
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBackbone-driven host–dopant miscibility modulates molecular doping in NDI conjugated polymers†
Diego
Rosas Villalva
 a,
Saumya
Singh
b,
Luke A.
Galuska
c,
Anirudh
Sharma
a,
Jianhua
Han
a,
Jian
Liu
d,
Md Azimul
Haque
a,
Soyeong
Jang
a,
Abdul Hamid
Emwas
e,
L. Jan Anton
Koster
d,
Xiaodan
Gu
c,
Bob C.
Schroeder
*b and
Derya
Baran
*a
a,
Saumya
Singh
b,
Luke A.
Galuska
c,
Anirudh
Sharma
a,
Jianhua
Han
a,
Jian
Liu
d,
Md Azimul
Haque
a,
Soyeong
Jang
a,
Abdul Hamid
Emwas
e,
L. Jan Anton
Koster
d,
Xiaodan
Gu
c,
Bob C.
Schroeder
*b and
Derya
Baran
*a
aKing Abdullah University of Science and Technology (KAUST), Physical Sciences and Engineering Division (PSE), KAUST Solar Center (KSC), Thuwal 23955, Saudi Arabia. E-mail: derya.baran@kaust.edu.sa
bDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: b.c.schroeder@ucl.ac.uk
cSchool of Polymer Science and Engineering, University of Southern Mississippi, Hattiesburg, MS 39406, USA
dZernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG, Groningen, The Netherlands
eKing Abdullah University of Science and Technology (KAUST), Core Labs, Thuwal 23955, Saudi Arabia
First published on 20th December 2021
Abstract
Molecular doping is the key to enabling organic electronic devices, however, the design strategies to maximize doping efficiency demands further clarity and comprehension. Previous reports focus on the effect of the side chains, but the role of the backbone is still not well understood. In this study, we synthesize a series of NDI-based copolymers with bithiophene, vinylene, and acetylenic moieties (P1G, P2G, and P3G, respectively), all containing branched triethylene glycol side chains. Using computational and experimental methods, we explore the impact of the conjugated backbone using three key parameters for doping in organic semiconductors: energy levels, microstructure, and miscibility. Our experimental results show that P1G undergoes the most efficient n-type doping owed primarily to its higher dipole moment, and better host–dopant miscibility with N-DMBI. In contrast, P2G and P3G possess more planar backbones than P1G, but the lack of long-range order, and poor host–dopant miscibility limit their doping efficiency. Our data suggest that backbone planarity alone is not enough to maximize the electrical conductivity (σ) of n-type doped organic semiconductors, and that backbone polarity also plays an important role in enhancing σ via host–dopant miscibility. Finally, the thermoelectric properties of doped P1G exhibit a power factor of 0.077 μW m−1 K−2, and ultra-low in-plane thermal conductivity of 0.13 W m−1K−1 at 5 mol% of N-DMBI, which is among the lowest thermal conductivity values reported for n-type doped conjugated polymers.
New conceptsIn recent studies, modulation of host–dopant miscibility led to enhanced molecular doping of organic semiconductors (OSCs), thus enabling their applicability several organic electronic devices such as organic thermoelectrics. The most common approach includes the addition of oligoethylene glycol side chains to the OSC to improve their miscibility with polar dopants such as N-DMBI. Herein we focus on the conjugated polymer (CP) backbone and its impact on the molecular doping efficiency of naphthalene diimide (NDI) based CPs, with various degrees of backbone planarity, rigidity, and dipole moments. Our data show a strong correlation between the backbone dipole moment and the maximum value of the electrical conductivity achieved upon molecular doping with N-DMBI. The most polar backbone in the series (P1G) showed the highest electrical conductivity at the lowest doping concentrations. On the contrary, P3G possessing the least polar backbone showed the lowest electrical conductivity, requiring larger dopant amounts to reach its maximum conductivity. This suggests a backbone-driven miscibility effect that can enable or limit molecular doping efficiency. Consequently, we measured the temperature-dependent thermoelectric properties of P1G, finding an ultra-low in-plane thermal conductivity value of 0.13 W m−1 K−1 enabled by the branched side chains. This opens a new possible strategy to further decrease the thermal conductivity of CPs. |
Introduction
Organic semiconductors (OSCs) hold the potential to enable a whole new generation of electronics such as flexible and wearable devices.1 Unlike their inorganic counterparts, OSCs can be solution-processed and possess inherent flexibility.2,3 However, they are still limited by their low mobility. Molecular doping has become a major enabler of several technologies such as transport layers in organic photovoltaics and organic light-emitting diodes, organic photodiodes, organic field-effect transistors, and most recently in organic electrochemical transistors and organic thermoelectrics (OTEs).4–11 While p-type doped OSCs have achieved σ in a range of 103 to 104 S cm−1, n-type doped OSCs possess σ in the order of 100 to 101 S cm−1.12–16 Functional organic thermoelectric devices require both n-type and p-type materials. Hence, the design of new n-type conjugated polymers (CPs) and, the understanding of the rules to design them, is of uttermost importance to overcome these challenges.17–19Naphthalenediimide (NDI) based n-type OSCs have been explored, mainly through modular modification of P(NDIOD-2T) (N2200), such as side chain alterations to increase dopant miscibility and enhance electrical conductivity.20–22 Additionally, the dipole moment of polar side chains screens the backbone and dopant charges, thus reducing Coulombic binding and improving the fraction of mobile charge carriers.23 Other design approaches include backbone modification such as substitution of bithiophene (2T) units by bithiazole.24,25 These modifications increased the backbone planarity, leading to better charge delocalization along the polymer backbone and enhanced molecular packing for better charge transport. Further modifications include backbone halogenation which can induce deeper LUMO levels to improve doping efficiency, and introducing rigid bonds to increase the backbone planarity, improve film crystallinity, and provide enhanced charge transport properties.26,27
In this contribution, we explore the backbone modification of NDI-based CPs and its effect on molecular doping. The synthesized polymers substituted with bithiophene, vinylene, and acetylenic groups, are named P(NDI4TEG-2T), P(NDI4TEG-V), and P(NDI4TEG-A) respectively. The synthetic route and chemical structure of the three polymers is depicted in Scheme 1. For simplicity, these will be referred to as P1G, P2G, and P3G respectively throughout the text. All the synthesized CPs contain branched TEG side chains motivated from previous reports to improve host–dopant miscibility and reduce Coulomb interaction, allowing to study the effect of the backbone on molecular doping and the electronic properties of CPs.20,21,28
 | ||
| Scheme 1 Synthetic routes towards polymers P1G, P2G and P3G, and chemical structure of N-DMBI. | ||
Experimental results showed that all three polymers can be doped by N-DMBI, however, the conductivity of P1G outperformed the other two polymers owed to a higher crystallinity and good miscibility with N-DMBI. While P2G exhibits good miscibility with N-DMBI, the lack of long-range order limits its σ. P3G shows poor host–dopant miscibility and a low degree of molecular order, which make it the polymer with the lowest σ. Finally, we analysed the temperature-dependent thermoelectric properties of P1G as its electrical conductivity is the largest of the three CPs. Our results show that OTEs can benefit from branched TEG side chains, as only 5 mol% of N-DMBI suffices to maximize the power factor (PF). Remarkably, P1G shows an in-plane thermal conductivity (κ‖) of only 0.13 W m−1 K−1, one of the lowest reported values for CPs. This suggests that branched TEG side chains are a promising strategy to reduce the thermal conductivity of CPs, in addition to improving their miscibility with polar dopants.
Results and discussion
Copolymers were synthesized via Stille coupling polymerization in chlorobenzene (Scheme 1). Synthesis of NDI monomer 1 and polymer P2G is reported in our previous work and synthetic protocols for polymers P1G and P3G are described in the supporting information.29 The molecular weight analysis of the polymers P1G (Mn = 21.4 kg mol− 1, Đ = 1.5) and P2G (Mn = 6.9 kg mol− 1, Đ = 1.8) was carried out by high-temperature gel permeation chromatography (GPC) using dichlorobenzene as a solvent. The molecular weight of P3G could not be obtained by GPC analysis due to the strong aggregation of P3G in solution, owing to its planar backbone.Previous reports suggest that the bithiophene-induced backbone twist in N2200 and perylene dimiide–bithiophene CPs limits the polaron delocalization length and the electrical conductivity, showing that more planar backbones possess higher σ.24,30 This makes backbone planarization an attractive approach to improve σ of n-type doped CPs. Hence, the designs of P2G and P3G are aimed at increasing the backbone planarity. Previous reports have used non-bonding conformational locks to improve molecular packing and charge transport properties.31–33 To better understand the role of the polymer conjugated backbone on molecular doping, we considered three parameters. First, the energetics of the polymers (considering lowest unoccupied molecular orbitals (LUMO)), second, the backbone planarity and rigidity, and third, the dipole moment of the backbone, which we shall define as the local charge separation induced by the atoms within the backbone of a polymer while disregarding the contributions of the atoms in the side chains.
The energy levels of the frontier molecular orbitals for the tetramers of the CPs were calculated by DFT (Fig. S4, ESI†), the energy levels are represented in Fig. 1a. P1G shows the shallowest LUMO (−3.46 eV) followed by P2G (−3.72 eV) and P3G (−3.98 eV), suggesting that all three polymers should be efficiently doped by the singly occupied molecular orbital (SOMO) of N-DMBI at −2.36 eV (Fig. S5, ESI†). For P1G, the HOMO is mainly delocalized on the bithiophene donor subunit, and the LUMO is delocalized on the NDI acceptor subunit, similarly to previously reported D–A CPs.34 In contrast, P2G and P3G show both HOMO and LUMO delocalized along the whole tetramer backbone, such delocalization might be beneficial for charge transport by extending the probability of both intra- and intermolecular hopping, consequently improve σ.35,36 Additionally, we calculated the torsion potential for all three polymers (Fig. 1b). P1G showed a twisted backbone with an angle of ∼48° for the bond between the NDI and 2T subunit, along with a moderate torsion energy barrier of 0.13 eV. P2G showed a more planar backbone with an angle of ∼25° between NDI and vinylene subunits, it also possesses the largest torsion energy barrier (0.27 eV) making it the most rigid backbone of the three CPs, owing to the interaction of the carbonyl group in the NDI and the hydrogen of the vinylene which work as a non-bonding conformational lock.32,37 P3G shows the most planar backbone, stabilizing ∼0°, however, its rather low torsion energy barrier (0.07 eV) implies that several conformers might be possible for this CP and hinder its charge transport properties. Last, we computed the dipole moments of the tetramers through DFT (Fig. 1c). The arrows are vectors depicting the magnitude and direction of the backbone dipole moment with respect to the conjugated backbone. P1G shows the largest dipole moment of 6.58 D, followed by P2G with 3.13 D, and P3G has the smallest dipole of 1.50 D. Although these CPs contain TEG side chains, we are interested in observing if the CP backbone will still influence the miscibility with the dopant, and consequently its electronic properties. N-DMBI, being a polar molecule, should have better miscibility with P1G due to its larger dipole moment.
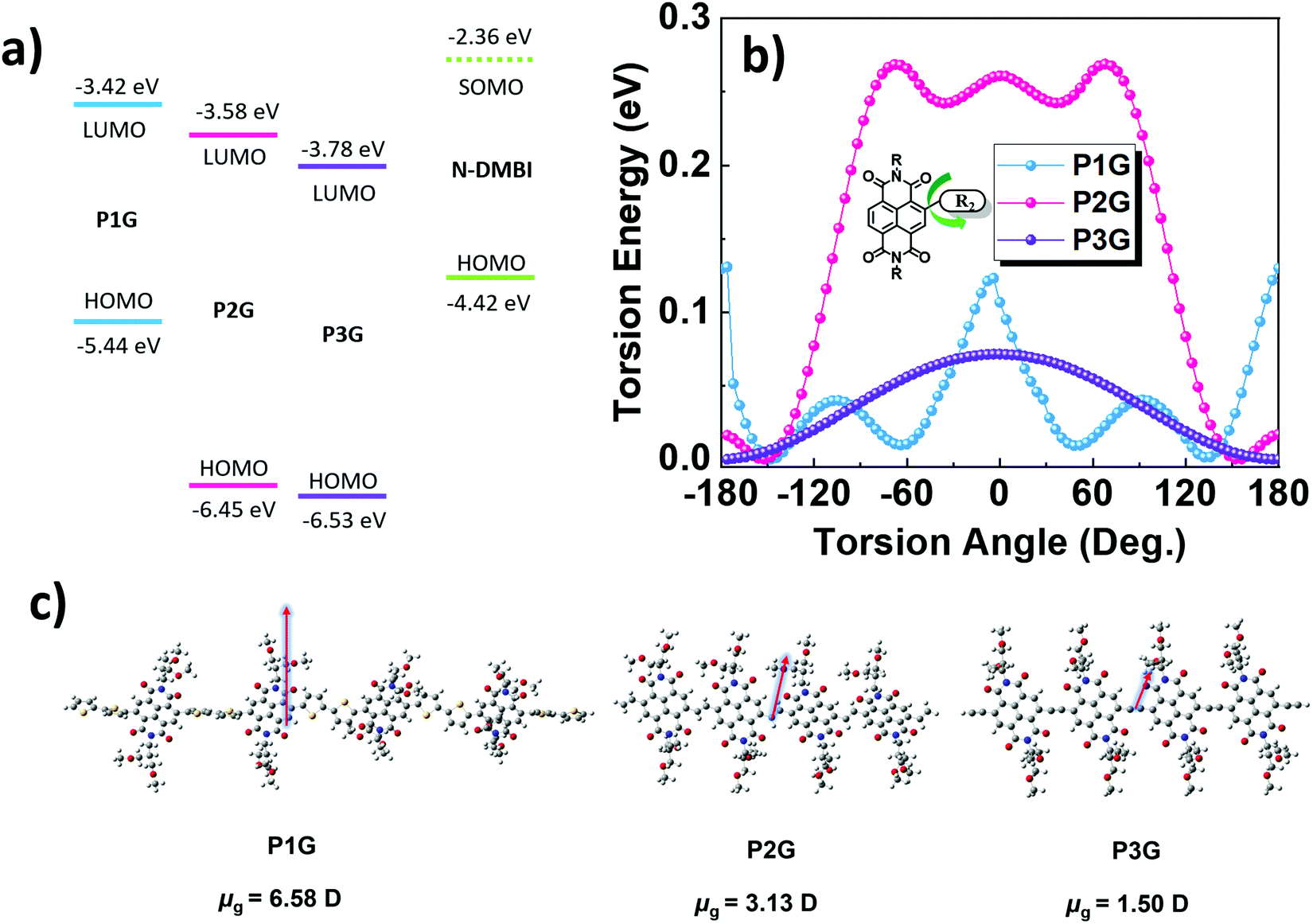 | ||
| Fig. 1 Results from DFT calculations. (a) energy levels of CPs and N-DMBI (b) torsion energy profiles of the NDI–R2 bond. (c) Energy minimized 3D structure and dipole moments of the backbone of tetramer models for CPs, arrows are vectors depicting the magnitude and direction of the dipole moment. | ||
After carrying out the DFT calculations, we proceeded to characterize the CP films. Pristine and doped polymer films were spin-coated from stock solutions in chloroform with a concentration of 10 mg mL−1. Solution doping was performed by mixing aliquots of stock chloroform solutions of polymer and N-DMBI dopant at corresponding amounts to achieve the desired molar ratio. This was calculated by considering the ratio of dopant molecules to repeat units of each polymer. Fig. S6 (ESI†) shows the absorption spectra of the pristine and doped polymer films. P1G shows similar absorption features compared to other N2200 analogue copolymers, with an intramolecular charge transfer (ICT) peak at ∼725 nm.38 Addition of N-DMBI resulted in the bleaching of this peak as a result of the charge transfer from N-DMBI to P1G. On the other hand, P2G and P3G lack the ICT feature and show the peaks of the π–π* transition at 640 and 555 nm, respectively. Addition of the N-DMBI results in bleaching of the π–π* transition and formation of new spectral features in the UV-vis spectrum with peaks in ∼690 nm (P2G) and 665 nm (P3G). An additional broadband feature appears in the near-infrared region for both polymers. This set of features can be correlated to the polaronic and bipolaronic transitions showing successful reduction of P2G and P3G by N-DMBI.
The electrical conductivity of pristine and doped polymers was compared by spin coating films on top of glass substrates with pre-patterned gold electrodes (Fig. 2a–c). All samples showed improved σ upon addition of N-DMBI, however, this change is more significant in the case of P1G, where σ of the doped films is about three orders of magnitude higher compared to neat polymer. In the case of P2G and P3G, the increment was about two orders of magnitude for each polymer. Additionally, P1G reached its maximum σ at rather low dopant concentrations, i.e. 10 to 15 mol% N-DMBI. The addition of higher dopant concentrations led to a reduction of σ, possibly due to a decrease in mobility (μ) originated by carrier-carrier scattering.39 In contrast P2G needed 25 mol%, and P3G more than 30 mol% dopant concentration to achieve their maximum conductivity. Further increasing the dopant ratio for P3G proved too challenging owed to the poor solubility of P3G, which limited the formation of continuous films and the accurate measurement of σ. These observations correlate with the calculated dipole moments. The polymer with the largest dipole moment (P1G) requires the least amount of N-DMBI to reach its maximum conductivity, and the one with the smallest dipole moment (P3G) requires more than twice the amount to achieve its maximum conductivity. Previous studies in fullerene systems have shown that increasing host–dopant miscibility can decrease the amount of dopant needed to achieve maximum conductivity.40 We further investigated these phenomena using AFM topography and phase imaging. In the case of P1G and P2G (Fig. S8 and S9, ESI†), no dopant clustering appeared, even at high doping concentrations. In the case of P3G, the topography images (Fig. S10, ESI†) reveal the formation of several aggregates only present in the doped polymer films. These aggregates show a strong contrast in the AFM phase image indicating such clusters are formed by phase-segregated dopant and implying poor host–dopant miscibility. Such phase segregation explains in part the much lower conductivity observed in P3G. Additionally, we measured the contact angle between water and the undoped CPs, N-DMBI, as well as the blended host–dopant polymer films, to determine their hydrophobic properties. Results are shown in Fig. S11 (ESI†). P1G and P2G show values of 54.1° and 31.6° respectively. These values are closer to the contact angle of N-DMBI (41.6°) than that of P3G (64.9°) suggesting better miscibility in the systems P1G:N-DMBI and P2G:N-DMBI, than in P3G:N-DMBI. This further confirms the observations carried out in AFM. Finally, the addition of N-DMBI to either P1G or P2G resulted in a decrease of the contact angle, whereas P3G remained unchanged.
 | ||
| Fig. 2 σ of P1G (a), P2G (b), and P3G (c) films doped with N-DMBI. Representative I–V curves are shown in Fig. S7 (ESI†). (d) Shows a summary of the spin densities obtained from EPR. | ||
To further elaborate on the differences in electronic properties of the three CPs, we proceeded to perform advanced spectroscopic analysis. Electron paramagnetic resonance (EPR) provides a quantitative analysis of unpaired electrons in the form of radical anions within a material, which can be correlated to the charge carrier density and offer an understanding of the differences observed in σ. We performed EPR in pristine and doped films of the NDI copolymers, the resulting spectra are shown in Fig. S12a–c (ESI†). The undoped polymer films show very weak EPR signals. The addition of N-DMBI leads to the generation of radical anions and thus large increments of the signal intensity in all polymers. Quantitative analysis of the signals provided the spin density of the samples (Fig. 2d). We observe that the undoped P1G film has a larger spin density (5 × 1018 cm−3) than the undoped films of P2G and P3G (1 × 1018 cm−3). As N-DMBI is added to the samples, the spin density increases, and both P1G and P2G show spin densities of about 1 × 1020 cm−3 while P3G shows only 3 × 1019 cm−3. Although this partially explains the lower σ of P3G, it does not explain the difference in σ of P1G and P2G at their respective maximums.
The ionization energy (IE) and electron affinity (EA) of all polymers P1G, P2G, and P3G were directly measured on thin films using ultraviolet photoelectron spectroscopy (UPS) and low-energy inverse photoelectron spectroscopy (LE-IPES) (Fig. 3a and b respectively). The IE and EA of P1G and P2G were found to be 5.58 eV/3.62 eV and 5.67/3.38 eV, respectively. Thin films of P3G were found to have an IE of 5.41 eV and the deepest EA of 3.73 eV among all three polymers. It must be noted that the DFT calculations estimated the orbital energies of tetramers, whereas the spectroscopic studies were carried out in thin solid films of polymers. The orbital energies of the isolated molecules and those in thin solid films are different,41 and would explain the differences in the calculated HOMO/LUMO and the measured IE/EA values. Among all the three polymers, P1G was found to have the most prominent peak at the onset of both UPS and IPES spectra, corresponding to the density of states (Fig. 3a and b) in the IE and EA, respectively. We attribute these features to an additional density of states (DOS) introduced by the ICT between the NDI and 2T subunits of P1G. These features can also be correlated to the peak at 725 nm in the absorption spectrum of P1G.38 Since the electronic states in the IE and EA regions contribute to the electrical conductivity of the materials, P1G is expected to have the highest electrical conductivity among all the polymers. These features are significantly weaker in the case of P2G and P3G and explain in part their lower σ.
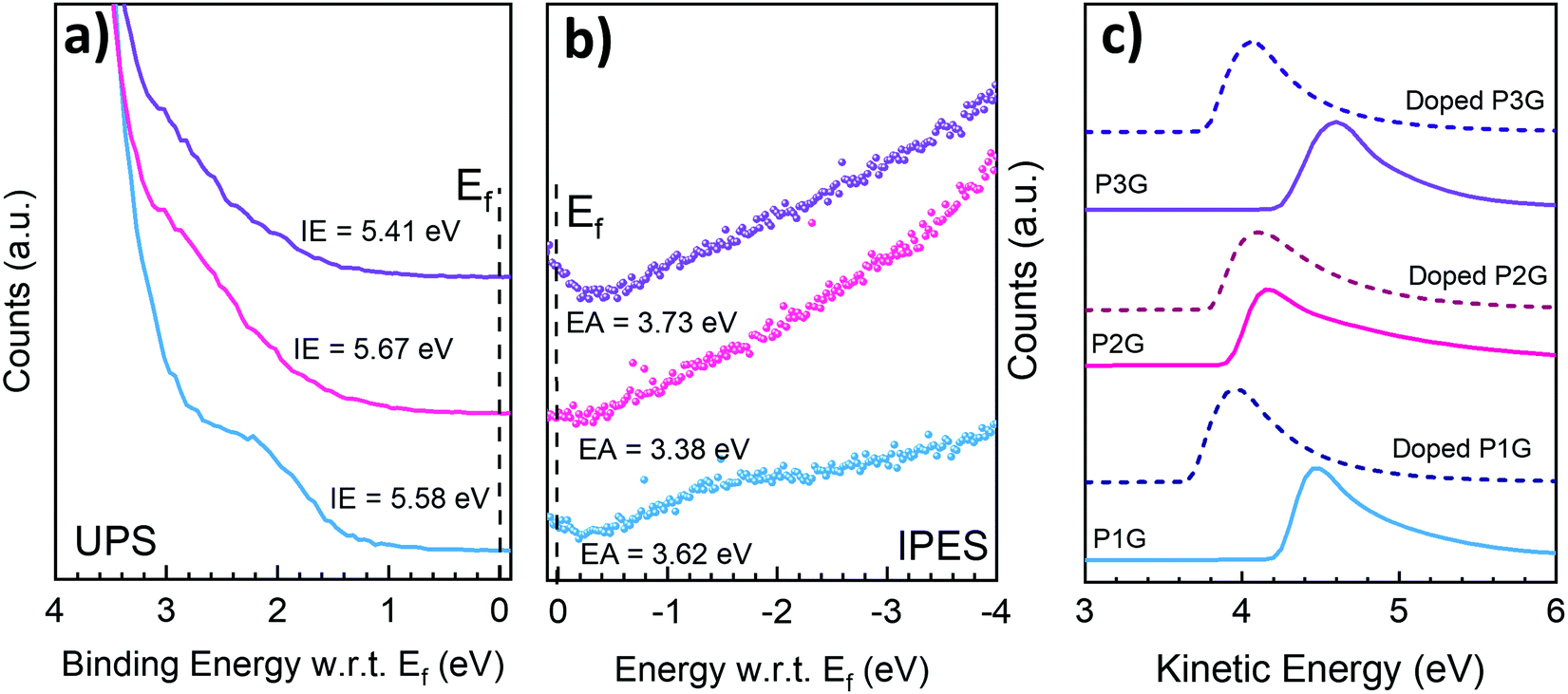 | ||
| Fig. 3 Photoelectron spectroscopy of the CPs. (a) shows the IE, (b) corresponds to the EA. (c) SECO showing the work function of the undoped films (continuous lines) and their shift upon doping (dashed lines). | ||
The secondary electron cut-off (SECO) of the UPS spectra displayed in Fig. 3c, provides direct observation of the surface work function of pristine and doped polymer films. Upon n-type doping, the SECO shifts toward vacuum level. The film of P1G demonstrates a significant reduction of over 0.56 eV in the work function from 4.23 eV to 3.67 eV, upon the addition of 20 mol% N-DMBI. Fig. S13a (ESI†) shows the IE from the UPS spectra, depicting the shift of 0.3 eV in the IE towards higher binding energy (Fermi level moves away from IE), confirming n-type doping of P1G with N-DMBI. Undoped P2G was found to have a low work function of 3.91 eV, which reduced further to 3.8 eV with the addition of N-DMBI. The ability of undoped P2G to modify the work function of a range of electrode surfaces enabled its use as an efficient electron transport layer in organic solar cells.29 The IE of P2G demonstrated a small shift of only 0.09 eV towards higher binding energy (Fig. S13b, ESI†) with 20 mol% of N-DMBI, again confirming n-type doping of P2G. Finally, the work function of P3G was also found to significantly reduce from 4.25 eV to 3.76 eV, upon the addition of 20 mol% N-DMBI accompanied by a 0.45 eV shift of the IE towards higher binding energy (Fig. S13c, ESI†), confirming n-type doping. The IE of P2G (5.39 eV) and P3G (5.63 eV) was found to be largely unchanged with the N-DMBI doping, whereas the IE of P1G was reduced from 5.58 eV to 5.32 eV. This shows that P1G undergoes the most efficient doping of the three polymers.
GIWAXS was carried out to study the morphology of the CP films. The 2D data and linecuts are displayed in Fig. 4, (see Fig. S14 for all 2D GIWAXS scattering results and Fig. S15 for their linecuts, ESI†) and a summary of the peaks are displayed in Tables S1 and S2 (ESI†). P1G (Fig. 4a) shows a clear face-on orientation, which is similar to that of N2200, with the lamellar and backbone scattering appearing along the in-plane direction, whereas the π–π stacking appears along the out-of-plane direction. P2G and P3G on the other hand have an amorphous nature, displaying isotropic diffused rings (Fig. 4b and c respectively). Despite the higher planarity of P3G and higher rigidity of P2G, their films did not develop long-range ordering, leading to lower electrical conductivities. The linecuts in Fig. 4d and e showcase the higher degree of molecular packing for P1G, e.g., prominent (200) scattering peaks at q = 0.5 A−1, compared to P2G and P3G. The addition of N-DMBI did not produce major changes on the d-spacing of either P1G or P2G, showing good miscibility with N-DMBI in the amorphous region of the film. Lastly, P3G did not undergo major d-spacing changes either, but additional diffraction peaks appeared on the heavily doped samples suggesting strong phase segregation and formation of dopant crystalline domains, which explains why P3G has the lowest electrical conductivity amongst others.
 | ||
| Fig. 4 2D GIWAXS from pristine (a) P1G, (b) P2G and (c) P3G. (d) Are in-plane linecuts, (e) are out-of-plane linecuts for pristine polymer films. (f) Shows the change of d-spacing upon the addition of N-DMBI. | ||
Thermoelectric properties of P1G
P1G shows higher σ at very low dopant loading concentrations suggesting its suitability for thermoelectric applications. We measured its temperature-dependent thermoelectric (TE) properties with a chip-based technique. σ and Seebeck coefficient (α) were measured using Van der Pauw configuration to measure temperature-dependent σ and α; temperature-dependent κ‖ was measured in the same chip using 3ω method. In the case of P2G and P3G, σ is lower than the measurement limit of the instrument. The temperature-dependent σ (Fig. 5a) shows that P1G follows typical semiconductor behaviour i.e. increasing proportionally with temperature. The highest α was measured at 5 mol% N-DMBI, at higher dopant concentrations this value decreases rapidly, as increased dopant concentrations reduce the average energy of the charge carriers, also known as transport energy (Et).42α shows negligible dependency on the temperature. The highest power factor for P1G was also achieved at this doping concentration (0.077 μW K−2 m−1 at room temperature and 0.25 μW K−2 m−1 at 130 °C). Finally, P1G shows an ultra-low κ‖ of 0.13 W m−1 K−1, which is one of the lowest values known for CPs so far. N2200, the alkyl-substituted analogue of P1G, possesses an in-plane thermal conductivity of 0.27 W m−1K−1.43κ‖ of P1G shows a low dependency on temperature within the studied temperature window. However, increasing the dopant concentration led to increased κ‖, hence, the highest figure of merit (ZT) of 2 × 10−4 was achieved at 5 mol% of N-DMBI (Fig. S16, ESI†). This suggests that TEG side chains are a promising approach to reduce the thermal conductivity of CPs, a key factor to improve their figure of merit for thermoelectric applications.44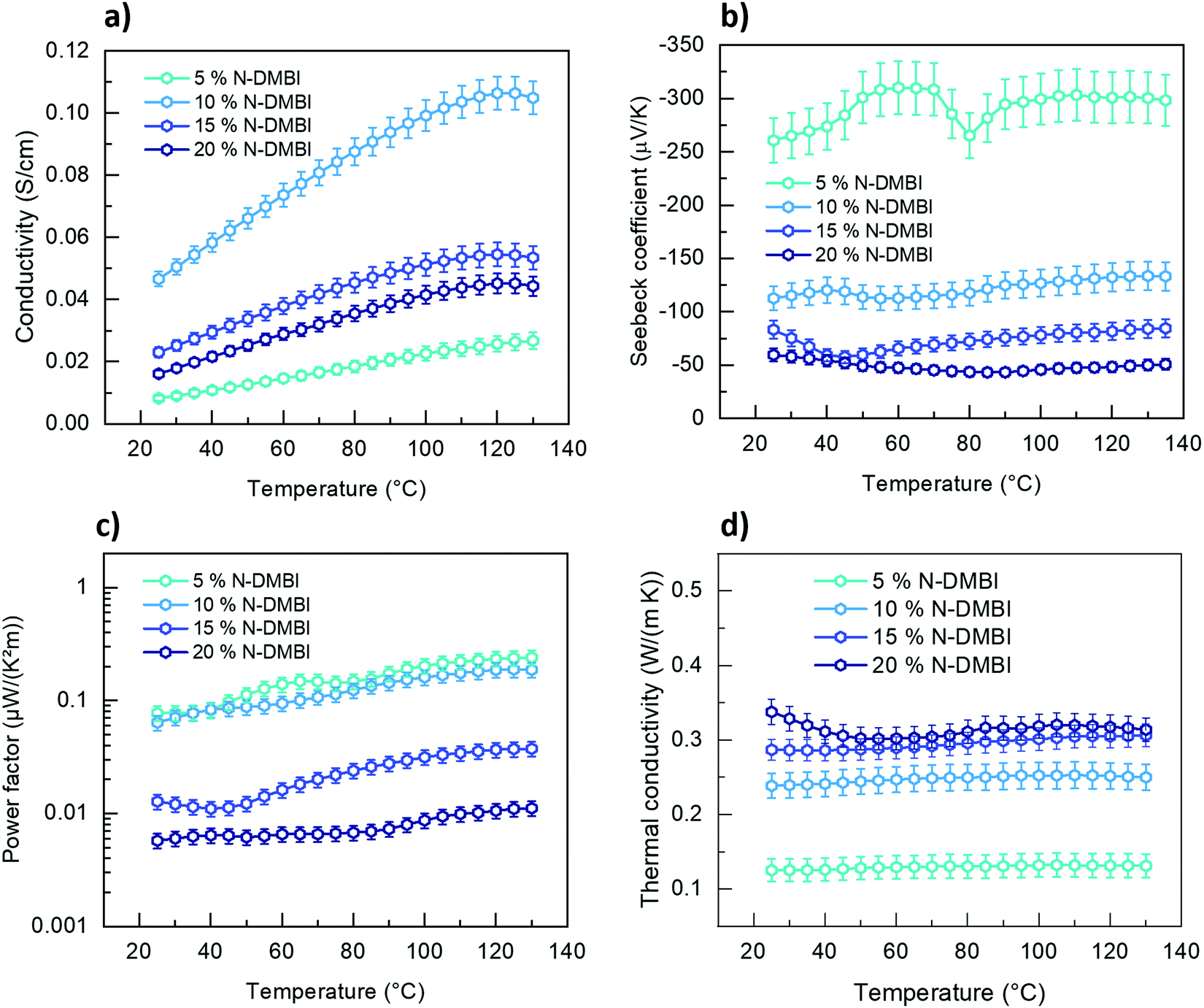 | ||
| Fig. 5 Temperature-dependent thermoelectric parameter of P1G (a) σ, (b) α, (c) PF, and (d) κ‖. | ||
Conclusions
A series of NDI-based copolymers with branched TEG side chains were synthesized, proposing conjugated vinylene and acetylenic linkers to reduce steric hindrance and improve backbone planarity and rigidity. While these spacer groups can effectively produce planar backbones, only P1G results in oriented molecular packing, P2G and P3G on the other hand are mostly amorphous suggesting backbone planarization alone is not enough to produce long-range ordering. While n-type molecular doping was demonstrated for all three polymers, P1G outperformed its analogues owing to its more ordered and long-range molecular packing and better host–dopant miscibility. P2G showed lower electrical conductivity values, with good host–dopant miscibility, but no long-range molecular packing. Finally, N-type doped P3G films showed the lowest conductivity of the three polymers due to its amorphous nature and poor host-dopant miscibility. This study showcases the impact of the backbone orientation on the efficiency of molecular doping and in the miscibility of molecular dopants where low polarity and miscibility of P3G overcame the presence of branched TEG side chains, limiting molecular doping with N-DMBI. Finally, the thermoelectric properties of P1G showed a maximum power factor at 5 mol% of N-DMBI, with a particularly low in-plane thermal conductivity. We believe this low value is enabled by the disordered nature of the branched TEG side chains, which opens a new path to discovering new conjugated polymers with ultra-low thermal conductivity values.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This publication is based upon work supported by the King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) under Award No. OSR-CRG2018-3737 and British Council Newton Fund Institutional Links (ref: 337067). B.C.S. acknowledges the UK Research and Innovation for Future Leaders Fellowship no. MR/S031952/1 and EPSRC grant (EP/P007767/1). L. G. and X. G. thanks U.S. Department Energy provides partial support for the scattering experiments in this work under award No. DE-SC0022050. The work by J. L. and L. J. A. K. was supported by a grant from STW/NWO (VIDI 13476).References
- F. Zhang, Y. Zang, D. Huang, C. A. Di and D. Zhu, Nat. Commun., 2015, 6, 8356 CrossRef CAS PubMed
.
- R. Kroon, D. A. Mengistie, D. Kiefer, J. Hynynen, J. D. Ryan, L. Yu and C. Muller, Chem. Soc. Rev., 2016, 45, 6147–6164 RSC
- O. Bubnova and X. Crispin, Energy Environ. Sci., 2012, 5, 9345–9362 RSC
- I. E. Jacobs and A. J. Moule, Adv. Mater., 2017, 29, 1703063 CrossRef PubMed
- Y. Xu, H. Sun, A. Liu, H. H. Zhu, W. Li, Y. F. Lin and Y. Y. Noh, Adv. Mater., 2018, 30, e1801830 CrossRef PubMed
- B. Russ, A. Glaudell, J. J. Urban, M. L. Chabinyc and R. A. Segalman, Nat. Rev. Mater., 2016, 1, 16050 CrossRef CAS
- L. M. Cowen, J. Atoyo, M. J. Carnie, D. Baran and B. C. Schroeder, ECS J. Solid State Sci. Technol., 2017, 6, N3080–N3088 CrossRef CAS
- W. Zhao, J. Ding, Y. Zou, C. A. Di and D. Zhu, Chem. Soc. Rev., 2020, 49, 7210–7228 RSC
- X. Peng, L. Hu, F. Qin, Y. Zhou and P. K. Chu, Adv. Mater. Interfaces, 2018, 5, 1701404 CrossRef
- A. F. Paterson, A. Savva, S. Wustoni, L. Tsetseris, B. D. Paulsen, H. Faber, A. H. Emwas, X. Chen, G. Nikiforidis, T. C. Hidalgo, M. Moser, I. P. Maria, J. Rivnay, I. McCulloch, T. D. Anthopoulos and S. Inal, Nat. Commun., 2020, 11, 3004 CrossRef CAS PubMed
- M. Kang, S.-M. Ko, J. Kim, S. Z. Hassan, D.-W. Jee and D. S. Chung, Mater. Horiz., 2020, 7, 3034–3042 RSC
- O. Bubnova, Z. U. Khan, A. Malti, S. Braun, M. Fahlman, M. Berggren and X. Crispin, Nat. Mater., 2011, 10, 429–433 CrossRef CAS PubMed
- V. Vijayakumar, Y. Zhong, V. Untilova, M. Bahri, L. Herrmann, L. Biniek, N. Leclerc and M. Brinkmann, Adv. Energy Mater., 2019, 9, 1900266 CrossRef
- C. Y. Yang, Y. F. Ding, D. Huang, J. Wang, Z. F. Yao, C. X. Huang, Y. Lu, H. I. Un, F. D. Zhuang, J. H. Dou, C. A. Di, D. Zhu, J. Y. Wang, T. Lei and J. Pei, Nat. Commun., 2020, 11, 3292 CrossRef CAS PubMed
- M. Xiong, X. Yan, J. T. Li, S. Zhang, Z. Cao, N. Prine, Y. Lu, J. Y. Wang, X. Gu and T. Lei, Angew. Chem., Int. Ed., 2021, 60, 8189–8197 CrossRef CAS PubMed
- J. Liu, B. van der Zee, R. Alessandri, S. Sami, J. Dong, M. I. Nugraha, A. J. Barker, S. Rousseva, L. Qiu, X. Qiu, N. Klasen, R. C. Chiechi, D. Baran, M. Caironi, T. D. Anthopoulos, G. Portale, R. W. A. Havenith, S. J. Marrink, J. C. Hummelen and L. J. A. Koster, Nat. Commun., 2020, 11, 5694 CrossRef CAS PubMed
- M. Goel, C. D. Heinrich, G. Krauss and M. Thelakkat, Macromol. Rapid Commun., 2019, 40, e1800915 CrossRef PubMed
- F. Zhang and C.-a. Di, Chem. Mater., 2020, 32, 2688–2702 CrossRef CAS
- H. Jia and T. Lei, J. Mater. Chem. C, 2019, 7, 12809–12821 RSC
- J. Liu, L. Qiu, R. Alessandri, X. Qiu, G. Portale, J. Dong, W. Talsma, G. Ye, A. A. Sengrian, P. C. T. Souza, M. A. Loi, R. C. Chiechi, S. J. Marrink, J. C. Hummelen and L. J. A. Koster, Adv. Mater., 2018, 30, 1704630 CrossRef PubMed
- D. Kiefer, A. Giovannitti, H. Sun, T. Biskup, A. Hofmann, M. Koopmans, C. Cendra, S. Weber, L. J. Anton Koster, E. Olsson, J. Rivnay, S. Fabiano, I. McCulloch and C. Muller, ACS Energy Lett., 2018, 3, 278–285 CrossRef CAS PubMed
- J. Liu, G. Ye, H. G. O. Potgieser, M. Koopmans, S. Sami, M. I. Nugraha, D. R. Villalva, H. Sun, J. Dong, X. Yang, X. Qiu, C. Yao, G. Portale, S. Fabiano, T. D. Anthopoulos, D. Baran, R. W. A. Havenith, R. C. Chiechi and L. J. A. Koster, Adv. Mater., 2021, 33, e2006694 CrossRef PubMed
- J. Liu, L. Qiu, G. Portale, S. Torabi, M. C. A. Stuart, X. Qiu, M. Koopmans, R. C. Chiechi, J. C. Hummelen and L. J. Anton Koster, Nano Energy, 2018, 52, 183–191 CrossRef CAS
- S. Wang, H. Sun, T. Erdmann, G. Wang, D. Fazzi, U. Lappan, Y. Puttisong, Z. Chen, M. Berggren, X. Crispin, A. Kiriy, B. Voit, T. J. Marks, S. Fabiano and A. Facchetti, Adv. Mater., 2018, 30, e1801898 CrossRef PubMed
- J. Liu, G. Ye, B. V. Zee, J. Dong, X. Qiu, Y. Liu, G. Portale, R. C. Chiechi and L. J. A. Koster, Adv. Mater., 2018, 30, e1804290 CrossRef PubMed
- K. Shi, F. Zhang, C. A. Di, T. W. Yan, Y. Zou, X. Zhou, D. Zhu, J. Y. Wang and J. Pei, J. Am. Chem. Soc., 2015, 137, 6979–6982 CrossRef CAS PubMed
- C. Y. Yang, W. L. Jin, J. Wang, Y. F. Ding, S. Nong, K. Shi, Y. Lu, Y. Z. Dai, F. D. Zhuang, T. Lei, C. A. Di, D. Zhu, J. Y. Wang and J. Pei, Adv. Mater., 2018, 30, e1802850 CrossRef PubMed
- J. Liu, L. Qiu, G. Portale, M. Koopmans, G. Ten Brink, J. C. Hummelen and L. J. A. Koster, Adv. Mater., 2017, 29, 1701641 CrossRef PubMed
- A. Sharma, S. Singh, X. Song, D. Rosas Villalva, J. Troughton, D. Corzo, L. Toppare, G. Gunbas, B. C. Schroeder and D. Baran, Chem. Mater., 2021, 33, 8602–8611 CrossRef CAS
- B. D. Naab, X. Gu, T. Kurosawa, J. W. F. To, A. Salleo and Z. Bao, Adv. Electron Mater., 2016, 2, 1600004 CrossRef
- T. Lei, J. H. Dou, X. Y. Cao, J. Y. Wang and J. Pei, J. Am. Chem. Soc., 2013, 135, 12168–12171 CrossRef CAS PubMed
- H. Huang, L. Yang, A. Facchetti and T. J. Marks, Chem. Rev., 2017, 117, 10291–10318 CrossRef CAS PubMed
- Y. Wang, T. Hasegawa, H. Matsumoto and T. Michinobu, J. Am. Chem. Soc., 2019, 141, 3566–3575 CrossRef CAS PubMed
- T. Erdmann, S. Fabiano, B. Milian-Medina, D. Hanifi, Z. Chen, M. Berggren, J. Gierschner, A. Salleo, A. Kiriy, B. Voit and A. Facchetti, Adv. Mater., 2016, 28, 9169–9174 CrossRef CAS PubMed
- Y. Kim, H. Hwang, N. K. Kim, K. Hwang, J. J. Park, G. I. Shin and D. Y. Kim, Adv. Mater., 2018, 30, e1706557 CrossRef PubMed
- M. Moser, A. Savva, K. Thorley, B. D. Paulsen, T. C. Hidalgo, D. Ohayon, H. Chen, A. Giovannitti, A. Marks, N. Gasparini, A. Wadsworth, J. Rivnay, S. Inal and I. McCulloch, Angew. Chem., Int. Ed., 2021, 60, 7777–7785 CrossRef CAS PubMed
- T. Lei, X. Xia, J. Y. Wang, C. J. Liu and J. Pei, J. Am. Chem. Soc., 2014, 136, 2135–2141 CrossRef CAS PubMed
- L. Gao, Z. G. Zhang, L. Xue, J. Min, J. Zhang, Z. Wei and Y. Li, Adv. Mater., 2016, 28, 1884–1890 CrossRef CAS PubMed
- M. Koopmans, M. A. T. Leiviska, J. Liu, J. Dong, L. Qiu, J. C. Hummelen, G. Portale, M. C. Heiber and L. J. A. Koster, ACS Appl. Mater. Interfaces, 2020, 12, 56222–56230 CrossRef CAS PubMed
- L. Qiu, J. Liu, R. Alessandri, X. Qiu, M. Koopmans, R. W. A. Havenith, S. J. Marrink, R. C. Chiechi, L. J. Anton Koster and J. C. Hummelen, J. Mater. Chem. A, 2017, 5, 21234–21241 RSC
- J. Sworakowski, Synth. Met., 2018, 235, 125–130 CrossRef CAS
- Z. Liang, H. H. Choi, X. Luo, T. Liu, A. Abtahi, U. S. Ramasamy, J. A. Hitron, K. N. Baustert, J. L. Hempel, A. M. Boehm, A. Ansary, D. R. Strachan, J. Mei, C. Risko, V. Podzorov and K. R. Graham, Nat. Mater., 2021, 20, 518–524 CrossRef CAS PubMed
- S. Kommandur and S. Yee, Rev. Sci. Instrum., 2018, 89, 114905 CrossRef PubMed
- D. Scheunemann and M. Kemerink, Phys. Rev. B, 2020, 101, 075206 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1mh01357b |
| This journal is © The Royal Society of Chemistry 2022 |