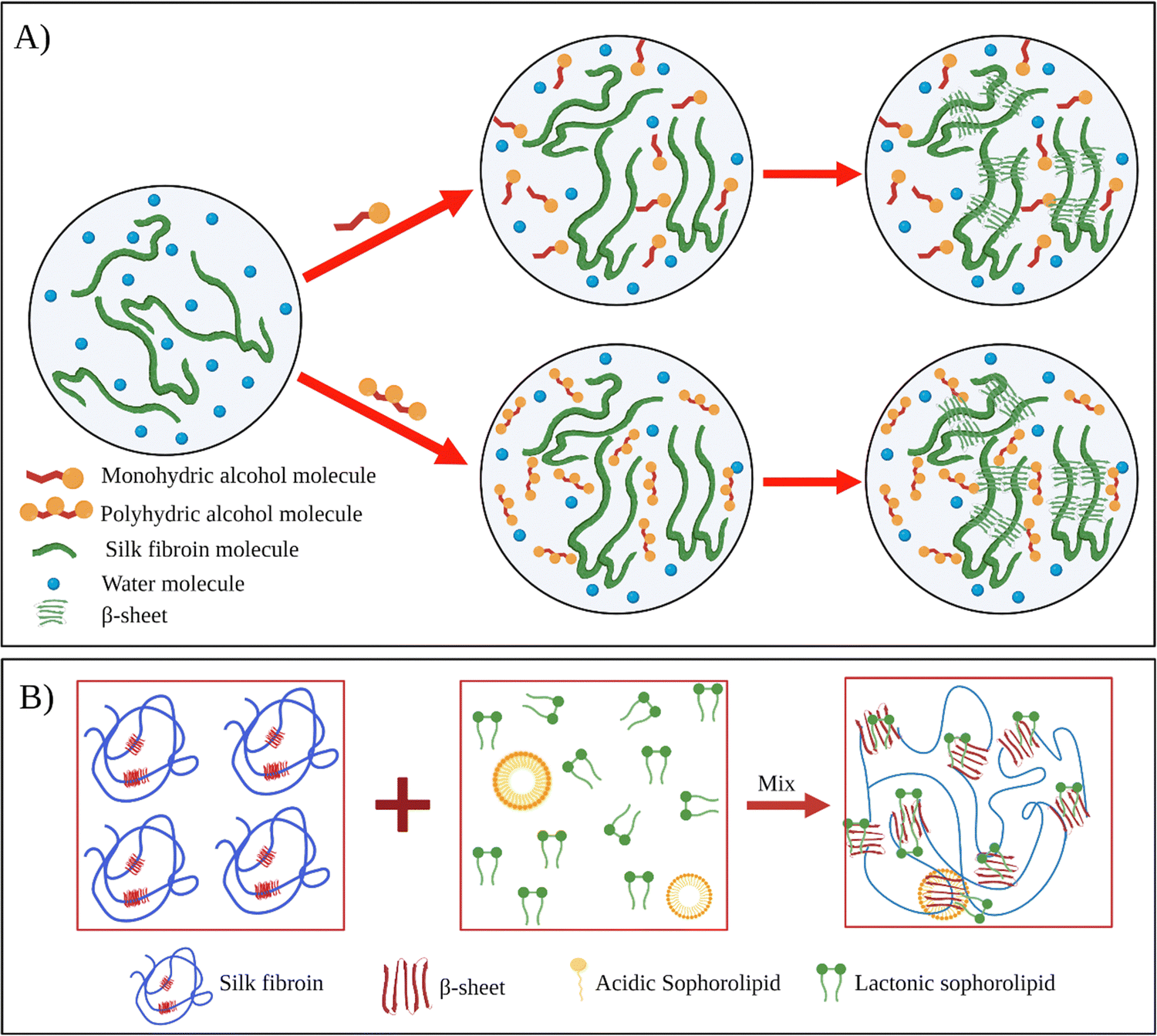 | ||
| Fig. 5 (A) Schematic illustration of silk fibroin gelation via monohydric and polyhydric alcohols. Reproduced with permission from Elsevier,84 (B) the proposed mechanism of SF gelation induced by sophorolipid molecules. Reproduced with permission from Wiley-VCH, copyright 2015.91 | ||
Moares et al. prepared self-assembled SF hydrogels via ethanol-induced self-assembly.86 They investigated the gel formation process in solutions with different ethanol contents. It was found that the gelation time decreased with increasing ethanol content. Without ethanol, it took 72 h for the regenerated fibroin solution to form a gel at 37 °C. The addition of ethanol significantly decreased the gelation time. Depending on the ethanol content, gelation of the solutions took place between 8 min and 7 h. The drug release profile of the hydrogels was also studied by using diclofenac sodium as a model compound. The drug compound was both soluble in water and ethanol. And it was loaded either by dissolving it in the regenerated fibroin solution directly or in ethanol prior to the induction of self-assembly. It was found that the presence of drug molecules did not alter the gelation kinetics significantly. A sustained release profile was obtained for hydrogels loaded in both ways.
Li et al. prepared curcumin functionalized SF hydrogels for enhancing adipogenic differentiation of bone marrow-derived human mesenchymal stem cells (hBMSC).87 Curcumin is a natural phenolic compound obtained from Curcuma longa. In various studies, it has been shown that curcumin has antioxidant, anti-inflammatory, and anticarcinogenic effects. To prepare the composite material, curcumin was dissolved in ethanol, and the resulting solution was mixed with the regenerated SF solution. Then the mixture was incubated overnight to obtain hydrogels. Curcumin was distributed homogenously in the SF matrix. The strong binding of curcumin molecules to the hydrophobic β-sheet domains of SF resulted in the entrapment and stabilization of curcumin molecules. Therefore, the curcumin release from the SF matrix was very low, and the composites retained the high antioxidant ability for at least one month. It was found that the SF film-associated curcumin significantly inhibited the proliferation of hBMSC while promoting adipogenic differentiation. The effect was dose-dependent, with a critical curcumin concentration of 0.125–0.250 mg mL. However, the presence of free curcumin in the cell culture medium inhibited both the proliferation and the adipogenesis of hBMSCs at a concentration range of 5–10 μM, which is almost 1000 times higher than the released curcumin from the composites. These findings showed that the curcumin delivery mode differentially affects the adipogenic differentiation of hBMSC, thus providing a tool to functionalize biomaterial scaffolds with antioxidants for tissue regeneration.
Ribeiro et al. prepared SF-nanohydroxyapatite (nano-HA) composite hydrogels for bone tissue engineering.88 The regenerated fibroin solutions were found to gel within 67 and 48 h, at 37 and 50 °C, respectively. This time was too long to prepare nanocomposites without hydroxyapatite aggregation. Therefore, ethanol was used to disperse nano-HA and facilitate the gelation via self-assembly. During the composite preparation, the use of 70% ethanol was found to be optimum. Because, at higher ethanol concentrations, fibroin precipitation was observed. At lower ethanol concentrations, hydroxyapatite deposition was observed due to increased gelation time. To prevent deposition, gelation should be achieved within 15 min. Aggregation was observed when hydroxyapatite content is greater than 15% relative to SF. Therefore, the composites were prepared with 10 and 15% hydroxyapatite. The incorporation of hydroxyapatite decreased the swelling degree of SF hydrogel. And compressive properties of the hydrogels were improved with increased hydroxyapatite content. The metabolic and alkaline phosphatase activities of osteoblastic cells were improved by incorporating hydroxyapatite into the SF matrix, making this composite a promising material for bone tissue engineering.
Rojas et al. prepared SF–porphyrin composite hydrogels for photodynamic therapy applications.89 Ethanol was used to induce SF hydrogelation. Porphin was dissolved in ethanol, and this solution was mixed with the regenerated SF solution to produce composite hydrogels. Rheological studies were performed with hydrogels with and without porphyrin. It was found that the presence of porphyrin did not interfere with the gel formation. In addition, the encapsulation of porphyrins in the self-assembled polypeptide network prevented their aggregation, thereby significantly increasing the generation of singlet oxygen. It was also shown that porphyrins could diffuse out of the hydrogel and permeate the outer skin layers. Therefore, these composite hydrogels are suitable platforms for the sustained release of photosensitizers for photodynamic therapy applications.
Tomoda et al. produced a controlled-release device composed of SF microparticles embedded in SF hydrogel.90 First, they produced SF microparticles by an atomization method. Then dye molecules were incorporated into the microparticles by adsorption. The loaded microparticles were then mixed with regenerated SF solution. After treatment with 1–1 ethanol solution, gelation of the system was achieved. They studied the kinetics of dye release from the loaded microparticles and hydrogels containing loaded microparticles. It was found that the dye release from the microparticles reached equilibrium in approximately 90 min. Whereas for the hydrogels containing the loaded microparticles, the release persisted for 900 min, exhibiting a release 10 times longer. The dye release from the microparticles followed a Fickian diffusion mechanism. However, SF hydrogels containing SF microparticles followed an anomalous diffusion mechanism, showing that the release was correlated with the network degradation. Therefore, the used strategy holds the potential for developing prolonged-release devices, especially for drugs and bioactive compounds.
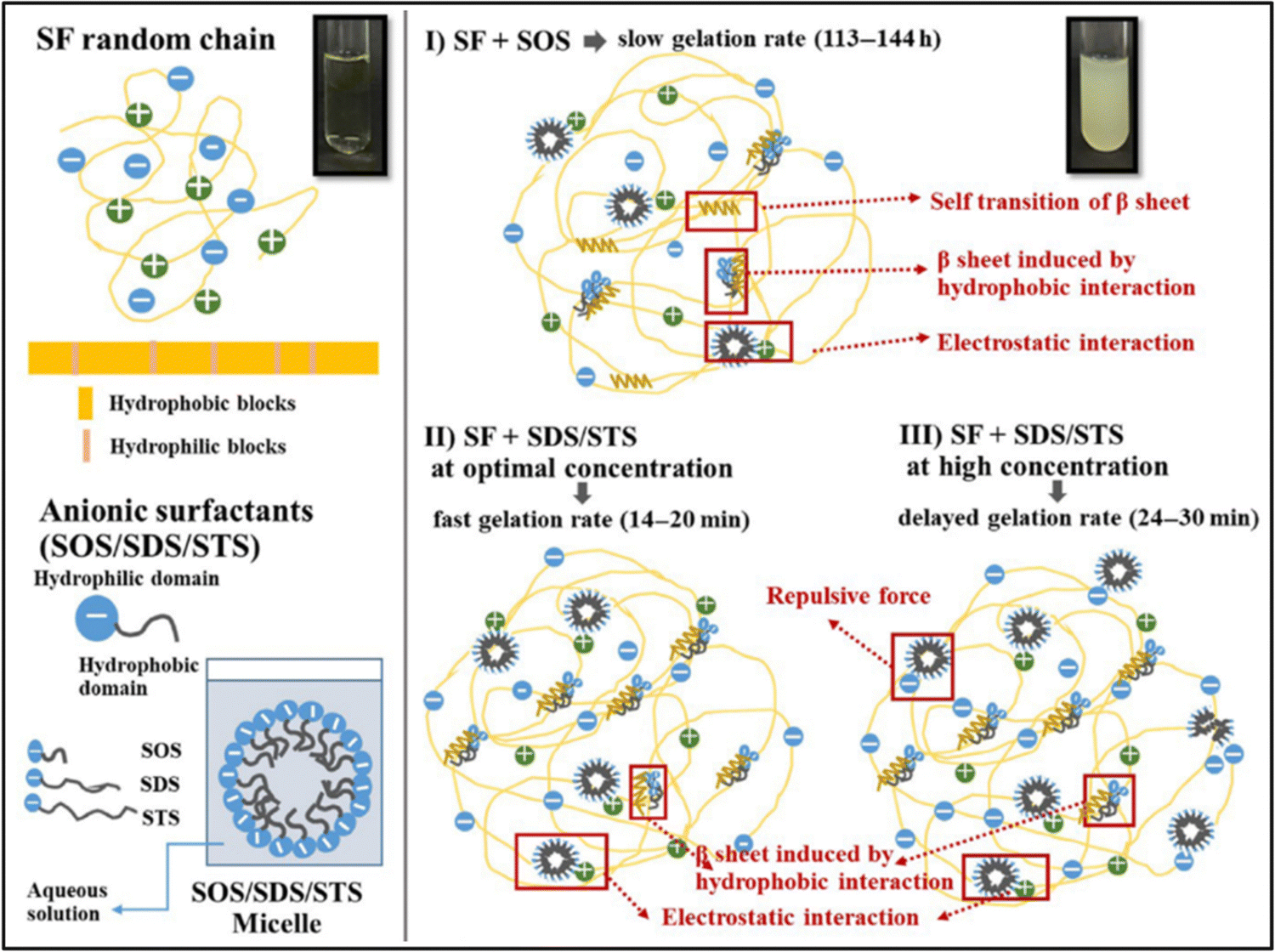 | ||
| Fig. 6 Proposed mechanisms of SF gelation induced by anionic surfactants. Reproduced with permission from Wiley-VCH, copyright 2019.99 | ||
Sun et al. prepared fibroin hydrogels using a cationic surfactant, Dodecyltrimethylammonium bromide (DTAB), as a gelation agent and studied the antimicrobial activity of the hydrogels.94 In the absence of surfactant, gelation of fibroin solution took place over 10 days at 37 °C. Upon surfactant addition, the gelation took place within an hour. To assess the antibacterial activity, zone of inhibition tests were performed with Escherichia coli and Staphylococcus aureus bacteria. The inhibition zone increased with the DTAB content of the hydrogels for both bacteria types, and virgin hydrogels failed to produce an inhibition zone.
Park et al. studied the effect of various surfactant types on the gelation of fibroin solution through self-assembly.95 They used neutral Triton X-100, anionic sodium dodecylbenzene sulfonate (SDBS), and several cationic alkyl ammonium bromide surfactants. In the absence of any surfactant, the fibroin gelation took place within 70 h. In the presence of neutral surfactant, the gelation time was gradually decreased with increasing surfactant concentration up to 50 mM. At 50 mM surfactant, the solution gelled in 15 h. With further increase in surfactant concentration, the gelation time was leveled off. At 100 mM, the gelation time was around 12 h. In the presence of anionic surfactant, the gelation was significantly faster, and at 100 mM surfactant, the solution was gelled within an hour. They used three different cationic surfactants: octyltrimethylammonium bromide (OTAB), dodecyltrimethyl ammonium bromide (DTAB), and hexadecyltrimethyl ammonium bromide (HTAB). HTAB is the most hydrophobic one among these three, and it was not soluble in the fibroin solution beyond 20 mM. The aggregate formation was observed at concentrations lower than 20 mM instead of gelation. Fibroin chains have an overall negative charge at neutral pH values, and the strong electrostatic interactions between surfactant and fibroin molecules are believed to cause this precipitation. In the case of DTAB, which has a shorter alkyl tail, at concentrations higher than 20 mM, aggregate formation was observed. However, below 20 mM, gelation took place, but aggregate formation and phase separation of the solution were also observed. When OTAB was used, which has the shortest alkyl chain, above 20 mM gelation was accompanied by aggregation and phase separation. Below 20 mM, gelation was achieved without aggregate formation or phase separation. However, the gelation time was very long, around 60 h. In the study mentioned previously, the authors obtained different results when the DTAB surfactant was used to induce gelation. This might be due to the difference in SF concentrations and the purity of the fibroin solution after the dialysis process. Anionic surfactants were the mostly used surfactants to induce fibroin gelation in general.
Zhang et al. used anionic sodium N-lauroyl sarcosinate (SNS) surfactant to produce SF hydrogels.96 It is an amino acid-based surfactant, and it is widely used in a variety of cosmetic and medical applications. They systematically studied the effects of fibroin concentration, surfactant concentration, and temperature on gelation behavior via optical density measurements. The native SF solution took 8–10 days to form a gel at 37 °C. At increased SNS concentrations, the gelation time decreased. A distinct reduction appeared at SNS levels below 4 g L−1, while a steady curve was observed for SNS concentrations above 4 g L−1. The shortest gelation time (20 min) was observed when the SNS concentration was 3–5 g L−1, and the SF concentration was 10–15 g L−1. And, at a given fibroin and SNS concentration, the gelation time decreased with increasing temperature. L929 cells were cultured on the surface of hydrogels for seven days. The cells grew and proliferated steadily on the surfaces. However, it was observed that the viability of cells was lower in hydrogels containing SNS compared to native fibroin gels due to the low oral toxicity of SNS.
Dubey et al. used sophorolipid (SL) as a gelling agent.91 SLs are carbohydrate-based amphiphilic biosurfactants derived from the non-pathogenic yeast Candida bombicola. SLs have several advantages over other surfactants. SLs are natural compounds derived using a completely biological process. Thus, SLs have improved biocompatibility and reduced environmental impact. The addition of SL to fibroin solution accelerated SF's gelation near physiological pH, and gelation time was reduced from a few weeks to a few hours (Fig. 5B). Fibroin hydrogels thus obtained were lyophilized to form 3D scaffolds. Moreover, it was shown that L929 cells were successfully adhered to and proliferated on these constructs.
In their follow-up study, Dubey et al. investigated the mechanism of SL-induced gelation of fibroin solutions.97 SLs are extracellularly produced by the yeast as a mixture that comprises two forms: acidic and lactonic form. When the carboxylic end of the fatty acid tail remains free, it is known as acidic SL (ASL), whereas internal esterification of this carboxylic group with the 4′ OH of the sugar molecule results in a closed ring structure, that is, lactonic SL (LSL). It was found that upon mixing fibroin solution and SL, the gelation time and mechanism differed depending upon the type of ASL and LSL content of the sophorolipid. For example, when the fibroin solution was mixed with ASL, the gelation started after 10 h, prior to which both the components were found to exist in the solution without any interaction. The shift in equilibrium between the ASL micelles and non-associated molecules led to the diffusion-dependent onset of gelation. However, in the presence of LSL, the fibroin gelation was faster. NMR studies confirmed that the reason for faster gelation was the preferential binding of LSL to fibroin chains, leading to the unfolding of the protein and eventual formation of β-sheets.
Li et al. produced SF hydrogels with improved mechanical properties.98 They used SDS as the surfactant to induce gelation, and the compressive and tensile modulus of the hydrogels were as high as 3.0 and 3.3 MPa when the fibroin concentration was about 15 wt%. It was shown that the mechanical properties mainly depended on the fibroin concentration, while the type and concentration of the surfactant had little effect. They also prepared virgin fibroin hydrogel and ethanol-induced fibroin hydrogel and compared their conformation and morphology with SDS-induced one. It was found that, although the total β-sheet content in the hydrogel almost did not change, the surfactant speeded up the formation of β-sheet structures, more importantly, confined their size. This confinement resulted in smaller average pore size and a more homogenous morphology, which were thought to contribute to the enhanced mechanical properties. In the same study, they also used neutral and cationic surfactants. In the presence of SDS, the gelation time was 35 min. When Triton neutral surfactant was used, the gelation time increased to 2 h. In the presence of cationic surfactants, gelation was not observed. Instead, aggregation or precipitation took place. Through in situ nanoparticle formation, they produced Fe3O4-loaded fibroin hydrogels. With this material, they efficiently catalyzed H2O2-related reactions.
Phosphorylglycerols are negatively charged amphipathic lipids. They are biologically important as pulmonary surfactants in mammals and as membrane components in microorganisms. They can induce conformational changes in several proteins, such as α-lactalbumin, and β-lactoglobulin. Laomeephol et al. used a natural sodium salt (DMPG), to induce fibroin assembly.100 Electrostatic and hydrophobic interactions between the DMPG and SF chains induced the structural transition of SF chains to β-sheet, and consequently, a rapid gel formation was observed within 50 min. The gelation time was controlled by varying the lipid concentration. To assess the potential of the hydrogels as cell carriers, several mammalian cell lines, including L929, NIH/3T3, SaOS-2, and CaSki, were encapsulated into the hydrogel. The SF-based hydrogels supported the normal growth of fibroblasts, corroborating their cytocompatibility. In addition, inhibition in the growth of cancer-derived cell lines was observed. Therefore, these DMPG-induced fibroin hydrogels were shown to be suitable for 3D platforms for biomedical applications, such as cell therapies and tissue regeneration.
 | ||
| Fig. 7 Gelation process after mixing equal volume of SF and PEG400 solutions at room temperature. Reproduced with permission from Elsevier.101 | ||
The same group further investigated the anti-adhesion ability of PEG–SF hydrogels.102 They evaluated the adhesion prevention of these hydrogels after laminectomies in New Zealand rabbits. Laminectomy is a standard operation in spine surgery to reduce spinal cord and nerve pressure. However, scar tissues often form in the spinal canal and adhere to the dura surface, resulting in low back pain post-surgery. For this reason, anti-adhesion materials are needed to isolate the surface of dura from the surrounding tissues physically. They demonstrated that the hydrogels reduced scar tissue formation around the dura and thus reduced epidural adhesion in rabbit models. The hydrogels fully degraded within 8 weeks post-surgery, but the adhesion prevention effect persisted past 8 weeks. The unique material properties of SF and the local, temporal release of PEG were thought to be responsible for this effective adhesion prevention.
The most used additives for the induction of SF assembly were summarized in this section. In addition, tannic acid,103–105 peptide gelators,106–108 carbon dioxide,109,110 and various copolymers111,112 were also employed as additives for the preparation of self-assembled SF hydrogels in various biomedical applications.
2.3 Shear-induced self-assembly
Shear forces are known to stretch polymer chains in solution.113 The stretching of the chains can dissociate intramolecular interactions and make chains more available for intermolecular interactions. Shear-induced gelation has been reported for amphiphilic or associating polymers in poor solvents.114 For associating polymers, increased intermolecular interactions between self-associating chains that undergo non-Gaussian stretching due to flow were argued to lead to shear-induced gelation. The SF protein is an amphiphilic block copolymer that consists of segments with predominantly hydrophobic domains that are phase-separated in the nanometer scale to enable solubilization in water. Therefore, in the regenerated fibroin solution, the chains are found in a metastable state. The shear gradient may cause the unfolding of SF molecular domains and the formation of macromolecule clusters rich in β-sheet content due to increased exposure of hydrophobic domains to water. The subsequent increase in the concentration and overall lifetime of inter-cluster crosslinks and the physical entanglements between dangling fibroin chains could eventually lead to a percolation-like transition into a permanent, physical hydrogel network. Sonication- and vortex-induced self-assembly of fibroin solutions have emerged as practical and gentle methods for preparing SF hydrogels.Sonication-induced gelation method of SF solutions was firstly introduced by Wang et al.119 It was shown that the sol–gel transition of fibroin solution was accelerated by ultrasonication as illustrated in Fig. 8. Since its introduction, this method has found extensive use for various biomedical applications because of several advantages compared to other methods to induce self-assembly. The gelation time can be controlled from minutes to hours based on the sonication parameters such as energy output, duration, and SF concentration. In the time frame between the end of sonication and the onset of gelation, cells or biomolecules such as drugs and growth factors can be mixed with the fibroin solution, and after the gelation, 100% encapsulation can be achieved.120,121 This approach is sufficiently gentle to maintain cell viability during encapsulation and avoids the use of harsh chemical treatments.122,123 It allows accurate cell seeding and homogenous distribution of cells throughout the hydrogel. The use of harsh chemical treatments is avoided. Very importantly, injection into the target tissue or site is possible as shown in Fig. 9.
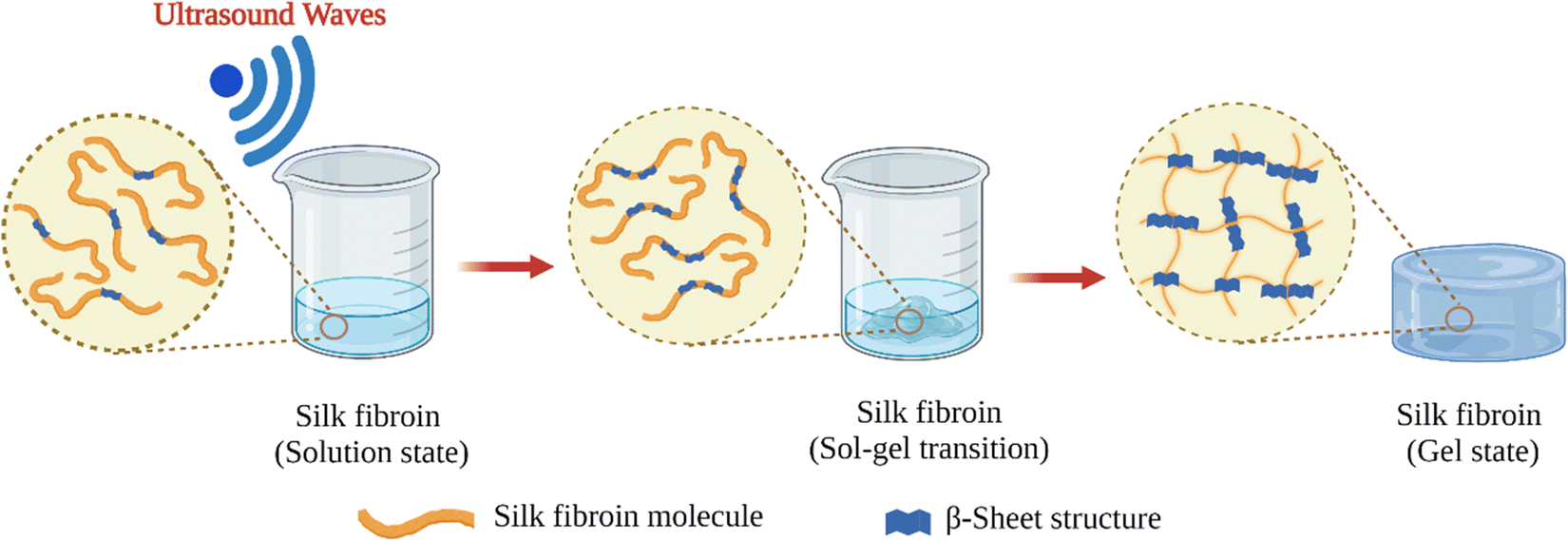 | ||
| Fig. 8 Schematic illustration of sonication-induced SF gelation. | ||
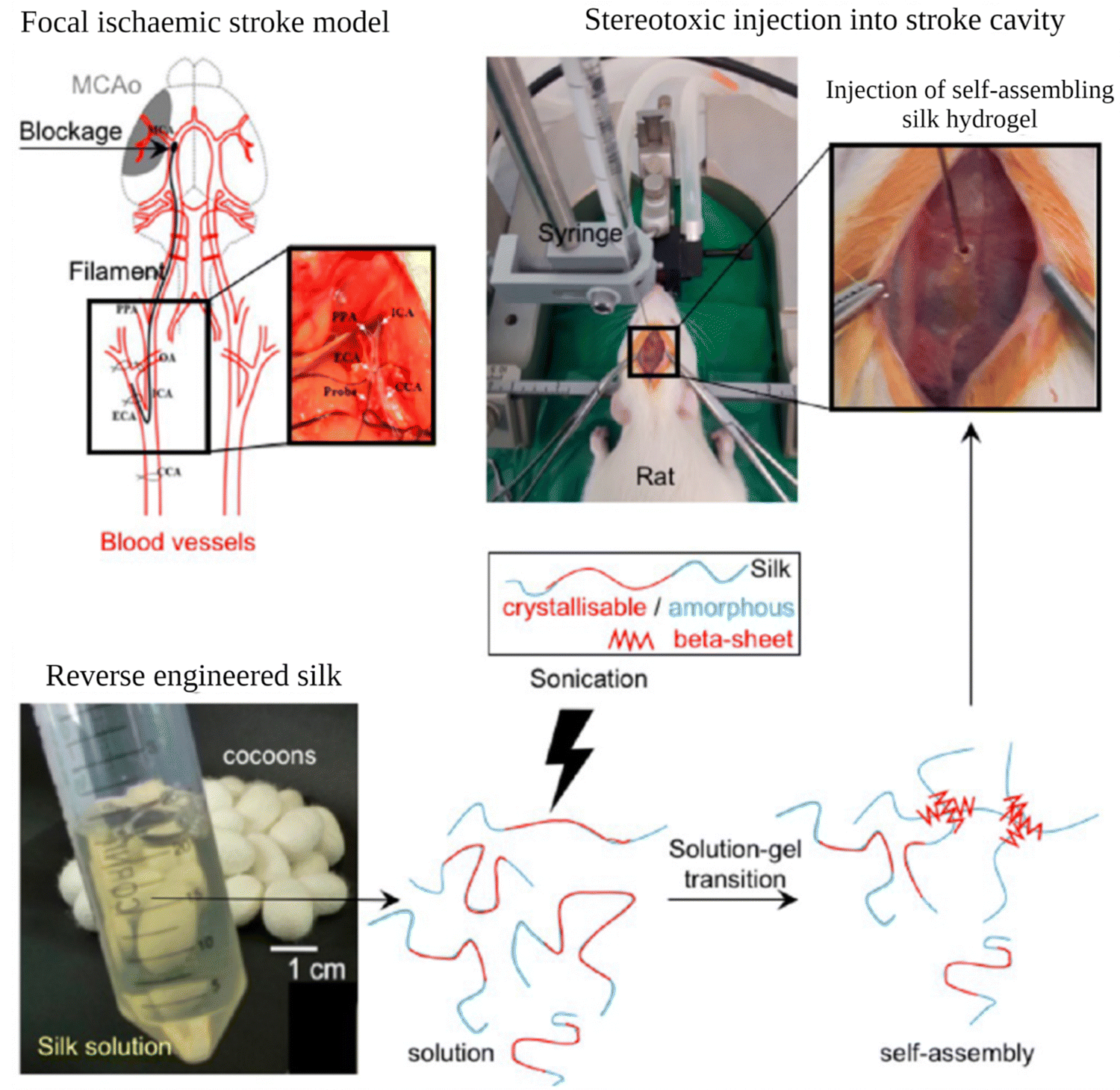 | ||
| Fig. 9 Preparation and administration of self-assembling SF hydrogels into rats with established focal cerebral ischemic stroke. Reproduced from ref. 132. | ||
In their pioneering study, Wang et al. investigated the effects of sonication parameters, fibroin concentration, ion type, and pH on gelation kinetics of fibroin solutions.119 They used 1, 2, 6, and 20% (w/v) fibroin solutions, and sonication was performed at 20% amplitude for 5, 10, or 30 s. Gelation time decreased with increasing concentration and duration of sonication, except for the 20% case where the gelation was slower compared to 6% due to the solution's high viscosity, which may hinder the propagation of sound waves. For example, gelation time decreased from around 40 h to less than an hour, when sonication time increased from 5 to 30 s for 6% SF solution. It was also found that lower concentrations of K+ ions (20–50 mM) and low pH (4) accelerated gelation, whereas the presence of Ca2+ ions and higher concentrations of K+ ions slowed down the gelation process. The mechanical properties of the gels were improved with increasing concentration. For instance, the modulus rose from 500 to 1500 kPa, when concentration increased from 4 to 12%. Cell culture studies were performed using hMSCs. Hydrogels with less than 4% fibroin concentration were found to be too weak to be manipulated during the cell culture studies. Sonicated fibroin solutions at 4, 8, and 12% (w/v) were mixed with hMSCs. Proliferation and differentiation of the cells were assessed by histological analysis and DNA quantification. The cells grew rapidly and proliferated over 21 days in the 4% hydrogel. However, the survival was lower in the gels with higher concentrations, likely due to limitations in the mass transport due to decreased mesh size of the hydrogel.124
SF hydrogels produced via this method are popular biomaterials for bone and cartilage tissue engineering applications. Ding et al. used fibroin hydrogels as scaffolds to deliver rat bone marrow stem cells into bone defects.125 4 wt% fibroin solutions were sonicated at 25% amplitude for 30 s. Then, cell suspensions at different final concentrations of 1.0 × 105, 1.0 × 106, and 1.0 × 107 cells per mL were mixed with sonicated fibroin solution before gel formation. The osteogenic potential of the as-obtained cell/fibroin constructs was investigated by in vitro and in vivo studies. The 1.0 × 107 group exhibited the highest ALP activity, the most calcium deposition, and the strongest osteogenic-related gene expression in vitro. That group also showed a clear increase in bone formation in vivo with superior performance with respect to both new bone volume and trabecular number in the defect area. More importantly, only in the 1.0 × 107 group, the function of the encapsulated stem cells guaranteed a continuous bone formation process in the long term. In their another study, the same group utilized fibroin hydrogels as a delivery vehicle for rabbit maxillary sinus floor augmentation.126 5 wt% fibroin solutions were sonicated at 25% amplitude for 30 s. Then the solutions were mixed with (i) vascular endothelial growth factor (VEGF165), (ii) bone morphogenic protein-2 (BMP-2), and (iii) both. Prior to gelation, the solutions were injected into elevated cavities. It was found that SF gel alone did not promote sufficient bone formation alone in the rabbit model. Nevertheless, angiogenic and osteogenic factors released from SF gel promoted tissue invasion and new bone formation. At the same time, the delivery of a single angiogenic agent was not sufficient for bone regeneration.
Roohaniesfahani et al. studied the effects of simultaneous release of Mg, Sr, and Si ions from injectable SF hydrogels to treat bone defects in mouse models.127 For this purpose, a triphasic ceramic, MSM-10, was used as a delivery agent. Ceramic powders were manually mixed with 4 wt% SF solutions at various concentrations. The solutions were sonicated at 45% amplitude and 25 Hz frequency. The sonication time was adjusted to achieve gelation within 5–10 min. Because faster gelation (<5 min) was impractical for handling and longer gelation (>10 min) resulted in phase separation of MSM-10 particles and SF. The presence of Mg, Sr, and Si ions significantly increased the gelation time of SF solutions and decreased the β-sheet content of the resulting gels. However, the presence of those ions improved the injectability and in vitro osteogenic properties of the gels. The implantation of ceramic/SF hydrogels into subcutaneous defects of mouse models showed reduced immune reaction and increased neovascularization compared to pure SF hydrogels.
Yodmuang et al. prepared SF microfiber-reinforced SF hydrogels as scaffolds for cartilage repair.128 4 wt% SF solutions were sonicated at 12% amplitude for 15 s and then mixed with SF fibers and chondrocytes. In addition, SF fiber-reinforced agarose gels were also prepared for comparison since agarose has been labeled as the gold-standard biomaterial for in vitro cartilage tissue formation. It was found that, while the addition of microfibers significantly increased the moduli of SF hydrogels, it contributed to a lesser extent for agarose hydrogels, likely due to weaker interfacial bonding between the SF microfibers and agarose. Fiber-reinforced SF hydrogels exhibited a 50% improvement in equilibrium modulus and a 60% increase in dynamic modulus at day 42 compared with SF hydrogels without fibers. In addition, higher glycosaminoglycan staining around the fibers was revealed after histological evaluation of fiber-reinforced SF hydrogels. This suggested that newly synthesized proteoglycan was localized to SF microfibers. In contrast, glycosaminoglycan staining was not localized to SF microfibers in fiber-reinforced agarose hydrogels since SF microfibers failed to form a physical bond with the agarose matrix. Excellent chondrocyte compatibility, glycosaminoglycan and collagen matrix deposition make these materials a feasible alternative to agarose for cartilage tissue engineering.
Self-assembled SF hydrogels have also found use in brain repair and regeneration applications. Nieto et al. examined the short- and long-term compatibility of injectable SF hydrogels in the striatum of mouse brains.129 Their preliminary studies showed that a gelation time of 10 min after sonication is required to inject the solution into the target area successfully. Therefore, several sonication conditions were screened by changing the amplitude, sonication time, number of sonication pulses, and concentration. Desired gelation time was obtained when 2 wt% fibroin solutions were subjected to two sonication pulses of 30 s at 60% amplitude. In vivo studies indicated that the animals reasonably tolerated the striatal injection of SF. Because, similar to the saline group, the survival rate exceeded 90%. After the injection, fibroin hydrogels exhibited acceptable biocompatibility with the brain tissue. The gradual degradation of hydrogel resulted in minor immune system activation. After two weeks, hydrogel deposits did not elicit immune reactions or behavioral deficits at all. The degradation of SF was also histologically confirmed in the brain parenchyma of rats one month after intracerebral injection due to the action of brain proteases or myeloid cells and microglia. One month after transplantation, cognitive and sensorimotor deficits were not evident by behavioral or electrophysiological analysis. Overall, the analysis of cell death, inflammatory response, and brain function examined by behavioral tests and electrophysiological analysis confirmed the tolerability of SF by the brain tissue.
Osama et al. explored the use of self-assembling SF hydrogels as a matrix for the minimally invasive delivery of MSCs to the brain.130 In that study, they tried to optimize the conditions for efficient delivery of MSCs. To induce self-assembly, fibroin solutions (1–5 wt%) were exposed to 3 to 6 sonication cycles on ice (one cycle consisted of 30 s on and 30 s off) at 30% amplitude. 1% hydrogel was found to be too soft for the application. Increasing SF concentration improved the stiffness of the gels. 2, 3, and 4 wt%, hydrogels exhibited matrix elasticity similar to the brain tissue. With these hydrogels, cell viability tests were performed in vitro. An initial drop in MSC viability was observed for all the gels. Then it stabilized on day 3 and then steadily increased over the remaining culture period (14 days). They also investigated the effect of the addition time of the cells to the sonicated solution, and it was found that a more homogeneous distribution of cells was obtained when cells were mixed with the solution just 10 min after sonication, where the mixture was still a solution. In their another study, the same group explored the use of self-assembling fibroin hydrogels as injectable platforms for regeneration in the stroke cavity. In vivo studies were performed with rats. It was found that the hydrogels demonstrated stability throughout the study (7 weeks), excellent space-filling and conformity, and excellent biocompatibility with minimum immune response. The gels also interacted with the glial scar and supported endogenous cell proliferation after the experimental stroke.
Self-assembled SF hydrogels produced via sonication have been used as sustained delivery platforms in various biomedical applications. Guziewicz et al. developed a system for the sustained delivery of monoclonal antibodies.131 It was found that protein stability was maintained during encapsulation. After encapsulation, some of the loaded hydrogels were lyophilized. The impact of lyophilization was evaluated by comparing antibody release rates between lyogels and the parent hydrogel material. Lyophilization of antibody-loaded SF hydrogels significantly improved long-term antibody release properties over the parent hydrogel material due to enhanced interactions between SF and antibody molecules after water removal.
Han et al. investigated the effect of delivery of microRNA-675 (miRNA-675), originating from the long non-coding RNA H19, to prevent aging-induced vascular dysfunction in mouse hindlimb.133 They explored the use of a combination of exosomes and SF hydrogel as a delivery platform. miRNA-675 is known to promote skeletal muscle regeneration and inhibit cardiomyocyte hypertrophy. However, naked miRNA is susceptible to degradation by RNase in the extracellular environment. In order to avoid the loss of miRNA during delivery, stem cell-derived exosomes were used to deliver miRNA-675. To induce self-assembly, 4 wt% SF solutions were sonicated three times for 30 s at 30% amplitude. To the freshly sonicated solution, exosome solution was added, and the mixture was kept at 37 °C to allow for gelation. It was shown that mice treated with miRNA-675 exosomes encapsulated in SF hydrogel showed significantly enhanced blood perfusion compared to miRNA-675 exosomes alone. The improved effects resulted from the enhanced retention and stability of miRNA-675 by exosomes and SF hydrogel. Thus, SF hydrogel-based delivery of exosomes represented an effective way to achieve sustained delivery of miRNA for tissue protection.
Zheng et al. incorporated 3-mercaptopropionic acid (MPA)-coated CdTe quantum dots (QD) into SF hydrogels via physical adsorption and entrapment.134 The fluorescence of the QDs remained stable for more than 96 h in PBS buffer or when subcutaneously injected into mice. The cytotoxicity of QDs was significantly reduced when the QDs were incorporated into SF hydrogels. This system was useful for tracking the degradation and distribution of SF biomaterials and can be used as a potential diagnostic tool. He et al. investigated the use of nanographene oxide (NGO) and lanthanide-doped rare-earth up-conversion nanoparticles (UCNPs) loaded SF hydrogels for tumor upconversion luminescence (UCL) imaging and photothermal therapy. NGO absorbs near-infra-red (NIR) radiation and transforms it into thermal energy. Therefore, it has been widely used in photothermal therapy applications. Furthermore, UCNPs can act as luminescent nanoprobes. These two compounds were mixed with sonicated SF solution, and the resulting hybrid hydrogels showed outstanding UCL imaging properties and a photothermal therapy effect under NIR laser irradiation. The in vivo studies presented excellent suppression of tumor growth in the 4T1 tumor-bearing mice. Therefore, this system was shown to hold great potential for combining tumor imaging and antitumor therapy for clinical applications.
Davis et al. used self-assembled SF hydrogels as transplantation devices to treat type-1 diabetes.136 For this purpose, isolated pancreatic islet cells were mixed with freshly vortexed SF solutions, and the mixture was allowed to gel for 2 h at 37 °C. In addition, to enhance islet cell graft survival and function, islets were encapsulated along with extracellular proteins (laminin and collagen IV) and mesenchymal stromal cells. It was found that, after a 7 day in vitro encapsulation, islets remained viable and maintained insulin secretion in response to glucose stimulation. And when islets were encapsulated with collagen IV or laminin, the insulin secretion was increased on day 2 and day 7, respectively. A 3.2-fold synergistic improvement in islet insulin secretion was observed when islets were co-encapsulated with stromal cells and ECM proteins. Furthermore, encapsulated islets had increased gene expression of functional genes; insulin I, insulin II, glucagon, somatostatin, and PDX-1, and lower expression of the de-differentiation genes cytokeratin 19 and vimentin compared to non-encapsulated cells. Therefore, the suitability of self-assembled SF hydrogels was demonstrated for co-encapsulation of various cell types and biomolecules for the treatment of type 1 diabetes.
Managing infection in ischemic, avascular, and necrotic tissues is a significant challenge because drug penetration into abscesses is limited by the lack of vascularity and the dense capsule wall. Therefore, high plasma concentrations of antibiotics are required to overcome this penetration barrier. Moreover, this is problematic due to dose-limiting tissue toxicities and high inter-individual variability in abscess permeability. Kojic et al. proposed an alternative treatment for focal infections, which is based on laser-mediated heating of Au-NP suspended in an injectable and degradable SF hydrogel.137 The use of Au-NP-loaded hydrogels was first evaluated in vitro. The effects of power and duration of laser application on temperature change and bactericidal performance were assessed for various nanoparticle concentrations. Then, in vitro studies were performed with mice infected with S. aureus. It was found that even single 10 min laser treatment of a subcutaneous infection in mice preserved the general tissue architecture while achieving a bactericidal effect. In addition, this procedure allows for case-specific treatment by controlling the nanoparticle concentration, laser exposure, and power. The self-assembled SF hydrogel was found to be very useful for this treatment since, apart from its biocompatibility and biodegradability, it prevented nanoparticle aggregation and ensured spatial stabilization at the injection site.
2.4 Self-assembly induced by the concentration-dilution method
As we have seen so far, the self-assembly of fibroin chains was accelerated via several methods, resulting in the formation of hydrogels. The as-obtained structure is not water-soluble, and this process is irreversible due to the hydrophobicity of the hierarchically assembled structure. Kaplan et al. developed a so-called “concentration-dilution method” to make this assembly process reversible.138 In the first step, the freshly prepared 6 wt% SF solution was concentrated at 60 °C up to 20 wt%. During this process, metastable nanoparticles were formed. After that, the solution was diluted below 2 wt% to induce disassembly. After incubation at 60 °C, the diluted solution was transformed into a hydrogel. Dynamic oscillatory studies proved that the solution and hydrogel states were entirely reversible and could be recycled many times. It was found that solutions produced this way had a higher zeta potential (above −50 mV) than previous SF materials, which tend to be below −30 mV. Due to their amphiphilic nature, the fibroin molecules can form micelles or aggregates in water. It was thought that these structures can migrate to the surface of the assemblies to reduce the free energy of the SF–water system, thus resulting in the increase in negative charge on the surface of the assemblies. It was proposed that β-sheet-rich nanofibers provide enough hydrophobicity for hydrogel formation, while high electrostatic repulsion restrains physical crosslinks, a combination of which allows a reversible sol–gel behavior. More importantly, hydrogels obtained in this way could still flow, unlike typical self-assembled SF hydrogels, due to the restricted aggregation of β-sheet domains. And these hydrogels were found the retain flowability for long times. Unlike self-assembled SF hydrogels, where physical crosslinking kinetics need to be tuned carefully for a suitable injection, these thixotropic SF nanofiber hydrogels can flow on demand once injected and solidify rapidly after shearing is ceased. These features can prevent unwanted leakage from the target site due to insufficient gelation rates. Therefore, the thixotropic property of these hydrogels resulted in better injectability when compared with other forms of injectable self-assembled SF hydrogels where there is a tight time window for the injection. This interesting self-assembly behavior was exploited for various applications by various groups.139,140Wu et al. used this methodology to prepare a fibroin-based injectable and local delivery platform for cancer treatment.141 6 wt% regenerated fibroin solution was first concentrated to above 20 wt% slowly over 24 h at 60 °C to form metastable nanoparticles and then diluted to 0.5 wt%. The diluted SF solutions were incubated for 24 h at 60 °C to induce nanofiber hydrogel formation. Then, doxorubicin (DOX) was directly dissolved into the SF hydrogel to yield DOX-loaded hydrogel. These thixotropic SF hydrogels allowed tunable pH-responsive and sustained DOX delivery. The sustained and tunable release of DOX continued over 8 weeks from these hydrogels and provided long-term control of cytotoxicity of cancer cells. Injection in a breast cancer model showed that the hydrogels improved anticancer capacity, suggesting that this approach is promising for localized chemotherapies.
Ding et al. prepared SF-hydroxyapatite hydrogels as injectable platforms for regenerating irregular bone defects.142 First, SF-coated HA nanoparticles were prepared via an aqueous precipitation reaction using SF as a template and a surface stabilizer. The 6 wt% regenerated fibroin solution was slowly concentrated to 20 wt% over 24 h at 60 °C to form metastable nanoparticles and then diluted to 2 wt% with distilled water. The diluted SF solution was incubated for 24 h at 60 °C to induce the formation of nanofiber hydrogel. These thixotropic SF nanofiber hydrogels and initially prepared SF-coated HA nanoparticles were blended to form injectable nanoscale systems with a homogeneous distribution of HA. The ratio of HA nanoparticles and SF nanofibers was tuned to achieve mechanical properties and organic–inorganic compositions similar to those of natural bone while retaining the ability to be injected. The composite hydrogels supported improved osteogenesis compared to that of SF nanofiber hydrogels. The optimized SF–HA hydrogels demonstrated good cytocompatibility and osteogenic differentiation capacity in vitro and supported bone formation in vivo, suggesting its potential to be used in bone tissue engineering and regeneration.
The same group further extended this work to improve osteogenesis activity.143 Desferrioxamine (DFO) is known to simultaneously induce the secretion of multiple angiogenesis-related factors and cytokines to facilitate more stable vascularization in bone regeneration. Bone morphogenetic protein-2 (BMP-2) is known to enhance osteoinduction. SF nanofibers and HA nanoparticles were used as nanocarriers to load DFO and BMP-2, forming injectable composite hydrogels with multiple angiogenic and osteogenic cues. It was found that the angiogenesis and osteogenesis capacity could be regulated independently by tuning the release behaviors of DFO and BMP-2 to achieve microenvironments for bone tissue regeneration. Both in vitro and in vivo results revealed that these hydrogels created suitable niches to accelerate the regeneration of vascularized bone compared with other SF-based hydrogels reported.
3. Self-assembled SF in interpenetrating network hydrogels
An interpenetrating polymer network (IPN) is a combination of two different polymers, at least one of which is synthesized or crosslinked in the immediate presence of the other.144 These networks are at least partially interlaced on a molecular scale but not covalently bonded to each other and cannot be separated unless chemical bonds are broken. In this way, the properties and structures of different polymers are combined into a new hybrid material. Because of this combination of favorable properties, IPN hydrogels have been used in numerous applications, including drug delivery, wound healing, and tissue engineering.145 Due to its favorable physico-chemical properties and self-assembly behavior, SF was used along with various biopolymers such as gelatin, collagen, hyaluronic acid, and PNIPAM to prepare IPN hydrogels for various applications as listed in Table 2.146,147| Other IPN component | Crosslinking method of the other network | SF network assembly induced by | Ref. |
|---|---|---|---|
| Gelatin | Thermal | Alcohol | 148–150 |
| Photocrosslinking | 151 | ||
| Enzymatic crosslinking | 152 | ||
| Photocrosslinking | Sonication | 153 | |
| Hyaluronic acid | Chemical crosslinking | Thermal | 154 |
| Photocrosslinking | Sonication | 155 | |
| Collagen | Thermal | Sonication | 156 and 157 |
| Chitin | Chemical crosslinking | Alcohol | 158 |
| Cellulose | Thermal | Alcohol | 159 |
| HPMC | Thermal | Thermal | 160 |
| PNIPAM | Chemical crosslinking | Alcohol | 161 and 162 |
| Poly(AC-co-SA) | Photocrosslinking | Thermal | 163 |
| PEG | Chemical crosslinking | Sonication | 146 and 147 |
3.1 SF-Gelatin IPN hydrogels
Gelatin is obtained by breaking the triple-helix conformation of collagen into single-stranded macromolecules. Upon cooling below 30 °C in an aqueous environment, its chains undergo a thermally induced reversible transformation from random-coil to triple-helix conformation.164 This introduces physical crosslinks, which induce gelation at sufficiently high gelatin concentrations. At temperatures near body temperature, 37 °C, its chains attain random-coil conformations, thus exhibiting unacceptably weak mechanical properties, and even can be dissolved away. To improve its mechanical and biological properties, SF was blended with gelatin, and IPN hydrogels were prepared (Fig. 10).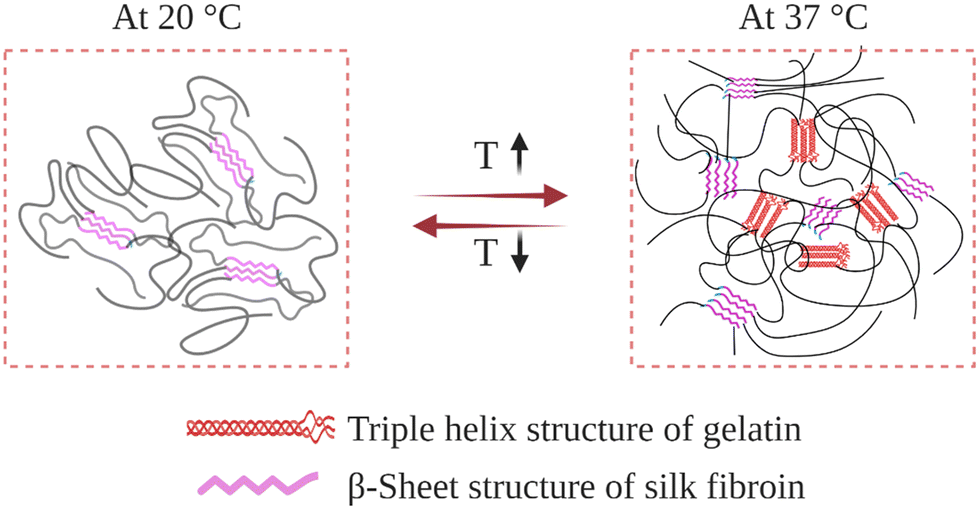 | ||
| Fig. 10 Schematic illustration of the gelatin–SF IPN hydrogel network as it responds to a temperature change. | ||
In their preliminary studies, Gil et al. prepared physical gelatin-SF IPN hydrogels.148 For this purpose, 4 wt% gelatin and SF aqueous solutions were mixed at varying ratios at 40 °C. Then, the mixtures were cast in Petri dishes and kept at 10 °C to induce gelatin triple-helix formation. After that, the mixtures were dried under vacuum, and films were obtained containing 0, 10, 25, 40, and 60 wt% SF. After that, the dried films were immersed in a 3–1 methanol–water solution to induce the assembly of fibroin chains and form β-sheet structures, which resulted in the formation of a second physically crosslinked network. As-obtained films were immersed in water at 5 °C to induce swelling. DSC analysis was performed to discern the relative importance of the triple-helix conformation of gelatin and the β-sheet conformation of SF on the thermal properties of IPN hydrogels. For the hydrogels containing 0, 10, 25, and 40 wt% SF, the helix-coil transition temperature of gelatin was around 34 °C. When SF content was increased to 60 wt%, transition temperature was increased to 46 °C. In the case of hydrogel containing 75 wt% SF, no transition was observed in the DSC analysis. This suggests that most of the crystallized SF network effectively alters the extent to which the gelatin chains rearrange. Since SF in β-sheet conformation is more hydrophobic than gelatin, at sufficiently high concentrations, they may shield gelatin chains from water molecules, thereby restricting their mobility and ability to accomplish conformational change. By performing DSC cooling scans, it was also found that in the presence of an established SF crystalline network, the helix-coil conformational transition of gelatin is thermally reversible between ambient and body temperature. Complementary dynamic rheological measurements were also performed to reveal the effect of the SF physical network on the mechanical properties of the IPN hydrogels. The formation of fibroin β-sheet crystals increased the dynamic elastic modulus of the hydrogels and stabilized the hydrogels to much higher temperatures than the corresponding untreated gels. Even when the hydrogels were heated above the gel–sol transition, they did not liquefy and remained intact. It was also found that the introduction of the β-sheet forming SF network; (i) improved the thermal stability of blends at elevated temperatures and (ii) enhanced the mechanical properties, such as tensile modulus, elongation, and tensile strength.
In a follow-up study, they analyzed the swelling and protein release kinetics of these IPN hydrogels at varying compositions and temperatures below and above the helix-coil transition of gelatin.149 It was found that the swelling degree was significantly higher at 37 °C than 20 °C, indicating that the random-coil conformation of gelatin permitted greater water sorption. It was also found that the IPN hydrogels underwent slight mass loss at 20 °C. However, at 37 °C, gelatin molecules were slowly released into the surrounding medium. Therefore, at 37 °C, the β-sheet crystals of SF were solely responsible for stabilizing the hydrogel. Due to their tailorable composition- and temperature-dependent swelling and release properties, these IPN hydrogels are thought to be attractive biomaterials for thermally responsive biomedical and pharmaceutical applications.
Xiao et al. synthesized a series of gelatin-SF IPN hydrogels with tunable features for potential biomedical applications.151 To form the gelatin 3D network, this time, they performed chemical crosslinking instead of physical crosslinking via the formation of triple-helix structures. For this purpose, they functionalized gelatin chains with methacrylate groups (Gel-MA). Then regenerated SF solution was mixed with Gel-MA solution at various concentrations to obtain compositions containing 0, 8, 14, and 25 wt% SF. These solutions were then poured into PDMS molds, and photo-crosslinking was performed under UV radiation to produce semi-IPN hydrogels. Then, the resulting gels were detached from the molds and treated with aqueous methanol solution to trigger the physical association of fibroin chains to form full-IPN hydrogels. Swelling properties of both semi- and full-IPN hydrogels were studied, and it was found that the introduction of SF chains decreased the swelling degree of semi-IPN gels compared to pure chemically crosslinked Gel-MA gels. After the methanol treatment, the swelling degree was significantly decreased compared to semi-IPN hydrogels since the formation of the β-sheet fibroin network increases the hydrophobicity of the system. The mechanical properties of the gels were studied by compression tests. Before the methanol treatment, for all compositions, the modulus was around 5 kPa. After the treatment, the modulus significantly increased and became 15, 30, and 75 kPa, for IPNs containing 8, 14, and 25 wt% SF, respectively. The enzyme-mediated degradation profiles of full-IPN hydrogels were investigated using collagenase solutions in vitro. The presence of β-sheet domains of SF decreased the degradation rate. An almost complete degradation was observed for the pure Gel-MA sample after 48 h. However, for the IPN samples, the degradation rate decreased significantly, around 25% weight loss was observed after 72 h. It was speculated that the presence of β-sheet structures could act as water-insoluble backbones, thus hindering the permeation of collagenase into the hydrogel. For the full-IPN samples, cell culture studies were performed using NIH-3T3 fibroblasts. It was found that the sample containing 8 wt% SF was the most suitable for cell spreading and proliferation. However, at higher contents of SF, the cell-biomaterial interactions became worse, and the cell proliferation rate decreased. In another study, Xiao and coworkers utilized a similar approach to prepare gelatin–SF IPN hydrogels. However, this time they used sonication to induce the assembly of fibroin chains instead of alcohol treatment (Fig. 11). It was found that obtained hydrogels display high swelling degrees, excellent compressive properties, and resistance to enzymatic degradation. Cell culture studies were performed with osteoblast cells. The cell attachment and viability studies demonstrated that the synthesis process was not toxic for cells, and the cell growth behavior in the hydrogels could be tuned by adjusting the ratio of fibroin to Gel-MA.
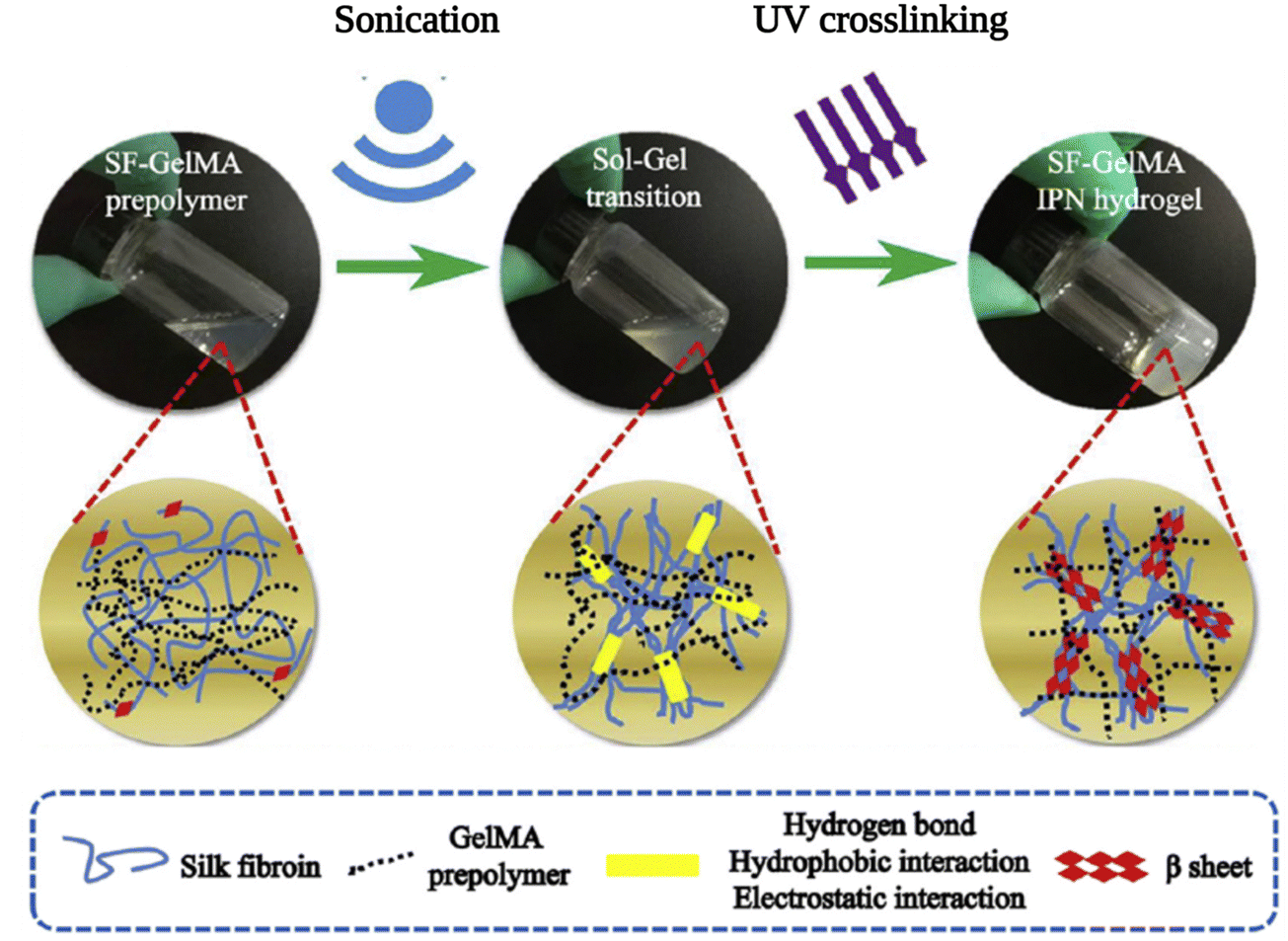 | ||
| Fig. 11 The schematic diagram of the preparation of the SF–GelMA IPN hydrogels. Reproduced with permission from Elsevier.153 | ||
In their studies, Park et al. produced gelatin-SF IPN hydrogels using a different approach to crosslink gelatin chains.152 They first treated an aqueous mixture of gelatin and SF with microbial transglutaminase that forms crosslinks between glutamine and lysine amino acids in gelatin molecules. Then, the physical assembly of SF chains was induced by immersing the hydrogel in aqueous ethanol. The resulting IPN hydrogel was swollen in water and lyophilized, and the second layer of the IPN was formed by repeating the dual crosslinking procedure again. The obtained IPN hydrogels were found to be highly elastic and tough. Compressive studies revealed that IPN hydrogels were much stiffer than pure SF or pure gelatin hydrogels. It was also found that adding another layer of the IPN further enhanced the mechanical properties. The obtained gels were non-cytotoxic and allowed adhesion and proliferation of human dermal fibroblasts.
3.2 SF-Collagen IPN hydrogels
Collagen is the major structural component of the extra-cellular matrix (ECM) found in connective tissues and internal organs.165 There are at least 28 members belonging to the collagen superfamily; however, Type I collagen is one of the most abundant molecules in the body. It assembles into fibers that form the structural and mechanical matrix of bone, skin, tendons, cornea, blood vessel walls, and other connective tissues. Collagen I has a typical chain composition of GPX, where X is any amino acid other than glycine, proline, or hydroxyproline.166 It is soluble in slightly acidic buffers and typically associates after neutralization by adjusting the pH around 7.5. As a result, Collagen I can assemble into a triple-helical structure, which can be hierarchically bundled and physically crosslinked in the form of fibrous networks.167 The mechanical properties of these physically assembled collagen hydrogels are very low. In order to obtain hydrogels with desired physical and biological properties, several groups produced collagen-SF IPN hydrogels.Buitrago et al. produced SF-collagen IPN hydrogels as cell encapsulating platforms.156 During preparation, neutralization at pH 7.0 was performed to induce the association of collagen chains; and sonication was used to trigger SF self-assembly as presented in Fig. 12. In their preliminary experiments, collagen and SF were not subjected to any treatments, and the blended solution did not gel as expected. When collagen was neutralized but SF was not sonicated, a very unstable and weak gel was obtained. When SF was sonicated, but collagen was not neutralized, coagulation of SF was observed without gelation. Only when both the collagen neutralization and the SF sonication were applied, the gelation took place. Hydrogels with controlled compressive moduli were obtained by tuning the SF/collagen ratio and their concentration, which are considered to find a wide range of applications for soft and hard tissues. In addition, the hydrogels were able to be molded easily into various shapes and display elasticity, which is difficult to achieve in the case of pure SF gels (too brittle) or collagen gels (easily losing water then yielded). It was shown that hBMSCs were successfully encapsulated during the gelation process. The encapsulated cells rapidly anchored and spread throughout the network. In addition, shrinkage of the hydrogel was not observed during culture studies. This is a significant improvement since substantial cell-mediated contraction is generally observed for pure collagen gels.
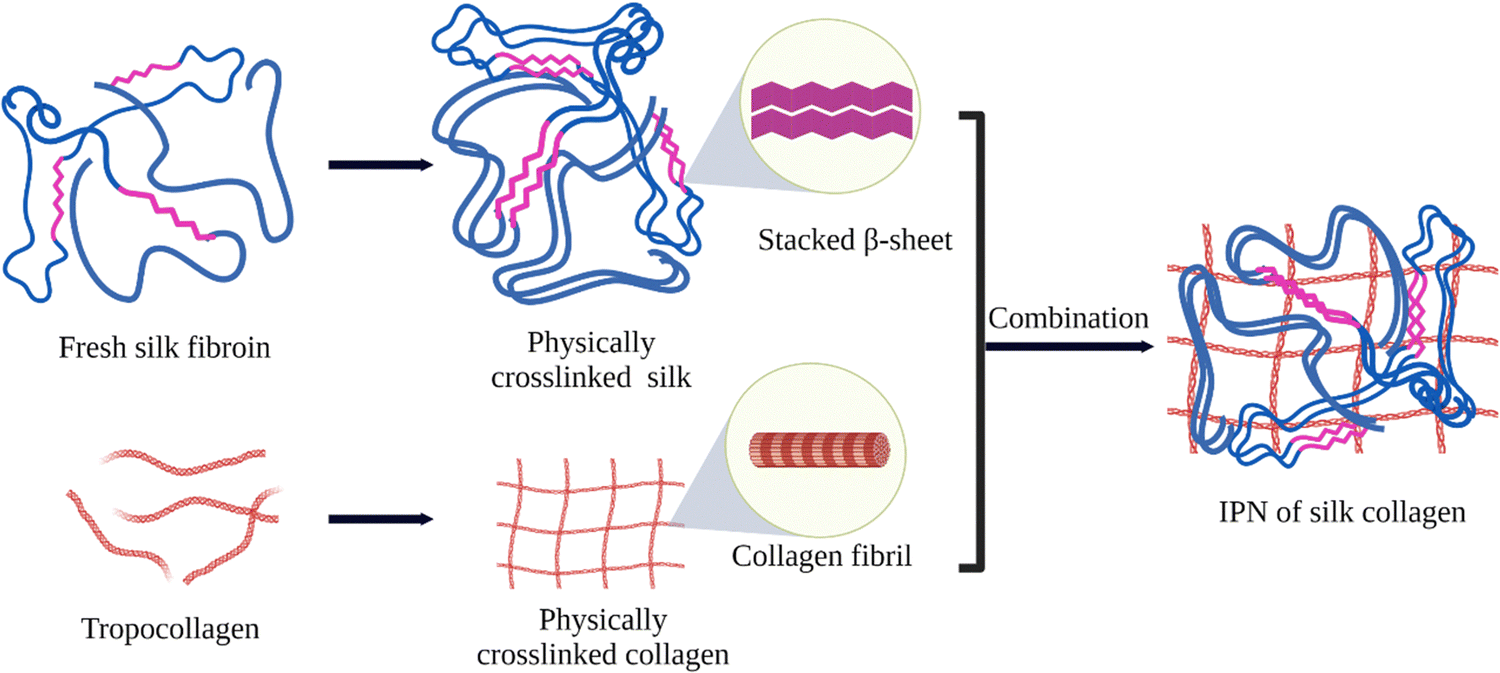 | ||
| Fig. 12 Schematic illustration of the formation of physically crosslinked collagen–SF IPN. Reproduced with permission from Elsevier.156 | ||
Khoo and coworkers prepared SF-collagen IPN hydrogels as biomaterials to elucidate breast cancer migration.157 Firstly, regenerated SF solution was sonicated at low amplitude to prevent quick gelation during preparation. After sonication, the collagen solution was added, and the solution pH was adjusted to 7.4 by adding sodium hydroxide to trigger collagen assembly and then lightly vortexed. IPN hydrogels were obtained after incubation. By changing the concentration of SF while keeping the collagen concentration constant, the compressive modulus of gels was tuned between 30 and 300 Pa. It was found that the invasion of breast cancer cells depended on both SF concentration and pore size. The maximum invasion was observed at intermediate SF concentration, which produced gels with a loss modulus of 100 Pa. At higher SF concentrations, the invasion decreased due to decreased pore size of the IPN network. It was further shown that the cancer cells could be biased toward mesenchymal migration by treatment with exogenous human epidermal growth factor since it increases cell contractility and enhances cell-matrix adhesions. Whereas, after treatment with taxol, the cancer cells were biased toward amoeboid migration since it disrupts cytoskeletal function by stabilizing microtubules.
3.3 SF-Hyaluronic acid IPN hydrogels
Hyaluronic acid (HA) is a naturally occurring, non-immunogenic glycosaminoglycan that plays a vital role in cell–ECM interactions, wound healing, inflammation and angiogenesis.168 HA is highly hydrophilic, thus can provide the hydration of tissues. However, its poor biomechanical performance and rapid degradation limit its applications. Therefore, HA-SF IPN hydrogels were prepared to expand the range of properties and hence applications available to both SF and HA individually. In this way, the hydrophilicity of HA can be combined with the strength and slow degradation of SF in a single material.Tavsanli et al. prepared mechanically robust and stretchable HA-SF IPN hydrogels.154 For this purpose, hydroxyl groups of HA were functionalized with methacrylate units at different degrees between 4 and 25 mol%. First, polymerization of methacrylated HA (MeHA) was performed in the presence of SF at 50 °C. Under these conditions, physical association of SF chains might be expected. However, no gel formation was observed in MeHA and SF aqueous solutions, which is ascribed to the steric hindrance effect of SF chains. Therefore, N,N-dimethyl acrylamide monomer was included in the reaction system as a spacer to connect methacrylated pendant groups of HA chains. The reaction was performed at 50 °C for two days to produce composite hydrogels. FTIR and XRD analyses confirmed the formation of β-sheet domains in the SF network. It is known that SF chains can self-assemble to form these domains even in the absence of any trigger, such as sonication or alcohol addition, at high temperatures. This procedure takes more time, but it would not be problematic in this case since a long time is already required for the chemical crosslinking of HA pendant groups. It was found that the presence of a physical SF network significantly enhanced the mechanical strength and toughness of MeHA hydrogels by providing a pathway for energy dissipation under load. Furthermore, mechanical and swelling properties of the IPN hydrogels were controlled by varying the methacrylation degree of MeHA and concentration of the polymers.
In their studies, Xiao et al. produced cell-laden IPN hydrogels based on SF and MeHA.155 They developed a one-pot, biocompatible approach for synthesizing cell-laden IPN hydrogels via sonication and photocrosslinking (Fig. 13). Methacrylation was achieved by reacting HA with methacrylic anhydride at pH 8. Then SF and MeHA solutions were mixed at room temperature to obtain prepolymer solutions. Then the prepolymer solution was sonicated and exposed to UV radiation sequentially to produce IPN hydrogels. For the cell encapsulation, sterilized prepolymer solution was sonicated and immediately mixed with the cultured MC3T3-E1 mouse pre-osteoblast cells. Then, the suspension was UV crosslinked and incubated at 37 °C. The sol–gel transition time of SF was regulated by adjusting the ultrasonic energy inputs, such as amplitude and time, to allow for further processing and cell encapsulation. The obtained IPN hydrogels were found to possess significantly enhanced mechanical strength compared to the neat SF hydrogels. Moreover, the introduction of the HA network increased the water content of the gels. Cellular fluorescence staining revealed that the osteoblasts could successfully attach to the IPN network. In addition, the cell viability analyses indicated that the synthetic process was biocompatible, and the IPN hydrogel supported the proliferation and spreading of osteoblast cells throughout the hydrogel network, showing that these gels are promising biomaterials for regenerative medicine.
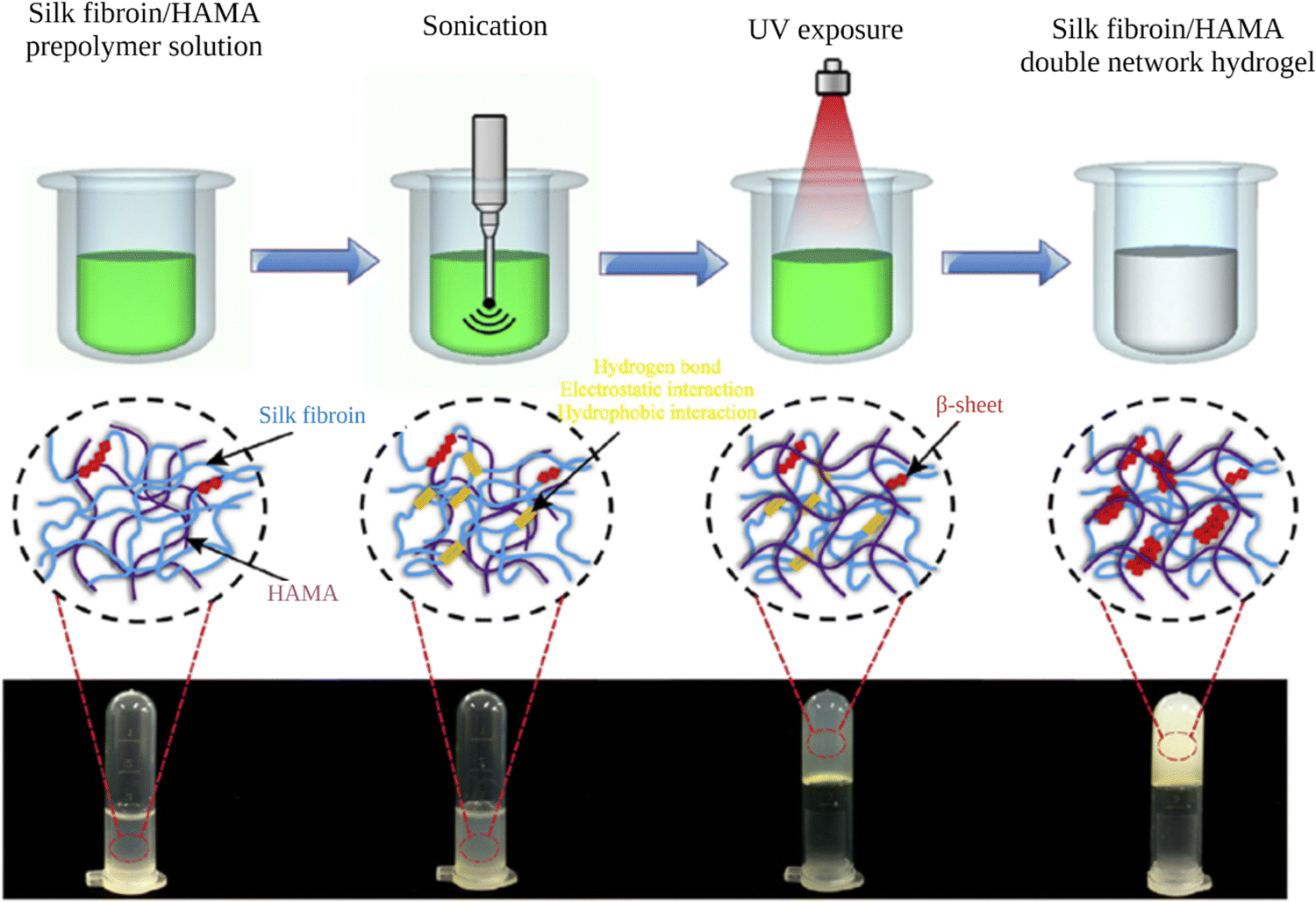 | ||
| Fig. 13 Preparation of HA–SF IPN hydrogels. Reproduced with permission from Elsevier.155 | ||
3.4 SF-Poly(N-isopropylacrylamide) IPN hydrogels
Poly(N-isopropylacrylamide) (PNIPAM) is one of the most widely studied temperature-responsive polymers. PNIPAM chains in aqueous solutions exhibit a coil to globule transition when the temperature increases above a lower critical solution temperature (LCST) of 32 °C.169 Correspondingly, in PNIPAM hydrogels, a volume transition takes place around this temperature. The fact that the LCST of PNIPAM lies close to body temperature and can easily be tuned above and below 37 °C renders PNIPAM-based materials very suitable for biomedical applications. PNIPAM hydrogels provide reversible expansion/contraction in aqueous environments upon thermal stimulus; however, these systems suffer from slow swelling/deswelling response, weak mechanical properties, and limited biocompatibility. To address these concerns, PNIPAM-SF IPN hydrogels were prepared by various research groups.In their studies, Gil et al. prepared PNIPAM-SF IPN hydrogels and investigated their viscoelastic and swelling/deswelling properties.161 For this purpose, aqueous NIPAAM and SF solutions were mixed at different proportions, and free radical polymerization was performed in the presence of N,N′-Methylenebis(acrylamide) crosslinker for 1 day at 10 °C. Then, the obtained semi-IPN gels were immersed in methanol solution to induce the association of SF chains to form a full-IPN. The viscoelastic properties of both semi- and full-IPN hydrogels (before/after methanol treatment) were investigated by dynamic shear oscillation measurements. For the semi-IPN hydrogels, both storage and loss moduli were decreased with the increasing SF content. Whereas, for the full-IPN hydrogels, the moduli values were increased with the SF content, indicating the effect of the physical SF network on the viscoelastic properties. The volume phase transition behavior of semi- and full-IPN hydrogels towards changes in the temperature and ionic strength was analyzed by swelling tests. It was found that the introduction of a physical SF network did not interfere with the volume phase transition of the PNIPAM network. Deswelling kinetics were determined by measuring the swelling degree first at 20 °C (swollen), then at various time intervals after placing them at 45 °C. With increasing SF content deswelling rate was initially increased, exhibited a maximum at 30 wt%, then started to decline. The rate increase in lower SF contents was ascribed to the reduced skin layer effects due to the ability of the physical SF network to act as water-draining channels. As the SF content increases, the tendency of PNIPAM chains to associate via hydrophobic interactions decreases above the transition temperature. This resulted in a decrease in the deswelling rate since the mobility of the PNIPAM network is responsible for the gel contraction above LCST. The introduction of the SF physical network brings about two effects, and there is a trade-off between them. This was thought to be the reason for the decreased deswelling rate at higher SF contents.
Kaplan and coworkers produced macroporous PNIPAM-SF IPN hydrogels and explored their potential as biocompatible actuating devices.162 Aqueous NIPAAM and SF solutions were mixed at different proportions, and free radical polymerization was performed in the presence of N,N′-methylenebis(acrylamide), and N,N,N′,N′-tetramethylethylenediamine crosslinkers for one day at room temperature. The formation of macropores was induced by freeze-drying. The dried gels were dipped in methanol solution to trigger the self-assembly of SF chains to create a physical network. The swelling degree of hydrogels with varying SF content was analyzed at 20 and 45 °C. At 20 °C, it was found to be constant regardless of IPN composition. However, at 45 °C, the swelling degree decreased with increasing SF content. The incorporation of the SF network enhanced the mechanical properties and actuating force. The swelling/deswelling behavior of IPN gels was studied. The introduction of the SF physical network enhanced both swelling and deswelling rates due to reduced skin layer formation. For example, the sample containing 30 wt% SF showed rapid and stable shrinking and swelling at a constant rate by switching the temperature at 40 s intervals between 20 and 45 °C. This shows the applicability of these IPN hydrogels as dynamic soft materials for various sensing and related functions.
3.5 Other self-assembled SF containing IPN hydrogels
Luo et al. prepared IPN hydrogels composed of hydroxypropyl methylcellulose (HPMC) and SF.160 HPMC is a cellulose derivative characterized by its LCST of 62 °C. To prepare the hydrogels, aqueous solutions of HPMC and SF were mixed in different proportions (HPMC/SF w/w, 9/1, 7/3, 5/5) at room temperature. Then, gelation of the mixtures was induced by storing them for 2 h at 70 °C. Aqueous solutions of HPMC are known to gel when the temperature is increased above its LCST, due to dehydration and increasing hydrophobic interactions between pendant groups. Even though no secondary treatment was performed to induce self-assembly of SF chains, the SF chains were aggregated to form β-sheet domains while being kept at 70 °C for 2 h. It was speculated that the exposure of hydrophobic side chains of HPMC, induced by heating above its LCST, drives the association of SF chains via hydrophobic interactions. The morphologies of the hydrogels with different proportions were investigated by confocal laser scanning microscopy. In the 9/1 sample, both networks were able to mix homogeneously. However, with increasing SF content, the degree of phase separation increased. Compressive analyses showed that all samples display elastomeric behavior. The 9/1 sample exhibited the most robust mechanical performance among all. It was the toughest sample with high flexibility and excellent elasticity due to the lack of significant phase separation. In vitro cytotoxicity tests were performed with L929 mouse fibroblasts cells. There was no significant difference in cell viability between the 9/1 hydrogel and the pristine SF hydrogel, with survival rates over 95%. These results demonstrated that the 9/1 IPN hydrogel holds great promise for biomedical applications where both high strength and biocompatibility are required.Kim et al. prepared cellulose-SF IPN hydrogels.159 Cellulose is the greatest renewable natural source of organic materials. Cellulose is insoluble in conventional solvents because of the strong H-bonding intermolecular interactions. Therefore, a LiBr aqueous solution was used to dissolve cellulose. Firstly, aqueous LiBr solutions of cellulose and SF were mixed at 120 °C for 2 min. The obtained mixture was cooled down to 70 °C, where gelation of cellulose took place. After that, methanol treatment was performed to induce the association of SF chains, resulting in the formation of IPN structure. Obtained gels were dipped in water to remove LiBr salts and methanol. Morphologies of the hydrogels were characterized by SEM after freeze-drying. Both cellulose and cellulose-SF samples were composed of long, interconnected nanofibrils which form a 3D network. The introduction of the SF network increased the specific surface area from 199 to 236 m2 g−1. While the porosity and swelling ratio of the gels were significantly reduced with increasing SF content, all the gels possessed a high water-uptake value. By adjusting the cellulose/SF proportion, the enzymatic degradation rate was controlled. The introduction of the SF network improved cell adhesion and proliferation, making these IPN systems valuable candidates for tissue engineering applications.
Zhao et al. produced tough IPN hydrogels composed of SF and poly(acrylamide-co-stearyl acrylate).163 Their unique approach includes the introduction of SDS surfactant, heat-induced SF β-sheet formation, and photoinitiated polymerization of the acrylamide (AM)–stearyl acrylate (SA) network as shown in Fig. 14. Firstly, SDS with variable contents was introduced to the mixture of SA and AM monomers. During the formation of micelles for 2 h at 40 °C, the hydrophobic SA monomers and hydrophilic AM monomers lie respectively inside and outside of the micelles. Afterwards, SF was introduced under stirring. Initially, a physical SF network was formed with β-sheet crosslinks around the shells of the micelles with hydrophobic cores due to the hydrophobic nature of the β-sheet structure. Then, photo-polymerization was performed to synthesize the second network, which has a structure similar to amphiphilic block polymers such that hydrophobic poly(SA) blocks lie in micelles, and hydrophilic poly(AM) blocks stay outside the micelles. The as-prepared IPN hydrogels were found to be mechanically weak due to the absorption of the large amount of water by the hydrophilic poly(AM) network, which can destroy the β-sheet domains. Therefore, an SDS immersion post-treatment was performed to prevent the breakdown of hydrogels by promoting the hydrophobic interactions and electrostatic repulsive forces. The stability of post-treated RSF IPN hydrogels also significantly increased. Mechanical properties of both single network hydrogels and IPN hydrogels were studied by tensile tests. It was found that SF single network hydrogel was too brittle to be clamped during the measurement. Moreover, poly(AM-co-SA) single network hydrogel was too soft. In comparison, all IPN hydrogels exhibited better mechanical properties with both high strength and large elongation at break. The tensile strength of equilibrium IPN hydrogels was between 0.26 and 0.44 MPa, depending on the SDS content. The optimal SDS content was found to be around 1/16 (SDS/AM molar ratio) for the highest tensile strength. This IPN hydrogel also displayed the greatest fracture strain and tensile elongation of 800%. The toughness of these IPN hydrogels was attributed to the presence of both the rigid RSF/SDS network and the soft poly(SA-co-AM) network. The elongation at break values of these IPN hydrogels was greater than the ones in previous works. Such physically interacted IPN hydrogels with robust mechanical and biocompatible features are expected to broaden the applications of SF in biomedical fields.
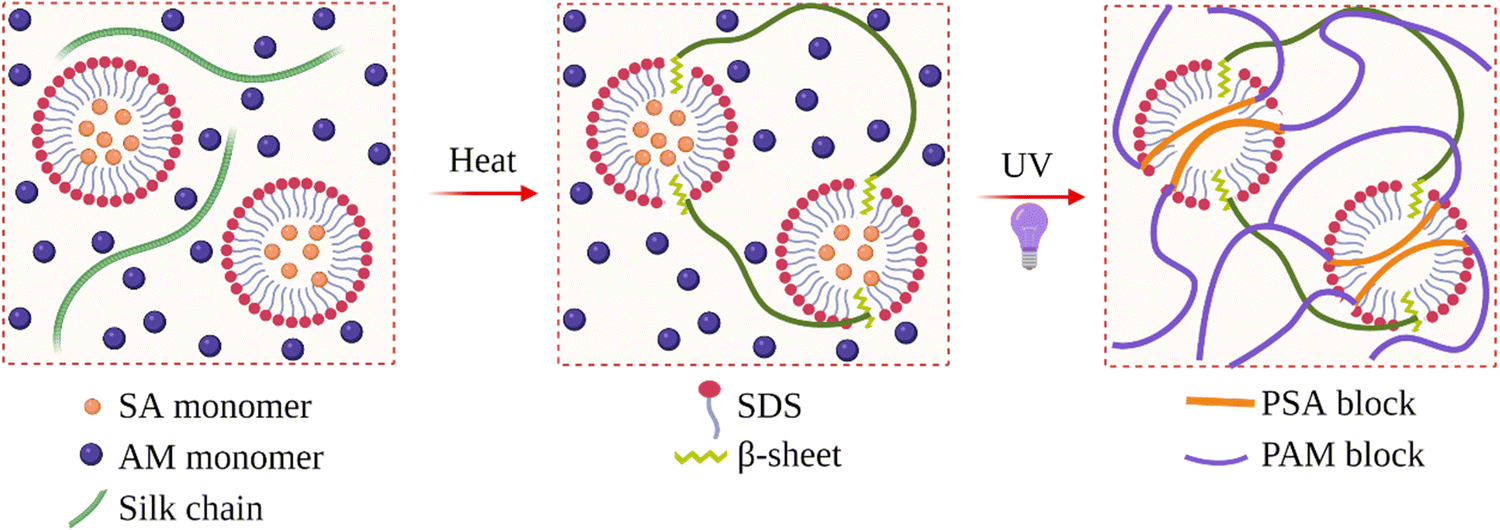 | ||
| Fig. 14 Preparation of IPN hydrogels composed of SF and poly(acrylamide-co-stearyl acrylate). Reproduced with permission from Wiley-VCH, copyright 2019.163 | ||
Yang et al. prepared chitin/SF IPN hydrogels for bile duct restoration application.158 First chitin/SF blend solutions were prepared by mixing each solution at 0 °C at predetermined amounts. Then epichlorohydrin was added to form a crosslinked chitin network. After gelation took place, the samples were taken out from the molds and dipped in ethanol solution to induce the formation of a physical SF network. The obtained IPN hydrogels were found to have high compressive strength (1.36 MPa) and toughness (0.71 MJ m−2 of fracture energy) which is required to be used as a bile duct cannula at the wound site. The IPN hydrogels effectively reduced bile leakage and accelerated the regeneration of the extrahepatic bile duct. In addition, the composite cannula also performed intriguing responsive degradation behavior in different bile concentrations. In the case of bile leakage, they were able to provide a low-bile ambience favor for fibroblast growth. This could be eliminated quickly in enterocoelia in the case of no leakage. These results indicate that chitin/SF IPN hydrogels can be used for the clinical treatment and prevention of postoperative biliary leakage.
4. Self-assembled SF hydrogels in bioink formulations for 3D printing
3D bioprinting is an extended application of additive manufacturing that involves building a tissue or organ layer-by-layer using a bottom-up approach.170,171 3D bioprinting aims to mimic the natural cellular architecture by depositing materials and cells together to restore the structure and functionality of complex tissues. In 3D bioprinting, cells or biomolecules are printed directly onto a substrate in a specific pattern such that the cells can hold together to form the required 3D construct. These biological printable materials are called bioinks. Bioinks are core to 3D bioprinting technology by providing a stable and biocompatible environment in which cells can proliferate and differentiate.172 Traditional inks used for 3D printing are mainly designed for non-biological applications. Therefore, the processing conditions are not suitable for cells.173 For example, 3D printing of thermoplastics including acrylonitrile butadiene styrene and polylactic acid (PLA), requires high temperatures, which are biologically deleterious.Hydrogels are widely applied as bioinks for cell encapsulation for 3D bioprinting. An ideal bioink consists of ECM-like features supporting cell and tissue growth and appropriate mechanical strength.174 Since cells require mild environments, processing and crosslinking of bioink during printing should be compatible. Decellularized ECM (dECM), synthetic and natural polymers are commonly used materials for 3D bioprinting. Although synthetic polymers have advantages due to their consistency in composition. Their use in biomedical applications is often limited due to the need for organic solvents for processing and limited bio-instructive features. In contrast, natural polymers are quite hydrophilic and contain inherent biological properties and functions that resemble the ECM. SF has been widely investigated and used in bioink formulations due to its biocompatibility, cytocompatibility, remarkable mechanical strength, and biodegradability.175,176 More importantly, the physical association of SF chains in aqueous solutions renders SF very useful in bioink formulations since the use of ultraviolet light or deleterious chemical crosslinkers can be avoided during gelation. In their studies, Ghosh et al. performed 3D printing of regenerated SF solution by direct-writing into a coagulating alcohol bath (86% methanol), such that the deposited fibers underwent β-sheet crystallization to form complex 3D micro-periodic architectures with diameters as low as 5 μm.177 However, the methanol bath cannot be used for the deposition of alive cells. Das and coworkers observed that during the printing process, upon applying shear, clogging of micronozzle was observed due to the association of SF chains induced by shear stress, obstructing a smooth flow of the printed filaments.178 It was found that printing SF solution alone without additives is very challenging. Therefore, to develop suitable bioink formulations, SF molecules were used along with non-toxic molecules that induce the self-assembly of SF chains and act as bulking agents.
Kaplan et al. developed SF-based bioinks for 3D printing.179 Several non-toxic polyols such as glycerol, 1,3-propanediol, 1,2,6-hexanetriol, adonitol, or erythritol were used to induce SF assembly. Furthermore, gelatin was used as a bulking agent to facilitate faster layer stacking and print height retention without contributing to the crystallization of SF chains. Bioink formulations containing 10% gelatin/5% SF/1% glycerol (w/v) produced the most robust structures, while SF alone without any additives were the weakest. In their follow-up study, Kaplan and coworkers used the same SF/gelatin/glycerol system to prepare 3D printed soft tissue constructs.180 They optimized the formulation by modulating concentrations of each component to generate materials under optimal printing conditions. These formulations were designed to yield under extrusion forces, undergo rapid gelation upon deposition, be biocompatible in vivo, and show volume retention over time. It was found that a gelatin concentration above 10% (w/v) was optimal for printing in physiological conditions, and a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 of SF to gelatin was needed to maintain a homogenous stable structure. This bioink formulation was used to print patient-specific cheek geometry to reconstruct soft tissue resected due to reconstructive surgical procedures such as tumor removal. The in vivo response of the printed implants was tested in a mouse model. They retain their shape for up to 3 months with minimal inflammatory response and tissue integration.
:1 of SF to gelatin was needed to maintain a homogenous stable structure. This bioink formulation was used to print patient-specific cheek geometry to reconstruct soft tissue resected due to reconstructive surgical procedures such as tumor removal. The in vivo response of the printed implants was tested in a mouse model. They retain their shape for up to 3 months with minimal inflammatory response and tissue integration.
Huang et al. also used a similar approach and prepared bioinks composed of SF, gelatin, and glycerol.181 For this purpose, SF and gelatin were mixed at a fixed ratio of 1/2 (w/w), and glycerol was added at a fixed ratio of 5/1 (w/w) SF to glycerol to induce the association of SF chains. Bacterial cellulose nanofibers (BCNF) were also included in the composition to improve the structural resolution and enhance the mechanical properties of composite hydrogel scaffolds. Following 3D printing of the bioink, freeze-drying was performed to introduce hierarchical porosity to the material via phase separation.182,183 It was found that the tensile strength of the printed sample increased significantly with the addition of BCNFs in the bioink. In vitro and in vivo biological studies were performed with L929 cell lines. This demonstrated that the hierarchical pore structure was beneficial to tissue ingrowth. The pores ranging from 10 to 20 μm served as hosts for cellular infiltration, while the pores with a diameter from 300 to 600 μm ensured sufficient mass transfer and nutrient supply.
Singh et al. prepared bioink formulations composed of SF and gelatin.184 Instead of using glycerol to induce the association of SF chains, they blended SF from B. mori and Philosamia ricini (P. ricini) to form a self-blended hydrogel in a crosslinker-free approach (Fig. 15). They have chosen this approach not only to induce β-sheet formation but also because of the unique properties of SF from P. ricini. For instance, its primary structure contains the cell-binding arginine-glycine-aspartic acid (RGD) units and poly(alanine) sequences, which impart enhanced mechanical properties. The components were blended at various concentrations to rheologically and structurally optimize the formulation and improve the printing efficiency. In that way, anatomical structures such as the human ear were printed with good fidelity and adequate resolution. The obtained constructs supported the growth and proliferation of encapsulated chondrocytes leading to the cartilaginous ECM formation, indicated by the high sulfated glycosaminoglycan and collagen levels, and upregulation of chondrogenic gene expression with minimal hypertrophy of chondrocytes.
 | ||
| Fig. 15 Scheme of bioink formulation composed of B. mori and P. ricini SF. Reprinted with permission from ref. 184. Copyright 2019 American Chemical Society. | ||
Zheng et al. studied the use of SF-PEG hydrogels as self-standing bioinks for 3D printing for tissue engineering.173 As mentioned previously, mixing PEG with SF induces the assembly of SF chains leading to its gelation. They explored the use of non-cellularized SF-PEG gels (PEG removed after printing) and cellularized SF-PEG hydrogels without PEG removal (Fig. 16). The gels with a SF concentration greater than 5% (w/v) showed better shape fidelity during printing, especially after the 3D printed constructs were incubated in phosphate-buffered saline (PBS) over 24 h. However, when the concentration was 10%, the gel was too stiff to be injected through the nozzle. Therefore, 7.5% (w/v) concentration was selected for the remainder of the study. To determine the amount of PEG that can be used safely, cytotoxicity studies were performed with hMSCs. When PEG concentration was 0.1% and 0.5%, the total cell metabolic activity was similar to that of the controls. At 1%, the cell metabolic activity was significantly retarded. When the PEG concentration increased to 2% and 5%, cells stopped growing completely. Animal studies were performed by subcutaneous implantation of 7.5% bioink gels in mice. The cells survived and proliferated in the gel matrix for at least 6 weeks post-implantation, revealing the suitability of these materials for tissue engineering applications.
 | ||
| Fig. 16 Preparation and 3D printing of bioink composed of SF and PEG. Reproduced with permission from Wiley-VCH, copyright 2017.173 | ||
In order to improve biological activity, Li et al. produced growth factor-loaded SF-based bioinks for cartilage tissue engineering.185 Platelet-rich plasma (PRP) was used as an autologous resource of growth factors. The inks were prepared by mixing an equal volume of 80% PEG solution with 10% SF solution, along with an appropriate PRP to obtain final 12.5%, 25%, and 50% (v/v) PRP concentrations. It was revealed that the obtained structures possess proper internal pore structure, good biomechanical properties, and a suitable degradation rate for cartilage regeneration. The cell culture studies were performed by using rabbit chondrocytes. Hematoxylin and eosin staining showed that the 50% PRP group formed cartilage-like lacunae structures at four weeks. Safranin-O and Masson's trichrome staining showed cartilage-specific ECM deposition, especially in 25% and 50% PRP groups. Immunohistochemistry revealed that the production of collagen II was higher in the 50% PRP group than in other constructs. These findings suggested that the samples containing 50% PRP could find uses in cartilage tissue engineering.
In their study, Zhang et al. used a similar approach to develop bioink formulations containing dECM and TGF-β3 growth factor for cartilage tissue engineering.186 For this purpose, SF and dECM were blended at various amounts to obtain final concentrations of 0–7.5% and 0–3%, respectively. The obtained mixtures were then mixed with an equal volume of 80% PEG400 to induce gelation. It was found that for an optimum printing, the concentrations of SF and dECM should be greater than 2.5 and 2.0%, respectively. When the dECM concentration was above 3% and SF concentration was above 7.5%, the bioink was highly viscous and required higher printing pressures that might decrease the viability of the encapsulated cells. Whereas, when the dECM concentration was below 2% and SF concentration was below 2.5%, the resulting low viscosity caused issues during printing due to sagging or collapse. After optimizing the formulation, the BMSCs were encapsulated within the bioink, and 3D printed. The chondrogenic differentiation of BMSCs in the constructs was assessed by the mRNA levels of several markers such as SOX-9, collagen I and II, and aggrecan via rt-PCR. After 28 days, the expression levels of these markers significantly increased, suggesting a successful differentiation of BMSCs. The chondrogenesis potential of the constructs was further assessed in vivo. 3D printed scaffolds were transplanted into nude mice in the subcutaneous region. The deposition of collagen and glycosaminoglycans was shown by Masson's trichrome, safranin-O and immunohistological staining. Also, characteristic cartilage lacunas and round chondrocyte-like cells were observed after 28 days.
They further extended this study and prepared bilayered structures consisting of decellularized extracellular matrix (dECM), PCL, and SF for osteochondral repair.187 For this purpose, two types of bioinks were prepared using cartilage dECM (DCM) and bone dECM (DBM). For each bioink, regenerated SF and PEG were added to the corresponding dECM. Then, BMSCs were encapsulated in both bioinks. To fabricate the bone layer, PCL was first extruded to print the outline, and then the DBM/SF bioink was printed to fill the space. Then, DCM/SF bioink was printed as the cartilage layer on top of the bone layer. First, the delivery capacity of growth factors and the potential of constructs for chondrogenesis or osteogenesis were measured in vitro. In vivo experiments demonstrated that the scaffolds loaded with growth factors promoted osteochondral regeneration in the rabbit knee joint model. This shows the suitability of these bilateral materials for applications in osteochondral regeneration.
5. Concluding remarks and future perspectives
SF is emerging as an essential protein biomaterial of broad biomedical applicability. It is already an established textile fiber, and nearly 1000 metric tons of silk are produced and processed annually. SF-based systems are useful for applications where slow biodegradation and good mechanical properties are critically required. In addition, it is economically advantageous to use silk for biomedical applications because of the available large-scale processing infrastructure of traditional silk textile industries. Over the past decades, SF-based hydrogels have emerged as valuable platforms for various biomedical applications spanning from cell culture and tissue engineering to controlled drug delivery.SF chains in aqueous solutions have a great tendency to self-assemble and form 3D physical networks. This physical crosslinking process is very suitable for biomedical applications because it is cheaper and safer, and the use of toxic chemical agents is not required. Also, the presence of residual chemical crosslinkers after purification is not a concern. In addition, shear-thinning platforms can be developed for minimally invasive operations by utilizing the tendency of SF chains to associate in aqueous solutions. The progress described here has demonstrated the feasibility of using self-assembled SF hydrogels in biomedical applications. The methods used to produce self-assembled SF hydrogels were summarized along with their pros and cons.
Despite the progress made, there is still room for improvements regarding the preparation of physical SF hydrogels. The degumming procedures and batch-to-batch variations of SF must be considered for making reasonable comparisons between different studies. A deeper understanding of the detailed physical gelation mechanisms may help researchers in the future to produce SF hydrogels with greater mechanical strength and modulus. The functionality of the SF-based hydrogels should be improved in the future to increase their role in drug delivery, bioelectronics, and regenerative medicine applications. In order to expand the use of SF-based hydrogels from basic biomedical research into clinical practice, a significant effort should be made to develop construction processes that offer slight batch-to-batch variations in an eco-friendly and time-saving way.
Author contributions
Ozgun Can Onder: writing – original draft, writing – review and editing, conceptualization, methodology, investigation, validation, supervision, project administration. Syeda Rubab Batool: validation, visualization, review and editing. Muhammad Anwaar Nazeer: validation, visualization, review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The schematics are created by using a paid version of BioRender®.References
- C. Belbéoch, J. Lejeune, P. Vroman and F. Salaün, Environ. Chem. Lett., 2021, 19, 1737–1763 CrossRef PubMed.
- A. M. Giacomin, J. B. Garcia, W. F. Zonatti, M. C. Silva-Santos, M. C. Laktim and J. Baruque-Ramos, IOP Conf. Ser. Mater. Sci. Eng., 2017, 254, 192008 CrossRef.
- Y. Cao, F. Liu, Y. Chen, T. Yu, D. Lou, Y. Guo, P. Li, Z. Wang and H. Ran, Sci. Rep., 2017, 7, 11913 CrossRef PubMed.
- M. A. Nazeer, E. Yilgor and I. Yilgor, Polymer, 2019, 168, 86–94 CrossRef CAS.
- Y. Lyu and H. S. Azevedo, Molecules, 2021, 26, 873 CrossRef CAS PubMed.
- O. C. Onder, P. Utroša, S. Caserman, M. Podobnik, E. Žagar and D. Pahovnik, Macromolecules, 2021, 54, 8321–8330 CrossRef CAS.
- S. R. Batool, M. A. Nazeer, D. Ekinci, A. Sahin and S. Kizilel, Int. J. Biol. Macromol., 2020, 150, 315–325 CrossRef CAS PubMed.
- M. A. Nazeer, S. R. Batool and S. Kizilel, Stimuli-responsive Drug Delivery Hydrogels, in Soft Matter for Biomedical Applications, ed. H. S. Azevedo, J. F. Mano and J. Borges, The Royal Society of Chemistry, London, 2021, ch. 22, pp. 542–565 Search PubMed.
- T. I. Mredha and I. Jeon, Prog. Mater. Sci., 2022, 124, 100870 CrossRef.
- M. A. Nazeer, I. C. Karaoglu, O. Ozer, C. Albayrak and S. Kizilel, APL Bioeng., 2021, 5, 021503 CrossRef CAS PubMed.
- P. Ghaderinejad, N. Najmoddin, Z. Bagher, M. Saeed, S. Karimi, S. Simorgh and M. Pezeshki-Modaress, Chem. Eng. J., 2021, 420, 130465 CrossRef CAS.
- Y. Ziai, F. Petronella, C. Rinoldi, P. Nakielski, A. Zakrzewska, T. A. Kowalewski, W. Augustyniak, X. Li, A. Calogero, I. Sabała, B. Ding, L. De Sio and F. Pierini, NPG Asia Mater., 2022, 14, 18 CrossRef CAS.
- C. Rinoldi, M. Lanzi, R. Fiorelli, P. Nakielski, K. Zembrzycki, T. Kowalewski, O. Urbanek, V. Grippo, K. Jezierska-Woźniak, W. Maksymowicz, A. Camposeo, R. Bilewicz, D. Pisignano, N. Sanai and F. Pierini, Biomacromolecules, 2021, 22, 3084–3098 CrossRef CAS PubMed.
- Z. Zhao, Z. Wang, G. Li, Z. Cai, J. Wu, L. Wang, L. Deng, M. Cai and W. Cui, Adv. Funct. Mater., 2021, 31, 2103339 CrossRef CAS.
- F. Valente, M. S. Hepburn, J. Chen, A. A. Aldana, B. J. Allardyce, S. Shafei, B. J. Doyle, B. F. Kennedy and R. J. Dilley, Bioprinting, 2022, 25, e00183 CrossRef.
- W. Sun, D. A. Gregory, M. A. Tomeh and X. Zhao, Int. J. Mol. Sci., 2021, 22, 1499 CrossRef CAS PubMed.
- S. S. S. Aye, Z. H. Zhang, X. Yu, H. Yu, W. D. Ma, K. Yang, X. Liu, J. Li and J. L. Li, Langmuir, 2022, 38, 50–61 CrossRef CAS PubMed.
- M. Farokhi, M. Aleemardani, A. Solouk, H. Mirzadeh, A. H. Teuschl and H. Redl, Biomed. Mater., 2021, 16, 022004 CrossRef CAS PubMed.
- J. Liu, X. Ge, L. Liu, W. Xu and R. Shao, Mater. Adv., 2022, 3, 2291 RSC.
- Y. Zhao, Z. S. Zhu, J. Guan and S. J. Wu, Acta Biomater., 2021, 125, 57 CrossRef CAS PubMed.
- S. Dhawan and K. P. Gopinathan, Dev. Genes Evol., 2003, 213, 435 CrossRef CAS PubMed.
- S. Inoue, K. Tanaka, F. Arisaka, S. Kimura, K. Ohtomo and S. Mizuno, J. Biol. Chem., 2000, 275, 40517 CrossRef CAS PubMed.
- Y. Takasu, T. Hata, K. Uchino and Q. Zhang, Insect Biochem. Mol. Biol., 2010, 40, 339 CrossRef CAS PubMed.
- Z. Dong, P. Zhao, Y. Zhang, Q. Song, X. Zhang, P. Guo, D. Wang and Q. Xia, Sci. Rep., 2016, 6, 21158 CrossRef CAS PubMed.
- R. I. Kunz, R. M. C. Brancalhão, L. D. F. C. Ribeiro and M. R. M. Natali, Biomed. Res. Int., 2016, 8175701 Search PubMed.
- T. Katashima, A. D. Malay and K. Numata, Curr. Opin. Chem. Eng., 2019, 24, 61 CrossRef.
- D. J. Andrew and M. M. Myat, Salivary Gland Development and Programmed Cell Death, in Comprehensive Molecular Insect Science, ed. L. I. Gilbert, Elsevier, Amsterdam, 2005, ch. 2.10, pp. 347–368 Search PubMed.
- Y. Qi, H. Wang, K. Wei, Y. Yang, R. Y. Zheng, I. S. Kim and K. Q. Zhang, Int. J. Mol. Sci., 2017, 18, 237 CrossRef PubMed.
- T. Asakura, Molecules, 2021, 26, 3706 CrossRef CAS PubMed.
- C. Z. Zhou, F. Confalonieri, M. Jacquet, R. Perasso, Z. G. Li and J. Janin, Proteins: Struct., Funct., Genet., 2001, 44, 119 CrossRef CAS PubMed.
- C. Z. Zhou, F. Confalonieri, N. Medina, Y. Zivanovic, C. Esnault, T. Yang, M. Jacquet, J. Janin, M. Duguet, R. Perasso and Z. G. Li, Nucleic Acids Res., 2000, 28, 2413 CrossRef CAS PubMed.
- D. Kaplan, W. W. Adams, B. Farmer and C. Viney, Silk Polymers: Materials Science and Biotechnology, ACS, 1993 Search PubMed.
- L. Lamboni, M. Gauthier, G. Yang and Q. Wang, Biotechnol. Adv., 2015, 33, 1855 CrossRef CAS PubMed.
- P. Aramwit, S. Kanokpanont, W. De-Eknamkul and T. Srichana, J. Biosci. Bioeng., 2009, 107, 556 CrossRef CAS PubMed.
- N. Lin and B. Zuo, J. Biomater. Sci. Polym. Ed., 2021, 32, 1983 CrossRef CAS PubMed.
- D. Chouhan and B. B. Mandal, Acta Biomater., 2020, 103, 24 CrossRef CAS PubMed.
- A. Z. Siavashani, J. Mohammadi, M. Rottmar, B. Senturk, J. Nourmohammadi, B. Sadeghi, L. Huber and K. Maniura-Weber, Int. J. Biol. Macromol., 2020, 153, 317 CrossRef CAS PubMed.
- T. Yucel, M. L. Lovett and D. L. Kaplan, J. Controlled Release, 2014, 190, 381 CrossRef CAS PubMed.
- D. N. Rockwood, R. C. Preda, T. Yücel, X. Wang, M. L. Lovett and D. L. Kaplan, Nat. Protoc., 2011, 6, 1612 CrossRef CAS PubMed.
- D. Ma, Y. Wang and W. Dai, Mater. Sci. Eng., C, 2018, 89, 456 CrossRef CAS PubMed.
- H. Chang, L. Meng, C. Shao, C. Cui and J. Yang, ACS Sustainable Chem. Eng., 2019, 7, 13324 CrossRef CAS.
- Y. Huang, K. Bailey, S. Wang and X. Feng, React. Funct. Polym., 2017, 116, 57 CrossRef CAS.
- S. H. Kim, Y. S. Nam, T. S. Lee and W. H. Park, Polym. J., 2003, 35, 185 CrossRef CAS.
- S. S. Silva, A. Motta, M. T. Rodrigues, A. F. M. Pinheiro, M. E. Gomes, J. F. Mano, R. L. Reis and C. Migliaresi, Biomacromolecules, 2008, 9, 2764 CrossRef CAS PubMed.
- D. Su, M. Yao, J. Liu, Y. Zhong, X. Chen and Z. Shao, ACS Appl. Mater. Interfaces, 2017, 9, 17489 CrossRef CAS PubMed.
- R. Yu, Y. Yang, J. He, M. Li and B. Guo, Chem. Eng. J., 2021, 417, 128278 CrossRef CAS.
- Q. Yu, Z. Meng, Y. Liu, Z. Li, X. Sun and Z. Zhao, Polymers, 2021, 13, 2302 CrossRef CAS PubMed.
- T. P. Nguyen, Q. V. Nguyen, V. H. Nguyen, T. H. Le, V. Q. N. Huynh, D. V. N. Vo, Q. T. Trinh, S. Y. Kim and Q. Van Le, Polymers, 2019, 11, 1933 CrossRef CAS PubMed.
- A. Tutar, R. Yüce-Erarslan, E. İzbudak and B. Bal-Öztürk, J. Mater. Chem. B, 2022, 10, 2912 RSC.
- S. Chen, M. Liu, H. Huang, L. Cheng and H. P. Zhao, Mater. Des., 2019, 181, 108077 CrossRef.
- T. Giesa, M. Arslan, N. M. Pugno and M. J. Buehler, Nano Lett., 2011, 11, 5038 CrossRef CAS PubMed.
- F. Vollrath and D. Porter, Soft Matter, 2006, 2, 377 RSC.
- T. Wongpinyochit, B. F. Johnston and F. P. Seib, ACS Biomater. Sci. Eng., 2018, 4, 942 CrossRef CAS PubMed.
- C. Guo, C. Li and D. L. Kaplan, Biomacromolecules, 2020, 21, 1678 CrossRef CAS PubMed.
- C. Holland, K. Numata, J. Rnjak-Kovacina and F. P. Seib, Adv. Healthcare Mater., 2019, 8, 1800465 CrossRef PubMed.
- Y. Hu, Q. Zhang, R. You, L. Wang and M. Li, Adv. Mater. Sci. Eng., 2012, 185905 Search PubMed.
- R. L. Horan, K. Antle, A. L. Collette, Y. Wang, J. Huang, J. E. Moreau, V. Volloch, D. L. Kaplan and G. H. Altman, Biomaterials, 2005, 26, 3385 CrossRef CAS PubMed.
- Y. Wang, D. D. Rudym, A. Walsh, L. Abrahamsen, H. J. Kim, H. S. Kim, C. Kirker-Head and D. L. Kaplan, Biomaterials, 2008, 29, 3415 CrossRef CAS PubMed.
- Ö. C. Önder, E. Yilgör and I. Yilgör, Polymer, 2016, 107, 240 CrossRef.
- O. C. Onder, E. Yilgor and I. Yilgor, Polymer, 2018, 136, 166 CrossRef CAS.
- P. Utroša, O. C. Onder, E. Žagar, S. Kovačič and D. Pahovnik, Macromolecules, 2019, 52, 9291–9298 CrossRef.
- P. Utroša, Š. Gradišar, O. C. Onder, E. Žagar and D. Pahovnik, Macromolecules, 2022, 55, 5892–5900 CrossRef.
- W. Shi, M. Sun, X. Hu, B. Ren, J. Cheng, C. Li, X. Duan, X. Fu, J. Zhang, H. Chen and Y. Ao, Adv. Mater., 2017, 29, 1701089 CrossRef PubMed.
- L. N. Woodard and M. A. Grunlan, ACS Macro Lett., 2018, 7, 976 CrossRef CAS PubMed.
- M. W. Clemens, S. Downey, F. Agullo, M. R. Lehfeldt, G. M. Kind, H. Palladino, D. Marshall, M. L. Jewell, A. B. Mathur, B. P. Bengtson and M. W. Clemens, Plast. Reconstr. Surg. – Glob. Open, 2014, 2, e246 CrossRef PubMed.
- N. Kasoju and U. Bora, Biomed. Mater., 2012, 7, 045004 CrossRef PubMed.
- L. Meinel, S. Hofmann, V. Karageorgiou, C. Kirker-Head, J. McCool, G. Gronowicz, L. Zichner, R. Langer, G. Vunjak-Novakovic and D. L. Kaplan, Biomaterials, 2005, 26, 147 CrossRef CAS PubMed.
- A. E. Thurber, F. G. Omenetto and D. L. Kaplan, Biomaterials, 2015, 71, 145 CrossRef CAS PubMed.
- M. Fini, A. Motta, P. Torricelli, G. Giavaresi, N. Nicoli Aldini, M. Tschon, R. Giardino and C. Migliaresi, Biomaterials, 2005, 26, 3527 CrossRef CAS PubMed.
- G. M. Nogueira, M. A. De Moraes, A. C. D. Rodas, O. Z. Higa and M. M. Beppu, Mater. Sci. Eng., C, 2011, 31, 997 CrossRef CAS.
- D. Wilson, R. Valluzzi and D. Kaplan, Biophys. J., 2000, 78, 2690 CrossRef CAS PubMed.
- Y. Ma, Q. Feng and X. Bourrat, Mater. Sci. Eng., C, 2013, 33, 2413 CrossRef CAS PubMed.
- Y. Jin, B. Kundu, Y. Cai, S. C. Kundu and J. Yao, Colloids Surf., B, 2015, 134, 339 CrossRef CAS PubMed.
- X. Li, S. Yan, J. Qu, M. Li, D. Ye, R. You, Q. Zhang and D. Wang, Int. J. Biol. Macromol., 2018, 117, 691 CrossRef CAS PubMed.
- N. Ul, Biochim. Biophys. Acta, Protein Struct., 1971, 229, 567 CrossRef.
- S. R. Batool, M. A. Nazeer, E. Yildiz, A. Sahin and S. Kizilel, Int. J. Biol. Macromol., 2021, 185, 165 CrossRef CAS PubMed.
- K. Hirabayashi, Z. H. Ayub and Y. Kume, sen’i gakkaishi, 1990, 46, 521 CrossRef CAS.
- Z. H. Ayub, M. Arai and K. Hirabayashi, Biosci., Biotechnol., Biochem., 1993, 57, 1910 CrossRef CAS.
- S. Nagarkar, A. Patil, A. Lele, S. Bhat, J. Bellare and R. A. Mashelkar, Ind. Eng. Chem. Res., 2009, 48, 8014 CrossRef CAS.
- Y. Kambe, A. Murakoshi, H. Urakawa, Y. Kimura and T. Yamaoka, J. Mater. Chem. B, 2017, 5, 7557 RSC.
- Z. L. Wang, Y. H. Shen, X. Sun, Z. H. Li, X. Y. Wang and Z. Zhao, Microchem. J., 2020, 157, 105036 CrossRef CAS.
- T. Shu, K. Zheng, Z. Zhang, J. Ren, Z. Wang, Y. Pei, J. Yeo, G. X. Gu and S. Ling, Biomacromolecules, 2021, 22, 1955 CrossRef CAS PubMed.
- K. Numata, T. Katashima and T. Sakai, Biomacromolecules, 2011, 12, 2137 CrossRef CAS PubMed.
- K. Kaewprasit, T. Kobayashi and S. Damrongsakkul, Int. J. Biol. Macromol., 2018, 118, 1726 CrossRef CAS PubMed.
- K. Kaewprasit, T. Kobayashi and S. Damrongsakkul, J. Appl. Polym. Sci., 2019, 137, 48731 CrossRef.
- M. A. De Moraes, C. R. A. Mahl, M. F. Silva and M. M. Beppu, J. Appl. Polym. Sci., 2015, 132, 41802 CrossRef.
- C. Li, T. Luo, Z. Zheng, A. R. Murphy, X. Wang and D. L. Kaplan, Acta Biomater., 2015, 11, 222 CrossRef CAS PubMed.
- M. Ribeiro, M. A. De Moraes, M. M. Beppu, M. P. Garcia, M. H. Fernandes, F. J. Monteiro and M. P. Ferraz, Eur. Polym. J., 2015, 67, 66 CrossRef CAS.
- J. E. U. Rojas, B. B. Gerbelli, A. O. Ribeiro, I. L. Nantes-Cardoso, F. Giuntini and W. A. Alves, Biopolymers, 2019, 110, e23245 Search PubMed.
- B. T. Tomoda, M. S. Pacheco, Y. B. Abranches, J. Viganó, F. Perrechil and M. A. De Moraes, Polymers, 2021, 13, 798 CrossRef CAS PubMed.
- P. Dubey, L. Nawale, D. Sarkar, A. Nisal and A. Prabhune, RSC Adv., 2015, 5, 33955 RSC.
- J. Youn, J. H. Choi, S. Lee, S. W. Lee, B. K. Moon, J. E. Song and G. Khang, Materials, 2021, 14, 1287 CrossRef CAS PubMed.
- X. Wu, J. Hou, M. Li, J. Wang, D. L. Kaplan and S. Lu, Acta Biomater., 2012, 8, 2185 CrossRef CAS PubMed.
- S. Sun, F. Zhang, S. Zhang, T. Xing and S. Lu, Biotechnology, 2013, 12, 128 CrossRef CAS.
- J. H. Park, M. H. Kim, L. Jeong, D. Cho, O. H. Kwon and W. H. Park, J. Sol–Gel Sci. Technol., 2014, 71, 364 CrossRef CAS.
- F. Zhang, J. Li, T. Zhu, S. Zhang, S. C. Kundu and S. Lu, J. Biomater. Sci., Polym. Ed., 2015, 26, 780 CrossRef CAS PubMed.
- P. Dubey, S. Kumar, V. K. Aswal, S. Ravindranathan, P. R. Rajamohanan, A. Prabhune and A. Nisal, Biomacromolecules, 2016, 17, 3318 CrossRef CAS PubMed.
- Z. Li, Z. Zheng, Y. Yang, G. Fang, J. Yao, Z. Shao and X. Chen, ACS Sustainable Chem. Eng., 2016, 4, 1500 CrossRef CAS.
- N. Chantong, S. Damrongsakkul and J. Ratanavaraporn, J. Surfactants Deterg., 2019, 22, 1395 CrossRef CAS.
- C. Laomeephol, M. Guedes, H. Ferreira, R. L. Reis, S. Kanokpanont, S. Damrongsakkul and N. M. Neves, J. Tissue Eng. Regen. Med., 2020, 14, 160 CrossRef CAS PubMed.
- X. Wang, B. Partlow, J. Liu, Z. Zheng, B. Su, Y. Wang and D. L. Kaplana, Acta Biomater., 2015, 12, 51 CrossRef CAS PubMed.
- Y. Wang, M. Liang, Z. Zheng, L. Shi, B. Su, J. Liu, D. L. Kaplan, B. Zhang and X. Wang, Adv. Healthcare Mater., 2015, 4, 2120 CrossRef CAS PubMed.
- S. Bai, X. Zhang, P. Cai, X. Huang, Y. Huang, R. Liu, M. Zhang, J. Song, X. Chen and H. Yang, Nanoscale Horiz., 2019, 4, 1333 RSC.
- S. Bin Bae, E. Kim, K. Chathuranga, J. S. Lee and W. H. Park, Polymer, 2021, 230, 124090 CrossRef.
- J. Zhu, J. Zhong, K. Zong, Y. Wang, S. Yang, H. Zhen, L. Tao, S. Sun, L. Yang and J. Li, Mater. Des., 2022, 213, 110347 CrossRef CAS.
- L. Wang, Z. Chen, Y. Yan, C. He and X. Li, Chem. Eng. J., 2021, 418, 129308 CrossRef CAS.
- J. L. Aye, S. S. S. Zhang, Z. H. Yu, X. Ma, W. D. Yang, K. Yuan, B. Liu and X. Li, ACS Biomater. Sci. Eng., 2022, 8, 89 CrossRef PubMed.
- B. Cheng, Y. Yan, J. Qi, L. Deng, Z. W. Shao, K. Q. Zhang, B. Li, Z. Sun and X. Li, ACS Appl. Mater. Interfaces, 2018, 10, 12474 CrossRef CAS PubMed.
- M. L. Floren, S. Spilimbergo, A. Motta and C. Migliaresi, Biomacromolecules, 2012, 13, 2060 CrossRef CAS PubMed.
- M. Floren, W. Bonani, A. Dharmarajan, A. Motta, C. Migliaresi and W. Tan, Acta Biomater., 2016, 31, 156 CrossRef CAS PubMed.
- T. Zhong, C. Deng, Y. Gao, M. Chen and B. Zuo, J. Biomed. Mater. Res. Part A, 2012, 100, 1983 CrossRef PubMed.
- T. Zhong, Z. Xie, C. Deng, M. Chen, Y. Gao and B. Zuo, J. Appl. Polym. Sci., 2013, 127, 2019 CrossRef CAS.
- G. L. He, R. Messina, H. Löwen, A. Kiriy, V. Bocharova and M. Stamm, Soft Matter, 2009, 5, 3054 RSC.
- J. Wang, L. Benyahia, C. Chassenieux, J. F. Tassin and T. Nicolai, Polymer, 2010, 51, 1964 CrossRef CAS.
- N. S. M. Yusof, B. Babgi, Y. Alghamdi, M. Aksu, J. Madhavan and M. Ashokkumar, Ultrason. Sonochem., 2016, 29, 568 CrossRef CAS PubMed.
- Y. Seida, K. Takeshita and Y. Nakano, J. Appl. Polym. Sci., 2003, 90, 2449 CrossRef CAS.
- K. Isozaki, H. Takaya and T. Naota, Angew. Chem., Int. Ed., 2007, 46, 2855 CrossRef CAS PubMed.
- C. Han, Z. Zhang, J. Sun, K. Li, Y. Li, C. Ren, Q. Meng and J. Yang, Int. J. Nanomed., 2020, 15, 10257 CrossRef CAS PubMed.
- X. Wang, J. A. Kluge, G. G. Leisk and D. L. Kaplan, Biomaterials, 2008, 29, 1054 CrossRef CAS PubMed.
- L. Zhu and L. Chen, Polym. Bull., 2021, 1–16 Search PubMed.
- Y. Fu, X. Xie, Y. Wang, J. Liu, Z. Zheng, D. L. Kaplan and X. Wang, ACS Biomater. Sci. Eng., 2021, 7, 2734 CrossRef CAS PubMed.
- Y. Zhang, M. Liu and R. Pei, Mater. Adv., 2021, 2, 4733 RSC.
- T. Yuan, Z. Li, Y. Zhang, K. Shen, X. Zhang, R. Xie, F. Liu and W. Fan, Tissue Eng., Part A, 2021, 27, 1213 CrossRef CAS PubMed.
- O. C. Onder, P. Utroša, S. Caserman, M. Podobnik, M. T. Žnidarič, J. Grdadolnik, S. Kovačič, E. Žagar and D. Pahovnik, Polym. Chem., 2020, 11, 4260 RSC.
- X. Ding, G. Yang, W. Zhang, G. Li, S. Lin, D. L. Kaplan and X. Jiang, Sci. Rep., 2017, 7, 2175 CrossRef PubMed.
- T. Diab, E. M. Pritchard, B. A. Uhrig, J. D. Boerckel, D. L. Kaplan and R. E. Guldberg, J. Mech. Behav. Biomed. Mater., 2012, 11, 123 CrossRef CAS PubMed.
- I. Roohaniesfahani, J. Wang, Y. J. No, C. de Candia, X. Miao, Z. Lu, J. Shi, D. L. Kaplan, X. Jiang and H. Zreiqat, Mater. Sci. Eng., C, 2019, 94, 976 CrossRef CAS PubMed.
- S. Yodmuang, S. L. McNamara, A. B. Nover, B. B. Mandal, M. Agarwal, T. A. N. Kelly, P. H. G. Chao, C. Hung, D. L. Kaplan and G. Vunjak-Novakovic, Acta Biomater., 2015, 11, 27 CrossRef CAS PubMed.
- L. Fernández-García, N. Marí-Buyé, J. A. Barios, R. Madurga, M. Elices, J. Pérez-Rigueiro, M. Ramos, G. V. Guinea and D. González-Nieto, Acta Biomater., 2016, 45, 262 CrossRef PubMed.
- I. Osama, N. Gorenkova, C. M. McKittrick, T. Wongpinyochit, A. Goudie, F. P. Seib and H. V. O. Carswell, Sci. Rep., 2018, 8, 13655 CrossRef CAS PubMed.
- N. Guziewicz, A. Best, B. Perez-Ramirez and D. L. Kaplan, Biomaterials, 2011, 32, 2642 CrossRef CAS PubMed.
- N. Gorenkova, I. Osama, F. P. Seib and H. V. O. Carswell, ACS Biomater. Sci. Eng., 2019, 5, 859 CrossRef CAS PubMed.
- C. Han, J. Zhou, B. Liu, C. Liang, X. Pan, Y. Zhang, Y. Zhang, Y. Wang, L. Shao, B. Zhu, J. Wang, Q. Yin, X. Y. Yu and Y. Li, Mater. Sci. Eng., C, 2019, 99, 322 CrossRef CAS PubMed.
- Z. Z. Zheng, M. Liu, S. Z. Guo, J. B. Wu, D. S. Lu, G. Li, S. S. Liu, X. Q. Wang and D. L. Kaplan, RSC Adv., 2015, 3, 6509 CAS.
- T. Yucel, P. Cebe and D. L. Kaplan, Biophys. J., 2009, 97, 2044 CrossRef CAS PubMed.
- N. E. Davis, L. N. Beenken-Rothkopf, A. Mirsoian, N. Kojic, D. L. Kaplan, A. E. Barron and M. J. Fontaine, Biomaterials, 2012, 33, 6691 CrossRef CAS PubMed.
- N. Kojic, E. M. Pritchard, H. Tao, M. A. Brenckle, J. P. Mondia, B. Panilaitis, F. Omenetto and D. L. Kaplan, Adv. Funct. Mater., 2012, 22, 3793 CrossRef CAS PubMed.
- S. Bai, X. Zhang, Q. Lu, W. Sheng, L. Liu, B. Dong, D. L. Kaplan and H. Zhu, Biomacromolecules, 2014, 15, 3044 CrossRef CAS PubMed.
- K. Wang, W. Cheng, Z. Ding, G. Xu, X. Zheng, M. Li, G. Lu and Q. Lu, J. Mater. Sci. Technol., 2021, 63, 172 CrossRef CAS.
- D. Yao, M. Li, T. Wang, F. Sun, C. Su and T. Shi, ACS Biomater. Sci. Eng., 2021, 7, 636 CrossRef CAS PubMed.
- H. Wu, S. Liu, L. Xiao, X. Dong, Q. Lu and D. L. Kaplan, ACS Appl. Mater. Interfaces, 2016, 8, 17118 CrossRef CAS PubMed.
- Z. Ding, H. Han, Z. Fan, H. Lu, Y. Sang, Y. Yao, Q. Cheng, Q. Lu and D. L. Kaplan, ACS Appl. Mater. Interfaces, 2017, 9, 16913 CrossRef CAS PubMed.
- W. Cheng, Z. Ding, X. Zheng, Q. Lu, X. Kong, X. Zhou, G. Lu and D. L. Kaplan, Biomater. Sci., 2020, 8, 2537 RSC.
- L. H. Sperling and V. Mishra, Polym. Adv. Technol., 1996, 7, 197 CrossRef CAS.
- E. S. Dragan, Chem. Eng. J., 2014, 243, 572 CrossRef CAS.
- Y. Zhang, Y. Cao, L. Zhang, H. Zhao, T. Ni, Y. Liu, Z. An, M. Liu and R. Pei, J. Mater. Chem. B, 2020, 8, 5845 RSC.
- H. Chen, Y. Zhang, T. Ni, P. Ding, Y. Zan, X. Cai, Y. Zhang, M. Liu and R. Pei, ACS Appl. Bio Mater., 2021, 4, 406 CrossRef CAS PubMed.
- E. S. Gil, R. J. Spontak and S. M. Hudson, Macromol. Biosci., 2005, 5, 702 CrossRef CAS PubMed.
- E. S. Gil, D. J. Frankowski, R. J. Spontak and S. M. Hudson, Biomacromolecules, 2005, 6, 3079 CrossRef CAS PubMed.
- E. S. Gil, D. J. Frankowski, M. K. Bowman, A. O. Gozen, S. M. Hudson and R. J. Spontak, Biomacromolecules, 2006, 7, 728 CrossRef CAS PubMed.
- W. Xiao, J. He, J. W. Nichol, L. Wang, C. B. Hutson, B. Wang, Y. Du, H. Fan and A. Khademhosseini, Acta Biomater., 2011, 7, 2384 CrossRef CAS PubMed.
- S. Park, S. Edwards, S. Hou, R. Boudreau, R. Yee and K. J. Jeong, Biomater. Sci., 2019, 7, 1276 RSC.
- W. Xiao, J. Li, X. Qu, L. Wang, Y. Tan, K. Li, H. Li, X. Yue, B. Li and X. Liao, Mater. Sci. Eng., C, 2019, 99, 57 CrossRef CAS PubMed.
- B. Tavsanli and O. Okay, Carbohydr. Polym., 2019, 208, 413 CrossRef CAS PubMed.
- W. Xiao, X. Qu, J. Li, L. Chen, Y. Tan, K. Li, B. Li and X. Liao, Eur. Polym. J., 2019, 118, 382 CrossRef CAS.
- J. O. Buitrago, K. D. Patel, A. El-Fiqi, J. H. Lee, B. Kundu, H. H. Lee and H. W. Kim, Acta Biomater., 2018, 69, 218 CrossRef CAS PubMed.
- A. S. Khoo, T. M. Valentin, S. E. Leggett, D. Bhaskar, E. M. Bye, S. Benmelech, B. C. Ip and I. Y. Wong, ACS Biomater. Sci. Eng., 2019, 5, 4341 CrossRef CAS PubMed.
- Y. Yang, S. Zhang, X. Bian, T. Xia, A. Lu, L. Zhang, Y. Wang and B. Duan, Chem. Eng. J., 2021, 422, 130088 CrossRef CAS.
- H. J. Kim, Y. J. Yang, H. J. Oh, S. Kimura, M. Wada and U. J. Kim, Cellulose, 2017, 24, 5079 CrossRef CAS.
- K. Luo, Y. Yang and Z. Shao, Adv. Funct. Mater., 2016, 26, 872 CrossRef CAS.
- E. S. Gil and S. M. Hudson, Biomacromolecules, 2007, 8, 258 CrossRef CAS PubMed.
- E. S. Gil, S. H. Park, L. W. Tien, B. Trimmer, S. M. Hudson and D. L. Kaplan, Langmuir, 2010, 26, 15614 CrossRef CAS PubMed.
- Y. Zhao, J. Guan and S. J. Wu, Macromol. Rapid Commun., 2019, 40, 1900389 CrossRef CAS PubMed.
- L. Guo, R. H. Colby, C. P. Lusignan and T. H. Whitesides, Macromolecules, 2003, 36, 9999 CrossRef CAS.
- L. E. Cote, E. Simental and P. W. Reddien, Nat. Commun., 2019, 10, 1592 CrossRef PubMed.
- M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2009, 78, 929 CrossRef CAS PubMed.
- P. Bunyaratavej and H.-L. Wang, J. Periodontol., 2001, 72, 215 CrossRef CAS PubMed.
- Z. Z. Khaing and S. K. Seidlits, J. Mater. Chem. B, 2015, 3, 7850 RSC.
- M. Podewitz, Y. Wang, P. K. Quoika, J. R. Loeffler, M. Schauperl and K. R. Liedl, J. Phys. Chem. B, 2019, 123, 8838 CrossRef CAS PubMed.
- S. Agarwal, S. Saha, V. K. Balla, A. Pal, A. Barui and S. Bodhak, Front. Mech. Eng., 2020, 6, 589171 CrossRef.
- M. A. Nazeer, O. C. Onder, I. Sevgili, E. Yilgor, I. H. Kavakli and I. Yilgor, Mater. Today Commun., 2020, 25, 101515 CrossRef CAS.
- N. Ashammakhi, S. Ahadian, C. Xu, H. Montazerian, H. Ko, R. Nasiri, N. Barros and A. Khademhosseini, Mater. Today Bio., 2019, 1, 100008 CrossRef CAS PubMed.
- Z. Zheng, J. Wu, M. Liu, H. Wang, C. Li, M. J. Rodriguez, G. Li, X. Wang and D. L. Kaplan, Adv. Healthcare Mater., 2018, 7, 1701026 CrossRef PubMed.
- A. Veiga, I. V. Silva, M. M. Duarte and A. L. Oliveira, Pharmaceutics, 2021, 13, 1444 CrossRef CAS PubMed.
- T. Ni, M. Liu, Y. Zhang, Y. Cao and R. Pei, Bioconjugate Chem., 2020, 31, 1938 CrossRef CAS PubMed.
- M. Sun, J. Cheng, J. Zhang, N. Wu, F. Zhao, Z. Li, H. Yu, X. Duan, X. Fu, X. Hu, H. Chen and Y. Ao, ACS Biomater. Sci. Eng., 2021, 7, 916 CrossRef CAS PubMed.
- S. Ghosh, S. T. Parker, X. Wang, D. L. Kaplan and J. A. Lewis, Adv. Funct. Mater., 2008, 18, 1883 CrossRef CAS.
- S. Das, F. Pati, Y. J. Choi, G. Rijal, J. H. Shim, S. W. Kim, A. R. Ray, D. W. Cho and S. Ghosh, Acta Biomater., 2015, 11, 233 CrossRef CAS PubMed.
- R. R. Jose, J. E. Brown, K. E. Polido, F. G. Omenetto and D. L. Kaplan, ACS Biomater. Sci. Eng., 2015, 1, 780 CrossRef CAS PubMed.
- M. J. Rodriguez, J. Brown, J. Giordano, S. J. Lin, F. G. Omenetto and D. L. Kaplan, Biomaterials, 2017, 117, 105 CrossRef CAS PubMed.
- L. Huang, X. Du, S. Fan, G. Yang, H. Shao, D. Li, C. Cao, Y. Zhu, M. Zhu and Y. Zhang, Carbohydr. Polym., 2019, 221, 146 CrossRef CAS PubMed.
- O. C. Onder, E. Yilgor and I. Yilgor, J. Polym. Sci., Part B: Polym. Phys., 2019, 57, 98 CrossRef CAS.
- O. C. Onder, M. A. Nazeer, E. Yilgör and I. Yilgör, Prog. Org. Coat., 2018, 125, 249 CrossRef CAS.
- Y. P. Singh, A. Bandyopadhyay and B. B. Mandal, ACS Appl. Mater. Interfaces, 2019, 11, 33684 CrossRef CAS PubMed.
- Z. Li, X. Zhang, T. Yuan, Y. Zhang, C. Luo, J. Zhang, Y. Liu and W. Fan, Tissue Eng., Part A, 2020, 26, 886 CrossRef CAS PubMed.
- X. Zhang, Y. Liu, C. Luo, C. Zhai, Z. Li, Y. Zhang, T. Yuan, S. Dong, J. Zhang and W. Fan, Mater. Sci. Eng., C, 2021, 118, 111388 CrossRef CAS PubMed.
- X. Zhang, Y. Liu, Q. Zuo, Q. Wang, Z. Li, K. Yan, T. Yuan, Y. Zhang, K. Shen, R. Xie and W. Fan, Int. J. Bioprint., 2021, 7, 401 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2022 |