
3D-printed, configurable, paper-based, and autonomous multi-organ-on-paper platforms†
Hongbin
Li‡
 ab,
Feng
Cheng‡
a,
Zixuan
Wang
a,
Wanlu
Li
a,
Juan Antonio
Robledo-Lara
ac and
Yu Shrike
Zhang
*a
ab,
Feng
Cheng‡
a,
Zixuan
Wang
a,
Wanlu
Li
a,
Juan Antonio
Robledo-Lara
ac and
Yu Shrike
Zhang
*a
aDivision of Engineering in Medicine, Brigham and Women's Hospital, Department of Medicine, Harvard Medical School, Cambridge, MA 02139, USA. E-mail: yszhang@research.bwh.harvard.edu
bCollege of Light Industry and Textile, Qiqihar University, Qiqihar, Heilongjiang 161000, P. R. China
cDepartamento de Ingeniería Mecatrónica y Eléctrica, Escuela de Ingeniería y Ciencias, Tecnológico de Monterrey, CP78211, San Luis Potosí, Mexico
First published on 24th August 2022
Abstract
We report the development of a class of 3D-printed, configurable, paper-based organ-on-paper platforms where autonomous and continuous delivery of media to engineered microtissue models is readily achieved without requiring external electrical power during device operations. Here, a passive flow with well-controlled flow rates was initiated along the flow path by capillary and evaporation-driven forces. Cell types representing the vasculature (human umbilical vein endothelial cells), the liver (HepG2 hepatocyte-like cells), the tumor (A549 lung cancer cells), and the kidney (HK-2 kidney proximal tubular cells) were cultured in the different, pre-configured, 3D-printed organ-on-paper platforms. We adopted cisplatin and the prodrug capecitabine as model drugs, which exhibited varying cytotoxicity and metabolism-dependent efficacy outcomes in the various single- or multi-organ models demonstrated. These 3D-printed, configurable, paper-based, cost-effective, and autonomous multi-organ-on-paper platforms would enable convenient generation of shelf-storable organ-mimicking structures, allowing for point-of-care construction of high-content in vitro microphysiological systems to rapidly evaluate both on-target efficacies and/or off-target toxicities, for potential applications in preclinical drug screening and personalized therapeutic selection.
Design, System, Application3D printing enables freeform spatial patterning of materials into sophisticated architectures towards various basic and translational applications. In particular, cellulose-based materials have shown great potential in serving as cell culture and point-of-care devices due to their cost-effectiveness as well unconventional performances provided by their nanostructural morphologies. Here, we report the unique combination of 3D extrusion printing with an ink composed of bacterial cellulose, cellulose nanocrystals, and nanoclays to create customizable multi-organ-on-a-chip systems. Such a paper-based multi-organ-on-a-chip system could be kept in its dry state for maintaining essentially an unlimited shelf-life for on-demand cell seeding at the bedside or in laboratory settings, therefore achieving point-of-care utility. Due to the use of the paper material as the substrate, it was further able to simultaneously create a passive, powerless micropump to generate autonomous, controlled flows and transfer medium and other fluids from one organ to another in continuous perfusion within the chip device. Our design concept of 3D-printed, configurable, paper-based, cost-effective, and autonomous multi-organ-on-paper technology will enable convenient generation of shelf-storable, high-content in vitro microphysiological systems to rapidly evaluate both on-target efficacies and off-target toxicities, achieving potential applications in preclinical drug screening and personalized therapeutic selection. |
Introduction
The current drug discovery process, which requires extensive pre-clinical testing and validation of protocols, is usually expensive and time-consuming.1,2 Pre-clinical evaluations of drug candidates rely on the utilities of in vitro cell culture platforms and in vivo animal models.3In vitro cell culture assays with conventional planar, static configurations oftentimes lack the complexities of living systems, which cannot sufficiently simulate the communications between tissues/organs, and are thus unable to predict the effects of sophisticated drug metabolisms and toxic effects on non-target tissues.4–7 On the other hand, while animal models have significantly improved our understanding of certain complex diseases and provided readouts of body-level drug effects in vivo,8 these models frequently fail to represent the human responses to drug treatments, and may sometimes lead to unwanted toxicities during clinical translation of preclinical results, due to interspecies differences.9Since the early 2000s, a class of microfluidic chip systems have been developed to capture tissue- and organ-level functions, termed organ-on-chips.10–12 The success of human physiology and pathophysiology tissue cultures on the individual microfluidic chip devices has prompted researchers to challenge the designs to come up with more systemic-level emulations of physicochemical microenvironments of multiple tissues in vitro through “multi-organ-on-a-chip”,13,14 “body-on-a-chip”,15,16 and “human-on-a-chip” platforms.17 These linked organ models are shown to accurately recapitulate the complex multi-axis human tissue and organ interactions to study systemic functions and to assess systemic drug effects.18,19 For instance, metabolism-dependent toxicity and anti-cancer efficacy of compounds have been shown to combine liver–small intestine models.20 Multi-organ-on-a-chip research has also seen some major breakthroughs in recent years such as the creation of up to eight- and ten-organ-on-a-chip configurations.21,22
However, most organ-on-a-chip devices have historically been made from the polydimethylsiloxane (PDMS) material using soft lithography,18,23 which is good for rapid prototyping yet is a generally expensive microfabrication technique that can be relatively slow and requires a collection of dedicated high-end instruments.24 Importantly, most of these devices are hand-made and would further involve multiple assembly steps, and therefore, they tend to be labor-intensive and have low yields.25,26 Injection-molding is a conventional technique for mass production of simple plastic parts in the manufacturing industry.27 Unfortunately, devices featuring sophisticated patterns and volumetric structures will likely face additional post-processing challenges, increasing the manufacturing costs and extending the processing time if they can be produced at all.28
In addition, syringe pumps,25,29 peristaltic pumps,30–32 and other active micropumps33,34 have been commonly used to achieve dynamic flows and connections of microfluidic tissue and organ cultures. However, continuous driving of the medium within a microfluidic device remains challenging due to the need for pumps that are usually bulky and necessitate external power sources. Passive microflow-generating systems, including osmotic pressure-driven micropumps,35 medium reservoir-enabled flow-control systems,36 and droplet-based surface tension-driven micropumps,37 are also frequently used. However, the existing passive systems are limited to short-term use only, and flow rates are typically not finely adjustable in these systems, and cannot maintain continuous, controllable, and long-term delivery of media. Therefore, a passive paper-based micropump with a siphon effect and porous structure may be considered as an effective device for generating a controllable flow rate similar to the interstitial flow.38 The functioning of such a micropump is attributed to the paper material consisting primarily of a network of cellulose micro- and/or nanofibers that essentially form a three-dimensional (3D) multi-porous structure, providing a variety of controllable surface morphologies, internal microstructures, and physicochemical properties for powerless liquid-wicking and movement. In addition, paper-based materials possess good biocompatibility due to their natural origins and the fact that they can be maintained with a sufficient supply chain at low production costs using mature processing procedures.
3D printing, or termed as additive manufacturing, has gained increasing attention in both laboratory and commercial applications to automate and scale up the production of numerous biomedical and medical devices with sophisticated architectures.39,40 In particular, complex multi-layered devices consisting of a variety of materials of different structures could be manufactured in single steps using 3D printing. This technology minimizes requirements for manual operations in the preparation process, and can effectively save processing time, reduce production cost, and exhibit high reproducibility.11 For instance, we successfully 3D-printed volumetric paper-based vascularized tumor models by taking advantage of hydrophobic fugitive inks directly extruded within a bacterial cellulose hydrogel matrix, featuring high content in tissue functions yet simplicity in fabrication, cost-effectiveness, and ability of extended shelf-availability.41,42 As such, we anticipate that the 3D printing technology, when combined with paper-based devices, may provide good opportunities for additional developments of point-of-care human multi-organ-on-a-chip platforms.43
Accordingly, here we report a new strategy of extrusion 3D printing for constructing in vitro multi-organ-on-a-chip systems using cellulose-based multi-component inks continuously patterned in a single step to rapidly fabricate organ-like shapes (Fig. 1A). Upon drying the printed ink pattern to form the porous multi-organ cellulose substrate hosted within a customized, non-drug-adsorbing plastic (polymethylmethacrylate, PMMA) holder, the interconnected cellulose-based multi-component ink pattern could be further connected with the medium reservoir in one corner and the volatilization chamber in another (Fig. 1B). Such a cellulose nanofiber-based multi-organ-on-a-chip system could be kept in its dry state for maintaining essentially an unlimited shelf-life for on-demand cell seeding at the bedside or in laboratory settings, therefore achieving point-of-care utility. Due to the use of the paper-based material as the substrate, we were able to simultaneously create a passive, powerless micropump that generated autonomous, controlled flows, using the siphon effect of the continuous cellulose network to transfer medium and other fluids from one organ to another in continuous perfusion within the chip. Such a passive liquid-transport not only was powerless, but also replaced the need for direct fluid plumbing (such as through tubing or microfluidic channels), avoiding complex engineering while providing complete refactorability provided by the 3D printing technology. Importantly, the flow rate of the micropump was readily adjustable by the height of the medium reservoir and opening diameter of the volatilization chamber, where the continuous flow was attained by capillary action of the porous paper substrate material as well as evaporation. Finally, relevant cell types when introduced into the respective printed organ-like structures in a multi-organ-on-a-chip format allowed convenient monitoring of drug efficacy or/and side toxicity (Fig. 1C).
 | ||
| Fig. 1 Fabrication of the 3D-printed, configurable, paper-based, autonomous multi-organ-on-paper platforms for drug screening in vitro. (A) Concept of the multi-organ-on-paper platforms fabricated via the 3D printing strategy by extruding a multi-organ pattern of the BC/CNC/LAPONITE® hydrogel ink. (B) Schematics showing the procedure of fabricating a representative human liver–lung chip connected with vasculature. (C) Process for medium circulation by the siphon effect of the paper-based platform in the two-organ system for studying cell–drug interactions and screening therapeutics. Black arrows indicate the direction of the medium flow in the platform. | ||
Results and discussion
Rheological evaluations of inks
One of the aims of this work was to determine whether the multi-component inks of the cellulose-based hydrogels could be used to print the multi-organ-on-a-chip substrates. To understand the printability of our multi-component inks, we first conducted a rheological study of bacterial cellulose (BC), cellulose nanocrystals (CNC), LAPONITE® nanoclay, BC/CNC, and BC/CNC/LAPONITE® inks at 37 °C using a rheometer (Fig. 2A). All these individual components or their combinations showed shear-thinning behaviors, which would allow their extrusion from the nozzle and adjustment of the deposition rate, improving the printing fidelity. The viscosity values of the materials decreased as the shear rate was increased. It was clearly observed that the viscosity of the BC/CNC combination became much higher than those of the pure BC or CNC formulations at the same shear rates. Meanwhile, viscosities of the inks that combined all the BC/CNC/LAPONITE® components were increased with elevated concentration of LAPONITE®, mainly attributed to the electrostatic attraction from the surficial charges of LAPONITE®, and intermolecular forces among BC/CNC macromolecules that further reinforced the BC/CNC network structure.44 Therefore, these results indicated that the LAPONITE® interactions can greatly improve the viscoelastic modulus of BC/CNC, and also play a part in the regulation of the printability of the BC/CNC inks. | ||
| Fig. 2 Rheological assessments, morphology observations, and printing performances of the BC/CNC/LAPONITE® inks. (A) Viscosity assessments of pure BC (1 w/v%), CNC (3 w/v%), and LAPONITE® (10 w/v%), as well as the mixture of BC (1 w/v%) and CNC (3 w/v%) with various LAPONITE® concentrations of 5, 10, and 15 w/v% (BC/CNC/5% LAPONITE®, BC/CNC/10% LAPONITE®, and BC/CNC/15% LAPONITE®). (B) SEM images showing cross-sectional morphologies of BC (1 w/v%), CNC (3 w/v%), LAPONITE® (10 w/v%), and BC/CNC/10% LAPONITE® after freeze-drying. (C–E) Photographs showing the printing performances of BC/CNC/LAPONITE® inks (BC: 0.5–2 w/v%; CNC: 2–4 w/v%; LAPONITE®: 5–15 w/v%) through extrusion printing, before (left column) and after (right column) freeze-drying, at 10 PSI of extrusion pressure and 6 mm s−1 of nozzle moving speed. Open circles: printable; filled circles: non-printable. | ||
Morphologies of BC/CNC/LAPONITE® materials
The cross-sectional morphologies of BC (1 w/v%), CNC (3 w/v%), LAPONITE® (10 w/v%), and BC/CNC/10% LAPONITE® inks were investigated by scanning electron microscopy (SEM) (Fig. 2B). The images revealed that BC or CNC had highly interconnected porous networks, while pure LAPONITE® had a laminated layered structure. Nevertheless, the BC/CNC/10% LAPONITE® material also revealed a well-ordered porous structure, which could be helpful for cell attachment and for enhancing the capillary effect of the medium in powerless pumping. The pore sizes of the BC, CNC, LAPONITE®, and BC/CNC/10% LAPONITE® inks in their dry states were quantified in the range of 30–200 μm (Fig. S1†), which might further provide sufficient space for cellular functions as well as oxygen and nutrient transport.Printability of extrusion-printed multi-organ structures
In extrusion printing, an ink filament should be continuously and uniformly extruded through the nozzle to achieve optimal printability.45 We evaluated the printability of the BC/CNC/LAPONITE® inks through changing the printing parameters such as the concentrations of the ink components, pressure, and/or nozzle moving speed. We first assessed the printability of BC/CNC/LAPONITE® inks (BC: 0.5–2 w/v%; CNC: 2–4 w/v%; LAPONITE®: 5–15 w/v%) in extrusion printing by depositing liver–lung organ-like patterns on the surfaces of cover glasses and compared the consistencies of the printed structures after freeze-drying (Fig. 2C–E). Our printability mapping suggested that the printing performance of the BC/CNC/LAPONITE® inks was improved by increasing the concentration of BC or CNC, at a given LAPONITE® concentration. Moreover, the printability of low-BC/CNC-concentration inks was also enhanced by including more LAPONITE®. However, the low-viscosity characteristic of the BC/CNC/LAPONITE® inks at a low concentration of LAPONITE® (5 w/v%) resulted in inefficient shape-retention and unpredictable aesthetic outcomes after printing (Fig. 2C). On the other hand, both low concentration (2%) and high concentration (4%) of CNC in the BC/CNC/LAPONITE® inks with a high LAPONITE® concentration (10 w/v%) also exhibited imperfect printed lines in some regions of the patterns (Fig. 2D), which further confirmed that the viscosity of the BC/CNC/LAPONITE® ink played an important role in the proper construction of 3D-printed paper-based patterns. Therefore, the ink with a too low or too high viscosity would not be conducive to its smooth extrusion from the nozzle. In addition, the BC/CNC/LAPONITE® inks with an exceedingly high LAPONITE® concentration (15 w/v%) exhibited discontinuous printed lines in some regions of the patterns (Fig. 2E), mainly attributable to the relatively low fluidity of LAPONITE® leading to ineffective extrusion of filaments. Therefore, we chose an optimal BC/CNC/LAPONITE® ink formulation of 1 w/v% BC and 3 w/v% CNC supplemented with 10 w/v% LAPONITE®, which resulted in the excellent printability and shape-retention ability of the printed structures during printing and after freeze-drying for 48 h (Fig. 2D). Shape-retention indicates whether the material could maintain its desired shape post-printing, which is of utmost importance to ensure the long-term success of multi-organ structure reconstruction.46We further investigated the relationship between the nozzle moving speed and extrusion pressure on the printing performances of our optimized BC/CNC/10% LAPONITE® ink. The printability diagram revealed that a balance between the nozzle moving speed and extrusion pressure reached appropriate structural fidelity to obtain the printed multi-organ models (Fig. S2†). The results suggested that the printing fidelity could be improved by extruding the appropriate amount of ink within a specific time range. When the nozzle moving speed was too fast or the pressure was too high, the extruded microfibers were too thin or too thick and easy to disperse or stack, resulting in lower printing resolutions and poor shape-retention capacities. Therefore, we selected 6 mm s−1 and 15 pounds per square inch (psi) as the optimized nozzle moving speed and extrusion pressure, respectively, along with the optimized ink formulation, for the subsequent experiments when printing multi-organ models.
Powerless flow characterization of multi-organ-on-paper platforms
The representative multi-organ-on-a-chip device contained two interconnected organ-mimics of the liver and the lung (Fig. 1B). The two organs were connected by vascular-like channels in between as well as a medium reservoir in one corner while a volatilization chamber in another. The advantage of our multi-organ-on-a-chip platform comes from the separation of the cell culture areas through 3D-printed, compartmentalized patterns, and the printed paper-based organ-mimics and vascular connections feature capillary force- and evaporation-driven passive, powerless perfusion flowing through the organs unidirectionally. Such a multi-organ-on-paper platform thus would enable convenient seeding of multiple respective cell types into desired locations in the chip, and may also be easily expanded to include more organ types with rapid, configurable, and shelf-storable printing capacities.Fig. 3 shows the flow-movement characteristics of the multi-organ-on-a-chip device. The capillary force-induced wicking rate was dependent on the height of the medium reservoir (i.e., the initial amount of medium present), the opening diameter of the volatilization reservoir, and the printed paper-based features without or with the infiltrated gelatin methacryloyl (GelMA) hydrogel (3 w/v%) (the latter simulating cell seeding in the presence of surrounding matrices). The actual flow conditions of the medium with various heights of the medium reservoir are shown in Fig. 3A and S3–S5,† where the opening was kept at 3 mm in diameter. Fig. 3B shows the quantified medium-wicking rates. The wicking rate clearly increased as the height of the medium reservoir was elevated. However, there was no significant difference for the reservoirs with heights of 6 and 10 cm. These results suggested that the height of the medium reservoir had some influence on the flow speed of the medium in our multi-organ-on-paper platforms.
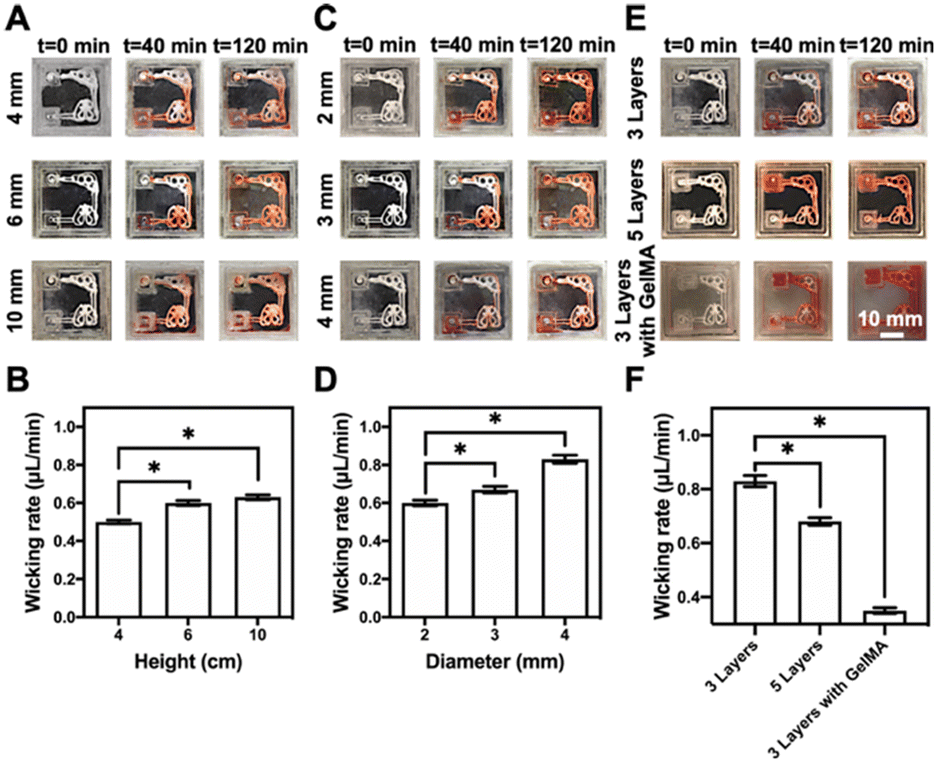 | ||
| Fig. 3 The siphon characteristics of the multi-organ-on-paper platforms. (A, C and E) Photographs showing the siphon characteristics of the multi-organ-on-paper platforms (A) with three layers of the printed structure at the height (4–10 mm) of the medium reservoir and 3 mm of diameter of the evaporation opening of the volatilization chamber, (C) with three layers of the printed structure, 2–4 mm of opening diameter of the volatilization reservoir, and 6 mm of height of the medium reservoir (E) with three layers of the printed structure without or with the infiltrated GelMA hydrogel and five layers of the printed structure without the infiltrated GelMA hydrogel, 3 mm of diameter of the evaporation opening of volatilization chamber, and 6 mm height of the medium reservoir at different time points after starting the medium flow. (B, D and F) The comparison of mean wicking rates of the culture media in the multi-organ-on-paper platforms (B) with three layers of the printed structure, 4–10 mm of height of the medium reservoir, and 3 mm of diameter of the evaporation opening of volatilization chamber, (D) with three layers of the printed structure, 2–4 mm of opening diameter of the volatilization reservoir, and 6 mm of height of the medium reservoir, and (F) with three layers of the printed structure without or with the infiltrated GelMA hydrogel and five layers of the printed structure without the infiltrated GelMA hydrogel, 3 mm of diameter of the evaporation opening of the volatilization chamber, and 6 mm of height of the medium reservoir, at 120 min of wicking. *P < 0.05. | ||
In addition, the powerless flow characteristics of the liver–lung-on-a-chip platforms with three layers of the printed structure prepared at the 2–4 mm of opening diameters of the volatilization reservoir and 6 mm height of the medium reservoir were studied (Fig. S4, S6, and S7†). It was shown that the larger diameter of the opening could effectively improve the wicking rate of the medium in our setup (Fig. 3C and D). The wicking rate was also related to the substrate itself, and the auxiliary materials, such as the additional incorporation of the GelMA hydrogel into the cellulose-based multi-component ink patterns (Fig. 3E and S7–S9†). The results indicated that more printed layers of these patterns and the addition of GelMA into the patterns could both impede the flow of the medium through the organ-mimics to different degrees (Fig. 3F).
To date, numerous configurations have been adopted to provide medium flows for microfluidic cell cultures, where conventional syringe pumps and roller-type pumps are typically used. Despite the diverse advantages of these active pumps, their extensive applications are limited in certain scenarios by the large pump sizes and the external power sources required for operations. Alternatively, several simple and cost-effective passive micropump systems have been developed to facilitate cell cultures.35,36,47 However, they can still be bulky, in addition to other disadvantages such as disintegration with the substrates used for cellular culture. Our proposed system seamlessly integrates all components into a self-operating, small-form factor system in a single 3D printing step to overcome the above-mentioned challenges. In particular, the fabrication process is also simple, cost-effective, configurable, and scalable, and the wicking rate could be precisely controlled over a reasonable range that is sufficient for our intended applications.
Biological performances of cell-laden multi-organ-on-paper platforms
Considering that our final goal is to generate the functional multi-organ-on-paper platforms, the devices should be able to perform a range of studies, from the autonomous culture of single-organ systems for assessing efficacy or toxicity to the more complex multi-organ interactional analyses. As examples, we created multi-organ-on-a-chip systems such as the liver–lung chip, liver–kidney chip, and kidney–lung chip where medium flows were realized by the aforementioned powerless perfusion enabled by a combinatory effects of capillary force, evaporation, and gravity (Fig. S10†).To analyze the compatibility of the printed cellulose-based multi-component materials with cells, several cell types were seeded into the corresponding organ-mimics for evaluating the viabilities, morphologies, and functions of each single organ during the 1 week of culture. As revealed in Fig. 4A, fluorescence micrographs showed that all the types of cells tested were uniformly distributed on the surfaces and within the boundaries of the respective organ-mimic structures and proliferated during the culture period, meanwhile the printed organ-mimic patterns could maintain their original structures after seeding with cells as well. The quantified cell viability analyses suggested that the percentages of live cells in all cases, i.e., HepG2 human hepatocyte-like cells, human umbilical vein endothelial cells (HUVECs), A549 human lung cancer cells, and HK-2 human proximal tubular cells, were close to 100% at 7 days of culture (Fig. 4B), indicating the favorable cytocompatibility of the cellulose-based ink that we used for printing the organ-like patterns. In addition, the proliferation rates of the HepG2, A549, and HK-2 cells were all higher than that of HUVECs (Fig. 4C), which might be attributed to the stronger growth abilities of these cell lines than the primary cells.
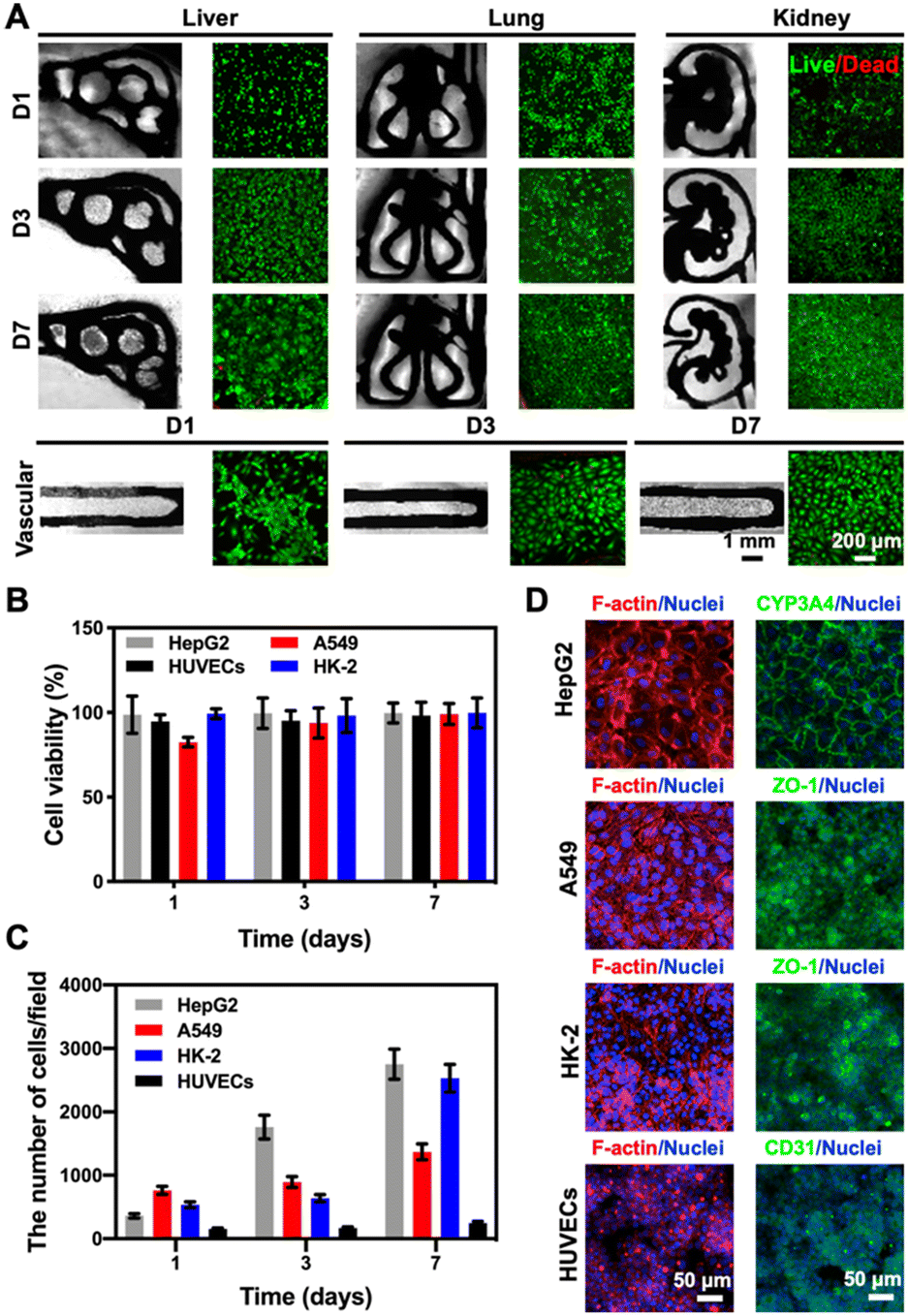 | ||
| Fig. 4 Compatibility of the printed cellulose-based multi-component materials with cells cultured in the corresponding organ-mimics. (A) Fluorescence images of live/dead staining after 1, 3, and 7 days of culture for the 3D-printed liver, lung, kidney, and vascular organ-mimics. (B) Quantitative analyses of cell viability (HepG2, A549 and HK-2 cells, and HUVECs) within the corresponding organ-mimics. (C) Quantitative analyses of the numbers of HepG2, A549 and HK-2 cells and HUVECs at 1, 3, and 7 days of culture in the corresponding organ-mimics. The area of each image field was 1.21 mm2. (D) Confocal projection views of F-actin and nuclei staining of the cells cultured in the corresponding organ-mimics, and the expressions of different cell-specific biomarkers, CYP3A4 (green) by HepG2, ZO-1 (green) by A549 and HK-2, and CD31 (green) by HUVECs, at 7 days of culture. | ||
We finally confirmed the biological functions of the corresponding organ-mimics by examining the behaviors of the HepG2, A549 and HK-2 cells, and HUVECs by immunostaining. As shown in Fig. 4D, the confocal projection views of the different biomarkers (CYP3A4 for the liver, zonula occludens (ZO)-1 for A549 and HK-2, and CD31 for HUVECs) verified their desirable tissue-specific distributions and strong expressions, revealing the functionality of the respective cell types over the 7 day culture period. These results suggested that our customizable, printed, paper-based organ-mimics created favorable microenvironments for the seeded cells to attach, grow, and function, potentially allowing further generation of multi-organ-on-a-chip systems for more sophisticated interactional studies.
Although culture of a single cell type can mimic some facets of the tissue microenvironment on chips, it is not usually sufficient to generate organ-level functionality. Further, multi-organ-on-a-chip platforms including the liver–lung chip (Fig. 5A), the kidney–lung chip (Fig. 5D), and the liver–kidney chip (Fig. 5G) were created by placing different cells in specific areas towards replicating organ-level interactions, respectively. As revealed in Fig. 5, the corresponding cell types were seeded into the pre-configured, 3D-printed multi-organ-on-paper platforms and the fluorescence images showed that all the cell types co-cultured within their respective multi-organ-mimics in the different configurations were again homogeneously distributed at 1, 3, and 7 days of culture (Fig. 5B, E and H). Live/dead staining showed that the cell viabilities of the co-culture in each multi-organ-on-a-chip setup were approaching 100% (Fig. 5C, F and I), not much different from those in single-organ cultures (Fig. 4B). Interestingly, the numbers of HUVECs at 7 days of culture in the kidney–lung chip and the liver–kidney chip were significantly higher compared with that for the liver–lung chip (Fig. S11†), suggesting that the unique combinations of the organ types might lead to differential cellular interactions and their activities.10 Our results indicated that the 3D-printed paper-based multi-organ-on-a-chip devices were amenable to cell culture, potentially allowing rapid, on-demand creation of single- or multi-organ configurations for on-demand drug screening and point-of-care therapeutic screening.
 | ||
| Fig. 5 Evaluations of the multi-organ-on-a-chip devices. (A, D and G) 3D-printed cellulose-based structures simulating multi-organ-on-a-chip: (A) liver–lung, (D) kidney–lung, and (G) liver–kidney configurations. (B, E and H) Fluorescence micrographs showing corresponding live/dead staining results after 1, 3, and 7 days of culture for (B) liver–lung, (E) kidney–lung, and (H) liver–kidney configurations. (C, F and I) Quantitative analyses of viable cells at 1, 3, and 7 days of culture for (C) liver–lung, (F) kidney–lung, and (I) liver–kidney configurations. | ||
Integrated multi-organ-on-paper platforms for therapeutic screening
To evaluate the potential applications of our 3D-printed, paper-based chip devices towards cell toxicity assays, we analyzed the side-toxicity of cisplatin on HepG2 cells, A549 cells, and HUVECs within the liver–lung chip configuration (Fig. 6A). Indeed, cisplatin has been shown to inhibit the growth of cancer cells;48 however, it also has certain toxic side-effects to healthy tissues such as myeloid toxicity and nephrotoxicity.49 To examine the responses of our liver–lung multi-organ-on-a-chip device, cisplatin concentrations from 0 to 80 μM were added into the reservoir and incubated for 48 h. The fluorescence images of live/dead staining suggested that cisplatin could significantly reduce the viability values of all cell types involved (Fig. 6B), mainly attributed to its interference with the DNA-synthesis process. As revealed in Fig. 6C–E, cisplatin at a high concentration (80 μM) has a greater toxic impact on HUVECs, where the decrease in their viability was faster than those for HepG2 and A549 cells; the inhibition rate of HUVECs induced by cisplatin reached 97.4% (Fig. S12;† compared to 91.4% and 86.8% for HepG2 and A549 cells, respectively). In addition, the dose–response curves for the cultured HepG2 cells, A549 cells, and HUVECs to cisplatin in the chip configurations, respectively, are shown in Fig. 6F–H. The 50% inhibitory concentration (IC50) values of the drug for HepG2 cells, A549 cells, and HUVECs were calculated at 1.038, 1.343, and 1.022 μM, respectively. Therefore, cisplatin at similarly lower concentrations could more effectively inhibit the growth of the various cell types showing both efficacy and toxicity (Fig. 6F–H). | ||
| Fig. 6 Effect of cisplatin on the viability of different target cells in the liver–lung multi-organ-on-a-chip device. (A) Schematic showing the seeding procedure of cells and addition of cisplatin into the liver–lung chip. (B) Fluorescence micrographs showing corresponding live/dead staining after HepG2 cells, A549 cells, and HUVECs were treated with different concentrations of cisplatin (0–80 μM). (C–E) Quantitative analyses of the viability values of HepG2 cells, A549 cells, and HUVECs at different concentrations of cisplatin (0–80 μM). (F–H) Dose–response curves of HepG2 cells, A549 cells, and HUVECs to cisplatin in the liver–lung chip configuration. | ||
In addition to the effects of drug side-toxicity on the cells, we also analyzed the potential applications of our 3D-printed, paper-based chip devices in drug-metabolism studies. We first employed capecitabine (CAP), a prodrug that can be activated by enzymes in hepatic cells to form the active 5-fluorouracil (5-FU), which is a thymidylate synthase-inhibitor,50 to demonstrate its liver-activated toxicity. In the liver–lung chip configuration, the continuous cell culture of the HepG2 cells, A549 cells, and HUVECs into the designated areas was exposed to CAP at different concentrations (0–200 μM) in the medium (Fig. 7A). Then, the toxicity was analyzed by live/dead viability staining after 24 h of culture, where quantitative PrestoBlue cell metabolic activity assay was performed at 48 h. The fluorescence micrographs suggested the reduced viability values of both A549 cells and HUVECs in this setup, which was mainly attributed to the metabolic capacity of the liver module in transforming 5-deoxy-5-fluorouridine (5-DFUR) into 5-FU,51 leading to toxic effects (Fig. 7B and C). The results suggested that 5-FU was generated by the secondary metabolism in the target cells under the primary metabolism in the hepatic organ, accordingly resulting in obvious cytotoxicity to the target cells.
 | ||
| Fig. 7 Effect of CAP metabolism on the viability of different target cells in the liver–lung multi-organ-on-a-chip device. (A) Schematic showing the seeding procedure of cells and CAP into the liver–lung chip. (B) Fluorescence micrographs showing the corresponding live/dead staining results after A549 cells and HUVECs were treated with different concentrations of CAP (0–200 μM) metabolized or not metabolized by the HepG2 cells. (C and D) Quantitative analyses of CAP metabolized or not metabolized by the HepG2 cells on the viability values of A549 cells and HUVECs. (E and F) Dose–response curves of A549 cells and HUVECs to CAP metabolized or not metabolized by the HepG2 cells in the liver–lung chips. | ||
It was further observed that the A549 cells were more sensitive to 5-FU than the HUVECs (Fig. S13†), indicating the differences in CAP activities between the two cell types. We also observed that the CAP not metabolized by the HepG2 cells had no significant toxic effects on the viabilities of A549 cells and HUVECs (Fig. 7D), as expected. In addition, the dose–response curves for the cultured A549 cells and HUVECs to CAP, pristine or metabolized, in the chip configurations in the absence or presence of the liver module, respectively, are shown in Fig. 7E and F. The IC50 values of the drug for A549 cells and HUVECs were calculated at 72.78 and 192.20 μM, respectively, when the liver organ was a part of the system. In contrast, the IC50 values of unmetabolized CAP for the cells could not be obtained within the range of doses that we assessed. Therefore, our 3D-printed, paper-based multi-organ-on-a-chip device demonstrated the advantages of the multi-step primary and secondary metabolic processes for the co-culture featuring the liver module and other desired cell types, potentially enabling early assessment and efficacy/toxicity-prediction of anticancer compounds in a point-of-care manner.
Conclusions
We have developed a continuous flow-generating, pumpless multi-organ-on-paper platforms by taking advantage of the siphon effect and paper-based materials. The 3D-printed multi-organ-on-paper platforms could be easily integrated into laser-engraved chip devices, where the entire systems are simple, cost-effective, configurable, shelf-storable, and autonomous in operations. This methodology will likely enable enhanced point-of-care multi-organ cultures for widespread applications in drug screening and personalized therapeutic selection in the pharmaceutical industry, in laboratory settings, and at the bedside. However, limitations associated with these paper-based devices still exist. For example, the possible non-specific adsorption of various biomolecules and drugs by the paper biomaterials52,53 oftentimes interferes with fluorescence imaging of the cells and tissues residing within these devices, potentially needing careful selection of the fluorophores to minimize such background fluorescence. Such an adsorption phenomenon also potentially biases the drug-dosing information that may need to be calibrated. Therefore, while the paper-based materials serve as an enabling platform for our intended applications, ways to improve their performances shall be further addressed to promote their applications in the future.Experimental
Materials
The BC hydrogels were purchased from Hainan Yide Industry Co. Ltd. (China). CNC powders were purchased from Guilin Qihong Technology Co. Ltd. (China). Nanosilicates (LAPONITE® XLG) were obtained from Southern Clay Products, Inc. (USA). No reagent was further purified. Phosphate-buffered saline (PBS) and other solutions were prepared with deionized water (18.2 MΩ cm, Millipore, USA).Cellular analysis reagents including Live/Dead® Viability/Cytotoxicity Kit, Alexa Fluor® 594-phalloidin, 4′,6-diamidino-2-phenylindole (DAPI), rabbit anti-mouse ZO-1, mouse anti-human CD31, and mouse anti-human CYP3A4 primary antibodies were purchased from Abcam (USA), and Alexa Fluor® 488-conjugated goat anti-rabbit or goat anti-mouse secondary antibodies were purchased from ThermoFisher (USA). Other chemicals were obtained from Sigma-Aldrich, unless otherwise specified.
Preparation of the multi-component inks
The BC hydrogel was prepared by the method used in our previous work.41 A certain amount of CNC powder was dispersed in deionized water to obtain a hydrogel using a mechanical homogenizer (Hamilton Beach Brands, USA) at a speed of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. Various hydrogel concentrations (w/v) of BC (0.5%, 1%, and 2%), CNC (2%, 3%, and 4%), and LAPONITE® (5%, 10%, and 15%) were used to produce the multi-component inks through a homogenizer and the bubbles were eliminated by ultrasonication at room temperature.
000 rpm. Various hydrogel concentrations (w/v) of BC (0.5%, 1%, and 2%), CNC (2%, 3%, and 4%), and LAPONITE® (5%, 10%, and 15%) were used to produce the multi-component inks through a homogenizer and the bubbles were eliminated by ultrasonication at room temperature.
The multi-component inks optimization
The 3D multi-organ structures were obtained by extruding the BC/CNC/LAPONITE® hydrogel ink directly onto the cover glass using a commercially available bioprinter (Allevi 2, 3D Systems, USA). The 3D multi-organ patterns were modeled using SolidWorks (Dassault Systèmes, France), saved as STL files, and imported into bioprinter slicer software (Repetier-Host V1.6.1, Hot-World GmbH & Co. KG, Germany) to convert to corresponding G-codes. The ink printability measurement was based on whether the inks could be smoothly extruded through the nozzle to form uniform lines and layer by layer. The multi-organ structures formed by the different compositions of the inks, extrusion pressures, and translocation speeds of the nozzle were examined for their morphologies before and after freeze-drying to optimize the 3D-printing conditions.Rheological properties of the multi-component inks
The rheological properties of BC (1 w/v%), CNC (3 w/v%), LAPONITE® (10 w/v%), as well as mixtures of BC (1 w/v%) and CNC (3 w/v%) with various LAPONITE® concentrations of 5, 10, and 15 w/v% (i.e., BC/CNC/5% LAPONITE®, BC/CNC/10% LAPONITE®, and BC/CNC/15% LAPONITE®, respectively) were assessed using a hybrid rheometer (HR-3, Waters, USA) in a shear-ramp mode running from 0.01 to 1000 s−1 with a 1000-μm gap size, at 25 °C.Morphological characterization of the multi-component inks
The BC (1 w/v%), CNC (3 w/v%), LAPONITE® (10 w/v%), and BC/CNC/10% LAPONITE® samples were broken upon freezing using liquid nitrogen, freeze-dried, fixed, and sputter-coated with gold for morphology analyses through SEM (Zeiss, Germany) at 20 kV.Fabrication of multi-organ-on-paper platforms
The multi-organ-on-paper platforms were fabricated by a simple, cost-effective method by combining the laser-engraving machine (VLS 3.50, Universal Laser Systems Inc., USA) and the 3D bioprinter (Allevi 2), each of which composed of a desired 3D-printed multi-organ paper-based system fixed into a laser-cut PMMA chip device (34 mm × 34 mm × (4–10) mm) containing a medium reservoir and a volatilization chamber (both 5 mm × 5 mm × (4–10) mm) (Fig. 1B). The opening diameter (2–4 mm) of the volatilization chamber was further varied.To initiate the siphon-driven flow, the culture medium (100–400 μL) was injected into the medium reservoir at an 8-h interval to ensure the consistent flow of the culture medium, which is directly connected to the 3D-printed paper structure, and a passive flow was generated by the capillary force and evaporation force to move the medium from the reservoir towards the volatilization chamber. The siphon characteristics were further evaluated by calculating the wicking rate in a standard cell-culture incubator in the multi-organ-on-paper configuration.
In addition, GelMA was synthesized according to previously reported methods.54 Then, 3% (w/v) GelMA aqueous solution containing 0.5% (w/w) photoinitiator (2-hydroxy-1-[4-(2-hydroxyethoxy)phenyl]-2-methyl-1-propanone, Irgacure 2959, CIBA Chemicals, Switzerland) was used to cover the surface of the 3D-printed region in the multi-organ-on-paper configuration and subsequently photocrosslinked under UV irradiation (∼10 mW cm−2, 360–480 nm, OmniCure, USA) for 30 s, and the siphon effects of the multi-organ-on-paper platforms with or without GelMA hydrogel coating were further studied.
Cell culture
In this study, four different cell types were selected to fulfill the experimental requirements, i.e., HepG2 hepatocyte-like cells (American Type Culture Collection (ATCC), USA), A549 human non-small cell lung cancer cells (ATCC), HK-2 human renal tubular epithelial cells (ATCC), and HUVECs (green fluorescent protein (GFP)-tagged or unlabeled, Angio-Proteomie, USA). HUVECs were cultured in endothelial growth medium-2 (EGM-2, Lonza, USA), where all the other cells were cultured in Dulbecco's modified Eagle's medium (DMEM, ThermoFisher) containing 10 v/v% fetal bovine serum (FBS, ThermoFisher) and 1 v/v% antibiotic–antimycotic (ThermoFisher). All cell types were inoculated in tissue culture polystyrene flasks at 37 °C and 5 v/v% CO2 in the incubator.Different cell types seeded into the multi-organs-on-a-chip platforms were cultured in a common medium with a volume ratio of 1:1 between EGM-2 and DMEM for the experiments. The chips were autoclaved for 15 min for sterilization before the cells were seeded. HepG2 cells (5 × 106 cells per mL), A549 cells (1 × 106 cells per mL), HK-2 cells (1 × 106 cells per mL) and HUVECs (1 × 106 cells per mL) were seeded into their corresponding patterns in the chips for 1 h before adding the mix medium.
Cell viability and morphology analyses
A Live/Dead Cell Viability/Cytotoxicity Kit was used to assess the cell viability according to the manufacturer's instructions at days 1, 3, and 7 after cell seeding in the single- or multi-organ-on-a-chip devices. The samples were treated with calcein-AM/ethidium homodimer-1 at desired concentrations in PBS at 37 °C for 10–15 min. Then, the samples were gently washed with PBS and observed using an inverted fluorescence microscope (Nikon Ti-E, Japan). For each condition, at least six images from different regions were randomly selected to quantify the numbers of live (green) and dead (red) cells using the ImageJ software (National Institutes of Health, USA) and the cell viability values were expressed as the percentage of the live cell number to the total number of cells. Cell proliferation of an organ or multiple organs was measured using the PrestoBlue Cell Viability Reagent (10 v/v%) to evaluate their metabolic activities. The protocol followed those reported previously.55To evaluate the different cell morphologies and the biological functions of the different microtissues, we performed immunofluorescence staining of F-actin/nuclei and cell-specific markers/nuclei, respectively. Briefly, at the desired time points of culture, the samples were fixed with 4 w/v% paraformaldehyde for 30 min, and then placed in 0.1 v/v% Triton X-100 in PBS for 30 min to permeabilize the cell membranes. The samples were blocked with 1 w/v% bovine serum albumin (BSA) in PBS for another 1 h at room temperature. For F-actin cytoskeleton staining, the samples were then cultured with Alexa Fluor® 594-phalloidin at 1:40 dilution in 0.1 w/v% BSA overnight at 4 °C, and the nuclei of cells were counterstained with DAPI (1:1000) for 5 min at room temperature.
For biomarker staining, CD31, ZO-1, or CYP3A4 primary antibody was used to characterize HUVECs, A549/HK-2 cells, or HepG2 cells, respectively. The samples were stained using the corresponding primary antibodies (1:200 dilution in 5 v/v% goat serum and 0.2 v/v% Triton X-100 in PBS) overnight at 4 °C. After washing three times with DPBS, corresponding second antibodies (Alexa Fluor® 488-conjugated goat anti-rabbit or goat anti-mouse) at 1:200 dilution were incubated overnight at 4 °C. The samples were washed three times with PBS and then counterstained for nuclei using DAPI for 5 min at room temperature. Finally, the samples were observed using a Zeiss Confocal Microscope (Zeiss LSM 880 with Airyscan, Zeiss, Germany), and the images were visualized using the ImageJ software.
Drug treatment
The cell culture medium containing 0–80 μM of cisplatin was added into the reservoir of the liver–lung chips and incubated for 48 h to evaluate the side-toxicity of cisplatin on HepG2 cells, A549 cells, and HUVECs. Drug toxicity was first analyzed using the Live/Dead Cell Viability/Cytotoxicity Kit in their single cultures. Quantitative cell proliferation data were measured using the PrestoBlue Cell Viability Reagent to determine the IC50 values. After optimizing the initial drug concentrations, the liver–lung chips were further used to measure the interactional drug effects.For prodrug CAP treatment, the continuous cell culture of the liver–lung chips with HepG2 cells, A549 cells, and HUVECs into the designated areas was treated with a series of CAP concentrations (0–200 μM) in the medium for 24 h. Then, those containing the liver organ were designated as the “HepG2 cells (+)” group, where toxicity was analyzed by live/dead viability staining after 24 h of culture and quantitative PrestoBlue cell metabolic activity assay at 48 h. In the “HepG2 cells (−)” group with only A549 cells and HUVECs, the cell culture medium containing 0–200 μM of CAP was added into the medium reservoir of the liver–lung chip without HepG2 cells. Cell viability and IC50 values were subsequently measured as described above.
Statistical analyses
All data are presented as means ± standard deviations of at least three independent experiments with each experiment including at least triplicates. Student's t-test was used for comparison among various experimental and control groups. P-values of less than 0.05 were considered statistically significant.Author contributions
Conceptualization: Y. S. Z. Methodology: F. C., H. L., Z. W., W. L., and J. A. R.-L. Investigation: F. C., H. L., Z. W., and J. A. R.-L. Supervision: Y. S. Z. Writing – original draft: F. C., H. L., and Y. S. Z. Writing – review and editing: F. C., H. L., Z. W., W. L., J. A. R.-L., and Y. S. Z.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Brigham Research Institute for the support.Notes and references
- C. Chen, Y. Gu, J. Philippe, P. Zhang and T. J. Huang, Nat. Commun., 2021, 12, 1118 CrossRef CAS PubMed.
- A. Sertkaya, H. H. Wong, A. Jessup and T. Beleche, Clin. Trials, 2016, 13, 117–126 CrossRef PubMed.
- M. S. F. Roeten, J. V. Meerloo, Z. J. Kwidama, G. T. Huizen and J. Cloos, Cell, 2021, 10, 665 CrossRef CAS PubMed.
- M. Danhof, E. Lange, O. Pasqua, B. A. Ploeger and R. A. Voskuyl, Trends Pharmacol. Sci., 2008, 29, 186–191 CrossRef CAS PubMed.
- S. Xiao, J. R. Coppeta, H. B. Rogers, B. C. Isenberg, J. Zhu, S. A. Olalekan, K. E. Mckinnon, D. Dokic, A. S. Rashedi and D. J. Haisenleder, Nat. Commun., 2017, 8, 14584 CrossRef CAS PubMed.
- J. R. Coppeta, M. J. Mescher, B. C. Isenberg, A. J. Spencer, E. S. Kim, A. R. Lever, T. J. Mulhern, R. Prantil-Baun, J. C. Comolli and J. T. Borenstein, Lab Chip, 2017, 17, 134–144 RSC.
- D. Cook, D. Brown, R. Alexander, R. March, P. Morgan, G. Satterthwaite and M. N. Pangalos, Nat. Rev. Drug Discovery, 2014, 13, 419–431 CrossRef CAS PubMed.
- K. Reczyńska, P. Tharkar, S. Y. Kim, Y. Wang, E. Pamua, H. K. Chan and W. Chrzanowski, Adv. Drug Delivery Rev., 2018, 123, 107–134 CrossRef PubMed.
- M. Dickson and J. P. Gagnon, Nat. Rev. Drug Discovery, 2004, 3, 417–429 CrossRef CAS PubMed.
- S. N. Bhatia and D. E. Ingber, Nat. Biotechnol., 2014, 32, 760–772 CrossRef CAS PubMed.
- L. A. Low, C. Mummery, B. R. Berridge, C. P. Austin and D. A. Tagle, Nat. Rev. Drug Discovery, 2021, 20, 345–361 CrossRef CAS PubMed.
- T. Ching, Y. C. Toh, M. Hashimoto and Y. S. Zhang, Trends Pharmacol. Sci., 2021, 42, 715–728 CrossRef CAS PubMed.
- S. N. Bhatia and D. E. Ingber, Nat. Biotechnol., 2014, 32, 760–772 CrossRef CAS PubMed.
- B. Zhang, K. Anastasia, B. Lai and R. Milica, Nat. Rev. Mater., 2018, 3, 257–278 CrossRef.
- J. H. Sung, Y. I. Wang, N. N. Sriram, M. Jackson, C. Long, J. J. Hickman and M. L. Shuler, Anal. Chem., 2018, 91, 330–351 CrossRef PubMed.
- A. Skardal, J. Aleman, S. Forsythe, S. Rajan and A. Atala, Biofabrication, 2020, 12, 025017 CrossRef CAS PubMed.
- M. T. Nelson, M. R. Charbonneau, H. G. Coia, M. J. Castillo and C. A. Mauzy, Nat. Commun., 2021, 12, 2805 CrossRef CAS PubMed.
- A. Herland, B. M. Maoz, D. Das, M. R. Somayaji, R. Prantil-Baun, R. Novak, M. Cronce, T. Huffstater, S. S. F. Jeanty, M. Ingram, A. Chalkiadaki, D. B. Chou, S. Marquez, A. Delahanty, S. Jalili-Firoozinezhad, Y. Milton, A. Sontheimer-Phelps, B. Swenor, O. Levy, K. K. Parker, A. Przekwas and D. E. Ingber, Nat. Biomed. Eng., 2020, 4, 421–436 CrossRef CAS PubMed.
- Z. Xu, E. Li, Z. Guo, R. Yu, H. Hao, Y. Xu, Z. Sun, X. Li, J. Lyu and Q. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 25840–25847 CrossRef CAS PubMed.
- M. Trapecar, C. Communal, J. Velazquez, C. Maass, Y. Huang, K. Schneider, C. Wright, V. Butty, G. Eng, O. Yilmaz, D. Trumper and L. Griffith, Cell Syst., 2020, 10, 223–239 CrossRef CAS PubMed.
- R. Novak, M. Ingram, S. Marquez, D. Das, A. Delahanty, A. Herland, B. Maoz, S. Jeanty, M. Somayaji, M. Burt, E. Calamari, A. Chalkiadaki, A. Cho, Y. Choe, D. Chou, M. Cronce, S. Dauth, T. Divic, J. Fernandez-Alcon, T. Ferrante, J. Ferrier, E. FitzGerald, R. Fleming, S. Jalili-Firoozinezhad, T. Grevesse, J. Goss, T. Hamkins-Indik, O. Henry, C. Hinojosa, T. Huffstater, K. Jang, V. Kujala, L. Leng, R. Mannix, Y. Milton, J. Nawroth, B. Nestor, C. Ng, B. O'Connor, T. Park, H. Sanchez, J. Sliz, A. Sontheimer-Phelps, B. Swenor, G. Thompson, G. Touloumes, Z. Tranchemontagne, N. Wen, M. Yadid, A. Bahinski, G. Hamilton, D. Levner, O. Levy, A. Przekwas, R. Prantil-Baun, K. Parker and D. Ingber, Nat. Biomed. Eng., 2020, 4, 407–420 CrossRef PubMed.
- K. Ronaldson-Bouchard, D. Teles, K. Yeager, D. N. Tavakol, Y. Zhao, A. Chramiec, S. Tagore, M. Summers, S. Stylianos, M. Tamargo, B. M. Lee, S. P. Halligan, E. H. Abaci, Z. Guo, J. Jackow, A. Pappalardo, J. Shih, R. K. Soni, S. Sonar, C. German, A. M. Christiano, A. Califano, K. K. Hirschi, C. S. Chen, A. Przekwas and G. Vunjak-Novakovic, Nat. Biomed. Eng., 2022, 6, 351–371 CrossRef CAS PubMed.
- D. Huh, H. J. Kim, J. P. Fraser, D. E. Shea, M. Khan, A. Bahinski, G. A. Hamilton and D. E. Ingber, Nat. Protoc., 2013, 8, 2135–2157 CrossRef CAS PubMed.
- K. N. Ren, J. H. Zhou and H. K. Wu, Acc. Chem. Res., 2013, 46, 2396–2406 CrossRef CAS PubMed.
- T. Satoh, S. Sugiura, K. Shin, R. Onuki-Nagasaki, S. Ishida, K. Kikuchi, M. Kakiki and T. Kanamori, Lab Chip, 2017, 18, 115–125 RSC.
- C. W. McAleer, C. J. Long, D. Elbrecht, T. Sasserath, L. R. Bridges, J. W. Rumsey, C. Martin, M. Schnepper, Y. Wang, F. Schuler, A. B. Roth, C. Funk, M. L. Shuler and J. J. Hickman, Sci. Transl. Med., 2019, 11, eaav1386 CrossRef CAS PubMed.
- J. H. Day, T. M. Nicholson, X. J. Su, T. L. van Neel, I. Clinton, A. Kothandapani, J. Lee, M. H. Greenberg, J. K. Amory, T. J. Walsh, C. H. Muller, O. E. Franco, C. R. Jefcoate, S. E. Crawford, J. S. Jorgensen and A. B. Theberge, Lab Chip, 2020, 20, 107–119 RSC.
- J. Yu, S. Lee, J. Song, S. R. Lee, S. Kim, H. Choi, H. B. Kang, Y. C. Hwang, Y. K. Hong and N. L. Jeon, Nano Convergence, 2022, 9, 16 CrossRef CAS PubMed.
- P. J. Hung, P. J. Lee, P. Sabounchi, N. Aghdam, R. Lin and L. P. Lee, Lab Chip, 2004, 5, 44–48 RSC.
- K. Domansky, W. Inman, J. G. Serdy, A. Dash and L. G. Griffith, Lab Chip, 2010, 10, 51–58 RSC.
- U. Sarkar, D. Rivera-Burgos, E. M. Large, D. J. Hughes and K. C. Ravindra, Drug Metab. Dispos., 2015, 43, 1091–1099 CrossRef CAS PubMed.
- J. M. Prot, L. Maciel, T. Bricks, F. Merlier, J. Cotton, P. Paullier, F. Y. Bois and E. Leclerc, Biotechnol. Bioeng., 2015, 111, 2027–2040 CrossRef PubMed.
- H. Lee, D. S. Kim, S. K. Ha, I. Choi, J. M. Lee and J. H. Sung, Biotechnol. Bioeng., 2017, 114, 432–443 CrossRef CAS PubMed.
- H. Nakayama, H. Kimura, T. Fujii and Y. Sakai, J. Biosci. Bioeng., 2014, 117, 756–762 CrossRef CAS PubMed.
- J. Y. Park, S. J. Yoo, C. M. Hwang and S. H. Lee, Lab Chip, 2009, 9, 2194–2202 RSC.
- O. C. Amadi, M. L. Steinhauser, Y. Nishi, S. Chung and R. T. Lee, Biomed. Microdevices, 2010, 12, 1027–1041 CrossRef CAS PubMed.
- J. Ju, J. Y. Park, K. C. Kim, H. Kim, E. Berthier, D. J. Beebe and S. H. Lee, J. Micromech. Microeng., 2008, 18, 87002–87005 CrossRef.
- G. S. Jeong, J. Oh, S. B. Kim, M. R. Dokmeci, H. Bae, S. H. Lee and A. Khademhosseini, Lab Chip, 2014, 14, 4213–4219 RSC.
- S. V. Murphy and A. Atala, Nat. Biotechnol., 2014, 32, 773–785 CrossRef CAS PubMed.
- A. Lee, A. R. Hudson, D. J. Shiwarski, J. W. Tashman and A. W. Feinberg, Science, 2019, 365, 482–487 CrossRef CAS PubMed.
- F. Cheng, X. Cao, H. Li, T. Liu, X. Xie, D. Huang, S. Maharjan, H. P. Bei, A. Gómez, J. Li, H. Zhan, H. Shen, S. Liu, J. He and Y. S. Zhang, Nano Lett., 2019, 19, 3603–3611 CrossRef CAS PubMed.
- H. Li, F. Cheng, W. Li, X. Cao, Z. Wang, M. Wang, J. A. Robledo-Lara, J. Liao, C. Chavez-Madero, S. Hassan, J. Xie, G. Trujillo-de Santiago, M. M. Alvarez, J. He and Y. S. Zhang, Biofabrication, 2020, 12, 045027 CrossRef CAS PubMed.
- H. Li, F. Cheng, J. A. Robledo-Lara, J. Liao, Z. Wang and Y. S. Zhang, Bio-Des. Manuf., 2020, 3, 252–265 CrossRef.
- D. Chimene, C. W. Peak, J. L. Gentry, J. K. Carrow, L. M. Cross, E. Mondragon, G. B. Cardoso, R. Kaunas and A. K. Gaharwar, ACS Appl. Mater. Interfaces, 2018, 10, 9957–9968 CrossRef CAS PubMed.
- Z. Y. Shrike, G. Haghiashtiani, T. Hübscher, D. J. Kelly, J. M. Lee, M. Lutolf, M. C. McAlpine, W. Y. Yeong, M. Zenobi-Wong and J. Malda, Nat. Rev. Methods Primers, 2021, 1, 75 CrossRef.
- S. Van Belleghem, L. Torres Jr., M. Santoro, B. Mahadik, A. Wolfand, P. Kofinas and J. P. Fisher, Adv. Funct. Mater., 2020, 30, 1907145 CrossRef CAS PubMed.
- J. Y. Park, C. M. Hwang, S. H. Lee and S. H. Lee, Lab Chip, 2007, 7, 1673–1680 RSC.
- P. M. Bruno, Y. P. Liu, G. Y. Park, J. Murai, C. E. Koch, T. J. Eisen, J. R. Pritchard, Y. Pommier, S. J. Lippard and M. T. Hemann, Nat. Med., 2017, 23, 461–471 CrossRef CAS PubMed.
- R. Oun, Y. E. Moussa and N. J. Wheate, Dalton Trans., 2018, 47, 6645–6653 RSC.
- D. B. Longley, D. P. Harkin and P. G. Johnston, Nat. Rev. Cancer, 2003, 3, 330–338 CrossRef CAS PubMed.
- Z. Li, Y. Guo, Y. Yu, C. Xu, H. Xu and J. Qin, Integr. Biol., 2016, 8, 1022–1029 CrossRef CAS PubMed.
- N. Carragher, F. Piccinini, A. Tesei, O. J. Trask, M. Bickle and P. Horvath, Nat. Rev. Drug Discovery, 2018, 17, 606 CrossRef CAS PubMed.
- A. C. Rios and H. Clevers, Nat. Methods, 2018, 15, 24–26 CrossRef CAS PubMed.
- S. Maharjan, J. Alva, C. Cámara, A. Rubio and Y. S. Zhang, Matter, 2020, 4, 217–240 CrossRef PubMed.
- S. Maharjan, D. Bonilla, P. Sindurakar, H. Li, W. Li, S. Duarte, A. Zarrinpar and Y. S. Zhang, Bio-Des. Manuf., 2021, 4, 157–170 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2me00142j |
| ‡ Hongbin Li and Feng Cheng contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |