
Enhanced antibacterial function of a supramolecular artificial receptor-modified macrophage (SAR-Macrophage)†
Qian
Cheng
a,
Meng
Xu
a,
Chen
Sun
a,
Kuikun
Yang
a,
Zhiqing
Yang
a,
Junyan
Li
a,
Jun
Zheng
b,
Ying
Zheng
ab and
Ruibing
Wang
 *ab
*ab
aState Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Taipa, Macau 999078, China. E-mail: rwang@um.edu.mo
bDepartment of Pharmaceutical Sciences, Faculty of Health Sciences, University of Macau, Taipa, Macau 999078, China
First published on 17th January 2022
Abstract
Bacterial infection has become a global concern owing to the significant morbidity and mortality. Although the phagocytosis of bacteria by immune cells acts as the front line to protect human body from invading pathogens, the relatively slow encounter and insufficient capture of bacteria by immune cells often lead to an inefficient clearance of pathogens. Herein, a supramolecular artificial receptor-modified macrophage (SAR-Macrophage) was developed to enhance the recognition and latch of bacteria in the systemic circulation, mediated via strong and multipoint host–guest interactions between the artificial receptors (cucurbit[7]uril) on the macrophage and the guest ligands (adamantane) selectively anchored on Escherichia coli (E. coli). As a result, the SAR-Macrophage could significantly accelerate the recognition of E. coli, catch and internalize more pathogens, which subsequently induced the M1 polarization of macrophages to generate ROS and effectively kill the intracellular bacteria. Therefore, the SAR-Macrophage represents a simple, yet powerful anti-bacterial approach.
New conceptsReminiscent of CAR-T and CAR-Macrophage, a supramolecular artificial receptor-modified macrophage (namely, SAR-Macrophage) was developed and applied for the first time to enhance the recognition and latch of bacteria, mediated via strong, multipoint host–guest interactions between the artificial receptors (cucurbit[7]uril) on the macrophage, and the guest ligands (adamantane) remotely labeled on Escherichia coli (E. coli). As a result, the SAR-Macrophage could significantly accelerate the recognition of E. coli, catch and internalize more pathogens, which subsequently induced the M1 polarization of macrophages to generate ROS and effectively killed the intracellular bacteria. However, most of the previous works relied on the upregulation of natural surface receptors of macrophages, which may imbalance their innate immunity and inevitably lead to undesirable side effects. Herein, the bioorthogonal recognition of bacteria via artificial receptors may mitigate such risks. Therefore, the SAR-Macrophage not only represents a simple cell-engineering approach for antibacterial applications, but also provides important new insights to design and develop cell-based biomaterial therapeutics for diverse biomedical applications via an enhanced, bioorthogonal, host–guest chemistry-mediated recognition. |
Bacterial infection has attracted increasing attention due to the acquired resistance of bacteria to various antibiotics, which poses a serious threat to public health.1,2 To avoid antibiotic resistance, increasing studies are turning to the natural immune system to combat bacterial infection.3 Macrophages play a key role in immune protection from bacterial pathogens by recognizing, engulfing and digesting bacteria through a phagocytosis process.4,5 Although the macrophage endocytic process is a natural response to pathogenic bacterial infection, it is often difficult for a macrophage to efficiently encounter, latch and catch pathogens, resulting in low sensitivity and elimination efficiency of macrophages towards bacteria, thus inevitably leading to a high infection rate.6 Various strategies and mechanisms have been explored to improve the antibacterial activity of macrophages. For instance, Casanova et al. reported that the overexpression of BAI1 (brain angiogenesis inhibitor 1) enhanced the macrophage‘s encounter, engulfment and clearance of Gram-negative bacteria through the recognition of their surface lipopolysaccharides.7 Similarly, Chinetti-Gbaguidi et al. found that the pretreatment of macrophages with Liver X Receptor agonists resulted in an enhanced recognition of lipopolysaccharides and an increased reactive oxygen species (ROS) generation to exert antibacterial activities.8 In another study, Biswal et al. discovered that the activation of Nrf2 (nuclear erythroid-related factor 2) upregulated its downstream MARCO (the scavenger receptor) of defective macrophage, which was suppressed in chronic obstructive pulmonary disease, leading to restored bacterial recognition and phagocytosis in alveolar macrophages.9 In spite of these successes, all of these approaches relied on the upregulation of natural surface receptors of macrophages, which may imbalance their innate immunity and inevitably lead to undesirable side effects.10,11 On the other hand, numerous nanomaterials have been extensively studied as antibiotic carriers or to generate ROS to directly combat with bacteria.12–16 In spite of the promising antibacterial activity, these nanomaterials still face a series of obstacles, including nonspecific toxicity and increased risk of drug resistance. Herein, through simply engineering the natural macrophage with artificial receptors that have specific, bioorthogonal recognition of bacteria may mitigate various risks, such as drug-resistance and undesirable side effects.
Indeed, artificial host–guest interactions offer a new paradigm for biomedical applications, displaying advantages in enabling bio-orthogonal, specific recognition, targeting and latching in vitro and in vivo.17–21 For instance, Kim et al. leveraged an ultrastable synthetic host–guest pair between cucurbit[7]uril (CB[7]) and adamantyl-(ADA) guests for the specific and bio-orthogonal protein imaging and bioimaging in vivo.22 Wang et al. demonstrated that CB[7]-grafted hyaluronic acid could recognize, latch and aggregate ADA-modified mitochondria to further induce mitochondrial aggregation and fusion intracellularly.23 On the basis of such a bio-orthogonal, high specificity and binding affinity between CB[7] and ADA in the complex biological environment,24 and inspired by the clinical success of CAR-T (Chimeric Antigen Receptor T-cell) and CAR-Macrophage,25,26 here, we designed a facile, yet powerful SAR-Macrophage (supramolecular artificial receptor macrophage), reminiscent of CAR-T and CAR-Macrophage, by anchoring CB[7] as an artificial receptor on the surface of macrophage, thus enhancing the recognition, latching and internalization of E. coli, which was specifically decorated with ADA as an artificial ligand, mediated via strong and multipoint host–guest interactions for significantly improving the antibacterial activity of the macrophage against E. coli (Scheme 1).
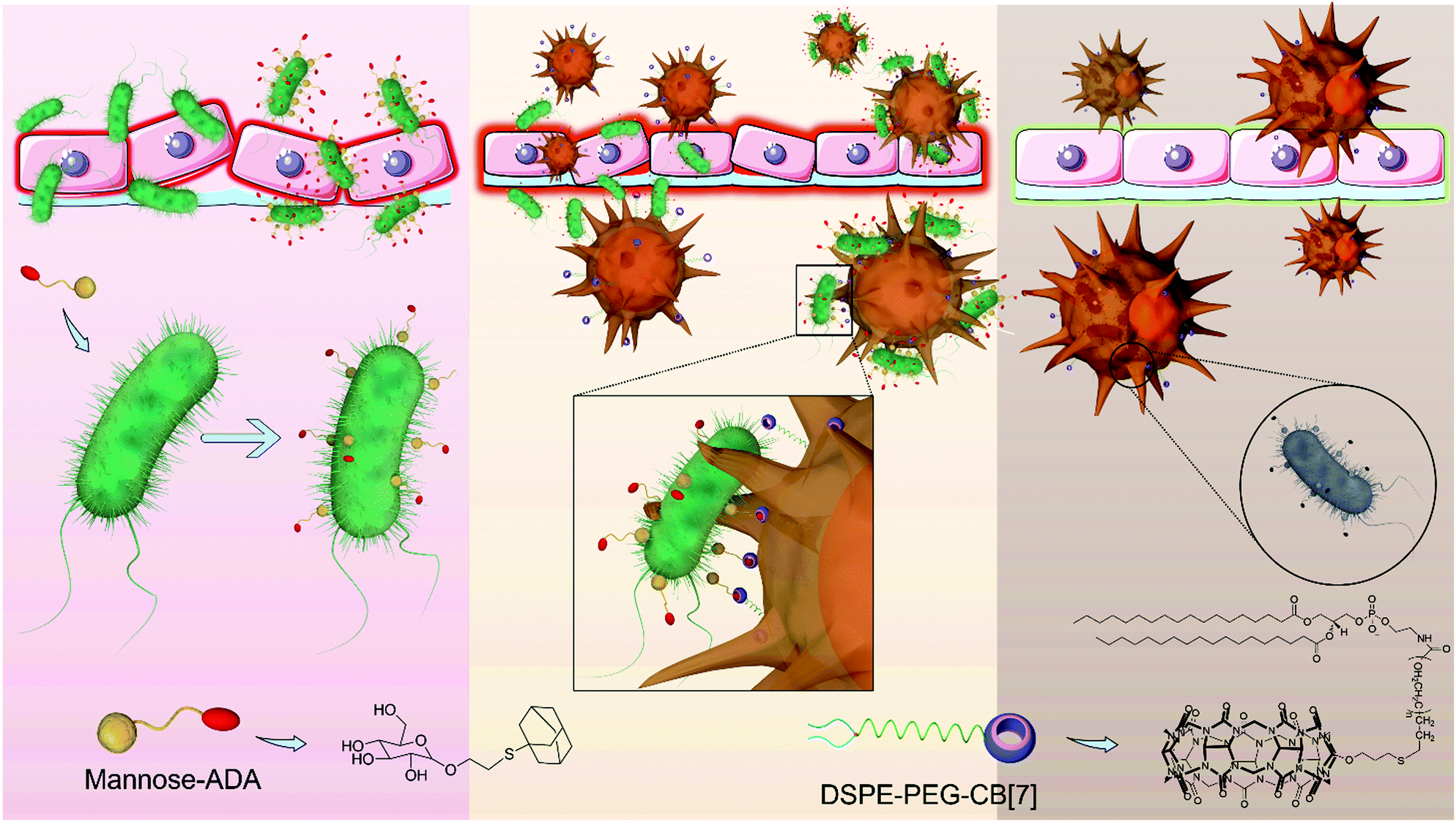 | ||
| Scheme 1 Supramolecular artificial receptor (SAR)-Macrophages rapidly and highly specifically recognize E. coli through strong and multipoint host–guest interactions, thus improving the latching and internalization of E. coli, and inducing M1 polarization of macrophages to generate ROS, and effectively kill the intracellular bacteria. | ||
First, as D-mannose can be recognized and adsorbed onto E. coli owing to its strong and specific interactions with FimH (Type 1 fimbrin D-mannose specific adhesion protein) on the pathogens,19 a mannose-ADA conjugate was designed and synthesized via a thiol–ene click reaction between allyl-α-D-mannopyranoside and 1-adamantanethiol to decorate the surface of E. coli with ADA via mannose-FimH interactions. The synthetic route of mannose-ADA is illustrated in Fig. S1 (ESI†). The successful synthesis was confirmed by 1H and 13C NMR spectroscopy (Fig. S2–S4, ESI†). As shown in Fig. S5 (ESI†), mannose-ADA (up to 500 μg mL−1) exhibited negligible cytotoxicity against E. coli after incubation for 24 h via MTT assays. Cy5-conjugated CB[7] (CB[7]–Cy5) was subsequently applied to investigate the successful decoration of E. coli with ADA. As shown in Fig. S6 (ESI†), after incubation with CB[7]–Cy5 for 10 min, E. coli pre-incubated with mannose-ADA (100 μg mL−1, 20 min) exhibited strong red fluorescence of Cy5 on the surface, attributed to the strong host–guest interactions between CB[7] of CB[7]–Cy5 and ADA from the E. coli surface. In contrast, no fluorescence was observed on the E. coli without pre-incubation with mannose-ADA, indicating that the successful recognition of CB[7]–Cy5 by E. coli was achieved via the CB[7]–ADA host–guest interaction.
SAR-Macrophage was constructed by decorating macrophage (Raw 264.7 cells) with CB[7] on the surface via simply inserting DSPE-PEG-CB[7] (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-poly(ethylene glycol)-CB[7]) into the surface membrane of the cells.27,28 The amount of CB[7] on the cell surface was quantitatively determined to be approximately 0.43 × 10−12 mol per cell, by incubation of the SAR-Macrophage with ferrocene (as a strong guest molecule of CB[7]) that was quantified via ICP-MS analysis. In addition, DSPE-PEG-CB[7] exhibited negligible cytotoxicity on RAW 264.7 cells after incubation for 24 h at a concentration up to 50 μM (Fig. S7, ESI†). Fluorescein isothiocyanate (FITC)-modified ADA (ADA-FITC) was utilized to evaluate the availability and stability of the surface CB[7] decorated on the macrophage. As shown in Fig. S8 (ESI†), RAW 264.7 cells incubated with DSPE-PEG-CB[7] for 1, 2, 4, 12 and 24 h, respectively, were subsequently treated with ADA-FITC for 5 min, and the green fluorescence of FITC maintained on the cell membrane for up to 12 h, indicating the good stability of DSPE-PEG-CB[7] in the cell membrane. The rapid recognition and stable host–guest interactions between CB[7] and ADA may ensure efficient recognition and latch of ADA-decorated E. coli by the SAR-Macrophage.
We subsequently investigated the recognition and internalization of ADA-modified E. coli by the SAR-Macrophage. In this study, the following treatment groups were prepared: macrophages were incubated with E. coli (CB[7](−), ADA(−)); SAR-Macrophages were incubated with E. coli (CB[7](+), ADA(−)); macrophages were incubated with ADA-decorated E. coli (CB[7](−), ADA(+)) and SAR-Macrophages were incubated with ADA-decorated E. coli (CB[7](+), ADA(+)) at different multiplicity of infection (MOI) ratios ranging from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 10:1. When the macrophages were incubated with E. coli at MOI = 1, strong green fluorescence from GFP-expressing E. coli was observed in the cytoplasm of RAW 264.7 cells in the (CB[7](+), ADA(+)) group, in contrast to the rather weak green fluorescence observed in the other three groups (Fig. 1A and Fig. S9A, ESI†), suggesting that the efficient recognition and internalization of ADA-decorated E. coli was realized by the SAR-Macrophage via strong host–guest interactions between CB[7] from the surface of the macrophage and ADA of E. coli. As shown in Fig. 1B, the SAR-Macrophages exhibited a significantly improved recognition of E. coli, ca. 14-fold of that recognized by regular macrophages (quantitied according to the fluorescence intensity).
:1 to 10:1. When the macrophages were incubated with E. coli at MOI = 1, strong green fluorescence from GFP-expressing E. coli was observed in the cytoplasm of RAW 264.7 cells in the (CB[7](+), ADA(+)) group, in contrast to the rather weak green fluorescence observed in the other three groups (Fig. 1A and Fig. S9A, ESI†), suggesting that the efficient recognition and internalization of ADA-decorated E. coli was realized by the SAR-Macrophage via strong host–guest interactions between CB[7] from the surface of the macrophage and ADA of E. coli. As shown in Fig. 1B, the SAR-Macrophages exhibited a significantly improved recognition of E. coli, ca. 14-fold of that recognized by regular macrophages (quantitied according to the fluorescence intensity).
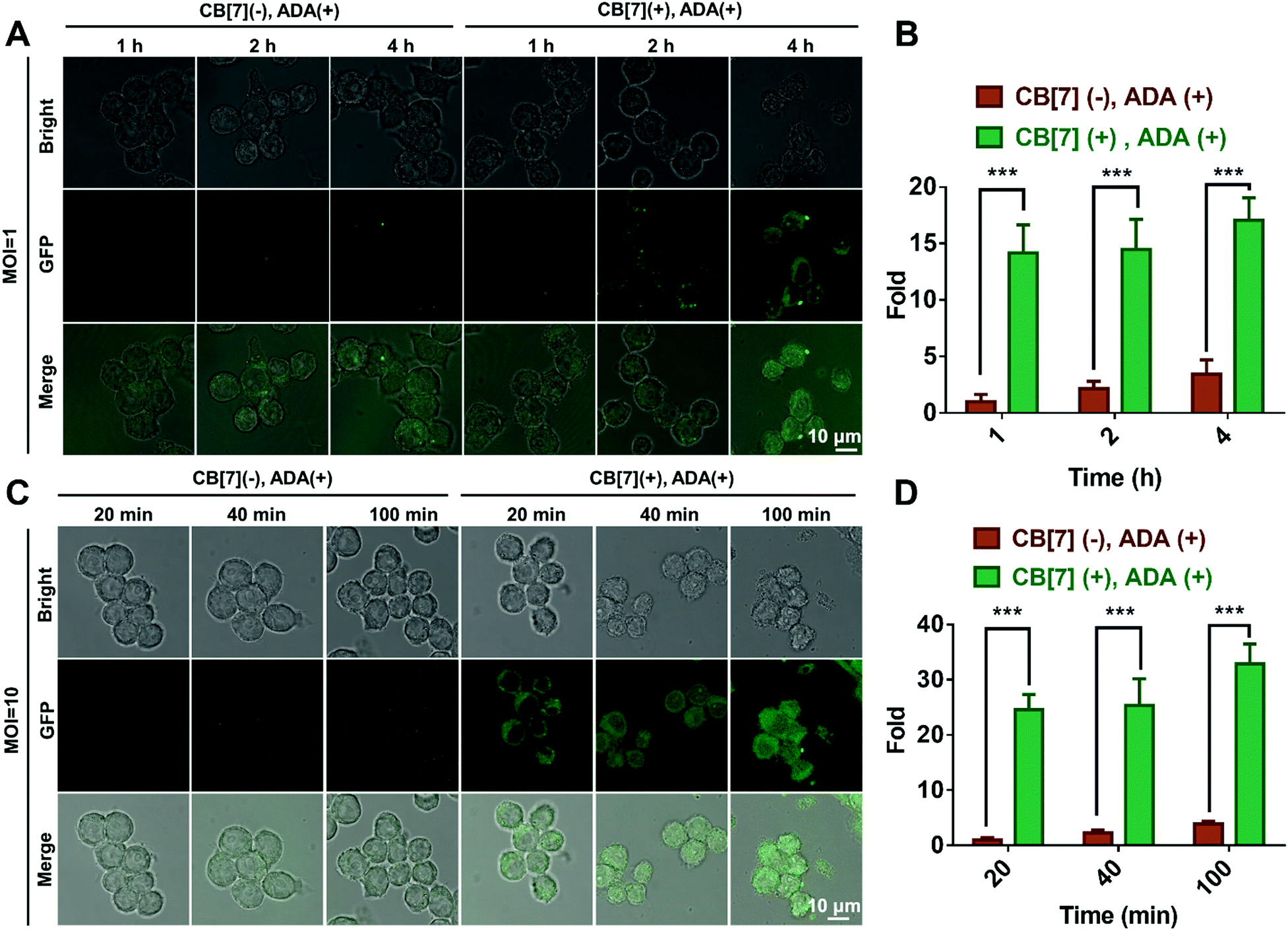 | ||
| Fig. 1 (A) CLSM images of the macrophage infected with E. coli (MOI = 1) in the (CB[7] (−), ADA (+)) and (CB [7] (+), ADA (+)) groups for 1, 2 and 4 h, respectively. (B) Quantitative GFP (E. coli) fluorescence from A. (C) CLSM images of macrophage infected with E. coli (MOI = 10) in the (CB[7] (−), ADA (+)) and (CB[7] (+), ADA (+)) group for 20, 40 and 100 min, respectively. (D) Quantitative GFP (E. coli) fluorescence from C. | ||
When the infected E. coli was increased to MOI = 10, the recognition of E. coli by the macrophages was significantly accelerated. As shown in Fig. 1C and Fig. S9B (ESI†), after incubation with various formulations for 20, 40 and 100 min, the GFP fluorescence (of E. coli) increased more rapidly in the (CB[7](+), ADA(+)) group of macrophages, resulting in a 32-fold increasement when compared with regular macrophages in 100 min, indicating that host–guest interactions indeed promoted the recognition and uptake of E. coli by the SAR-Macrophages (Fig. 1D). In both MOI = 1 and MOI = 10 CLSM (confocal laser scanning microscopy) images, the SAR-Macrophages were found to be morphologically deformed along with the formation of pseudofeet, indicating the polarization of macrophages to M1 would be beneficial to elimination of the intracellular bacteria. In contrast, the macrophages of other groups maintained the regular spherical morphology. The bacterial recognition and morphology of macrophages incubated with different formulations under MOI = 10 were further examined under scanning electron microscopy (SEM). As shown in Fig. 2A, abundant ADA-decorated E. coli were observed on the surface of the SAR-Macrophage, accompanied by the appearance of pseudopodia. In comparison, the rest three groups of macrophages showed relatively low efficiency in catching E. coli and exhibited a smooth cell membrane surface and a spherical shape, consistent with the observations under CLSM.
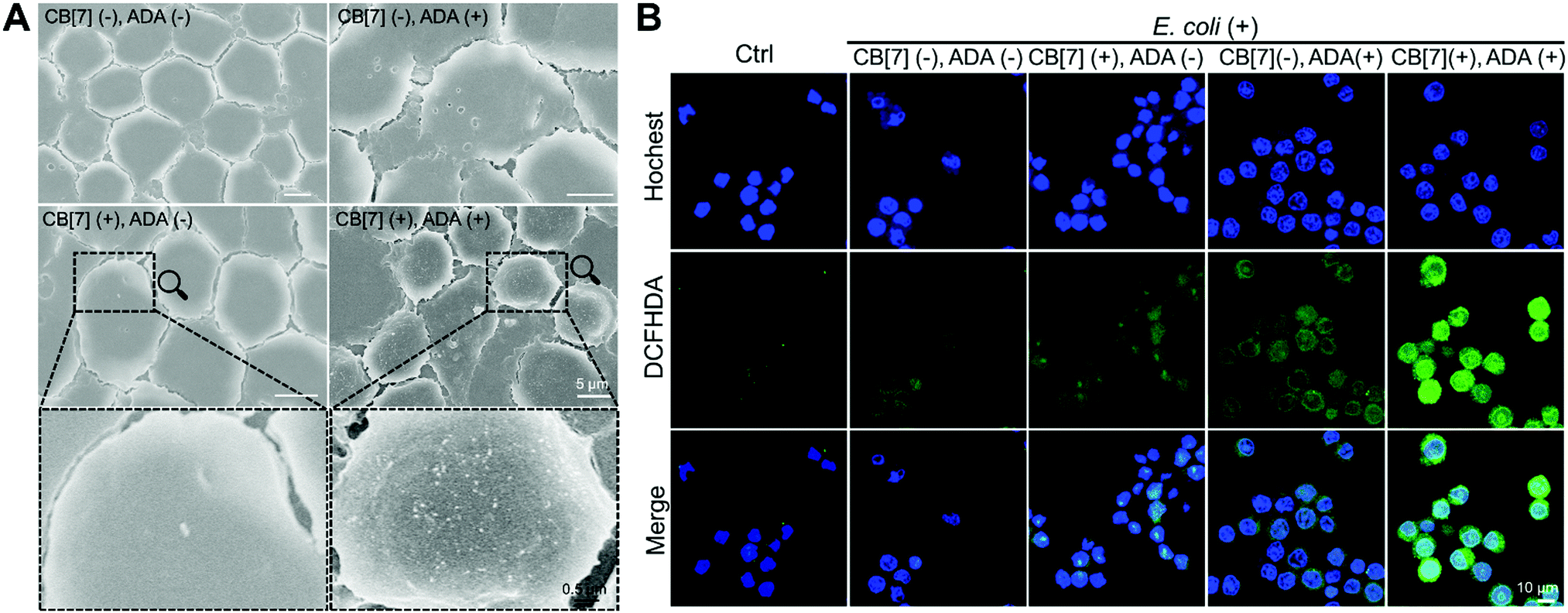 | ||
| Fig. 2 (A) SEM images of macrophages and E. coli with different formulations (CB[7](−), ADA(−)); (CB[7](+), ADA(−)); (CB[7](−), ADA(+)) and (CB[7](+), ADA(+)) (infected with MOI = 10, 100 min). (B) Intracellular ROS generation of different formulations of the macrophage after infecting E. coli including (CB[7](−), ADA(−)); (CB[7](−), ADA(+)); (CB[7](+), ADA(−)) and (CB[7](+), ADA(+)), respectively, followed by the evaluation of the intracellular ROS by DCFHDA straining and imaging under CLSM. | ||
The M1 polarization of macrophages was evaluated via flow cytometry assays after the internalization of E. coli (MOI = 10). As shown in Fig. S10 (ESI†), in the (CB[7](+), ADA(+)) group, the M1 polarization ratio (CD11c+ cells) increased to 40.88% in comparison with 1.71%, 4.07%, 8.43% and 10.27% observed in free macrophage, (CB[7](−), ADA(−)) group, (CB[7](+), ADA(−)) group and (CB[7](−), ADA(+)) group, respectively, indicating that the M1 polarization of the SAR-Macrophages was induced by the promoted recognition and arrest of E. coli. In addition, no M2 polarization (CD206+) was observed in all of these groups.
To further confirm the antibacterial efficacy of the M1-polarized macrophages, we infected different formulations of macrophages with E. coli at MOI = 10, followed by the evaluation of the intracellular ROS by straining with 2′,7-dichlorofluorescenindiacetate (DCFHDA).29 According to the CLSM results (Fig. 2B), the (CB[7](+), ADA(+)) group of macrophages showed obvious intracellular green fluorescence due to the host–guest mediated recognition and arrest of E. coli, which promoted the intracellular ROS production. Conversely, the macrophages of other groups showed rather weak intracellular fluorescence, indicating the insufficient internalization of E. coli into the macrophages. The intracellular ROS generation was further quantitatively analyzed via flow cytometry upon DCFHDA staining. As shown in Fig. S11 (ESI†), the fluorescence intensity of (CB[7](+), ADA(+)) was 2.1-fold higher than that of the control group, while the (CB[7](−), ADA(−)) group, the (CB[7](+), ADA(−)) group and the (CB[7](−), ADA(+)) group showed modestly elevated fluorescence (1.3–1.5-fold that of the control group), consistent with the results observed under CLSM. As intracellular ROS play a key role in killing the internalized E. coli, the viability of the intracellular bacteria was evaluated by using the bacterial live/dead staining assays at 18 h after infection. Intense red signals were found inside the macrophages of all groups, suggesting that most of the intracellular E. coli were dead. Of a significant note, more red fluorescence, indicative of dead bacteria, was observed in the SAR-Macrophage (Fig. S12, ESI†). These results indicated that the SAR-Macrophages exhibited an enhanced recognition and arrest of extracellular bacteria and effectively killed bacteria via M1 polarization.
Plate counting assay was subsequently adopted to confirm the antibacterial activities of the SAR-Macrophage. It was shown that after treatment with (CB[7](+), ADA(+)), plate counting results confirmed the remarkable antibacterial activity of the SAR-Macrophage towards E. coli, which was comparable to, but not as good as, the first-line antibiotics, such as ampicillin and ofloxacin, which was not unexpected because here E. coli is a regular strain sensitive to antibiotics (Fig. S13, ESI†). To assess the effective therapeutic dose range, different quantities of SAR-Macrophages were added to E. coli, as shown in Fig. S14 (ESI†), and the SAR-Macrophage (CB[7](+),ADA(+)) exhibited decent antibacterial effects even at a concentration of 103 mL−1. In contrast, in the presence of the same quantity of regular macrophages, there were much more bacteria that survived, confirming that the strong and specific host–guest interaction was the main reason for the enhanced antibacterial properties.
Furthermore, in order to show the unique advantages of the SAR-Macrophage against drug-resistant bacteria, Uropathogenic Escherichia coli Y9 (UPEC), which is a type of clinically isolated drug-resistant bacterium,30 was employed in our anti-bacterial studies and compared against a couple of classic antibiotics. As shown in Fig. S15 (ESI†), the first-line antibiotics, such as ampicillin and ofloxacin, exhibited little antibacterial activities against UPEC due to the well-developed antibiotic resistance of UPEC. In contrast, after treatment with the SAR-Macrophage (CB[7](+),ADA(+)), the survival of UPEC decreased significantly, indicating the effective antibacterial capability of the SAR-Macrophage to the drug-resistant strains, which was otherwise not achievable by regular macrophages.
The recognition and arrest of E. coli by the SAR-Macrophages were further investigated in zebrafish in vivo, as immune-capture of bacteria usually takes place in the circulative system, and this transparent animal modal allows a direct visualization of the arrest process in the fish body at a single cell level.31 After sequentially injecting DiD-labeled RAW 264.7 cells into the common cardinal vein (CCV) of zebrafish larvae (48 hpf (hours post fertilization)) and GFP-expressed E. coli into the dorsal vein for free circulation, the red-fluorescence macrophages could not catch the green-fluorescence E. coli efficiently in the fast-circulating blood system (Fig. 3). In dramatic contrast, the SAR-Macrophages caught ADA-decorated E. coli in a rather efficient manner, as was evidenced by the overlapped DiD and GFP fluorescence in vivo. These data showed that the DiD-labeled SAR-Macrophages efficiently recognized and captured pathogens in living zebrafish.
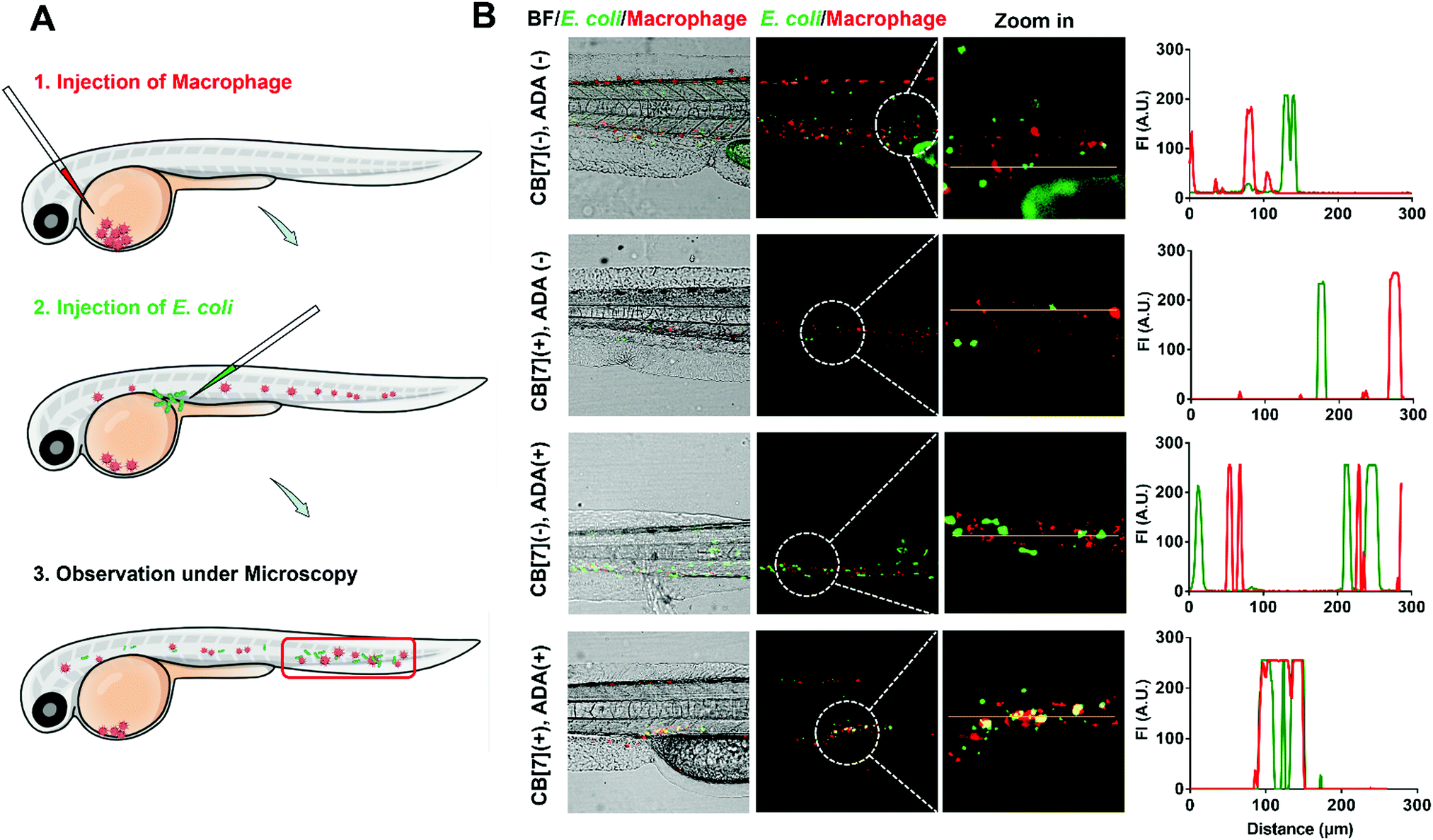 | ||
| Fig. 3 (A) Scheme showing a two-step injection of the macrophage and E. coli into zebrafish. (B) Representative CLSM images and corresponding quantification of GFP- (green) and DiD- (red) co-localization in zebrafish, showing the binding between DiD-loaded RAW 264.7 and GFP-expressed E. coli, with different formulations, including (CB[7](−), ADA(−)), (CB[7](+), ADA(−)), (CB[7](−), ADA(+)) and (CB[7](+), ADA(+)), respectively. For the macrophage, approximately 100–200 cells with DiD fluorescent labeling were injected into the common cardinal vein (CCV) of the fish larvae (48 hpf). For bacteria, approximately 1000–2000 CFU were injected into the vein to allow circulation. | ||
To further evaluate the applicability of the SAR-Macrophage in the fight against pathogen infection in vivo, a mouse wound infection model was established, where full-thickness wounds were created with a 6 mm biopsy punch on the back of mice and were subsequently infected with E. coli (10 μL, 1 × 109 CFU per mL). After the mice underwent infection for 4 h, the wound area was treated with mannose-ADA and the SAR-Macrophage (105 cells) sequentially, with a 1 h gap between the two injections for the (CB[7](+), ADA(+)) group (Fig. 4A). As shown in Fig. 4B and C, the (CB[7](+), ADA(+)) group of mice exhibited a rapid wound healing process, with the scabbed wound of only 22% that of the original area on Day 5, attributed to the fast recognition and elimination of mannose-ADA labeled E. coli by the SAR-Macrophage. In contrast, in the other treatment groups, the wounds changed from a clear raw appearance to yellowish dense mucus, and recovered slowly. The wound tissues from the infected areas were removed on Day 5 for the quantitative analysis of viable bacteria. As shown in Fig. 4D and Fig. S16 (ESI†), (CB[7](+), ADA(+)) exhibited superior antibacterial effects and significantly lowered the number of bacteria in the infectious tissue, when compared with other groups. In addition, dramatically reduced inflammatory infiltration and more newly formed collagen and blood vessels were observed in the (CB[7](+), ADA(+)) treated group, when compared with other groups (Fig. 4E). Major organs of the mice showed no obvious damage, indicating the decent biosafety of this therapeutic approach (Fig. S17, ESI†). Collectively, these results demonstrated that the sequential injection of mannose-ADA and the SAR-Macrophage afforded excellent antibacterial effects in a mammal model, suggesting a significant potential for clinical translation.
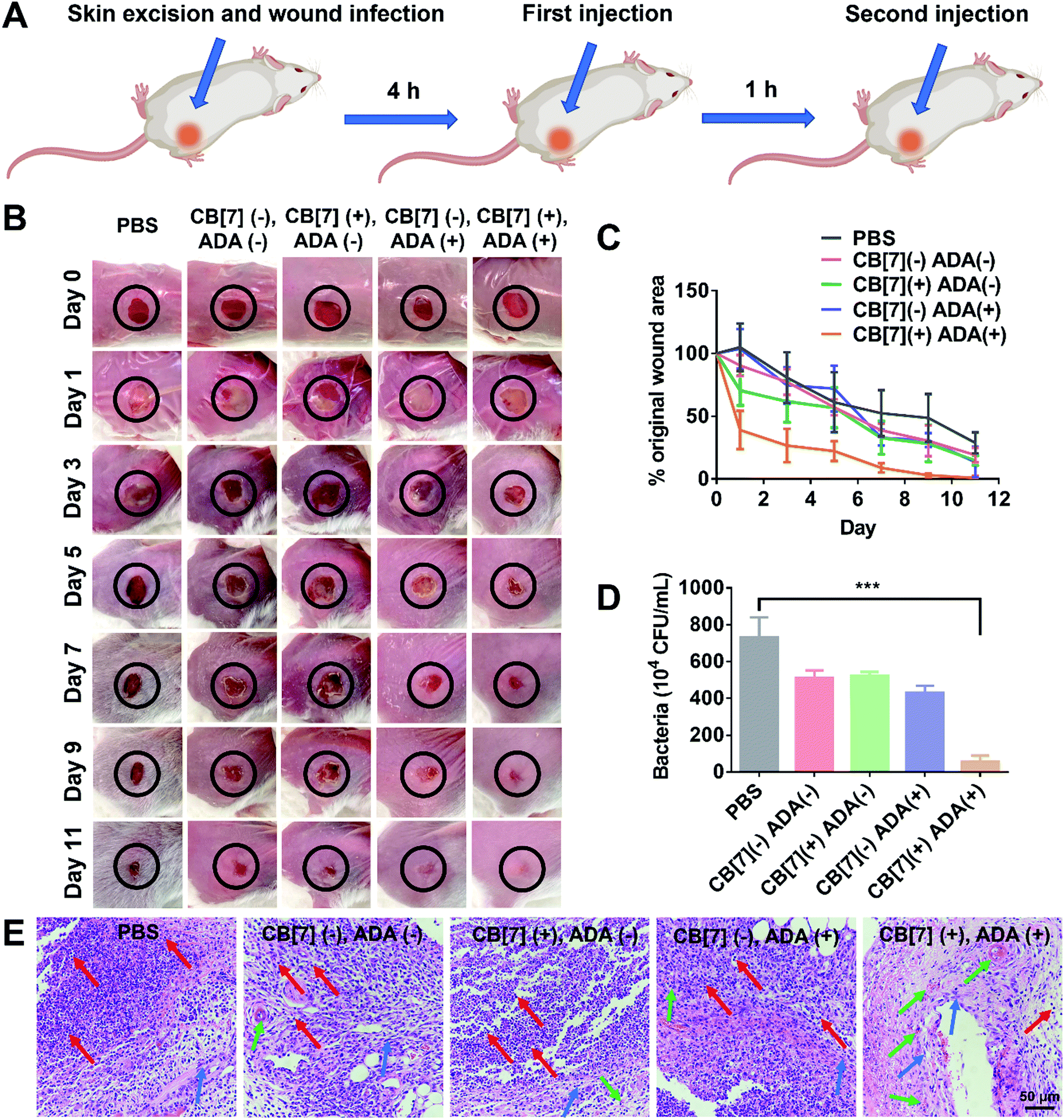 | ||
| Fig. 4 (A) Scheme showing E. coli infection (107 CFU, 1 × 109 CFU per mL for 10 μL of E. coli) in a skin wound on mouse and the two-step injection therapeutic process (first injection of mannose-ADA (50 μg mL−1) and second injection of the SAR-Macrophage (105, 106 mL−1 for 100 μL)) (n = 3 in each group). (B) Photographs of the wounds of mice infected with E. coli after treatment with PBS, (CB[7](−), ADA(−)), (CB[7](+), ADA(−)), (CB[7](−), ADA(+)) and (CB[7](+), ADA(+)), respectively on Day 1, 3, 5, 7, 9 and 11. (C) The evolving wound area in mice skin during the treatment in each group. (D) Quantitative analysis of the bacterial colony in the wound tissue after treatment for 5 days in each group. (E) The H&E staining of wound tissue sections after treatment with PBS, (CB[7](−), ADA(−)), (CB[7](+), ADA(−)), (CB[7](−), ADA(+)) and (CB[7](+), ADA(+)), respectively (red arrows indicate inflammatory cells; blue arrows indicate collagenous fiber; green arrows indicate new blood vessels). | ||
Conclusions
A SAR-Macrophage was developed and applied for the first time to enhance the recognition and latching of bacteria, mediated via strong and multipoint host–guest interactions between the artificial receptors (CB[7]) on the macrophage and the guest ligands (ADA) on E. coli. The SAR-Macrophage was shown to rapidly recognize, efficiently internalize and exterminate guest-anchored E. coli both in vitro and in vivo, resulting in M1 polarization and ROS generation for the effective intracellular bacteria inhibition. More importantly, the SAR-Macrophage showed powerful antibacterial effects in drug-resistant E. coli strain, UPEC, where common antibiotics exhibited little antibacterial activities. In this approach, mannose-ADA was designed and employed to specifically pre-target and anchor ADA on the surface of E. coli, facilitating the targeted recognition and arresting by the SAR-Macrophage. Although mannose-ADA could not be extended to other bacterial strains than E. coli, the SAR-Macrophage could be applied to a variety of other pathogens, in principle, as long as pathogens could be remotely modified with ADA via other ligand-receptor interactions, similar to mannose-FimH interactions. Nevertheless, the SAR-Macrophage, reminiscent of CAR-Macrophage, not only represents a simple cell-engineering approach for potential antibacterial applications, but also provides important new insights to design and develop cell-based therapeutics for diverse biomedical applications via an enhanced target-recognition.Author contributions
This project was conceptually designed by QC and RW. The majority of the experiments were conducted by QC, assisted by MX, CS, KY, ZY and JL. Data analysis was conducted by QC, JZ, YZ and RW. The manuscript was prepared by QC and RW. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Science and Technology Development Fund (FDCT), Macau SAR (file no.: 0065/2021/A2), the National Natural Science Foundation of China (21871301 and 22071275), and Dr. Stanley Ho Medical Development Foundation (SHMDF-OIRFS/2021/002) are gratefully acknowledged for providing financial support to this work.Notes and references
- R. Partridge Sally, M. Kwong Stephen, N. Firth and O. Jensen Slade, Clin. Microbiol. Rev., 2018, 31, e00088–e00017 Search PubMed
.
- M. Tyers and G. D. Wright, Nat. Rev. Microbiol., 2019, 17, 141–155 CrossRef CAS PubMed
- A. J. Wolf and D. M. Underhill, Nat. Rev. Immunol., 2018, 18, 243–254 CrossRef CAS PubMed
- P. J. Murray, Annu. Rev. Physiol., 2017, 79, 541–566 CrossRef CAS PubMed
- G. Weiss and U. E. Schaible, Immunol. Rev., 2015, 264, 182–203 CrossRef CAS PubMed
- K. B. R. Belchamber, R. Singh, C. M. Batista, M. K. Whyte, D. H. Dockrell, I. Kilty, M. J. Robinson, J. A. Wedzicha, P. J. Barnes and L. E. Donnelly, Eur. Respir. J., 2019, 54, 1802244 CrossRef CAS PubMed
- S. Das, K. A. Owen, K. T. Ly, D. Park, S. G. Black, J. M. Wilson, C. D. Sifri, K. S. Ravichandran, P. B. Ernst and J. E. Casanova, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2136–2141 CrossRef CAS PubMed
- C. Fontaine, E. Rigamonti, A. Nohara, P. Gervois, E. Teissier, J.-C. Fruchart, B. Staels and G. Chinetti-Gbaguidi, Circ. Res., 2007, 101, 40–49 CrossRef CAS PubMed
- C. J. Harvey, R. K. Thimmulappa, S. Sethi, X. Kong, L. Yarmus, R. H. Brown, D. Feller-Kopman, R. Wise and S. Biswal, Sci. Transl. Med., 2011, 3, 78ra32 Search PubMed
- S. Watanabe, M. Alexander, A. V. Misharin and G. R. S. Budinger, J. Clin. Investig., 2019, 129, 2619–2628 CrossRef PubMed
- D. G. DeNardo and B. Ruffell, Nat. Rev. Immunol., 2019, 19, 369–382 CrossRef CAS PubMed
- Y. Sun, Y. Liu, B. Zhang, S. Shi, T. Zhang, D. Zhao, T. Tian, Q. Li and Y. Lin, Bioact. Mater., 2021, 6, 2281–2290 CrossRef CAS PubMed
- Y. Yang, X. Wu, L. Ma, C. He, S. Cao, Y. Long, J. Huang, R. D. Rodriguez, C. Cheng, C. Zhao and L. Qiu, Adv. Mater., 2021, 33, 2005477 CrossRef CAS PubMed
- X. Fan, X. Wu, F. Yang, L. Wang, K. Ludwig, L. Ma, A. Trampuz, C. Cheng and R. Haag, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202113833
- L. Li, L. Cao, X. Xiang, X. Wu, L. Ma, F. Chen, S. Cao, C. Cheng, D. Deng and L. Qiu, Adv. Funct. Mater., 2021, 31, 2107530 Search PubMed
- X. Fan, F. Yang, C. Nie, L. Ma, C. Cheng and R. Haag, Adv. Mater., 2021, 33, 2100637 CrossRef CAS PubMed
- M. J. Webber and R. Langer, Chem. Soc. Rev., 2017, 46, 6600–6620 RSC
- J. Zhou, G. Yu and F. Huang, Chem. Soc. Rev., 2017, 46, 7021–7053 RSC
- W. Li, K. Dong, H. Wang, P. Zhang, Y. Sang, J. Ren and X. Qu, Biomaterials, 2019, 217, 119310 CrossRef CAS PubMed
- X. Ma and Y. Zhao, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS PubMed
- X. Li, H. Bai, Y. Yang, J. Yoon, S. Wang and X. Zhang, Adv. Mater., 2019, 31, 1805092 Search PubMed
- K. L. Kim, G. Sung, J. Sim, J. Murray, M. Li, A. Lee, A. Shrinidhi, K. M. Park and K. Kim, Nat. Commun., 2018, 9, 1712 CrossRef PubMed
- C. Sun, Z. Wang, L. Yue, Q. Huang, Q. Cheng and R. Wang, J. Am. Chem. Soc., 2020, 142, 16523–16527 CrossRef CAS PubMed
- W. Liu, S. K. Samanta, B. D. Smith and L. Isaacs, Chem. Soc. Rev., 2017, 46, 2391–2403 RSC
- K. Newick, S. O'Brien, E. Moon and S. M. Albelda, Annu. Rev. Med., 2017, 68, 139–152 CrossRef CAS PubMed
- M. Klichinsky, M. Ruella, O. Shestova, X. M. Lu, A. Best, M. Zeeman, M. Schmierer, K. Gabrusiewicz, N. R. Anderson, N. E. Petty, K. D. Cummins, F. Shen, X. Shan, K. Veliz, K. Blouch, Y. Yashiro-Ohtani, S. S. Kenderian, M. Y. Kim, R. S. O’Connor, S. R. Wallace, M. S. Kozlowski, D. M. Marchione, M. Shestov, B. A. Garcia, C. H. June and S. Gill, Nat. Biotechnol., 2020, 38, 947–953 CrossRef CAS PubMed
- C. Gao, Q. Cheng, J. Li, J. Chen, Q. Wang, J. Wei, Q. Huang, S. M. Y. Lee, D. Gu and R. Wang, Adv. Funct. Mater., 2021, 31, 2102440 CrossRef CAS
- C. Gao, Q. Cheng, J. Wei, C. Sun, S. Lu, C. H. T. Kwong, S. M. Y. Lee, Z. Zhong and R. Wang, Mater. Today, 2020, 40, 9–17 CrossRef CAS
- Z. Wang, Y. Zhang, E. Ju, Z. Liu, F. Cao, Z. Chen, J. Ren and X. Qu, Nat. Commun., 2018, 9, 3334 CrossRef PubMed
- Z.-x. Cheng, C. Guo, Z.-g. Chen, T.-c. Yang, J.-y. Zhang, J. Wang, J.-x. Zhu, D. Li, T.-t. Zhang, H. Li, B. Peng and X.-x. Peng, Nat. Commun., 2019, 10, 3325 CrossRef PubMed
- X. He, X. Yin, J. Wu, S. L. Wickström, Y. Duo, Q. Du, S. Qin, S. Yao, X. Jing, K. Hosaka, J. Wu, L. D. Jensen, A. Lundqvist, A. I. Salter, L. Bräutigam, W. Tao, Y. Chen, R. Kiessling and Y. Cao, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 22910–22919 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1mh01813b |
| This journal is © The Royal Society of Chemistry 2022 |